 Lijun Zheng1†
Lijun Zheng1†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 17 March 2023
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1128160
Introduction: Candida albicans is a prevalent opportunistic human fungal pathogen. However, there are currently very few antifungal treatments available. Inositol phosphoryl ceramide synthase is an essential and fungal-specific protein that also provides a novel and promising antifungal target. Aureobasidin A is a widely used inhibitor of inositol phosphoryl ceramide synthase, however the mechanism of resistance to aureobasidin A is largely unknown in pathogenic fungi.
Methods: Here we investigated how C. albicans adapted to low and high concentrations of aureobasidin A.
Results and discussions: We identified trisomy of chromosome 1 as the predominant mechanism of rapid adaptation. Resistance to aureobasidin A was unstable because of the inherent instability of aneuploids. Importantly, chromosome 1 trisomy simultaneously regulated genes which were associated with aureobasidin A resistance that are on this aneuploid chromosome as well as on other chromosomes. Furthermore, the pleiotropic effect of aneuploidy caused altered resistance not only to aureobasidin A but also to other antifungal drugs including caspofungin and 5-flucytosine. We posit aneuploidy provides a rapid and reversible mechanism of development of drug resistance and cross resistance in C. albicans.
In recent years, the increase in advanced transplant procedures, the use of immunosuppressive therapies and the pandemic spread of HIV has resulted in an increase in the population of immunocompromised patients (Benedict et al., 2019), and the frequency of opportunistic fungal infections, especially invasive mycoses, has also increased significantly (Pfaller and Diekema, 2004). The most prevalent fungal infections are aspergillosis and candidemia. Candida species are the fourth most common cause of nosocomial bloodstream infections in the United States (Pfaller and Diekema, 2007, 2010). Candida albicans is the predominant cause of candidiasis (Pappas et al., 2018).
Fungi are eukaryotes and share many similar metabolic pathways and essential cellular machinery with humans. These inherent similarities have severely limited the antifungal drugs to only four classes: echinocandins, azoles, polyenes and flucytosine (Fisher et al., 2022). Alarmingly, multi-drug resistance in C. glabrata and C. auris has been reported (Arendrup and Patterson, 2017). In this context, identifying novel antifungal targets and the development of new antifungal agents are urgently needed.
Sphingolipids are complex lipids that have a sphingoid base as a backbone, to which a fatty acid and variable head groups are attached. Sphingolipids are found on both the inner and outer membrane of eukaryotic cells. They are essential components of eukaryotic cell membranes and play a pivotal role in a variety of biological processes such as cell division, heat stress response, acid/alkaline tolerance, morphogenesis, signal transduction, endocytosis and apoptosis (Singh and Del Poeta, 2016; McEvoy et al., 2020). The major fungal sphingolipids are inositol phosphoryl-ceramides (IPCs) and glucosylceramide (GlcCer). IPCs are the product of inositol phosphoryl ceramide synthase (Ipc1). Ipc1 is an essential fungal-specific enzyme for fungal cell growth and has no mammalian homolog (Denny et al., 2006). Therefore, Ipc1 is an ideal potential antifungal target.
Aureobasidin A (AbA) is one of the most widely studied IPC inhibitors. AbA is a cyclic depsipeptide isolated from the fungus Aureobasidium pullulans R106 (Ikai et al., 1991). It is fungicidal. AbA strongly inhibits Ipc1 almost exclusively in yeasts, including some notorious pathogenic fungi such as C. albicans, and Cryptococcus neoformans (Tan and Tay, 2013; Teymuri et al., 2021). In yeasts, Ipc1 is encoded by the gene AUR1. The mechanisms of resistance to AbA have been best studied in the model yeast Saccharomyces cerevisiae. In most cases, resistance to AbA is caused by mutation or overexpressoin of AUR1 (Heidler and Radding, 1995; Hashida-Okado et al., 1996). Overexpression of PDR16 also confers resistance to AbA putatively via reducing the effectiveness of AbA against Ipc1 (Katsuki et al., 2018). In addition to AUR1, deletion of other genes encoding sphingolipid-metabolizing enzymes also causes AbA resistance (Oh et al., 1997; Schorling et al., 2001; Tani and Kuge, 2010). AbA is likely a substrate of the ATP-binding cassette (ABC) transporters in S. cerevisiae. Overexpression of YOR1, which encodes plasma membrane ABC transporter, confers resistance to AbA (Ogawa et al., 1998). However, reports of genetic mechanisms of resistance to AbA in pathogenic fungi are still lacking.
In this study, we investigated genetic mechanisms of adaptation to both sub-inhibitory and lethal amount of AbA in C. albicans. We found formation of Chromosome 1 trisomy (Chr1 × 3) was the predominant adaptation strategy. In the absence of stress, Chr1 × 3 mutants were unstable and spontaneously reverted to euploids with resistance to AbA concomitantly lost. We found C. albicans PDR16 and AUR1 genes were haploinsufficient. Heterozygous deletions of PDR16 and AUR1 caused hypersensitivity to AbA. Chr1 × 3 simultaneously upregulated expression of genes on Chr1 including PDR16, as well as genes on other chromosomes including AUR1. Furthermore, Chr1 × 3 conferred pleiotropic effects on susceptibility to other antifungals including decreased resistance to caspofungin and increased resistance to 5-flucytosine. Therefore, we posit Chr1 × 3 provides a rapid mechanism of reversible adaptation to AbA via simultaneously upregulating genes on and outside of the aneuploid chromosome.
Strains used in this study are listed in Supplementary Table 1. Primers are listed in Supplementary Table 2. Drugs were dissolved in dimethyl sulfoxide (DMSO) and stored at –20°C.
C. albicans lab strain SC5314 was used as wild type in this study. Stock culture was preserved in 25% glycerol and maintained at –80°C. Cells were grown in Yeast extract-Peptone-Dextrose (YPD) media (1% [w/v] yeast extract, 2% [w/v] peptone and 2% [w/v] D-glucose) at 37°C in a shaking incubator at 150–200 rpm. For solid medium, 2% [w/v] agar was added. For the selection of gene knockout strains, YPD agar containing 400 μg/ml nourseothricin (Werner BioAgents) medium was used (YPD + NAT).
