
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci., 17 February 2022
Sec. Marine Fisheries, Aquaculture and Living Resources
Volume 8 - 2021 | https://doi.org/10.3389/fmars.2021.817701
 Inmaculada Varó1‡María Prado-Álvarez2‡Ignacio Ortea3Amalia E. Morales4Pablo García-Fernández2†Pedro Domingues5Ricardo Tur5†Sonia Dios2*Camino Gestal2*
Inmaculada Varó1‡María Prado-Álvarez2‡Ignacio Ortea3Amalia E. Morales4Pablo García-Fernández2†Pedro Domingues5Ricardo Tur5†Sonia Dios2*Camino Gestal2*The common octopus is the most demanded cephalopod species for human consumption. Despite important advances realized recently, the main bottleneck for commercial production of the common octopus, Octopus vulgaris, is the mass mortality of paralarvae in the first 15–20 days post-hatching (dph), with the main responsible factors still unknown. Thus, the identification of the limiting culture factors is, therefore, crucial for their aquaculture. This study investigates proteomic and transcriptomic responses of octopus paralarvae fed on an improved live preys-mixed diet (M) compared to an Artemia-based (A) reference diet. M diet resulted in the highest paralarvae specific growth rate obtained to date under culture conditions. This is supported by most of the proteins and genes over-expressed in the M group being linked to the cell cycle and replication, production of structural components, and development of the nervous system. Furthermore, the differential nutritional regulation of several genes and proteins seems to indicate that, instead of fatty acids, the preferred fuels for cephalopods would be proteins and carbohydrates. Also, M diet provides a better nutrient balance, which has allowed carrying out this comparative study in paralarvae under optimal conditions at a more advanced stage of growth (20 dph) than in previous studies. Moreover, the paralarvae culture extended up to 40 dph showed for the first time a proper pre-settlement behavior.
The common octopus (Octopus vulgaris) is, nowadays, the most demanded cephalopod species for human consumption. This species was postulated for aquaculture diversification to supply its increasing demand in the market worldwide, which only relies on continuously declined field captures (Iglesias and Fuentes, 2014; Rodhouse et al., 2014). Despite important advances made recently, the main bottleneck for the commercial production of the common octopus is still the massive mortalities in paralarvae within the first 15–20 days post-hatching (dph).
Indeed, the real causes of these massive mortalities remain still unknown, and various factors related with the zootechnical production (i.e., temperature, water quality, and photoperiod, or light condition) have been referred as possible sources of stress that can negatively affect the viability of the octopus paralarvae rearing (Iglesias and Fuentes, 2014; Garrido et al., 2018). Besides, the use of inadequate/imbalanced diets, particularly in essential lipid content, such as phospholipids and highly unsaturated fatty acids [HUFA: eicosapentaenoic (EPA) and docosahexaenoic (DHA)], has been pointed out as one of the major factors of nutritional stress (Guinot et al., 2013; Navarro et al., 2014).
It is well known that crustacean zoeae are a better live prey than Artemia nauplii or metanauplii for paralarvae culture (Iglesias and Fuentes, 2014; Garrido et al., 2018). In the last years, important progress in terms of paralarvae growth and survival has been achieved by improving the feeding protocols. The supply of different species of decapod crustacean zoeae (i.e., Maja brachydactyla, Pagurus sp, and Grapsus adscensionis) and amphipods in co-feeding with Artemia, and also the use of Artemia metanauplii enriched with marine lecithin (LC60, rich in phospholipids and long-chain polyunsaturated fatty acids (LC-PUFAs) have notably improved paralarvae culture performance (Iglesias and Fuentes, 2014; Reis et al., 2015, 2021; Garrido et al., 2016, 2018). However, the use of these diets did not produce the rearing outputs needed for the scale-up and establishment of industrial production.
On the other hand, co-feeding with wild live preys such as the previously mentioned crustaceans seems to be a good strategy to balance the quality of diet, achieving best growth and survival until settlement at 30-40 dph. However, this approach might not be feasible in terms of massive production due to logistic reasons. Among them is the dependence on sesonal availablity of preys, their handling and capture from the natural environment, which is not sustainable or economically viable to supply the increasing demand for hatcheries. As a result, research is now focused on the production of marine zooplankton (i.e., amphipods, copepods) as an alternative to wild preys, and also on the development of new Artemia enrichments with nutritional profiles closer to those of crustacean zoeae and wild zooplankton (Iglesias and Fuentes, 2014; Garrido et al., 2016; Tur et al., 2020). Moreover, the design of inert diets to complement the enriched mixed diets to feed older paralarvae next to the settling stage is acquiring high interest (Seixas et al., 2010a; Navarro et al., 2014; Roo et al., 2017; de Moraes Lenz et al., 2019).
Nutritionally related molecular biomarkers can help to determine the effectiveness of new or improved diets on paralarvae aquaculture. In this context, proteomic and transcriptomic analyses have been used as effective strategies to reveal the molecular mechanism, responding to nutritional and environmental stress of octopus paralarvae in captivity. In previous studies, we have investigated the basic processes involved in nutritional deficiencies of common octopus paralarvae using high-throughput technology. Using proteomic approaches, (Varó et al., 2017) analyzed the effect of fasting during the first 4 days of paralarvae development, as well as the response of two diets, one based on co-feeding with crustacean zoeae and Artemia as inadequate diet, in paralarvae until 16 dph. The results obtained allowed defining characteristic proteome profiles of paralarvae, depending on diet, as well as the differential expression of some proteins like S-crystallin, arginine kinase, and NAD+-specific isocitrate dehydrogenase, as potential molecular biomarkers specifically related to starvation.
Transcriptomic approaches combined with proteomic approaches were also applied to study the influence of two diets, Artemia in co-feeding with crustacean zoeae of M. brachydactyla, and only Artemia, in 10-dph paralarvae. These findings have allowed the identification of genes related to lipid metabolism whose expression is modulated by diet, presenting higher transcriptional levels in zoeae co-feeding groups. Also, these findings indicated that carbohydrate and amino acid metabolism, as well as immune and stress responses, were upregulated in paralarvae co-fed on zoeae (García-Fernández et al., 2019). Overall, these studies have made possible the identification of genes and proteins as potential biomarkers to study development and health in the common octopus paralarvae under culture conditions. However, despite of this substantial advance, the range of molecular biomarkers available to study the nutritional impact of the diet is still limited and strongly focused on the early stages of development. Therefore, it seems of high interest to evaluate the benefits of co-feeding protocols based on mixed diets of live preys to study the underlying molecular dietary response from early to late stages of paralarvae development.
This study aims to investigate through a proteogenomic approach the nutritional responses of O. vulgaris paralarvae fed on an improved mixed diet of live preys (M) composed of four crustacean species, specifically spider crab zoeae (M. brachydactyla), velvet crab (Necora puber), amphipods gammarids (Jassa sp.), and Artemia in co-feeding, which was compared with Artemia-based diet (A) as a conventional reference diet. Paralarvae culture was performed following an adaptation of the protocol patented by the Spanish Institute of Oceanography (Tur et al., 2020), which allowed the optimal results in terms of paralarvae growth and survival. To this end, proteomic and transcriptomic analyses of 20-dph paralarvae were carried out. We used the specific protein sequences database built from the enhanced de novo reference transcriptome sequencing (RNA seq) of early-life-cycle stages of the common octopus grown in captivity under different conditions, previously generated in our laboratory (Prado-Álvarez et al., under review).
O. vulgaris paralarvae were obtained from a broodstock kept in captivity and reared following the conditions described by Iglesias and Fuentes (2014) and Iglesias et al. (2014). Paralarvae culture was carried out following an adaptation of the protocol patented by the Spanish Institute of Oceanography (Tur et al., 2020), and as described in (Prado-Álvarez et al., under review). In brief, octopus paralarvae were reared in 500-L black cylindrical tanks with central aeration at an initial density of 4 individuals L−1. The flow-through seawater system was equipped with 10-, 5-, and 1-μm filter cartridges. The rearing conditions were: 35 PSU salinity, 20.8 ± 1.1°C temperature, oxygen concentration 6 ± 0.5 ppm, and 600 lx of light intensity with 14L:10D photoperiod. For green water, Nannochloropsis sp and Isochrysis aff. galbana (T-Iso) were used (3·105 cells ml−1 in a 3:1 proportion).
Two diets were tested: Artemia-based diet (A), consisted of paralarvae fed three times per day on Artemia metanauplii (Sep-Art EG, INVE Aquaculture, Belgium), growth for 5–7 days (1.5 mm size) with the microalgae Tetraselmis chuii and T-Iso (50:50), and then fed on T-Iso for the last 24 h. Paralarvae were fed ad libitum with Artemia metanauplii, and concentration was daily adjusted according to consumption, which ranged between 0.14 and 2.25 μg dry weight/ml (density equivalent to 0.03-0.5 preys/ml). Mixed diet (M) consisted of four live preys, supplied three times per day to the culture tank according to consumption as follows: zoeae of spider crab (M. brachydactyla) at 0.20–0.99 μg dry weight/ml (0.002–0.01 preys/ml), zoeae of velvet crab (N. puber) at 0.20–0.99 μg dry weight/ml (0.001–0.005 preys/ml), juveniles of amphipods gammarids (Jassa sp.) at 0.08–0.17 μg dry weight/ml (0.0005–0.001 preys/ml), and Artemia at 0.23–0.9 μg dry weight/ml (0.05–0.2 preys/ml). Samples of 20- dph paralarvae were collected for massive transcriptomic RNA-seq and proteomic SWATH analysis. A total of 10 paralarvae (5 biological samples per dietary group) and 12 paralarvae (6 biological samples per dietary group) were used for RNA-seq analysis and proteomic study, respectively.
Before any processing, paralarvae had been anesthetized in chilled sea water (lower than 2°C) for 5 s, following the procedure described by Nande et al. (2016). Lack of spontaneous movement was immediately observed. No response to a noxious stimulus and no stressful reactions were observed, and decrease of ventilation was confirmed (Fiorito et al., 2015). Then, paralarvae samples for protein isolation were euthanized by freezing in liquid nitrogen and individually stored at −80°C until analysis, while samples for RNA extraction were stored in RNA later (QIAGEN, GmbH, Germany) following the manufacturer's instructions for fixation and preservation at −80°C until use.
A set of paralarvae (n = 20) from the same experimental culture was also used to determine the individual dry weight after oven drying for 24 h at 80°C (Iglesias et al., 2014). The percentage of the standard growth rate (SGR %) was calculated following the formula: (% BW·day−1) = [(LnDWf-LnDWi) ×100/(tf-ti)], where BW is the body weight and DWf and DWi are the paralarvae dry weight at final time (tf) and initial time (ti), respectively.
The animal study was reviewed and approved by the Ethics Committee of the National Competent Authority (Project No. CEIBA 2017-0249) in compliance with the Spanish law 53/2013 within the framework of the European Union directive on animal welfare (Directive 2010/63/EU) for the protection of animals employed for scientific purposes, following the Guidelines for the care and welfare of cephalopods proposed by Fiorito et al. (2015).
Total RNA from whole individual paralarvae of 20 dph fed on the two diets (M and A) was extracted using Trizol (Thermo Fisher Scientific®, Waltman, MA, USA) following the manufacturer's instructions. Once the quality and the integrity of total RNA were confirmed by bioanalyzer (Agilent 2100), samples were processed for sequencing using the Illumina NovaSeq 6000 platform and the paired-end reads mode (Novogene Co., Ltd). A de novo transcriptome was used as reference in this study (Prado-Álvarez et al., under review). Briefly, the de novo reference RNA-seq included 20-dph paralarvae of both dietary groups (M and A), paralarvae grown at optimal and suboptimal conditions, paralarvae sampled at different developmental stages (from embryo to paralarvae of 40 dph), and paralarvae (0 and 40 dph) challenged with different pathogens, which conformed a total of 12 libraries with 5 individual biological replicates each.
