 Robert J. Salmond
Robert J. Salmond
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 25 September 2018
Sec. Signaling
Volume 6 - 2018 | https://doi.org/10.3389/fcell.2018.00122
This article is part of the Research Topic The Warburg Effect Regulation Under Siege: The Intertwined Pathways in Health and Disease View all 12 articles
T cell activation, differentiation and effector function is intrinsically linked to the regulation of metabolic pathways. Evidence has shown that inflammatory T cell responses are dependent upon the adoption of aerobic glycolytic metabolism. Furthermore, activation and regulation of the mechanistic target of rapamycin signaling pathway serves a key determinant of T cell metabolism, with subsequent effects on T cell effector responses. In this mini-review, we discuss the mechanisms underpinning the function of the Warburg effect in T cell responses and the role of mTOR in these processes.
T cells serve as cellular effectors and orchestrators of adaptive immune responses during infection and cancer. In the past decade, a wealth of data has determined that T cell activation, clonal expansion, effector differentiation, and function is closely linked to and dependent upon the regulation of basic cellular metabolic processes. It has become clear that in effector T cells, the pyruvate produced by glycolysis is preferentially fermented to lactate even in the presence of oxygen; a classic example of the Warburg effect in non-transformed cells. In this review, we discuss how the engagement of aerobic glycolysis influences T cell activation and describe the role of the mechanistic target of rapamycin (mTOR) pathway in these processes.
Prior to encountering antigen, T cells are quiescent and lack effector function. These naïve T cells uptake low levels of glucose and amino acids, and rely on mitochondrial oxidative phosphorylation (OXPHOS) to maintain cellular ATP levels [reviewed in Geltink et al. (2018)]. Naïve T cells may survive for years circulating through the blood and lymph, only rarely undergoing cell division. Upon encounter with peptide antigen-major histocompatibility complexes (MHC) presented by antigen-presenting cells, the differentiation of naïve CD4+ T cells to a plethora of specialized helper T cell (Th) subsets enables the immune system to respond appropriately to a huge variety of pathogens, from extracellular parasitic worms to intracellular viruses and bacteria. In this regard, CD4+ Th cells modulate the activity and function of innate and adaptive immune cells by secreting cytokines. CD4+ Th1 cells promote cell-mediated immunity by secreting interleukin (IL)-2, interferon (IFN)-γ and tumor necrosis factor (TNF) whereas Th2 cells promote humoral immunity through the production of IL-4, IL-5 and IL-13 (Asnagli and Murphy, 2001). Th17 cells produce high levels of IL-17 and are important for maintenance of homeostasis and protection from pathogens at barrier sites, such as the intestine (Stockinger and Omenetti, 2017). By contrast, regulatory CD4+ T cells (Treg), characterized by expression of the transcription factor forkhead box P3 (FOXP3), have a key role in limiting inflammation and preventing autoimmunity by suppressing the activity of other immune cell types (Sakaguchi et al., 2010). Cytotoxic CD8+ T cells have the capacity to target and kill infected and transformed cells, and produce inflammatory cytokines such as IFNγ (Halle et al., 2017). Upon resolution of an immune response, a number of memory T cell populations capable of responding rapidly to a second antigenic encounter are retained, facilitating life-long protection from re-infection.
The processes of T cell activation are bioenergetically expensive; for example, it has been estimated that, during infection, virus-specific CD8+ T cells undergo rapid proliferation with a population doubling time of only ∼8 h (De Boer et al., 2003). Therefore, a key question in immunology is: how do T cells fuel the processes of activation, proliferation and differentiation? Whereas cytokines such as IL-7 maintain low level glycolytic metabolism in naïve T cells (Jacobs et al., 2010), triggering of the T cell antigen receptor (TCR) by cognate peptide antigen-MHC presented on the surface of antigen-presenting cells, results in the upregulation of anabolic biosynthetic pathways in order to facilitate T cell activation. The integration of TCR, CD28 co-stimulation and cytokine receptor signals determines T cell metabolism and subsequently impacts upon differentiation, and effector function (Fox et al., 2005; Cornish et al., 2006; Jacobs et al., 2010; Michalek et al., 2011; Shi et al., 2011; Finlay et al., 2012; Gubser et al., 2013; Ray et al., 2015; Richer et al., 2015; Tan et al., 2017; Geltink et al., 2018). The regulation of aerobic glycolysis is central to these fate decisions.
An important role for glucose uptake, and glycolysis in T cell function was suggested four decades ago by the demonstration that the glycolysis inhibitor 2-deoxyglucose (2-DG) impaired T cell cytotoxic capacity (MacDonald, 1977; MacDonald and Cerottini, 1979). Furthermore, studies indicated that 2-DG treatment selectively reduced the expression of key effector molecules, including IFNγ and granzymes, and cell cycle proteins in both mouse (Cham and Gajewski, 2005; Cham et al., 2008) and human (Renner et al., 2015) CD8+ T cells. At low doses that do not impact upon TCR-induced proliferation, 2-DG also inhibits CD4+ Th2 (Yang et al., 2016) and Th17 (Shi et al., 2011) cell differentiation, but promotes Treg differentiation (Shi et al., 2011). Together, these studies indicate that the regulation of glycolytic flux plays a central role in cell fate decisions, and T cell differentiation. In recent years, mass-spectrometry based proteomic analyses have further informed our understanding of the extent to which the regulation of metabolic pathways is prioritized by T cells. Thus, studies from the Cantrell lab have shown that 41 glycolytic proteins represent 7% of the total protein molecules in effector cytotoxic CD8+ T cells (Hukelmann et al., 2016).
Upon TCR triggering, expression of plasma membrane glucose transporters is enhanced as part of the general process of metabolic reprogramming. T cell-specific knockout of the glucose transporter SLC2A1/GLUT1 substantially inhibited the activation of mouse CD4+ T cells (Macintyre et al., 2014). Whilst the homeostasis and survival of naïve T cells was unaffected by the absence of GLUT1, TCR-induced CD4+ T cell growth, and proliferation were profoundly impaired. Furthermore, differentiation of Slc2a1−/− T cells to effector Th1, Th2 and Th17, but not Treg, lineages was blocked (Macintyre et al., 2014), consistent with the known effects on T cell differentiation of inhibiting glycolytic flux with 2-DG. As anticipated, T cell activation defects in TCR-stimulated Slc2a1−/− T cells were associated with reduced rates of glucose uptake, glycolysis and lactate production (Macintyre et al., 2014). The lack of a catastrophic impact of GLUT1-deficiency on glycolytic flux, and cell survival in naïve T cells is likely to be a consequence of expression of additional glucose transporters, including GLUT3, by T cells (Macintyre et al., 2014; Hukelmann et al., 2016). The importance of glucose uptake in T cell responses in vivo has been further highlighted by recent studies indicating that T cells and cancer cells directly compete for nutrients in the tumor microenvironment (Chang et al., 2015; Ho et al., 2015; Siska et al., 2017). Thus, highly glycolytic tumor variants suppress the activity of anti-tumor T cells, at least in part, by reducing the bioavailability of glucose.
