 Douglas R. Green
Douglas R. Green
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Cell Death , 11 September 2024
Sec. Cellular Stress and Survival: Crosslinks, Intersections, and Pathways
Volume 3 - 2024 | https://doi.org/10.3389/fceld.2024.1477346
This article is part of the Research Topic Editor’s Choice in Cell Death Research View all articles
For decades, we have assumed that all regulated cell death pathways have a dedicated “point of no return” that precedes death but after which cells are committed to die. The realization that this is often not the case represents what can be considered a paradigm shift in the field. Here we consider how cells can survive despite engagement of a cell death pathway and the consequences of a “near death experience” in cancer cells and possibly other physiological conditions.
How cells die is a fundamental aspect of the biology of the cell, and as such, it is realistic to assume that despite decades of research into the various modes and pathways of cell death, we continue to have a great deal to learn. Perhaps paradoxically, the field of cell death remains vibrantly alive, as evidenced by the frequent advances and publications in this realm of molecular and cellular biology.
From the outset, it is useful to consider three general types of cell death (which I distinguish from earlier descriptions of Type I, II, and III; terms I will not use herein). The first we can consider “murder,” that is, the cell is actively killed. Some examples of murder are freeze-thaw, hypotonic shock, complement-mediated lysis, and cytotoxic cell killing. These need not be simple (freeze-thaw) but can include the participation of the dying cell (cytotoxic cell killing), and can have important roles in organismal biology. The second type to consider is “sabotage,” that is, the disruption of a process necessary for the cell to survive. This, too, can involve the active participation of the cell—just as an industrial loom will not be sabotaged by a wooden shoe if the machine is not running. (While this image is often suggested to be the origin of the word “sabotage,” from sabot, or wooden shoe, the term actually derives from the effect of removing a sabot, or railroad tie, to damage a moving train. In either case, the effect, that of destroying something that actively participates in its own destruction amply conveys the point here). An example of cellular sabotage is the process of ferroptosis, and there are many other named cell death modalities that can be classified in this way. The third type can be considered cell “suicide,” that is, the activation of a pathway that at least appears to have evolved to result in cell death when engaged. These are the well described processes of apoptosis, necroptosis, and pyroptosis, and there may be others.
Many years ago, at one of the first meetings on cell death, Robert Horwitz noted that there are potentially a great many ways that a cell can be killed, and by extension, that a cell might kill itself. If we consider that many genes and their products are essential for cell survival, simply shutting off such a gene would be a mechanism whereby a cell could commit suicide on demand. From this perspective, it is surprising that so few mechanisms of cellular suicide seem to have evolved.
While I refer to these “types” of cell death herein, there are other ways in which cell death is often parsed. “Programmed” and “regulated” cell death are terms that are often used to distinguish a cell death modality from simple destruction, as are the traditional classifications of necrosis, apoptosis, and “autophagic” cell death. All of these are distinctions that blur as connections between the modalities come to light (e.g., the phenomenon of “secondary necrosis” in apoptosis (Rogers et al., 2017), “PANoptosis” (Lee et al., 2021)). More recently, there has been a move to define cell death modalities based on molecular mechanisms (Galluzzi et al., 2018), and for the most part, this is the tactic that I employ herein.
In any cell death, of nearly any type, there is a question of how cells can evade death and survive, and when, precisely a cell is committed to die. This is the question that concerns us herein. While most discussions of this sort provide overviews of the various cell death pathways under consideration, here I will assume that the reader is familiar with at least the outlines of these pathways, and if not, I refer that reader to the many excellent overviews of the cell death pathways we discuss herein.
Like any biological process, we can think of cell death as an example of stimulus-response. The stimulus in this case can be inherently damaging, such as radiation or a chemical insult, or not inherently so, such as ligation of a receptor. In response to this stimulus, a cell may die or may survive. If the pathway to cell death is complex, the decision to die or survive might occur at any step of the process. That is, signals that propagate from the stimulus may or may not proceed in the pathway to the response, i.e. death (Figure 1). If the signal does not propagate, the cell survives. At some point, however, we suspect that the cell has been committed to die, that is, it has no way to recover. Where is this point of no return?