Raw sequencing reads were assessed for quality using FASTQC v0.11.7 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) (Andrews, 2010) to remove Illumina adapters (TruSeqTM universal primer), sample indexes, and low-quality reads (Phred cutoff score ≥ 30). Hierarchical clustering was then carried out by Corset (v1.05) to reduce redundancies and select the longest transcript (unigene) within each cluster. Functional annotation of diet-related transcripts was performed, running NCBI BLAST algorithms v2.7.1 (Altschul et al., 1990) in the local mode on the subset of non-redundant contigs. The BLASTx algorithm (Altschul et al., 1990) was used to search against the NCBI nucleotide sequences (Nt, threshold E-value of 10−5). The software Diamond was used to map against the NCBI non-redundant protein sequences (Nr, threshold E-value of 10−5), the euKaryotic Orthologous Groups based on NCBI's gene orthologous relationships (KOG, threshold E-value of 10−3), and the Swiss-Prot protein database downloaded in February 2019 from UNIPROT (threshold E-value of 10−5). Prediction of the protein structure domain and families collection (Pfam) was carried out using the HMMER 3.0 package, with a threshold E-value of 0.01. The resulting annotations were mapped against the Gene Ontology (GO) using Blast2GO v2.5 software (Götz et al., 2008) with a threshold E-value of 10−6, and also loaded in the KAAS [Kyoto Encyclopedia of Genes and Genomes (KEGG) Automatic Annotation Server] with a threshold E-value of 10−10. Annotation results were not manually reviewed. Assembled transcriptome is available on NCBI under accession PRJNA754143.
To further identify transcripts exhibiting significant differences in expression levels corresponding to dietary experimental samples, Corset-filtered contigs were mapped against the de novo reference transcriptome (Prado-Álvarez et al., under review), and the expression level was quantified using RSEM (v1.2.26) (Li and Dewey, 2011). Pairwise comparisons between diet conditions (M vs. A) were performed, and the differentially expressed genes (DEGs) were extracted using the R package DESeq (1.10.1) (Anders and Huber, 2010). Significance was accepted at a false discovery rate (FDR) cutoff of 0.05. Volcano plot was represented using the “res” R function, hierarchical clustering was constructed on Heatmapper (Babicki et al., 2016), and Venn diagram was obtained from the web server (http://bioinformatics.psb.ugent.be/webtools/Venn/). The GOs enrichment analysis in the subset of DEGs was implemented by the GOSeq R packages based on Wallenius non-central hyper-geometric distribution (Young et al., 2010), following topGO v2.30.1 R package. KOBAS software (Mao et al., 2005) was used to test the statistical enrichment of DEGs in KEGG pathways.
To confirm differentially expressed genes (DEGs) obtained from the paralarvae transcriptomic profile, the gene expression level of 12 genes was quantified by RT-qPCR in a set of different samples (3 individual paralarvae) obtained from the same experimental condition. Sequences from the transcriptome assembly were used to design forward and reverse primers (Supplementary Table 1) using PrimerQuest (https://eu.idtdna.com/PrimerQuest/Home/Index), and amplification efficiencies were determined following the Pfaffl method (Pfaffl, 2001). RNA isolation was conducted as described above and quantified using NanoDrop ND200 spectrophotometer (Thermo Fisher Scientific®). First strand cDNA was synthesized using the Maxima First Strand cDNA Synthesis kit for RT-PCR (Thermo Fisher Scientific®) using 100 ng of total RNA, treated with DNAse (QIAGEN). RT-qPCR reactions were performed in a QuantStudio3 (Applied Biosystems) sequence detector. Each well contained 1 μl of cDNA (dilution 1/10), 12.5 μl of SYBR green PCR master mix (Thermo Fisher Scientific®), 6.5 μl of water, and 2.5 μl of each diluted primer (3 μM). Amplification of a single PCR product was monitored by a melting curve analysis. All reactions were performed as technical duplicates. The expression of the selected genes was normalized using the Ubiquitin as a reference gene (García-Fernández et al., 2016), and analyzed following the Pfaffl method (Pfaffl, 2001). Results were expressed as the mean ± standard error. Fold units were calculated by dividing the normalized expression values between conditions (M vs. A). Data were evaluated using parametric test Student's T-test. Significant differences were considered at p < 0.05.
Total proteins from whole individual paralarvae of 20 dph fed on the two diets (M and A) were extracted after mechanical cell lysis in a glass potter using a lysis buffer (100-mM Tris-HCl pH 7.5, 150-mM NaCl 1% n-Dodecyl-beta-D-maltoside and phenil-metasulfonil-fluoridic as a proteinase inhibitor). Samples were incubated for 5 min on ice, and lysates were centrifuged at 16,000 xg at 4°C for 30 min. Proteins obtained in the supernatant were quantified following the Bradford method, using BSA as a standard (Brooks, 1975).
Protein extracts were subjected to protein precipitation with TCA/acetone for removing of contaminants. Protein content was solubilized in 0.2% RapiGest SF (Waters, Milford, MA, USA), and total protein content was quantified in each sample using the Qubit Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). For each sample, 25-μg protein was subjected to trypsin digestion as described in González-Fernández et al. (2019). Briefly, protein samples were incubated with 5-mM DTT at 60°C for 30 min, and then with 10-mM iodoacetamide at room temperature and darkness for 30 min. Sequencing Grade Modified Trypsin (Promega, Madison, WI, USA) was added (ratio, 1:40, trypsin:protein) in two steps, incubating at 37°C for 2 h in the first step and for 15 h at the second step. RapiGest was then precipitated by centrifugation after incubating with 0.5% TFA at 37°C for 1 h, and peptide concentration was adjusted to 0.25-μg peptide/μl. RePliCaliRT peptides (PolyQuant GmbH, Bad Abbach, Germany) were spiked to each sample according to the manufacturer's instructions in order to calibrate retention times later in the SWATH runs. Peptide digests were transferred to low-volume HPLC vials and frozen at −20°C until further analysis by liquid chromatography (LC) - mass spectrometry (MS).
The peptide solutions were analyzed by a shotgun data-dependent acquisition (DDA) approach by nano-LC-MS. To obtain a good representation of the peptides and proteins present in all the samples, several pooled vials were prepared from equal mixtures of the original peptide digests. Each pooled sample (4 μl, meaning 1-μg protein equivalent in column) was injected and analyzed on a nano-LC system Ekspert nLC415 (Eksigent, Dublin, CA, USA), coupled to hybrid quadrupole-TOF mass spectrometer Triple TOF 5600+ (Sciex, Redwood City, CA, USA). LC parameters were: gradient from 5 to 30% B in 120 min, with water and ACN, both containing 0.1% formic acid, as solvents A and B, respectively; a flow rate of 300 nl/min; an Acclaim PepMap C18 column (75 μm x 25 cm, 3 μm, 100 Å) (Thermo Fisher Scientific) was used. MS acquisition parameters were: positive mode; 2,600 V voltage at the ion source (NanoSpray III ESI source, Sciex); a 250-ms survey MS scan from 350 to 1,250 m/z, followed by an MS/MS scan from 230 to 1,700 m/z (60 ms acquisition time, rolling collision energy) of the top 65 precursor ions from the survey scan, this making a total cycle time of 4.2 s; the fragmented precursors were added to a dynamic exclusion list for 15 s; any singly charged ions were excluded from the MS/MS analysis.
In order to perform the protein identification, a paralarvae protein database was previously generated in our lab, with a total of 88,035 peptide sequences obtained for the final candidates ORFs predicted by Blast and Swiss-Prot, obtained from de novo transcriptomics. Unigenes with no hits in Blast were analyzed with ESTScan (3.0.3) to predict their coding regions and determine their sequence direction (Prado-Álvarez et al., under review).
The peptide and protein identifications were performed using Protein Pilot software (v5.0.1, Sciex) with the specific protein database as specified above. The concatenated target-reverse decoy database approach was used in order to calculate FDR; iodoacetamide was specified as Cys alkylation; trypsin as an enzyme; TripleTOF 5600 as an instrument; and thorough ID as a search effort. FDR was set to 0.01 for both peptides and proteins. The MS/MS spectra of the identified peptides were then used to generate the spectral library for SWATH peak extraction using the add-in for PeakView Software (version 2.1, Sciex) MS/MSALL with SWATH Acquisition MicroApp (v2.0, Sciex). Peptides obtained from the Protein Pilot database search with a confidence score above 99% were included in the spectral library.
Each individual peptide digest was then analyzed by a SWATH data-independent acquisition (DIA) LC-MS approach. Each sample (4 μl, 1-μg protein in a column) was analyzed using the same nano-LC system, LC gradient, and MS equipment as before for the DDA runs, but using a SWATH-MS acquisition method. This method consisted of repeating a cycle with 60 TOF MS/MS scans (from 230 to 1,500 m/z, 90-ms acquisition time) of overlapping sequential precursor isolation windows of variable width (1 m/z overlap), covering the 350 to 1,200 m/z mass range, with a previous MS scan (350 to 1,250 m/z, 50-ms acquisition time), thus making a 5.5-s total cycle time. The width of the 60 variable windows was optimized according to the ion density found in previous DDA runs.
Protein quantitative data for the proteins contained in the peptide library were obtained from the SWATH runs by extracting the corresponding fragment ion chromatograms using the MS/MSALL with SWATH Acquisition MicroApp (v.2.0, Sciex). Peptide retention times were calibrated in all the SWATH runs using the RePliCaliRT peptides, spiked into each sample. The area under the extracted chromatogram peaks for up to seven fragment ions per peptide, and 10 peptides per protein, was added to obtain the protein quantitation. To be confident about the proteins being identified and quantified, only those showing confidence scores above 95% and FDR below 1% were included in the analysis. Quantitative data were normalized for inter-run variability using the Total Area Sums normalization in MarkerView software (Sciex). Mass spectrometry proteomics data are available via ProteomeXchange with identifier PXD027907.
For each protein, fold-changes were calculated between the averages of the relative protein amount on each experimental group, and differences in protein abundance between groups were statically assessed by applying a Mann–Whitney test, checking for multiple kinds of testing underestimation of p-values by obtaining a q-value estimation for FDR using the q-value R package (http://github.com/jdstorey/qvalue).
Principal Components Analysis was performed with normalized protein abundance using all proteins identified in samples of both dietary groups (M and A), and also with the differentially expressed proteins by diet. Score plots of the generated components were used to visualize expression patterns between experimental groups (M and A). As a further objective test of group (dietary treatment) separation, a t-test was applied to the scores of PC1. PCA analysis was performed using IBM SPSS Statistic, and p < 0.05 value was considered statistically significant. Hierarchical clustering using all differentially expressed proteins was performed to establish differences between experimental groups. Statistical analyses were carried out using R, and differences were considered statistically significant at p < 0.01.
Additionally, protein functional analysis of all proteins identified in both dietary groups (M and A) was performed with the PANTHER program (http://www.pantherdb.org, v14.0), for the Gene Ontology (GO) classification based on protein class (PC), biological process (BP), molecular functions (MF), cellular components (CC), and pathways (P).
For proteins differentially expressed by diet, Quick GO terms for BP, MF, and CC were searched in UniProtKB (protein knowledge base). UniProt IDs were used as input in DAVID for enrichment analyses (Huang et al., 2009).