In addition to upregulating glycolytic metabolism, activated T cells also increase uptake and hydrolysis of amino acids such as glutamine, and modulate mitochondrial, and lipid metabolism [reviewed in Geltink et al. (2018)]. Distinct T cell populations differ in their utilization, and dependence upon these metabolic programs. Effector CD8+ T cells, and Th1, Th2 and Th17 CD4+ T cells are highly glycolytic, whereas Tregs are dependent upon fatty acid oxidation (FAO) (Michalek et al., 2011; Shi et al., 2011 Berod et al., 2014) (Figure 1). Based on the use of chemical inhibitors, FAO has also been suggested to be important for the development of memory T cells (reviewed in (Lochner et al., 2015)); although recent evidence using genetic mouse models suggest that the requirement for FAO is not absolute (Pan et al., 2017; Raud et al., 2018). The use of electron microscopy has determined that memory T cells have altered mitochondrial morphology with fused cristae, that appears to favor OXPHOS and FAO (Buck et al., 2016). Furthermore, a recent study identified a crucial role for CD28 co-stimulatory signals during initial T cell activation to ‘prime’ mitochondria with elevated spare respiratory capacity, that is necessary for the rapid recall responses of memory T cells (Klein Geltink et al., 2017). The ability of quiescent memory T cells to re-acquire effector function rapidly upon TCR triggering is also dependent upon immediate re-engagement of glycolysis (Gubser et al., 2013; Klein Geltink et al., 2017). Therefore, in general terms, a highly glycolytic metabolism is associated with T cell effector responses, whereas low level glycolysis and lipid metabolism is associated with memory and regulatory T cell responses.
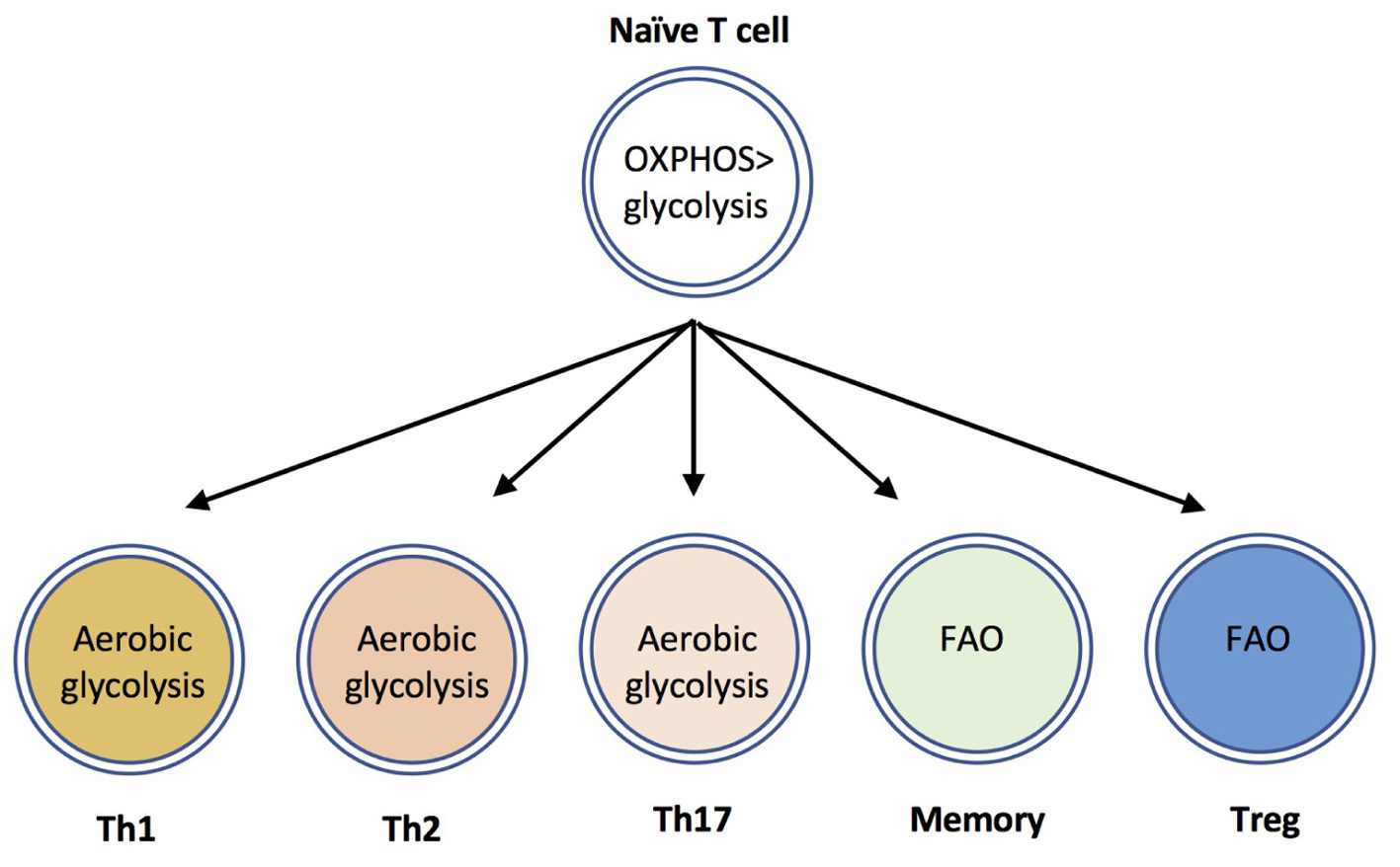
FIGURE 1. CD4+ T cell subsets use distinct metabolic programs. Naïve T cells uptake low levels of glucose and primarily utilize mitochondrial oxidative phosphorylation to maintain homeostasis. The differentiation and effector functions of inflammatory Th1, Th2, and Th17 cells relies on engagement of aerobic glycolysis. By contrast, memory T cells and Tregs are dependent upon fatty acid oxidation (FAO) pathways.
