 Parvinder Kaur1*Ramya V. K.1Naveenkumar C. N.1Bharathkumar K.1Mayas Singh1Sven N. Hobbie2†‡Radha Krishan Shandil1†Shridhar Narayanan1†
Parvinder Kaur1*Ramya V. K.1Naveenkumar C. N.1Bharathkumar K.1Mayas Singh1Sven N. Hobbie2†‡Radha Krishan Shandil1†Shridhar Narayanan1†- 1Foundation for Neglected Disease Research, Bengaluru, India
- 2University Hospital Basel, Basel, Switzerland
Introduction: Mycobacterium tuberculosis (Mtb) has the capability to dodge the immune system by escaping into alternate physiological forms by forming drug tolerant populations under the immune pressure in the host. New drugs are urgently needed to treat these non-replicating persisters. In the past, aminoglycoside antibiotics have played a pivotal role in TB chemotherapy.
Methods: Here, we explored the therapeutic potential of a monosubstituted deoxystreptamine aminoglycoside, apramycin (APR) which is different in its chemical structure from the other clinically relevant aminoglycoside antibiotics that are all disubstituted, e.g., amikacin (AMI). We determined the APR MIC as 0.25-1 µg/ml for sensitive and multidrug-resistant Mtb (MDRTB), including amikacin (AMI) resistant strains.
Results: In standard time-kill kinetic assays, the bactericidal activity of APR was similar to that of AMI demonstrating dose-dependent killing of planktonic Mtb. However, in biofilm and macrophage intracellular killing assays, APR appeared significantly more potent than AMI. Further, APR monotherapy was efficacious in a mouse chronic TB lung infection model (~0.92 log10 CFU/lung reduction). APR combination therapy with the current 1st line standard of care (SoC) antibiotic combination of isoniazid (H), rifampicin (R), ethambutol (E), and pyrazinamide (Z) was found to be additive (HREZ=1.88 vs. HREZ-APR=2.78 log10CFU/lung reduction).
Discussion: The results indicate the potential of apramycin-based combinations for the treatment of human tuberculosis.
Introduction
Mycobacterial infections are relatively complicated to treat and remain one of the major global health problems (40). This is partly due to the ability of Mycobacterium tuberculosis (Mtb) to exist in multiple phenotypes in the human host under immune or drug pressure. The non-replicating phenotypes are relatively drug-tolerant and refractory to treatment, which is why the treatment of tuberculosis (TB) is complex and involves the use of multidrug regimens (40). The emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) strains has further derailed the treatment outcomes in TB. The first- and second-line TB drug regimens have nearly failed and have led to MDR- and XDR-TB (1). A few years ago, data on the treatment success rates of approximately 54% in MDR-TB and only 13% in XDR-TB patients were alarming (2). Hence, there is a definitive need to develop new therapeutic options with better differentiated mechanism of action (MOA) to tackle MDR-TB (42). In this study, we explored the potential of a veterinary aminoglycoside drug, apramycin (APR), which is an aminocyclitol antibiotic (Supplementary Figure S1) produced by Streptomyces tenebrarius (3). Aminoglycosides such as streptomycin have been a mainstay as the first-line treatment for TB, while amikacin (AMI) and kanamycin (KAN) are the second-line regimens for the treatment of MDR-TB. The caveat is that they are injectables and are not good for long-term treatment. In addition, irreversible ototoxicity issues in 90% of patients under long-term treatment remain a major clinical challenge (40, 3, 4). Moreover, TB is also rampant in immunocompromised hosts with underlying lung diseases, such as chronic obstructive pulmonary disease (COPD), HIV/viral diseases, leukemia, and organ transplant (5). TB drug resistance is rapidly evolving (42, 40). The new all-oral and shorter drug combination of bedaquiline, pretomanid, and linezolid (BPaL) has been developed and proven to treat 91% of MDR-TB cases in a mere 6 months vs. 18–24 months (42, 6). However, toxicity still remains an issue (6). Another regimen, which comprises bedaquiline, pretomanid, linezolid, and moxifloxacin (BPaLM), is recommended programmatically in place of 9- or 18-month regimens in patients with MDR-/RR-TB (rifampicin-resistant TB) (7). The development of the BPaL-based combination has certainly revolutionized the treatment of TB in recent years and has opened the possibility of a significantly shortened TB treatment by targeting new mechanisms. In addition to new drugs, the repurposing of old drugs from other indications with known safety profiles is an alternate viable option (8, 9) for the treatment of TB, particularly for drug-resistant cases.
APR is a different aminoglycoside, isolated in 1967, from nebramycin, an aminoglycoside antibiotic complex (3, 10) approved for the treatment of infections in veterinary medicine. APR is not orally bioavailable. It exhibits broad-spectrum activity and is currently being explored for the treatment of human infections, including Gram-negative infections and pneumonia (6). We explore the possibility of developing APR as an option for the treatment of TB. APR has a unique structure compared with other clinically relevant aminoglycosides. It has a bicyclic sugar moiety and a monosubstituted deoxystreptamine (unlike AMI and other aminoglycosides, which are disubstituted). It binds to the deep groove of 16S ribosomal RNA and blocks translocation to inhibit protein synthesis in bacteria, including Mtb. In addition, APR could also bind to the eukaryotic decoding site despite several differences in the key residues required for APR recognition by the bacterial target. Resistance to aminoglycosides is driven by the chemical inactivation of the drug by aminoglycoside-modifying enzymes (AMEs), reducing its affinity for the bacterial ribosome. However, in the case of Mtb, mutations in the 16S ribosomal RNA and rpsL gene have been reported, leading to “pan-aminoglycoside resistance” (11–15). APR is an aminoglycoside with a unique octa-diose core [molecular weight (MW) = 539.58] (Supplementary Figure S1) and, thus, works against most aminoglycoside resistance mechanisms known so far. Its distinct structure is not affected by any of the pathogen’s innate aminoglycoside resistance mechanisms or the multidrug resistance regulator (e.g., WhiB7) (25). Subcellular fractionation of several aminoglycosides such as streptomycin, gentamycin, kanamycin, and AMI has shown their preferential localization into lysosomes, but not into phagosomes where intracellular pathogens reside (16). This was attributed to be the reason for the poor intracellular bacterial killing of aminoglycosides. However, APR has previously been shown to be efficacious in an acute TB lung infection mouse model, while AMI was merely bacteriostatic (17). Furthermore, APR showed a better dose–response efficacy than AMI and linezolid (LNZ) in the blood and kidney in a Staphylococcus aureus septicemia model (17). The efficacy of APR in mouse infection models of TB and S. aureus has illustrated its potential clinical utility.
In the present study, we investigated the anti-TB potential of the APR drug formulation against i) the “standard” bacterial phenotypes (e.g., planktonic Mtb and drug-sensitive Mtb); ii) phenotypic resistance/drug tolerance associated with specific bacterial physiological states and lifestyles (e.g., biofilm and intracellular Mtb); and iii) genotypically resistant strains with mutations (i.e., drug-resistant Mtb). We further substantiated the in vitro results by demonstrating the in vivo efficacy of APR against chronic Mtb infection in a mouse model of TB. These studies provided preclinical evidence that APR can be repurposed for the development of a new parenteral combination regimen for the treatment of TB. However, the detailed pharmacokinetic–pharmacodynamic (PK-PD) relationship of APR needs to be explored further.
Materials and methods
Mtb strains and growth conditions
The Mtb strain H37Rv (ATCC 27294) and various drug-resistant Mtb strains were used in the present study (Supplementary Table S1). All of the Mtb strains were cultured in Middlebrook 7H9 broth or 7H10 agar complete medium [albumin, dextrose, and catalase (ADC)/oleic acid plus ADC (OADC) supplemented], 0.2% glycerol and 0.1% Tween-80, at 37°C with 5% CO2. All of the experiments with Mtb were conducted at the Foundation for Neglected Diseases Research (FNDR) in a BSL-3 facility. The biofilm of Mtb H37Rv and the planktonic cultures of different strains of Mtb were grown at 37°C in the presence of 5% CO2. A tight and consistent biofilm formation was observed only when the cultures were grown in the presence of 5% CO2.
