 Yawen Liu
Yawen Liu- 1Department of Gastroenterology, Affiliated Hospital of Jiangsu University, Jiangsu University, Zhenjiang, China
- 2Laboratory of Cellular Metabolism and Metaboli Regulation, VIB-KU Leuven Center for Cancer Biology, VIB, Leuven, Belgium
- 3Laboratory of Cellular Metabolism and Metabolic Regulation, Department of Oncology, KU Leuven and Leuven Cancer Institute (LKI), Leuven, Belgium
Malignant growth is defined by multiple aberrant cellular features, including metabolic rewiring, inactivation of tumor suppressors and the activation of oncogenes. Even though these features have been described as separate hallmarks, many studies have shown an extensive mutual regulatory relationship amongst them. On one hand, the change in expression or activity of tumor suppressors and oncogenes has extensive direct and indirect effects on cellular metabolism, activating metabolic pathways required for malignant growth. On the other hand, the tumor microenvironment and tumor intrinsic metabolic alterations result in changes in intracellular metabolite levels, which directly modulate the protein modification of oncogenes and tumor suppressors at both epigenetic and post-translational levels. In this mini-review, we summarize the crosstalk between tumor suppressors/oncogenes and metabolism-induced protein modifications at both levels and explore the impact of metabolic (micro)environments in shaping these.
Introduction
Cancer initiation and progression are characterized by a series of genetic and epigenetic alterations (1). As such, mutations inducing a gain-of-function in oncogenes or a loss-of-function in tumor suppressor genes (TSGs) are able to adjust the cell cycle boundaries and transform a mammalian cell into its malignant derivative (2, 3). As a result, the cells maintain continuous growth thanks to aberrant oncogenic signaling and the inactivation of cell cycle suppressors (4, 5).
To fulfill the energy and bio-mass demands of continuous proliferation, malignant cells acquire the capacity to rewire their metabolism (6). During this reprogramming, tumors rely on specific metabolic pathways depending on extrinsic factors like the site of tumor growth, as well as intracellular signals (7). Previously, mutations in oncogenes and TSGs have been linked to an altered metabolic phenotype, with the Warburg effect – the preference for glycolysis even under sufficient oxygen supply – as an example of metabolic reprogramming regulated by oncogenic signaling (8–12). Nevertheless, while oncogenic signaling reprograms cellular metabolism, also metabolites and metabolic enzymes themselves are able to modulate the activity of oncogenic proteins in a bi-directional feedback loop (13).
One way by which the function of an (onco)protein can be affected by metabolites is through protein modifications, a process in which a small chemical substrate (such as lactyl-CoA, palmitoyl-CoA, acetyl-CoA, succinyl-CoA) is covalently bound to a protein of interest (14). Depending on the type and dynamics of the modification, this changes the transcription of oncogenes or their protein stability, interaction, localization and overall activity (14). Many of the attached substrates are derived from central carbon metabolites, hereby linking the metabolic phenotype to downstream protein modifications (13). Consequently, changes in the metabolism and its derived metabolites can alter the modifications during cancer progression and thus the downstream regulation of the protein signaling cascade.
A tumor does not only consist of transformed cancer cells, but contains a complex entity of heterogeneous cell types which has been referred to as the “tumor microenvironment” (TME) (15). This TME displays a differing nutritional and metabolite composition depending on the intracellular metabolism of the various cell types, the cell-cell interactions and the specific tumor location in the body (16, 17). This change in nutrient availability will not only directly affect the fuels used for cellular proliferation, but will also alter metabolite-derived protein modifications. Therefore, the type and extent of the modifications will depend on the environment in which the cancer cells site (18).
In this mini-review, we highlight a select number of established and emerging metabolite-derived protein modifications by which epigenetic changes related to oncogenes/TSGs or modulation of oncogenic protein products can be induced; and explore the role of the metabolic microenvironment in shaping these modifications.
Lactylation
While a tumor grows, the inner center becomes deprived of oxygen. This phenomenon, called hypoxia, induces direct metabolic, transcriptomic and genetic changes within multiple cell types in the TME (8, 19). Both the lack of oxygen and the Warburg effect push the cancer cells to mainly rely on glycolysis, produce high levels of lactate and even excrete the metabolite into the extracellular space. This lactate has long been considered a metabolic waste product, but gained more notice after the discovery of several novel physiological functions in past years. Indeed, lactate is now known as a fuel for mitochondrial metabolism and as immune regulator through impairing cytokine production and polarization (20–25). However, the study of lactate function at molecular level is still a largely unexplored area.
Next to its conventional role in central carbon metabolism, lactate and its derived acyl-coenzyme A (CoA) - lactyl-CoA - have been shown to modify histone proteins in a process called lactylation (26). In this novel protein modification, lactic acid from exogenous and glucose-dependent endogenous sources is covalently bound to a lysine residue in histone proteins and directly affects gene transcription in multiple cancer cell types and macrophages under hypoxic conditions (26). Over the past few years, a growing number of studies show a strong association between histone lactylation and oncogenic signaling to induce the progression of human cancers (27, 28). Accordingly, elevated levels of global and specific H3K18 histone lactylation in ocular melanoma tissues have been associated with poor patient prognosis, which could be related to the transcriptional levels of the novel oncogene YTH N6 methyladenosine RNA binding protein (YTHDF2) in tumor-initiating myeloid cells (Figure 1) (29). Lactylated YTHDF2 recognizes efficiently the m6A modification sites in the mRNAs of TSGs such as PER1 and P53 and then induces their degradation to accelerate tumor progression of ocular melanoma. Genetically or pharmacologically impairing this histone lactylation of YTHDF2 not only inhibit proliferation and migration in vitro, but also reduces tumor size of orthotopic xenografts of ocular melanoma, reflecting the relevance of this modification in vivo (29).
