
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 30 November 2021
Sec. Breast Cancer
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.782766
This article is part of the Research Topic Drug Resistance in Breast Cancer – Mechanisms and Approaches to Overcome Chemoresistance View all 15 articles
 Andrew M. K. Law1,2†
Andrew M. K. Law1,2† Laura Rodriguez de la Fuente1,2,3†
Laura Rodriguez de la Fuente1,2,3† Thomas J. Grundy4
Thomas J. Grundy4 Guocheng Fang5
Guocheng Fang5 Fatima Valdes-Mora3,6*
Fatima Valdes-Mora3,6* David Gallego-Ortega1,2,5*
David Gallego-Ortega1,2,5*Over 90% of potential anti-cancer drug candidates results in translational failures in clinical trials. The main reason for this failure can be attributed to the non-accurate pre-clinical models that are being currently used for drug development and in personalised therapies. To ensure that the assessment of drug efficacy and their mechanism of action have clinical translatability, the complexity of the tumor microenvironment needs to be properly modelled. 3D culture models are emerging as a powerful research tool that recapitulates in vivo characteristics. Technological advancements in this field show promising application in improving drug discovery, pre-clinical validation, and precision medicine. In this review, we discuss the significance of the tumor microenvironment and its impact on therapy success, the current developments of 3D culture, and the opportunities that advancements that in vitro technologies can provide to improve cancer therapeutics.
Uncontrolled division of neoplastic cells results in the development of a tumour mass composed of a large variety of cellular and non-cellular components, including the heterogeneous population of cancer cells, infiltrating and resident normal cells, extracellular matrix (ECM) proteins and secreted factors. This complex and highly heterogeneous conglomerate of multiple cell types and extracellular components inside of the tumour mass is known as the tumour microenvironment (TME) (1). The interacting networks established in the TME among cancer cells and the other cell types are the key contributors to the hallmarks of cancer and determine the aggressiveness of the tumour (2–4). Furthermore, this tumour heterogeneity within the TME widely contributes to the extent of patient responses to anti-cancer therapies (5). Resembling the network and the heterogeneity involved in every type of cancer is considered one of the most challenging practices among oncology researchers globally. However, understanding the molecular features in the TME of each cancer is fundamental for the successful development of clinically translatable anti-cancer drugs.
The complexity within the TME is propagated by the heterogeneous nature of different tumor entities; that is each individual tumor harbors its own unique intricacies comprised of structural, cellular, genetic, and molecular composition. Our continuous effort to improve our understanding of oncology has led to the development of more effective diagnostic and therapeutic approaches. However, we are also simultaneously unravelling the anomalous disease complexities within cancer that challenges clinical success. In a comprehensive survey of clinical success rates by Hay et al., oncology drugs were found to have only a 6.7% success rate of being approved (6), with other studies estimating as low as 3.4% (7). There are various reasons that contribute to this high rate of failure including 1) inadequate efficacy from poor biodistribution and metabolism of the drug – unsatisfactory therapeutic index; 2) safety concerns associated with significant side effects and off-target toxicities; 3) financial or commercial issues such as insufficient funding or patient recruitment and retention (8–11). Ineffectiveness of therapies is the most common factor (57%) attributed to failure during clinical development (10, 11). Unfortunately, most experimental drugs that were designed through using pre-clinical models to therapeutically target known molecular components are poorly translated to clinical practice.
During the pre-clinical phase, the most commonly employed cancer models are 2D cell cultures before transitioning to in vivo mice models (Figure 1) (12). Drug testing in animals prior to clinical trials have been a mainstay for determining drug efficacy and toxicity; however, there are also various issues associated with animal models, from increased costs, logistic demand, limited bioavailability, and an increasing ethical concern (13–15). Although these models have provided us with better insights into tumor biology and have made a significant impact on approaches to cancer healthcare, they do not accurately recapitulate the complex TME and molecular features within a human tumor (16, 17). The dismal results of clinical translatability of drugs developed from pre-clinical models highlight the limitations of our current understanding (16). Currently, one of the major obstacles for delivering better cancer patient cares is associated with accurate diagnosis and prediction to therapeutic responses (18). As such, the importance of developing more accurate, cost-effective, and efficient pre-clinical technologies for better in vitro and in vivo models are crucial to creating more efficacious therapies, predicting therapeutic outcomes, and guiding clinical practice.
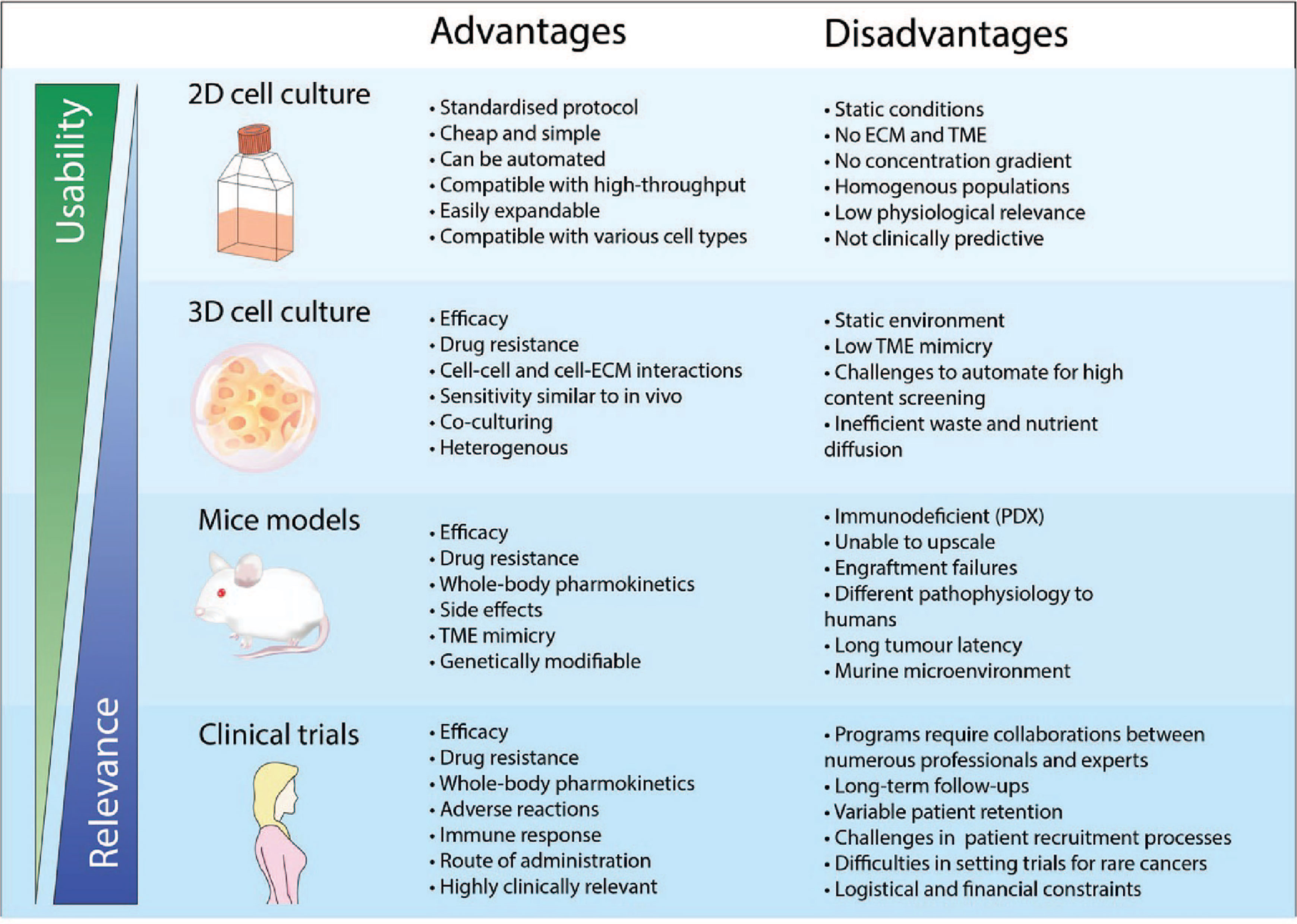
Figure 1 Advantages and disadvantages of drug development using different pre-clinical models and clinical trials. The physical features when using a pre-clinical model is crucial to ensure physiological relevance. 2D cell cultures is a widely adopted and well-established model that has been used consistently in drug discovery and high throughput screening. However, cancer cells cultured in 2D do not recapitulate the biology of an in vivo tumor and thus has very poor performance for clinical prediction. As such, the use of more complex models such as 3D cell culture and mice models has been more representative of clinical cases compared to 2D cell culture. However, the standardized implementation of these models for applications in high content screening and personalised medicine remains a challenge.
Many researchers use 2D cell cultures as the in vitro pre-clinical model for testing anti-tumor drugs before proceeding with in vivo trials (13). This is primarily due to the convenience, simplicity and cost-effectiveness of using a 2D cell culture as a model (Figure 2). However, it is evident that results attained from 2D in vitro models have almost no clinical translatability to human tumors (13). The 2D monolayer cultures have been optimized to grow on rigid plastic surfaces and thus fail to capture the crucial elements that make up the complex 3D tissue architecture of the TME, which ultimately affects the cellular response of cells to drugs and the off-target effects. While 2D cultures are still predominantly used for drug discovery due to its simplicity and compatibility with high-content screening platforms, 3D culture systems have numerous advantages over 2D cell culture. Thus the transition to 3D preclinical models have become more appealing as improvement in tissue engineering technology has made 3D cell culture more adaptable and tunable over the microenvironmental factors to better reflect the functional pathology of in vivo tumors.
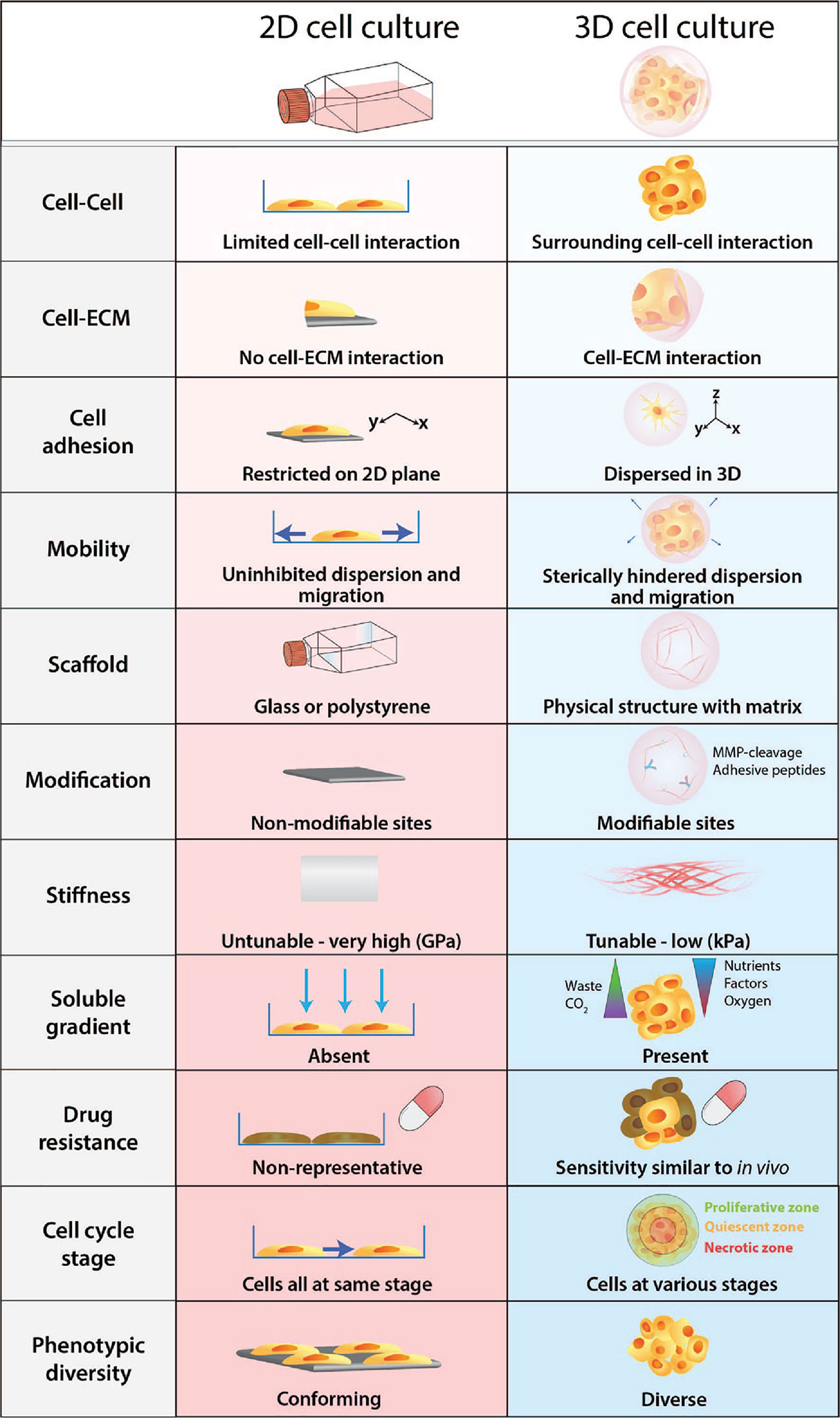
Figure 2 Physiological differences between 2D cell culture and 3D cell culture. Cells develop as a 2D monolayer adopt an apical-basal polarity when plated on a culture flask or a petri dish. The environment that cells are exposed to within the culture flask is a poor representation and does not accurately recapitulate physiological conditions. Comparatively, 3D cultures provide greater biological relevance and cellular response to perturbations are more reflective of in vivo.