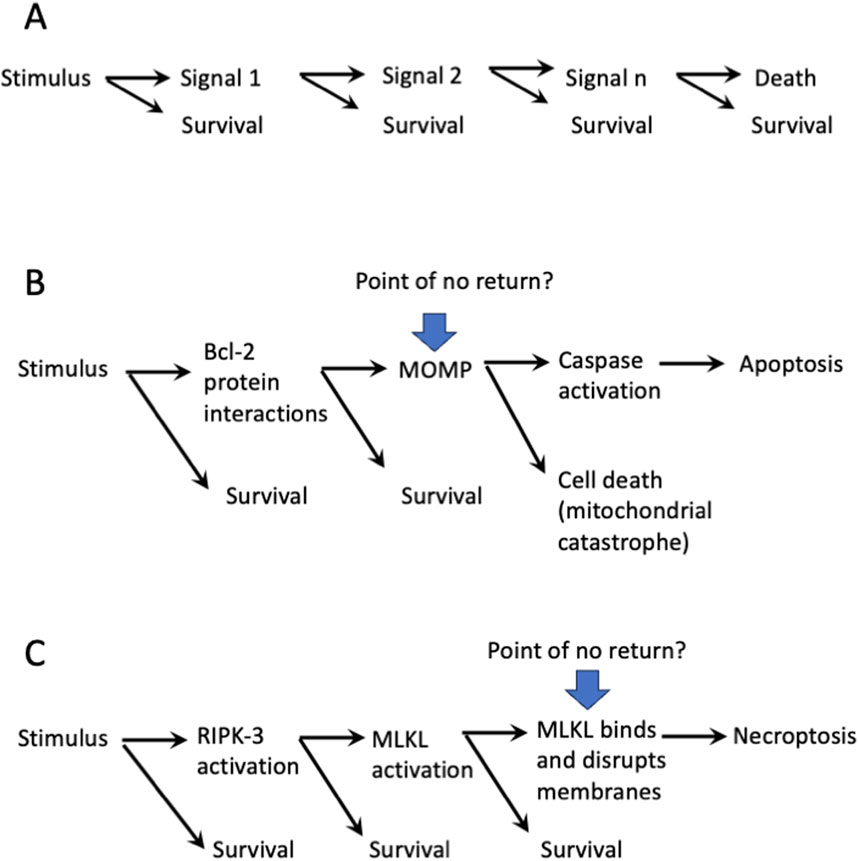
Figure 1. Points of no return in cell death pathways? (A). The “one-way” ticket to cell death has several stops to get off. (B). The mitochondrial pathway of apoptosis: Is MOMP a point of no return? (C). The necroptosis pathway: Is MLKL-mediated membrane disruption a point of no return.?
Until recently, we were comfortable with the idea that there was a point of no return in each of the defined cell death pathways. As an example, during one molecular pathway of apoptosis, the outer membranes of the mitochondria are permeabilized by the action of the BCL2 family effector proteins (mitochondrial outer membrane permeabilization, MOMP). Even if caspases were not subsequently activated as a consequence of MOMP, the cell would succumb to a “mitochondrial catastrophe” as the essential functions of the mitochondria are impaired (Lartigue et al., 2009). MOMP, then, was seen as a point of no return (Figure 1B).
Another example is in the process of necroptosis. In this case, the stimulus proceeds via signals to the activation of Receptor Interacting Kinase-3 (RIPK3), which in turn phosphorylates and thereby activates Mixed Lineage Kinase-Like (MLKL). MLKL then oligomerizes and disrupts cellular membranes, including the plasma membrane, being a point of no return (Figure 1C).
It is easy to envision similar points of no return for other cell death pathways and modalities. However, as I discuss below, we now know that our assumptions were wrong. Before going into why we were wrong, it may be useful to consider the problem from another perspective.
When a population of genetically identical cells are treated with a death-inducing stimulus, it often happens that not all cells die. If there were a threshold at which the amount of stimulus engages the relevant cell death pathway, then we might expect a dose response to the stimulus to resemble a step function (Figure 2A). Instead, however, most dose responses to a death-inducing stimulus resemble a sigmoidal curve (Figure 2A). Given the considerations discussed above, this is likely due to variations in the levels of the signal transduction components that result in cell death for that stimulus. Indeed, there is experimental evidence that cells with nearly identical levels of proteins (i.e., two daughter cells following mitosis) are more likely to die synchronously in response to a death-inducing stimulus than are cells that have diverged in such levels (Spencer et al., 2009).
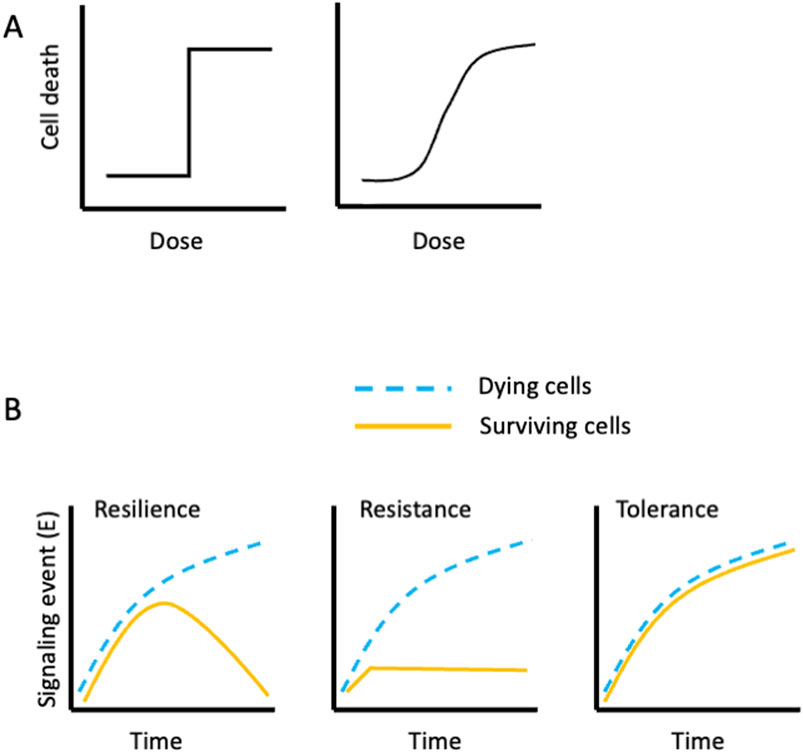
Figure 2. Idealized cell death responses to a death-inducing agent. (A) (Left) If all cells are identical, we might expect a dose threshold at which all cells die in response to the agent. (Right) A more typical pattern; cell death follows a sigmoidal dose response. (B). Reaction norms of cells evading cell death. At some doses of a death-inducing agent, a fraction of cells survive. If we measure a signaling event (E) along the cell death pathway, we can envision different ways the signal manifests in the surviving cells (yellow lines; dying cells, blue lines). (Left) The signal increases over time in both the surviving and the dying population, but then declines in the survivors. The surviving cells display resilience with respect to the signaling event, (E). (Center) The signal (E) appears in the dying population but not in the surviving population. The surviving cells thus display resistance with respect to the signaling event, (E). (Right) The signal manifests similarly in both the dying and the surviving populations. The surviving cells thus display tolerance with respect to the signaling event, (E).