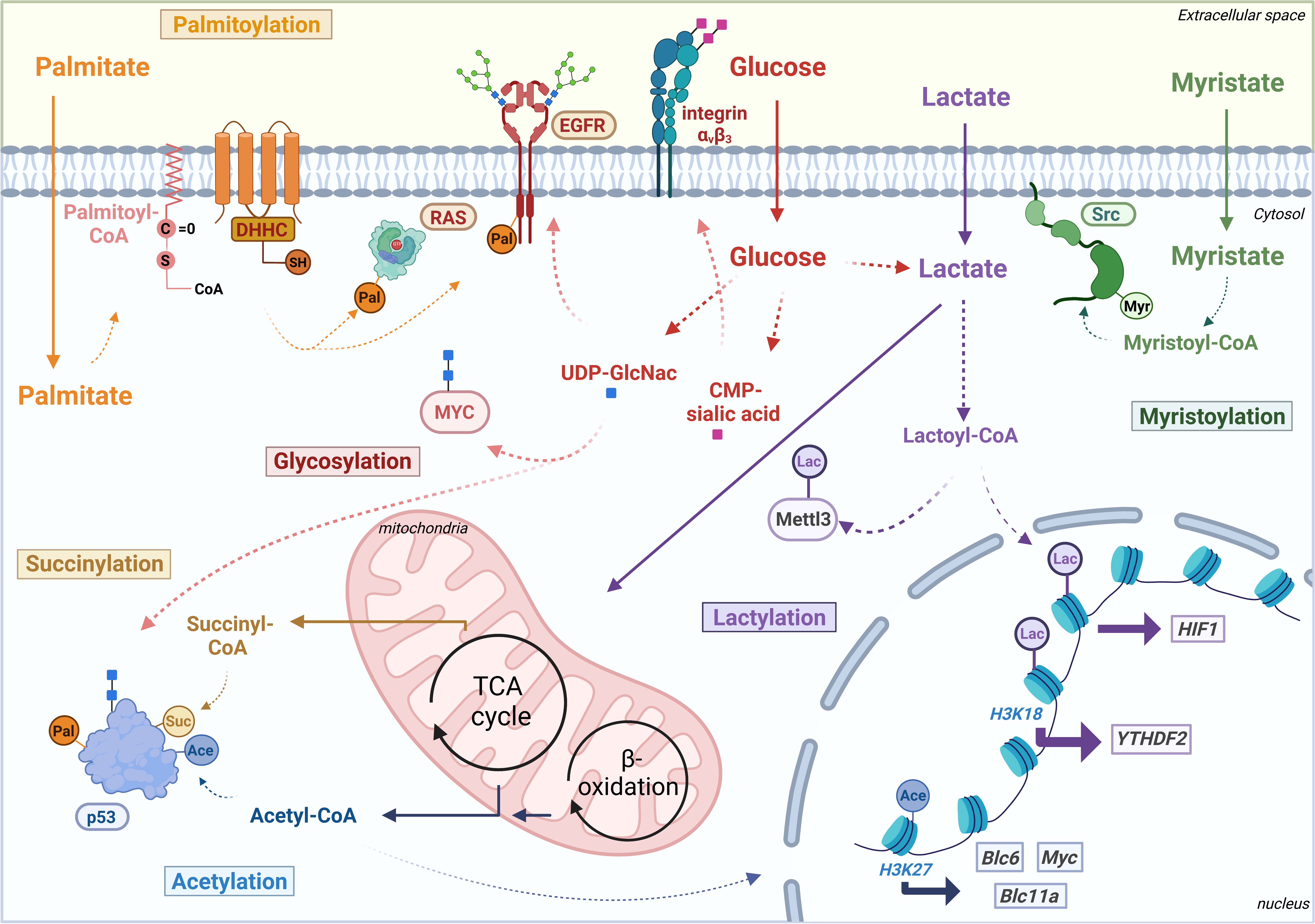
Figure 1 Schematic overview of metabolite-derived modifications palmitoylation (Pal), succinylation (Suc), glycosylation: O-GlcNAcylation (blue squares) and sialylation (pink squares), acetylation (Ace), lactylation (Lac) and myristoylation (Myr) affecting various oncogenes and TSGs at epigenetic level or post-translational level. Transcriptionally regulated oncogenes are put in squares and oncoproteins or tumor suppressor proteins in ovals. Thick arrows indicate transcriptional induction. B-cell lymphoma 6 protein (Bcl6), B-cell lymphoma 11a protein (Bcl11a), cytidine monophosphate sialic acid (CMP-sialic acid), Coenzyme A (CoA), Epidermal growth factor receptor (EGFR), hypoxia-inducible factor 1 (HIF1), Methyltransferase 3 (Mettl3), tricarboxylic acid (TCA), uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), YTH domain-containing family protein 2 (YTHDF2). Figure created with Biorender.
Not only deprivation of oxygen, also the general availability of exogenous lactate can induce histone lactylation in a dose-dependent manner, highlighting the role of the metabolic microenvironment in inducing this protein modification (26, 27, 30). Indeed, in tumor-infiltrating myeloid cells – a key population involved in tumor immune escape – environmental lactate was shown to increase the transcriptional expression of oncogene methyltransferase like 3 (METTL3) through lactylation of histone H3K18. Remarkably, they found that lactate also directly targets two lysine residues in the METTL3 protein, which is highly essential for its activity to target RNAs and add m6A-modifications (Figure 1). By inserting these modifications in the mRNA of JAK1, lactylated METTL3 strengthens the pro-tumoral JAK/STAT3 pathway and enhances tumor progression of colon cancer both in vitro and in vivo (31).
In non-small cell lung carcinoma (NSCLC), which has been shown to highly take up lactate for energetic purposes in vitro and in patients, this metabolite can act in two ways to aid cell survival (28). First of all, lactate fuels the tricarboxylic acid (TCA) cycle in the mitochondria to generate energy in terms of ATP. Next to that, lactate can activate the downstream transcription of hypoxia inducible factor 1 (HIF-1) through lactylating the promoter regions of glycolytic enzymes like hexokinase 1 (HK-1). As HIF-1 signaling is known to support the glycolytic phenotype and induce lactate generation, lactate thus stimulates its own regeneration by acting as transcriptional regulator (28, 32). All of this illustrates the link between a nutritionally enriched environment and epigenetic regulation of oncogenic signaling through nutrient-derived protein modification. However, more research will be needed to gain a comprehensive understanding of how environmental lactate stimulates cancer growth through (histone) lactylation of oncogenes.
Protein lipidation
Upon measuring interstitial fluid of mice and patient-derived melanoma xenografts, marked increases in various free fatty acid (FA) species were seen, suggesting dynamic changes in lipid composition in the TME (33). Indeed, numerous studies have indicated the uptake of exogenous FAs and lipids by cancer cells to boost their tumor growth in vitro and in vivo (33–37). It is widely accepted that lipids and FA metabolism provide an important source of energy in cancer cells. However, other roles for fatty acids and their by-products are emerging in literature (38). In that regard, fatty acids palmitate and myristate have been shown to associate with proteins in order to enhance their hydrophobicity, protein-protein interactions and improve protein folding and stability (39). This attachment of lipid species is termed protein lipidation and comprises an essential class amongst the metabolic post-translational modifications (PTMs) (40).
S-Palmitoylation
Attachment of palmitoyl-CoA - the acyl-CoA derived from palmitate - occurs through a thioester bond with a cysteine residue in the protein of interest (39, 40). This process called S-palmitoylation is a key feature for protein functionality and its imbalance can induce detrimental consequences in terms of cellular malignancy (41, 42). Transfer of the palmitoyl-CoA occurs enzymatically through a family of zinc finger DHHC-containing palmitoyl S-acyltransferases (DHHCs), of which multiple have been implicated in various cancer types like renal, pancreatic, ovarian and gastric cancer (40, 43, 44).
As S-palmitoylation drives protein stability, this lipid-derived protein modification can affect oncogenic and tumor suppressor protein products to promote cancer growth. For example, signaling of tyrosine kinase EGFR – one of the essential drivers of oncogenic signaling in various types of lung cancer – is amplified and prolonged thanks to its palmitoylation by transferase DHHC20 (Figure 1) (45). Since EGFR is known to mitigate its signaling through PI3K-Akt and RAS, loss of DHHC20 or a palmitoyl-resistant EGFR significantly reduces their oncogenic signaling and downstream Myc in a mouse model of oncogenic KRAS-driven lung adenocarcinoma and subsequently affects tumor growth both in vitro and in vivo (45, 46).
Other well-known example of oncogenes regulated by S-palmitoylation is the GTPase RAS (Figure 1), which is more explicitly discussed in the mini-review from Busquets-Hernandez et al (47). With RAS proteins being amongst the most frequently altered oncogenes in various human cancers, targeting their palmitoylation might be an effective therapy for multiple cancer types. As such, mice transplanted with bone marrow cells expressing the palmitoylation-deficient NRAS mutant remained without malignant growth for two years, while the wild-type oncogene limited survival to a maximum of 3 months upon developing a fatal myeloid leukemia-like disease (48). Also, recently, it was shown that members of the α/β hydrolase domain 17 family (ABHD17) family efficiently depalmitoylate NRAS (removal of the palmitoyl-CoA group). Reducing the expression of the ABHD17 family or treatment with a potent new pan-inhibitor – ABD957 – which targets all family members both impairs N-RAS signaling and hereby tumor growth in AML and immortalized kidney cells (49, 50). Therefore, aiming at the palmitoylation/depalmitoylation cycle of oncogenes provide a novel opportunity to target cancer cells that are dependent on NRAS for their growth (50).
Tumor suppressors can also be affected by S-palmitoylation. It was shown that S-palmitoylation of tumor suppressor p53 promotes its nuclear translocation and subsequent signaling (51). However, multiple in vitro cancer cells carrying a wild-type p53 are able to circumvent this PTM by recruiting epigenetic regulators to the promoter region of palmitoyltransferase DHHC1 (51). By doing this, cancer cells can overcome the tumor suppressive activity of p53 and promote their own progression (51). In addition, palmitoylation of protein GNA13 – a TSG commonly mutated in germinal center B-cell-like diffuse large B-cell lymphoma (GCB-DLBCL) and Burkitt’s lymphoma – was shown to be essential for its protein stability, membrane association and tumor suppressor activity (52). Interestingly, it was recently shown that GNA13 negatively correlates with the expression of cell death regulator BCL2 in a palmitoylation-dependent manner in these GCB-DLBL cells. By affecting the palmitoylation of GNA13, these cancer cells with a wild-type GNA13 protein became again sensitized to a BCL2 inhibitor, suggesting GNA13 palmitoylation as potential target for combinatory therapy with BCL2 inhibitors (53).