Antibiotics, drugs, chemicals, and media
Pharmaceutical grade APR was manufactured as a crystalline monohydrate (EBL-1003) (18). APR was assessed in the in vitro assays and for in vivo efficacy in a chronic TB infection mouse model. As reference antibiotics, AMI, rifampicin (RIF), moxifloxacin (MOX), isoniazid (INH), ethambutol (EMB), pyrazinamide (PZA), ofloxacin (OFX), and thioacetazone (THZ), pretomanid (Pa), LNZ and menadione (MEN) were procured from Sigma-Aldrich (Merck, Rahway, NJ, USA) or were received as gifts from Lupin Pharmaceuticals (Mumbai, India). Bedaquiline (BDQ) was obtained through the National Institutes of Health (NIH), as HIV Reagent Program, Division of AIDS, NIAID, NIH: Bedaquiline Fumarate (BDQ) ARP-12702, contributed by Janssen Pharmaceuticals. The media and the supplements used in this study included Middlebrook 7H9 broth base and Middlebrook 7H10 agar base, ADC supplements obtained from BD/Difco (Franklin Lakes, NJ, USA), and Tween-80 purchased from Merck (Rahway, NJ, USA). The stock solutions (12.8 mg/ml) of the test compounds and the reference drug controls (e.g., RIF and MOX) were prepared separately in dimethyl sulfoxide (DMSO), while INH was prepared in water. Working solutions were freshly prepared at the time of experimentation. The choice of antibiotics was dictated by the drug resistance profiles of the clinical isolates and the aminoglycoside comparator of APR (i.e., AMI). APR was compared for biofilm inhibition potential against the first-line (HRE) and new regimen (BPaLM) drug treatments for TB. MEN was used as a known cytotoxic control drug.
Minimal inhibitory concentration of APR against planktonic Mtb
The minimal inhibitory concentration (MIC) of APR was determined against a replicating/planktonic population of Mtb as recommended by the Clinical and Laboratory Standards Institute guidelines M24 (36, 19, 41) and the previously described procedures for Mtb, with appropriate modifications (20). Briefly, the test compounds were dissolved in DMSO and serially double-diluted in a 10-concentration dose–response (10c-DR) ranging from 64 to 0.125 μg/ml in 96-well plates. Middlebrook 7H9 broth (supplemented with 10% ADC) complete medium was used for the assay. Mtb cultures were added at 200 μl in each well to all columns, except for the medium control (200 μl of medium was added) column to give a final inoculum of 3–7 × 105 colony forming unit (CFU)/ml. The quality control (QC) included the following: medium controls, growth controls, and the reference antibiotics e.g., AMI, RIF, and MOX). The assay plates were incubated at 37°C, and the results were noted on day 6 for Mtb using a turbidometric readout. Clear wells indicate inhibition of growth, while turbid wells indicate uninhibited growth. MIC is the minimum concentration of molecules that completely inhibited the turbidometric growth of bacteria/colorimetric readout. The MIC assays were conducted in triplicate.
Determination of the minimum bactericidal concentration of APR
The minimum bactericidal concentration (MBC) was determined using a procedure we have previously reported (19, 21, 41) from a parallel set of MIC plates. Each dilution of the inhibited culture wells from the MIC plate (25 µl) was plated in triplicate onto Middlebrook 7H9 agar supplemented with 10% OADC and incubated at 37°C for different Mtb strains. The CFU was enumerated after the respective incubation period. MBC was taken as the lowest concentration that killed 99% of the initial Mtb inoculum.
Killing kinetics of APR against Mtb
The killing kinetics on the replicating Mtb phenotype was determined as described previously (22, 23). The Mtb (H37Rv) culture was inoculated at ~3–8 × 107 CFU/ml in fresh Middlebrook 7H9 complete medium containing varying concentrations of APR and the reference drug AMI (1, 4, 8, 32, and 128 μg/ml). The cultures were incubated at 37°C at different time intervals. CFU enumeration was conducted by plating aliquots of cultures (from 10−1 to 10−8 dilutions) containing different concentrations of the compounds on days 3, 7, and 14. Data were analyzed and plotted as log10 CFU per milliliter on days 3, 7, and 14 as a function of the concentration of APR using Prism v9.0 software to calculate the killing potential of APR.
Cytotoxicity against mammalian cell lines
The cytotoxicity of APR was evaluated against different cell lines: the HepG2, A549, Vero, and PMA-activated THP-1 macrophage cell lines (22). Briefly, HepG2 (ATCC HB-8065), A549 (ATCC CCL-185), Vero (ATCC CCL-81), or THP-1 monocytes (ATCC TIB-202) were maintained in RPMI 1640 medium supplemented with 2 mM l-glutamine and 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a humidified atmosphere of 5% CO2. FBS was obtained from Life Technologies (Carlsbad, CA, USA). Resazurin and trypan blue were purchased from Sigma-Aldrich (St. Louis, MO, USA). Furthermore, THP-1 cells in the RPMI medium were activated using 50 nM of phorbol 12-myristate 13-acetate (PMA) for 48–72 h at 37°C with 5% CO2. HepG2 and A549 cells were grown as a monolayer. Post-maturation, THP-1 macrophages, along with the HepG2, Vero, and A549 cells, were exposed to the test compound APR (twofold dilutions, 256–0.5 µg/ml) and incubated at 37°C with 5% CO2 for 48 h. Post-incubation, resazurin dye was added at 0.125 mg/ml concentration with an equal volume of the RPMI medium and further incubated for 24 h. The colorimetric readings were taken 24 h after the addition of resazurin dye (22, 23).
Intracellular efficacy of APR in THP-1 macrophages
The THP-1 (tumor macrophage-derived) cell line was used to examine the efficacy of APR against Mtb in the intracellular compartment. The assay was performed as biological triplicates. THP-1 cells were grown in RPMI medium (Gibco-BRL Life Technologies, Gaithersburg, MD, USA) and supplemented with 100 mM sodium pyruvate, 200 mM l-glutamine, 3.7 g/L sodium bicarbonate (Sigma, St. Louis, MO, USA), and 10% fetal calf serum (Gibco-BRL Life Technologies, Gaithersburg, MD, USA) without any antibiotics. The viable macrophages were seeded (approximately 5 × 105 cells/well) in 96-well plates (Nunc, Roskilde, Denmark) with complete RPMI medium, incubated overnight, induced with 50 nM PMA to achieve macrophage differentiated phenotypes, and incubated again at 37°C for 48–72 h in 5% CO2 atmosphere. After 48 h of activation, the THP-1 macrophages were infected with the Mtb H37Rv strain at a multiplicity of infection (MOI) of 1:10 and then incubated for 2 h at 37°C with 5% CO2 (22, 23). The macrophage monolayers were washed with 3 ml of phosphate-buffered saline (PBS) (Ca2+ and Mg2+) to remove the free bacteria. These monolayers were further treated with AMI (15 µg/ml) for 2 h to kill extracellular bacteria (24), washed again, and then replenished with fresh complete RPMI medium.
APR (at 32–2 µg/ml) and the control drugs MOX and AMI (at 32–0.5 µg/ml) were added to the infected THP-1 macrophages in triplicate. Cultures were sampled from the untreated infection control, and the wells were treated with different concentrations of APR, MOX, and AMI. These were plated on D-0 (infection control), D-3, and D-7 and were enumerated for residual viable numbers of intracellular Mtb. The intracellular mycobacterial killing curves were generated by plotting the log10 CFU per milliliter against different drug concentrations (in micrograms per milliliter).
MIC of APR against the clinical isolates of Mtb strains
The MIC of APR was determined against a total of 10 clinical isolates of Mtb with known sensitivity and drug resistance profiles in parallel to a wild-type (WT) strain, as per the method described in the “Minimal inhibitory concentration of APR against planktonic Mtb” (36, 19–21) and the CLSI breakpoints. The 10 TB clinical isolates included strains that were WT-sensitive (n = 1), clinical isolate-sensitive (n = 1), double drug-resistant (DDR; n = 3), and MDR (n = 6).
