
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Sustain. Food Syst. , 12 February 2025
Sec. Crop Biology and Sustainability
Volume 9 - 2025 | https://doi.org/10.3389/fsufs.2025.1505001
This article is part of the Research Topic Innovative Solutions For Next-Generation Fertilizers View all 13 articles
 Maura Santos Reis de Andrade da Silva1,2
Maura Santos Reis de Andrade da Silva1,2 Lucas Amoroso Lopes de Carvalho1
Lucas Amoroso Lopes de Carvalho1 Carlos Henrique Barbosa Santos3
Carlos Henrique Barbosa Santos3 Edvan Teciano Frezarin1
Edvan Teciano Frezarin1 Cleudison Gabriel Nascimento da Silva4
Cleudison Gabriel Nascimento da Silva4 Daniel Guariz Pinheiro1
Daniel Guariz Pinheiro1 Everaldo Zonta2
Everaldo Zonta2 Olubukola Oluranti Babalola5
Olubukola Oluranti Babalola5 Everlon Cid Rigobelo1*†
Everlon Cid Rigobelo1*†Introduction: The effect of co-inoculation with plant growth-promoting bacteria on the microbiome of soybean roots was investigated in a field experiment. Soybean plants were inoculated with Bacillus subtilis, Bacillus aryabhattai, Streptomyces sp., and Saccharopolyspora spinosa and compared to a control treatment that received mineral fertilization.
Methods: The yield parameters and endophytic microbiome of soybean roots were evaluated.
Results: No significant differences in yield were observed among the treatments, suggesting that microbial inoculation can serve as an alternative to mineral fertilization without compromising productivity. Among the most abundant genera, there was a high prevalence of members of the phylum Proteobacteria (21 of the top 25 genera). Overall, the genera of these phyla represented 88.61% of the samples on average. There were also genera in the phyla Bacteroidetes (2/25), Actinobacteria (1/25), and Firmicutes (1/25). The massive presence of Bradyrhizobium, which represented 71.22% of the sequences at the genus level, was remarkable. Bradyrhizobium was the most abundant genus in all samples, except for Saccharopolyspora spinosa (ST treatment), whose abundance was only 12.66%. Co-occurrence network analysis revealed changes in the microbial community structure and genera considered as hubs.
Discussion: These findings demonstrate the potential of co-inoculation with plant growth-promoting bacteria to modulate the root microbiome and enhance the colonization of B. japonicum, which may contribute to improving the efficiency of this symbiont in promoting plant growth. Further research is required to elucidate the mechanisms underlying these interactions and their implications for soybean productivity.
Plant growth-promoting rhizobacteria (PGPR) are beneficial soil bacteria that inhabit the rhizosphere and colonize the root surface and surrounding soil (Khoso et al., 2024). They play crucial roles in enhancing plant growth and development through various direct and indirect mechanisms. PGPR directly promote plant growth by assisting in resource acquisition, such as nitrogen fixation, phosphate solubilization, and essential mineral uptake, and by modulating plant hormone levels (Paulus and Tooy, 2024). They act as biocontrol agents, thereby reducing the inhibitory effects of pathogens on plant growth and development (Gu et al., 2020).
Interestingly, PGPR can form biofilms at nutrient- and moisture-rich sites, which further enhances their ability to promote plant growth, improve nutrient and water uptake, and increase plant stress tolerance (Kumar et al., 2024). Additionally, PGPR have been found to alleviate various environmental stresses in plants, including salinity, flooding, heavy metals, drought, and cold, through mechanisms such as the production of osmolytes, exopolysaccharides, and activation of antioxidant defense systems in plants (Mun et al., 2024). Notably, these effects on plant growth may be attributed to alterations in the plant microbiome caused by bacterial inoculation.
Bacterial inoculation has significant effects on the soybean plant microbiome, influencing both the rhizosphere and root-associated bacterial communities. Inoculation with rhizobia and plant growth-promoting bacteria (PGPB) can alter the diversity and composition of soybean root endophytic bacteria throughout the growing season (Verma et al., 2024). Co-inoculation with multiple bacterial strains can promote synergistic effects on plant growth and stress tolerance (Wang et al., 2024). The origin of the microbial inoculants also plays a role in shaping the rhizosphere bacterial community structure and influencing plant growth (Khosravi et al., 2024). There are several mechanisms by which bacterial inoculation changes the plant microbiome, including competition for resources (Verma et al., 2024; Wahab et al., 2024). Introduced bacteria compete with existing microorganisms for nutrients and space, potentially altering microbiome composition (Ikiz et al., 2024). Modification of root exudates by ample-inoculated bacteria can influence plant physiology, leading to changes in root exudates. This, in turn, attracts or repels different microbial species and reshapes the microbiome. Members of the plant microbiome can interact with each other in a synergistic manner. Synergistic interactions may form beneficial relationships with existing microbes, enhancing their collective effects on plant health and microbiome structure (Gu et al., 2020).