Another example of a tumor suppressor protein whose palmitoylation is essential for its activity is Scribble (SCRIB), a protein critical in cell polarity and cell-cell junctions (40, 54). S-palmitoylation by DHHC7 is essential for its efficient plasma-membrane targeting and subsequent tumor suppressive activities. Indeed, upon loss of its palmitoyl group, downstream oncogenic pathways of the Hippo pathway such as Yes-associated protein (YAP), MAPK and PI3K/Akt take over and promote tumorigenesis in immortalized kidney, ovarian and breast cancer cell lines (40, 54).
With palmitate being one of the most abundant FAs, an increased abundance and uptake within the TME could have a direct impact on the S-palmitoylation of target proteins in the various cell types. Even though this environmental link has not been described yet in cancer models, an increase in palmitate abundance due to diet or disease induces hyper-palmitoylation in hippocampal neurons and hepatocytes of mice (55–57). Therefore, considering the key role of palmitoylation in regulating protein localization and function and a marked increase of FAs in TME, further work investigating the relationship between these two features will arise.
Myristoylation
Like palmitate, myristate and its derived myristoyl groups (14-carbon saturated fatty acyl groups) can be covalently attached to N-terminal glycine residues of proteins (58, 59). This lipidation called myristoylation is also critical for protein localization and stability, and it has been linked to various malignancies such as ovarian, lung cancer and leukemia (60, 61).
Lipidomics analysis of metastasizing ovarian cancer cells identified specifically myristic acid as being highly enriched compared to non-metastatic cells (60). Indeed, the increased myristic acid abundance was seen to enhance ovarian cancer in both mice and patients’ samples by inducing myristoylation of the oncogenic SRC pathway (Figure 1) (62, 63). Src and Src family kinases (SFKs) are proto-oncogenes that play a key role in regulating various cell surface signaling, and SFK myristoylation was shown to induce attachment to the cytoplasmic membrane and improvement their kinase activity (62, 63). Next to ovarian cancer, also prostate cancer progression can be affected through inhibition of SRC myristoylation, highlighting its importance in various cancer types (64). Myristoylation plays an important role in the in vivo growth of lung cancer, by affecting both tumor suppressor FUS1 and oncogene methyl transferase EZH2 (65, 66). While myristoylation of FUS1 is highly essential for its ability to suppress in vivo tumor growth, myristoylated EZH2 was shown to efficiently bind with STAT3 and promote in vivo lung cancer (67).
Acetylation
Multiple nutritional sources (glucose, fatty acids, acetate, amino acids, etc) can generate acetyl-CoA, a metabolic intermediate carbon source with a key role in mitochondrial energetic purposes and lipid biosynthesis (68). Next to its metabolic role, acetyl-CoA can act as a substrate for acetylation, a reversible protein modification in which an acetyl group from acetyl-CoA is linked to lysine residues in histone or non-histone proteins. This modification, catalyzed by lysine acetyltransferases (KATs, formerly termed histone acetyltransferases or HATs), has been shown to contribute to cancer development and progression through the regulation of gene transcription (69–71). Indeed, cancer cells maintain their oncogenic signaling through acetylation of their epigenome, with the aberrant expression of MYC, BCL6 and BCL11A induced by acetylation of histone H3 lysine 27 (H3K27) in lymphoma as an example (Figure 1) (72).
While histone acetylation has been proven to regulate the expression of some oncogenes (72–74), recent studies have shown that KATs are also capable of acetylating non-histone proteins at post-translational level, including a large number of oncogenes or TSGs (75, 76). It has been long known that acetylation is an essential regulator of p53 protein to stabilize and support its DNA-binding activities (77, 78). While its importance is extensively described in other reviews (77, 79, 80), Cao et al. recently observed an unexpected role for acetylated p53 in promoting PD-1 (programmed cell death protein 1) expression in tumor cell lines of different origins, including lung cancer, osteosarcoma, melanoma, and pancreatic cancer (81). Once acetylated, p53 facilitates PD-1 transcription by recruiting acetyltransferases onto its promoter. This regulation seems to be dependent on the two specific acetylation sites (K120/164) in the p53 protein, suggesting that acetylation at specific sites can determine the activity of a protein.
Regarding oncogenes, studies showed evidence of both direct and indirect acetylation of c-Myc signaling. For example, the oncoprotein MYC in T-cell leukemia is directly acetylated, while acetylation through histone H3K9 also indirectly elevated MYC transcription in neuroblastoma cells and hepatocarcinoma (82, 83). Indeed, many interacting cofactors of MYC possess acetyltransferase activity and modify different lysine residues in the oncoprotein during their interaction (84–86). As lysine can be a binding dock for both ubiquitinylation and acetylation, these modifications can interfere with one another. Therefore, direct acetylation prevents MYC from ubiquitin-mediated degradation and stabilizes the protein (85–87).
Remarkably, protein acetylation is highly sensitive to changes in acetyl-CoA levels and local changes in its intracellular compartmentalized abundance can impact acetylation at the specific intracellular compartments of the cell (cytosol versus nucleus) (88, 89). In this regard, even though acetyl-CoA itself cannot be taken up from the environment, differing availabilities from its sources can directly impact the intracellular acetyl-CoA abundance and its derived modification in vivo (90–92). Accordingly, acetate can not only act as bioenergetic substrate but can also function as an epigenetic regulator by enhancing histone acetylation of Twist - an inducer of EMT - in hepatocellular cell lines and patient samples, suggesting a plausible link between environmental acetate and intracellular acetylation (93–95). However, further studies are needed to evaluate the direct regulation of oncogenic signaling by environmentally induced acetylation. It is interesting to mention that immortalized astrocytes with tumor suppressor deficiency have marked upregulated expression of nucleo-cytosolic acetyl CoA synthetase (ACSS) enzymes that oxidize acetate to acetyl-CoA, suggesting the usage of acetate as carbon source in oncogenic settings (94). However, if they use acetate-derived acetyl-CoA for its protein modification remains elusive.
Succinylation
Gastrointestinal stromal tumors, renal, thyroid, testicular tumors and neuroblastomas are often characterized by germline mutations in succinate dehydrogenase (SDH) enzyme. As a key metabolic enzyme in the TCA cycle, its deficiency results in an accumulation of succinate both intra- and extracellularly (96). As succinate can be immediately interconverted to succinyl-CoA, its increase will significantly affect succinyl-CoA levels as well as its derived succinylation (97). Succinylation is a PTM, in which succinyl groups are being dynamically and reversibly attached to lysine residues of proteins (98). The major substrate for succinylation is succinyl-CoA – a cofactor that can be generated from the mitochondrial TCA cycle, lipid and amino acid metabolism. This PTM has been shown to play an important regulatory role in the progression of a variety of tumors like thyroid, gastric and breast cancer (98, 99). In short, succinylation was seen to boost proliferation of both thyroid and breast cancer cells and promote metastasis in gastric cancer. Remarkably, while this PTM can promote tumorigenesis in those types of cancer, it has a tumor suppressive effect on liver cancer, lung cancer and osteosarcoma (100–102).
Mass spectrometry identified more than 500 succinylation sites in about 300 proteins in gastric cancer tissues, highlighting its importance in regulating cancer progression (99, 103). Next to the above-mentioned modifications of TSG p53, succinylation was added very recently to the list of its regulatory PTMs. Using mass spectrometric analysis, p53 was seen to be succinylated at lysine 120 (K120) which is also a common acetylation site and key modulatory residue (Figure 1). Loss of this modification by SIRT5 highly affects the p53 response to DNA damage and all gene expressions related to apoptosis and cell cycle arrest (p21, MDM2, TIGAR, SFN), indicating the importance of this PTM for its functionality (104). Nevertheless, whether this is related to any specific type of cancer remains elusive.