Development of Mtb biofilms
There are several methods available in the literature for the production of biofilms of different microbes. We developed stable and visibly strong biofilms of Mtb in a 96-well format (37). The Mtb strain H37Rv (ATCC 27294) was grown for biofilm formation in Sauton’s medium (+10% ADC) at 37°C for 3 weeks to reproducibly obtain well-formed biofilms every time (37). The cultures were diluted to 3–7 × 106 CFU/ml in the respective media and were dispensed into assay plates (96-well plates, 300 µl/well). The 96-well plates containing the respective cultures were tightly wrapped in parafilm so that the culture plates were airtight. The plates were incubated for 4–6 weeks for biofilm formation.
Activity of APR on Mtb biofilm
Once biofilm formation was achieved, the compounds were diluted as a four-concentration DR (fourfold) with final concentrations of 64×, 16×, 4×, and 1× the MIC values of the planktonic Mtb. The MICs of APR and the reference drugs (e.g., AMI, HRE, and BPaLM) were evaluated by exposing the preformed biofilms of Mtb to different concentrations of APR in a dose–response manner. The plates were again wrapped airtight and incubated at 37°C for 3 weeks. At the end of the incubation time, Tween-80 (0.1%) was added to the wells and the plates incubated at room temperature (RT) for 15 min. The biofilm and the entire contents were transferred into 15-ml tubes and were pelleted by centrifugation (3,000 rpm for 20 min). The cell pellet was resuspended in fresh media, and the residual CFU was enumerated on 7H10 agar plates. The MIC was taken as the first concentration showing a CFU reduction ≥0.2 log10 reduction in the drug-treated samples compared with the growth control (without drug treatment). The MICs of APR and the reference drugs against the biofilm and planktonic forms were plotted as CFU log10 reduction for comparison. However, the biofilm biomass could not be estimated during this study due to technical challenges.
In vivo efficacy of APR in a mouse model of Mtb infection
Animal studies
An animal infection and efficacy study was carried out in strict accordance with the recommendations of the Institutional Animal Ethics Committee (IAEC), registered with the Committee for the Control and Supervision (CCSEA), Government of India (registration no. 48/GO/Re-SL/BiS/99/CPCSEA). All of the experimental protocols involving the use of animals were reviewed and approved in advance by the IAEC.
BALB/c mice aged between 6 and 8 weeks, with an average body weight of 25–30 g, were used. The animals were housed in individually ventilated cages (IVCs) in the BSL-3 facility and were randomly assigned to cages and allowed to acclimatize for 1 week before the experiments. Feed and water were provided ad libitum.
In vivo efficacy of APR: chronic (non-replicating) model of Mtb infection
BALB/c mice were infected via aerosol inhalation in a Glas-Col chamber (Glas-Col, Terre Haute, IN, USA) calibrated to deliver ~100–200 CFU of Mtb H37Rv (ATCC 27294) in the BSL3 facility (22). Infected mice were housed in IVC isolators (Allentown Technologies, Allentown, PA, USA) during the entire period of experimentation. Treatment began 4 weeks post-infection. APR (200 mg/kg) was administered once daily through the subcutaneous (S/C) route in PBS 6 days a week, for a period of 4 weeks. A standard-of-care (SoC) four-drug combination regimen (H15R5E100Z150) formulated in 0.5% HPMC + 0.1% Tween-80 was administered orally either without APR (H15R5E100Z150) or in combination with APR (H15R5E100Z150 + APR200). All of the groups had n = 5, except for the post-drug control (n = 3). On completion of dosing, the animals were sacrificed 48 h later through CO2 narcosis. The lungs were then removed, homogenized, and plated for the enumeration of Mtb CFU/lung on Middlebrook 7H11 plates supplemented with OADC and PANTA (polymyxin B, amphotericin B, nalidixic acid, trimethoprim, and azilocillin BD-245114). The plates were incubated at 37°C with 5% CO2 for 3 weeks prior to reading of the bacterial CFU counts. The data were analyzed and the graphs were plotted using Prism v9.0 software.
Results
MIC of APR against clinical Mtb isolates
The MIC and MBC of APR in wild-type H37Rv were found to be 0.5–1 and 4 µg/ml, respectively (Table 1). APR demonstrated MIC activity against all 10 clinical isolate strains of Mtb (MIC = 0.25–0.5 µg/ml) with different sensitivity and resistance profiles, including the MDR strains (Table 1). The antibiogram of the drug-resistant strains was confirmed by respective QC drugs. More specifically, APR inhibited the aminoglycoside-resistant Mtb strains such as the streptomycin- and AMI-resistant strains, thus confirming that APR is a different aminoglycoside drug and should work on aminoglycoside-resistant strains.
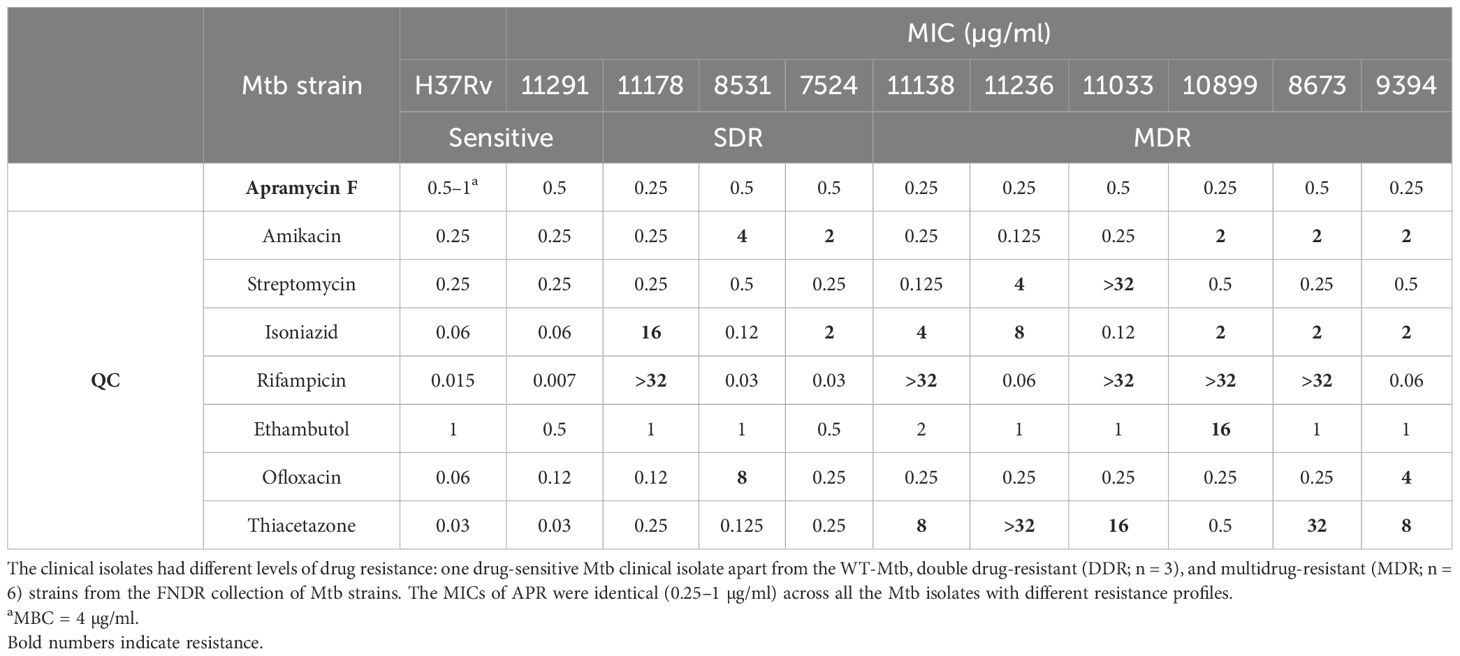
Table 1. Minimal inhibitory concentration (MIC) of apramycin (APR) against Mycobacterium tuberculosis (Mtb) wild type (WT) and Mtb clinical isolates.
Killing kinetics of APR against replicating Mtb
Studies on the in vitro killing kinetics determine the PK-PD drivers of antibacterial activity by measuring the maximum efficacy (Emax) upon exposure to various drug concentrations (22, 23). The killing kinetics were investigated over 14 days of exposure to the drugs using concentrations between 1 and 128 μg/ml against Mtb H37Rv. The bacterial CFU were enumerated, the data were compiled, and the kill curve graphs were generated by plotting the log10 CFU per milliliter values against time (in hours) with GraphPad Prism v9.0 (Figure 1). APR displayed strong bactericidal effects, and the Emax values were 5.20 and 5.28 log10 CFU/ml for APR and AMI at 32 µg/ml, respectively (Figures 1A, B). APR demonstrated an increased kill with increasing concentrations and longer time durations and displayed an exposure-dependent killing profile.