Approximately 2.5 × 103 cells/ml of SC5314 in 150 μl YPD with or without AbA were incubated in a 96 well plate at 37°C. OD595 was monitored in a Tecan plate reader (Infinite F200 PRO, Tecan, Switzerland) at 15 min time intervals for 24 h. Data are represented as the mean ± SD of three biological replicates.
Cells were suspended in distilled water and adjusted to 1 × 107 cells/ml. 3 μl of 10-fold serial dilutions were spotted on YPD plates with or without drugs (control) at 37°C and photographed after 2 days.
Cells were suspended in distilled water and adjusted to 1 × 107 cells/ml. 100 μl of cell suspension were on YPD plates supplemented with 20 ng/ml AbA. The plates were incubated at 37°C for 3 days. 18 mutants were randomly chosen. The mutants were streaked onto YPD plates and incubated at 37°C for 36 h. For each mutant, 4–6 colonies of similar size were selected and frozen in 1 ml of 25% glycerol at –80°C.
Approximately 1 × 103 cells/ml of SC5314 was grown in YPD broth (control) or YPD broth supplemented with 5 ng/ml AbA (test). After 24 h of shaking at 37°C, 10 μL of the culture was transferred to 1 ml YPD broth containing the same AbA concentration. After 24 h, the culture was centrifuged at 3,000 rpm for 1 min, and resuspended in distilled water. Then it was diluted with distilled water. Approximately 200 cells were spread onto YPD plates. The plates were incubated at 37°C for 24 h. Randomly 40 colonies from each plate were tested for resistance to AbA.
Gene deletions were constructed using the NAT1 flipper cassette as described previously (Yang et al., 2019). Briefly, approximately 500 bp of upstream and downstream region of the target gene was amplified using SC5314 genomic DNA as template. The 3’ end of the upstream forward primers overlapped with the 5’ end of the NAT1 flipper; the 5’ end of the downstream reverse primers overlapped with the 3’ end of the NAT1 flipper. The amplicon and plasmid pJK863 were used as templates to fuse the upstream region to the 5’ region of the cassette, and the downstream region to the 3’ region of the cassette. The fusion products were transformed into C. albicans following the lithium acetate method (Wilson et al., 2000), and transformants were selected on YPD plates supplemented with 400 μg/ml nourseothricin (NAT). Positive transformants were confirmed by diagnostic PCRs with primers that annealed outside the flanking homology regions. All primer sequences are listed in Supplementary Table 2.