The values for dry weight and SGR of paralarvae of 20 dph fed on A diet were 1.2 ± 0.2 mg of dry weight and SGR of 7.1%, whereas the group fed on M diet reached 2.7 ± 0.3 mg and 11.1% SGR. The dry weight paralarvae fed on mixed diet (M) were significantly higher (p < 0.05, t-test) compared to the group fed only on Artemia (A diet). It should be noted that the survival of the group fed on A diet dropped after 30 dph, while no decline in survival was observed in the paralarvae fed on the improved mixed diet (M), showing pre-settlement behavior at 40 dph.
Two libraries (OPM and OPA corresponding to 20-dph paralarvae fed on M diet and A diet, respectively) with five biological replicates each were obtained after RNA sequencing and compared for differentially gene expression. Percentage of mapping against the de novo reference transcriptome showed an average mapping rate of 71 and 71.6% in OPM and OPA libraries, respectively (Prado-Álvarez et al., under review).
After read assembly and contig clustering, an average of 130,809 and 130,869 unigenes were obtained in OPM and OPA libraries, respectively. Similar values of annotation percentage were obtained in each replicated sample. An average of 47% of all unigenes were annotated against the Nt database, 35% of unigenes mapped against sequences deposited in the Nr database, 33% were annotated against Pfam and GO databases, whereas 28% had homology to manually deposited protein sequences in Swiss-Prot. The lowest percentage of annotation was obtained against KOG and KO databases, with average mean values of 17 and 2%, respectively. More than half sequences were annotated in at least one of the databases, and a mean percentage of 0.26 and 0.38% of total contigs was annotated in all databases in OPM and OPA libraries, respectively.
A total of 22,852 unigenes were exclusively expressed in paralarvae fed on mixed diet (M), and 24,337 genes were uniquely expressed on Artemia-based diet (A), while 123,873 genes were observed co-expressed in both dietary groups (Figure 1A). Among the total numbers of unigenes (171,072), 918 resulted differentially expressed, 250 of which were upregulated and 668 were downregulated in the M diet group compared to the A diet group (M vs. A, comparison). Fold change gene expression is represented against the p-adjusted value in a volcano plot shown in Figure 1B. The representation of the normalized expression [log10 (FPKM+1)] of DEGs in a heatmap clustered the unigenes according to the diet group (Supplementary Figure 1), showing the overall similarity among the 5 biological replicates at the DEG level. All DEGs (up- and downregulated) are listed in the Supplementary Table 2, including the specific fold change gene expression and the corresponding complete annotation against the databases described above.
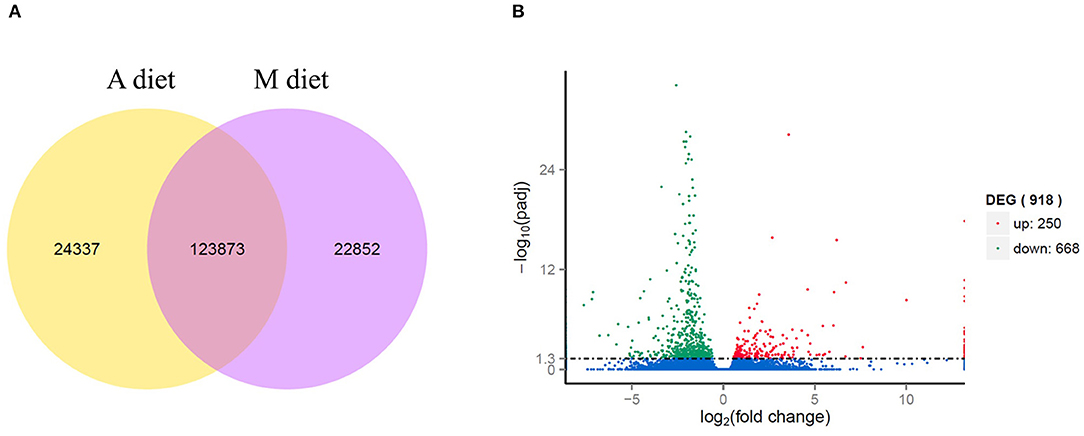
Figure 1. A Venn diagram showing the number of co-expressed genes and uniquely expressed genes between M and A diets (A). Overall distribution of differentially expressed genes (DEGs) represented by a volcano plot (B). The normalized gene expression values and the corresponding normalized p-value are presented by dots. The statistically significant upregulated genes are represented by red dots, down-regulated genes are represented by green dots, and blue dots show no difference in expression.
Functional enrichment analyses by GO terms of downregulated genes (M vs. A, comparison) are shown in Figure 2. In both categories, biological process (BP) and molecular function (MF), the most represented GO terms (corrected p < 0.05) were proteolysis and hydrolase activity, with 43 and 128 DEGs annotated for each term, respectively. Terms related with peptidase activity (55 DEGs), peptidase activity acting on L-amino acid peptides (50 DEGs), and endopeptidase activity (45 DEGs) were also downregulated by M diet. A complete report of differential GO enrichment analysis is given in Supplementary Table 2.
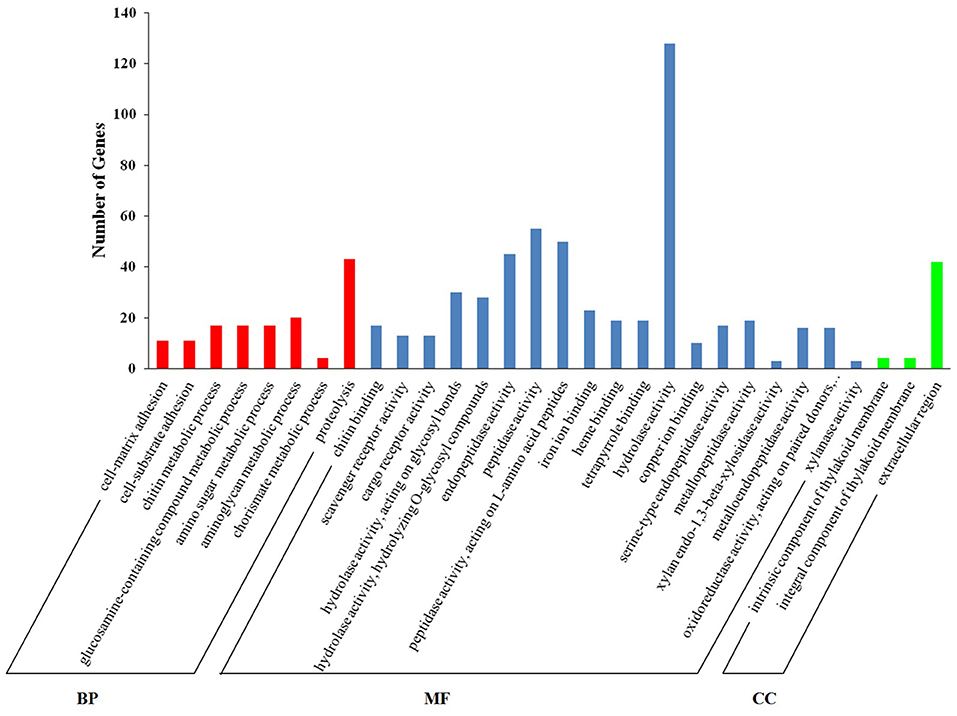
Figure 2. Enriched GO terms in downregulated genes (M vs. A, comparison). The X-axis shows Gene Ontology (GO) terms in the sub-level of the GO three main domains. The Y-axis shows the number of DEGs annotated for each term.
Among the eight enriched BP terms, four categories corresponded to metabolic processes of carbohydrates (chitin, GO:00006030; glucosamine-containing compounds, GO:1901071; amino sugars, GO:0006040; and aminoglycans, GO:0006022). Unigenes with homology chitotriosidase (Cluster-58773.106392, Cluster-58773.106395, and Cluster-58773.113564) and chitinase (Cluster-58773.163428, Cluster-58773.163431) were the most abundant and appeared in the four categories together with peritrophin-44 (Cluster-58773.180067), peritrophin-1 (Cluster-58773.191042), and vitrin (Cluster-58773.114933). The unigenes with homology to pyruvate carboxylase (Cluster-58773.135223), glutamine synthetase (Cluster-58773.160657), and peptidoglycan recognition protein (Cluster-58773.98587) appeared uniquely in the aminoglycan metabolic process term.
Additionally, two categories among the enriched BP terms corresponded to the cell adhesion process (cell matrix, GO:0007160); and (cell-substrate adhesion, GO:0031589). Unigenes with homology to mucin-like (Cluster-58773.76317) appeared in the cell-matrix adhesion category.
One category corresponding to dicarboxylic acid metabolism (the chorismate metabolic process, GO:0046417) was also represented with unigenes showing homology to chorismate mutase type II, SRP-54-type protein (Cluster-58773.162110; Cluster-58773.160866), and apolipoprotein (Cluster-58773.162111; Cluster-58773.160865).
The enriched BP term corresponding to proteolysis (GO:0006508) included unigenes comprising mainly proteases from the hydrolase class, such as digestive cysteine proteinase (Cluster-58773.155311), legumain (Cluster-58773.179190), or cathepsins B/L (Cluster-58773.153403, Cluster-58773.132042, respectively). A class of serine proteases was also represented by neuroendocrine convertase 1 (Cluster-58773.157033), chymotrypsin (Cluster-58773.126901), and serine protease 1 (Cluster-58773.135700). Among the class metalloprotease, homology to matrix metalloproteinase-14 (Cluster-58773.40511), myosinase-I (Cluster-58773.166547), a disintegrin and metalloproteinase with thrombospondin motifs 5 (Cluster-58773.39611) was also displayed. Carboxypeptidases such as carboxypeptidase A1 (Cluster-58773.158098) and glutamate carboxypeptidase 2 (Cluster-58773.49739), were also included within the enriched term proteolysis.
A KEGG pathway enrichment analysis was also carried out to identify significantly enriched metabolic or signal transduction pathways associated with DEGs compared to the whole genome background. A total of 8 pathways with corrected p-value < 0.05 were considered significant. Four pathways were identified as downregulated in M diet (Figure 3A), including routes associated with lysosomes, antigen processing and presentation, the amino sugar and nucleotide sugar metabolism, and the circadian rhythm. Three genes coding for chitinases and UDP-N-acetylglucosamine/UDP-N-acetylgalactosaminediphosphorylase appeared downregulated by diet in the amino sugar and a nucleotide sugar metabolism route (Supplementary Figure 2). Also, four routes were identified as upregulated in M diet (Figure 3B), including DNA replication and three pathways related to the cell cycle, particularly with the MCM complex (Mini-Chromosome Maintenance) represented in Figure 4.