The emergence of 3D cell culture models as research tools plays a vital role during early pre-clinical drug development. Recently there has been a paradigm shift in the way researchers study the TME; 3D models are able to better mimic the in vivo microenvironment compared to 2D cell culture and their applications are simpler, more efficient, versatile, and cost-effective compared to using animal models (13). Currently, intense efforts are taken to generate new cell lines that represent the vast heterogeneity of tumors. Three-dimensional cultures offer a higher chance to represent the genomic diversity and allow testing of new drugs targeting specific signaling pathways. Additionally, 3D culture is a more efficient way to generate new patient-derived cell lines that fail to grow in 2D. For example, in breast cancer and melanoma, tumor circulating cells derived from patients are successfully grown under hypoxia conditions in suspension cultures (19, 20). And in prostate cancer, organoid models from patient-derived xenografts can be also used to assay drug sensitivity (21).
Furthermore, cells embedded within a 3D matrix self-assemble to form structures more similar to their organisation in vivo and enable better intercellular contact and communication. Recent advancement in 3D culture has led to the development of new technologies that can generate more complex 3D cell models that aim to bridge the gap between 2D cell culture and animal models. The improved biological relevance of 3D models is due to several key features: dimensionality, presence of ECM, and concentration gradients (Figure 2).
3D culture models cultivate a more relevant pathophysiological microenvironment that allows cells to aggregate, proliferate, and display phenotypes as they do within the body. The complex cellular interactions between other cells and the 3D matrix are crucial for maintaining regular cell structure, function and mobility. Since cell migration occurs in three dimensions the matrix provides a topology that mimics the 3D architecture of a tissue, allowing cells attach and interact with their surrounding environment (22). The dynamic tensile forces from the matrix play a crucial role in cell migration and are involved in activating pathological mechanisms associated with invasion, ECM remodeling, and metastasis (23, 24). Kock et al. had conducted a study investigating the biomechanical tractions utilized by various carcinoma cells to invade through a collagen gel. Interestingly, the level of matrix contraction was not associated with invasiveness, but rather the cellular adoption of an elongated spindle-like morphology and the complexity of the collagen deformation (24). Furthermore, fibroblasts were reported to migrate more rapidly on a 3D matrix and maintained a more spindle-like characteristic compared to those that were cultured in 2D (25). Contrarily, cells grown on a 2D plane have much less physical hindrance as they move across a planar surface that is only impeded by surface inhibition (23). As such, 3D cultures have been used to elucidate the mechanisms that drive cancer invasion and metastasis. For example, matrix degradation and ECM remodeling are key factors involved in invasive malignancy and have been studied in ex vivo models to identify potential targets for cancer therapies, such as inhibiting matrix metalloproteinases and invadopodia formation (26–28).
The ECM has been well established to influence cell behaviour and response to external factors (29, 30). Cellular phenotypes and functions are dictated by a complex network of signaling that occurs within the context of the microenvironment through cell-cell communications, cell-ECM interactions, soluble factors, and small molecules (29). The importance of these dynamic interactions between cells and its surrounding ECM becomes apparent as cells grown in 3D adopt physical and genetic properties more akin to in vivo, such as morphology, phenotype, and expression profiles; whereas 2D monolayers have more vastly different characteristics forced by the unnatural plastic environment (31–33). Additionally, the biomechanical properties of the ECM can modify the signal transductions that occurs within the microenvironment via the spatial organisation of cells, stiffness of the matrices, and physical constraints to hinder cell mobility (29, 34). During tumorigenesis, the stiffness of the ECM causes compressive stress that increases the mechanical pressure as the tumor grows and expand. This increased ECM resistance promotes cell-ECM and cell-cell within the tumor communications that can induce hyperactivated mechanotransduction pathways such as RHO/ROCK (35). Consequently, this upregulation of ROCK can increase cancer cell proliferation, migration, epithelial-mesenchymal transition (EMT), and cancer-associated fibroblast (CAF) reprogramming to promote tumor progression (35, 36). Within the ECM various molecules can also regulate the behaviour, differentiation, migration, and phenotypic fates of cells (37). These can include: glycoproteins such as laminin and fibronectin that connects structural molecules together or with cells to orchestrate cell attachment and migration through the ECM; ECM fibres such as collagen and elastin to provide structural elements of tensile strength and elasticity; and proteoglycans such as hyaluronic acid, keratan sulphate, and chondroitin sulphate, that can regulate structural and adhesive properties of the ECM, angiogenesis, and sequester growth factors (37, 38). Additionally, drug sensitivity in cells can be variable based on cell-ECM interactions and spatial positioning of cells relative to the ECM (30, 39, 40). Changes in ECM composition and its biophysical properties do not only alter cell phenotype but can also regulate the cellular response to drugs, such as promoting acquired resistance or reducing drug accumulation within the tumor (41, 42).
Soluble metabolites, oxygen concentration, and pH throughout the TME can strongly affect the tumor pathophysiology and the efficacy of therapies (39–41). These components exist as a gradient within the tumor; peripheral cells in closer proximity to blood vessels have more access to soluble constituents and oxygen, which decreases as it diffuses through the ECM to the tumor core. The concentration gradients of growth factors, nutrients, wastes, and gases compounds to the intratumoral heterogeneity and influences the signaling within the microenvironment including cell function, proliferation, morphogenesis, and chemotaxis (30). As such, cells grown in larger 3D aggregates also mimic the in vivo condition by existing in various proliferative states based on nutritional access that is restricted by the concentration gradient. From the peripheral to the core of the spheroid is composed of the outer proliferative zone, semi-peripheral quiescent zone, and the central necrotic zone, where each region is in different cell cycle stages (34). This difference in cell cycle stage amongst cancer cells in 3D cultures also contributes to the variable sensitivity of drugs and tumor recurrence from quiescent cells (32, 34). Since blood vessels are unevenly distributed throughout the tumor, regions with low or absent vasculature are hypoxic and acidic and contain high interstitial oncotic pressure (43). In the context of pharmacokinetic, the concentration gradient limits the penetrance of drugs through the tumor and attains a dosage sufficient to exert their therapeutic effects on all the cancer cells (44). In addition, the half-life of drugs also determine the distribution of the agent throughout the tumor; drugs with a long half-life will have more uniform distribution across the tumor even if the rate of the diffusion is low, whereas drugs with a short half-life will have a nonuniform distribution (45). Most research also focuses on the role of mechanisms of action for drugs or therapy resistance, however the physiochemistry of drugs is often neglected (44). As a result, the impeded distribution and diffusion of pharmaceutical agents through the tumor still remains one of the major challenges in anti-cancer treatments. This important, yet often overlooked, the property makes 3D cultures a more accurate model to study the impact of pharmacokinetics and even bacterial biodiversity (46) from concentration gradients (47); compared to cells in 2D cultures which are all homogenously exposed to nutrients and agents (30).
The clinical research on the association of microbiota and cancer started in 1868 by William Busch. After centuries of research, increasing evidence implicates that microbiota influences the TME, tumor metabolism, and tumor immunotherapy response (48). For instance, gut microbiota dysbiosis may induce breast tumorigenesis (49). The influence of microbiota in tumourigenesis and tumor progression may differentially impact different types of tumors, as it has been demonstrated the existence of tumour type-specific intracellular bacteria (50). This tumour microbiome diversity, specificity and relevancy provide both challenges and possibilities for tumour treatment (51). Modelling the interactions of microbiota and tumour offers an efficient method to understand the inner correlations and evaluate the microbiota-target drugs.
Compared to the 2D cell models, the 3D culture can replicate the mechanical cues of solid tumors and the chemical gradience (pH, hypoxia, lactate, etc.), which influence the microbiota proliferation, distribution, movement, variety, and metabolism. This, in turn, could affect the metabolite levels in the TME, for instance, by regulating the gene expression (52). Stem-cell derived organoids, relying on 3D culture, have become indispensable tools to investigate the host-microbiota interactions (53). For instance, intestinal organoids usually form luminal structures within the hydrogel’s matrix where the bacteria of interest can be microinjected (54). As such, stomach organoids were modelled with Helicobacter Pylori (55). The organ-on-chip approach could also mimic the complexity of 3D tissues or tumors, which attracts more attention to the study of microbiome and disease, an example of this approach has been applied to the gut-microbiome on a chip (56, 57). The bidirectional interactions of drugs with local microbiota manipulate the host response to chemotherapeutic drugs (49, 51, 58, 59), which potentially highlights the importance of 3D cultured models in pharmomicrobiomics. In addition, with the fast development of engineered microbial therapies, 3D cultures become a good candidate for more reliable screening, enabling parallel and long-term monitoring (60).
Anti-cancer drug screening and the development of new personalised therapies are primarily conducted in 2D cultures of cancer cell lines (30). Researchers have generated the Cancer Cell Line Encyclopedia to help provide predictive modelling of anticancer drug sensitivity (61, 62). 2D cultures are a mainstay in biological research and have provided us with a deeper insight and understanding of cancer mechanisms, biomarker discovery, and stratification of tumour profiles. From a drug-development perspective, the improvement of more predictive preclinical models is essential to permit the earlier dismissal of drug candidates from clinical trials and reduce pharmaceutical cost – the development of a new drug is estimated to be $2.6billion (63). The disparate response to therapies observed in 2D cultures and in mouse models becomes evident in clinical trials, in which oncology drugs are known to have as low as 3.4% success rate (7). For example, the drug Palifosfamide was a DNA alkylating agent used as a first-line treatment for metastatic soft tissue sarcoma that had failed in Phase III PICASSO 3 trial due to not being able to meet its primary endpoint of progression-free survival in patients (NCT01168791). Within the lab, Palifosfamide demonstrated cytotoxicity in sarcoma cell lines with an IC50 range of 0.5-1.5ug/mL and treatment in xenograft SCID mice resulted in tumour growth inhibition and improved event-free survival (64).
Recapitulation of the fundamental tissue environment within the human body is essential for the proper evaluation of drug effectiveness. From both the cellular populations to the acellular compositions, such as the ECM, pre-clinical models aim to replicate both pathophysiological and healthy bodily functions. Mimicking the complexities of all the biological processes in a single model is highly challenging. Therefore, researchers are developing new techniques to make 3D culture more applicable and easier to implement (Figure 3). As such, 3D cell cultures are becoming more convenient and accessible while allowing researchers to improve upon the traditional in vitro 2D cultures, aiming to model more native-like interactions of tissues to study their mechanisms.