Test strains were grown on YPD-agar plates. Randomly 25–30 colonies with similar sizes were collected. Total DNA extraction and genomic DNA library preparation were performed as described previously (Yang et al., 2021d). The final libraries were sequenced by BGISEQ-500. Raw fastq files were uploaded to YMAP (version 1.0)1 (Abbey et al., 2014). Read depth was plotted as a function of chromosome position using the Assembly 22 version of the SC5314 reference genome.2
RNA-seq was performed as described previously (Yang et al., 2021b). Strains were streaked onto YPD plates from the –80°C freezer. After 36 h incubation at 37°C, several colonies of similar sizes were chosen. Colonies were suspended in distilled water and adjusted to 1 × 104 cells/ml. 100 μl of cell suspension were spread on YPD plates. The plates were incubated at 37°C for 36 h. Cells were collected by centrifugation, washed and flash frozen in liquid nitrogen. Total RNA extraction and purification, library construction, and sequencing were performed as described in Yang et al. (2013).
Susceptibility of the C. albicans lab strain SC5314 to AbA was measured in YPD broth and on YPD agar supplemented with AbA. In the liquid medium, growth in the presence of 5 ng/ml or 10 ng/ml of AbA did not significantly inhibit growth compared to the in YPD broth without AbA [p > 0.05, one-way ANOVA (ANalysis Of VAriance) with post hoc Tukey HSD (Honestly Significant Difference)]. 20 ng/ml of AbA completely inhibited growth (Figure 1A). On the plates, growth was also inhibited by 20 ng/ml of AbA (Figure 1B). Therefore, we chose 5 ng/ml and 20 ng/ml of AbA as sub-inhibitory and inhibitory concentrations, respectively, for further studies.
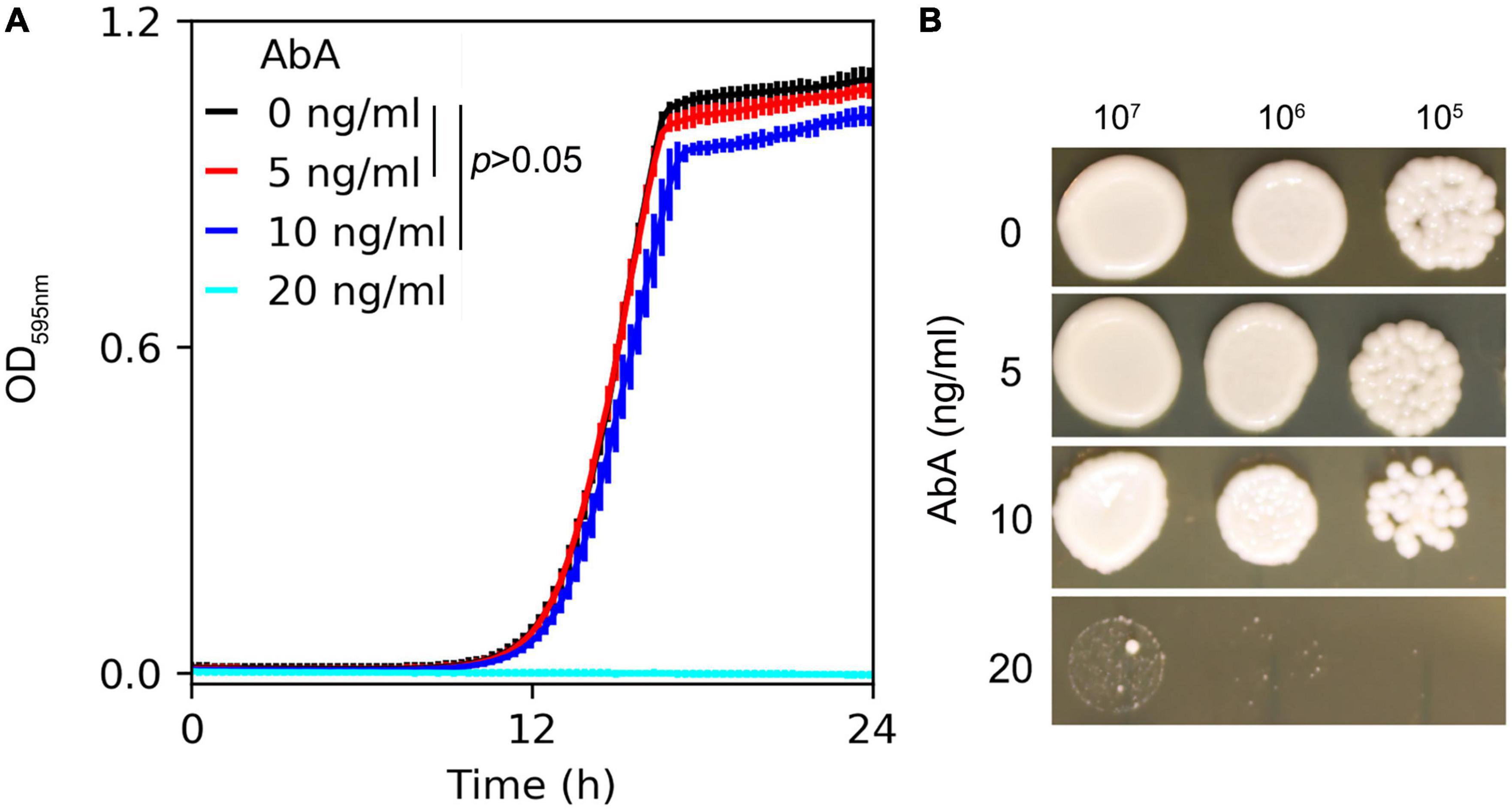
Figure 1. Susceptibility of C. albicans to aureobasidin A. C. albicans lab strain SC5314 was grown in YPD broth (A) or on YPD plates (B) supplemented with aureobasidin A. In panel (A) optical density at 595 nm (OD595) was measured every 15 min for 24 h at 37°C using a Tecan plate reader (Infinite F200 PRO, Tecan, Switzerland). Data are represented as the mean ± SD of three biological repeats. In panel (B), 3 μl of 10-fold serial dilutions were spotted on YPD plates. Drug concentrations are shown in the figure. The plates were incubated at 37°C for 48 h then photographed.
Approximately 1 million cells of SC5314 were spread on YPD plate supplemented with 20 ng/ml AbA. After 3 days of incubation at 37°C, randomly 18 colonies (mutants) were chosen (Figure 2A). Spot assay indicated only 2 (#1 and #3) of the 18 mutants (#1-#18) grew better than the parent in the presence of AbA (Figure 2B). However, when grown on YPD plates in the absence of AbA, these 2 mutants were unstable, exhibiting mostly small sized colonies (indicated by cyan arrows) and a few