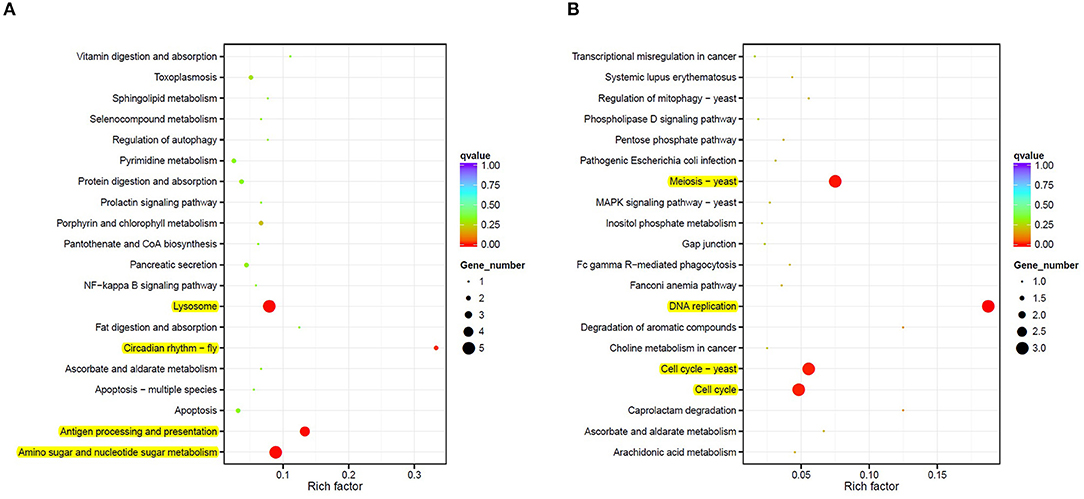
Figure 3. A scattered plot of downregulated (A) and upregulated (B) enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in the mix-based diet samples (M diet). The degree of KEGG enrichment is measured by the rich factor, the p-value, and the number of genes enriched in each pathway. The rich factor refers to the ratio of the DEGs number in the pathway and the number of all genes annotated in the pathway. Significant pathways are highlighted in yellow.
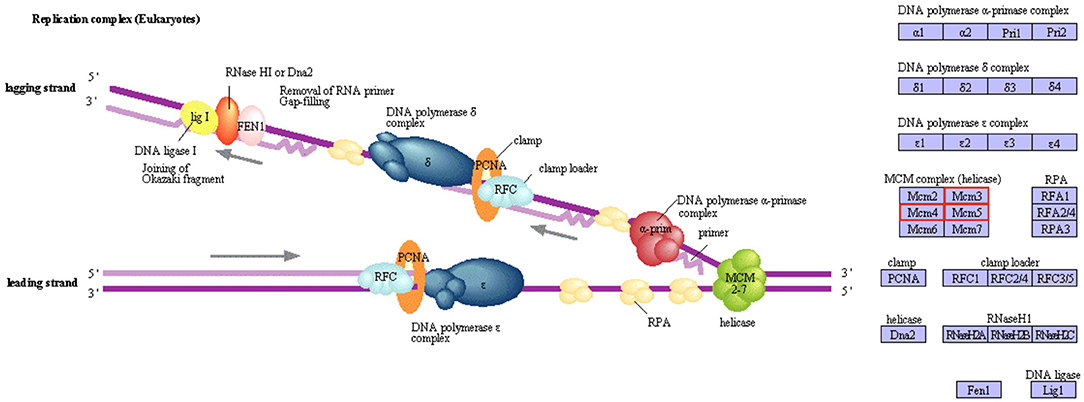
Figure 4. The DNA replication pathway enriched in paralarvae fed on M diet based on KEGG pathways enrichment analysis. Upregulated genes involved in this route are marked in red.
Figure 5 shows the relative expression (M vs. A) of a selection of differentially expressed genes obtained by RNA-seq. The results obtained by qPCR validated the tendency in expression by RNAseq. Significant differences in expression (p-value < 0.05) were observed in all genes analyzed except the glutamine synthetase and microphthalmia-TF-coding genes, although they showed the same trend as RNA-seq results. Regarding upregulated genes in M diet, the highest expression was obtained in the MCM5 gene, with a value of 1.9-fold change (log2) compared to A diet. Four join-box-coding genes and a setmar-coding gene reached a mean fold change (log2) values, ranging from 0.6 and 0.78, respectively, and the S1 peptidase reached a mean fold induction of 1.1 compared to A diet. The dual-tyrosine PR kinase was slightly upregulated with a mean fold change (log2) of 0.3 compared to A diet. The five significantly downregulated genes, piruvate carboxilase, cytosolic-phospholipase, lysozyme, chitinase 3, and carnitine-O-palmitoyltransferase 1, reached −1.6, −1.5, −2.4, −0.64, and −0.98-fold change (log2) values, respectively, in M diet.
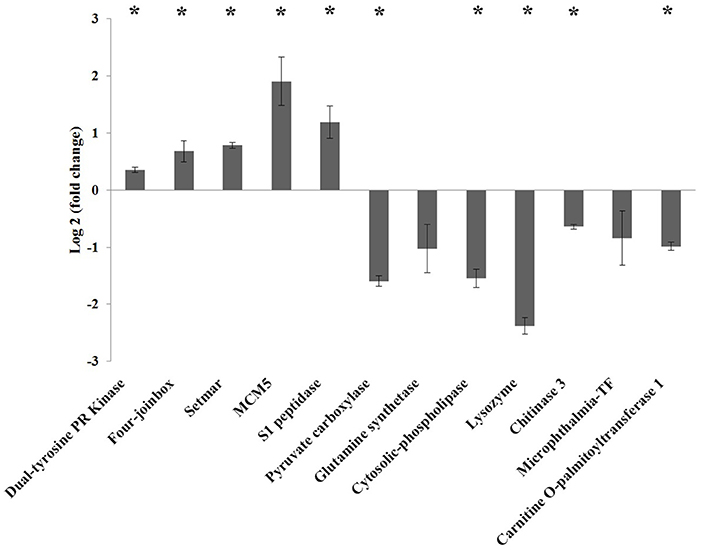
Figure 5. Gene expression by qPCR on a selection of differentially expressed genes induced by diet (M vs. A, comparison). Results are presented as mean ± standard error of log2 fold change. Significant differences compared to controls are indicated with an asterisk (p < 0.05).
A total of 2,341 proteins were quantified through all 12 samples (two dietary groups x 6 biological samples) in the SWATH-MS analysis, with an FDR < 1%, using the specific protein database of O. vulgaris paralarvae previously generated in our lab. The quantitative values for all quantified proteins for all paralarvae samples fed on both diets are presented in Supplementary Material (Supplementary Table 3A).
Statistical analysis of the total quantified proteins revealed that a subset of 43 proteins showed significant differential abundance related to diet; among them, 12 were upregulated and 31 downregulated in paralarvae fed on M diet. The protein identities differentially expressed in paralarvae fed on M diet compared to those fed on A diet (M vs. A, comparison), the fold changes (FC), and the Mann–Whitney p-values, as well as the GO annotations on Quick GO for BP and MF function, are given in Supplementary Table 4.
Also, complete information of FC, Mann–Whitney p-values and FDR estimated q-values for all the quantified proteins in paralarvae fed on both diets (M vs. A, comparison) is presented in Supplementary Table 3B.
PCA analysis of all proteins (2,341) of paralarvae quantified by SWATH-MS analysis allowed to separate protein expression profiles related to diet. Both PC1 and PC2 explained 33.3% of the total variance (Figure 6A). Although the scores of both experimental groups are close, they can be easily segregated in PC1, and the t-test further applied to the means of the scores in this component is at the verge of conventional (95%) significance (p = 0.064). Moreover, PCA using only those proteins differentially expressed (43) showed even a clearer separation in PC1. Both PC1 and PC2 explain 39% of the total variance (Figure 6B), and the means of the scores of the dietary groups in PC1 are significantly different (t-test; p < 0.05).
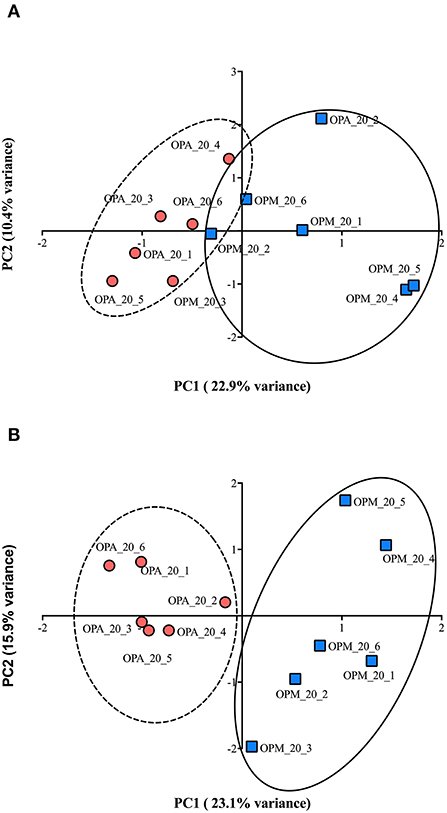
Figure 6. (A) Principal Components Analysis for 20 days post-hatching paralarvae of O. vulgaris fed on improved mixed diet (M) and Artemia-based diet A. Representation of the similarity of all quantified proteins (2,341) among the 6 individual per diet analyzed by SWATH-MS. (B) Representation of the similarity of 43 proteins with significant changes in abundance related to diet.
The hierarchical clustering (heatmap Euclidean Ward) analyses using the 43 differentially expressed proteins in paralarvae showed also a good separation of experimental groups according to the diet (Figure 7).
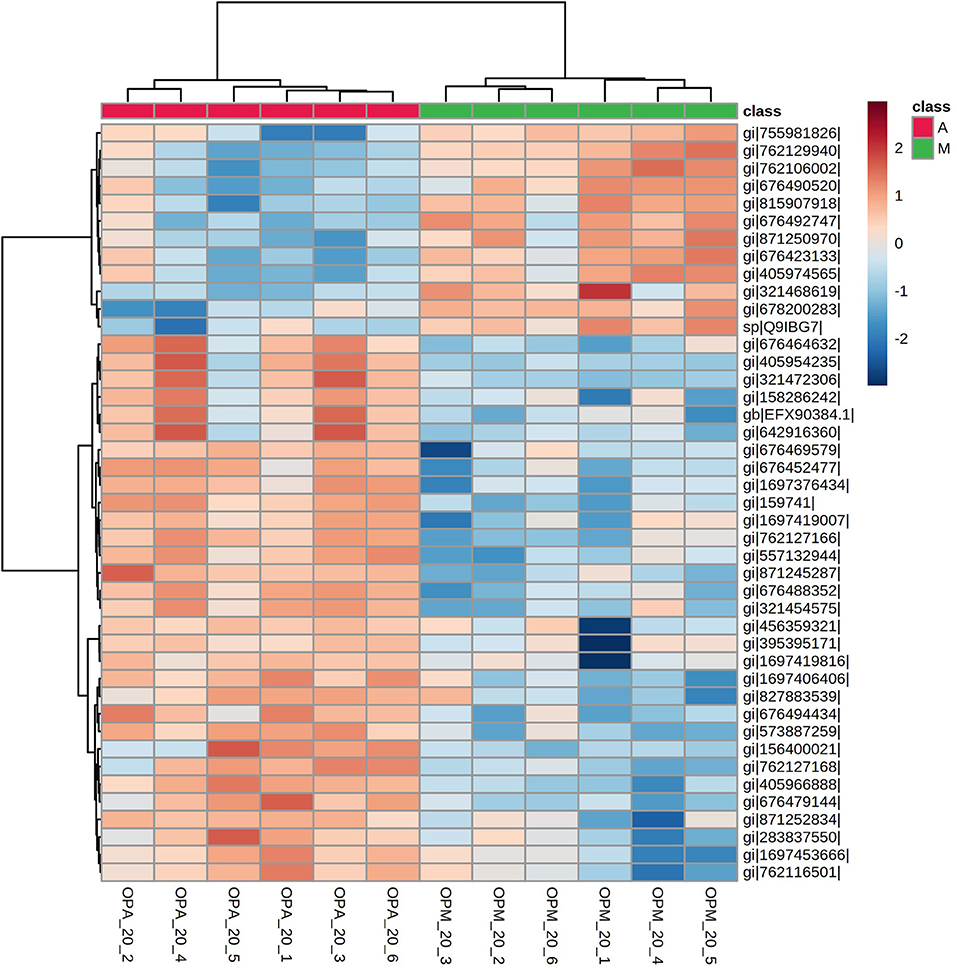
Figure 7. A heatmap showing hierarchical clustering of proteins with significant changes in abundance related to diet found in the SWATH-MS proteomics analysis in O. vulgaris paralarvae of 20 days post-hatching fed on improved mixed diet (M) and Artemia-based diet (A). This subset of 43 proteins showing changes in abundance (fold-change > 2 and p-value < 0.01) allowed the complete separation of both experimental groups. See Supplementary Table 4 for complete information.