Imagine a clonal cell population that is treated with a death-inducing stimulus that results in half of the cells dying and half surviving (LD50). If we measure a signaling event in the pathway to death, E, we can envision three ways in which the surviving and dying cells may engage E (Figure 2B). In effect, these are reaction norms (a term from evolutionary biology). My considerations of these reaction norms and their interpretations are strongly influenced by seminal studies in host-microbe interactions (Ayres, 2020), and have application to the discussion of cell death. Examples of E in the apoptotic pathways include assessment of caspase activation in single cells, mitochondrial permeabilization, and morphological changes preceding death. Similar examples of E for other cell death modalities are MLKL-dependent phosphatidylserine externalization prior to plasma membrane disruption during necroptosis (Gong et al., 2017) and lipid peroxidation during ferroptosis.
Measurements of E in our dying and surviving cells following LD50 exposure to a death-inducing stimulus may follow three possible, idealized trajectories over time. In the first, both populations engage E at similar rates but then the levels of E decline in the surviving cells (Figure 2B). This pattern represents resilience, as homeostatic mechanisms restore the levels of E to pre-treatment levels. If E represents a form of damage, then we can think of this as repair, but the effects are not limited to damage (e.g. loss of phospho-MLKL in “resuscitated” cells in necroptosis (Gong et al., 2017)). Alternatively, we might observe that E does not manifest in our surviving cells (or manifests to lower levels). This is resistance, that is, the signal E, does not occur (or less so) in the surviving cells, and thus cell death does not occur. Finally, we may see that E occurs to the same extent in both populations, and therefore the surviving cells show tolerance of the signal E. Tolerance can occur when the signal transduction pathway is disrupted or countered downstream of E, prior to death. Of course, in each case, another E may show distinctly different reaction norms for these two populations, which helps us understand why a cell ultimately survived the treatment.
Our discussion here is overly simplistic, of course. Not all death-inducing stimuli are binary in their effects. For example, agents that cause DNA damage can induce death or senescence, as well as resulting in survival of some cells. Further, the signals generated by a death-inducing stimulus can be much more complex than in our illustrations. DNA damage can stabilize p53 such that it increases until cell death or senescence occurs, or can result in cyclic levels of p53 that are associated with p53-induced cell cycle arrest and DNA repair (Purvis et al., 2012). Thus, the survival-death bifurcation that occurs at an LD50 dose of a death-inducing signal can be much more complex than our simple models describe.
The ways in which a cell survives a death-inducing signal can have consequences for the cell and its subsequent behavior. Before diving into these consequences, however, it will be useful to consider the strategies that allow cells to survive such signals.
If a cell receiving a cell death-inducing signal has not reached its “point of no return,” and the cell subsequently survives, this may be due to a disruption in the signals downstream of whatever we measure as E, as we’ve discussed. This disruption might be due to deficient levels of the downstream signaling molecules, but can also be a result of mechanisms that counteract this signal. Some of these counteracting mechanisms are triggered by the upstream signal itself, representing a negative feedback in the cell death pathway. Until recently, such negative feedback remained undescribed, and indeed, was thought not to occur.
For example, a cell that survives a stimulus that promotes apoptosis can do so by virtue of the action of anti-apoptotic BCL2 family proteins (such as BCL2, BCLxL, and MCL1) that prevent the activation and function (MOMP) of the pro-apoptotic BCL2 effector proteins (e.g., BAX, BAK). They can also survive if a minority of mitochondria in the cell engage MOMP, due to differential distribution of the anti-apoptotic proteins or dynamics of the mitochondrial reticulum (Cao et al., 2022). However, there is no evidence that engagement of apoptosis directly causes a negative feedback to promote anti-apoptotic BCL2 family protein levels or function (although some stressors can promote transcriptional upregulation, independent of apoptosis engagement). Similarly, although caspase activity is inhibited by X-linked Inhibitor of Apoptosis Protein (XIAP), there is no evidence that the levels or functions of XIAP are increased as a result of caspase activation.
A recent exception is the ability of Cytochrome c, released to the cytosol upon MOMP, which triggers caspase activation, but also allows the kinase, HRI (Heme-regulated eIF2α kinase), to function in promoting an integrated stress response (ISR), leading to synthesis of the transcription factor, ATF4 (Kalkavan et al., 2022). The ISR appears to promote survival of cells, despite the occurrence of MOMP. In principle, caspase activity can also promote the ISR via cleavage and activation of another kinase, Protein kinase R (PKR) (Kalai et al., 2007), although the role of this effect in cell survival has not been studied. Nevertheless, cells can sustain a level of caspase activation, sufficient to produce changes in cell morphology, and yet survive (Nano et al., 2023; Sun et al., 2017).