The function of the Warburg effect in activated effector T cells is incompletely understood. Aerobic glycolysis is an inefficient means of energy production, producing only 2 molecules of ATP as compared to between 30 and 36 produced by OXPHOS. Furthermore, experiments using the ATP synthase inhibitor oligomycin demonstrated that mitochondrial ATP production via OXPHOS is required for initial stages of T cell activation and proliferation (Chang et al., 2013). By contrast, data indicate that expression of the rate-limiting glycolytic enzyme hexokinase 2 (HK2) is actually dispensable for early stages of T cell activation (Tan et al., 2017). It has been suggested that a key advantage of the Warburg effect for cancer cells, and presumably for all proliferating cells, is that it allows the metabolic flexibility required to build biomass (Vander Heiden et al., 2009). Thus, a key function of a switch to aerobic glycolysis might be to enable T cells to use glucose for the generation of biosynthetic precursors for amino acids and nucleic acids, critical for rapid growth, and population expansion, via the pentose phosphate pathway (PPP) (reviewed in (Lunt and Vander Heiden, 2011)). Carbon tracing experiments indicate that, in activated T cells, up to 85% of glucose is excreted as lactate (Fox et al., 2005), indicating that only a minor proportion of glucose-derived carbon is used to fuel biosynthetic pathways. Rather, the NADPH generated by the PPP is rate-limiting in the production of amino acids, nucleic acids, and fatty acids in T cells, and it is likely that aerobic glycolysis allows a faster flux through this pathway as compared to mitochondrial respiration (Vander Heiden et al., 2009). Indeed, blockade of lactate excretion using pharmacological inhibitors of the monocarboxylate transporter MCT1 inhibits T cell proliferation (Murray et al., 2005). Furthermore, a number of recent studies have provided evidence that elevated glucose uptake, and engagement of aerobic glycolysis modulates T cell effector responses through additional mechanisms.
Chang et al. (2013) demonstrated an important role for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in regulating effector T cell function via a post-transcriptional mechanism. These researchers showed that, in addition to functioning as a glycolytic enzyme, GAPDH binds to the 3′-untranslated region (UTR) of IFNγ mRNA and prevents efficient translation (Chang et al., 2013). By engaging glycolysis, effector T cells sequester GAPDH away from IFNγ mRNA and thereby enhance cytokine production. Further mechanistic insight into the role of glycolysis in inflammatory cytokine production comes from studies of lactate dehydrogenase A (LDHA) function in T cells. LDHA expression is enhanced in activated T cells and is required to support aerobic glycolysis (Peng et al., 2016). Furthermore, IFNγ production was reduced in LDHA-deficient CD4+ Th1 cells as compared to control cells, consistent with the known role of glycolysis in T cell effector function. This effect was independent of the Ifng 3′-UTR, indicating a distinct effect on cytokine production from that mediated by GAPDH. In the absence of LDHA, histone 3 acetylation at lysine 9 (H3K9Ac) and lysine 27 (H3K27Ac) within the Ifng promoter region was substantially decreased (Peng et al., 2016). This glycolysis-dependent epigenetic regulation of IFNγ expression via histone acetylation was mediated by LDHA-dependent maintenance of high levels of acetyl-CoA in effector Th1 cells (Peng et al., 2016).
Studies from the Kaech laboratory revealed a further role for glycolytic flux in T cell activation. Thus, production of the glycolytic metabolite phosphoenolpyruvate (PEP) via enolase promotes prolonged Ca2+ responses and activation of the transcription factor nuclear factor of activated T cells (NFAT) (Ho et al., 2015). Nuclear translocation and the transcriptional activity of NFAT regulates the expression of key effector molecules such as IL-2, IFNγ and CD40L in T cells (Hogan, 2017). PEP binds and inhibits the activity of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), preventing transfer of Ca2+ from the cytosol to the SR and prolonging NFAT activation (Ho et al., 2015). Importantly, defects in Ca2+/NFAT signaling and T cell activation under conditions of low glucose could be partially corrected by restoration of PEP levels following enforced expression of the gluconeogenesis enzyme PEP carboxykinase 1 (PEPCK1) (Ho et al., 2015). Furthermore, PEPCK1-overexpressing CD4+ (Ho et al., 2015) and CD8+ (Ma et al., 2018) T cells had elevated anti-tumor responses as compared to control cells, indicating that PEP production serves as a metabolic checkpoint in vivo. In NFATc1-deficient T cells, transcript levels of glycolytic proteins such as GLUT1, GLUT3 and HK2 were substantially reduced with a concomitant impairment of glycolytic flux, an effect that could be rescued partially by IL-2 (Klein-Hessling et al., 2017). Therefore NFATc1 regulates T cell activation and upregulation of the glycolytic pathway, which in turn acts in a positive-feedback loop to prolong NFATc1 signaling via PEP.
A novel role for the glycolytic enzyme enolase-1 (Eno1) in inducible Treg function was recently described by Materese and colleagues. These researchers showed that inhibition of glycolysis using 2-DG limited FOXP3 gene splicing and expression in human Tregs (De Rosa et al., 2015). In Tregs treated with 2-DG, a substantially increased proportion of Eno1 was recruited to the FOXP3 promoter and regulatory elements, whilst shRNA knockdown of Eno1 expression restored FOXP3 expression (De Rosa et al., 2015). These data suggest that nuclear Eno1 regulates FOXP3 splicing and that engagement of the glycolytic function of Eno1 interferes with this nuclear role, thereby stabilizing the Treg phenotype and function.
A further key role for glucose in T cell activation is to fuel protein O-GlcNacylation. In this pathway, glucose is diverted from the glycolytic pathway (at the level of fructose-6-phosphate) into the hexosamine biosynthetic pathway, which ultimately provides the donor substrate for O-GlcNacylation (Yang and Qian, 2017). TCR triggering results in a substantial increase in the pool of intracellular UDP-GlcNac, resulting in post-translational modification of Ser / Thr residues, and modifying the activity or stability of key proteins, including c-Myc (Swamy et al., 2016). Experiments investigating the impact of T cell-specific deletion of O-GlcNAc transferase (OGT) demonstrated a requirement for this pathway in T cell development in the thymus as well as the clonal expansion of mature T cells (Swamy et al., 2016). Supplementation of in vitro T cell cultures with GlcNAc favors Treg differentiation, at the expense of inflammatory Th17 cells, by promoting IL-2R signaling (Araujo et al., 2017). Thus, it is possible that aerobic glycolysis might impinge on Treg differentiation by limiting the supply of metabolites to the hexosamine and O-GlcNAc biosynthetic pathways.
In summary, it is clear that engagement of aerobic glycolysis impacts on T cell function through a number of distinct mechanisms: (i) glycolysis provides a source of ATP and enables the production of biosynthetic precursors to enable proliferation and cell growth; (ii) engagement of the glycolytic pathway and enzymes such as Eno1 and GAPDH diverts their function away from non-glycolytic functions that impinge on T cell gene expression; (iii) glycolytic metabolites such as PEP have additional signaling functions in T cells; (iv) the engagement of glycolysis interacts in a complex network with additional metabolic pathways such as the hexosamine pathway and glutaminolysis to regulate T cell behavior. Further investigation into the function of the Warburg effect in T cells will, no doubt, add to this list of mechanisms in the coming years.