Interestingly, bacterial inoculation can also help soybean plants cope with environmental stresses. For instance, co-inoculation with bacteria improved plant growth and nodulation under arsenic stress, while also reducing As translocation to the aerial parts (Armendariz et al., 2019). Additionally, composite bacterial inoculants containing multiple PGPR strains can coordinately modulate rhizosphere microbial community structure, improve soil nutrient availability, and enhance soybean growth (Paulus and Tooy, 2024).
Although some studies have demonstrated changes induced by bacterial inoculation, there is limited information regarding the effects of these specific bacterial strains on the plant microbiome. Therefore, the objective of this study was to evaluate the effects of inoculation with four different bacterial species on the microbiome of soybean plants.
The experiment was carried out in an agricultural area belonging to the Teaching, Research and Extension Farm (FEPE) of UNESP—Campus of Jaboticabal, starting in November 2019 and ending in March 2020. The climate of the Jaboticabal region is Cwa, according to the Köppen classification, with wet summers and dry winters. The UNESP unit is located in the northwestern region of São Paulo state (21° 15′ 22″ S, 48° 18′ 58″ W, and an altitude of 605 m).
Plots in the field condition of 30 m2 (6 m × 5 m) were established, with a spacing of 0.5 m between rows and 0.1 m between plants. The soil of the area was of the latosol type, and sowing was carried out mechanically at a depth of 5 cm. Chemical analysis of the soil was previously carried out to evaluate its acidity and nutrient availability. The nitrogen dose, urea corresponding to the control treatment was applied manually by repetitions, row by row, after the emergence of the plants. Due to the history of having successive crops with soybean inoculated with strains of Bradyrhizobium, there was no need for reinoculation of this inoculant in this area.
The microorganisms used in this study were sourced from diverse sources. Specifically, Bacillus subtilis was obtained from soil and identified through sequencing and was deposited in the bank under the accession number MZ133755. Bacillus aryabhattai CCT4005 and Streptomyces sp. 244 were provided by André Tosello, and Saccharopolyspora spinosa ATCC 49460 was obtained from the American Type Culture Collection (ATCC). The bacterial strains belong to Laboratory of Soil Microbiology (LSM) of FCAV/UNESP in Jaboticabal. Microorganisms were inoculated in suitable culture media for growth. The inoculants B. subtilis and B. aryabhattai were prepared in nutrient broth, Streptomyces sp. in SM medium (1% glucose, 3% casein peptone, 1.0% yeast extract, 0.05% potassium dihydrogen phosphate, and 0.2% magnesium sulfate heptahydrate) and S. spinosa in Jensen’s medium (2% sucrose, 0.1% dipotassium phosphate, 0.05% magnesium sulfate, 0.05% sodium chloride, 0.01% ferrous sulfate, 0.0005% sodium molybdate, 0.2% calcium carbonate and 1.5% agar). All the strains were grown in a BOD chamber for 72 h, after which a final concentration of 1 × 108 colony forming units (CFU) mL−1 was applied. In total, two applications were carried out, the first 15 days after planting (DAP) and the second 30 days after planting. The applications of the inoculants were carried out using 1000 mL ha−1 backpack sprayers via foliar way.
In the field, the plots were arranged in randomized blocks, with five treatments and four replications. The treatments are detailed in Table 1. Description of control treatment.
