EDITORIAL
Published on 29 Jun 2022
Editorial: Chemoinformatics Approaches to Structure- and Ligand-Based Drug Design, Volume II
doi 10.3389/fphar.2022.945747
- 1,023 views
22k
Total downloads
92k
Total views and downloads
EDITORIAL
Published on 29 Jun 2022
ORIGINAL RESEARCH
Published on 29 Jun 2022
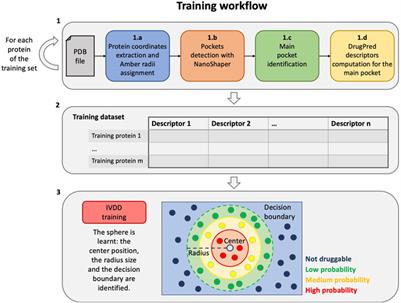
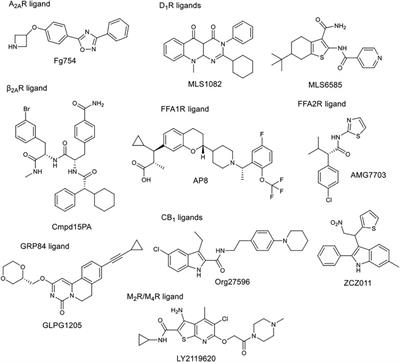
ORIGINAL RESEARCH
Published on 03 May 2022
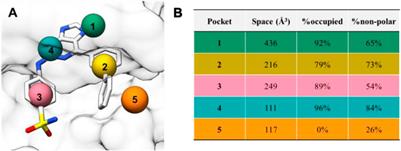
REVIEW
Published on 03 May 2022
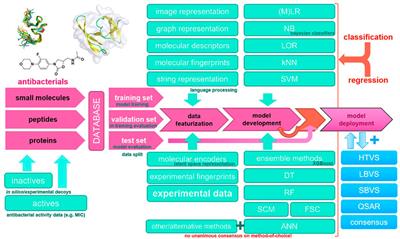
TECHNOLOGY AND CODE
Published on 26 Apr 2022

ORIGINAL RESEARCH
Published on 21 Apr 2022
ORIGINAL RESEARCH
Published on 18 Mar 2022
ORIGINAL RESEARCH
Published on 18 Mar 2022
ORIGINAL RESEARCH
Published on 11 Mar 2022
REVIEW
Published on 10 Mar 2022
ORIGINAL RESEARCH
Published on 10 Mar 2022
REVIEW
Published on 09 Mar 2022