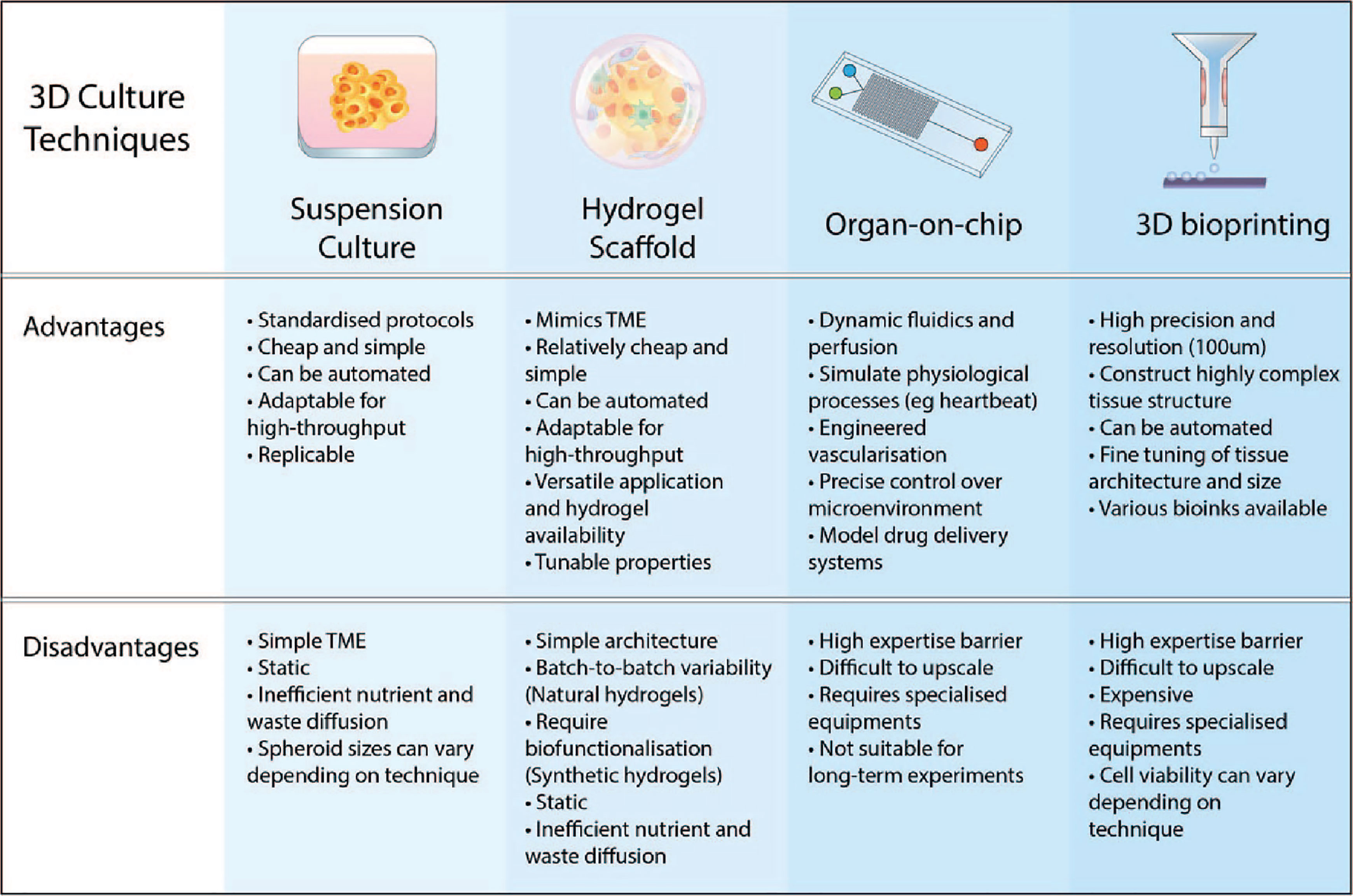
Figure 3 Advantages and disadvantages of various 3D culture approaches. The key features of 3D culturing aim to improve the biomimicry and predictive value of pre-clinical models. Suspension cultures and scaffold-based approaches are easier to implement in the lab and upscale for high-throughput. Advancements in microfabrication technology such as microfluidic chips and 3D bioprinting have resulted in more complex and physiologically-relevant models that can be generated.
Spheroids are grown as aggregates in suspension and have been applied in various cell types, such as cancer cells, hepatocytes, and stem cells (65). Additionally, they can be grown as a monoculture or together as a co-culture with other cell types to provide more physiologically relevant interactions. In a study by Courau et al., colon cancer cells were co-cultured with T cells and NK cells to evaluate tumour-lymphocyte communication and test immunomodulatory antibodies (66). Spheroids are able to recapitulate the in vivo characteristics of intercellular communications, cell-ECM interaction, and behaviour. The size of spheroids are dictated by the initial seeding cell number; thus it is crucial to optimize the culture conditions to ensure that the spheroids do not become too large and suffer from hypoxia and necrosis from poor nutrient diffusion (30). Spheroids can be generated through 1) hanging drop; 2); low adhesion plates 3) magnetic levitation.
The hanging drop technique is one of the earliest methods of developing 3D cell culture (67). This technique uses specialized hanging drop plates that contain a bottomless well where the droplet of media forms. Cells aggregate within the small droplet of culture media to generate the spheroid over several days. Co-culturing can be conducted by adding cells during the initial dispensing or from consecutive addition of the cells (65). However, transfer of the spheroids from the hanging drop plate to another non-attachment plate will be necessary if growing larger spheroids or downstream assays. The hanging drop technique is relatively facile and efficient and has been adapted for use in various cell lines for toxicity testing and drug screening (68, 69). This technique has very high reproducibility with consistent size spheroids (70).
Low adhesion plates have a low attachment coating on the surface of the wells that reduces cell adherence and promotes cell aggregation into spheroids. The coating can include the non-adherent poly-HEMA or agarose (30). Larger volumes of media can also be used in the low adhesion plate allowing a more efficient generation of tumour spheroids. Furthermore, low adhesion plates are designed for high-throughput screening, allowing 3D cell culturing and assaying within the same, unlike the hanging drop technique (65).
Magnetic levitation generates spheroids through the use of magnetic nanoparticles. Cells are incubated with the nanoparticles for several hours to overnight and are then loaded in a low adhesion plate. The low adhesion plate minimizes cell adhesion to the plate while the application of a magnetic field above the plate incites cells to aggregate and produce the spheroids, which can be maintained without requiring a continuous magnetic force. The spheroids can then be subsequently manipulated using other magnetic tools, such as to accelerate cell migration (71). Magnetic levitation can be scalable for use in high throughput screening and drug discovery (72).
Biomimetic scaffolds that model the ECM have been developed over the past few decades to develop microenvironments that can overcome the limitations of traditional 2D cell cultures. In particular, hydrogels have gained interest as physical support that provides the architecture, topology, and biomechanical properties which enables more in vivo-like cellular behaviour and communication. Hydrogels can be used to generate various natural and synthetic ECMs that simulate the microenvironment and stiffness of most soft tissues (29). The internal structures of hydrogels consist of networks of cross-linked polymers that can be moulded through mild gelation conditions that have minimal cytotoxicity (13). Furthermore, hydrogels can be chemically modified to tailor matrix stiffness and viscoelasticity (73–75). Integrin interaction (76, 77), growth factor binding (78), and the 3D organisation of the cells (79) can be tuned through the decoration of hydrogel with a variety of peptides (80, 81). ECM remodeling and cell migration can be facilitated through the inclusion of degradable MMP cleavage sites (76, 77), while the synthetic ECM environment can be enriched with matrix proteins including collagens (82), laminins (83), and fibronectin (84), as well as critical matrix molecules such as hyaluronic acid (hyaluronan) (85). This customizability allows hydrogels to have extensive application and versatility in biological research by offering a range of physical and biochemical characteristics.
Natural hydrogels are derived from sources that are inherently biocompatible (29). Various ECM constituents have been derived from materials such as collagen, fibrin, hyaluronic acid, alginate, and the commercial product Matrigel, a reconstituted basement membrane extracted from murine sarcoma cells (86). These hydrogels have various endogenous factors that promote bioactivity and sustain natural cell function, proliferation, and differentiation. For example, collagen is a widely used ECM that orchestrate controlled cell migration, proliferation, and response to therapies through alteration in stiffness and collagen concentration (87, 88). A study from Puls et al. had studied the progression of metastasis in pancreatic cancer using 3D matrices created with type I collagen and found that exposure to fibrillar collagen induced EMT (89). Increased density of collagen fibril resulted in closer arrangements of cell clusters and matrix stiffness (89). Alginate is also a natural polymer derived from brown algae that can gelate via ionic crosslinking of the polysaccharide backbone by divalent cations, such as calcium, magnesium, or barium (73, 90). The stiffness of alginate hydrogel can be modified based on the level of cross-linking that is dictated by the concentration of the crosslinking agent. Importantly, alginate gels are inert as they do not contain any mammalian cell adhesion ligands, and with their low protein adsorption, makes them ideal as a matrix for the encapsulation of cells and tissue (91). Additionally, alginate gels under neutral pH and room temperature, resulting in minimal cellular disruption under gelation conditions (90). Alginate can be biofunctionalized with the addition of adhesive and hydrolytic moieties and has been used as a matrix for various biomedical applications (92–94). A key advantage of alginate matrices is that cells can be easily recovered by dissolving the alginate with a chelating agent, such as sodium citrate. Recently, alginate matrices have been proposed for drug screening in breast cancer tumoroids derived from tumour pieces that retain luminal mechanics (95). Hyaluronic acid is another natural hydrogel that has major biomedical applications due to its high moisture retention and viscoelasticity (96). Hyaluronic acid is a non-immunogenic polysaccharide that is found ubiquitously in the ECM in epithelial and connective tissues and is involved in wound healing, inflammation, and embryonic development (96). It can be modified with functional groups allowing for a diverse range of applications in regenerative medicine, oncology, and bioengineering (97–101). However, some drawbacks of natural hydrogels can include poor control over the gelation condition, uncontrolled polymer network structures, lower mechanical integrity, and lower experimental reproducibility due to batch-to-batch variations (65, 86).
Synthetic hydrogels are inert scaffolds that permit a higher degree of modification for desired biological or physical conditions, such as biodegradability, porosity, functionalization with adhesive peptide sequences, growth factors or cleavage sites (29, 30). Compared to natural hydrogels, synthetic gels are cheaper and add improved experimental reproducibility as it has a lower batch to batch variation during manufacturing and can be adapted to suit the research need. However, the disadvantage of most synthetic hydrogels is that they act as a minimalistic matrix and have a less complex microenvironment due to the lack of endogenous factors that are generally present in natural hydrogels (29). As such adhesive moieties and catalytic sites need to be crosslinked into the synthetic scaffold to improve their biofunctionality, such as peptides that can mimic fibronectin or laminin-integrin binding (102, 103). Various non-natural sources can be derived to produce these matrices, such as polyethylene glycol (PEG) (104, 105), polyvinyl alcohol, and polylactic acid (PA) (30, 106). PEG has been used for various 3D culturing and tissue engineering applications. For example, PEG has been cultured with breast cancer cells and CAFs to evaluate drug resistance through pathways associated with tumour-stromal interactions (107, 108). In another study, Caiazzo et al. found that PEG can facilitate pluripotency by manipulating the microenvironment of the matrix to create a “reprogramming niche” that promotes MET and increased epigenetic remodeling capable of shifting the somatic cell fate (109). Biomechanical strain and tension induced by the matrix have been reported to modulate the epigenetic and transcriptomic state of cells as a response to their surrounding environment (110–112).
The advancement in microfabrication technology has led to the development of microfluidics systems that provides more dynamic microenvironments. These systems are designed with specific structures and scaffolds that can be manufactured through patterning techniques such as soft lithography, photolithography, and micro-contact printing (65). Microfluidics permits precise control over small volumes of fluid through hollow channels that can be smaller than 1µm in diameter (13). These devices or chips have been an essential development in microsystems technology that can generate and manipulate the fluid flow and spatiotemporal gradients to improve the biological relevance of in vitro models (113). Nutrients, drugs, and wastes can be readily delivered or removed via continuous perfusion through the microchannel (114). Within the microfluidic system, spheroids can be generated at high throughput and with a precision that are uniform in size for both monocultures and co-cultures (115–117).