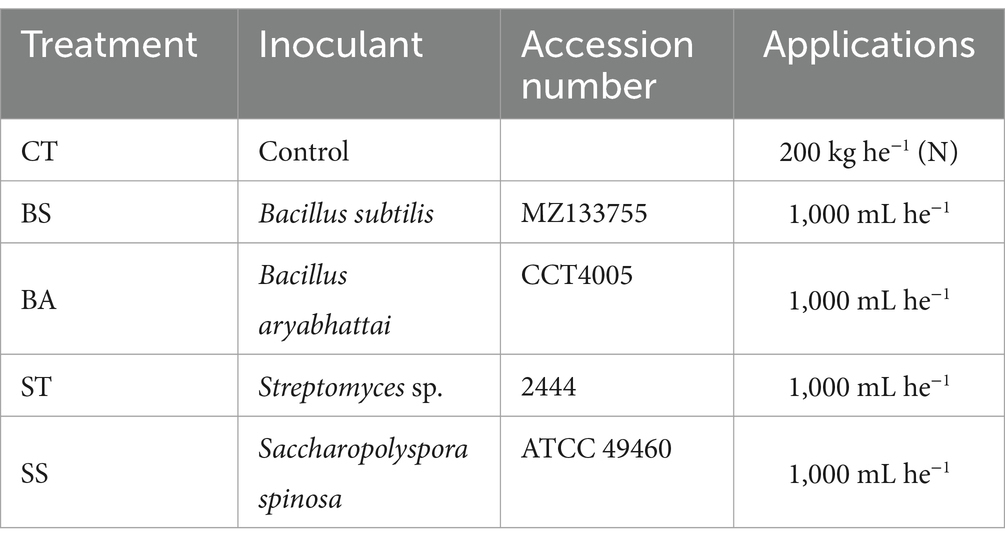
Table 1. Description of the treatments carried out with several microorganisms inoculated into soybean under field conditions.
The control treatment received the same fertilization as the other treatments, with the addition of 200 kg ha−1 of urea (a nitrogen source). No bacterial inoculation was performed in the control group. In contrast, the other treatments were subjected to bacterial inoculation, but did not receive urea supplementation.
After the physiological maturity of the grains, which were obtained at 120 days after sowing, the soybean was harvested by means of a specialized parcel harvester, and the grains were then packed in paper bags and stored at room temperature until drying.
The dry mass of the aerial part was determined at the beginning of the flowering of the soybean crop. Five randomly chosen plants were collected from each plot during this period, placed in paper bags and dried in an oven with forced air circulation at 65°C for 72 h. After this period, the dry matter was obtained by weighing the material on a semianalytical balance (Lobo et al., 2019).
Five hundred micrograms of dried and ground plant samples were weighed and placed into 50 mL digestion tubes, which were left to decouple at room temperature for 1.5 h. The tubes were then positioned in a digestion block, initially heated to 80°C for 20 min before the temperature was increased to 160°C. The tubes were monitored and removed once the material ascended the tube walls, and most of the HNO3 evaporated, leaving a clear solution. After cooling, 1.3 mL the concentrated HClO4 was added to each tube. The tubes were returned to the block, the temperature was increased to 210°C, and digestion was deemed complete when the solution turned colorless with dense white vapor of HClO4 and H2O formed above the dissolved material. The tubes were cooled and the contents were diluted to 25 mL with water in a snap-cap glass (Wan et al., 2020).
For phosphorus analysis, 1 mL of the digested sample was transferred to a test tube, to which 4 mL of water and 2 mL of reagent mix (comprising equal parts of 5% ammonium molybdate and 0.25% vanadate) were added. The mixture was allowed to rest for 15 min before measuring the absorbance at 420 nm using a UV VIS spectrophotometer (Meier et al., 2020).
Using a box with holes adapted for the simultaneous counting of 100 grains of the crop, the mass of 1000 grains was determined by weighing eight subsamples with 100 grains each on a semianalytical scale.
At the end of the field experiment, the soybeans were harvested by a specialized plot harvester, stored at room temperature for drying and later weighed on a semianalytical scale to determine the total grain yield. The productivity in kg ha−1 relative to the production per unit of area was also determined.
Data normality and heteroscedasticity were assessed by the Shapiro–Wilk and Bartlett tests, respectively. Analysis of variance (ANOVA) was performed for each experiment to detect differences, and Tukey’s multiple comparison test was used to compare the means at the 5% significance level. PCA was performed after data standardization (to avoid the influence of different units for the means of response variables), and all the statistical analyses were performed with R software (R Core Team, 2023).
Roots were manually harvested using surgical gloves and immediately stored in sterilized plastic containers. To dislodge the rhizospheric soil, roots were placed in a 50 mL conical tube containing 35 mL of phosphate buffer and 0.02% Tween 20 and vortexed for 2 min. The roots were then transferred to sterile paper towels using sterilized forceps and subsequently transferred to 50 mL centrifuge tubes for superficial sterilization. The sterilization involved treating the plant tissues were sterilized with 100% ethanol for 3 min, 2% sodium hypochlorite for 2 min, and 70% ethanol for 3 min, based on a modified protocol from Cao et al. (2005). To confirm the sterilization efficacy, the final wash was cultured on nutrient agar plates and checked for the absence of microbial growth. Following sterilization, the roots were pooled by treatment and macerated using a sterile mortar and pestle in liquid nitrogen. Genomic DNA was extracted from these samples using the PowerMax Soil DNA Extraction Kit (Mo Bio Laboratories, Carlsbad, CA, United States), according to the manufacturer’s specifications. DNA concentration and purity were assessed using fluorometry (Qubit™ 3.0, Invitrogen) and spectrophotometry (NanoDrop™ 1000, Thermo Fisher Scientific), respectively, to determine the A260/A280 ratio.