large colonies (indicated by magenta arrows) (Figure 2C). When tested for resistance to AbA, only the small colonies were resistant. The large colonies were not (Figure 2D). Whole genome sequencing indicated in both mutants the small colonies were Chr1 × 3 and both had the B homolog duplicated (ABB), and the large colonies were euploid (Figure 2E). Therefore, exposure to lethal amount of AbA selected for resistant mutants which were Chr1 × 3. The resistance was unstable. Loss of aneuploidy caused concomitant loss of AbA resistance.
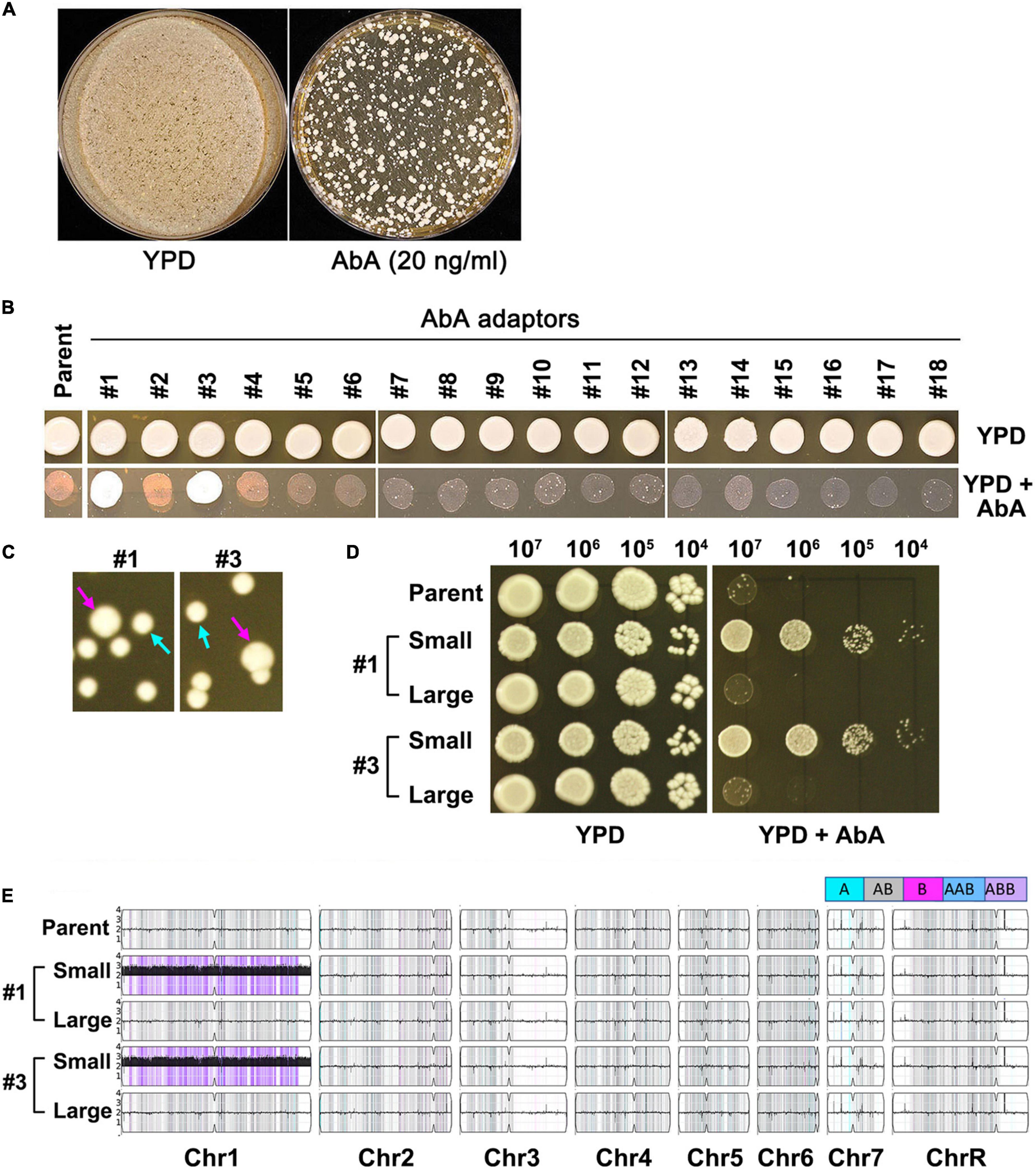
Figure 2. Lethal amount of aureobasidin A selects unstable aneuploid mutants. Approximately 1 million cells of SC5314 were spread on YPD plate supplemented with 20 ng/ml aureobasidin A or without drug (control). The plates were incubated at 37°C for 3 days (A). Randomly 18 mutants were chosen. Spot assay was performed to compare level of resistance between mutants and the parent (B). Two resistant mutants (#1 and #3) were spread on YPD plates. Cyan arrows indicate small colonies and magenta arrows indicate large colonies (C). Both small and large colonies were tested for resistance to aureobasidin A (D) and were sequenced (E). The karyotypes were generated using Ymap (Abbey et al., 2014). Allele frequencies are color coded: homolog “a” is cyan, homolog “b” is magenta and heterozygous alleles are gray.
We asked if short term growth in sub-inhibitory AbA was sufficient to select for resistant mutants. SC5314 was grown in YPD broth or YPD broth supplemented with 5 ng/ml AbA. After 48 h, the cultures were washed, diluted and plated onto YPD. Surprisingly, the colonies (mutants) from the AbA-evolved culture showed obvious size variations: 117 mutants were large (indicated by magenta arrows) and 38 mutants were small (indicated by cyan arrows). In contrast, mutants from the YPD-evolved culture did not show colony size variations and the size was similar to the large mutants from the AbA-evolved culture (Figure 3A).
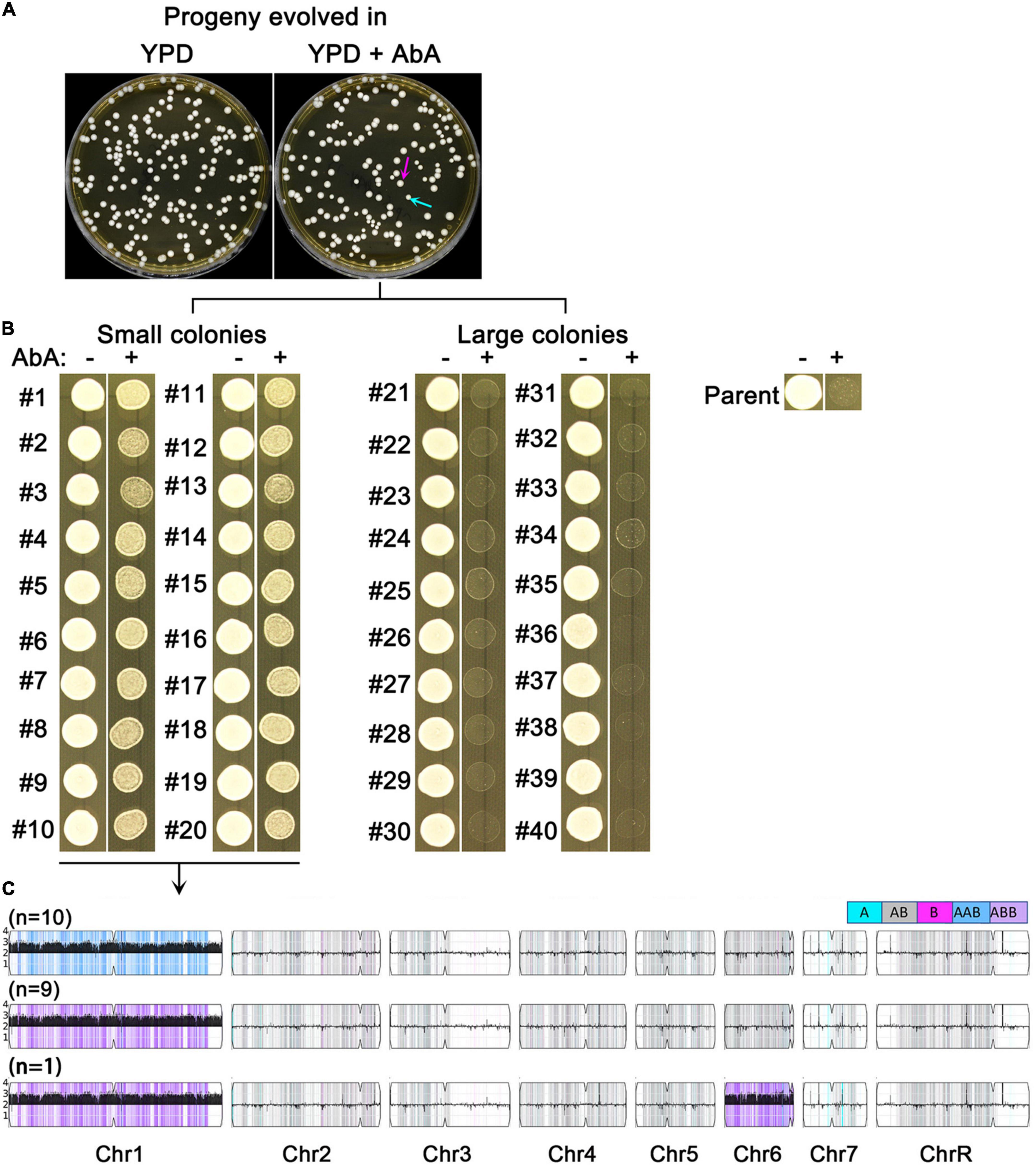
Figure 3. Sub-inhibitory amount of aureobasidin A selects aneuploid mutants. SC5314 was grown in YPD broth supplemented with or without 5 ng/ml aureobasidin A. After 48 h, the cultures were washed and diluted using distilled water. Approximately 200 cells were spread on YPD. The plates were photographed after 24 h incubation at 37°C. Cyan arrow indicates small colonies and magenta arrow indicates large colonies (A). Randomly 20 small and 20 large colonies from the drug evolved culture were tested for resistance to aureobasidin A (B), and all the 20 resistant mutants were sequenced (C).