Glycosylation
Glycosylation the addition of sugar chains to proteins or lipids - is one of the main features of malignancy and shows a glycosylation-specific gene expression during cancer progression (105–108). The enzyme O-linked N-acetylglucosamine transferase (OGT) involved in attaching the substrate to the protein and modification of the substrate O-linked β-N-acetylglucosamine (O-GlcNAc) itself are known to be necessary for tumorigenesis and metastasis abilities of multiple cancer types, like papillary thyroid cancer in vivo, breast, colon, liver and lung cancer (109–112).
Similar to the other PTMs described above, oncogenes and tumor suppressors can also be glycosylated to alter their activity (113, 114): EGFR family, estrogen receptor (ER) family, c-MYC, YAP, B-catenin, TSG protein retinoblastoma (Rb), p53 and others (Figure 1) (114–120). Indeed, O-glycosylation at threonine 58 in oncogene c-Myc was shown to be a major mutation site in human lymphomas and was shown to stabilize the protein to promote proliferation and tumorigenesis of hepatocellular carcinoma cells in vivo and in vitro (121–123). Moreover, oncoprotein YAP is stabilized by O-GlcNAcylation in both in vitro and in vivo models to promote high glucose-induced liver tumorigenesis (120). Similar, to other PTMs, oncogenes themselves can induce O-GlcNAcylation and OGT expression directly as part of their oncogenic signaling, indicating the existence of a bi-directional feedback loop between glycosylation and oncogenic expression (124).
Often, glycans and glycosylated proteins are located at the extracellular side of the plasma membrane. Also, any changes in the intracellular and microenvironmental metabolite pools can have a direct effect on O-GlcNAcylation, of which its substrate UDP-GlcNAc is produced through the hexosamine biosynthetic pathway. As glucose, amino acid, fatty acid and nucleotide metabolism are all linked to this pathway and therefore the synthesis of the substrate, any difference in metabolite levels from these pathways might influence O-GlcNAcylation (110). This highlights the possibility of a direct role for the microenvironment in the interplay between glycosylation and various cell types of the TME. Indeed, the glycolytic phenotype induced by loss of p53 was shown to elevate O-GlcNAc levels and its derived PTM in mouse embryonic fibroblasts and human transformed fibroblasts in vitro (125). A more detailed description of these dynamics can be found in the review Peixoto et al. (2019) (126).
Interestingly, very recently it was shown that glycosylation can be modulated by the heterogeneous protein expression of the metabolic enzyme phosphoglycerate dehydrogenase (PHGDH), which affects breast cancer-derived metastasis formation (111). Mechanistically, loss of PHGDH was shown to induce the activation of the hexosamine-sialic acid pathway and hereby the sialylation – a specific type of glycosylation - of integrin αvβ3 (Figure 1). Doing this, the PHGDHlow breast cancer cells promote the migration and metastatic dissemination from the primary tumor (111). However, more research will be needed to not only fully elucidate the environmental role in inducing O-GlcNAcylation of oncogenes, but also the role of non-clonal metabolic heterogeneity in alterating glycosylation in general.
Conclusion
Metabolites are not only intermediate products of intracellular metabolic reactions, but also important regulators for the PTMs of cancer-related proteins like oncogenes and tumor suppressors. Emerging research demonstrates the existence of a metabolic niche within the TME, in which differing nutrient availability can mediate an indirect crosstalk between cell types and oncogenic intracellular signaling through shaping the metabolic PTMs. Targeting this novel interchange between PTMs and the environment offers new opportunities to target cancer growth in an organ-specific and more efficient manner. So far, drugs targeting modification-related enzymes such as deacetylase (HDAC) inhibitors have shown significant potential to slow down tumor growth in therapeutic settings (Table 1). However, at the moment, less explored PTMs as well as the interplay between the modifications and their environment lack sufficient knowledge to develop specific inhibitors. Investigating the complexity of these environmentally-driven metabolic PTMs and their dependence on tumor localization will allow us to gain a deeper mechanistic understanding and ultimately exploit these for future cancer therapies.

Table 1 List of oncogenes and tumor suppressors that can be regulated by post-translational modifications.
Author contributions
YL, AV and PA-M: Conceptualization and Writing of the first draft. YL, AV, PA-M, S-MF: Review and editing. YL, AV, PA-M, MX, S-MF: Conceptualization and Review. All authors contributed to the article and approved the submitted version.
Funding
AV receives funding from Fonds Wetenschappelijk Onderzoek (FWO 1176823N), YL receives funding from China Scholarship Council (CSC). S-MF acknowledges funding from the European Research Council under the ERC Consolidator Grant Agreement n. 771486–MetaRegulation, FWO – Research Projects (G0B4122N), KU Leuven – FTBO and Internal Funding, King Baudouin Foundation, Beug Foundation, Stichting tegen Kanker and Fonds Baillet Latour. PA-M acknowledges funding from Marie Sklodowska-Curie Actions Grant Agreement n. 839896 MetaTarGet and Beug Foundation.
Conflict of interest
S.-MF has received funding from Bayer AG, Merck, Black Belt Therapeutics and Alesta Therapeutics; has consulted for Fund+; and serves on the advisory board of Alesta Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Takeshima H, Ushijima T. Accumulation of genetic and epigenetic alterations in normal cells and cancer risk. NPJ Precis Oncol (2019) 3(1):7. doi: 10.1038/s41698-019-0079-0
2. Zhu K, Liu Q, Zhou Y, Tao C, Zhao Z, Sun J, et al. Oncogenes and tumor suppressor genes: Comparative genomics and network perspectives. BMC Genomics (2015) 16 Suppl 7(Suppl 7):S8–8. doi: 10.1186/1471-2164-16-S7-S8
3. Kontomanolis EN, Koutras A, Syllaios A, Schizas D, Mastoraki A, Garmpis N, et al. Role of oncogenes and tumor-suppressor genes in carcinogenesis: A review. Anticancer Res (2020) 40(11):6009–15. doi: 10.21873/anticanres.14622
4. Hanahan D, Weinberg RA, Francisco S. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9
5. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
6. Hanahan D, Weinberg RA. Review hallmarks of Cancer: The next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
7. Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol Cell (2021) 81(4):691–707.e6. doi: 10.1016/j.molcel.2020.12.012
8. Brandon F, Ashley S, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science (2020) 368(6487):eaaw5473. doi: 10.1126/science.aaw5473
9. Nagarajan A, Malvi P, Wajapeyee N. Oncogene-directed alterations in cancer cell metabolism. Trends Cancer (2016) 2(7):365–77. doi: 10.1016/j.trecan.2016.06.002
10. Iurlaro R, León-Annicchiarico CL, Muñoz-Pinedo C. Chapter three - regulation of cancer metabolism by oncogenes and tumor suppressors; Methods in Enzymology, Academic Press (2014) 542:59–80. doi: 10.1016/B978-0-12-416618-9.00003-0
11. Min HY, Lee HY. Oncogene-driven metabolic alterations in cancer. Biomol Ther (2018) 26(1):45–56. doi: 10.4062/biomolther.2017.211
12. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
13. Lorendeau D, Christen S, Rinaldi G, Fendt S-M. Metabolic control of signalling pathways and metabolic auto-regulation. Biol Cell (2015) 107(8):251–72. doi: 10.1111/boc.201500015
14. Chen L, Liu S, Tao Y. Regulating tumor suppressor genes: post-translational modifications. Signal Transduct Target Ther (2020) 5(1):90. doi: 10.1038/s41392-020-0196-9
15. Anderson NM, Simon MC. The tumor microenvironment. Curr Biol (2020) 30(16):R921–5. doi: 10.1016/j.cub.2020.06.081
16. Elia I, Haigis MC. Metabolites and the tumour microenvironment: From cellular mechanisms to systemic metabolism. Nat Metab (2021) 3(1):21–32. doi: 10.1038/s42255-020-00317-z
17. Comito G, Ippolito L, Chiarugi P, Cirri P. Nutritional exchanges within tumor microenvironment: Impact for cancer aggressiveness. Front Oncol (2020) 10:396. doi: 10.3389/fonc.2020.00396
18. Li W, Li F, Zhang X, Lin H-K, Xu C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal Transduct Target Ther (2021) 6(1):422. doi: 10.1038/s41392-021-00825-8
19. Egeblad M, Nakasone ES, Werb Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev Cell (2010) 18(6):884–901. doi: 10.1016/j.devcel.2010.05.012
20. Brooks GA. Lactate as a fulcrum of metabolism. Redox Biol (2020) 35:101454. doi: 10.1016/j.redox.2020.101454
21. Romero-Garcia S, Moreno-Altamirano MMB, Prado-Garcia H, Sánchez-García FJ. Lactate contribution to the tumor microenvironment: Mechanisms, effects on immune cells and therapeutic relevance. Front Immunol (2016) 7:52. doi: 10.3389/fimmu.2016.00052
22. Certo M, Tsai C-H, Pucino V, Ho P-C, Mauro C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat Rev Immunol (2021) 21(3):151–61. doi: 10.1038/s41577-020-0406-2
23. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate metabolism in human lung tumors. Cell (2017) 171(2):358–371.e9. doi: 10.1016/j.cell.2017.09.019
24. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
25. Geeraerts X, Fernández-Garcia J, Hartmann FJ, de Goede KE, Martens L, Elkrim Y, et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. (Cambridge), Cell Rep (2021) 37(13):110171. doi: 10.1016/j.celrep.2021.110171
26. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
27. Chen A-N, Luo Y, Yang Y-H, Fu J-T, Geng X-M, Shi J-P, et al. Lactylation, a novel metabolic reprogramming code: Current status and prospects. Front Immunol (2021) 12:688910. doi: 10.3389/fimmu.2021.688910
28. Jiang J, Huang D, Jiang Y, Hou J, Tian M, Li J, et al. Lactate modulates cellular metabolism through histone lactylation-mediated gene expression in non-small cell lung cancer. Front Oncol (2021) 11:647559. doi: 10.3389/fonc.2021.647559
29. Yu J, Chai P, Xie M, Ge S, Ruan J, Fan X, et al. Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol (2021) 22(1):85. doi: 10.1186/s13059-021-02308-z
30. Wang Z-H, Peng W-B, Zhang P, Yang X-P, Zhou Q. Lactate in the tumour microenvironment: From immune modulation to therapy. eBioMedicine (2021) 73:103627. doi: 10.1016/j.ebiom.2021.103627
31. Xiong J, He J, Zhu J, Pan J, Liao W, Ye H, et al. Lactylation-driven METTL3-mediated RNA m6A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol Cell (2022) 82(9):1660–77. doi: 10.1016/j.molcel.2022.02.033
32. Nagao A, Kobayashi M, Koyasu S, Chow CCT, Harada H. HIF-1-Dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int J Mol Sci (2019) 20(2):238. doi: 10.3390/ijms20020238
33. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell (2017) 32(3):377–91. doi: 10.1016/j.ccell.2017.08.004
34. Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab (2017) 5(1):1. doi: 10.1186/s40170-016-0163-7
35. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med (2011) 17(11):1498–503. doi: 10.1038/nm.2492
36. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS-O, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature (2017) 541(7635):41–5. doi: 10.1038/nature20791
37. Xu S, Chaudhary O, Rodríguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity (2021) 54(7):1561–1577.e7. doi: 10.1016/j.immuni.2021.05.003
38. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer (2020) 122(1):4–22. doi: 10.1038/s41416-019-0650-z
39. Chen B, Sun Y, Niu J, Jarugumilli GK, Wu X. Protein lipidation in cell signaling and diseases: Function, regulation, and therapeutic opportunities. Cell Chem Biol (2018) 25(7):817–31. doi: 10.1016/j.chembiol.2018.05.003
40. Ko P-J, Dixon SJ. Protein palmitoylation and cancer. EMBO Rep (2018) 19(10):e46666. doi: 10.15252/embr.201846666
41. Yeste-Velasco M, Linder ME, Lu Y-J. Protein s-palmitoylation and cancer. Biochim Biophys Acta - Rev Cancer (2015) 1856(1):107–20. doi: 10.1016/j.bbcan.2015.06.004
42. Anderson AM, Ragan MA. Palmitoylation: A protein s-acylation with implications for breast cancer. NPJ Breast Cancer (2016) 2(1):16028. doi: 10.1038/npjbcancer.2016.28
43. Yuan M, Chen X, Sun Y, Jiang L, Xia Z, Ye K, et al. ZDHHC12-mediated claudin-3 s-palmitoylation determines ovarian cancer progression. Acta Pharm Sin B (2020) 10(8):1426–39. doi: 10.1016/j.apsb.2020.03.008
44. Oo HZ, Sentani K, Sakamoto N, Anami K, Naito Y, Uraoka N, et al. Overexpression of ZDHHC14 promotes migration and invasion of scirrhous type gastric cancer. Oncol Rep (2014) 32(1):403–10. doi: 10.3892/or.2014.3166
45. Kadry YA, Lee J-Y, Witze ES. Regulation of EGFR signalling by palmitoylation and its role in tumorigenesis. Open Biol (2021) 11(10):210033. doi: 10.1098/rsob.210033
46. Akriti K, Walter DM, Gudiel AA, Schek N, Feldser DM, Witze ES. Blocking EGFR palmitoylation suppresses PI3K signaling and mutant KRAS lung tumorigenesis. Sci Signal (2020) 13(621):eaax2364. doi: 10.1126/scisignal.aax2364
47. Busquets-Hernández C, Triola G. Palmitoylation as a key regulator of ras localization and function. Front Mol Biosci (2021) 8:659861. doi: 10.3389/fmolb.2021.659861
48. Cuiffo B, Ren R. Palmitoylation of oncogenic NRAS is essential for leukemogenesis. Blood (2010) 115(17):3598–605. doi: 10.1182/blood-2009-03-213876
49. Lin DTS, Conibear E. ABHD17 proteins are novel protein depalmitoylases that regulate n-ras palmitate turnover and subcellular localization. Elife (2015) 4:e11306. doi: 10.7554/eLife.11306
50. Remsberg JR, Suciu RM, Zambetti NA, Hanigan TW, Firestone AJ, Inguva A, et al. ABHD17 regulation of plasma membrane palmitoylation and n-ras-dependent cancer growth. Nat Chem Biol (2021) 17(8):856–64. doi: 10.1038/s41589-021-00785-8
51. Tang J, Peng W, Feng Y, Le X, Wang K, Xiang Q, et al. Cancer cells escape p53’s tumor suppression through ablation of ZDHHC1-mediated p53 palmitoylation. Oncogene (2021) 40(35):5416–26. doi: 10.1038/s41388-021-01949-5
52. Xia Z, Zhang X, Liu P, Jiao B, Ren R. Palmitoylation of GNA13 is essential for its tumor suppressor function and its inhibition confers hypersensitivity of b-cell lymphoma to BCL2 inhibitors. Blood (2019) 134(Supplement_1):3787. doi: 10.1182/blood-2019-126368
53. Xia Z, Zhang X, Liu P, Zhang R, Huang Z, Li D, et al. GNA13 regulates BCL2 expression and the sensitivity of GCB-DLBCL cells to BCL2 inhibitors in a palmitoylation-dependent manner. Cell Death Dis (2021) 12(1):54. doi: 10.1038/s41419-020-03311-1
54. Chen B, Zheng B, DeRan M, Jarugumilli GK, Fu J, Brooks YS, et al. ZDHHC7-mediated s-palmitoylation of scribble regulates cell polarity. Nat Chem Biol (2016) 12(9):686–93. doi: 10.1038/nchembio.2119