PANTHER functional analysis of all identified proteins (2,341 proteins) in paralarvae of 20 dph enabled to find 170 annotated proteins and obtain GO terms related to protein classes, BP, CC, MF, and pathways (Supplementary Figure 3). The analyses revealed the presence of 16 different protein classes, where a metabolite interconversion enzyme (35.4%), a protein-modifying enzyme (18.9%), a transporter (8.7%), a cytoskeletal protein (7.9%), and a translational protein (7.9%) were the main classes. Regarding the biological process category, the cellular process (40.3%) and the metabolic process (30.9%) were the most representative processes, followed by biological regulation (8.9%), localization (8.9%) and response to stimulus (5.2%), as well as other diverse categories e.g., the multicellular organismal process, the developmental process, signaling, or locomotion. The molecular function of proteins was mainly related to catalytic activity (48.2%) and binding (32.5%), followed by a transporter (7.9%), structural molecule activity (6.1%), a molecular function regulator (3.5%), molecular transducer activity (0.9%), and translational regulator activity (0.9%). Concerning cellular components, proteins were associated mainly with a cellular anatomical entity (42%) and intracellular (39.9%) and protein-containing complexes (18.1%). Regarding the number of pathways, our results showed that proteins were involved in a total of 51 different pathways. Among them, the ubiquitin proteasome pathway (7.2%), Huntington disease (5.8%), cytoskeletal regulation by Rho GTPase (4.3%), and glycolysis (4.3%) were the most representative, with many other pathways with lower representation (between 2.9 and 1.4%).
Quick GO annotations for BP of proteins differentially expressed by diet in M vs. A comparison showed that the main terms were related to biological regulation (22%), the cellular process (19.5%), the metabolic process (14.6%), and the rhythmic process (7.3%). However, the MF of proteins differentially expressed by diet was mainly associated with catalytic activity (52.6%), transporter activity (26.3%), and binding (21.1%). Concerning CC, proteins were mainly related to cellular anatomical entity (51.7%), intracellular anatomical structure (34.5%), and protein-containing complex (13.8%).
For metabolic and cellular processes, specific GO terms of proteins upregulated in paralarvae fed on M diet were mainly involved in proteolysis with serine-endopeptidase activity (i.e., a chymotrypsin-like protein), phosphorylation related to activation of protein kinase activity, regulation of cell migration, regulation of mitotic cell, neuron projection morphogenesis (i.e., PREDICTED: serine/threonine-protein kinase mig-15 isoform X10), as well as RNA processing (hypothetical protein LOTGIDRAFT_133731). For binding classification, specific GO terms were involved in ATP binding, nucleotide binding, RNA binding, and metal ion binding.
Specific GO terms of proteins downregulated in paralarvae fed on diet M, for the metabolic process, were involved in carbohydrate derivate metabolism (i.e., hypothetical protein LOTGIDRAFT_94382, partial; PREDICTED: glutamine–fructose-6-phosphate aminotransferase [isomerizing] 1-like isoform X5 and X6, protein metabolism related to protein biosynthesis (PREDICTED: tyrosine-tRNA ligase, cytoplasmic like), the amino acid biosynthetic process (i.e., putative delta 1-pyrroline-5-carboxylate synthetase), and the nucleotide metabolic process (AGAP007120-PA), and also with glycolysis (i.e., enolase). Nevertheless, proteins involved in proteolysis (i.e., caspase 3, serine protease 1) were also found.
Also remarkable was the presence of one protein upregulated related with lipid transport activity (vitellogenin-2, partial), and other protein downregulated involved in lipid droplet stabilization and lipolysis (adipophilin). Besides, a protein downregulated involved in localization and transport of calcium ion (calcium-transporting ATPase). Full details are given in Figure 8 and Supplementary Table 5.
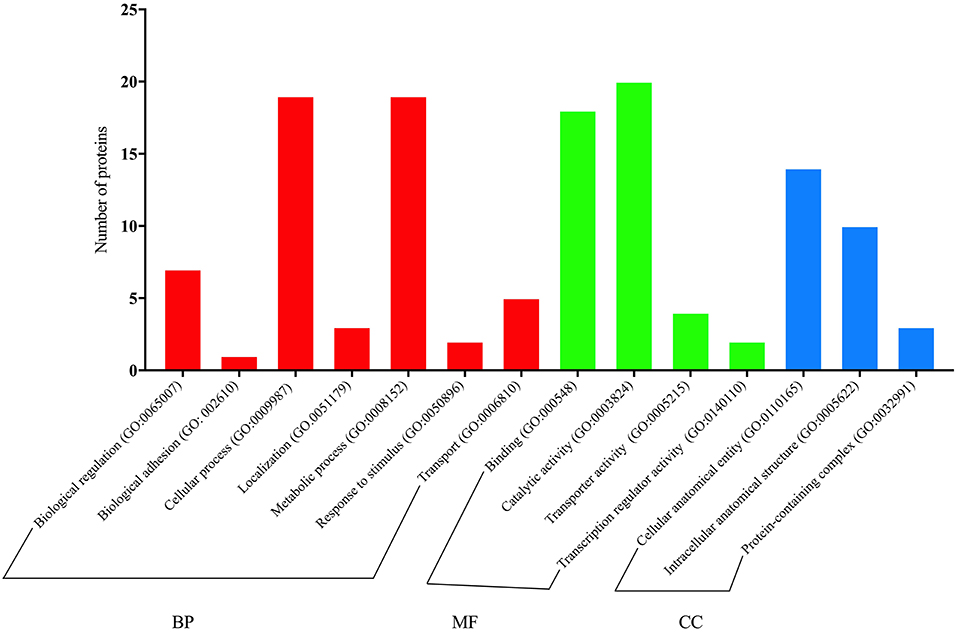
Figure 8. Quick GO classification for biological process (BP), cellular components (CC), and molecular function (MF) of proteins with significant changes in abundance related to diet (M vs. A, comparison) identified by SWATH-MS proteomics analysis.
The KEGG analysis revealed that the pathway related to amino sugar and nucleotide sugar metabolism was enriched in proteins downregulated by M diet, including two representatives of chitinases (Supplementary Figure 2).
In the present study, the combined use of shotgun proteomics (SWATH-MS) and RNAseq transcriptomics analyses allowed the successful identification of 2,341 proteins and 918 differentially expressed genes in 20-dph O. vulgaris paralarvae fed on two live prey-based diets. The number of identified proteins increased 1.8-fold compared with our previous study in 10-dph paralarvae fed on spider crab zoea (M. brachydactyla) in co-feeding with Artemia (García-Fernández et al., 2019), using the new customized protein database generated from de novo transcriptome library built up ad hoc (Prado-Álvarez et al., under review). We also obtained a significantly higher number of differentially expressed genes compared to our previous analysis (García-Fernández et al., 2019), whereas the average mapping rate was slightly lower (71.3%). This might be related to the higher coverage of our reference transcriptome, which included not only diet conditions but also different development stages, infections, and suboptimal culturing conditions (Prado-Álvarez et al., under review). Moreover, the quality of the sequencing was very successful, obtaining a Phred quality score over 20 in 96% of the reads, which revealed 99% of base call accuracy. The appropriate environmental parameters and the improved diet applied to the paralarvae cultured in this study have favored good development and proper settlement of the early-life stage of the octopus in the benthic media, which has allowed us to analyze the paralarvae at a more advanced stage of growth than in our previous studies. This has been reflected in the transcriptomic and proteomic results obtained, and highlighted the benefits of combining both analyses, particularly in a non-model organism (Cogne et al., 2020).
The multivariate analyses, PCA, and heatmap carried out in identified proteins showed that changes in the proteome profiles of octopus paralarvae associated with diet allowed separating both dietary groups. The two dietary groups split particularly well when the differentially expressed proteins and genes were combined in the analyses. These results are in line with our previous studies in octopus at earlier days post-hatchling, in which changes in a specific sub-set of protein expression were related to diet, allowing to define specific proteomes, depending on the octopus paralarvae diet (Varó et al., 2017; García-Fernández et al., 2019).
Panther analysis of all identified proteins (2,341) allowed annotating a small set of proteins (175), accounting for 7.5% of total proteins. However, the GO-terms classification showed a wide variety of protein classes, biological processes, molecular functions, cellular components, and pathways, which could be considered as a good representation of the global octopus paralarvae proteome. Regarding the expressed genes, the highest level of annotation was found against the Nt database (47% of all unigenes). Despite the high and recent increase in cephalopod databases (Albertin et al., 2015; Kim et al., 2018; Belcaid et al., 2019; García-Fernández et al., 2019; da Fonseca et al., 2020), a significant percentage of differentially expressed genes, 34 and 39% in up- and downregulated genes, respectively, remained still unidentified or uncharacterized based on Nt database search. Additionally, transcriptomic results obtained by RNA-seq were validated by analyzing the gene expression of a number of genes in paralarvae samples from the same diet experiment. Relative gene expression corroborated the results obtained by the massive gene analysis, confirming the suitability of the technique for obtaining a transcriptomic overview in this species. We observed the same effect after protein identification, on which the percentage of predicted, predicted/uncharacterized, and hypothetical proteins exceeds 75% of the total. This fact revealed the high complexity of working on massive data in non-model organisms, and highlighted the need of proper annotation to not miss valuable and biologically relevant information in the future studies.
Overall analyses showed a sub-set of genes and proteins differentially expressed by diet, mostly composed by downregulated genes (668 out of 918) and down-expressed proteins (31 out of 43) in paralarvae fed on M diet compared to A diet. These results differed from our previous study (García-Fernández et al., 2019), on which octopus paralarvae fed for 10 days on Artemia showed more downregulated genes and proteins than those fed with spider crab zoea in co-feeding with Artemia. This difference may be related to the composition of the improved mixed diet used in the present study (M diet), but also to the improvement of the zootechnical processes based on the methodology described by Tur et al. (2020), which could also have an influence on the development and survival of paralarvae. Additionally, we cannot ignore possible differences related to the age of paralarvae or culture temperature. In the present study, growth performance of 20-dph paralarvae fed on M diet (2.7-mg dry weight; 11.1% SGR) was 2-fold higher compared to 15-dph paralarvae (dry weight, 1.2 mg; 6% SGR) fed on spider crab zoea in co-feeding with Artemia (García-Fernández et al., 2019). Indeed, no decline in survival was observed in paralarvae fed on M diet. Moreover, animals showed pre-settlement behavior at 40 dph, tending to remain attached to the bottom and walls of the tank to stay there for a time, and only showing swimming movements to capture preys in the water column, which would indicate that the benthic life would be approaching (Vidal et al., 2014). On the other hand, the Artemia-fed group showed high mortality after 30 dph, making its cultivation unviable beyond this age. These results are in agreement with those previously reported for octopus paralarvae fed only with Artemia, which showed lower growth and survival rates compared to that fed using single species of crustacean zoeae generally in co-feeding with Artemia (Reis et al., 2015; Roo et al., 2017; Varó et al., ; Garrido et al., 2018; García-Fernández et al., 2019), or even when zoeae of G. adscensionis were used as sole live prey (Reis et al., 2021). The improved mixed diet used in this study, with the incorporation of amphipods as live prey, suggests that their intensive farming could lead to a viable and sustainable culture of the common octopus.