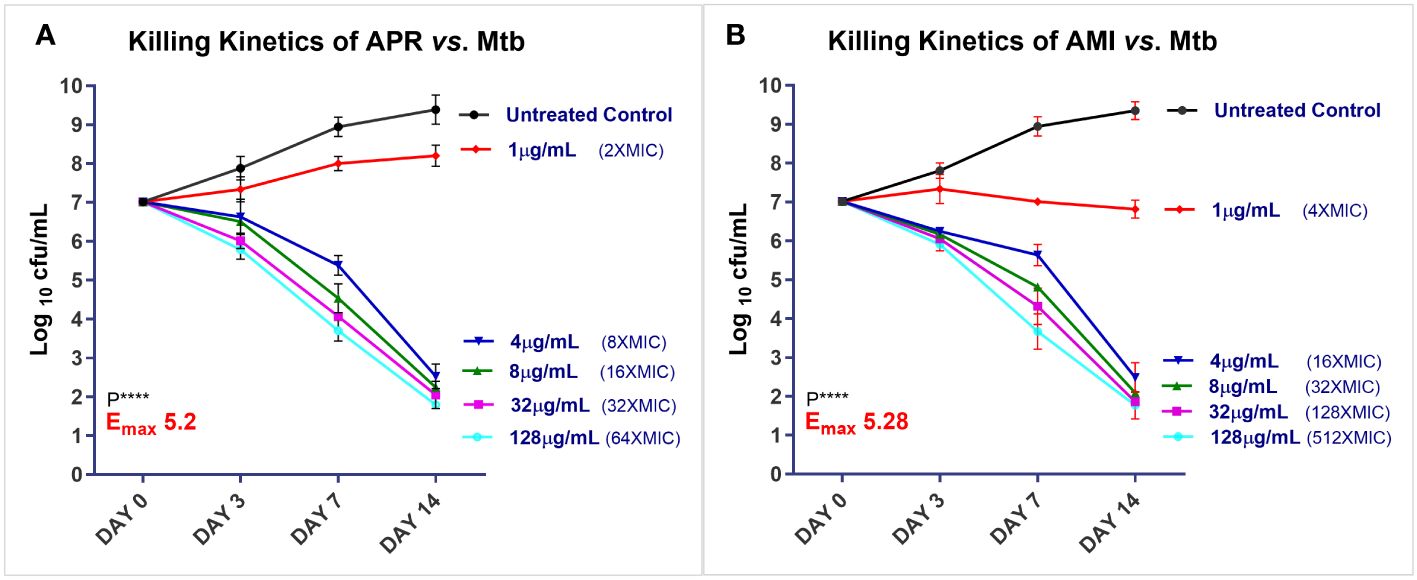
Figure 1. Time kill kinetics of apramycin (APR) and amikacin (AMI) against Mycobacterium tuberculosis (Mtb) H37Rv. Symbols [residual log10 colony forming unit (CFU) per milliliter] are uniform across the graphs of APR (A) and AMI (B). Filled black circles denote day 0 (D-0) control culture unexposed to the drug; filled red diamonds represent drug exposure at 1 µg/ml; blue inverted triangle denotes 4 µg/ml; green upright triangle represents 8 µg/ml; filled pink square represents 32 µg/ml; and cyan open circles denote exposure to 128 µg/ml. The assay was performed with three biological replicates (n = 3). Data represent the mean ± SD of three biological replicates. Emax (log10 CFU reduction per milliliter at the maximum concentration tested) upon APR exposure vs. the D-0 control culture: APR Emax128 = 5.2; AMI Emax128 = 5.28. Statistical significance was evaluated using one-way ANOVA (Dunnett’s test) compared with the untreated (bacteria only) control. ****p < 0.0001.
Intracellular efficacy of APR in THP-1 macrophages
APR exhibited a good dose-dependent intracellular efficacy (IE) against Mtb in THP-1 macrophages. The bacterial kill increased with time from day 3 to day 7. APR exhibited a net Emax of 1.15 log10 CFU/ml (Figure 2A) reduction. The positive control drug RIF showed a 1.81 log10 CFU/ml reduction, while AMI did not show any kill of intracellular TB, as expected (Figure 2B).
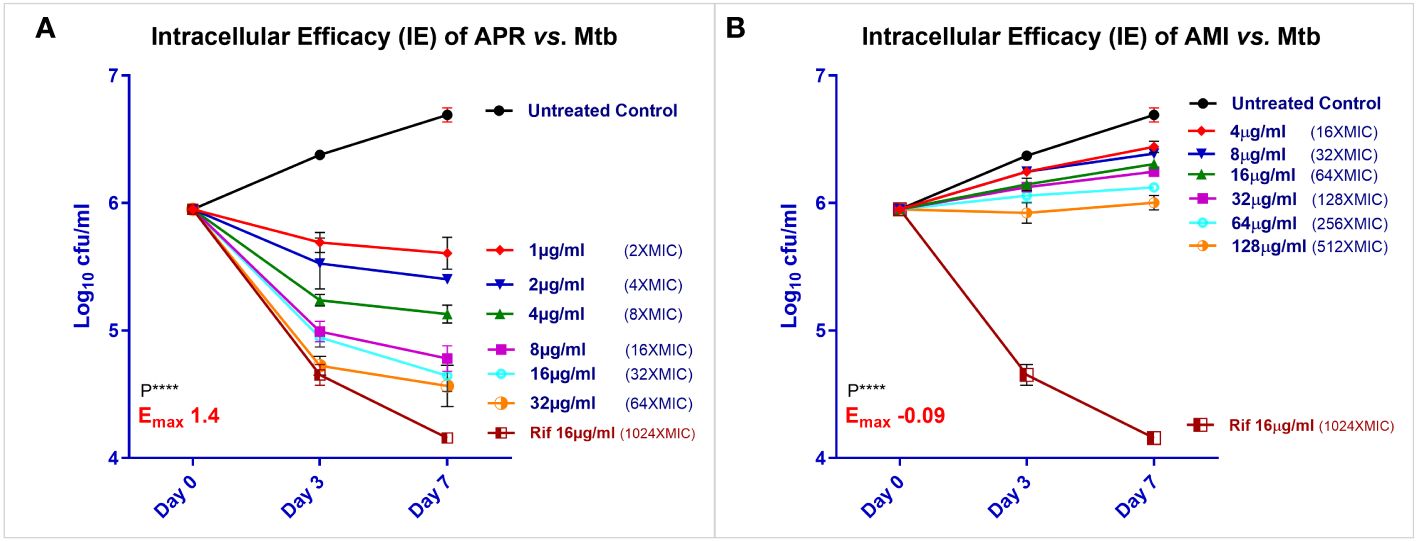
Figure 2. Intracellular efficacy (IE) of apramycin (APR) and amikacin (AMI) against Mycobacterium tuberculosis (Mtb) H37Rv in THP-1 macrophages. IE was determined for APR (A) along with the reference drugs rifampicin (RIF) and AMI (B) as biological triplicates from 32 μg/ml as twofold of a six-concentration dose–response (DR) (1, 2, 4, 8, 16, and 32 µg/ml) vs. the no drug control in THP-1 macrophages infected with Mtb. Symbols [residual log10 colony forming unit (CFU) per milliliter after days 0, 3, and 7) are uniform across the graphs of APR, AMI, and RIF. Filled black circles denote the day 0 (D-0) control culture unexposed to drugs, while filled red diamonds represent drug exposure at the first concentration, blue inverted triangle the second concentration, green upright triangle the third, filled pink square the fourth, cyan open circles the fifth, and orange half open circle the sixth highest concentration tested. Brown half-open square with a brown line denotes exposure to RIF at 16 µg/ml. Data represent the mean ± SD of three biological replicates. The Emax (log10 CFU reduction per milliliter at the maximum concentration tested) for APR was 1.4 at 64× minimal inhibitory concentration (MIC) and for AMI was −0.09 (growth) until 512× MIC compared with the D-0 control culture. Statistical significance was evaluated using one-way ANOVA (Dunnett’s test) compared with the untreated (bacteria only) control. ****p < 0.0001.