Microfluidic technology has been used to create more cost-effective and accurate biomedical models to test the pharmacokinetics, efficacy, and toxicity of treatments. The internal dimensions of a microfluidic chip can be composed of multiple channels – depending on the design and application – where the size of structures can be between the micrometer to millimeter range (118). Generally, microfluidic chips are manufactured using an inert and non-toxic polymer as a base material, such as poly-dimethylsiloxane (PDMS) (23). The microfluidics control and miniaturization of the whole system present several key benefits: 1) high-throughput capabilities; 2) cost-efficient and low consumption of reagents – within the nanoliter to picolitre range; 3) fine-tuning of conditions and automation (118, 119).
A major approach of the microfluidic system is developing organ-on-chip which is able to create a complex in vitro model that recapitulates more organ-specific microenvironments. Organ-on-chip focuses on capturing the critical aspects of the normal biological functions or disease states of the organ of interest. This allows researchers to investigate disease phenotypes and pharmacological responses that are clinically relevant and provide more accurate predictions of treatment efficacy (65, 120). Nutrients, growth factors, oxygen, and drugs can be circulated through the chip as a continuous supply via dynamic perfusion which can be automated – in addition to waste removal (12). The controlled fluidic motions can also be used to mimic various mechanical signals including shear stress; compressive forces; physiological flow, such as blood flow; and tissue-specific motions, such as cardiac rhythms and respiratory (120–122). Consequently, microfluidics chips have been used to recapitulate aspects of the TME for anti-cancer drug developments, circulating cancer cell detection in blood samples, and personalised organ-on-chips (123–126). The simplest tumour-on-chip models have been applying 3D spheroids within a microfluidics system (127–129). However, more sophisticated tumour-on-chips platforms have been developed that utilizes the dynamic flow of microfluidics. In a study by Chen et al, an in vitro breast tumour model was created on a chip to evaluate nanoparticle-based drug delivery systems (130). This chip included a layer of endothelium that lined a microvessel wall, the ECM and tumour spheroids to generate a real-time drug delivery model. Treatments such as doxorubicin – a standard of care therapy for breast cancer – was loaded in carbon dots to study the penetrance of the treatment through the endothelium to the spheroids, where the efficacy and cytotoxicity of the drug delivery were assessed using in situ assays within the same system (130). Tumour-on-chips can also contain engineered vascularization as part of the model using perfusable system to imitate the flow of blood vessels to more closely mimic other mechanisms within the TME, including metastasis, angiogenesis, and drug metabolism (131–134). Argwal et al. discovered that vascularized in vitro 3D breast tumors exhibited significantly higher resistance to doxorubicin compared to avascular 3D tumors (4.7 times) and 2D culture cells (139.5 times) (135). Interestingly, this high drug resistance could also be overcome via a nanoparticle-based drug delivery method (135). The inclusion of vascularization and dynamic flow has also allowed researchers to study the pathophysiology of blood-based cancer with in vitro models, such as lymphoma (136).
The development of in vitro 3D models that increase the probability of preclinical drug research representing patient outcomes in drug trials, and potentially remove the need for animal studies, may render preclinical cancer research more cost-effective and accessible. However, the use of novel 3D models in cancer research remains restricted by model reproducibility; a prerequisite for specialized training and limitations relating to throughput. The development and commercialization of 3D bioprinting technologies offer an exciting solution to these challenges. 3D bioprinting is an additive manufacturing process defined by the creation of a 3D structure through controlled and typically automated deposition of a biocompatible material or ‘bioink’. This advanced technology is capable of accurately constructing complex tissue structures that faithfully recapitulate native in vivo architecture (137). 3D structures can be created directly from highly viscous or shear-thinning bioinks, where the bioinks can be mixed with the cell suspension to generate functionalized cell models. Alternatively, printed bioinks that are less viscous can be solidified through the addition of other chemicals, cooling, or exposure to light or heat (138).
Bioinks are printable, biocompatible solutions that comprise the necessary elements of a desired 3D microenvironment. Bioinks vary greatly in their composition depending on the printing method and the application. Cells, native proteins, growth factors, and signaling molecules can be combined with synthetic compounds that are both printable and biomimetic. Synthetic molecules can likewise be decorated with peptide sequence (139), MMP degradable ligands and drug molecules (140) so that they are more biocompatible, biodegradable or bioactive. Modifications to the bioink properties and bioprinting methods can be tuned to tailor to the desired applications and studies. For example concentrated bioinks may be necessary for creating dense, stiff structures such as bone biomimetics (141), or dense tumour microenvironment models (142). However, concentrated bioinks are highly viscous and result in increased cell death during printing due to high shear forces. As such, it is also important to optimize these modifications to ensure compatibility with the cell types.
Most 3D bioprinting strategies involve droplet, extrusion, and stereolithographic-based structure creation – for an extensive review on the methods refer to the reviews (138, 143). Commonly employed 3D bioprinting processes include 1) droplet-based 3D bioprinting (DBB), which uses sequential depositions of discrete bioink droplets to create structures (144); 2) drop-on-demand bioprinting (DOD), a subcategory of DBB that controls droplet size and placement by regulating the position and ejection of bioink from a nozzle (145, 146); and 3) laser-assisted bioprinting, an alternative DBB technology that propels bioink droplets from an inverted ‘donor slide’ onto a receiving slide using localized heating of a substrate sensitive to laser radiation (147, 148).
Each 3D bioprinting strategy has various, often interlinked, tradeoffs and downstream applications. For instance, extrusion-based 3D bioprinters create structures by layering continuous beads of bioink from nozzles, whereas stereolithographic 3D bioprinting uses light to cure regions of bioink precursor within a bath, building a structure layer by layer (143). In this case, stereolithography limits printing to a single bioink at a time but is excellent for creating complex networked microarchitecture. For example, this has been used to create osteoblast and MSC-laden bone biomimetics (141) and replica microvasculature (149), which were seeded with invasive cancer cells to simulate metastasis and investigate cancer cell migration. Furthermore, the placement of ink on a printing surface is less complex in extrusion printing compared to droplet-based systems where droplet size, flight and placement vary with ink properties (150). However, printing with droplets offers an advantage in throughput and high-resolution patterning as the same nozzle set of a DOD system simultaneously creates multiple structures comprising many different bioinks. Extrusion printing has been used in the creation of large 3D structures to investigate glioblastoma-macrophage interactions (151), and meshes of cervical cancer (152), lung adenocarcinoma (153) and mammary epithelial cells (142) for 3D cancer modelling and drug screens. Conversely, the throughput advantage offered by DOD bioprinting has been exploited to create arrays of hepatic and brain cancer cell lines for drug screening (154), and co-culture patterning of ovarian cancer cells and fibroblasts for investigations of cell interactions and paracrine signaling (155).
There is currently a matter of contention in 3D bioprinting created by the conflicting practices of requiring printing processes to be completed quickly, and simultaneously allowing complex 3D models sufficient time to develop and mature. Bioprinting exposes cancer cells to reagents, processes and forces that fall outside their typical environmental niche. As such, reducing the time for which cells are exposed to the reagents and forces improves cell viability and preserves the in vivo biology critical to accurate tumour model creation (138). However, the biological processes central to the development of histological micro-architecture are rarely static, proceed slowly and require time to develop. There is a tendency within 3D bioprinting to emphasize time reduction and to prioritize the rapid completion of printing procedures (143). Yet incorporation of time-related factors and processes will be critical as our general understanding and mastery of 3D bioprinting progresses and becomes further integrated into cancer research.
The term ‘4D bioprinting’ has been used to describe 3D bioprinting strategies that integrate the changing of printed structures over time (156). These strategies may rely on organically occurring biological processes such as matrix deposition, tissue self-organisation and cell differentiation (25). Brassard et al. relied on biological dynamics to create complex macro-structures reminiscent of vascular, connective and gastrointestinal tissues (157). These structures were self-assembled from concentrated cell solutions printed into an ECM hydrogel prior to gelation. The creation of in vitro organoids is critical for translatable studies into cancer cell behaviour and drug toxicity. Similar concepts are also being embraced to replicate and investigate the tumour microenvironment directly. For example, Yi et al. created an advanced glioblastoma brain cancer model with initial depositions of silicone ink, endothelial and tumour cells (158). Maturation of the model led to the formation of various features typical of glioblastoma including necrotic foci and pseudopalisades within the tumour cell mass, and leaky endothelial microvessels (159, 160).
In addition to internal biological drivers, externally controlled stimuli can be used to modulate cell behaviour and the printed material surrounding them. The creation of dynamic 3D printed structures is critical for studying the ECM remodeling integral to tumour growth, cell metastasis and drug permeability. Studies have used various stimuli including temperature (161), pH, osmolarity (162), light (163, 164), humidity, magnetic force (165) and electrical charge, to affect material stiffness, size, density, binding affinity (166) and molecular organization (166) of responsive ‘smart’ materials. Stimuli may cause unidirectional irreversible material responses, or they may be bidirectional and reversible (161). Responses can also be stacked, allowing multiple different material states. In an example of this, Tabriz et al. enabled a multistage crosslinking of printed alginate structures through the addition of sequential Ca2+ and Ba2+ solutions (167). Each stage further increased the printed structures’ durability, facilitating both the initial printability of the bioink, as well as its long-term stability under culture conditions. Aside from material properties, external stimuli can be used to alter the shape of printed structures. Gladman et al., used anisotropic swelling to create complex dimensionality, folds and curvature in 3D planar printed shapes (168). A similar concept was used in a ductal carcinoma study to create geometric mimicry of mammary ducts and acini (169). The impact of responsive bioinks on cancer research is yet to be fully realized. However burgeoning developments in stimulus-responsive geometry and embracing temporal biochemical and biophysical dynamics offer the potential for 3D bioprinted models to be shaped by factors outside of printing complexity (170).
The ability to create representative in vitro models is progressing and our understanding of 3D cellular biology continues to grow. To leverage the advances made in these areas within cancer research, the throughput and reproducibility made possible through 3D bioprinting will be critical. Economically viable cancer research requires in vitro models that are not only representative of physiological and pathological conditions, but that can be created quickly and efficiently. For this to be possible, we require 3D advanced bioprinting techniques that exploit both intrinsic cell behaviors and innovative biomaterial developments. Synthesis within these areas offers interesting future opportunities for complex 3D model development and the attainment of critical cancer research goals.
Although 3D culture has been demonstrated to show great promise as a pre-clinical model, a major drawback of 3D cultures is in their implementation for high-throughput screening; a vital aspect for high-content screening and drug development (171, 172). In particular, three significant technical challenges hamper the adoption of 3D culture technology for high-throughput screening: 1) the automation of liquid handling in 3D culture; 2) culture optimization and assay variability; and 3) automated imaging and visualization of 3D structures. The automation of liquid handling can be conducted in suspension cultures such as through the use of ultra-low-attachment microplates or hanging drop technique (30). However, the application of automated liquid handling translates poorly when using hydrogel-based techniques, such as Matrigel. This is primarily due to the undefined compositions between batches that impact reproducibility and consistency and require highly controlled working environments and rapid processing due to their temperature-sensitive gelation conditions (173). Additionally, this batch-to-batch variation in natural hydrogels considerably impacts cell culture conditions and assay quality and reproducibility; as such it is crucial to ensure consistency between batches when conducting high-throughput screening, such as ECM composition and protein content (103). Finally, 3D models permit co-culturing of multiple cell types and provide a higher morphological complexity compared to 2D cultures; allowing improved multiparametric analysis of cell response to drugs. The additional parameters are particularly valuable as they provide a more accurate evaluation of the efficacy and mechanisms of pharmaceutical agents (174). However, this dimensionality also poses a difficulty in computational image analysis and visualization. The complex topology and thickness of 3D models make them it incompatible with most automated imaging systems due to low light penetration and absorption across the multi-layered structures (103). As a result, this can introduce an imaging bias in which only the exterior cells – the layer where cells are exposed to the highest concentration gradient for nutrients and drugs – are imaged and the internal cells are excluded. Despite these challenges, new culture platforms and imaging systems are being developed that aim to overcome these technical difficulties to create 3D cultures that are amenable for high-throughput screening. These developments include using synthetic hydrogels to generate more consistent 3D cell cultures; automated high-resolution imaging using light-sheet microscopy; and integrated computational platforms for data analysis and visualization of 3D cultures (175–177).