Randomly, 40 mutants of the YPD-evolved progeny were tested by spot assay for resistance to AbA. None of them gained resistance (data not shown). Randomly 20 small mutants and 20 large mutants from the AbA-evolved progeny were tested. All the 20 small mutants (#1-#20) were more resistant than the parent to AbA. None of the large mutants (#21 – #40) were resistant (Figure 3B).
All the 20 resistant mutants were sequenced, and all had Chr1 × 3: 19 had Chr1 × 3 only, and 1 mutant had Chr1 × 3 + Chr6 × 3. Among them, 10 had AAB and 10 had ABB of Chr1. Therefore, there was no homolog bias in the Chr1 × 3 mutants (Figure 3C).
Thus, 48 h exposure to sub-inhibitory AbA was sufficient to select for resistant mutants. The resistance was due to amplification of Chr1. There was no biased homolog duplication in the Chr1 × 3 mutants.
In the C. albicans genome, PDR16 is on Chr1 and AUR1 resides on Chr5. We asked why Chr1 × 3 caused resistance to AbA. The transcriptome of one Chr1 × 3 mutant was compared to the wild type strain. The expression of genes on Chr1 were generally up-regulated in the Chr1 × 3 mutant, including PDR16 (Figure 4A and Supplementary Table 3). Chr1 × 3 also up-regulated genes on other chromosomes, including AUR1 (Figure 4A and Supplementary Table 3). We found PDR16 and AUR1 were haploinsufficient. Deletion of one allele of PDR16 or AUR1 in SC5314 caused hypersensitivity to AbA. Deletion of one allele of PDR16 or AUR1 in a Chr1 × 3 mutant also caused loss of AbA resistance (Figure 4B). Therefore, Chr1 × 3 caused resistance to AbA via simultaneously up-regulating genes on the aneuploid chromosome (such as PDR16) as well as genes on euploid chromosomes (such as AUR1) which were associated with AbA resistance.
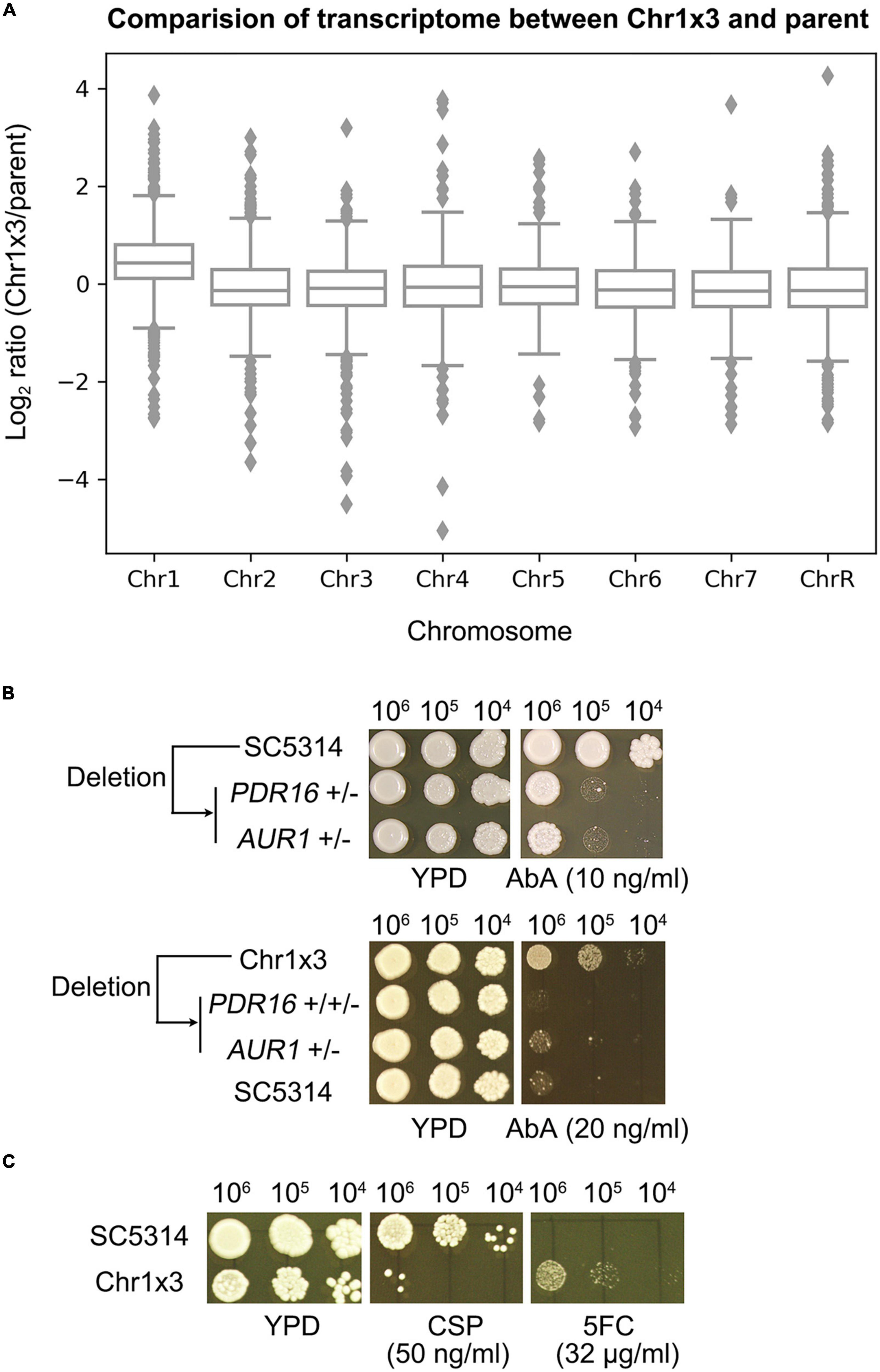
Figure 4. Chromosome 1 trisomy has pleiotropic effect on whole genome transcription and antifungal resistance profile. Transcriptome of one Chr1 trisomy mutant was compared to the diploid wild type strain. Cells were grown on YPD plates at 37°C. Log2 ratio of genes expression levels were plotted as a function of chromosome position (A). Strains with deletions of one allele of PDR16 or AUR1 were compared to wild type diploid strain parent SC5314, or a Chr1 × 3 parent, for resistance to aureobasidin A (Ab) (B). Pleiotropic effect of Chr1 trisomy on antifungal resistance was tested by spot assay on YPD plates supplemented with caspofungin (CSP) or 5-flucytosine (5FC) (C). In panels (B,C), the plates were incubated at 37°C for 48 h then photographed.