55. Spinelli M, Fusco S, Mainardi M, Scala F, Natale F, Lapenta R, et al. Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a. Nat Commun (2017) 8(1):2009. doi: 10.1038/s41467-017-02221-9
56. Zhao L, Zhang C, Luo X, Wang P, Zhou W, Zhong S, et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J Hepatol (2018) 69(3):705–17. doi: 10.1016/j.jhep.2018.04.006
57. Spinelli M, Fusco S, Grassi C. Nutrient-dependent changes of protein palmitoylation: Impact on nuclear enzymes and regulation of gene expression. Int J Mol Sci (2018) 19(12):3820. doi: 10.3390/ijms19123820
58. Yuan M, Song Z-H, Ying M-D, Zhu H, He Q-J, Yang B, et al. N-myristoylation: from cell biology to translational medicine. Acta Pharmacol Sin [Internet] 2020/03/18 (2020) 41(8):1005–15. doi: 10.1038/s41401-020-0388-4
59. Fhu CW, Ali A. Protein lipidation by palmitoylation and myristoylation in cancer. Front Cell Dev Biol (2021) 9:1–15. doi: 10.3389/fcell.2021.673647
60. Zhang Q, Zhou W, Yu S, Ju Y, To SKY, Wong AST, et al. Metabolic reprogramming of ovarian cancer involves ACSL1-mediated metastasis stimulation through upregulated protein myristoylation. Oncogene (2021) 40(1):97–111. doi: 10.1038/s41388-020-01516-4
61. Liang J, Xu Z-X, Ding Z, Lu Y, Yu Q, Werle KD, et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat Commun (2015) 6(1):7926. doi: 10.1038/ncomms8926
62. Patwardhan P, Resh MD. Myristoylation and membrane binding regulate c-src stability and kinase activity. Mol Cell Biol (2010) 30(17):4094–107. doi: 10.1128/MCB.00246-10
63. Ortiz MA, Mikhailova T, Li X, Porter BA, Bah A, Kotula L. Src family kinases, adaptor proteins and the actin cytoskeleton in epithelial-to-mesenchymal transition. Cell Commun Signal (2021) 19(1):67. doi: 10.1186/s12964-021-00750-x
64. Kim S, Alsaidan OA, Goodwin O, Li Q, Sulejmani E, Han Z, et al. Blocking myristoylation of src inhibits its kinase activity and suppresses prostate cancer progression. Cancer Res (2017) 77(24):6950–62. doi: 10.1158/0008-5472.CAN-17-0981
65. Uno F, Sasaki J, Nishizaki M, Carboni G, Xu K, Atkinson EN, et al. Myristoylation of the Fus1 protein is required for tumor suppression in human lung cancer cells. Cancer Res (2004) 64(9):2969–76. doi: 10.1158/0008-5472.CAN-03-3702
66. Zhang J, Zeng Y, Xing Y, Li X, Zhou L, Hu L, et al. Myristoylation-mediated phase separation of EZH2 compartmentalizes STAT3 to promote lung cancer growth. Cancer Lett (2021) 516:84–98. doi: 10.1016/j.canlet.2021.05.035
67. Kang J-H, Jang Y-S, Lee HJ, Lee C-Y, Shin DY, Oh SH. Inhibition of STAT3 signaling induces apoptosis and suppresses growth of lung cancer: Good and bad. Lab Anim Res (2019) 35(1):30. doi: 10.1186/s42826-019-0030-0
68. Lei I, Tian S, Gao W, Liu L, Guo Y, Tang P, et al. Acetyl-CoA production by specific metabolites promotes cardiac repair after myocardial infarction via histone acetylation. Elife (2021) 10:e60311. doi: 10.7554/eLife.60311
69. Zhang B, Li Y, Wu Q, Xie L, Barwick B, Fu C, et al. Acetylation of KLF5 maintains EMT and tumorigenicity to cause chemoresistant bone metastasis in prostate cancer. Nat Commun (2021) 12(1):1714. doi: 10.1038/s41467-021-21976-w
70. Zhang Y, Jing Y, Wang Y, Tang J, Zhu X, Jin W-L, et al. NAT10 promotes gastric cancer metastasis via N4-acetylated COL5A1. Signal Transduct Target Ther (2021) 6(1):173. doi: 10.1038/s41392-021-00489-4
71. Slaughter MJ, Shanle EK, Khan A, Chua KF, Hong T, Boxer LD, et al. HDAC inhibition results in widespread alteration of the histone acetylation landscape and BRD4 targeting to gene bodies. Cell Rep (2021) 34(3):108638. doi: 10.1016/j.celrep.2020.108638
72. Sungalee S, Liu Y, Lambuta RA, Katanayeva N, Donaldson Collier M, Tavernari D, et al. Histone acetylation dynamics modulates chromatin conformation and allele-specific interactions at oncogenic loci. Nat Genet (2021) 53(5):650–62. doi: 10.1038/s41588-021-00842-x
73. Hogg SJ, Motorna O, Cluse LA, Johanson TM, Coughlan HD, Raviram R, et al. Targeting histone acetylation dynamics and oncogenic transcription by catalytic P300/CBP inhibition. Mol Cell (2021) 81(10):2183–2200.e13. doi: 10.1016/j.molcel.2021.04.015
74. Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr (1996) 5(4–5):245–53.
75. Xia C, Tao Y, Li M, Che T, Qu J. Protein acetylation and deacetylation: An important regulatory modification in gene transcription (Review). Exp Ther Med (2020) 20(4):2923–40. doi: 10.3892/etm.2020.9073
76. Zhao D, Li F-L, Cheng Z-L, Lei Q-Y. Impact of acetylation on tumor metabolism. Mol Cell Oncol (2014) 1(3):e963452. doi: 10.4161/23723548.2014.963452
77. Reed SM, Quelle DE. p53 acetylation: Regulation and consequences. Cancers (Basel) (2014) 7(1):30–69. doi: 10.3390/cancers7010030
78. Xia Z, Kon N, Gu AP, Tavana O, Gu W. Deciphering the acetylation code of p53 in transcription regulation and tumor suppression. Oncogene (2022) 41(22):3039–50. doi: 10.1038/s41388-022-02331-9
79. Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell (2008) 133(4):612–26. doi: 10.1016/j.cell.2008.03.025
80. Liu Y, Tavana O, Gu W. p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol (2019) 11(7):564–77. doi: 10.1093/jmcb/mjz060
81. Cao Z, Kon N, Liu Y, Xu W, Wen J, Yao H, et al. An unexpected role for p53 in regulating cancer cell-intrinsic PD-1 by acetylation. Sci Adv (2021) 7(14):eabf4148. doi: 10.1126/sciadv.abf4148
82. Romeo MM, Ko B, Kim J, Brady R, Heatley HC, He J, et al. Acetylation of the c-MYC oncoprotein is required for cooperation with the HTLV-1 p30(II) accessory protein and the induction of oncogenic cellular transformation by p30(II)/c-MYC. Virology (2015) 476:271–88. doi: 10.1016/j.virol.2014.12.008
83. Wu H, Yang T-Y, Li Y, Ye W-L, Liu F, He X-S, et al. Tumor necrosis factor receptor–associated factor 6 promotes hepatocarcinogenesis by interacting with histone deacetylase 3 to enhance c-myc gene expression and protein stability. Hepatology (2020) 71(1):148–63. doi: 10.1002/hep.30801
84. Vervoorts J, Lüscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, et al. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep (2003) 4(5):484–90. doi: 10.1038/sj.embor.embor821
85. Vervoorts J, Lüscher-Firzlaff J, Lüscher B. The ins and outs of MYC regulation by posttranslational mechanisms*. J Biol Chem (2006) 281(46):34725–9. doi: 10.1074/jbc.R600017200
86. Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen C-J, et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol Cell Biol (2004) 24(24):10826–34. doi: 10.1128/MCB.24.24.10826-10834.2004
87. Faiola F, Liu X, Lo S, Pan S, Zhang K, Lymar E, et al. Dual regulation of c-myc by p300 via acetylation-dependent control of myc protein turnover and coactivation of myc-induced transcription. Mol Cell Biol (2005) 25(23):10220–34. doi: 10.1128/MCB.25.23.10220-10234.2005
88. Boon R. Metabolic fuel for epigenetic: Nuclear production meets local consumption. Front Genet (2021) 12:768996. doi: 10.3389/fgene.2021.768996
89. Sivanand S, Viney I, Wellen KE. Spatiotemporal control of acetyl-CoA metabolism in chromatin regulation. Trends Biochem Sci (2018) 43(1):61–74. doi: 10.1016/j.tibs.2017.11.004
90. Ho C-L, Yu SCH, Yeung DWC. 11C-acetate PET imaging in hepatocellular carcinoma and other liver masses. J Nucl Med (2003) 44(2):213–21.