The improvement in 3D culture technology has led to the generation of in vitro models that can encompass more physiological and tissue-specific microenvironments with the aim to overcome the drawbacks observed in other pre-clinical models and have better predictive value for clinical outcomes. 3D culture models allow researchers to recreate specific pathophysiological conditions and tumorigenic processes to identify potential biomarkers for therapeutic targeting or assessing cell response to therapies and drug efficacy. Currently, there has been significant interest in using primary clinical samples in 3D culture for personalised drug screening platforms to improve clinical outcomes and reduce side effects (178, 179). Although there are still practical challenges in the widespread adoption of 3D cultures, advancements in this field will provide researchers with a powerful tool to dissect disease mechanisms, identify new biomarkers, provide valuable data in drug development, and realize the potential in the next generation of personalised medicine.
Writing— original draft preparation, review and editing by AL, LR, TG, GF, FV-M, and DG-O. Figures designed by AL. Supervision by FV-M and DG-O. All authors contributed to the article and approved the submitted version.
AL is supported by a UPA Scholarship from UNSW. LR is supported by a UIPA Scholarship from UNSW Sydney and a PhD top-up award from Kids Cancer Alliance (KCA). FV-M holds a Cancer Institute New South Wales Fellowship (CDF181218). DG-O is holds the Elaine Henry Fellowship from the National Breast Cancer Foundation (NBCF) of Australia (IIRS-21-096) and is supported by a Cancer Council New South Wales (CCNSW) grant (RG18-03).
TG is employed by Inventia Life Science Pty Ltd as stated in affiliations.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the members of the Tumour Development Laboratory at Garvan Institute of Medical Research and the Cancer Epigenetics Biology and Therapeutics group at Children’s Cancer Institute for helpful discussions.
1. Whiteside TL. The Tumor Microenvironment and Its Role in Promoting Tumor Growth. Oncogene (2008) 27(45):5904–12. doi: 10.1038/onc.2008.271
2. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
3. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of Tumor Microenvironment in Tumorigenesis. J Cancer (2017) 8(5):761–73. doi: 10.7150/jca.17648
4. Hanahan D, Coussens LM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell (2012) 21(3):309–22. doi: 10.1016/j.ccr.2012.02.022
5. Dagogo-Jack I, Shaw AT. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat Rev Clin Oncol (2018) 15(2):81–94. doi: 10.1038/nrclinonc.2017.166
6. Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical Development Success Rates for Investigational Drugs. Nat Biotechnol (2014) 32(1):40–51. doi: 10.1038/nbt.2786
7. Wong CH, Siah KW, Lo AW. Estimation of Clinical Trial Success Rates and Related Parameters. Biostatistics (2019) 20(2):273–86. doi: 10.1093/biostatistics/kxx069
8. Fogel DB. Factors Associated With Clinical Trials That Fail and Opportunities for Improving the Likelihood of Success: A Review. Contemp Clin Trials Commun (2018) 11:156–64. doi: 10.1016/j.conctc.2018.08.001
9. Alavijeh MS, Palmer AM. The Pivotal Role of Drug Metabolism and Pharmacokinetics in the Discovery and Development of New Medicines. IDrugs (2004) 7(8):755–63.
10. Hwang TJ, Carpenter D, Lauffenburger JC, Wang B, Franklin JM, Kesselheim AS. Failure of Investigational Drugs in Late-Stage Clinical Development and Publication of Trial Results. JAMA Intern Med (2016) 176(12):1826–33. doi: 10.1001/jamainternmed.2016.6008
11. Arrowsmith J, Miller P. Trial Watch: Phase II and Phase III Attrition Rates 2011-2012. Nat Rev Drug Discovery (2013) 12(8):569.
12. Tomas-Bort E, Kieler M, Sharma S, Candido JB, Loessner D. 3D Approaches to Model the Tumor Microenvironment of Pancreatic Cancer. Theranostics (2020) 10(11):5074–89. doi: 10.7150/thno.42441
13. Lv D, Hu Z, Lu L, Lu H, Xu X. Three-Dimensional Cell Culture: A Powerful Tool in Tumor Research and Drug Discovery. Oncol Lett (2017) 14(6):6999–7010. doi: 10.3892/ol.2017.7134
14. Gould SE, Junttila MR, de Sauvage FJ. Translational Value of Mouse Models in Oncology Drug Development. Nat Med (2015) 21(5):431–9. doi: 10.1038/nm.3853
15. Joffe AR, Bara M, Anton N, Nobis N. The Ethics of Animal Research: A Survey of the Public and Scientists in North America. BMC Med Ethics (2016) 17:17. doi: 10.1186/s12910-016-0100-x
16. Ellis LM, Fidler IJ. Finding the Tumor Copycat. Therapy Fails, Patients Don't. Nat Med (2010) 16(9):974–5.
17. Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J. Patient-Derived Xenografts Undergo Mouse-Specific Tumor Evolution. Nat Genet (2017) 49(11):1567–75. doi: 10.1038/ng.3967
18. Day CP, Merlino G, Van Dyke T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. Cell (2015) 163(1):39–53. doi: 10.1016/j.cell.2015.08.068
19. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, et al. Cancer Therapy. Ex Vivo Culture of Circulating Breast Tumor Cells for Individualized Testing of Drug Susceptibility. Science (2014) 345(6193):216–20.
20. Hong X, Roh W, Sullivan RJ, Wong KHK, Wittner BS, Guo H, et al. The Lipogenic Regulator SREBP2 Induces Transferrin in Circulating Melanoma Cells and Suppresses Ferroptosis. Cancer Discovery (2021) 11(3):678–95. doi: 10.1158/2159-8290.CD-19-1500
21. Karkampouna S, La Manna F, Benjak A, Kiener M, De Menna M, Zoni E, et al. Patient-Derived Xenografts and Organoids Model Therapy Response in Prostate Cancer. Nat Commun (2021) 12(1):1117.
22. Gjorevski N, Piotrowski AS, Varner VD, Nelson CM. Dynamic Tensile Forces Drive Collective Cell Migration Through Three-Dimensional Extracellular Matrices. Sci Rep (2015) 5:11458. doi: 10.1038/srep11458
23. Duval K, Grover H, Han LH, Mou Y, Pegoraro AF, Fredberg J, et al. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiol (Bethesda) (2017) 32(4):266–77.
24. Koch TM, Munster S, Bonakdar N, Butler JP, Fabry B. 3d Traction Forces in Cancer Cell Invasion. PloS One (2012) 7(3):e33476. doi: 10.1371/journal.pone.0033476
25. Hakkinen KM, Harunaga JS, Doyle AD, Yamada KM. Direct Comparisons of the Morphology, Migration, Cell Adhesions, and Actin Cytoskeleton of Fibroblasts in Four Different Three-Dimensional Extracellular Matrices. Tissue Eng Part A (2011) 17(5-6):713–24. doi: 10.1089/ten.tea.2010.0273
26. Mishra DK, Sakamoto JH, Thrall MJ, Baird BN, Blackmon SH, Ferrari M, et al. Human Lung Cancer Cells Grown in an Ex Vivo 3D Lung Model Produce Matrix Metalloproteinases Not Produced in 2D Culture. PloS One (2012) 7(9):e45308. doi: 10.1371/journal.pone.0045308
27. Wang S, Li E, Gao Y, Wang Y, Guo Z, He J, et al. Study on Invadopodia Formation for Lung Carcinoma Invasion With a Microfluidic 3D Culture Device. PloS One (2013) 8(2):e56448. doi: 10.1371/journal.pone.0056448
28. Goertzen C, Eymael D, Magalhaes M. Three-Dimensional Quantification of Spheroid Degradation-Dependent Invasion and Invadopodia Formation. Biol Proced Online (2018) 20:20. doi: 10.1186/s12575-018-0085-6
29. Tibbitt MW, Anseth KS. Hydrogels as Extracellular Matrix Mimics for 3D Cell Culture. Biotechnol Bioeng (2009) 103(4):655–63. doi: 10.1002/bit.22361
30. Langhans SA. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front Pharmacol (2018) 9:6. doi: 10.3389/fphar.2018.00006
31. Birgersdotter A, Sandberg R, Ernberg I. Gene Expression Perturbation In Vitro–A Growing Case for Three-Dimensional (3D) Culture Systems. Semin Cancer Biol (2005) 15(5):405–12. doi: 10.1016/j.semcancer.2005.06.009
32. Kapalczynska M, Kolenda T, Przybyla W, Zajaczkowska M, Teresiak A, Filas V, et al. 2D and 3D Cell Cultures - A Comparison of Different Types of Cancer Cell Cultures. Arch Med Sci (2018) 14(4):910–9.
33. Yamada KM, Cukierman E. Modeling Tissue Morphogenesis and Cancer in 3D. Cell (2007) 130(4):601–10. doi: 10.1016/j.cell.2007.08.006
34. Edmondson R, Broglie JJ, Adcock AF, Yang L. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. Assay Drug Dev Technol (2014) 12(4):207–18. doi: 10.1089/adt.2014.573
35. Boyle ST, Kular J, Nobis M, Ruszkiewicz A, Timpson P, Samuel MS. Acute Compressive Stress Activates RHO/ROCK-Mediated Cellular Processes. Small GTPases (2020) 11(5):354–70. doi: 10.1080/21541248.2017.1413496
36. Boyle ST, Poltavets V, Kular J, Pyne NT, Sandow JJ, Lewis AC, et al. ROCK-Mediated Selective Activation of PERK Signalling Causes Fibroblast Reprogramming and Tumour Progression Through a CRELD2-Dependent Mechanism. Nat Cell Biol (2020) 22(7):882–95. doi: 10.1038/s41556-020-0523-y
37. Frantz C, Stewart KM, Weaver VM. The Extracellular Matrix at a Glance. J Cell Sci (2010) 123(Pt 24):4195–200. doi: 10.1242/jcs.023820
38. Yue B. Biology of the Extracellular Matrix: An Overview. J Glaucoma (2014) 23(8 Suppl 1):S20–3. doi: 10.1097/IJG.0000000000000108
39. Lovitt CJ, Shelper TB, Avery VM. Doxorubicin Resistance in Breast Cancer Cells Is Mediated by Extracellular Matrix Proteins. BMC Cancer (2018) 18(1):41. doi: 10.1186/s12885-017-3953-6
40. Nunes AS, Barros AS, Costa EC, Moreira AF, Correia IJ. 3D Tumor Spheroids as In Vitro Models to Mimic In Vivo Human Solid Tumors Resistance to Therapeutic Drugs. Biotechnol Bioeng (2019) 116(1):206–26. doi: 10.1002/bit.26845
41. Sebens S, Schafer H. The Tumor Stroma as Mediator of Drug Resistance–a Potential Target to Improve Cancer Therapy? Curr Pharm Biotechnol (2012) 13(11):2259–72. doi: 10.2174/138920112802501999
42. Sun Y. Tumor Microenvironment and Cancer Therapy Resistance. Cancer Lett (2016) 380(1):205–15. doi: 10.1016/j.canlet.2015.07.044
43. Di Paolo A, Bocci G. Drug Distribution in Tumors: Mechanisms, Role in Drug Resistance, and Methods for Modification. Curr Oncol Rep (2007) 9(2):109–14. doi: 10.1007/s11912-007-0006-3
44. Minchinton AI, Tannock IF. Drug Penetration in Solid Tumours. Nat Rev Cancer (2006) 6(8):583–92. doi: 10.1038/nrc1893