Plasma membrane damage, which can lead to cell death, can be repaired by the action of the ESCORT machinery (Endosomal sorting complex required for transport), especially the ESCRTIII complex (Jimenez et al., 2014). During necroptosis, the action of phosphorylated MLKL causes membrane damage, and ESCRTIII proteins can repair this damage, allowing cells to survive this otherwise catastrophic event (Gong et al., 2017). Similarly, during pyroptosis, membrane damage caused by the activation of Gasdermin D to create pores is offset by the function of ESCRT III (Ruhl et al., 2018). The same occurs when cytotoxic lymphocytes cause plasma membrane damage by the action of Perforin (Ritter et al., 2022). There is also evidence that ESCRTIII offsets membrane damage caused by lipid peroxidation in ferroptosis (Dai et al., 2020; Pedrera et al., 2021).
Is the function of ESCRTIII a negative feedback? This repair mechanism is triggered by an influx of calcium ions when the plasma membrane is damaged (Scheffer et al., 2014), and therefore the answer may be “yes.” But calcium influx upon plasma membrane disruption also generates additional signals that affect the behavior of the surviving cells, discussed in more detail below.
Another example of negative feedback in a pathway leading to cell death is the ability of reactive oxygen species, such as are involved in ferroptosis and other oxidative cell death modalities, to promote the anti-oxidant response. One way this occurs is via the inactivation of Keap1 (Kelch-like ECH-associated protein 1) by oxidation of key cysteine residues, releasing the transcription factor, NRF2, to promote the expression of genes involved in the anti-oxidant response.
There are stresses that can induce apoptosis and also induce resolution of the stress. These include the p53 response, as discussed above, as well as stress that engages the ISR, such as endoplasmic reticulum stress. In the latter case, the translational reprogramming can enhance the synthesis of the anti-apoptotic protein, MCL1 (Dong et al., 2011). Unlike the negative feedback discussed here, these effects are not a result of engaging the cell death pathway, but rather represent an alternative signaling pathway related to the level of stress.
We do not know if there are other negative feedback mechanisms in cell death pathways. That is, whether other signals in a pathway to cell death promote signals for survival. Nearly all other cellular biological pathways, including signal transduction, metabolism, cell cycle, and others feature such negative feedback. It would be surprising if the pathways leading to cell death do not.
If a cell engages a cell death pathway and survives, this can affect the behavior of the surviving cell. We have used the term, “flatliner,” to describe such near (cell) death experiences (Kalkavan et al., 2023). Further, it appears that such survival has consequences for the subsequent behavior of the cell, with potential ramifications for pathology and normal physiology.
During apoptosis, the engagement of the executioner caspases, caspase-3 and -7, results in the cleavage of Inhibitor of Caspase-Activated DNase (iCAD). This allows the nuclease, CAD, to translocate into the nucleus where it cuts DNA in inter-nucleosomal regions. Cells that survive a pro-apoptotic stimulus, then, can manifest iCAD-CAD dependent DNA damage. Indeed, studies have shown that cancer cells that survive a chemotherapeutic treatment sustain such CAD-dependent damage, and must engage DNA repair mechanisms to propagate (Ali et al., 2022). Such DNA damage and repair can lead to mutation, and a caspase- and CAD-dependent increase in mutation frequency has been observed in cells that survive a pro-apoptotic stimulus (Cao et al., 2022; Hawkins and Miles, 2021; Ichim et al., 2015).
BH3 mimetic drugs kill cells by specifically inhibiting anti-apoptotic BCL2 family proteins. Cancer cells that survive such treatment express ATF4 as a consequence of the MOMP-Cytochrome c-HRI-ISR response described above, and as a consequence these cells transiently display better intravasation, extravasation, and colonization than the untreated parental cells when introduced into animals (Kalkavan et al., 2022). These features are seen in the cells that have engaged MOMP (as detected with probes) and yet survived. The expression profiles of these flatliners are strikingly similar, regardless of the pro-apoptotic stimulus (BH3 mimetics, conventional therapeutics, targeted therapeutics); it appears that it is the near death experience that drives the transient, invasive state.
Cancer cells that survive a therapeutic treatment without selection for genetic variants have been termed “persister cells” (Shen et al., 2020). Such cells display a transient tolerance of not only the original treatment, but other treatments as well, and this tolerance reverts over time. Notably, persister cells generated from a large range of cancer lines, treated with a variety of therapeutic agents, become more sensitive to the induction of ferroptosis than their untreated brethren. Persister cells also display genomic instability (Shen et al., 2020; Killarney et al., 2024; McDonald and Dedhar, 2024), and multiple drug resistance mechanisms can emerge from a persister cell population that were not present in the original cancer. Persister cells also display a number of characteristic biological changes that may result in increased invasiveness and metastasis, among other changes (Shen et al., 2020; McDonald and Dedhar, 2024).