Sterilized roots were macerated using a sterile mortar and pestle with liquid nitrogen. The PowerMax Soil DNA Extraction Kit (Mo Bio Laboratories, Carlsbad, CA) was used to extract the genomic DNA according to the Manufacturer’s instructions. The concentration of the extracted DNA was determined by fluorometry (Qubit™ 3.0, Invitrogen), and purity was estimated by calculating the A260/A280 ratio via spectrophotometry (NanoDrop™ 1000, Thermo Fisher Scientific) (de Souza et al., 2016). The hypervariable region V4 of the 16S rRNA gene was amplified using the primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). Three forward primers were used for amplification. These were modified by adding degenerate nucleotides (Ns) to the 5′ region to increase the diversity of target sequences (de Souza et al., 2016). PCR was performed in 30 cycles using the HotStarTaq Plus Master Mix Kit (Qiagen) under the following conditions: 94°C for 3 min, followed by 28 cycles at 94°C for 30 s, 53°C for 40 s, and 72°C for 1 min, and a final elongation step at 72°C for 5 min. PNA clamp sequences (PNA Bio) were added to block amplification of the 16S rRNA gene from the ribosomes and mitochondria. The amplification products were analyzed on a 2% agarose gel to determine the amplification success and relative intensity of the bands. Amplicon sequencing was performed on the Illumina MiSeq platform (Lundberg et al., 2013).
The initial data quality was assessed using the “FastQC” program (Version: 0.11.9; Andrews, 2010). Then, the parameters for quality processing were determined through the “USEARCH” program (Version: 11.0.667), where the thresholds for quality pruning (“-fastx_info”) and size (“-fastq_eestats2”), as well as the position of the primers used for amplification of the target region (“-search_oligodb”). The detected primers (515F. 5′-GTGCCAGCMGCCGCGGTAA; 806R 5′-GGACTACHVGGGTWTCTAAT) were then removed by the “Atropos” program (Version: 1.1.31; Didion et al., 2017). The low-quality ends were pruned by the “PRINSEQ-lite” program (Version: 0.20.4; Schmieder and Edwards, 2011), removing bases whose average quality window was less than Q20 (read forward) or Q18 (read reverse) (“-trim_qual_window 3”; “-trim_qual_right 20/18”), as well as complete sequences with a mean quality lower than Q20 (“-min_qual_mean 20”). The processed reads were submitted to the “DADA2” pipeline (Version: 1.22.0; Callahan et al., 2016) through its package for the statistical program “R” (Version: 4.1.2; R Core Team, 2020). The reads were subjected to a quality control process (“filterAndTrim”), where size truncation (“truncLen = 250”) and quality filtering (“maxEE = 2”) were performed based on the values stipulated by the “USEARCH.” Then, the exact amplicon sequence variants (ASVs) were designated for each sample, which was later filtered to retain possible chimeric sequences (“removeBimeraDenovo”) and then taxonomically classified (“assignTaxonomy”) based on the NCBI RefSeq 16S rRNA database supplemented by RDP (Version: 16; Cole et al., 2014). The taxonomic annotations of the ASVs, as well as their per-sample counts, were exported into a “phyloseq” object (R package “phyloseq”; Version: 1.38.0; McMurdie and Holmes, 2013) and transformed into compositional data (function “phyloseq_standardize_otu_abundance” from the R package “metagMisc”; Version: 0.0.4; Tedersoo et al., 2022). The datasets generated for this study were deposited in the NCBI: Sequence Read Archive (SRA), under the accession number PRJNA1086858.