Furthermore, we asked if Chr1 × 3 had pleiotropic effects on the aneuploid cells. Susceptibility of Chr1 × 3 mutant was compared to parent by spot assay. Chr1 × 3 caused hypersensitivity to caspofungin (CSP). GSC1 and GSL1, which encode the target protein β-1,3-glucan synthase of CSP, are on Chr1. RNA-seq indicated expression of GSC1 was compensated to diploid level. GSL1 was significantly up-regulated in Chr1 × 3, however, expression of GSL2, which also encodes a subunit of the β-1,3-glucan synthase, was significantly down-regulated (Supplementary Table 3). Deletion of the PKC pathway kinase genes MKK2 and MKC1, and deletion of CMP1 and CNB1, which encode subunits of calcineurin, cause hypersensitivity to CSP (Yang et al., 2021b). Here we found expression of MKK2 and MKC1 were significantly down-regulated in Chr1 × 3, but expression of CMP1 or CNB1 was not significantly different (Supplementary Table 3). In addition to altered susceptibility to CSP, Chr1 × 3 also conferred resistance to 5-flucytosine (5FC) (Figure 4C). RNA-seq indicated FCA1 gene was significantly down-regulated in Chr1 × 3, other genes associated with 5FC resistance, such as FUR1, FCY2, FCY21, were not differentially expressed (Supplementary Table 3).
Gene copy number variation, mainly via aneuploidy, is a prevalent mechanism of rapid adaptation to stresses in C. albicans (reviewed in Tsai and Nelliat, 2019). Although C. albicans can exist as a haploid (Hickman et al., 2013), most C. albicans strains are diploids with 8 pairs of chromosomes (Jones et al., 2004). C. albicans is tolerant to aneuploidy. Each chromosome can be trisomic, but specific aneuploidy is always selected for by particular stressors and usually causes resistance to that stress (Yang et al., 2021d). Furthermore, at least in yeasts, aneuploidy typically causes proportional alterations of gene transcription and in most cases translation, therefore, aneuploidy has the potential of causing cross-resistance to unrelated stresses (Yang et al., 2013, 2019, 2021a,b,c).
In this study, we found Chr1 × 3 formation was the predominant mechanism of rapid adaptation to both sub-inhibitory and lethal amount of AbA in C. albicans. Genetic mutations of AUR1 and PDR16, which are known genes associated with resistance to AbA, were not detected in the mutants. The loss of the extra copy of Chr1 was accompanied by loss of AbA resistance. Therefore, copy number of Chr1 was the mechanism of altered AbA resistance. We also found PDR16 and AUR1 genes were dosage-sensitive genes. Deletion of one allele was sufficient to cause hypersensitivity to AbA. In the mutants, PDR16 is on the aneuploid chromosome and AUR1 is on euploid chromosome. Interestingly, Chr1 × 3 simultaneously up regulated both PDR16 and AUR1. Therefore, Chr1 × 3 had pleiotropic effects on the whole genome transcription and phenotypes. As a result, we found Chr1 × 3 not only caused AbA resistance, it also conferred resistance to 5FC and hypersensitivity to CSP.
In the C. albicans genome, three genes encode subunits of the target protein β-1,3-glucan synthase of CSP: GSC1, GSL1 and GSL2. GSC1 and GSL1 are on Chr1 and GSL2 is on ChrR. Compared to the euploid parent, in Chr1 × 3 mutant, transcription of GSL1 was up regulated and transcription of GSL2 was down regulated. Imbalanced expression level of genes encoding subunits of protein complex always leads to misassembly of the complex (Cardarelli et al., 2011). This mechanism might explain why Chr1 × 3 mutant was hypersensitive to CSP.
5FC is a prodrug. Upon uptake into fungal cells via the cytosine permease (encoded by FCY2 and FCY21), it is converted into toxic 5-fluorouracil (5FU) by cytosine deaminase (encoded by FCA1). 5FU is then further processed by uracil phosphoribosyltransferase (encoded by FUR1), and the product inhibits both DNA and protein synthesis. Loss of function mutations of genes involved in the uptake and intracellular metabolism of 5FC have been associated with resistance in clinical Candida (Hope et al., 2004; Papon et al., 2007) and Cryptococcus isolates (Whelan, 1987; Billmyre et al., 2020; Chang et al., 2021). Here we found FCA1 was down regulated in Chr1 × 3 mutant. FUR1, FCY2 and FCY21 were not differentially expressed. Therefore, decreased expression of FCA1 might be the cause of resistance to 5FC.