There are many possible explanations for the phenomenon of persistence, including upregulation of the Multi-Drug Resistance (MDR) transporters, differences in the developmental state of the cell, cell cycle dormancy, and others (Shen et al., 2020; McDonald and Dedhar, 2024). For example, stem cells, including cancer stem cells, express MDR transporters (Cho and Kim, 2020). In humans, only one gene encodes MDR1 (ABCB1), while in rodents there are two (MDR1a/b, ABCB1a/b). Another transporter, BCRP1 (Breast cancer resistance protein 1; ABCG2) plays similar roles in the efflux of drugs and other toxic compounds. This may not be surprising—MDR transporters would be expected to protect the developing embryo from environmental toxicants. That said, mice lacking MDR1a, MDR1b, and BCRP1 develop normally, although hematopoietic stem cells from these animals show increased sensitivity to xenobiotics (Zhou et al., 2003). Such considerations may explain why cancer stem cells appear to be resistant to conventional therapies. Thus, the presence of an embryonic or stem cell-like population of cells in a cancer may account for the persister cell effect. That said, clinical studies of MDR inhibitors in cancer therapy have been disappointing (Dong et al., 2023; Raderer and Scheithauer, 1993).
However, the persister cell phenomenon has been demonstrated in a large number of cell lines, and it has been shown that all cells have the ability to enter the persister cell state (Hangauer et al., 2017; Yang et al., 2021). It is therefore reasonable to question whether this is generally due to the presence of “cancer stem cells” in these lines, or whether the relative and transient drug tolerance of persister cells is always due to the expression of MDR transporters. Another proposed mechanism is the induction of a state resembling diapause, a quiescence that is observed across the animal kingdom (Easwaran and Montell, 2023). It is possible that this quiescent state is associated with a downregulation of cell death pathways, although this has not formally been addressed.
More recently, we (Kalkavan et al., 2023; Killarney et al., 2024) and others (McDonald and Dedhar, 2024) have proposed that flatliners represent a general mechanism for the generation of persister cells, that is, the persister phenotype manifests as a direct consequence of engaging the apoptotic pathway and surviving. Indeed, the flatliners display many of the described features of persister cells, including the increased sensitivity to induction of ferroptosis, and an increased ability to colonize, invade, and metastasize in vivo.
As we have seen, the flatliner phenomenon is not restricted to cells exposed to pro-apoptotic stimuli. Cells that have engaged necroptosis, with active MLKL, can survive, and as a consequence, transiently alter their behavior. In a recent study, plasma membrane damage caused by a chemically-activated MLKL (without RIPK3 (Receptor-Interacting Protein Kinase 3 (RIPK3) activation) led to the expression and secretion, by the surviving cells, of a number of chemokines and cytokines, molecules that profoundly affect surrounding cells (Wang et al., 2022). This was shown to be the result of the influx of calcium ions due to plasma membrane damage, which activated Protein Kinase C, leading in turn to signal transduction events culminating in gene expression. The effect was not restricted to necroptosis; plasma membrane damage caused by mild detergent, active Gasdermin D, or Perforin had similar effects.
The persister phenomenon, and by extension, flatliners, has important implications for minimal residual disease in cancer. However, clearly, the mechanisms whereby cells survive a death-inducing stimulus and take on new properties did not evolve to promote cancer relapse; there must be physiological roles for such processes.
At first glance, the idea that flatliner cells that are “instructed” to die might “struggle” to survive might sound obvious. Cells are living things, and the instinct for survival is an inherent feature of all living things. But our cells are part of an individual, and natural selection acts both at the level of cells and at the level of the individual; when these levels of selection are conflicting, the result can be disease (e.g., cancer, where selection for cell fitness decreases the fitness of the organism) or can promote health (e.g., affinity maturation in antibody responses, where selection of antibody producing B cells in the face of decreasing antigen levels promotes higher affinity antibody production). While it is conceivable that the processes that allow flatliners have no role in normal organismal physiology, this seems unlikely. But what, then, are flatliners “for?”
The general features of flatliners may give us a clue, as might asking where and under what circumstances flatliners might arise. For example, during an ischemia-reperfusion injury, there is a focal necrotic region. While the features of the cell death that occurs under such insult are controversial, evidence supports both necroptosis and ferroptosis (Belavgeni et al., 2020) as having roles in the demise of the cells (although the roles of these cell death modalities might vary with the tissue and features of the insult (e.g. (Newton et al., 2016)). Nevertheless, cells that engage these mechanisms to survive plasma membrane damage (flatliners) would be expected to secrete signals (chemokines and cytokines) as discussed above. This has been observed in kidney ischemia-reperfusion injury in human samples (Bonventre and Zuk, 2004). As a consequence, phagocytic cells such as macrophages are recruited to the injury where dead cells are engulfed (efferocytosis), and the engulfing cells further express signals important for wound healing (Boada-Romero et al., 2020).
Surrounding the necrotic foci in ischemia-reperfusion injury, there is often a penumbra of apoptotic cells (Choi, 1996; Gottlieb and Engler, 1999), presumably because the stress of the insult, diluting from the center, reaches a threshold where apoptosis rather than necrosis occurs. We can imagine (with no evidence) that at some distance from the center of the insult there is enough reduction in the stress that flatliners may emerge (i.e., when the death-inducing signal is below the lethal dose that kills 100% of the cells, LD100). These apoptotic flatliners, we suppose, would take on the features of persister cells (see above), with increased invasiveness, epithelial to mesenchymal transition, and a greater propensity to “colonize” the wound. It might be that flatliners function in wound repair.