Killing potential of apramycin against biofilm forms of Mtb
The Mtb biofilm was formed as a thick leathery pellicle layer in 96-well plates. The activity against biofilms was determined by measuring the reduction in bacterial CFU per milliliter following a 3-week drug exposure. The activity of APR and the SoC drugs AMI and MOX examined at 1×, 4×, 16×, and 64× MICs showed good dose-dependent bacterial reduction (Figures 3A, B). APR showed equally good potency against the Mtb biofilm (Supplementary Table S2 and Figure 3A), but the CFU reduction was better than that of AMI and MOX. The Emax (log10 CFU per milliliter reduction) was in the order of APR (1.07) > MOX (0.70) > AMI (0.28), as depicted in Supplementary Table S2 and Figures 3A, B.
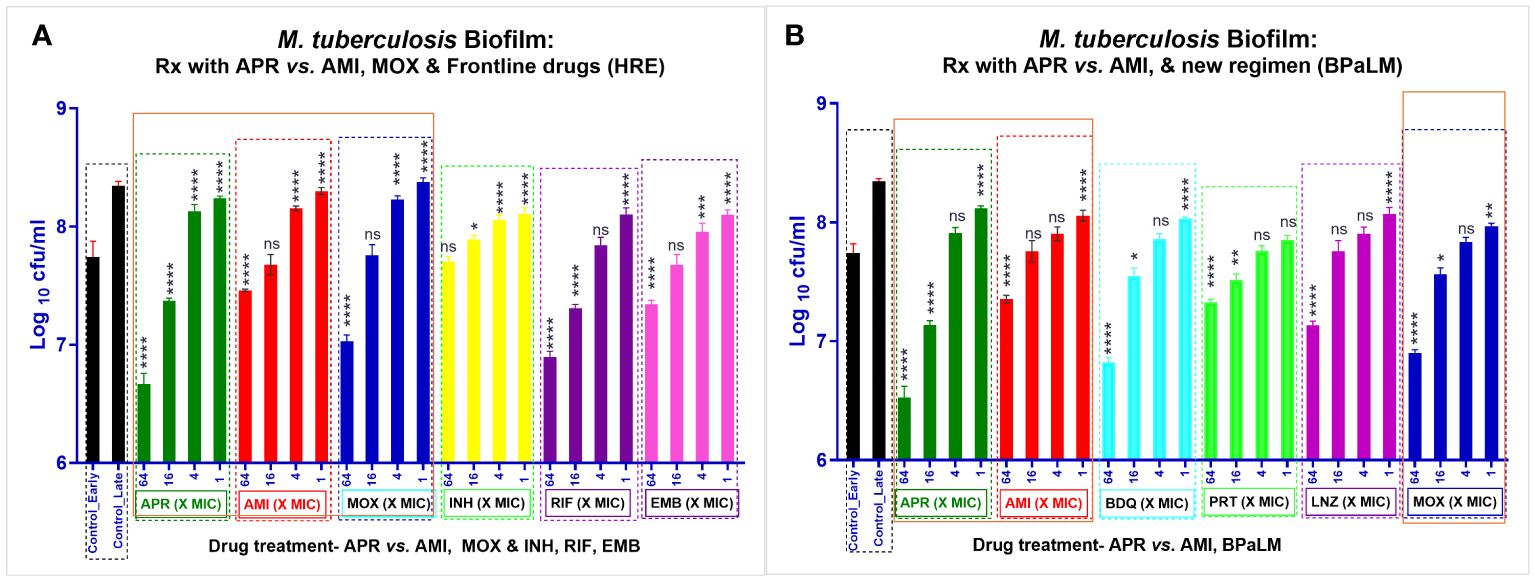
Figure 3. Activity of apramycin (APR), amikacin (AMI), and other reference drugs against Mycobacterium tuberculosis (Mtb) H37Rv biofilms. Preformed biofilms (A) were treated with APR, AMI, moxifloxacin (MOX), isoniazid (INH), rifampicin (RIF), and ethambutol (EMB) (biological replicates, n = 3). Black bars, growth control; dark green bars, APR; red bars, AMI; dark blue bar, MOX; yellow bars, INH; purple bars, RIF; pink bars, EMB. (B) Treatment with APR, AMI, bedaquiline (BDQ), pretomanid (PRT), linezolid (LNZ), and moxifloxacin (MOX) (biological replicates, n = 3). Dark green bars, APR; red bars, AMI; cyan blue bar, BDQ; light green bars, PRT; purple bars, LNZ; and dark blue bars, MOX. APR, AMI, and MOX (highlighted in orange box) were the technical replicates from (A, B). The drugs were tested at four different doses of × minimal inhibitory concentration (MIC) (1×, 4×, 16×, and 64×). The assay plates were incubated at 37°C along with an untreated control. This was followed by colony forming unit (CFU) counts post-treatment. The graphs (A, B) represent the residual log10 CFU per milliliter of Mtb. MICbiofilm is the first concentration showing growth inhibition or CFU reduction >0.2 log10 in the drug-treated samples vs. the growth control. Error bars indicate the mean ± SD of biological triplicates (n = 3). Statistical significance was evaluated using one-way ANOVA (Dunnett’s test) compared with the untreated (bacteria only) control. ****p < 0.0001. APR showed approximately 1.07 and 1.02 log10 CFU/ml reduction at 64× MIC vs. the untreated control. AMI did not show good activity against the Mtb biofilm (0.28 and 0.2 log10 CFU/ml). All the other drug controls showed CFU reduction as Emax: MOX = 0.76, INH = −0.09, RIF = 0.67, and EMB = 0.28 (A) and BDQ = 0.73, PRT = 0.22, LNZ = 0.4, and MOX = 0.65 (B) CFU log10 CFU/ml reduction of Mtb in biofilm. The order of CFU reduction is as follows: APR (1.07 and 1.02) > BDQ (0.73) > MOX (0.76 and 0.65) > RIF (0.67) > LNZ (0.4) > EMB (0.28) > AMI (0.28 and 0.2) > PRT (0.22) > INH (−0.09).
In vivo efficacy of APR in a chronic model of Mtb infection
APR was assessed in a chronic Mtb infection mouse model via the S/C dosing route for 4 weeks either alone or in combination with HREZ (Figure 4). APR at 200 mg/kg (S/C) was found to be efficacious, with a 0.92 log10 CFU/lung kill (p < 0.0001), while HREZ alone exhibited 1.88 log10 CFU/lung kill (p < 0.0001) vs. untreated control mice. Since the bacterial loads at the beginning of treatment (4 weeks post-infection) in the chronic infection model are usually as high as 106 CFU/lung, there was enough window following HREZ treatment to show the magnitude of kill. As envisaged, the addition of APR to the HREZ regimen significantly (p < 0.0001) increased the Mtb kill to 2.78 log10 CFU/lung. The combination regimen was well tolerated, and none of the animals exhibited any adverse effects during the 4 weeks of drug treatment. There was no antagonism observed. APR exhibited an additive killing in combination with HREZ, the SoC drug regimen for TB.