The extent of sequencing coverage per sample was inferred through rarefaction curves obtained by the “amp_rarecurve” function of the R package “ampvis2” (version 2.7.17; Andersen et al., 2004). Richness (Chao1) and diversity (Shannon and Gini-Simpson indices) indices were calculated using the “alpha” function of the R package “microbiome” (Version: 1.10.0). The means of these measurements were compared pairwise, using the Student/Wilcoxon t-test (depending on the adherence of the data to normality and homoscedasticity, measured by the Shapiro–Wilk and Bartlett tests, respectively). For beta diversity, the dissimilarities were calculated using the Bray–Curtis index (“distance” function of the R package “phyloseq”), which was used for principal coordinate analysis (PCoA). The statistical significance of the separation of the evaluated treatments was determined through a PERMANOVA, considering a p-value of 0.05. Finally, the taxa whose abundances significantly changed were identified. For this purpose, the “DESeq2” approach (Version: 1.34.0; Love et al., 2014), which models the data assuming a negative binomial distribution and implements the Wald test to compare the means (p-value adjusted <0.05), was used. The taxonomic profiles and other graphical representations were generated in R using the “ggplot2” package (Version 3.3.5). The networks of each treatment were established based on the classified genera of each treatment. A minimum abundance filter was used to avoid spurious correlations (minimum relative abundance of 0.1%). The relationships between genera were inferred through Pearson’s correlation coefficients. For the calculation of correlations, the tables of counts of ASVs aggregated at the sex level were submitted to the function “corr.test” of the “psych” R package (Version: 2.2.5), considering a minimum correlation threshold of 0.75 (strongly positive or negative relationships) at a confidence level of 95% (p-value <0.05). Correlation values were provided to the function “graph_from_data_frame” of the R package “igraph” (Version: 1.3.1; Csardi, 2005) to construct the network graph. Centrality and topological measures were obtained from the graphs. The numbers of co-occurring genera (nodes), relationships between them (edges), and the formation of modules were evaluated. The number of connections (degree), intermediation ability (betweenness), and tendency to form dense clusters (clustering coefficient) were also estimated. The most important genera (Hubs) were determined from the computation of Kleinberg’s hubbiness score (Kleinberg, 2000).
High-throughput sequencing of the 20 samples generated 1,639,843 reads, which were relatively well distributed across treatments (Table 2). The generated sequences were of good quality, reflecting a low loss during the quality control and processing steps (Table 2). However, there was high contamination with sequences from the host plant, such as mitochondria and chloroplasts. Filtering these contaminating sequences caused a considerable decrease in usable reads (average: 70.2%), which was particularly severe in the BA treatment (84.3%) (Table 2).
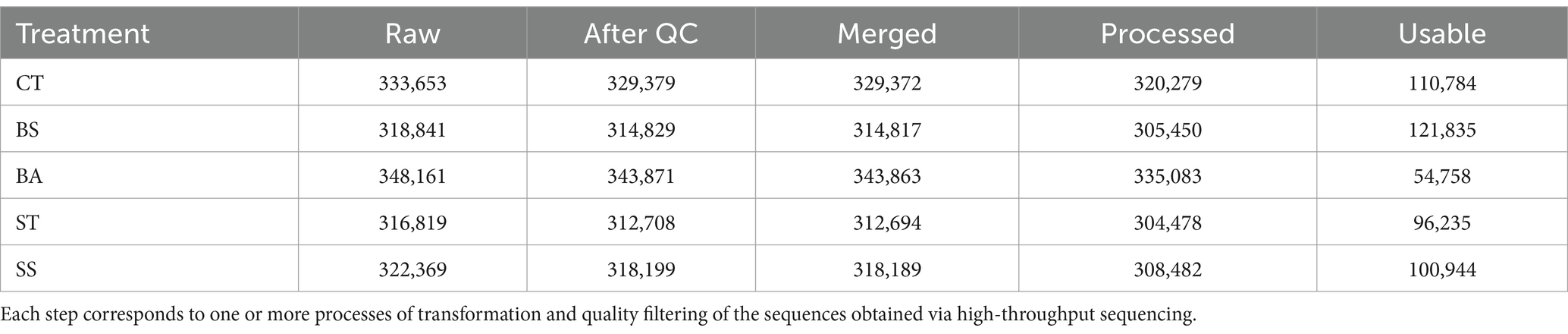
Table 2. The number of reads per processing step was counted.
Despite the substantial reduction in the number of sequenced reads, the rarefaction curves showed that the remaining usable reads had enough coverage to represent the bacterial communities present in the samples (Figure 1). The graphs show that in all samples, there was a tendency to form an asymptotic curve, thus reflecting the stabilization of the encounter of new sequences as the sampling effort increased (i.e., an increase in the number of reads).