The signaling pathways that regulate T cell metabolic reprogramming have been the subject of intense research in the past decade. mTOR is an evolutionarily conserved ser/thr kinase that, in T cells, integrates nutrient sensing and antigen-receptor signaling (reviewed in (Salmond and Zamoyska, 2010, 2011; Powell et al., 2012)). mTOR forms two main signaling complexes, mTORC1 and mTORC2, that differ in their sensitivity to the macrolide inhibitor rapamycin. mTORC1 is composed of mTOR in complex with the adapter protein raptor, mammalian lethal with SEC13 protein 8 (MLST8) and proline-rich Akt substrate (PRAS) 1, an endogenous regulator DEPTOR, and is sensitive to rapamycin. By contrast, mTORC2, composed of mTOR, rictor, GβL, and mammalian stress-activated protein kinase interacting protein 1 (mSIN1), is insensitive to acute inhibition by rapamycin. The pathways that regulate mTOR activation in T cells are summarized in Figure 2. In brief, mTORC1 activity is regulated by intracellular amino acids via the nutrient sensing Rag GTPases (Wolfson and Sabatini, 2017). Upon TCR stimulation, T cells upregulate the expression of plasma membrane transporters that enable the uptake of amino acids such as leucine and glutamine from the extracellular environment, that in turn sustain mTORC1 activation. Knockout mouse studies have shown that upregulation of the System L amino acid transporter SLC7A5 (Sinclair et al., 2013) and glutamine-transporter SLC1A5 (Nakaya et al., 2014) are both essential for mTORC1 activity in T cells. Glucose levels also regulate mTORC1 by influencing the activity of the negative regulator AMP kinase (AMPK) (Rolf et al., 2013). Furthermore, recent work has shown that, following TCR signaling, the kinase activity of mTORC1 is activated via the upstream kinase PDK1, in a PI3K/Akt-independent manner (Finlay et al., 2012). In addition, co-stimulation through CD28 and signaling mediated via cytokines such as IL-2 and IL-15 contribute to the magnitude of mTOR activation in T cells (Cornish et al., 2006; Ray et al., 2015). Key downstream targets / effectors of mTORC1 include the translational regulators 4E-binding proteins (4E-BPs) and ribosomal protein S6 kinases (S6Ks). The mechanism by which mTORC2 is activated is less well understood but likely involves PI3K/Akt activity (Zinzalla et al., 2011; Yang et al., 2015). mTORC2 targets include Akt and serum and glucocorticoid-induced protein kinase (SGK).
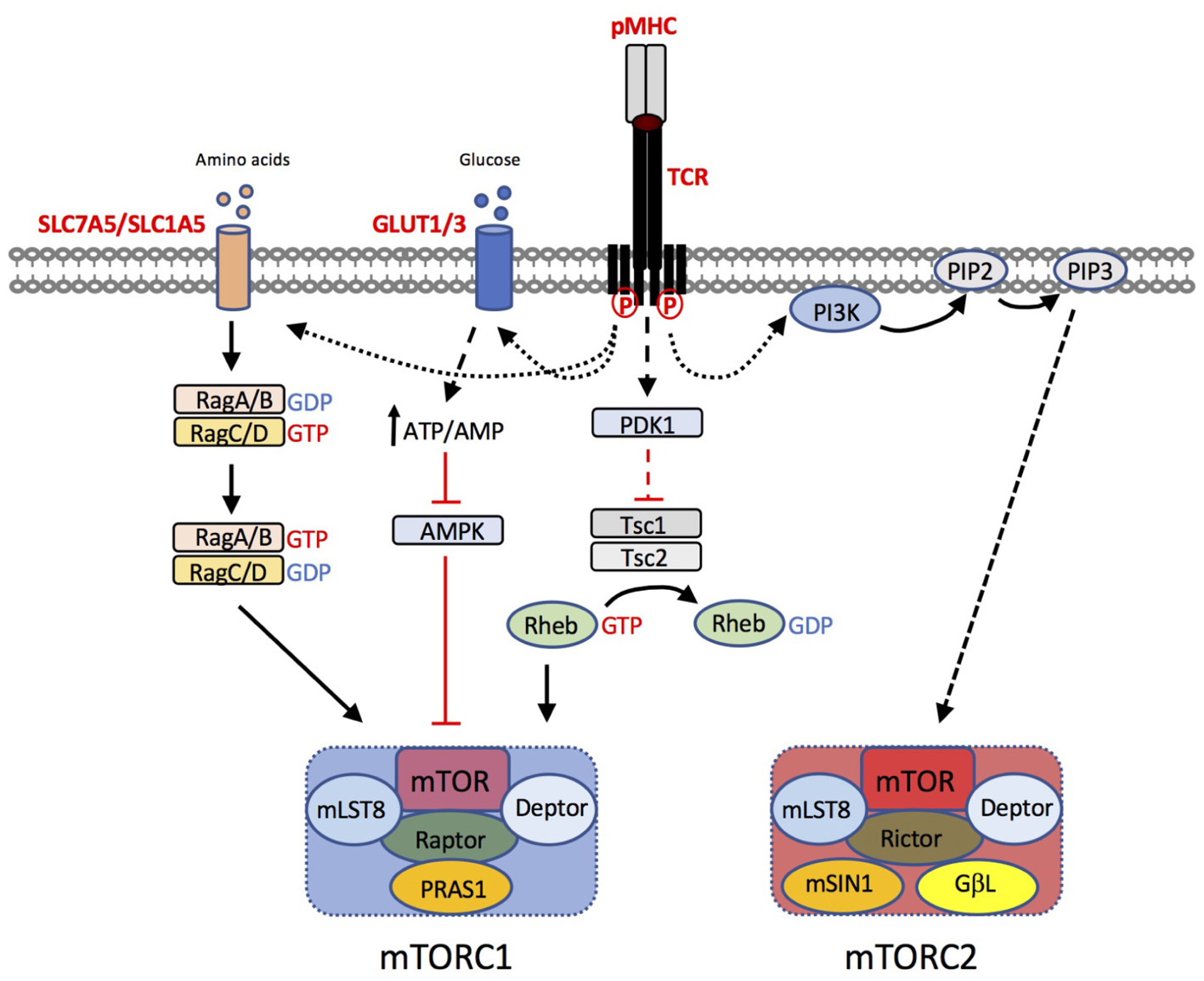
FIGURE 2. T cell pathways to mTORC1 and mTORC2 activation. T cell receptor (TCR) triggering by peptide (p)MHC complexes results in upregulation of amino acid transporters such as SLC7A5 and SLC1A5. Leucine and glutamine are critical amino acids for the activation of the Rag GTPases, that in turn regulate mTORC1 activation at the lysosome. TCR induced upregulation of glucose transporters GLUT1 and GLUT3 enhances glucose uptake. Increases in the intracellular ATP:AMP ratio as a consequence of increased glucose availability, suppresses the activity of AMP kinase (AMPK), preventing its inhibitory effect on mTORC1 activity. Furthermore, TCR-induced phosphoinositide-dependent kinase (PDK)1, is critical for mTOR activation. PDK1 dependent signals prevent the GTPase activating protein (GAP) activity of the Tsc1/Tsc2 complex. Loss of Tsc GAP activity enables Rheb-GTP to activate mTORC1 through an incompletely understood mechanism. The mechanism of mTORC2 activation is less well understood but is likely to be downstream of PI3K/PIP3-dependent pathways.