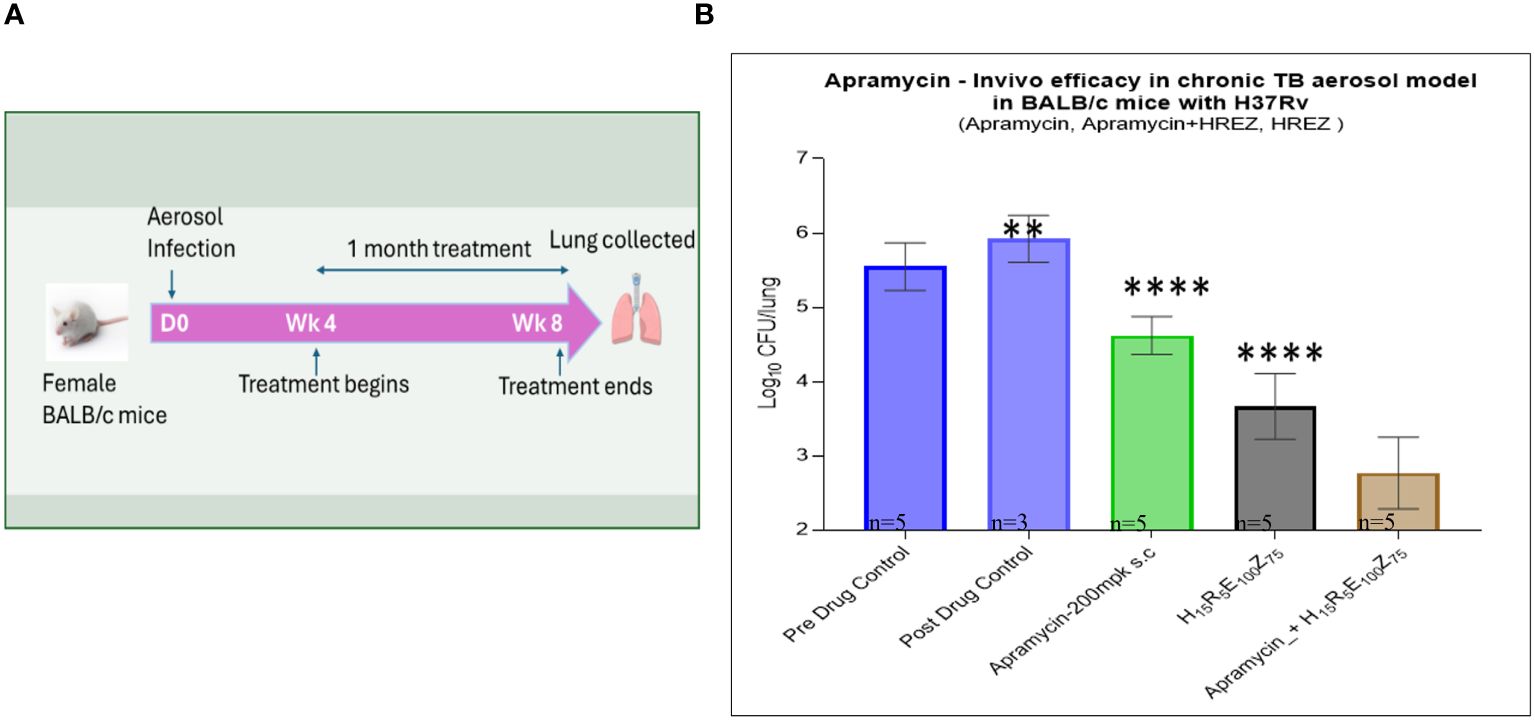
Figure 4. In vivo efficacy of apramycin in mouse models of Mycobacterium tuberculosis (Mtb) infection. (A) Schematic of the experimental design and timelines. (B) In vivo efficacy of apramycin tested against Mtb in a chronic lung infection BALB/c mouse model (all n = 5, except for the late control, n = 3) for inhibition and colony forming unit (CFU) reduction at different doses, along with an untreated control. Light blue bar with border denotes pre-drug growth control, while light blue bar represents post-drug growth control. Apramycin-200 mg/kg via the subcutaneous (S/C) route is shown as light green bar, HREZ (isoniazid, rifampicin, ethambutol, and pyrazinamide) is shown as gray bar, and apramycin-200 + HREZ is shown as light beige bar. In the chronic Mtb infection BALB/c mouse model, treatment with apramycin 200 mg/kg (S/C route) over 4 weeks showed a significant 0.92 log10 CFU kill, whereas apramycin in combination with HREZ exhibited 2.78 log10 CFU kill. HREZ alone exhibited 1.88 log10 CFU kill. The addition of apramycin significantly increased the kill (2.78 log10 CFU/lung). Apramycin did not demonstrate any antagonistic effect; rather, an additive effect was observed in combination with HREZ, the standard-of-care (SoC) drug regimen for tuberculosis (TB). Error bars indicate ±SD of the biological replicates. Statistical significance was evaluated using one-way ANOVA (Dunnett’s test) compared with the untreated (bacteria only) control. ****p < 0.0001. Symbol ** is P value < 0.001, indicates statistical significance.
Cytotoxicity against mammalian cell lines
APR did not demonstrate any toxicity to THP-1, HepG2, and A549 cells up to 256× MIC (256 µg/ml). MOX (MIC = 0.06 µg/ml) and AMI (MIC = 0.5 µg/ml), which were used as controls, also had no cytotoxicity up to 64× MIC, while MEN (the cytotoxic drug control) showed cytotoxicity at 8 µg/ml.
Discussion
Aminoglycosides are widely used as powerful antibiotics in the clinic. However, emerging drug resistance and the ototoxicity associated with this class have presented unsurmountable challenges (6). New drugs with a novel MOA and better safety profiles are needed. The antibacterial activity of APR has been successfully dissected from ototoxicity. The cytotoxicity data from a variety of cell lines in the present study clearly ruled out the cytotoxicity issues of APR up to >256 μg/ml, indicating that APR with a differentiated MOA and a lower cytotoxic potential deserves development for the treatment of many infections (25). APR works against almost all known preexisting aminoglycoside resistance mechanisms in Gram-negative and mycobacterial pathogens (Table 1) (13, 25–27).
APR was found to be highly potent in bloodstream Gram-negative pathogens resistant to other aminoglycosides, carbapenems, third-generation cephalosporins, and colistin from Southeast Asia (28). More importantly, all of the Acinetobacter baumannii and Pseudomonas clinical isolates tested were sensitive to APR, including ~94 colistin-resistant isolates. This suggested that APR is a potential aminoglycoside drug candidate for the treatment of MDR Gram-negative systemic infections. In addition, APR indeed exhibited much better efficacy than AMI and CLA in Mycobacteroides abscessus infection models (6, 17, 29), thus making it an ideal candidate for the treatment of difficult-to-treat non-tuberculous mycobacterial (NTM) lung infections. APR is currently in clinical development for the treatment of M. abscessus (6, 18). Juvabis has successfully completed phase 1 human safety clinical trials (6) and is now further exploring its development as an infusion (APRIVIN) and inhalation drug (APRINAH) for the treatment of M. abscessus infections.
Decades of impressive history of aminoglycoside use in the treatment of TB, despite their ototoxicity, prompted us to study a veterinary drug, i.e., APR, with a differentiated MOA and a lower ototoxic potential for the treatment of TB (25). APR is a broad-spectrum antibiotic. Several studies (e.g., MIC, MBC, activity against drug-resistant/MDR strains, and activity against biofilms) have been performed and reported on Mtb, NTM, and Gram-positive and Gram-negative pathogens (Supplementary Table S4) (27, 30–34). However, our studies have been extended in order to show activity against more complex models of replicating and non-replicating Mtb (e.g., in macrophages and biofilms). Furthermore, we demonstrated the efficacy of APR in combination with HREZ on non-replicating bacilli in chronic infection mouse models of Mtb. APR is a potent bactericidal agent and has resulted in a dose-dependent multi-log kill of Mtb in vitro and in macrophages (Figures 1A, 2A).
APR, as opposed to several other aminoglycosides such as AMI (Figure 2B), resulted in >1 log10 kill of intracellular TB in a concentration-dependent manner (Figure 2A). AMI is known to be ineffective against intracellular TB and is indeed used to pretreat cellular monolayers in order to kill extracellular bacteria in such experiments (38, 30). The differential cellular penetration of APR and the killing of intracellular TB in macrophages might be explained by its unique structural differences and its ability to localize in phagosomes compared with AMI (18).
Studies on the cellular penetration of aminoglycosides such as streptomycin, gentamycin, kanamycin, and AMI have shown that these drugs localize into lysosomes, but not into phagosomes where intracellular pathogens reside, thus explaining the poor intracellular bacterial killing of aminoglycosides in general (16, 24).
Keeping in mind the potent antibacterial activity of APR, we evaluated its activity against drug-sensitive and drug-resistant Mtb clinical isolates and its killing kinetics on replicating and non-replicating TB in macrophages and biofilms. Most of the reported studies on APR have been against planktonic mycobacteria, but none against Mtb in the biofilm phase (25, 26).