45. Tredan O, Galmarini CM, Patel K, Tannock IF. Drug Resistance and the Solid Tumor Microenvironment. J Natl Cancer Inst (2007) 99(19):1441–54. doi: 10.1093/jnci/djm135
46. Biagini F, Calvigioni M, Lapomarda A, Vecchione A, Magliaro C, De Maria C, et al. A Novel 3D In Vitro Model of the Human Gut Microbiota. Sci Rep (2020) 10(1):21499. doi: 10.1038/s41598-020-78591-w
47. Knitsch R, AlWahsh M, Raschke H, Lambert J, Hergenroder R. In Vitro Spatio-Temporal NMR Metabolomics of Living 3d Cell Models. Anal Chem (2021) 93(40):13485–94. doi: 10.1021/acs.analchem.1c02221
48. Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, Knight R. The Microbiome and Human Cancer. Science (2021) 371(6536). doi: 10.1126/science.abc4552
49. Alpuim Costa D, Nobre JG, Batista MV, Ribeiro C, Calle C, Cortes A, et al. Human Microbiota and Breast Cancer-Is There Any Relevant Link?-A Literature Review and New Horizons Toward Personalised Medicine. Front Microbiol (2021) 12:584332. doi: 10.3389/fmicb.2021.584332
50. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The Human Tumor Microbiome Is Composed of Tumor Type-Specific Intracellular Bacteria. Science (2020) 368(6494):973–80. doi: 10.1126/science.aay9189
51. Alexander JL, Wilson ID, Teare J, Marchesi JR, Nicholson JK, Kinross JM. Gut Microbiota Modulation of Chemotherapy Efficacy and Toxicity. Nat Rev Gastroenterol Hepatol (2017) 14(6):356–65. doi: 10.1038/nrgastro.2017.20
52. Kasper SH, Morell-Perez C, Wyche TP, Sana TR, Lieberman LA, Hett EC. Colorectal Cancer-Associated Anaerobic Bacteria Proliferate in Tumor Spheroids and Alter the Microenvironment. Sci Rep (2020) 10(1):5321. doi: 10.1038/s41598-020-62139-z
53. Min S, Kim S, Cho SW. Gastrointestinal Tract Modeling Using Organoids Engineered With Cellular and Microbiota Niches. Exp Mol Med (2020) 52(2):227–37. doi: 10.1038/s12276-020-0386-0
54. Puschhof J, Pleguezuelos-Manzano C, Martinez-Silgado A, Akkerman N, Saftien A, Boot C, et al. Intestinal Organoid Cocultures With Microbes. Nat Protoc (2021) 16(10):4633–49. doi: 10.1038/s41596-021-00589-z
55. Bartfeld S, Clevers H. Organoids as Model for Infectious Diseases: Culture of Human and Murine Stomach Organoids and Microinjection of Helicobacter Pylori. J Vis Exp (2015) (105):e53359. doi: 10.3791/53359
56. Jalili-Firoozinezhad S, Gazzaniga FS, Calamari EL, Camacho DM, Fadel CW, Bein A, et al. A Complex Human Gut Microbiome Cultured in an Anaerobic Intestine-on-a-Chip. Nat BioMed Eng (2019) 3(7):520–31. doi: 10.1038/s41551-019-0397-0
57. Poceviciute R, Ismagilov RF. Human-Gut-Microbiome on a Chip. Nat BioMed Eng (2019) 3(7):500–1. doi: 10.1038/s41551-019-0425-0
58. Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. Mapping Human Microbiome Drug Metabolism by Gut Bacteria and Their Genes. Nature (2019) 570(7762):462–7. doi: 10.1038/s41586-019-1291-3
59. Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, et al. Extensive Impact of non-Antibiotic Drugs on Human Gut Bacteria. Nature (2018) 555(7698):623–8. doi: 10.1038/nature25979
60. Harimoto T, Singer ZS, Velazquez OS, Zhang J, Castro S, Hinchliffe TE, et al. Rapid Screening of Engineered Microbial Therapies in a 3D Multicellular Model. Proc Natl Acad Sci USA (2019) 116(18):9002–7. doi: 10.1073/pnas.1820824116
61. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature (2012) 483(7391):603–7. doi: 10.1038/nature11003
62. Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature (2019) 569(7757):503–8. doi: 10.1038/s41586-019-1186-3
63. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the Pharmaceutical Industry: New Estimates of R&D Costs. J Health Econ (2016) 47:20–33. doi: 10.1016/j.jhealeco.2016.01.012
64. Hingorani P, Zhang W, Piperdi S, Pressman L, Lin J, Gorlick R, et al. Preclinical Activity of Palifosfamide Lysine (ZIO-201) in Pediatric Sarcomas Including Oxazaphosphorine-Resistant Osteosarcoma. Cancer Chemother Pharmacol (2009) 64(4):733–40. doi: 10.1007/s00280-008-0922-4
65. Fang Y, Eglen RM. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discovery (2017) 22(5):456–72.
66. Courau T, Bonnereau J, Chicoteau J, Bottois H, Remark R, Assante Miranda L, et al. Cocultures of Human Colorectal Tumor Spheroids With Immune Cells Reveal the Therapeutic Potential of MICA/B and NKG2A Targeting for Cancer Treatment. J Immunother Cancer (2019) 7(1):74. doi: 10.1186/s40425-019-0553-9
67. Simian M, Bissell MJ. Organoids: A Historical Perspective of Thinking in Three Dimensions. J Cell Biol (2017) 216(1):31–40. doi: 10.1083/jcb.201610056
68. Andersson TB. Evolution of Novel 3d Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Basic Clin Pharmacol Toxicol (2017) 121(4):234–8. doi: 10.1111/bcpt.12804
69. Tung YC, Hsiao AY, Allen SG, Torisawa YS, Ho M, Takayama S. High-Throughput 3D Spheroid Culture and Drug Testing Using a 384 Hanging Drop Array. Analyst (2011) 136(3):473–8. doi: 10.1039/C0AN00609B
70. Kelm JM, Timmins NE, Brown CJ, Fussenegger M, Nielsen LK. Method for Generation of Homogeneous Multicellular Tumor Spheroids Applicable to a Wide Variety of Cell Types. Biotechnol Bioeng (2003) 83(2):173–80. doi: 10.1002/bit.10655
71. Shen H, Tong S, Bao G, Wang B. Structural Responses of Cells to Intracellular Magnetic Force Induced by Superparamagnetic Iron Oxide Nanoparticles. Phys Chem Chem Phys (2014) 16(5):1914–20. doi: 10.1039/C3CP51435H
72. Timm DM, Chen J, Sing D, Gage JA, Haisler WL, Neeley SK, et al. A High-Throughput Three-Dimensional Cell Migration Assay for Toxicity Screening With Mobile Device-Based Macroscopic Image Analysis. Sci Rep (2013) 3:3000. doi: 10.1038/srep03000
73. Caliari SR, Burdick JA. A Practical Guide to Hydrogels for Cell Culture. Nat Methods (2016) 13(5):405–14. doi: 10.1038/nmeth.3839
74. Denisin AK, Pruitt BL. Tuning the Range of Polyacrylamide Gel Stiffness for Mechanobiology Applications. ACS Appl Mater Interfaces (2016) 8(34):21893–902. doi: 10.1021/acsami.5b09344
75. Tse JR, Engler AJ. Preparation of Hydrogel Substrates With Tunable Mechanical Properties. Curr Protoc Cell Biol (2010) Chapter 10:Unit 10 16. doi: 10.1002/0471143030.cb1016s47
76. Pradhan S, Slater JH. Tunable Hydrogels for Controlling Phenotypic Cancer Cell States to Model Breast Cancer Dormancy and Reactivation. Biomaterials (2019) 215:119177. doi: 10.1016/j.biomaterials.2019.04.022
77. Han WM, Anderson SE, Mohiuddin M, Barros D, Nakhai SA, Shin E, et al. Synthetic Matrix Enhances Transplanted Satellite Cell Engraftment in Dystrophic and Aged Skeletal Muscle With Comorbid Trauma. Sci Adv (2018) 4(8):eaar4008. doi: 10.1126/sciadv.aar4008
78. Nguyen EH, Daly WT, Le NNT, Farnoodian M, Belair DG, Schwartz MP, et al. Versatile Synthetic Alternatives to Matrigel for Vascular Toxicity Screening and Stem Cell Expansion. Nat BioMed Eng (2017) 1(0096):1–14. doi: 10.1038/s41551-017-0096
79. Wee Y, Moore AN, Jia S, Zhou J, Colombo JS, D'Souza RN. A Single-Step Self-Assembly Approach for the Fabrication of Aligned and Multilayered Three-Dimensional Tissue Constructs Using Multidomain Peptide Hydrogel. SLAS Technol (2019) 24(1):55–65. doi: 10.1177/2472630318777759
80. Sarwat M, Surrao DC, Huettner N, St John JA, Dargaville TR, Forget A. Going Beyond RGD: Screening of a Cell-Adhesion Peptide Library in 3D Cell Culture. BioMed Mater (2020) 15(5):055033. doi: 10.1088/1748-605X/ab9d6e
81. Brosicke N, Sallouh M, Prior LM, Job A, Weberskirch R, Faissner A. Extracellular Matrix Glycoprotein-Derived Synthetic Peptides Differentially Modulate Glioma and Sarcoma Cell Migration. Cell Mol Neurobiol (2015) 35(5):741–53. doi: 10.1007/s10571-015-0170-1
82. Ulrich TA, Jain A, Tanner K, MacKay JL, Kumar S. Probing Cellular Mechanobiology in Three-Dimensional Culture With Collagen-Agarose Matrices. Biomaterials (2010) 31(7):1875–84. doi: 10.1016/j.biomaterials.2009.10.047
83. Barros D, Conde-Sousa E, Goncalves AM, Han WM, Garcia AJ, Amaral IF, et al. Engineering Hydrogels With Affinity-Bound Laminin as 3D Neural Stem Cell Culture Systems. Biomater Sci (2019) 7(12):5338–49. doi: 10.1039/C9BM00348G
84. Licht C, Rose JC, Anarkoli AO, Blondel D, Roccio M, Haraszti T, et al. Synthetic 3d PEG-Anisogel Tailored With Fibronectin Fragments Induce Aligned Nerve Extension. Biomacromolecules (2019) 20(11):4075–87. doi: 10.1021/acs.biomac.9b00891
85. Wang C, Tong X, Yang F. Bioengineered 3D Brain Tumor Model to Elucidate the Effects of Matrix Stiffness on Glioblastoma Cell Behavior Using PEG-Based Hydrogels. Mol Pharm (2014) 11(7):2115–25. doi: 10.1021/mp5000828
86. Chaicharoenaudomrung N, Kunhorm P, Noisa P. Three-Dimensional Cell Culture Systems as an In Vitro Platform for Cancer and Stem Cell Modeling. World J Stem Cells (2019) 11(12):1065–83. doi: 10.4252/wjsc.v11.i12.1065
87. Doyle AD, Carvajal N, Jin A, Matsumoto K, Yamada KM. Local 3D Matrix Microenvironment Regulates Cell Migration Through Spatiotemporal Dynamics of Contractility-Dependent Adhesions. Nat Commun (2015) 6:8720. doi: 10.1038/ncomms9720
88. Holle AW, Young JL, Spatz JP. In Vitro Cancer Cell-ECM Interactions Inform In Vivo Cancer Treatment. Adv Drug Delivery Rev (2016) 97:270–9. doi: 10.1016/j.addr.2015.10.007
89. Puls TJ, Tan X, Whittington CF, Voytik-Harbin SL. 3D Collagen Fibrillar Microstructure Guides Pancreatic Cancer Cell Phenotype and Serves as a Critical Design Parameter for Phenotypic Models of EMT. PloS One (2017) 12(11):e0188870. doi: 10.1371/journal.pone.0188870
90. Lee KY, Mooney DJ. Alginate: Properties and Biomedical Applications. Prog Polym Sci (2012) 37(1):106–26. doi: 10.1016/j.progpolymsci.2011.06.003
91. Andersen T, Auk-Emblem P, Dornish M. 3d Cell Culture in Alginate Hydrogels. Microarrays (Basel) (2015) 4(2):133–61. doi: 10.3390/microarrays4020133
92. Bidarra SJ, Barrias CC. 3d Culture of Mesenchymal Stem Cells in Alginate Hydrogels. Methods Mol Biol 2019 (2002) p:165–80.