Are there other settings where flatliners may play physiological roles? Accumulating evidence supports the idea that the activation of executioner caspases promotes biological functions that are independent of cell death. For example, active Caspase-3 in mammals has been suggested to play roles in synaptic pruning in neurons (Geden et al., 2019), myoblast differentiation (Fernando et al., 2002), lens fiber differentiation (Zandy et al., 2005), macrophage polarization (Solier et al., 2023), and the differentiation of embryonic stem cells (Fujita et al., 2008). At present, we know of no way for Caspase-3 to be activated except by cleavage in the inter-subunit region, an event that occurs as a consequence of initiator Caspase (Caspase-8 or Caspase-9) function, but can occur with other proteases, such as Granzyme B. In most cases, a cell that activates Caspase-3 but does not die would fit the definition of a flatliner, if the activation of the caspase is as a result of engaging an apoptotic pathway. At present, we do not know if this is the case for any example of non-apoptotic roles for Caspase-3. Conversely, though, we may expect that cells that survive the activation of Caspase-3 as a consequence of the apoptosis machinery (flatliners) may engage similar consequences of Caspase-3 activation.
Cell survival versus death is, on the surface, a binary decision: Cells are either alive or dead. We know, of course, that this is a bit simplistic; how a cell dies can have consequences for the tissue in which the cell death occurred, influencing compensatory proliferation, recruitment of immune cells, and priming of an immune response. Further, cells that survive may differentiate, undergo senescence, and/or acquire biologically relevant features.
The idea that flatliners can engage a cell death pathway and yet survive is, on its own, a potentially interesting phenomenon, but its importance depends on several factors. One such factor is context; for example, a cancer cell that is induced to die but does not die has the potential to cause relapse. Another factor is consequence; if the engagement of the cell death pathway induces signaling events that manifest in the surviving cell (but are irrelevant to the dead cell), then flatliners can take on new properties. For example, flatliners that survive treatment with BH3 mimetics engage the ISR, which promotes transient tolerance of pro-apoptotic stimuli, increased invasiveness and metastasis, and an acquired vulnerability to the induction of ferroptosis (Kalkavan et al., 2022). Further, apoptotic flatliners can increase their frequency of mutation, owing to DNA damage resulting from the function of CAD (Cao et al., 2022; Hawkins and Miles, 2021; Ichim et al., 2015). A gene expression signature derived from BH3 mimetic-treated flatliners was identified in a dataset of minimal residual disease in treated lung cancer patients (Kalkavan et al., 2022).
Persister cells are defined as cancer cells that survive a therapeutic treatment. While flatliners may represent a subset of persister cells, there are certainly other mechanisms of persistence. For example, cells may be present in a developmental state that does respond to the treatment, exclude the therapeutic through drug efflux, and/or harbor increased inhibitors of the engagement of a cell death pathway. Such cells display resistance, according to our definitions (Figure 2B), and since they do not engage the cell death pathway induced by the therapeutic, they are not flatliners by our definition. Nevertheless, we suggest that the flatliner phenomenon represents a universal mechanism, agnostic to the cell type of origin, whereby persister cells may emerge.
Persister cells and flatliners have generally been studied in the context of transformed cells. Ultimately, our understanding of the role of flatliners in normal development and homeostasis will depend on our ability to identify flatliners and their progeny. Attempts to do this with sensors that detect caspase activation have been informative in Drosophila (Ding et al., 2016). We can envision using such approaches to permanently “mark” a cell that has survived caspase activation (Sun et al., 2023), and the successful use of such strategies in mammalian systems is likely to provide new insights into the physiological function of the flatliner phenomenon.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
DG: Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. DRG is supported by grants from the US NIH, CA231620 and AI44828.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ali, M., Lu, M., Ang, H. X., Soderquist, R. S., Eyler, C. E., Hutchinson, H. M., et al. (2022). Small-molecule targeted therapies induce dependence on DNA double-strand break repair in residual tumor cells. Sci. Transl. Med. 14, eabc7480. doi:10.1126/scitranslmed.abc7480