Whilst the anti-proliferative and immunosuppressive properties of rapamycin have been known for decades, seminal studies published in 2009 determined that mTOR activity also influences T cell effector-memory cell fate decisions in vivo (Araki et al., 2009). Thus, rapamycin treatment enhanced the quantity and quality of virus-specific CD8+ memory T cells in mice. Knockdown of mTORC1 targets S6K1 and 4E-BPs also impacted upon T cell memory differentiation (Araki et al., 2009). Consistent with these findings, activation of the AMPK1 pathway via metformin or via glucose-deprivation restrains mTOR activity (Pearce et al., 2009; Rolf et al., 2013) and enhances T cell memory. Furthermore, experiments have shown that IL-2 drives high levels of mTOR activation and effector CD8+ T cell differentiation, whereas IL-15 drives lower levels of mTOR activation and favors memory cell formation (Cornish et al., 2006; Pipkin et al., 2010; Ray et al., 2015; Richer et al., 2015). Recent studies examining daughter cells from the first cell division following TCR stimulation indicate that mTORC1 activity is asymmetrically inherited (Pollizzi et al., 2016). Importantly, the asymmetric inheritance of mTORC1 influences T cell metabolic capacity and cell fate. Thus, daughter cells with high mTORC1 activity had elevated glycolytic flux and generated T cell populations with enhanced effector capacity, whereas cells with lower mTORC1 activity generated long-lived memory cells (Pollizzi et al., 2016).
Studies using mice with T cell-specific deficiencies in mTOR or with selective ablation or hyperactivation of mTORC1 or mTORC2 signaling pathways have defined an important role for both of these signaling complexes in T cell activation, differentiation and effector function (Delgoffe et al., 2009, 2011; Lee et al., 2010; Yang et al., 2011; Zeng et al., 2013, 2016; Pollizzi et al., 2015). CD4+ T cells completely lacking mTOR fail to differentiate into Th1, Th2 or Th17 lineages and instead differentiate preferentially into FOXP3+ regulatory T cells (Delgoffe et al., 2009). Nonetheless, whilst mTORC1 signals negatively regulate de novo Treg differentiation, the suppressive function of fully differentiated Treg also requires mTORC1 activity (Zeng et al., 2013; Gerriets et al., 2016; Chapman et al., 2018; Sun et al., 2018). T cells deficient in the upstream activator of mTORC1, Rheb, are defective in Th1 and Th17 differentiation (Delgoffe et al., 2011) whilst raptor deficiency also impinges upon Th2 differentiation (Yang et al., 2013). By contrast, deletion of tuberous sclerosis 1 (Tsc1) or Tsc2 results in hyperactive mTORC1 and a subsequent loss of naïve T cell quiescence, indicating that restraining mTOR activity is important for the maintenance of immune homeostasis (Yang et al., 2011; Pollizzi et al., 2015). Rictor deficiency (i.e., loss of mTORC2) has a milder effect on Th1 cell activation in vivo as compared to loss of mTORC1 function (Yang et al., 2013) but compromises CD4+ Th2 differentiation (Lee et al., 2010). Similarly, Rheb/mTORC1-dependent signals are also required for CD8+ T cell differentiation whilst rictor/mTORC2-dependent signals regulate CD8+ T cell memory (Pollizzi et al., 2015; Zhang et al., 2016).
Thus, studies of knockout mouse models have shed significant insight into the multifarious roles of mTOR in T cell differentiation and effector function. In this regard, mTORC1 has dual and apparently opposing roles in Treg biology; on the one hand, elevated mTORC1 favors the differentiation of effector T cells at the expense of Tregs, whilst on the other, mTOR expression in Tregs is essential to prevent autoimmunity. Furthermore, these studies have suggested distinct roles for the mTORC1 and mTORC2 complexes in T cells; for example, in Th1 vs. Th2 differentiation. Importantly, evidence indicates that the regulation of T cell metabolism by mTOR complexes is central to these complex phenotypes.
In T cells, mTORC1 signaling serves to promote aerobic glycolysis and as a consequence impacts upon T cell differentiation and effector function. Rapamycin treatment substantially impairs the initial TCR-induced upregulation of glucose transporters, glucose uptake and glycolytic enzymes in both CD4+ and CD8+ T cells (Shi et al., 2011; Finlay, 2012). Similarly, genetic ablation of Rheb (Pollizzi et al., 2015) or raptor (Yang et al., 2013) impairs the upregulation of aerobic glycolysis in TCR-stimulated T cells, whilst hyperactivation of mTORC1 in Tsc1 or Tsc2-deficient T cells is associated with enhanced glycolytic metabolism. Furthermore, mTORC1 activity is required to sustain high levels of aerobic glycolysis in effector T cells (Finlay, 2012; Hukelmann et al., 2016). In this regard, rapamycin treatment caused an approximate 50% reduction in levels of GLUT1 and GLUT3 in IL-2 maintained effector CTLs and a proportional decrease in glucose uptake and lactate production (Hukelmann et al., 2016). By contrast, inhibition of mTORC2 activity actually increases the metabolic capacity of CD8+ T cells. Thus, Rictor-deficient CD8+ T cells have elevated glycolytic flux, spare respiratory capacity (SRC) and FAO (Pollizzi et al., 2015; Zhang et al., 2016). The mechanism by which deletion of mTORC2 results in increased metabolic fitness has not been fully elucidated but may involve stabilization of nuclear Foxo1 transcription factor (Zhang et al., 2016). Thus, knockdown of Foxo1 reverses the impact of Rictor-deficiency on T cell memory formation whilst expression of a constitutively active Foxo1 in CD8+ T cells results in elevated SRC and FAO (Zhang et al., 2016).
The molecular mechanisms by which mTORC1 signals regulate glycolytic pathways in T cells are also incompletely understood. Studies have identified transcription factors including Myc (Wang et al., 2011) and hypoxia inducible factor 1 alpha (HIF-1α) (Shi et al., 2011; Finlay et al., 2012) as key drivers of metabolic reprogramming in T cells. Myc-deficient T cells are defective in TCR-induced upregulation of glucose transporters and glycolytic enzymes and have substantially reduced glycolytic flux (Wang et al., 2011). HIF-1α is upregulated strongly in Th17 cells (Shi et al., 2011) and effector CD8+ T cells (Finlay et al., 2012) and, similar to Myc, is important for the upregulation of aerobic glycolysis in these T cell subsets. Importantly, rapamycin impairs the TCR-induced expression of both Myc and HIF-1α (Shi et al., 2011; Wang et al., 2011; Finlay et al., 2012; Pollizzi et al., 2016) indicating that mTOR serves to regulate aerobic glycolysis, at least in part, through regulation of Myc and HIF-1α expression and their subsequent downstream transcriptional programs. mTORC1 has been reported to regulate Myc expression via post-transcriptional mechanisms as levels of Myc protein, but not mRNA, were reduced in Raptor-deficient T cells as compared to controls (Yang et al., 2013).