We observed that APR penetrated the Mtb biofilms (non-replicating phenotype) and, at 64 µg/ml, showed an Emax of 1.07 (log10 CFU/ml reduction), which is a well-tolerated, safe, and efficacious concentration based on available data (cytotoxicity data from the present study). APR has a comparatively lower toxicity than other aminoglycosides of the disubstituted deoxystreptamine drug class (13). The ability of APR to penetrate the lung tissue by ~88% and kill replicating TB in an acute infection mouse model has been previously demonstrated (35, 17). APR monotherapy at the 200-mg/kg dose exhibited ~1 log10 CFU/lung reduction, while AMI was merely bacteriostatic. We further substantiated the in vivo efficacy of APR in a chronic infection mouse model (representing non-replicating TB) as a monotherapy and in combination with the HREZ regimen. APR reduced the Mtb bacterial load by 0.92 log10 CFU/lung at a 200-mg/kg dose given for 4 weeks. However, our study lacked the lung histopathology data. Furthermore, combination studies with HREZ showed that APR is safe, and it was well tolerated in the 4-week dosing regimen. The lung bacterial counts showed that APR acts additively with HREZ and increased the efficacy of HREZ alone from 1.88 to 2.78 log10 CFU/lung reduction. We hypothesized that the addition of APR to HREZ could present an opportunity to treat larger populations (about 90% of the drug-susceptible vs. only the drug-resistant patients). This unique combination regimen might help 1) prevent the development of resistance in Mtb by virtue of hitting yet another target mechanism and 2) possibly shorten the treatment duration from 6 months to a shorter period. The in vitro and preclinical results of APR demonstrated that it is suitable for the HREZ-based combination regimen.
However, the outcomes of the combination of APR with the second-line and new anti-TB drugs in the clinic/pipeline (bedaquiline, pretomanid, delamanid, and linezolid, among others) remain to be established.
Shortening the duration of therapy is a major goal of all TB discovery and development programs. However, no new drugs for TB have been discovered and reached the clinic between 1970 and 2010. The most recent therapies for TB, i.e., the bedaquiline-based regimens BPaL and BPaLM, have been proven to shorten the duration of TB treatment to 6 months, or lesser, vs. 24 months (7). Many more new clinical candidates, including pretomanid, delamanid, new oxazolidinones, BTZ-43 and TBA-7371, and others (39), are in various stages of clinical trials. We envision that there is a strong possibility that drug candidates such as APR, which have high bactericidal activity on replicating and non-replicating TB phenotypes including biofilms and MDR strains, could help in the development of the next level of combinations to further shorten the treatment of TB to less than 6 months. However, APR, being a broad-spectrum antibiotic and an injectable drug, might pose some challenges.
In summary, our in vitro and in vivo studies suggest that APR is a comparatively safer aminoglycoside drug candidate with potent bactericidal activity against replicating and non-replicating phenotypes of Mtb and against MDR-Mtb and Mtb in biofilms, macrophages, and in vivo in mice. APR holds significant promise for development as a therapeutic option for the exploration of more potent combinations that could further shorten the duration of TB treatment.
Data availability statement
The original contributions presented in the study are publicly available. The data can be found here: https://doi.org/10.6084/m9.figshare.26462791.v1.
Ethics statement
Animal infection and efficacy study was carried out in strict accordance with recommendations of the Institutional Animal Ethics Committee (IAEC), registered with the Committee for the Control and Supervision (CCSEA), Government of India (registration no. 48/GO/Re-SL/BiS/99/CPCSEA). All the experimental protocols involving the use of animals were reviewed and approved in advance by the IAEC. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
PK: Conceptualization, Formal Analysis, Investigation, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. RV: Data curation, Formal Analysis, Methodology, Software, Writing – original draft. NC: Data curation, Formal Analysis, Methodology, Software, Writing – original draft. BK: Data curation, Formal Analysis, Methodology, Software, Writing – original draft. MS: Project administration, Writing – review & editing. SH: Conceptualization, Supervision, Visualization, Writing – review & editing. RS: Funding acquisition, Investigation, Resources, Validation, Writing – original draft, Writing – review & editing. SN: Funding acquisition, Investigation, Resources, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the Foundation for Neglected Disease Research, Bangalore, India.
Conflict of interest
SH is a co-founder of Juvabis AG.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fitd.2024.1413211/full#supplementary-material
Supplementary Figure 1 | Structure of both aminoglycosides: a. Apramycin (MW=539.5), b. Amikacin (MW=585.6).
References
1. Kwonjune JS, Keshavjee S, Rich ML. Multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis. Cold Spring Harb Perspect Med. (2015) 5:a017863. doi: 10.1101/cshperspect.a0178633
2. Bridgen G, NyangWa T, du Cross P, Varaine F, Huges J, Rich M, et al. Principle for designing future regimens for multidrug resistant tuberculosis. Bull World Health Org. (2014) 92:68–74. doi: 10.2471/BLT.13.122028
3. O’Connor S, Lam LKT, Jones ND, Chaney M. Apramycin, a unique aminocyclitol antibiotic. J Org Chem. (1976) 41:2087. doi: 10.1021/jo00874a003
4. Global Tuberculosis Report. (2021). Geneva: World Health Organization. Available online at: https://www.who.int/publications/i/item/9789240037021.
5. Jain NK. Chronic obstructive pulmonary disease and tuberculosis. Lung India. (2017) 34:468–9. doi: 10.4103/lungindia.lungindia_183_17
6. Becker K, Cao S, Nilsson A, Erlandsson M, Hotop SK, Kuka J, et al. Antibacterial activity of apramycin at acidic pH warrants a wide therapeutic window in the treatment of complicated urinary tract infections and acute pyelonephritis. EBioMedicine. (2021) 73:103652. doi: 10.1016/j.ebiom.2021.103652
7. Francesca C. Treatment of highly drug-resistant pulmonary tuberculosis. New Eng.J.Med. (2020) 382:893–902. doi: 10.1056/NEJMoa2119430
8. Padmapriyadarsini C, Devaleenal B, Ponnuraja C, Ramraj B, Singla R, Parmar M, et al. Randomised trial to evaluate the effectiveness and safety of varying doses of linezolid with bedaquiline and pretomanid in adults with pre-extensively drug-resistant or treatment intolerant/non-responsive multidrug-resistant pulmonary tuberculosis: study protocol. BMJ Open. (2022) 12:e058606. doi: 10.1136/bmjopen-2021-058606
9. Sharma K, Ahmed F, Sharma T, Grover A, Agarwal M, Grover S. Potential repurposed drug candidates for tuberculosis treatment: progress and update of drugs identified in over a decade. ACS Omega. (2023) 8(20):17362–80. doi: 10.1021/acsomega.2c05511
10. Cohen KA, Stott KE, Munsamy V, Manson AL, Earl AM, Pym AS. Evidence for expanding the role of streptomycin in the management of drug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. (2020) 64:e00860–20. doi: 10.1128/AAC.00860-20
11. Ramirez MS, Tolmasky ME. Aminoglycoside modifying enzymes. Drug Resist Update. (2010) 13:151–71. doi: 10.1016/j.drup.2010.08.003
12. Doi Y, Wachino JI, Arakawa Y. Aminoglycoside resistance: the emergence of acquired 16S ribosomal RNA methyltransferases. Infect Dis Clin North Am. (2016) 30:523–37. doi: 10.1016/j.idc.2016.02.011