91. Oyama N, Akino H, Kanamaru H, Suzuki Y, Muramoto S, Yonekura Y, et al. 11C-acetate PET imaging of prostate cancer. J Nucl Med (2002) 43(2):181–6.
92. Kim S, Kim D, Kim SH, Park M, Chang JH, Yun M. The roles of 11C-acetate PET/CT in predicting tumor differentiation and survival in patients with cerebral glioma. Eur J Nucl Med Mol Imaging (2018) 45(6):1012–20. doi: 10.1007/s00259-018-3948-9
93. Lu M, Zhu W-W, Wang X, Tang J-J, Zhang K-L, Yu G-Y, et al. ACOT12-dependent alteration of acetyl-CoA drives hepatocellular carcinoma metastasis by epigenetic induction of epithelial-mesenchymal transition. Cell Metab (2019) 29(4):886–900.e5. doi: 10.1016/j.cmet.2018.12.019
94. Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell (2014) 159(7):1603–14. doi: 10.1016/j.cell.2014.11.025
95. Gao X, Lin S-H, Ren F, Li J-T, Chen J-J, Yao C-B, et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun (2016) 7(1):11960. doi: 10.1038/ncomms11960
96. Zhao Y, Feng F, Guo Q-H, Wang Y-P, Zhao R. Role of succinate dehydrogenase deficiency and oncometabolites in gastrointestinal stromal tumors. World J Gastroenterol (2020) 26(34):5074–89. doi: 10.3748/wjg.v26.i34.5074
97. Li F, He X, Ye D, Lin Y, Yu H, Yao C, et al. NADP+-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol Cell (2015) 60(4):661–75. doi: 10.1016/j.molcel.2015.10.017
98. Mu R, Ma Z, Lu C, Wang H, Cheng X, Tuo B, et al. Role of succinylation modification in thyroid cancer and breast cancer. Am J Cancer Res (2021) 11(10):4683–99.
99. Wang C, Zhang C, Li X, Shen J, Xu Y, Shi H, et al. CPT1A-mediated succinylation of S100A10 increases human gastric cancer invasion. J Cell Mol Med (2019) 23(1):293–305. doi: 10.1111/jcmm.13920
100. Chen X-F, Tian M-X, Sun R-Q, Zhang M-L, Zhou L-S, Jin L, et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep (2018) 19(5). doi: 10.15252/embr.201745124
101. Lin Z-F, Xu H-B, Wang J-Y, Lin Q, Ruan Z, Liu F-B, et al. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem Biophys Res Commun (2013) 441(1):191–5. doi: 10.1016/j.bbrc.2013.10.033
102. Yang X, Wang Z, Li X, Liu B, Liu M, Liu L, et al. SHMT2 desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Res (2018) 78(2):372–86. doi: 10.1158/0008-5472.CAN-17-1912
103. Song Y, Wang J, Cheng Z, Gao P, Sun J, Chen X, et al. Quantitative global proteome and lysine succinylome analyses provide insights into metabolic regulation and lymph node metastasis in gastric cancer. Sci Rep (2017) 7:42053. doi: 10.1038/srep42053
104. Liu X, Rong F, Tang J, Zhu C, Chen X, Jia S, et al. Repression of p53 function by SIRT5-mediated desuccinylation at lysine 120 in response to DNA damage. Cell Death Differ (2022) 29(4):722–36. doi: 10.1038/s41418-021-00886-w
105. Thomas D, Rathinavel AK, Radhakrishnan P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim Biophys Acta Rev Cancer (2021) 1875(1):188464. doi: 10.1016/j.bbcan.2020.188464
106. Varki A. Biological roles of glycans. Glycobiology (2017) 27(1):3–49. doi: 10.1093/glycob/cww086
107. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer (2015) 15(9):540–55. doi: 10.1038/nrc3982
108. Reily C, Stewart TJ, Renfrow MB, Novak J. Glycosylation in health and disease. Nat Rev Nephrol (2019) 15(6):346–66. doi: 10.1038/s41581-019-0129-4
109. Li X, Wu Z, He J, Jin Y, Chu C, Cao Y, et al. OGT regulated O-GlcNAcylation promotes papillary thyroid cancer malignancy via activating YAP. Oncogene (2021) 40(30):4859–71. doi: 10.1038/s41388-021-01901-7
110. Lee JB, Pyo K-H, Kim HR. Role and function of O-GlcNAcylation in cancer. Cancers (Basel) (2021) 13(21):5365. doi: 10.3390/cancers13215365
111. Rossi M, Altea-Manzano P, Demicco M, Doglioni G, Bornes L, Fukano M, et al. PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature (2022) 605(7911):747–53. doi: 10.1038/s41586-022-04758-2
112. Oliveira-Ferrer L, Legler K, Milde-Langosch K. Role of protein glycosylation in cancer metastasis. Semin Cancer Biol (2017) 44:141–52. doi: 10.1016/j.semcancer.2017.03.002
113. Hofmann BT, Schlüter L, Lange P, Mercanoglu B, Ewald F, Fölster A, et al. COSMC knockdown mediated aberrant O-glycosylation promotes oncogenic properties in pancreatic cancer. Mol Cancer (2015) 14(1):1–15. doi: 10.1186/s12943-015-0386-1
114. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu Rev Biochem (2011) 80:825–58. doi: 10.1146/annurev-biochem-060608-102511
115. Olivier-Van Stichelen S, Dehennaut V, Buzy A, Zachayus J-L, Guinez C, Mir A-M, et al. O-GlcNAcylation stabilizes β-catenin through direct competition with phosphorylation at threonine 41. FASEB J Off Publ Fed Am Soc Exp Biol (2014) 28(8):3325–38. doi: 10.1096/fj.13-243535
116. Kaszuba K, Grzybek M, Orłowski A, Danne R, Róg T, Simons K, et al. N-glycosylation as determinant of epidermal growth factor receptor conformation in membranes. Proc Natl Acad Sci (2015) 112(14):4334–9. doi: 10.1073/pnas.1503262112
117. Cheng X, Cole RN, Zaia J, Hart GW. Alternative O-Glycosylation/O-Phosphorylation of the murine estrogen receptor β. Biochemistry (2000) 39(38):11609–20. doi: 10.1021/bi000755i
118. Wells L, Slawson C, Hart GW. The E2F-1 associated retinoblastoma-susceptibility gene product is modified by O-GlcNAc. Amino Acids (2011) 40(3):877–83. doi: 10.1007/s00726-010-0709-x
119. Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, et al. Modification of p53 with O-linked n-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol (2006) 8(10):1074–83. doi: 10.1038/ncb1470
120. Zhang X, Qiao Y, Wu Q, Chen Y, Zou S, Liu X, et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat Commun (2017) 8:15280. doi: 10.1038/ncomms15280
121. Chou TY, Dang CV, Hart GW. Glycosylation of the c-myc transactivation domain. Proc Natl Acad Sci U S A (1995) 92(10):4417–21. doi: 10.1073/pnas.92.10.4417
122. Chou T-Y, Hart GW, Dang CV. C-myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas *. J Biol Chem (1995) 270(32):18961–5. doi: 10.1074/jbc.270.32.18961
123. Jiang T, Yang J, Yang H, Chen W, Ji K, Xu Y, et al. SLC35B4 stabilizes c-MYC protein by O-GlcNAcylation in HCC. Front Pharmacol (2022) 13:851089. doi: 10.3389/fphar.2022.851089
124. Sun L, Lv S, Song T. O-GlcNAcylation links oncogenic signals and cancer epigenetics. Discovery Oncol (2021) 12(1):54. doi: 10.1007/s12672-021-00450-5
125. Kawauchi K, Araki K, Tobiume K, Tanaka N. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-n-acetyl glucosamine modification. Proc Natl Acad Sci USA (2009) 106(9):3431–6. doi: 10.1073/pnas.0813210106
126. Peixoto A, Relvas-Santos M, Azevedo R, Santos LL, Ferreira JA. Protein glycosylation and tumor microenvironment alterations driving cancer hallmarks. Front Oncol (2019) 9:380. doi: 10.3389/fonc.2019.00380
127. Beauchamp E, Yap MC, Iyer A, Perinpanayagam MA, Gamma JM, Vincent KM, et al. Targeting n-myristoylation for therapy of b-cell lymphomas. Nat Commun (2020) 11(1):5348. doi: 10.1038/s41467-020-18998-1
128. Nebbioso A, Carafa V, Conte M, Tambaro FP, Abbondanza C, Martens J, et al. C-myc modulation and acetylation is a key HDAC inhibitor target in cancer. Clin Cancer Res (2017) 23(10):2542–55. doi: 10.1158/1078-0432.CCR-15-2388
129. Qin Z, Wang T, Su S, Shen L, Zhu G, Liu Q, et al. BRD4 promotes gastric cancer progression and metastasis through acetylation-dependent stabilization of snail. Cancer Res (2019) 79(19):4869–81. doi: 10.1158/0008-5472.CAN-19-0442
130. Ala M. Target c-myc to treat pancreatic cancer. Cancer Biol Ther (2022) 23(1):34–50. doi: 10.1080/15384047.2021.2017223
131. Mustachio LM, Roszik J, Farria AT, Guerra K, Dent SY. Repression of GCN5 expression or activity attenuates c-MYC expression in non-small cell lung cancer. Am J Cancer Res (2019) 9(8):1830–45.