In addition to the role of mTOR in promoting glycolytic metabolism in effector T cells, recent evidence has shown that mTOR also has a vital role in the regulation of mitochondrial metabolism. Gene expression and pathway analysis identified the regulation of both glycolysis and OXPHOS as being significantly impacted by hyperactive mTOR activity in Tsc1−/− T cells (Shrestha et al., 2014). Furthermore, mTOR catalytic site inhibitors reduced expression of mitochondrial and OXPHOS genes in activated Tregs (Chapman et al., 2018). In both cases, mTOR activity was required for the transcriptional programs driving OXPHOS in T cells, highlighting the dual role for this kinase in regulating mitochondrial and glycolytic metabolism.
The use of rapamycin/sirolimus as an immunosuppressive agent in the clinic was approved by the US FDA in 1999. Rapamycin and its derivatives have been used extensively in transplantation to limit organ rejection, however, recent evidence has shown that these compounds may have broader applicability in the treatment of cancers and autoimmunity, as well as in vaccine design (reviewed in Perl, 2015, 2016). Importantly, the clinical benefits of mTOR blockade in inflammatory diseases has been linked to the modulation of T cell metabolism. For example, inflammatory T cells from systemic lupus erythematosus (SLE) patients have substantially elevated glycolytic and mitochondrial metabolism (Yin et al., 2015) and mTOR activity (Kato and Perl, 2014), as compared to healthy controls. Importantly, in both mouse models and in human patients, T cell metabolism and inflammatory cytokine production could be normalized by reducing mTOR activity through metformin or rapamycin treatment (Kato and Perl, 2014; Yin et al., 2015). Furthermore, and consistent with an important role for mTOR-driven inflammatory T cells in the pathogenesis of lupus, a recent phase 1/2 trial reported that rapamycin had a beneficial impact on clinical disease scores in a cohort of 43 SLE patients (Lai et al., 2018). Improved disease outcomes were associated with decreased inflammatory T cell activity and increased Treg numbers (Lai et al., 2018), consistent with the known role of mTOR in regulating T cell metabolism and effector responses.
Our understanding of the close links between the regulation of aerobic glycolysis and T cell function has been transformed in the past decade. Furthermore, mTOR signals have emerged as a key driver of these processes. As our understanding of the molecular details and signaling pathways leading to metabolic reprogramming increases, then the opportunity to translate these findings into the clinic should emerge. In this regard, evidence for distinct roles for mTORC1 and mTORC2 in modulating T cell metabolism and activation gives scope for more precise manipulation of these pathways in the future.
RS wrote the manuscript and generated the figures.
Work in the Salmond lab was supported by a grant from Cancer Research United Kingdom (23269).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
RS wishes to thank Dr. R. Brownlie for critically reviewing the manuscript.
Araki, K., Turner, A. P., Shaffer, V. O., Gangappa, S., Keller, S. A., Bachmann, M. F., et al. (2009). mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112. doi: 10.1038/nature08155
Araujo, L., Khim, P., Mkhikian, H., Mortales, C. L., and Demetriou, M. (2017). Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. eLife 6:e21330. doi: 10.7554/eLife.21330
Asnagli, H., and Murphy, K. M. (2001). Stability and commitment in T helper cell development. Curr. Opin. Immunol. 13, 242–247. doi: 10.1016/S0952-7915(00)00210-7
Berod, L., Friedrich, C., Nandan, A., Freitag, J., Hagemann, S., Harmrolfs, K., et al. (2014). De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 20, 1327–1333. doi: 10.1038/nm.3704
Buck, M. D., O’Sullivan, D., Klein, Geltink RI, Curtis, J. D., Chang, C. H., Sanin, D. E., et al. (2016). Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76. doi: 10.1016/j.cell.2016.05.035
Cham, C. M., Driessens, G., O’Keefe, J. P., and Gajewski, T. F. (2008). Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 38, 2438–2450. doi: 10.1002/eji.200838289
Cham, C. M., and Gajewski, T. F. (2005). Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J. Immunol. 174, 4670–4677. doi: 10.4049/jimmunol.174.8.4670
Chang, C. H., Curtis, J. D., Maggi, L. B. Jr, Faubert, B., Villarino, A. V., O’Sullivan, D., et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. doi: 10.1016/j.cell.2013.05.016
Chang, C. H., Qiu, J., O’Sullivan, D., Buck, M. D., Noguchi, T., Curtis, J. D., et al. (2015). Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241. doi: 10.1016/j.cell.2015.08.016
Chapman, N. M., Zeng, H., Nguyen, T. M., Wang, Y., Vogel, P., Dhungana, Y., et al. (2018). mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat. Commun. 9:2095. doi: 10.1038/s41467-018-04392-5
Cornish, G. H., Sinclair, L. V., and Cantrell, D. A. (2006). Differential regulation of T-cell growth by IL-2 and IL-15. Blood 108, 600–608. doi: 10.1182/blood-2005-12-4827
De, Rosa V, Galgani, M., Porcellini, A., Colamatteo, A., Santopaolo, M., Zuchegna, C., et al. (2015). Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 16, 1174–1184. doi: 10.1038/ni.3269
De Boer, R. J., Homann, D., and Perelson, A. S. (2003). Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J. Immunol. 171, 3928–3935. doi: 10.4049/jimmunol.171.8.3928
Delgoffe, G. M., Kole, T. P., Zheng, Y., Zarek, P. E., Matthews, K. L., Xiao, B., et al. (2009). The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844. doi: 10.1016/j.immuni.2009.04.014
Delgoffe, G. M., Pollizzi, K. N., Waickman, A. T., Heikamp, E., Meyers, D. J., Horton, M. R., et al. (2011). The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 12, 295–303. doi: 10.1038/ni.2005
Finlay, D. K. (2012). Regulation of glucose metabolism in T cells: new insight into the role of Phosphoinositide 3-kinases. Front. Immunol. 3:247. doi: 10.3389/fimmu.2012.00247