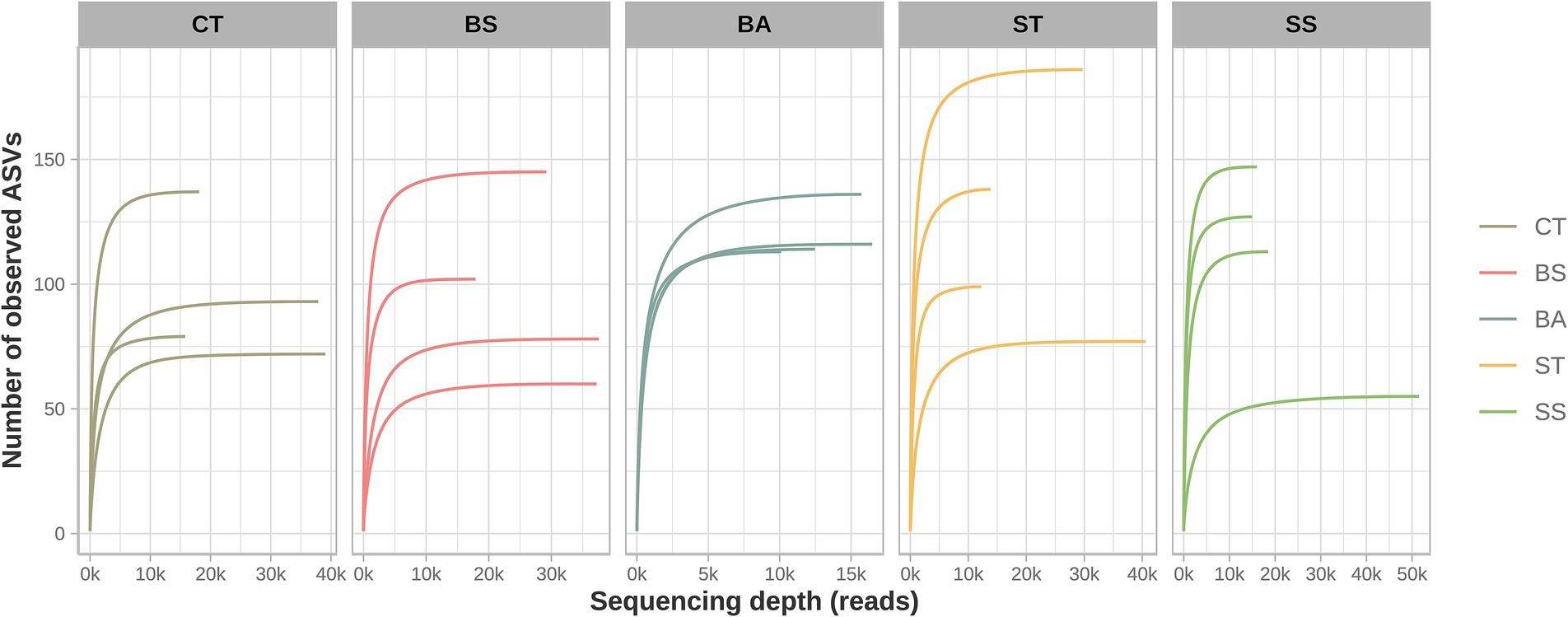
Figure 1. Rarefaction curves. The stabilization of the curves indicated a reduction in the detection of new ASVs as the number of reads increased.
Taxonomic assignment of the sequences revealed a total of 2 domains, 18 phyla, 35 classes, 68 orders, 115 families, 239 genera, and 94 species of unique prokaryotes. The vast majority of usable reads could be assigned to the genus level (average: 95.9%) (Table 3). The BA and ST treatments had greater numbers of unique genera and species (Table 3). The BA treatment also had greater numbers of families, classes, orders, and phyla than did the other treatments (Table 3). Members of the prokaryotic kingdom “Archaea” were observed only in samples from the BA, ST, and SS treatments but in very low abundance (mean relative abundance >0.01%).