Most of the over-expressed proteins and genes in the M diet group were involved in the cell cycle and replication, production of structural components, and nervous system development. Among them, the MCM5 gene appeared upregulated and codes for a protein involved in DNA replication, cell cycle regulation, and genome stability, including also transcription activation (Kontos et al., 2011). The protein coded by the MCM5 gene is a component of the MCM complex (Mini-Chromosome Maintenance complex), which is constituted in eukaryotes by 6 helicases (DNA replication licensing factors MCM2-7) and utilizes the energy from ATP hydrolysis to unwind double-stranded DNA during replication. This KEGG pathway was enriched in upregulated genes, which revealed the importance of the cell cycle regulation and the replication process in paralarvae feed on M diet. Similarly, LIM and SH3 domain protein 1, a protein involved in neuronal differentiation and development (Phillips et al., 2004; Myers et al., 2020; Pollitt et al., 2020), and the PREDICTED: SAFB-like transcription modulator isoform X1, an RNA-DNA-binding protein, which plays crucial roles in DNA repair, processing of mRNA and regulatory RNA, and in the interaction with chromatin-modifying complexes (Norman et al., 2016), were also upregulated. Altogether, over-representation of these genes and proteins showed that a proper development and correct growth were occurring in paralarvae fed on M diet. Another over-expressed protein was kielin/chordin-like protein, which plays a role in dorso-ventral patterning (Garrick et al., 2012). Additional genes, such as those that encode D-3-phosphoglycerate dehydrogenase and phosphoserine aminotransferase, both involved in the serine biosynthetic pathway, were upregulated in the M group. Serine (Ser) is a non-essential amino acid with key roles in cellular proliferation and differentiation (Jaeken et al., 1996). Ser is crucial for protein synthesis but is also a precursor for the synthesis of numerous compounds, such as glycine, cysteine, phosphatidylserine, sphingomyelin, and cerebrosides, all related to growth and development. In addition, Ser is considered the largest source of intermediates for purine and thymidine synthesis (Snell, 1984; Barret, 1994). Also, the upregulation of both the protein and the gene of chymotrypsin like in the M group (a protein related to the digestive process) would indicate a better digestive function in these paralarvae.
In general, these results support the higher growth of the paralarvae fed on M diet. In addition, this group showed better overall condition with a higher survival rate and settlement behavior after 40 days. In this sense, downregulation of several proteins and genes would support this fact. Among them, both proteins and genes coding for glucosamine fructose-6-phosphate aminotransferase (GFAT) and nucleoside diphosphate kinase (NDK) appeared downregulated in the M group. The GFAT controls the flux of glucose into the hexosamine pathway and is involved in the regulation of glycosylation of proteins; and the NDK protein may display multiple functions in diverse biological processes and is involved in the promotion of cancer. Transcripts coding for these proteins have been upregulated in response to several stressors, including viral infection in the white shrimp (Litopenaeus vannamei) (Liu et al., 2015, 2017), which would indicate that paralarvae fed on A diet would be in poorer condition than those fed on M diet. In fact, caspase 3 that is involved in the apoptotic process was over-expressed in the A group.
The immune system processes and the related lysozyme and peroxidase activities (represented, among others, by the TNF receptor-associated factor 4-like gene and the lysozyme gene) were down regulated in the M group, suggesting a better immune status. The over-expression of vitellogenin-2 in this group might be also related to these aspects. Although vitellogenin is present in the females of nearly all oviparous species as the precursor of major egg yolk proteins, its synthesis has been shown to occur in male and sexually immature animals, and accumulating data indicate that vitellogenin performs also non-nutritional functions in male and female fish, possessing both antibacterial and antioxidant activities (Shi et al., 2006; Sun and Zhang, 2015).
The expression of carbonic anhydrase was downregulated in the M group. This fact might indicate a reduced metabolic rate in these animals since this protein is primarily involved in the systemic transport and excretion of CO2 in animals (Maren, 1967), and its activity may reflect the metabolic rate (CO2 production) of an organism (Kochevar and Childress, 1996; Seibel and Walsh, 2003). Supporting such a reduced metabolic rate, superoxide dismutase, catalase, NADP+-dependent malic enzyme, and 6-phosphogluconate dehydrogenase-coding genes, related to enzymatic antioxidant defenses, were downregulated, which indicates lower generation of reactive oxygen species (ROS) and a better oxidative state of paralarvae fed on M diet. A lower metabolic rate might also indicate that paralarvae of the M group had less activity than those fed exclusively on Artemia. Given the exceptionally good growth performance on paralarvae fed on M diet, it may be reasonable to hypothesize that nutritional requirements were met with the combination of the four crustacean's species used in this diet, resulting in slowing down of metabolism in animals. It was also worth noting that the group fed on M diet displayed a relaxed swimming behavior compared to the group fed on A diet. This observation was based on the fact that explosive movements on swimming typical at the moment of feeding were more abundant on paralarvae fed on A diet even several hours after the food was provided. On the other hand, paralarvae fed on Artemia, considered a deficient diet, might need to continue feeding on the remaining food available in the tank in order to obtain adequate amount of nutrients for a correct development. Moreover, the downregulated expression of chitinase, both the protein and the coding gene, could also support the hypothesis of a lower intake in the M group. The increased energy expenditure that needed to maintain this sustained predatory activity would explain the lower growth achieved in the group fed on A diet. Indeed, GO terms involved in protein metabolism, such as hydrolysis of proteins into smaller polypeptides and/or amino acids by cleavage of their peptide bonds, were more represented in downregulated genes. Similarly, the KEGG significant pathways in downregulated genes also showed a relevant involvement of functions related to hydrolysis and protease activity represented by routes related to lysosomes and antigen-associated biological processes. In relation to the activity rate of paralarvae, ryanodine-sensitive calcium-release channel-coding gene, calcium load-activated calcium channel, and calcium-transporting ATPase proteins involved in the excitation-contraction process in muscle fiber were downregulated in the M group, while the upregulation observed in the phospholamban-coding gene, an integral membrane protein of the sarcoplasmic reticulum that inhibits the Ca2+-ATPase pump, has been associated with depressed rates of contraction and relaxation in mouse (Song et al., 2004). Finally, actin (both the protein and the related gene) that plays a fundamental role in muscle contraction was downregulated in the M group.
Overall activation of metabolism in paralarvae fed on A diet might explain the higher number of enriched terms in downregulated genes when the M group is compared to the A group. Increased food intake to compensate for dietary deficiencies might result in an excess intake of other nutrients that can only be partially retained by the animal, thus increasing its oxidation rate. In this context, several proteins involved in lipid transport and storage were upregulated in the A group. This is the case of adipophilin, a structural protein involved in lipid droplet stabilization and control of lipolysis (Brasaemle, 2007) and the gene encoding the lipid exporter ABCA1 and related proteins of the ABC superfamily, involved in lipoprotein production (Phillips, 2018). Increased levels of lipoproteins and lipid droplets are indicative of enhanced lipid transport and intracellular storage, respectively, which is related to increased lipid uptake (Walther and Farese, 2012). Inside the cell, fatty acids are incorporated into membranes as components of phospholipids, reassembled into triacylglycerols to form lipid droplets, or enter into β-oxidation (Loewen et al., 2004; Bartz et al., 2007). Fatty acid oxidation depends on the concentration and composition of fatty acids in the diet and the specificity of the enzymes. In fish, it has been reported that the higher intake of a fatty acid, the lower is its relative deposition, implying higher in vivo oxidation (Monroig et al., 2018). This could explain the upregulation of genes involved in lipid metabolism in paralarvae fed on A diet. The higher expression of the acyl-CoA synthetase gene might result in a higher rate of fatty acid activation to fatty acyl-CoA, the form in which the fatty acid must be transported into the mitochondria to be oxidized. This entry is regulated by the mitochondrial carnitine palmitoyltransferase system, which is composed of two enzymatic modules: carnitine O-acetyltransferase (CPT1), in the outer mitochondrial membrane, and carnitine/acylcarnitine carrier, in the inner mitochondrial membrane. The genes encoding both enzymatic modules were also upregulated in the A group. In addition, the protein delta 1-pyrroline-5-carboxylate synthetase (P5CS) was over-expressed in the A group, and increased P5CS activity has been reported to enhance expression of the fatty acid transporter CPT1 (Yang et al., 2021). In agreement with these results, a marked increase in expression of cpt1 has been reported in rainbow trout (Oncorhynchus mykiss) (Kolditz et al., 2008) and seabream (Sparus aurata) (Betancor et al., 2016) fed on high-fat diets. Carnitine O-acetyltransferase had been previously found upregulated in 10-dph paralarvae co-fed on mix zoea and Artemia, suggesting that, at that specific culture conditions and at this age (10 dph), the lipid catabolic pathway of paralarvae could be activated to obtain energy (García-Fernández et al., 2019). However, at 20 dph, the data obtained in the present study suggest a reduced use of fatty acids to obtain energy in paralarvae fed on M diet. Indeed, Matsubara et al. (2016) showed important depletion in L-carnitine levels from hatchling until 20-40-dph paralarvae of O. sinensis fed with Artemia and a low survival pattern, which also seems to support high dependence of β-oxidation to obtain energy. The increased fatty acid oxidation in the A group in the present study may reflect the already reported unbalanced lipid profile of Artemia as live prey for O. vulgaris (Navarro and Villanueva, 2000, 2003; Seixas et al., 2010a,b; Navarro et al., 2014; Reis et al., 2015), since an increased β-oxidation may be indicative of a high rate of dietary fatty acid bioconversion for essential fatty acid synthesis. The increased rate of β-oxidation found from 6 dph onwards in paralarvae fed on Artemia in a previous study would support these results (Morales et al., 2017). A high rate of de novo synthesis of shorter chain-length fatty acids from β-oxidation of docosahexaenoic acid (DHA) has been reported to take place in Artemia, thus reducing availability of such essential fatty acid for paralarvae. On the other hand, DHA is the fatty acid with the highest esterification rate in Artemia triacylglycerols, and paralarvae possess a low capacity to use prey triacylglycerols (Reis et al., 2019). The reduced rate of fatty acid oxidation in the M group could indicate a more adequate and balanced lipid composition of M diet for the provision of structural components for cells. This would be also supported by the lower expression of both the protein [3-oxoacyl-(acyl-carrier-protein) reductase FabG] and the coding genes related to lipid synthesis, such as the fatty acid synthase complex and the pentose phosphate shunt and the NADP+-dependent malic enzyme, responsible for the NADPH provision for fatty acid synthesis.
Most of the metabolic processes were downregulated in the group fed on M diet, which would agree with lower energy expenditure and higher performance of paralarvae than in the group fed on A diet. Indeed, the KEGG pathways in downregulated genes were mostly related to carbohydrate metabolism and generation and maintenance of physiological rhythms of the organism. However, the upregulated expression of several genes confirms that cephalopod metabolism is mainly based on protein and carbohydrate as fuels (Storey and Storey, 1983; O'Dor and Webber, 1986; Hochachka, 1995; Lee, 1995). Namely, genes encoding argininosuccinate lyase and fumarase, involved in the use of arginine (Arg) as an oxidative substrate, were upregulated. In this sense, Hochachka and Fields (1982) already reported that, in addition to their fundamental role supporting anaerobic activity as part of the phosphagen phosphoarginine, Arg is among the most metabolically active amino acids in cephalopods, and is highly oxidized in tissues of this group (Hochachka et al., 1983). Incorporation of Arg and other amino acid intermediates to the Krebs cycle would be guaranteed by the upregulated expression of genes coding for pyruvate kinase and triosephosphate isomerase in the M group. In this regard, a large protein to lipid ratio has been reported to be a critical factor in octopus paralarvae (Seixas et al., 2010b; Vidal et al., 2014). Also, Matsubara et al. (2016) concluded that amino acid deficiencies may be among the causes of the low growth and survival of O. sinensis fed on Artemia. In this sense, the prey combination in M diet also appears to provide a more appropriate amino acid profile to support the high growth rate of cephalopods, which is highly dependent on adequate assimilation of amino acid (Navarro et al., 2014). Paralarvae derived from this experiment and the diets used for feeding will be further analyzed at the biochemical level as it would be of high interest to know the composition of the M and A diets and also the biochemical profile of the animals fed on these diets to decipher the differences in the growth and development due to the diet supplied during the first stages.
