EDITORIAL
Published on 16 Nov 2022
Editorial: Neuronal ceroid lipofuscinosis: A multidisciplinary update
doi 10.3389/fneur.2022.1083113
- 890 views
12k
Total downloads
45k
Total views and downloads
Select the journal/section where you want your idea to be submitted:
EDITORIAL
Published on 16 Nov 2022
CASE REPORT
Published on 22 Aug 2022
REVIEW
Published on 12 Aug 2022
MINI REVIEW
Published on 18 Apr 2022
REVIEW
Published on 04 Apr 2022
ORIGINAL RESEARCH
Published on 29 Mar 2022
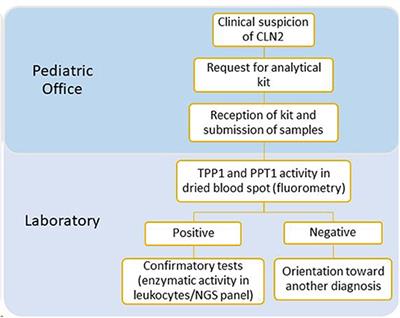
REVIEW
Published on 11 Mar 2022

ORIGINAL RESEARCH
Published on 10 Mar 2022
REVIEW
Published on 25 Feb 2022
MINI REVIEW
Published on 08 Feb 2022
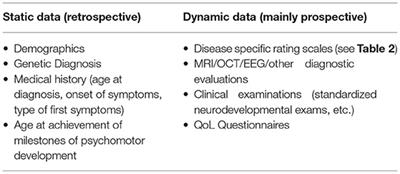
REVIEW
Published on 18 Oct 2021
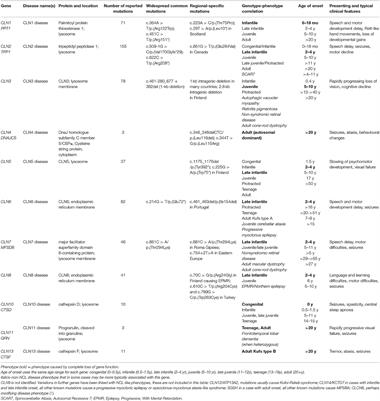
MINI REVIEW
Published on 25 Jun 2021

Frontiers in Pediatrics