In summary, this study indicates aneuploidy is the major mechanism of rapid adaptation to AbA in C. albicans. Aneuploidy directly regulates genes on the aneuploidy chromosome and indirectly regulates genes on the euploid chromosome. The pleiotropic effect of aneuploidy has the potential of causing cross resistance to antifungal drugs.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/arrayexpress/, E-MTAB-8942; https://www.ebi.ac.uk/arrayexpress/, E-MTAB-12400; and https://www.ebi.ac.uk/arrayexpress/, E-MTAB-12403.
FY and LG analyzed the data. FY wrote the manuscript. LZ and YX carried out the research. YD, XM, and CW helped to develop the experimental idea and design. YX and LG funded the experiments. All authors approved the submitted version.
This study was supported by the Science and Technology Development Plan of Suzhou (SLJ2022018), Scientific Research Project of Suzhou Commission of Health (GSWS2020028) to LG, and the National Natural Science Foundation of China (81402978) to YX.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YJ declared a shared affiliation with the author FY to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1128160/full#supplementary-material
Abbey, D. A., Funt, J., Lurie-Weinberger, M. N., Thompson, D. A., Regev, A., Myers, C. L., et al. (2014). YMAP: A pipeline for visualization of copy number variation and loss of heterozygosity in eukaryotic pathogens. Genome Med. 6:100. doi: 10.1186/PREACCEPT-1207699561372700
Arendrup, M. C., and Patterson, T. F. (2017). Multidrug-resistant Candida: Epidemiology, molecular mechanisms, and treatment. J. Infect. Dis. 216, S445–S451. doi: 10.1093/infdis/jix131
Benedict, K., Jackson, B. R., Chiller, T., and Beer, K. D. (2019). Estimation of direct healthcare costs of fungal diseases in the United States. Clin. Infect. Dis. 68, 1791–1797. doi: 10.1093/cid/ciy776
Billmyre, R. B., Applen Clancey, S., Li, L. X., Doering, T. L., and Heitman, J. (2020). 5-fluorocytosine resistance is associated with hypermutation and alterations in capsule biosynthesis in Cryptococcus. Nat. Commun. 11:127. doi: 10.1038/s41467-019-13890-z
Cardarelli, L., Maxwell, K. L., and Davidson, A. R. (2011). Assembly mechanism is the key determinant of the dosage sensitivity of a phage structural protein. Proc. Natl. Acad. Sci. U.S.A. 108, 10168–10173. doi: 10.1073/pnas.1100759108
Chang, Y. C., Lamichhane, A. K., Cai, H., Walter, P. J., Bennett, J. E., and Kwon-Chung, K. J. (2021). Moderate levels of 5-fluorocytosine cause the emergence of high frequency resistance in Cryptococci. Nat. Commun. 12:3418. doi: 10.1038/s41467-021-23745-1
Denny, P. W., Shams-Eldin, H., Price, H. P., Smith, D. F., and Schwarz, R. T. (2006). The protozoan inositol phosphorylceramide synthase: A novel drug target that defines a new class of sphingolipid synthase. J. Biol. Chem. 281, 28200–28209. doi: 10.1074/jbc.M600796200
Fisher, M. C., Alastruey-Izquierdo, A., Berman, J., Bicanic, T., Bignell, E. M., Bowyer, P., et al. (2022). Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol. 20, 557–571. doi: 10.1038/s41579-022-00720-1
Hashida-Okado, T., Ogawa, A., Endo, M., Yasumoto, R., Takesako, K., and Kato, I. (1996). AUR1, a novel gene conferring aureobasidin resistance on Saccharomyces cerevisiae: A study of defective morphologies in Aur1p-depleted cells. Mol. Gen. Genet. 251, 236–244. doi: 10.1007/BF02172923
Heidler, S. A., and Radding, J. A. (1995). The AUR1 gene in Saccharomyces cerevisiae encodes dominant resistance to the antifungal agent aureobasidin A (LY295337). Antimicrob. Agents Chemother. 39, 2765–2769. doi: 10.1128/AAC.39.12.2765
Hickman, M. A., Zeng, G., Forche, A., Hirakawa, M. P., Abbey, D., Harrison, B. D., et al. (2013). The ‘obligate diploid’ Candida albicans forms mating-competent haploids. Nature 494, 55–59. doi: 10.1038/nature11865
Hope, W. W., Tabernero, L., Denning, D. W., and Anderson, M. J. (2004). Molecular mechanisms of primary resistance to flucytosine in Candida albicans. Antimicrob. Agents Chemother. 48, 4377–4386. doi: 10.1128/AAC.48.11.4377-4386.2004
Ikai, K., Takesako, K., Shiomi, K., Moriguchi, M., Umeda, Y., Yamamoto, J., et al. (1991). Structure of aureobasidin A. J. Antibiot. (Tokyo) 44, 925–933. doi: 10.7164/antibiotics.44.925
Jones, T., Federspiel, N. A., Chibana, H., Dungan, J., Kalman, S., Magee, B. B., et al. (2004). The diploid genome sequence of Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 101, 7329–7334. doi: 10.1073/pnas.0401648101
Katsuki, Y., Yamaguchi, Y., and Tani, M. (2018). Overexpression of PDR16 confers resistance to complex sphingolipid biosynthesis inhibitor aureobasidin A in yeast Saccharomyces cerevisiae. FEMS Microbiol. Lett. 365:fnx255. doi: 10.1093/femsle/fnx255
McEvoy, K., Normile, T. G., and Poeta, M. D. (2020). Antifungal drug development: Targeting the fungal Sphingolipid pathway. J. Fungi (Basel) 6:142. doi: 10.3390/jof6030142