132. Xenaki G, Ontikatze T, Rajendran R, Stratford IJ, Dive C, Krstic-Demonacos M, et al. PCAF is an HIF-1alpha cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene (2008) 27(44):5785–96. doi: 10.1038/onc.2008.192
133. Lim J-H, Lee Y-M, Chun Y-S, Chen J, Kim J-E, Park J-W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol Cell (2010) 38(6):864–78. doi: 10.1016/j.molcel.2010.05.023
134. Yang MH, Nickerson S, Kim ET, Liot C, Laurent G, Spang R, et al. Regulation of RAS oncogenicity by acetylation. Proc Natl Acad Sci (2012) 109(27):10843–8. doi: 10.1073/pnas.1201487109
135. Song H, Biancucci M, Kang H, O’Callaghan C, Park S, Principe DR, et al. SIRT2 deletion enhances KRAS-induced tumorigenesis in vivo by regulating K147 acetylation status. Oncotarget. (2016) 7(49):80336–49. doi: 10.18632/oncotarget.12015
136. Markham D, Munro S, Soloway J, O’Connor DP, La Thangue NB. DNA-Damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO Rep (2006) 7(2):192–8. doi: 10.1038/sj.embor.7400591
137. Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat Cell Biol (2001) 3(7):667–74. doi: 10.1038/35083062
138. Kai L, Samuel SK, Levenson AS. Resveratrol enhances p53 acetylation and apoptosis in prostate cancer by inhibiting MTA1/NuRD complex. Int J Cancer (2010) 126(7):1538–48. doi: 10.1002/ijc.24928
139. Jeong M-H, Ko H, Jeon H, Sung G-J, Park S-Y, Jun WJ, et al. Delphinidin induces apoptosis via cleaved HDAC3-mediated p53 acetylation and oligomerization in prostate cancer cells. Oncotarget (2016) 7(35):56767–80. doi: 10.18632/oncotarget.10790
140. Li Q, Hao Q, Cao W, Li J, Wu K, Elshimali Y, et al. PP2Cδ inhibits p300-mediated p53 acetylation via ATM/BRCA1 pathway to impede DNA damage response in breast cancer. Sci Adv (2022) 5(10):eaaw8417. doi: 10.1126/sciadv.aaw8417
141. Wang J, Qian J, Hu Y, Kong X, Chen H, Shi Q, et al. ArhGAP30 promotes p53 acetylation and function in colorectal cancer. Nat Commun (2014) 5(1):4735. doi: 10.1038/ncomms5735
142. Cai W, Su L, Liao L, Liu ZZ, Langbein L, Dulaimi E, et al. PBRM1 acts as a p53 lysine-acetylation reader to suppress renal tumor growth. Nat Commun (2019) 10(1):5800. doi: 10.1038/s41467-019-13608-1
143. Chen H, Zhang W, Cheng X, Guo L, Xie S, Ma Y, et al. β2-AR activation induces chemoresistance by modulating p53 acetylation through upregulating Sirt1 in cervical cancer cells. Cancer Sci (2017) 108(7):1310–7. doi: 10.1111/cas.13275
144. De U, Son JY, Sachan R, Park YJ, Kang D, Yoon K, et al. A new synthetic histone deacetylase inhibitor, MHY2256, induces apoptosis and autophagy cell death in endometrial cancer cells via p53 acetylation. Int J Mol Sci (2018) 19(9):2743. doi: 10.3390/ijms19092743
145. Alhebshi H, Tian K, Patnaik L, Taylor R, Bezecny P, Hall C, et al. Evaluation of the role of p53 tumour suppressor posttranslational modifications and TTC5 cofactor in lung cancer. Int J Mol Sci (2021) 22(24):13198. doi: 10.3390/ijms222413198
146. Yu X, Li H, Zhu M, Hu P, Liu X, Qing Y, et al. Involvement of p53 acetylation in growth suppression of cutaneous T-cell lymphomas induced by HDAC inhibition. J Invest Dermatol (2020) 140(10):2009–2022.e4. doi: 10.1016/j.jid.2019.12.041
147. Zhang J, Wang P, Cui Y. Long noncoding RNA NEAT1 inhibits the acetylation of PTEN through the miR-524-5p/HDAC1 axis to promote the proliferation and invasion of laryngeal cancer cells. Aging (Albany, NY). (2021) 13(22):24850–65. doi: 10.18632/aging.203719
148. Meng Z, Jia L-F, Gan Y-H. PTEN activation through K163 acetylation by inhibiting HDAC6 contributes to tumour inhibition. Oncogene (2016) 35(18):2333–44. doi: 10.1038/onc.2015.293
149. Qian Y, Liu Z, Yan H, Yuan Y, Levenson AS, Li K. Pterostilbene inhibits MTA1/HDAC1 complex leading to PTEN acetylation in hepatocellular carcinoma. BioMed Pharmacother (2018) 101:852–9. doi: 10.1016/j.biopha.2018.03.022
150. Taparra K, Wang H, Malek R, Lafargue A, Barbhuiya MA, Wang X, et al. O-GlcNAcylation is required for mutant KRAS-induced lung tumorigenesis. J Clin Invest (2018) 128(11):4924–37. doi: 10.1172/JCI94844
151. Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell (2014) 54(5):820–31. doi: 10.1016/j.molcel.2014.04.026
152. Li H, Al-Japairai K, Tao Y, Xiang Z. RPN2 promotes colorectal cancer cell proliferation through modulating the glycosylation status of EGFR. Oncotarget (2017) 8(42):72633–51. doi: 10.18632/oncotarget.20005
153. Jia C, Li H, Fu D, Lan Y. GFAT1/HBP/O-GlcNAcylation axis regulates β-catenin activity to promote pancreatic cancer aggressiveness. BioMed Res Int (2020) 2020:1921609. doi: 10.1155/2020/1921609
Keywords: tumor suppressor gene, post-translational modification, metabolites, tumor microenvironment, oncogenic signaling
Citation: Liu Y, Vandekeere A, Xu M, Fendt S-M and Altea-Manzano P (2022) Metabolite-derived protein modifications modulating oncogenic signaling. Front. Oncol. 12:988626. doi: 10.3389/fonc.2022.988626
Received: 07 July 2022; Accepted: 23 August 2022;
Published: 23 September 2022.
Edited by:
Reinhold Schafer, Charité Universitätsmedizin Berlin, GermanyReviewed by:
Chunming Cheng, The Ohio State University, United StatesCopyright © 2022 Liu, Vandekeere, Xu, Fendt and Altea-Manzano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah-Maria Fendt, sarah-maria.fendt@kuleuven.be; Patricia Altea-Manzano, patricia.alteamanzano@kuleuven.be
†These authors have contributed equally to this work