In summary, the results of this study based on a proteogenomic approach showed that the use of the improved mixed diet (M diet), composed of three crustacean species co-fed with Artemia, has allowed, for the first time, to analyze older octopus paralarvae (20 dph) and with the highest specific growth rate (SGR) achieved in comparison with previous studies where paralarvae were fed on other live preys, or co-fed with Artemia. In addition, paralarvae grown until 40 dph showed pre-settlement behavior. In view of the higher growth rate and survival time shown by the M group, the live prey mix of M diet seems to provide a better balance of nutrients, which appears to have resulted in lower energy expenditure and higher performance in terms of growth and welfare of these paralarvae than of those in the A group. This is also supported by the fact that most of the proteins and genes over-expressed in the M group are linked to the cell cycle and replication, the production of structural components, and the development of the nervous system. Additionally, the decreased transcriptional levels of genes related to lipid metabolism found in the M group seem to confirm the hypothesis that, instead of fatty acids, the preferred fuels for cephalopod mitochondria would be proteins and carbohydrates. Ensuring adequate dietary intake of essential fatty acids would be of the utmost importance in preventing mitochondrial energy pathways from being saturated by the need for bioconversion of dietary lipids. Overall, the results obtained suggest that the appropriate environmental parameters and the improved diet favored good development and settlement of the paralarvae, which is indicative of the welfare state of the culture. At the industrial level, the sustainable culture of a species depends mainly on a continued supply of economically viable food. In this regard, intensive production of amphipods as live preys could contribute to pave the way for a viable and sustainable O. vulgaris culture, thereby reducing and/or avoiding the use of the offspring of commercially valuable crustacean species (zoeae) to feed the paralarvae.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA754143 and http://www.proteomexchange.org/, PXD027907.
The animal study was reviewed and approved by Comité de Ética de la Investigación y Bienestar Animal Vicerrectorado de Investigación y Transferencia de Conocimiento, Universidad de La Laguna, 38071, La Laguna, España.
Conceptualization and resources were contributed by IV and CG. Methodology was contributed by IV, MP-Á, IO, SD, and CG. Transcriptomic analysis was contributed by MP-Á, SD, and CG. SWATH analysis and validation were contributed by IO. Proteomic analysis was contributed by IV and IO. Data curation was contributed by IV, MP-Á, IO, SD, and CG. Paralarvae rearing was contributed by PG-F, RT, and PD. Collecting and processing samples for RNA-seq analysis were contributed by PG-F. Analysis, discussion of the results, and writing of the manuscript were contributed by IV, MP-Á, AEM, SD, and CG. All authors supervised and approved the final version of the manuscript.
This work was part of the I+D+i grant AGL2017-89475-C2-1 – OCTOMICS, funded by MCIN/AEI/10.13039/501100011033. Funds will be also received for open access publication fees from a collaboration agreement between Innovation Galician Agency and CSIC (Program Agreement 2021–2022).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.817701/full#supplementary-material
Albertin, C. B., Simakov, O., Mitros, T., Wang, Z. Y., Pungor, J. R., Edsinger-Gonzales, E., et al. (2015). The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature. 524, 220–224. doi: 10.1038/nature14668
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, 1–12. doi: 10.1186/gb-2010-11-10-r106
Andrews, S. (2010). FastQC. A quality control tool for high throughput sequence data. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Babicki, S., Arndt, D., Marcu, A., Liang, Y., Grant, J. R., Maciejewski, A., et al. (2016). Heatmapper: web-enabled heat mapping for all. Nucl. Acids Res. 44, 147–153. doi: 10.1093/nar/gkw419
Barret, A. (1994). Proteolytic Enzymes: Serine and Cysteine Peptidases. Methods in Enzymology vol 244., ed. A. Barrett Academic Press.
Bartz, R., Li, W. H., Venables, B., Zehmer, J. K., Roth, M. R., Welti, R., et al. (2007). Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J. Lipid Res. 48, 837–847. doi: 10.1194/jlr.M600413-JLR200
Belcaid, M., Casaburi, G., McAnulty, S. J., Schmidbaur, H., Suria, A. M., Moriano-Gutierrez, S., et al. (2019). Symbiotic organs shaped by distinct modes of genome evolution in cephalopods. Proc. Natl. Acad. Sci. U. S. A. 116, 3030–3035. doi: 10.1073/pnas.1817322116
Betancor, M. B., Sprague, M., Montero, D., Usher, S., Sayanova, O., Campbell, P. J., et al. (2016). Replacement of marine fish oil with de novo omega-3 oils from transgenic Camelina sativa in feeds for gilthead sea bream (Sparus aurata, L.). Lipids. 51, 1171–1191. doi: 10.1007/s11745-016-4191-4
Brasaemle, D. L. (2007). The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 48, 2547–2559. doi: 10.1194/jlr.R700014-JLR200
Cogne, Y., Gouveia, D., Chaumot, A., Degli-Esposti, D., Geffard, O., Pible, O., et al. (2020). Proteogenomics-Guided Evaluation of RNA-Seq Assembly and protein database construction for emergent model organisms. Proteomics. 20, 1900261. doi: 10.1002/pmic.201900261
da Fonseca, R. R., Couto, A., Machado, A. M., Brejova, B., Albertin, C. B., Silva, F., et al. (2020). A draft genome sequence of the elusive giant squid, Architeuthis dux. Gigascience. 9, 1–12. doi: 10.1093/gigascience/giz152
de Moraes Lenz, T., Escanéz Pérez, A., Tur, R., Delgado, A., Évora, C., and Almansa, E. (2019). First attempts of the use of intake tracers in encapsulated diets with chitosan for octopus paralarvae. Aquac. Res. 50, 3070–3073. doi: 10.1111/are.14234
Fiorito, G., Affuso, A., Basil, J., Cole, A., De Girolamo, P., D'Angelo, L., et al. (2015). Guidelines for the care and welfare of cephalopods in research –A consensus based on an initiative by CephRes, FELASA and the Boyd Group. Lab. Anim. 49, 1–90. doi: 10.1177/0023677215580006
García-Fernández, P., Castellanos-Martínez, S., Iglesias, J., Otero, J. J., and Gestal, C. (2016). Selection of reliable reference genes for RT-qPCR studies in Octopus vulgaris paralarvae during development and immune-stimulation. J. Invertebr. Pathol. 138, 57–62. doi: 10.1016/j.jip.2016.06.003
García-Fernández, P., Prado-Alvarez, M., Nande, M., García de la Serrana, D., Perales-Raya, C., Almansa, E., et al. (2019). Global impact of diet and temperature over aquaculture of Octopus vulgaris paralarvae from a transcriptomic approach. Sci. Rep. 9, 10312. doi: 10.1038/s41598-019-46492-2
Garrick, D. J., Baumgard, L. H., and Neibergs, H. L. (2012). Invited review: Genomic analysis of data from physiological studies. J. Dairy Sci. 95, 499–507. doi: 10.3168/jds.2011-4970
Garrido, D., Martín, M. V., Rodríguez, C., Iglesias, J., Navarro, J. C., Estévez, A., et al. (2018). Meta-analysis approach to the effects of live prey on the growth of Octopus vulgaris paralarvae under culture conditions. Rev. Aquac. 10, 3–14. doi: 10.1111/raq.12142
Garrido, D., Navarro, J. C., Perales-Raya, C., Nande, M., Martín, M. V., Iglesias, J., et al. (2016). Fatty acid composition and age estimation of wild Octopus vulgaris paralarvae. Aquaculture. 464, 564–569. doi: 10.1016/j.aquaculture.2016.07.034
González-Fernández, M., Fabrikov, D., Ramos-Bueno, R., Guil-Guerrero, J. L., and Ortea, I. (2019). SWATH Differential abundance proteomics and cellular assays show in vitro anticancer activity of arachidonic acid- and docosahexaenoic acid-basmonoacylglycerols in HT-29 colorectal cancer cells. Nutrient. 11, 2984. doi: 10.3390/nu11122984
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucl. Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Guinot, D., Monroig, Ó., Navarro, J. C., Var,ó, I., Amat, F., and Hontoria, F. (2013). Enrichment of Artemia metanauplii in phospholipids and essential fatty acids as a diet for common octopus (Octopus vulgaris) paralarvae. Aquac. Nutr. 19, 837–844. doi: 10.1111/anu.12048
Hochachka, P. W. (1995). Oxygen efficient design of cephalopod muscle metabolism. Mar. Freshw. Behav. Physiol. 25, 61–67. doi: 10.1080/10236249409378908
Hochachka, P. W., and Fields, J. H. A. (1982). Arginine, glutamate, and proline as substrates for oxidation and for glycogenesis in cephalopod tissues. Pacific Sci. 36, 325–335
Hochachka, P. W., Mommsen, T. P., Storey, J. M., Storey, K. B., Johansen, K., and French, C. J. (1983). The relationship between arginine and proline metabolism in cephalopods. Mar. BioIogy Lett. 4, 1–21
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Iglesias, J., and Fuentes, L. (2014). Octopus vulgaris. Paralarval Culture. In: Cephalopod Culture, eds. J. Iglesias, L. Fuentes, and R. Villanueva (Dordrecht: Springer Netherlands) p. 427–450. doi: 10.1007/978-94-017-8648-5_23
Iglesias, J., Pazos, G., Fernández, J., Sánchez, F. J., Otero, J. J., Domingues, P., et al. (2014). The effects of using crab zoeae (Maja brachydactyla) on growth and biochemical composition of Octopus vulgaris (Cuvier 1797) paralarvae. Aquac. Int. 22, 1041–1051. doi: 10.1007/s10499-013-9725-7
Jaeken, J., Detheux, M., Van Maldergem, L., Foulon, M., Carchon, H., and Van Schaftingen, E. (1996). 3-Phosphoglycerate dehydrogenase deficiency: an inborn error of serine biosynthesis. Arch. Dis. Child. 74, 542–545. doi: 10.1136/adc.74.6.542
Kim, B. M., Kang, S., Ahn, D. H., Jung, S. H., Rhee, H., Yoo, J. S., et al. (2018). The genome of common long-arm octopus Octopus minor. Gigascience 7, 1–7. doi: 10.1093/gigascience/giy119
Kochevar, R., and Childress, J. J. (1996). Carbonic anhydrase in deep-sea chemoautotrophic symbioses. Mar. Biol. 125, 375–383. doi: 10.1007/BF00346318
Kolditz, C., Borthaire, M., Richard, N., Corraze, G., Panserat, S., Vachot, C., et al. (2008). Liver and muscle metabolic changes induced by dietary energy content and genetic selection in rainbow trout (Oncorhynchus mykiss). Am. J. Physiol. - Regul. Integr. Comp. Physiol. 294, 1154–1164. doi: 10.1152/ajpregu.00766.2007
Kontos, C., Pavlou, M., and Giaginis, C. (2011). MCM5 (minichromosome maintenance complex component 5). Atlas Genet. Cytogenet. Oncol. Haematol. 14, 1045–1049. doi: 10.4267/2042/46072
Lee, P. G. (1995). Nutrition of cephalopods: fueling the system. Mar. Freshw. Behav. Physiol. 25, 35–51. doi: 10.1080/10236249409378906
Li, B., and Dewey, C. N. (2011). RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12, 323. doi: 10.1186/1471-2105-12-323
Liu, P. F., Liu, Q. H., Wu, Y., and Huang, J. (2017). Increased nucleoside diphosphate kinase activity induces white spot syndrome virus infection in Litopenaeus vannamei. PLoS ONE. 12, 1–14. doi: 10.1371/journal.pone.0175741
Liu, Y., Cai, D. X., Wang, L., Li, J. Z., and Wang, W. N. (2015). Glucosamine: fructose-6-phosphate amidotransferase in the white shrimp Litopenaeus vannamei: characterization and regulation under alkaline and cadmium stress. Ecotoxicology. 24, 1754–1764. doi: 10.1007/s10646-015-1480-2
Loewen, C. J. R., Gazpar, M. L., Jesch, S. A., Delon, C., Ktistakis, N. T., Henry, S. A., et al. (2004). Phospholipid metabolism regulated by a transcription factor sensing phosphatidic acid. Science. 304, 1644–1647. doi: 10.1126/science.1096083
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Matsubara, K., Morii, S., Abe, M., et al. (2016). Early feeding by cultured paralarvae of Octopus sinensis d'Orbigny 1841: Comparison of survival, and fatty acid and amino acid profiles, using two species of Artemia. Asian Fish. Sci. 29, 57–64. doi: 10.33997/j.afs.2016.29.1.005
Monroig, O., Tocher, D. R., and Castro, L. F. C. (2018). Polyunsaturated fatty acid biosynthesis and metabolism in fish. In: Polyunsaturated Fatty Acid Metabolism, ed. G. C. Burdge (AOCS Press) p. 31–60. doi: 10.1016/B978-0-12-811230-4.00003-X
Morales, A. E., Cardenete, G., Hidalgo, M. C., Garrido, D., Martín, M. V., and Almansa, E. (2017). Time course of metabolic capacities in paralarvae of the common octopus, Octopus vulgaris, in the first stages of life. Searching biomarkers of nutritional imbalance. Front. Physiol. 8, 1–8. doi: 10.3389/fphys.2017.00427
Myers, K. R., Yu, K., Kremerskothen, J., Butt, E., and Zheng, J. Q. (2020). The nebulin family LIM and SH3 proteins regulate postsynaptic development and function. J. Neurosci. 40, 526–541. doi: 10.1523/JNEUROSCI.0334-19.2019
Nande, M., Miranda, D., Gestal, G., Otero, J. J., Santamaría, F. J., and Almansa, E. (2016). Effect of hypothermia on sedation and recovery in the paralavae of Octopus vulgaris. In: Aquaculture Europe 2016. Edinburgh, Scotland 20-23 September, 2016.