93. Cavo M, Caria M, Pulsoni I, Beltrame F, Fato M, Scaglione S. A New Cell-Laden 3D Alginate-Matrigel Hydrogel Resembles Human Breast Cancer Cell Malignant Morphology, Spread and Invasion Capability Observed "In Vivo". Sci Rep (2018) 8(1):5333. doi: 10.1038/s41598-018-23250-4
94. Li Y, Kumacheva E. Hydrogel Microenvironments for Cancer Spheroid Growth and Drug Screening. Sci Adv (2018) 4(4):eaas8998. doi: 10.1126/sciadv.aas8998
95. Fang G, Lu H, Rodriguez de la Fuente L, Law AMK, Lin G, Jin D, et al. Mammary Tumor Organoid Culture in Non-Adhesive Alginate for Luminal Mechanics and High-Throughput Drug Screening. Adv Sci (Weinh) (2021) p:e2102418. doi: 10.1002/advs.202102418
96. Sze JH, Brownlie JC, Love CA. Biotechnological Production of Hyaluronic Acid: A Mini Review. 3 Biotech (2016) 6(1):67. doi: 10.1007/s13205-016-0379-9
97. Corradetti B, Taraballi F, Martinez JO, Minardi S, Basu N, Bauza G, et al. Hyaluronic Acid Coatings as a Simple and Efficient Approach to Improve MSC Homing Toward the Site of Inflammation. Sci Rep (2017) 7(1):7991. doi: 10.1038/s41598-017-08687-3
98. Gurski LA, Jha AK, Zhang C, Jia X, Farach-Carson MC. Hyaluronic Acid-Based Hydrogels as 3D Matrices for In Vitro Evaluation of Chemotherapeutic Drugs Using Poorly Adherent Prostate Cancer Cells. Biomaterials (2009) 30(30):6076–85. doi: 10.1016/j.biomaterials.2009.07.054
99. Gerecht S, Burdick JA, Ferreira LS, Townsend SA, Langer R, Vunjak-Novakovic G. Hyaluronic Acid Hydrogel for Controlled Self-Renewal and Differentiation of Human Embryonic Stem Cells. Proc Natl Acad Sci USA (2007) 104(27):11298–303. doi: 10.1073/pnas.0703723104
100. Ehlers EM, Behrens P, Wunsch L, Kuhnel W, Russlies M. Effects of Hyaluronic Acid on the Morphology and Proliferation of Human Chondrocytes in Primary Cell Culture. Ann Anat (2001) 183(1):13–7. doi: 10.1016/S0940-9602(01)80007-8
101. Suo A, Xu W, Wang Y, Sun T, Ji L, Qian J. Dual-Degradable and Injectable Hyaluronic Acid Hydrogel Mimicking Extracellular Matrix for 3D Culture of Breast Cancer MCF-7 Cells. Carbohydr Polym (2019) 211:336–48. doi: 10.1016/j.carbpol.2019.01.115
102. Weber LM, Hayda KN, Haskins K, Anseth KS. The Effects of Cell-Matrix Interactions on Encapsulated Beta-Cell Function Within Hydrogels Functionalized With Matrix-Derived Adhesive Peptides. Biomaterials (2007) 28(19):3004–11. doi: 10.1016/j.biomaterials.2007.03.005
103. Booij TH, Price LS, Danen EHJ. 3d Cell-Based Assays for Drug Screens: Challenges in Imaging, Image Analysis, and High-Content Analysis. SLAS Discovery (2019) 24(6):615–27. doi: 10.1177/2472555219830087
104. Nagahama K, Oyama N, Ono K, Hotta A, Kawauchi K, Nishikata T. Nanocomposite Injectable Gels Capable of Self-Replenishing Regenerative Extracellular Microenvironments for In Vivo Tissue Engineering. Biomater Sci (2018) 6(3):550–61. doi: 10.1039/C7BM01167A
105. Rezakhani S, Gjorevski N, Lutolf MP. Low-Defect Thiol-Michael Addition Hydrogels as Matrigel Substitutes for Epithelial Organoid Derivation. Adv Func Mater (2020) 30(48):1–12. doi: 10.1002/adfm.202000761
106. Raeber GP, Lutolf MP, Hubbell JA. Molecularly Engineered PEG Hydrogels: A Novel Model System for Proteolytically Mediated Cell Migration. Biophys J (2005) 89(2):1374–88. doi: 10.1529/biophysj.104.050682
107. Ham SL, Thakuri PS, Plaster M, Li J, Luker KE, Luker GD, et al. Three-Dimensional Tumor Model Mimics Stromal - Breast Cancer Cells Signaling. Oncotarget (2018) 9(1):249–67. doi: 10.18632/oncotarget.22922
108. Pape J, Emberton M, Cheema U. 3d Cancer Models: The Need for a Complex Stroma, Compartmentalization and Stiffness. Front Bioeng Biotechnol (2021) 9:660502. doi: 10.3389/fbioe.2021.660502
109. Caiazzo M, Okawa Y, Ranga A, Piersigilli A, Tabata Y, Lutolf MP. Defined Three-Dimensional Microenvironments Boost Induction of Pluripotency. Nat Mater (2016) 15(3):344–52. doi: 10.1038/nmat4536
110. Downing TL, Soto J, Morez C, Houssin T, Fritz A, Yuan F, et al. Biophysical Regulation of Epigenetic State and Cell Reprogramming. Nat Mater (2013) 12(12):1154–62. doi: 10.1038/nmat3777
111. Pickup KE, Pardow F, Carbonell-Caballero J, Lioutas A, Villanueva-Canas JL, Wright RHG, et al. Expression of Oncogenic Drivers in 3D Cell Culture Depends on Nuclear ATP Synthesis by NUDT5. Cancers (Basel) (2019) 11(9):1–17. doi: 10.3390/cancers11091337
112. Luca AC, Mersch S, Deenen R, Schmidt S, Messner I, Schafer KL, et al. Impact of the 3D Microenvironment on Phenotype, Gene Expression, and EGFR Inhibition of Colorectal Cancer Cell Lines. PloS One (2013) 8(3):e59689. doi: 10.1371/journal.pone.0059689
113. Huh D, Hamilton GA, Ingber DE. From 3D Cell Culture to Organs-on-Chips. Trends Cell Biol (2011) 21(12):745–54. doi: 10.1016/j.tcb.2011.09.005
114. Whitesides GM. The Origins and the Future of Microfluidics. Nature (2006) 442(7101):368–73. doi: 10.1038/nature05058
115. Torisawa YS, Mosadegh B, Luker GD, Morell M, O'Shea KS, Takayama S. Microfluidic Hydrodynamic Cellular Patterning for Systematic Formation of Co-Culture Spheroids. Integr Biol (Camb) (2009) 1(11-12):649–54. doi: 10.1039/b915965g
116. Torisawa YS, Chueh BH, Huh D, Ramamurthy P, Roth TM, Barald KF, et al. Efficient Formation of Uniform-Sized Embryoid Bodies Using a Compartmentalized Microchannel Device. Lab Chip (2007) 7(6):770–6. doi: 10.1039/b618439a
117. Patra B, Chen YH, Peng CC, Lin SC, Lee CH, Tung YC. A Microfluidic Device for Uniform-Sized Cell Spheroids Formation, Culture, Harvesting and Flow Cytometry Analysis. Biomicrofluidics (2013) 7(5):54114. doi: 10.1063/1.4824480
118. Cui P, Wang S. Application of Microfluidic Chip Technology in Pharmaceutical Analysis: A Review. J Pharm Anal (2019) 9(4):238–47. doi: 10.1016/j.jpha.2018.12.001
119. Kraly JR, Holcomb RE, Guan Q, Henry CS. Review: Microfluidic Applications in Metabolomics and Metabolic Profiling. Anal Chim Acta (2009) 653(1):23–35. doi: 10.1016/j.aca.2009.08.037
120. Bhatia SN, Ingber DE. Microfluidic Organs-on-Chips. Nat Biotechnol (2014) 32(8):760–72. doi: 10.1038/nbt.2989
121. Esch EW, Bahinski A, Huh D. Organs-On-Chips at the Frontiers of Drug Discovery. Nat Rev Drug Discovery (2015) 14(4):248–60. doi: 10.1038/nrd4539
122. El-Ali J, Sorger PK, Jensen KF. Cells on Chips. Nature (2006) 442(7101):403–11. doi: 10.1038/nature05063
123. Shang M, Soon RH, Lim CT, Khoo BL, Han J. Microfluidic Modelling of the Tumor Microenvironment for Anti-Cancer Drug Development. Lab Chip (2019) 19(3):369–86. doi: 10.1039/C8LC00970H
124. Gogoi P, Sepehri S, Zhou Y, Gorin MA, Paolillo C, Capoluongo E, et al. Development of an Automated and Sensitive Microfluidic Device for Capturing and Characterizing Circulating Tumor Cells (CTCs) From Clinical Blood Samples. PloS One (2016) 11(1):e0147400. doi: 10.1371/journal.pone.0147400
125. Caballero D, Kaushik S, Correlo VM, Oliveira JM, Reis RL, Kundu SC. Organ-On-Chip Models of Cancer Metastasis for Future Personalized Medicine: From Chip to the Patient. Biomaterials (2017) 149:98–115. doi: 10.1016/j.biomaterials.2017.10.005
126. van den Berg A, Mummery CL, Passier R, van der Meer AD. Personalised Organs-on-Chips: Functional Testing for Precision Medicine. Lab Chip (2019) 19(2):198–205. doi: 10.1039/C8LC00827B
127. Ruppen J, Cortes-Dericks L, Marconi E, Karoubi G, Schmid RA, Peng R, et al. A Microfluidic Platform for Chemoresistive Testing of Multicellular Pleural Cancer Spheroids. Lab Chip (2014) 14(6):1198–205. doi: 10.1039/C3LC51093J
128. Fang G, Lu H, Law A, Gallego-Ortega D, Jin D, Lin G. Gradient-Sized Control of Tumor Spheroids on a Single Chip. Lab Chip (2019) 19(24):4093–103. doi: 10.1039/C9LC00872A
129. Huang K, Boerhan R, Liu C, Jiang G. Nanoparticles Penetrate Into the Multicellular Spheroid-On-Chip: Effect of Surface Charge, Protein Corona, and Exterior Flow. Mol Pharm (2017) 14(12):4618–27. doi: 10.1021/acs.molpharmaceut.7b00726
130. Chen Y, Gao D, Wang Y, Lin S, Jiang Y. A Novel 3D Breast-Cancer-on-Chip Platform for Therapeutic Evaluation of Drug Delivery Systems. Anal Chim Acta 2018 (1036) p:97–106.
131. Trujillo-de Santiago G, Flores-Garza BG, Tavares-Negrete JA, Lara-Mayorga IM, Gonzalez-Gamboa I, Zhang YS, et al. The Tumor-On-Chip: Recent Advances in the Development of Microfluidic Systems to Recapitulate the Physiology of Solid Tumors. Materials (Basel) (2019) 12(18):1–40. doi: 10.3390/ma12182945
132. Shirure VS, Bi Y, Curtis MB, Lezia A, Goedegebuure MM, Goedegebuure SP, et al. Tumor-On-a-Chip Platform to Investigate Progression and Drug Sensitivity in Cell Lines and Patient-Derived Organoids. Lab Chip (2018) 18(23):3687–702. doi: 10.1039/C8LC00596F
133. Osaki T, Sivathanu V, Kamm RD. Vascularized Microfluidic Organ-Chips for Drug Screening, Disease Models and Tissue Engineering. Curr Opin Biotechnol (2018) 52:116–23. doi: 10.1016/j.copbio.2018.03.011
134. Bray LJ, Werner C. Evaluation of Three-Dimensional in Vitro Models to Study Tumor Angiogenesis. ACS Biomater Sci Eng (2018) 4(2):337–46. doi: 10.1021/acsbiomaterials.7b00139
135. Agarwal P, Wang H, Sun M, Xu J, Zhao S, Liu Z, et al. Microfluidics Enabled Bottom-Up Engineering of 3D Vascularized Tumor for Drug Discovery. ACS Nano (2017) 11(7):6691–702. doi: 10.1021/acsnano.7b00824
136. Mannino RG, Santiago-Miranda AN, Pradhan P, Qiu Y, Mejias JC, Neelapu SS, et al. 3D Microvascular Model Recapitulates the Diffuse Large B-Cell Lymphoma Tumor Microenvironment In Vitro. Lab Chip (2017) 17(3):407–14. doi: 10.1039/C6LC01204C
137. Deo KA, Singh KA, Peak CW, Alge DL, Gaharwar AK. Bioprinting 101: Design, Fabrication, and Evaluation of Cell-Laden 3d Bioprinted Scaffolds. Tissue Eng Part A (2020) 26(5-6):318–38. doi: 10.1089/ten.tea.2019.0298
138. Ma X, Liu J, Zhu W, Tang M, Lawrence N, Yu C, et al. 3D Bioprinting of Functional Tissue Models for Personalized Drug Screening and In Vitro Disease Modeling. Adv Drug Delivery Rev (2018) 132:235–51. doi: 10.1016/j.addr.2018.06.011
139. Chia HN, Vigen M, Kasko AM. Effect of Substrate Stiffness on Pulmonary Fibroblast Activation by TGF-Beta. Acta Biomater (2012) 8(7):2602–11. doi: 10.1016/j.actbio.2012.03.027
140. Sharma R, Smits IPM, de la Vega L, Lee C, Willerth SM. 3d Bioprinting Pluripotent Stem Cell Derived Neural Tissues Using a Novel Fibrin Bioink Containing Drug Releasing Microspheres. Front Bioeng Biotechnol (2020) 8:57. doi: 10.3389/fbioe.2020.00057
141. Zhou X, Zhu W, Nowicki M, Miao S, Cui H, Holmes B, et al. 3d Bioprinting a Cell-Laden Bone Matrix for Breast Cancer Metastasis Study. ACS Appl Mater Interfaces (2016) 8(44):30017–26. doi: 10.1021/acsami.6b10673
142. Swaminathan S, Hamid Q, Sun W, Clyne AM. Bioprinting of 3D Breast Epithelial Spheroids for Human Cancer Models. Biofabrication (2019) 11(2):025003. doi: 10.1088/1758-5090/aafc49
143. Kacarevic ZP, Rider PM, Alkildani S, Retnasingh S, Smeets R, Jung O, et al. An Introduction to 3D Bioprinting: Possibilities, Challenges and Future Aspects. Materials (Basel) (2018) 11(11):1–21. doi: 10.3390/ma11112199
144. Gudapati H, Dey M, Ozbolat I. A Comprehensive Review on Droplet-Based Bioprinting: Past, Present and Future. Biomaterials (2016) 102:20–42. doi: 10.1016/j.biomaterials.2016.06.012
145. Graham AD, Olof SN, Burke MJ, Armstrong JPK, Mikhailova EA, Nicholson JG, et al. High-Resolution Patterned Cellular Constructs by Droplet-Based 3d Printing. Sci Rep (2017) 7(1):7004. doi: 10.1038/s41598-017-06358-x
146. Utama RH, Atapattu L, O'Mahony AP, Fife CM, Baek J, Allard T, et al. A 3d Bioprinter Specifically Designed for the High-Throughput Production of Matrix-Embedded Multicellular Spheroids. iScience (2020) 23(10):101621.