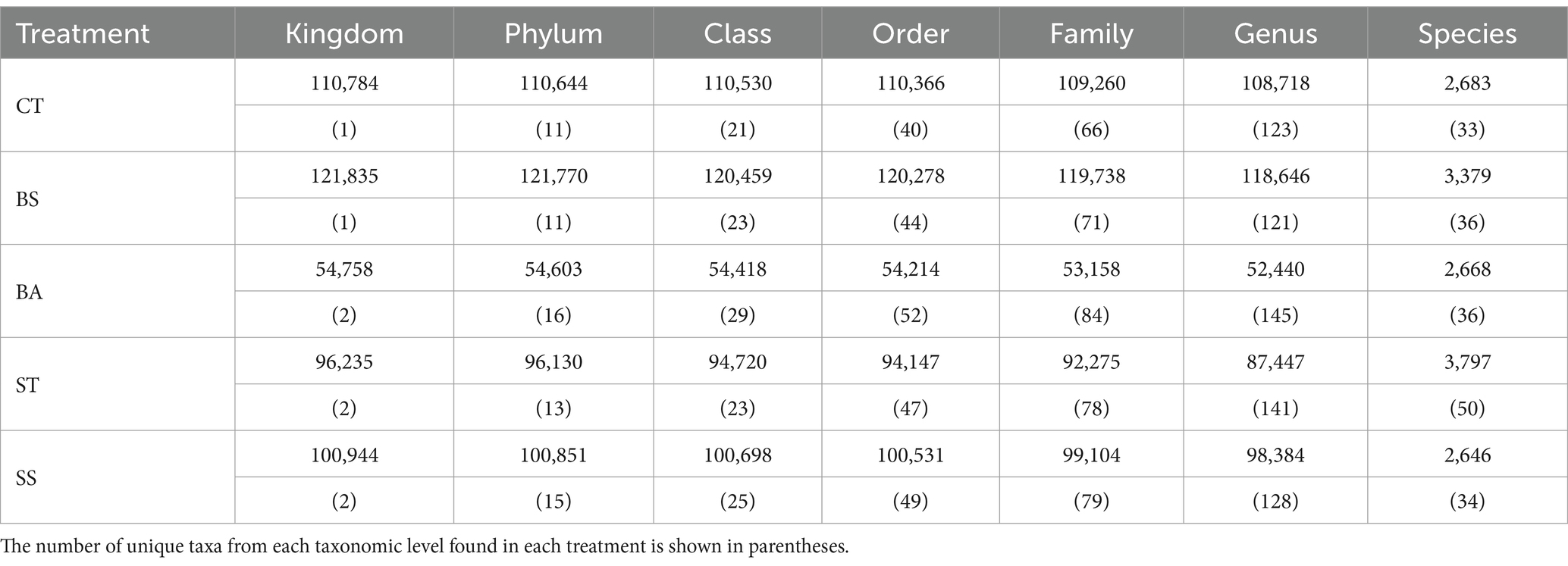
Table 3. The number of successful assignments up to each taxonomic rank was counted.
The microbial communities were mostly dominated by the phylum Proteobacteria (Figure 2A). This phylum represented, on average, 93.75% of the relative abundance. The BS treatment had the greatest relative abundance of Proteobacteria in the samples (average: 95.41%). Sequences derived from other phyla of lower abundance were concentrated in Bacteroidetes (average: 2.82%), Actinobacteria (average: 2.01%), and Firmicutes (average: 1.09%). On average, sequences unclassified at the phylum level represented 0.16% of the relative abundance of the samples.
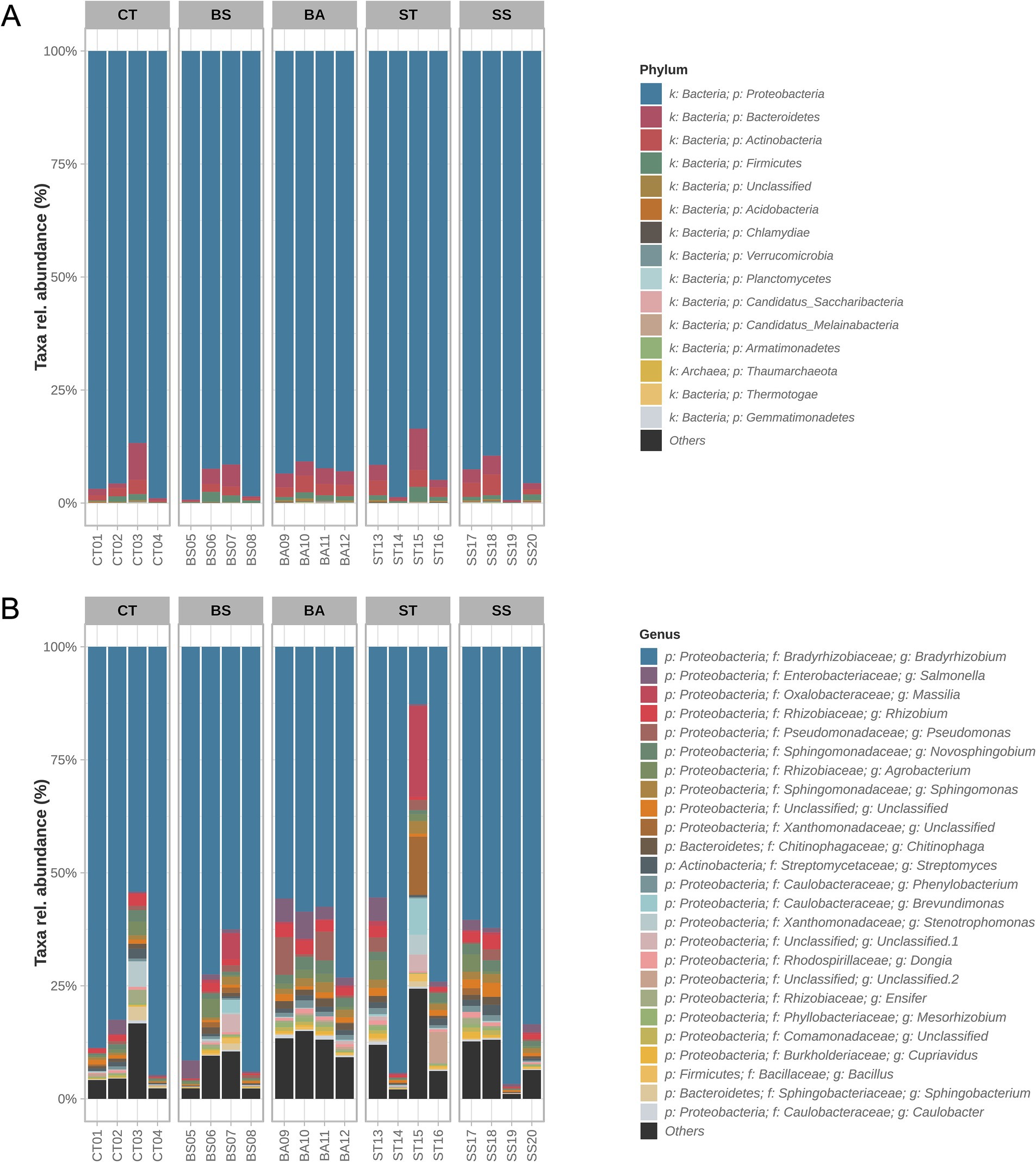
Figure 2. Taxonomic profiles of the samples. Relative abundances of the 15 most prevalent phyla (A) and 25 most prevalent genera (B). The taxa with the highest prevalence were aggregated in the “others” category.
Among the most abundant genera (Figure 2B), there was a high prevalence of members of the Proteobacteria phylum (21 of the top 25; Figure 2B). Together, the genera of these phyla represented, on average, 88.61% of the samples. There were also genera of the phyla Bacteroidetes (2/25), Actinobacteria (1/25), and Firmicutes (1/25). The massive presence of the genus Bradyrhizobium, which represented, on average, 71.22% of the sequences at the genus level, is remarkable. Bradyrhizobium was the most abundant genus in all the samples, except for sample ST (ST treatment), whose abundance was only 12.66%. Four ASVs of the genus Bradyrhizobium were detected; however, none of them were classified at the species level. The second most abundant genus throughout the samples was Salmonella (average: 1.94%), corresponding to a single ASV whose species was not classified. The third most abundant genus, Massilia (average: 1.59%), presented nine different ASVs, six of which were from unclassified species and three of which were assigned to the species M. agri strain K-3-1, M. phosphatilytica strain 12-OD1, and M. kyonggiensis strain TSA1.