Ayres, J. S. (2020). The biology of physiological health. Cell 181, 250–269. doi:10.1016/j.cell.2020.03.036
Belavgeni, A., Meyer, C., Stumpf, J., Hugo, C., and Linkermann, A. (2020). Ferroptosis and necroptosis in the kidney. Cell Chem. Biol. 27, 448–462. doi:10.1016/j.chembiol.2020.03.016
Boada-Romero, E., Martinez, J., Heckmann, B. L., and Green, D. R. (2020). The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 21, 398–414. doi:10.1038/s41580-020-0232-1
Bonventre, J. V., and Zuk, A. (2004). Ischemic acute renal failure: an inflammatory disease? Kidney Int. 66, 480–485. doi:10.1111/j.1523-1755.2004.761_2.x
Cao, K., Riley, J. S., Heilig, R., Montes-Gomez, A. E., Vringer, E., Berthenet, K., et al. (2022). Mitochondrial dynamics regulate genome stability via control of caspase-dependent DNA damage. Dev. Cell 57, 1211–1225 e6. doi:10.1016/j.devcel.2022.03.019
Cho, Y., and Kim, Y. K. (2020). Cancer stem cells as a potential target to overcome multidrug resistance. Front. Oncol. 10, 764. doi:10.3389/fonc.2020.00764
Choi, D. W. (1996). Ischemia-induced neuronal apoptosis. Curr. Opin. Neurobiol. 6, 667–672. doi:10.1016/s0959-4388(96)80101-2
Dai, E., Meng, L., Kang, R., Wang, X., and Tang, D. (2020). ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem. Biophys. Res. Commun. 522, 415–421. doi:10.1016/j.bbrc.2019.11.110
Ding, A. X., Sun, G., Argaw, Y. G., Wong, J. O., Easwaran, S., and Montell, D. J. (2016). CasExpress reveals widespread and diverse patterns of cell survival of caspase-3 activation during development in vivo. Elife 5, e10936. doi:10.7554/eLife.10936
Dong, J., Yuan, L., Hu, C., Cheng, X., and Qin, J. J. (2023). Strategies to overcome cancer multidrug resistance (MDR) through targeting P-glycoprotein (ABCB1): an updated review. Pharmacol. Ther. 249, 108488. doi:10.1016/j.pharmthera.2023.108488
Dong, L., Jiang, C. C., Thorne, R. F., Croft, A., Yang, F., Liu, H., et al. (2011). Ets-1 mediates upregulation of Mcl-1 downstream of XBP-1 in human melanoma cells upon ER stress. Oncogene 30, 3716–3726. doi:10.1038/onc.2011.87
Easwaran, S., and Montell, D. J. (2023). The molecular mechanisms of diapause and diapause-like reversible arrest. Biochem. Soc. Trans. 51, 1847–1856. doi:10.1042/BST20221431
Fernando, P., Kelly, J. F., Balazsi, K., Slack, R. S., and Megeney, L. A. (2002). Caspase 3 activity is required for skeletal muscle differentiation. Proc. Natl. Acad. Sci. U. S. A. 99, 11025–11030. doi:10.1073/pnas.162172899
Fujita, J., Crane, A. M., Souza, M. K., Dejosez, M., Kyba, M., Flavell, R. A., et al. (2008). Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell 2, 595–601. doi:10.1016/j.stem.2008.04.001
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541. doi:10.1038/s41418-017-0012-4
Geden, M. J., Romero, S. E., and Deshmukh, M. (2019). Apoptosis versus axon pruning: molecular intersection of two distinct pathways for axon degeneration. Neurosci. Res. 139, 3–8. doi:10.1016/j.neures.2018.11.007
Gong, Y. N., Guy, C., Olauson, H., Becker, J. U., Yang, M., Fitzgerald, P., et al. (2017). ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169, 286–300. doi:10.1016/j.cell.2017.03.020
Gottlieb, R. A., and Engler, R. L. (1999). Apoptosis in myocardial ischemia-reperfusion. Ann. N. Y. Acad. Sci. 874, 412–426. doi:10.1111/j.1749-6632.1999.tb09255.x
Hangauer, M. J., Viswanathan, V. S., Ryan, M. J., Bole, D., Eaton, J. K., Matov, A., et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. doi:10.1038/nature24297
Hawkins, C. J., and Miles, M. A. (2021). Mutagenic consequences of sublethal cell death signaling. Int. J. Mol. Sci. 22, 6144. doi:10.3390/ijms22116144
Ichim, G., Lopez, J., Ahmed, S. U., Muthalagu, N., Giampazolias, E., Delgado, M. E., et al. (2015). Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 57, 860–872. doi:10.1016/j.molcel.2015.01.018
Jimenez, A. J., Maiuri, P., Lafaurie-Janvore, J., Divoux, S., Piel, M., and Perez, F. (2014). ESCRT machinery is required for plasma membrane repair. Science 343, 1247136. doi:10.1126/science.1247136
Kalai, M., Suin, V., Festjens, N., Meeus, A., Bernis, A., Wang, X. M., et al. (2007). The caspase-generated fragments of PKR cooperate to activate full-length PKR and inhibit translation. Cell Death Differ. 14, 1050–1059. doi:10.1038/sj.cdd.4402110
Kalkavan, H., Chen, M. J., Crawford, J. C., Quarato, G., Fitzgerald, P., Tait, S. W. G., et al. (2022). Sublethal cytochrome c release generates drug-tolerant persister cells. Cell 185, 3356–3374 e22. doi:10.1016/j.cell.2022.07.025
Kalkavan, H., Ruhl, S., Shaw, J. J. P., and Green, D. R. (2023). Non-lethal outcomes of engaging regulated cell death pathways in cancer. Nat. Cancer 4, 795–806. doi:10.1038/s43018-023-00571-6
Killarney, S. T., Tait, S. W. G., Green, D. R., and Wood, K. C. (2024). Sublethal engagement of apoptotic pathways in residual cancer. Trends Cell Biol. 34, 225–238. doi:10.1016/j.tcb.2023.07.005