147. Koch L, Gruene M, Unger C, Chichkov B. Laser Assisted Cell Printing. Curr Pharm Biotechnol (2013) 14(1):91–7.
148. Guillotin B, Souquet A, Catros S, Duocastella M, Pippenger B, Bellance S, et al. Laser Assisted Bioprinting of Engineered Tissue With High Cell Density and Microscale Organization. Biomaterials (2010) 31(28):7250–6. doi: 10.1016/j.biomaterials.2010.05.055
149. Huang TQ, Qu X, Liu J, Chen S. 3D Printing of Biomimetic Microstructures for Cancer Cell Migration. BioMed Microdevices (2014) 16(1):127–32. doi: 10.1007/s10544-013-9812-6
150. Guillemot F, Souquet A, Catros S, Guillotin B. Laser-Assisted Cell Printing: Principle, Physical Parameters Versus Cell Fate and Perspectives in Tissue Engineering. Nanomed (Lond) (2010) 5(3):507–15. doi: 10.2217/nnm.10.14
151. Heinrich MA, Bansal R, Lammers T, Zhang YS, Michel Schiffelers R, Prakash J. 3d-Bioprinted Mini-Brain: A Glioblastoma Model to Study Cellular Interactions and Therapeutics. Adv Mater (2019) 31(14):e1806590. doi: 10.1002/adma.201806590
152. Zhao Y, Yao R, Ouyang L, Ding H, Zhang T, Zhang K, et al. Three-Dimensional Printing of Hela Cells for Cervical Tumor Model In Vitro. Biofabrication (2014) 6(3):035001. doi: 10.1088/1758-5082/6/3/035001
153. Wang X, Zhang X, Dai X, Wang X, Li X, Diao J, et al. Tumor-Like Lung Cancer Model Based on 3D Bioprinting. 3 Biotech (2018) 8(12):501. doi: 10.1007/s13205-018-1519-1
154. Zhang J, Chen F, He Z, Ma Y, Uchiyama K, Lin JM. A Novel Approach for Precisely Controlled Multiple Cell Patterning in Microfluidic Chips by Inkjet Printing and the Detection of Drug Metabolism and Diffusion. Analyst (2016) 141(10):2940–7. doi: 10.1039/C6AN00395H
155. Xu F, Celli J, Rizvi I, Moon S, Hasan T, Demirci U. A Three-Dimensional In Vitro Ovarian Cancer Coculture Model Using a High-Throughput Cell Patterning Platform. Biotechnol J (2011) 6(2):204–12. doi: 10.1002/biot.201000340
156. Ashammakhi N, Ahadian S, Zengjie F, Suthiwanich K, Lorestani F, Orive G. Advances and Future Perspectives in 4D Bioprinting. Biotechnol J (2018) 13(12):e1800148. doi: 10.1002/biot.201800148
157. Brassard JA, Nikolaev M, Hubscher T, Hofer M, Lutolf MP. Recapitulating Macro-Scale Tissue Self-Organization Through Organoid Bioprinting. Nat Mater (2021) 20(1):22–9. doi: 10.1038/s41563-020-00803-5
158. Yi HG, Jeong YH, Kim Y, Choi YJ, Moon HE, Park SH, et al. A Bioprinted Human-Glioblastoma-on-a-Chip for the Identification of Patient-Specific Responses to Chemoradiotherapy. Nat BioMed Eng (2019) 3(7):509–19. doi: 10.1038/s41551-019-0363-x
159. Alves TR, Lima FR, Kahn SA, Lobo D, Dubois LG, Soletti R, et al. Glioblastoma Cells: A Heterogeneous and Fatal Tumor Interacting With the Parenchyma. Life Sci (2011) 89(15-16):532–9. doi: 10.1016/j.lfs.2011.04.022
160. Castaneda CA, Casavilca S, Orrego E, Garcia-Corrochano P, Deza P, Heinike H, et al. Glioblastoma: Molecular Analysis and Its Clinical Implications. Rev Peru Med Exp Salud Publica (2015) 32(2):316–25.
161. Stroganov V, Al-Hussein M, Sommer JU, Janke A, Zakharchenko S, Ionov L. Reversible Thermosensitive Biodegradable Polymeric Actuators Based on Confined Crystallization. Nano Lett (2015) 15(3):1786–90. doi: 10.1021/nl5045023
162. Hribar KC, Finlay D, Ma X, Qu X, Ondeck MG, Chung PH, et al. Nonlinear 3D Projection Printing of Concave Hydrogel Microstructures for Long-Term Multicellular Spheroid and Embryoid Body Culture. Lab Chip (2015) 15(11):2412–8. doi: 10.1039/C5LC00159E
163. Booth MJ, Schild VR, Graham AD, Olof SN, Bayley H. Light-Activated Communication in Synthetic Tissues. Sci Adv (2016) 2(4):e1600056. doi: 10.1126/sciadv.1600056
164. Wang LL, Highley CB, Yeh YC, Galarraga JH, Uman S, Burdick JA. Three-Dimensional Extrusion Bioprinting of Single- and Double-Network Hydrogels Containing Dynamic Covalent Crosslinks. J BioMed Mater Res A (2018) 106(4):865–75. doi: 10.1002/jbm.a.36323
165. Kokkinis D, Schaffner M, Studart AR. Multimaterial Magnetically Assisted 3D Printing of Composite Materials. Nat Commun (2015) 6:8643. doi: 10.1038/ncomms9643
166. Aronsson C, Jury M, Naeimipour S, Boroojeni FR, Christoffersson J, Lifwergren P, et al. Dynamic Peptide-Folding Mediated Biofunctionalization and Modulation of Hydrogels for 4D Bioprinting. Biofabrication (2020) 12(3):035031. doi: 10.1088/1758-5090/ab9490
167. Tabriz AG, Hermida MA, Leslie NR, Shu W. Three-Dimensional Bioprinting of Complex Cell Laden Alginate Hydrogel Structures. Biofabrication (2015) 7(4):045012. doi: 10.1088/1758-5090/7/4/045012
168. Gladman AS, Matsumoto EA, Nuzzo RG, Mahadevan L, Lewis JA. Biomimetic 4D Printing. Nat Mater (2016) 15(4):413–8. doi: 10.1038/nmat4544
169. Kwag HR, Serbo JV, Korangath P, Sukumar S, Romer LH, Gracias DH. A Self-Folding Hydrogel In Vitro Model for Ductal Carcinoma. Tissue Eng Part C Methods (2016) 22(4):398–407. doi: 10.1089/ten.tec.2015.0442
170. Kuribayashi-Shigetomi K, Onoe H, Takeuchi S. Cell Origami: Self-Folding of Three-Dimensional Cell-Laden Microstructures Driven by Cell Traction Force. PloS One (2012) 7(12):e51085. doi: 10.1371/journal.pone.0051085
171. Ryan SL, Baird AM, Vaz G, Urquhart AJ, Senge M, Richard DJ, et al. Drug Discovery Approaches Utilizing Three-Dimensional Cell Culture. Assay Drug Dev Technol (2016) 14(1):19–28. doi: 10.1089/adt.2015.670
172. Montanez-Sauri SI, Beebe DJ, Sung KE. Microscale Screening Systems for 3D Cellular Microenvironments: Platforms, Advances, and Challenges. Cell Mol Life Sci (2015) 72(2):237–49. doi: 10.1007/s00018-014-1738-5
173. Ruel-Gariepy E, Leroux JC. In Situ-Forming Hydrogels–Review of Temperature-Sensitive Systems. Eur J Pharm Biopharm (2004) 58(2):409–26. doi: 10.1016/j.ejpb.2004.03.019
174. Horvath P, Aulner N, Bickle M, Davies AM, Nery ED, Ebner D, et al. Screening Out Irrelevant Cell-Based Models of Disease. Nat Rev Drug Discovery (2016) 15(11):751–69. doi: 10.1038/nrd.2016.175
175. Worthington P, Drake KM, Li Z, Napper AD, Pochan DJ, Langhans SA. Beta-Hairpin Hydrogels as Scaffolds for High-Throughput Drug Discovery in Three-Dimensional Cell Culture. Anal Biochem (2017) 535:25–34. doi: 10.1016/j.ab.2017.07.024
176. Eismann B, Krieger TG, Beneke J, Bulkescher R, Adam L, Erfle H, et al. Automated 3D Light-Sheet Screening With High Spatiotemporal Resolution Reveals Mitotic Phenotypes. J Cell Sci (2020) 133(11):1–11. doi: 10.1242/jcs.245043
177. Bilgin CC, Fontenay G, Cheng Q, Chang H, Han J, Parvin B. BioSig3D: High Content Screening of Three-Dimensional Cell Culture Models. PloS One (2016) 11(3):e0148379. doi: 10.1371/journal.pone.0148379
178. Fong ELS, Toh TB, Yu H, Chow EK. 3d Culture as a Clinically Relevant Model for Personalized Medicine. SLAS Technol (2017) 22(3):245–53. doi: 10.1177/2472630317697251
Keywords: 3D culture systems, personalised medicine, drug resistance prevention, tumor microenvironment, 3D bioprinting, extracellular matrix, microfluidics
Citation: Law AMK, Rodriguez de la Fuente L, Grundy TJ, Fang G, Valdes-Mora F and Gallego-Ortega D (2021) Advancements in 3D Cell Culture Systems for Personalizing Anti-Cancer Therapies. Front. Oncol. 11:782766. doi: 10.3389/fonc.2021.782766
Received: 24 September 2021; Accepted: 11 November 2021;
Published: 30 November 2021.
Edited by:
Maria Rosaria De Miglio, University of Sassari, ItalyReviewed by:
Diogo Alpuim Costa, CUF Oncologia, PortugalCopyright © 2021 Law, Rodriguez de la Fuente, Grundy, Fang, Valdes-Mora and Gallego-Ortega. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fatima Valdes-Mora, RlZhbGRlc01vcmFAY2NpYS5vcmcuYXU=; David Gallego-Ortega, RGF2aWQuR2FsbGVnb09ydGVnYUB1dHMuZWR1LmF1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.