13. Juhas M, Widlake E, Teo J, Huseby DL, Tyrrell JM, Polikanov YS, et al. In vitro activity of apramycin against multidrug-, carbapenem, and aminoglycoside-resistant Enterobacteriaceae and Acinetobacter baumannii. J Antimicrob Chemother. (2019) 74:944–52. doi: 10.1093/jac/dky546
14. Hu Y, Liu L, Zhang X, Feng Y, Zong Z. In vitro activity of neomycin, streptomycin, paromomycin and apramycin against carbapenem-resistant enterobacteriaceae clinical strains. Front Microbiol. (2019) 8:2275. doi: 10.3389/fmicb.2017.02275
15. Bader MS, Loeb M, Leto D, Brooks AA. Treatment of urinary tract infections in the era of antimicrobial resistance and new antimicrobial agents. Postgrad Med. (2020) 132:234–50. doi: 10.1080/00325481.2019.1680052
16. Tulkens PM. Intracellular distribution and activity of antibiotics. Eur J Clin Microbiol Infect Dis. (1991) 10:100–6. doi: 10.1007/BF01964420
17. Meyer M, Freihofer P, Scherman M, Teague J, Lenaerts A, Böttger EC. In vivo efficacy of apramycin in murine infection models. Antimicrob Agents Chemother. (2014) 58:6938–41. doi: 10.1128/AAC.03239-14
18. Zhao C, Chirkova A, Rosenborg S, Palma Villar R, Lindberg J, Hobbie SN, et al. Population pharmacokinetics of apramycin from first-in-human plasma and urine data to support the prediction of efficacious dose. J Antimicrob Chemother. (2022) 77:2718–28. doi: 10.1093/jac/dkac225
19. Kaur P, Ghosh A, Krishnamurthy RV, Bhattacharjee DG, Achar V, Datta S, et al. A high-throughput cidality screen for mycobacterium tuberculosis. PloS One. (2015) 10:e0117577. doi: 10.1371/journal.pone.0117577
20. Shirude PS, Shandil R, Sadler C, Naik M, Hosagrahara V, Hameed S, et al. Azaindoles: noncovalent DprE1 inhibitors from scaffold morphing efforts kill Mycobacterium tuberculosis and are efficacious in vivo. J Med Chem. (2013) 56:9701–8. doi: 10.1021/jm401382v
21. Kaur P, Potluri V, Ahuja VK, Naveenkumar CN, Krishnamurthy RV, Gangadharaiah ST, et al. A multi-targeting pre-clinical candidate against drug-resistant tuberculosis. Tuberculosis (Edinb). (2021) 129:102104. doi: 10.1016/j.tube.2021.102104
22. Shandil RK, Jayaram R, Kaur P, Gaonkar S, Suresh BL, Mahesh BN, et al. Moxifloxacin, Ofloxacin, Sparfloxacin, and Ciprofloxacin against Mycobacterium tuberculosis: Evaluation of in vitro and pharmacodynamic indices that best predict in vivo efficacy. Antimicrob Agents Chemother. (2007) 51:576–82. doi: 10.1128/AAC.00414-06
23. Jayaram R, Gaonkar S, Kaur P, Suresh BL, Mahesh BN, Jayashree R, et al. Pharmacokinetic-pharmacodynamic relationships for rifampicin in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother. (2003) 47:2118–24. doi: 10.1128/aac.47.7.2118-2124.200336
24. Ferro D, Mouton JW, van Ingen J, Gumbo T. Amikacin pharmacokinetics/pharmacodynamics in a novel hollow-fiber Mycobacterium abscessus disease model. Antimicrob Agents Chemother. (2016) 60:1242–8. doi: 10.1128/AAC.02282-15.
25. Matt T, Ng CL, Lang K, Sha SH, Akbergenov R, Shcherbakov D, et al. Dissociation of antibacterial activity and aminoglycoside ototoxicity in the 4-monosubstituted 2-deoxystreptamine apramycin. Proc Natl Acad Sci U S A. (2012) 109:10984–9. doi: 10.1073/pnas.1204073109
26. Boettger E, Vasella A. Treatment of bacterial Infectious Diseases Patent WO 2012/034955 A1, 2012. Switzerland: University of Zurich and UTH Zurich. (2012).
27. Bordeleau E, Stogios PJ, Evdokimova E, Koteva K, Savchenko A, Wright GD. ApmA is a unique aminoglycoside antibiotic acetyltransferase that inactivates apramycin. mBio. (2021) 12:e02705–20. doi: 10.1128/mBio.02705-20
28. Gysin M, Hon PY, Tan P, Sengduangphachanh A, Simmalavong M, Hinfonthong P, et al. Apramycin susceptibility of multidrug-resistant Gram-negative blood culture isolates in five countries in Southeast Asia. Int J Antimicrob Agents. (2022) 60:106659. doi: 10.1016/j.ijantimicag.2022.106659
29. Selchow P, Ordway DJ, Verma D, Whittel N, Petrig A, Hobbie SN, et al. Apramycin overcomes the inherent lack of antimicrobial bactericidal activity in mycobacterium abscessus. Antimicrob Agents Chemother. (2022) 66:e0151021. doi: 10.1128/aac.01510-21
30. Hermann T, Tereshko V, Skripkin E, Patel DJ. Apramycin recognition by the human ribosomal decoding site, Blood Cell. Molecules Dis. (2007) 38:193–8. doi: 10.1016/j.bcmd.2006.11.006
31. Kim M, Kim M, Kim KS. YmdB-mediated down-regulation of sucA inhibits biofilm formation and induces apramycin susceptibility in Escherichia coli. Biochem Biophys Res Commun. (2016). doi: 10.1016/j.bbrc.2016.12.157
32. Hao M, Shi X, Lv J, Niu S, Cheng S, Du H, et al. In vitro Activity of Apramycin Against Carbapenem-Resistant and Hypervirulent Klebsiella pneumoniae Isolates. Front Microbiol. (2020) 11:425. doi: 10.3389/fmicb.2020.00425
33. Di Bonaventura G, Lupetti V, Verginelli F, Giancristofaro S, Barbieri R, Gherardi G, et al. Repurposing the veterinary antibiotic apramycin for antibacterial and antibiofilm activity against pseudomonas aeruginosa from cystic fibrosis patients. Front Microbiol. (2022) 12:801152. doi: 10.3389/fmicb.2021.801152
34. Atlas N, Uzair B, Movellan J, Gracia R, Dupin D, Loinaz I, et al. In vitro activity of novel apramycin-dextran nanoparticles and free apramycin against selected Dutch and Pakistani Klebsiella pneumonia isolates. Heliyon. (2023) 9(12). doi: 10.1016/j.heliyon.2023.e22821
35. Becker K, Aranzana-Climent V, Cao S, Nilsson A, Shariatgorji R, Haldimann K, et al. Efficacy of EBL-1003 (apramycin) against Acinetobacter baumannii lung infections in mice. Clin Microbiol Infection. (2021) 27:1315–21. doi: 10.1016/j.cmi.2020.12.004
36. Woods GL, Brown-Elliott BA, Conville PS, Desmond PE, Hall GS, Lin G, et al. Wayne, PA. Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes [Internet]. 2nd edition. 940 West Valley Road, Suite 1400, Wayne, PA 19087 USA: Clinical and Laboratory Standards Institute; 2011 Mar. (CLSI publication / Clinical and Laboratory Standards Institute, No. 31.5.). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK544374/.
37. Kulka K, Hatfull G, Ojha AK. Growth of mycobacterium tuberculosis biofilms. J Vis Exp. (2012) 60:e3820. doi: 10.3791/3820
38. Tabrizi SN, Robins-Browne RM. Elimination of extracellular bacteria by antibiotics in quantitative assays of bacterial ingestion and killing by phagocytes. J Immunol Methods. (1993) 158:201–6. doi: 10.1016/0022-1759(93)90215-S
39. Working group on new TB drugs. (2024) Available online at: https://www.newtbdrugs.org/pipeline/discovery.
40. Global TB Report 2022. Geneva: World Health Organization. (2023) Available at: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/globaltuberculosis-report-2022.
41. Kaur P, Datta S, Shandil RK, Kumar N, Robert N, Sokhi UK, et al. Unravelling the secrets of mycobacterial cidality through the lens of antisense. PLoS One. (2016) 11(5):e0154513. doi: 10.1371/journal.pone.0154513
Keywords: Mycobacterium tuberculosis (Mtb), multidrug resistance (MDR), planktonic, biofilm (BF), apramycin, replicating, non-replicating
Citation: Kaur P, V. K. R, C. N. N, K. B, Singh M, Hobbie SN, Shandil RK and Narayanan S (2024) Apramycin kills replicating and non-replicating Mycobacterium tuberculosis. Front. Trop. Dis 5:1413211. doi: 10.3389/fitd.2024.1413211
Received: 24 April 2024; Accepted: 08 July 2024;
Published: 20 August 2024.
Edited by:
Jacob Lorenzo-Morales, University of La Laguna, SpainCopyright © 2024 Kaur, V. K., C. N., K., Singh, Hobbie, Shandil and Narayanan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Parvinder Kaur, parvinder.kaur@fndr.in
†These authors share senior authorship
‡ORCID: Sven N. Hobbie, orcid.org/0000-0002-9205-0384