Ogawa, A., Hashida-Okado, T., Endo, M., Yoshioka, H., Tsuruo, T., Takesako, K., et al. (1998). Role of ABC transporters in aureobasidin A resistance. Antimicrob. Agents Chemother. 42, 755–761. doi: 10.1128/AAC.42.4.755
Oh, C. S., Toke, D. A., Mandala, S., and Martin, C. E. (1997). ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J. Biol. Chem. 272, 17376–17384. doi: 10.1074/jbc.272.28.17376
Papon, N., Noel, T., Florent, M., Gibot-Leclerc, S., Jean, D., Chastin, C., et al. (2007). Molecular mechanism of flucytosine resistance in Candida lusitaniae: Contribution of the FCY2, FCY1, and FUR1 genes to 5-fluorouracil and fluconazole cross-resistance. Antimicrob. Agents Chemother. 51, 369–371. doi: 10.1128/AAC.00824-06
Pappas, P. G., Lionakis, M. S., Arendrup, M. C., Ostrosky-Zeichner, L., and Kullberg, B. J. (2018). Invasive candidiasis. Nat. Rev. Dis. Prim. 4:18026. doi: 10.1038/nrdp.2018.26
Pfaller, M. A., and Diekema, D. J. (2004). Rare and emerging opportunistic fungal pathogens: Concern for resistance beyond Candida albicans and Aspergillus fumigatus. J. Clin. Microbiol. 42, 4419–4431. doi: 10.1128/JCM.42.10.4419-4431.2004
Pfaller, M. A., and Diekema, D. J. (2007). Epidemiology of invasive candidiasis: A persistent public health problem. Clin. Microbiol. Rev. 20, 133–163. doi: 10.1128/CMR.00029-06
Pfaller, M. A., and Diekema, D. J. (2010). Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 36, 1–53. doi: 10.3109/10408410903241444
Schorling, S., Vallee, B., Barz, W. P., Riezman, H., and Oesterhelt, D. (2001). Lag1p and Lac1p are essential for the Acyl-CoA-dependent ceramide synthase reaction in Saccharomyces cerevisae. Mol. Biol. Cell 12, 3417–3427. doi: 10.1091/mbc.12.11.3417
Singh, A., and Del Poeta, M. (2016). Sphingolipidomics: An important mechanistic tool for studying fungal pathogens. Front. Microbiol. 7:501. doi: 10.3389/fmicb.2016.00501
Tan, H. W., and Tay, S. T. (2013). The inhibitory effects of aureobasidin A on Candida planktonic and biofilm cells. Mycoses 56, 150–156. doi: 10.1111/j.1439-0507.2012.02225.x
Tani, M., and Kuge, O. (2010). Defect of synthesis of very long-chain fatty acids confers resistance to growth inhibition by inositol phosphorylceramide synthase repression in yeast Saccharomyces cerevisiae. J. Biochem. 148, 565–571. doi: 10.1093/jb/mvq090
Teymuri, M., Shams-Ghahfarokhi, M., and Razzaghi-Abyaneh, M. (2021). Inhibitory effects and mechanism of antifungal action of the natural cyclic depsipeptide, aureobasidin A against Cryptococcus neoformans. Bioorg. Med. Chem. Lett. 41:128013. doi: 10.1016/j.bmcl.2021.128013
Tsai, H. J., and Nelliat, A. (2019). A Double-Edged Sword: Aneuploidy is a prevalent strategy in fungal adaptation. Genes (Basel) 10:787. doi: 10.3390/genes10100787
Whelan, W. L. (1987). The genetic basis of resistance to 5-fluorocytosine in Candida species and Cryptococcus neoformans. Crit. Rev. Microbiol. 15, 45–56. doi: 10.3109/10408418709104447
Wilson, R. B., Davis, D., Enloe, B. M., and Mitchell, A. P. (2000). A recyclable Candida albicans URA3 cassette for PCR product-directed gene disruptions. Yeast 16, 65–70. doi: 10.1002/(SICI)1097-0061(20000115)16:1<65::AID-YEA508>3.0.CO;2-M
Yang, F., Todd, R. T., Selmecki, A., Jiang, Y.-Y., Cao, Y.-B., and Berman, J. (2021d). The fitness costs and benefits of trisomy of each Candida albicans chromosome. Genetics 218:iyab056. doi: 10.1093/genetics/iyab056
Yang, F., Gritsenko, V., Slor Futterman, Y., Gao, L., Zhen, C., Lu, H., et al. (2021b). Tunicamycin potentiates antifungal drug tolerance via aneuploidy in Candida albicans. mBio 12:e0227221. doi: 10.1128/mBio.02272-21
Yang, F., Gritsenko, V., Lu, H., Zhen, C., Gao, L., Berman, J., et al. (2021a). Adaptation to fluconazole via aneuploidy enables cross-adaptation to amphotericin b and flucytosine in Cryptococcus neoformans. Microbiol. Spectr. 9:e0072321. doi: 10.1128/Spectrum.00723-21
Yang, F., Lu, H., Wu, H., Fang, T., Berman, J., and Jiang, Y. Y. (2021c). Aneuploidy underlies tolerance and cross-tolerance to drugs in Candida parapsilosis. Microbiol. Spectr. 9:e0050821. doi: 10.1128/Spectrum.00508-21
Yang, F., Kravets, A., Bethlendy, G., Welle, S., and Rustchenko, E. (2013). Chromosome 5 monosomy of Candida albicans controls susceptibility to various toxic agents, including major antifungals. Antimicrob. Agents Chemother. 57, 5026–5036. doi: 10.1128/AAC.00516-13
Keywords: Candida albicans, aureobasidin A, sphingolipid biosynthesis, aneuploidy, antifungal drugs
Citation: Zheng L, Xu Y, Dong Y, Ma X, Wang C, Yang F and Guo L (2023) Chromosome 1 trisomy confers resistance to aureobasidin A in Candida albicans. Front. Microbiol. 14:1128160. doi: 10.3389/fmicb.2023.1128160
Received: 20 December 2022; Accepted: 06 March 2023;
Published: 17 March 2023.
Edited by:
Rustam Aminov, University of Aberdeen, United KingdomReviewed by:
Koichi Tanabe, Ryukoku University, JapanCopyright © 2023 Zheng, Xu, Dong, Ma, Wang, Yang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liangsheng Guo, gls2135@sina.com
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.