Navarro, J. C., Monroig, Ó., and Sykes, A. V. (2014). “Nutrition as a key factor for cephalopod aquaculture. In: Cephalopod Culture, eds. J. Iglesias, L. Fuentes, and R. Villanueva (Springer Netherlands) p. 77–95. doi: 10.1007/978-94-017-8648-5_5
Navarro, J. C., and Villanueva, R. (2000). Lipid and fatty acid composition of early stages of cephalopods: an approach to their lipid requirements. Aquaculture. 183, 161–177. doi: 10.1016/S0044-8486(99)00290-2
Navarro, J. C., and Villanueva, R. (2003). The fatty acid composition of Octopus vulgaris paralarvae reared with live and inert food: deviation from their natural fatty acid profile. Aquaculture. 219, 613–631. doi: 10.1016/S0044-8486(02)00311-3
Norman, M., Rivers, C., Lee, Y. B., Idris, J., and Uney, J. (2016). The increasing diversity of functions attributed to the SAFB family of RNA-/DNA-binding proteins. Biochem. J. 473, 4271–4288. doi: 10.1042/BCJ20160649
O'Dor, R. K., and Webber, D. M. (1986). The constraints on cephalopods: why squid aren't fish. Can. J. Zool. 64, 1591–1605. doi: 10.1139/z86-241
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucl. Acids Res. 29, 16–21. doi: 10.1093/nar/29.9.e45
Phillips, G. R., Anderson, T. R., Florens, L., Gudas, C., Magda, G., Yates, J. R., et al. (2004). Actin-binding proteins in a postsynaptic preparation: Lasp-1 is a component of central nervous system synapses and dendritic spines. J. Neurosci. Res. 78, 38–48. doi: 10.1002/jnr.20224
Phillips, M. C. (2018). Is ABCA1 a lipid transfer protein? J. Lipid Res. 59, 749–763. doi: 10.1194/jlr.R082313
Pollitt, S. L., Myers, K. R., Yoo, J., and Zheng, J. Q. (2020). LIM and SH3 protein 1 localizes to the leading edge of protruding lamellipodia and regulates axon development. Mol. Biol. Cell. 31, 2718–2732. doi: 10.1091/mbc.E20-06-0366
Reis, D. B., Acosta, N. G., Almansa, E., Garrido, D., Andrade, J. P., Sykes, A. V., et al. (2019). Effect of Artemia inherent fatty acid metabolism on the bioavailability of essential fatty acids for Octopus vulgaris paralarvae development. Aquaculture. 500, 264–271. doi: 10.1016/j.aquaculture.2018.10.021
Reis, D. B., García-Herrero, I., Riera, R., Felipe, B. C., Rodríguez, C., Sykes, A. V., et al. (2015). An insight on Octopus vulgaris paralarvae lipid requirements under rearing conditions. Aquac. Nutr. 21, 797–806. doi: 10.1111/anu.12205
Reis, D. B., Shcherbakova, A., Riera, R., Martín, V. M., Domingues, P., Andrade, J. P., et al. (2021). Effects of feeding with different live preys on the lipid composition, growth and survival of Octopus vulgaris paralarvae. Aquac. Res. 52, 105–116. doi: 10.1111/are.14873
Rodhouse, P. G. K., Pierce, G. J., Nichols, O. C., Sauer, W. H. H., Arkhipkin, A. I., Laptikhovsky, V. V., et al. (2014). Environmental effects on cephalopod population dynamics. In: Advances in Marine Biology Book Series, ed. E. A. G. Vidal (Elsevier) 99–233. doi: 10.1016/B978-0-12-800287-2.00002-0
Roo, J., Estefanell, J., Betancor, M. B., Izquierdo, M., Fernández-Palacios, H., and Socorro, J. (2017). Effects of supplementation of decapod zoea to Artemia basal diet on fatty acid composition and digestive gland histology in common octopus (Octopus vulgaris) paralarvae. Aquac. Res. 48, 633–645. doi: 10.1111/are.12910
Seibel, B. A., and Walsh, P. J. (2003). Biological impacts of deep-sea carbon dioxide injection inferred from indices of physiological performance. J. Exp. Biol. 206, 641–650. doi: 10.1242/jeb.00141
Seixas, P., Otero, A., Valente, L. M. P., Dias, J., and Rey-Méndez, M. (2010a). Growth and fatty acid composition of Octopus vulgaris paralarvae fed with enriched Artemia or co-fed with an inert diet. Aquac. Int. 18, 1121–1135. doi: 10.1007/s10499-010-9328-5
Seixas, P., Rey-Méndez, M., Valente, L. M. P., and Otero, A. (2010b). High DHA content in Artemia is ineffective to improve Octopus vulgaris paralarvae rearing. Aquaculture. 300, 156–162. doi: 10.1016/j.aquaculture.2009.12.021
Shi, X., Zhang, S., and Pang, Q. (2006). Vitellogenin is a novel player in defense reactions. Fish Shellfish Immunol. 20, 769–772. doi: 10.1016/j.fsi.2005.09.005
Snell, K. (1984). Enzymes of serine metabolism in normal and neoplastic rat tissues. Adv. Enzyme Regul. 22, 325–400. doi: 10.1016/0065-2571(84)90021-9
Song, Q., Young, K. B., Chu, G., Gulick, J., Gerst, M., Grupp, I. L., et al. (2004). Overexpression of phospholamban in slow-twitch skeletal muscle is associated with depressed contractile function and muscle remodeling. FASEB J. 18, 974–976. doi: 10.1096/fj.03-1058fje
Storey, K. B., and Storey, J. M. (1983). Carbohydrate metabolism in cephalopod molluscs. In: Metabolic Biochemistry and Molecular Biomechanics, ed. P. W. Hochachka (Elsevier) p. 91–136. doi: 10.1016/B978-0-12-751401-7.50010-7
Sun, C., and Zhang, S. (2015). Immune-relevant and antioxidant activities of vitellogenin and yolk proteins in fish. Nutrients. 7, 8818–8829. doi: 10.3390/nu7105432
Tur, R., Domingues, P., Almansa, E., Lago, M., García-Fernández, P., and Pérez, E. (2020). ES2714930. Procedimiento para el cultivo de paralarvas del pulpo común Octopus vulgaris. Instituto Español de Oceanografía.
Varó, I., Cardenete, G., Hontoria, F., Monroig, Ó., Iglesias, J., Otero, J. J. J., et al. (2017). Dietary effect on the proteome of the common octopus (Octopus vulgaris) paralarvae. Front. Physiol. 8, 309. doi: 10.3389/fphys.2017.00309
Vidal, E. A. G., Villanueva, R., Andrade, J. P., Gleadall, I. G., Iglesias, J., Koueta, N., et al. (2014). Cephalopod culture: Current status of main biological models and research priorities. In: Advances in Cephalopod Science: Biology, Ecology, Cultivation and Fisheries. Advances in Marine Biology Book Series, ed. E. A. G. Vidal (Elsevier), p. 1–98. doi: 10.1016/B978-0-12-800287-2.00001-9
Walther, T. C., and Farese, R. V. J. (2012). Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 81, 687–714. doi: 10.1146/annurev-biochem-061009-102430
Yang, Z., Zhao, X., Shang, W., Liu, Y., Ji, J. F., Liu, J. P., et al. (2021). Pyrroline-5-carboxylate synthase senses cellular stress and modulates metabolism by regulating mitochondrial respiration. Cell Death Differ. 28, 303–319. doi: 10.1038/s41418-020-0601-5
Keywords: Octopus vulgaris, paralarvae, diet, proteomic, transcriptomic, growth, nutrition
Citation: Varó I, Prado-Álvarez M, Ortea I, Morales AE, García-Fernández P, Domingues P, Tur R, Dios S and Gestal C (2022) Proteogenomic Study of the Effect of an Improved Mixed Diet of Live Preys on the Aquaculture of Octopus vulgaris Paralarvae. Front. Mar. Sci. 8:817701. doi: 10.3389/fmars.2021.817701
Received: 18 November 2021; Accepted: 20 December 2021;
Published: 17 February 2022.
Edited by:
Benjamin Costas, University of Porto, PortugalReviewed by:
Graziano Fiorito, Zoological Station Anton Dohrn, ItalyCopyright © 2022 Varó, Prado-Álvarez, Ortea, Morales, García-Fernández, Domingues, Tur, Dios and Gestal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sonia Dios, sdios@iim.csic.es; Camino Gestal, cgestal@iim.csic.es
†Present address: Pablo García-Fernández and Ricardo Tur, Pescanova Biomarine Center, O Grove, Spain
‡These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.