Lartigue, L., Kushnareva, Y., Seong, Y., Lin, H., Faustin, B., and Newmeyer, D. D. (2009). Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol. Biol. Cell 20, 4871–4884. doi:10.1091/mbc.e09-07-0649
Lee, S., Karki, R., Wang, Y., Nguyen, L. N., Kalathur, R. C., and Kanneganti, T. D. (2021). AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 597, 415–419. doi:10.1038/s41586-021-03875-8
McDonald, P. C., and Dedhar, S. (2024). Persister cell plasticity in tumour drug resistance. Semin. Cell Dev. Biol. 156, 1–10. doi:10.1016/j.semcdb.2023.11.003
Nano, M., Mondo, J. A., Harwood, J., Balasanyan, V., and Montell, D. J. (2023). Cell survival following direct executioner-caspase activation. Proc. Natl. Acad. Sci. U. S. A. 120, e2216531120. doi:10.1073/pnas.2216531120
Newton, K., Dugger, D. L., Maltzman, A., Greve, J. M., Hedehus, M., Martin-McNulty, B., et al. (2016). RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 23, 1565–1576. doi:10.1038/cdd.2016.46
Pedrera, L., Espiritu, R. A., Ros, U., Weber, J., Schmitt, A., Stroh, J., et al. (2021). Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 28, 1644–1657. doi:10.1038/s41418-020-00691-x
Purvis, J. E., Karhohs, K. W., Mock, C., Batchelor, E., Loewer, A., and Lahav, G. (2012). p53 dynamics control cell fate. Science 336, 1440–1444. doi:10.1126/science.1218351
Raderer, M., and Scheithauer, W. (1993). Clinical trials of agents that reverse multidrug resistance. A literature review. Cancer 72, 3553–3563. doi:10.1002/1097-0142(19931215)72:12<3553::aid-cncr2820721203>3.0.co;2-b
Ritter, A. T., Shtengel, G., Xu, C. S., Weigel, A., Hoffman, D. P., Freeman, M., et al. (2022). ESCRT-mediated membrane repair protects tumor-derived cells against T cell attack. Science 376, 377–382. doi:10.1126/science.abl3855
Rogers, C., Fernandes-Alnemri, T., Mayes, L., Alnemri, D., Cingolani, G., and Alnemri, E. S. (2017). Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 8, 14128. doi:10.1038/ncomms14128
Ruhl, S., Shkarina, K., Demarco, B., Heilig, R., Santos, J. C., and Broz, P. (2018). ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960. doi:10.1126/science.aar7607
Scheffer, L. L., Sreetama, S. C., Sharma, N., Medikayala, S., Brown, K. J., Defour, A., et al. (2014). Mechanism of Ca²⁺-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 5, 5646. doi:10.1038/ncomms6646
Shen, S., Vagner, S., and Robert, C. (2020). Persistent cancer cells: the deadly survivors. Cell 183, 860–874. doi:10.1016/j.cell.2020.10.027
Solier, S., Mondini, M., Meziani, L., Jacquel, A., Lacout, C., Berghe, T. V., et al. (2023). Caspase inhibition modulates monocyte-derived macrophage polarization in damaged tissues. Int. J. Mol. Sci. 24, 4151. doi:10.3390/ijms24044151
Spencer, S. L., Gaudet, S., Albeck, J. G., Burke, J. M., and Sorger, P. K. (2009). Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459, 428–432. doi:10.1038/nature08012
Sun, G., Guzman, E., Balasanyan, V., Conner, C. M., Wong, K., Zhou, H. R., et al. (2017). A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 216, 3355–3368. doi:10.1083/jcb.201706134
Sun, L., Yao, C., Li, X., Wang, Y., Wang, R., Wang, M., et al. (2023). Anastasis confers ovarian cancer cells increased malignancy through elevated p38 MAPK activation. Cell Death Differ. 30, 809–824. doi:10.1038/s41418-022-01081-1
Wang, W., Prokopec, J. S., Zhang, Y., Sukhoplyasova, M., Shinglot, H., Wang, M. T., et al. (2022). Sensing plasma membrane pore formation induces chemokine production in survivors of regulated necrosis. Dev. Cell 57, 228–245 e6. doi:10.1016/j.devcel.2021.12.015
Yang, C., Tian, C., Hoffman, T. E., Jacobsen, N. K., and Spencer, S. L. (2021). Melanoma subpopulations that rapidly escape MAPK pathway inhibition incur DNA damage and rely on stress signalling. Nat. Commun. 12, 1747. doi:10.1038/s41467-021-21549-x
Zandy, A. J., Lakhani, S., Zheng, T., Flavell, R. A., and Bassnett, S. (2005). Role of the executioner caspases during lens development. J. Biol. Chem. 280, 30263–30272. doi:10.1074/jbc.M504007200
Keywords: apoptosis, necroptosis, pyroptosis, ferroptosis, integrated stress response (ISR), ESCRT (endosomal sorting complex required for transport)
Citation: Green DR (2024) Not dead yet: cell death and survival in cancer and normal physiology. Front. Cell. Death 3:1477346. doi: 10.3389/fceld.2024.1477346
Received: 07 August 2024; Accepted: 30 August 2024;
Published: 11 September 2024.
Edited by:
Lawrence M. Schwartz, University of Massachusetts Amherst, United StatesReviewed by:
Zhenggang Liu, National Institutes of Health (NIH)∗, United StatesCopyright © 2024 Green. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Douglas R. Green, douglas.green@stjude.org
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.