Finlay, D. K., Rosenzweig, E., Sinclair, L. V., Feijoo-Carnero, C., Hukelmann, J. L., Rolf, J., et al. (2012). PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 209, 2441–2453. doi: 10.1084/jem.20112607
Fox, C. J., Hammerman, P. S., and Thompson, C. B. (2005). Fuel feeds function: energy metabolism and the T-cell response. Nat. Rev. Immunol. 5, 844–852. doi: 10.1038/nri1710
Geltink, R. I. K., Kyle, R. L., and Pearce, E. L. (2018). Unraveling the complex interplay between T cell metabolism and function. Annu. Rev. Immunol. 36, 461–488. doi: 10.1146/annurev-immunol-042617-053019
Gerriets, V. A., Kishton, R. J., Johnson, M. O., Cohen, S., Siska, P. J., Nichols, A. G., et al. (2016). Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 17, 1459–1466. doi: 10.1038/ni.3577
Gubser, P. M., Bantug, G. R., Razik, L., Fischer, M., Dimeloe, S., Hoenger, G., et al. (2013). Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 14, 1064–1072. doi: 10.1038/ni.2687
Halle, S., Halle, O., and Forster, R. (2017). Mechanisms and dynamics of T cell-mediated cytotoxicity In Vivo. Trends Immunol. 38, 432–443. doi: 10.1016/j.it.2017.04.002
Ho, P. C., Bihuniak, J. D., Macintyre, A. N., Staron, M., Liu, X., Amezquita, R., et al. (2015). Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228. doi: 10.1016/j.cell.2015.08.012
Hogan, P. G. (2017). Calcium-NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium 63, 66–69. doi: 10.1016/j.ceca.2017.01.014
Hukelmann, J. L., Anderson, K. E., Sinclair, L. V., Grzes, K. M., Murillo, A. B., Hawkins, P. T., et al. (2016). The cytotoxic T cell proteome and its shaping by the kinase mTOR. Nat. Immunol. 17, 104–112. doi: 10.1038/ni.3314
Jacobs, S. R., Michalek, R. D., and Rathmell, J. C. (2010). IL-7 is essential for homeostatic control of T cell metabolism in vivo. J. Immunol. 184, 3461–3469. doi: 10.4049/jimmunol.0902593
Kato, H., and Perl, A. (2014). Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8- double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J. Immunol. 192, 4134–4144. doi: 10.4049/jimmunol.1301859
Klein Geltink, R. I., O’Sullivan, D., Corrado, M., Bremser, A., Buck, M. D., Buescher, J. M., et al. (2017). Mitochondrial priming by CD28. Cell 171, 385.e11–397.e11. doi: 10.1016/j.cell.2017.08.018
Klein-Hessling, S., Muhammad, K., Klein, M., Pusch, T., Rudolf, R., Flöter, J., et al. (2017). NFATc1 controls the cytotoxicity of CD8(+) T cells. Nat. Commun. 8:511. doi: 10.1038/s41467-017-00612-6
Lai, Z. W., Kelly, R., Winans, T., Marchena, I., Shadakshari, A., Yu, J., et al. (2018). Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet 391, 1186–1196. doi: 10.1016/S0140-6736(18)30485-9
Lee, K., Gudapati, P., Dragovic, S., Spencer, C., Joyce, S., Killeen, N., et al. (2010). Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32, 743–753. doi: 10.1016/j.immuni.2010.06.002
Lochner, M., Berod, L., and Sparwasser, T. (2015). Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 36, 81–91. doi: 10.1016/j.it.2014.12.005
Lunt, S. Y., and Vander Heiden, M. G. (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464. doi: 10.1146/annurev-cellbio-092910-154237
Ma, R., Ji, T., Zhang, H., Dong, W., Chen, X., Xu, P., et al. (2018). A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8(+) T cells. Nat. Cell Biol. 20, 21–27. doi: 10.1038/s41556-017-0002-2
MacDonald, H. R. (1977). Energy metabolism and T-cell-mediated cytolysis. II. selective inhibition of cytolysis by 2-deoxy-D-glucose. J. Exp. Med. 146, 710–719. doi: 10.1084/jem.146.3.710
MacDonald, H. R., and Cerottini, J. C. (1979). Inhibition of T cell-mediated cytolysis by 2-deoxy-D-glucose:dissociation of the inhibitory effect from glycoprotein synthesis. Eur. J. Immunol. 9, 466–470. doi: 10.1002/eji.1830090610
Macintyre, A. N., Gerriets, V. A., Nichols, A. G., Michalek, R. D., Rudolph, M. C., Deoliveira, D., et al. (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72. doi: 10.1016/j.cmet.2014.05.004
Michalek, R. D., Gerriets, V. A., Jacobs, S. R., Macintyre, A. N., MacIver, N. J., Mason, E. F., et al. (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303. doi: 10.4049/jimmunol.1003613
Murray, C. M., Hutchinson, R., Bantick, J. R., Belfield, G. P., Benjamin, A. D., Brazma, D., et al. (2005). Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat. Chem. Biol. 1, 371–376. doi: 10.1038/nchembio744
Nakaya, M., Xiao, Y., Zhou, X., Chang, J. H., Chang, M., Cheng, X., et al. (2014). Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40, 692–705. doi: 10.1016/j.immuni.2014.04.007
Pan, Y., Tian, T., Park, C. O., Lofftus, S. Y., Mei, S., Liu, X., et al. (2017). Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256. doi: 10.1038/nature21379
Pearce, E. L., Walsh, M. C., Cejas, P. J., Harms, G. M., Shen, H., Wang, L. S., et al. (2009). Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107. doi: 10.1038/nature08097
Peng, M., Yin, N., Chhangawala, S., Xu, K., Leslie, C. S., and Li, M. O. (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484. doi: 10.1126/science.aaf6284
Perl, A. (2015). mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann. N. Y. Acad. Sci. 1346, 33–44. doi: 10.1111/nyas.12756
Perl, A. (2016). Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat. Rev. Rheumatol. 12, 169–182. doi: 10.1038/nrrheum.2015.172
Pipkin, M. E., Sacks, J. A., Cruz-Guilloty, F., Lichtenheld, M. G., Bevan, M. J., and Rao, A. (2010). Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90. doi: 10.1016/j.immuni.2009.11.012
Pollizzi, K. N., Patel, C. H., Sun, I. H., Oh, M. H., Waickman, A. T., Wen, J., et al. (2015). mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J. Clin. Invest. 125, 2090–2108. doi: 10.1172/JCI77746
Pollizzi, K. N., Sun, I. H., Patel, C. H., Lo, Y. C., Oh, M. H., Waickman, A. T., et al. (2016). Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat. Immunol. 17, 704–711. doi: 10.1038/ni.3438
Powell, J. D., Pollizzi, K. N., Heikamp, E. B., and Horton, M. R. (2012). Regulation of immune responses by mTOR. Annu. Rev. Immunol. 30, 39–68. doi: 10.1146/annurev-immunol-020711-075024
Raud, B., McGuire, P. J., Jones, R. G., Sparwasser, T., and Berod, L. (2018). Fatty acid metabolism in CD8(+) T cell memory: challenging current concepts. Immunol. Rev. 283, 213–231. doi: 10.1111/imr.12655
Ray, J. P., Staron, M. M., Shyer, J. A., Ho, P. C., Marshall, H. D., Gray, S. M., et al. (2015). The Interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T Helper 1 and follicular B helper T cells. Immunity 43, 690–702. doi: 10.1016/j.immuni.2015.08.017
Renner, K., Geiselhöringer, A. L., Fante, M., Bruss, C., Färber, S., Schönhammer, G., et al. (2015). Metabolic plasticity of human T cells: preserved cytokine production under glucose deprivation or mitochondrial restriction, but 2-deoxy-glucose affects effector functions. Eur. J. Immunol. 45, 2504–2516. doi: 10.1002/eji.201545473
Richer, M. J., Pewe, L. L., Hancox, L. S., Hartwig, S. M., Varga, S. M., and Harty, J. T. (2015). Inflammatory IL-15 is required for optimal memory T cell responses. J. Clin. Invest. 125, 3477–3490. doi: 10.1172/JCI81261
Rolf, J., Zarrouk, M., Finlay, D. K., Foretz, M., Viollet, B., and Cantrell, D. A. (2013). AMPKalpha1: a glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 43, 889–896. doi: 10.1002/eji.201243008
Sakaguchi, S., Miyara, M., Costantino, C. M., and Hafler, D. A. (2010). FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 10, 490–500. doi: 10.1038/nri2785
Salmond, R. J., and Zamoyska, R. (2010). How does the mammalian target of rapamycin (mTOR) influence CD8 T cell differentiation? Cell Cycle 9, 2952–2957. doi: 10.4161/cc.9.15.12358
Salmond, R. J., and Zamoyska, R. (2011). The influence of mTOR on T helper cell differentiation and dendritic cell function. Eur. J. Immunol. 41, 2137–2141. doi: 10.1002/eji.201141523
Shi, L. Z., Wang, R., Huang, G., Vogel, P., Neale, G., Green, D. R., et al. (2011). HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376. doi: 10.1084/jem.20110278
Shrestha, S., Yang, K., Wei, J., Karmaus, P. W., Neale, G., and Chi, H. (2014). Tsc1 promotes the differentiation of memory CD8+ T cells via orchestrating the transcriptional and metabolic programs. Proc. Natl. Acad. Sci. U.S.A. 111, 14858–14863. doi: 10.1073/pnas.1404264111
Sinclair, L. V., Rolf, J., Emslie, E., Shi, Y. B., Taylor, P. M., and Cantrell, D. A. (2013). Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 14, 500–508. doi: 10.1038/ni.2556
Siska, P. J., Beckermann, K. E., Mason, F. M., Andrejeva, G., Greenplate, A. R., Sendor, A. B., et al. (2017). Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2:93411. doi: 10.1172/jci.insight.93411
Stockinger, B., and Omenetti, S. (2017). The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol. 17, 535–544. doi: 10.1038/nri.2017.50
Sun, I. H., Oh, M. H., Zhao, L., Patel, C. H., Arwood, M. L., Xu, W., et al. (2018). mTOR complex 1 signaling regulates the generation and function of central and effector foxp3(+) regulatory T cells. J. Immunol. 201, 481–492. doi: 10.4049/jimmunol.1701477
Swamy, M., Pathak, S., Grzes, K. M., Damerow, S., Sinclair, L. V., van, Aalten DM, et al. (2016). Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat. Immunol. 17, 712–720. doi: 10.1038/ni.3439
Tan, H., Yang, K., Li, Y., Shaw, T. I., Wang, Y., Blanco, D. B., et al. (2017). Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 46, 488–503. doi: 10.1016/j.immuni.2017.02.010
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. doi: 10.1126/science.1160809
Wang, R., Dillon, C. P., Shi, L. Z., Milasta, S., Carter, R., Finkelstein, D., et al. (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. doi: 10.1016/j.immuni.2011.09.021
Wolfson, R. L., and Sabatini, D. M. (2017). The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab. 26, 301–309. doi: 10.1016/j.cmet.2017.07.001
Yang, G., Murashige, D. S., Humphrey, S. J., and James, D. E. (2015). A positive feedback loop between Akt and mTORC2 via SIN1 phosphorylation. Cell Rep. 12, 937–943. doi: 10.1016/j.celrep.2015.07.016
Yang, J. Q., Kalim, K. W., Li, Y., Zhang, S., Hinge, A., Filippi, M. D., et al. (2016). RhoA orchestrates glycolysis for TH2 cell differentiation and allergic airway inflammation. J. Allergy Clin. Immunol. 137, 231.e4–245.e4. doi: 10.1016/j.jaci.2015.05.004
Yang, K., Neale, G., Green, D. R., He, W., and Chi, H. (2011). The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat. Immunol. 12, 888–897. doi: 10.1038/ni.2068
Yang, K., Shrestha, S., Zeng, H., Karmaus, P. W., Neale, G., Vogel, P., et al. (2013). T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity 39, 1043–1056. doi: 10.1016/j.immuni.2013.09.015
Yang, X., and Qian, K. (2017). Protein O-GlcNAcylation: emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 18, 452–465. doi: 10.1038/nrm.2017.22
Yin, Y., Choi, S. C., Xu, Z., Perry, D. J., Seay, H., Croker, B. P., et al. (2015). Normalization of CD4+ T cell metabolism reverses lupus. Sci. Transl. Med. 7:274ra218. doi: 10.1126/scitranslmed.aaa0835
Zeng, H., Cohen, S., Guy, C., Shrestha, S., Neale, G., Brown, S. A., et al. (2016). mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity 45, 540–554. doi: 10.1016/j.immuni.2016.08.017
Zeng, H., Yang, K., Cloer, C., Neale, G., Vogel, P., and Chi, H. (2013). mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499, 485–490. doi: 10.1038/nature12297
Zhang, L., Tschumi, B. O., Lopez-Mejia, I. C., Oberle, S. G., Meyer, M., Samson, G., et al. (2016). Mammalian target of rapamycin complex 2 controls CD8 T cell memory differentiation in a foxo1-dependent manner. Cell Rep. 14, 1206–1217. doi: 10.1016/j.celrep.2015.12.095
Keywords: T cell, aerobic glycolysis, mTOR, metabolism, immune responses
Citation: Salmond RJ (2018) mTOR Regulation of Glycolytic Metabolism in T Cells. Front. Cell Dev. Biol. 6:122. doi: 10.3389/fcell.2018.00122
Received: 19 June 2018; Accepted: 06 September 2018;
Published: 25 September 2018.
Edited by:
Concetta Bubici, Brunel University London, United KingdomReviewed by:
Andras Perl, Upstate Medical University, United StatesCopyright © 2018 Salmond. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert J. Salmond, ci5qLnNhbG1vbmRAbGVlZHMuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.