The taxonomic profiles revealed that the BA treatment had greater consistency in the presence of different genera. This can be seen in the high average abundance of genera other than Bradyrhizobium (average 38.77%) and in the higher percentage of less abundant genera (“Others”; average 12.67%) (Figure 2B). In the other treatments, there were occasional samples with less dominance of the genus Bradyrhizobium; however, this was not consistent throughout the entire treatment (Figure 2B).
Despite the noticeable differences observed in the proportions of the taxonomic profiles (Figure 2), alpha diversity measures were similar between treatments since there was no significant difference in pairwise comparisons between the different indices evaluated (Table 4). In the measurement of the richness of ASVs, inferred by the Chao1 index, there was an increase in the values of the treatments inoculated with microorganisms (BS, BA, ST, and SS; Table 1); however, the variance of these values in the samples did not allow us to identify statistical differences when compared with the control. Similarly, for the diversity indices (Shannon and Gini-Simpson), the BA and ST treatments had greater values but were not significantly different from the control treatment (Table 4).

Table 4. Alpha diversity measures.
The similarity in sample compositions was also reinforced by beta diversity analysis. The Bray–Curtis distances between the samples did not show any separation between the treatments (PERMANOVA: p-value >0.05), although the composition can explain most of the variation found within them (first two axes = 78.64% of the variation; Figure 3).
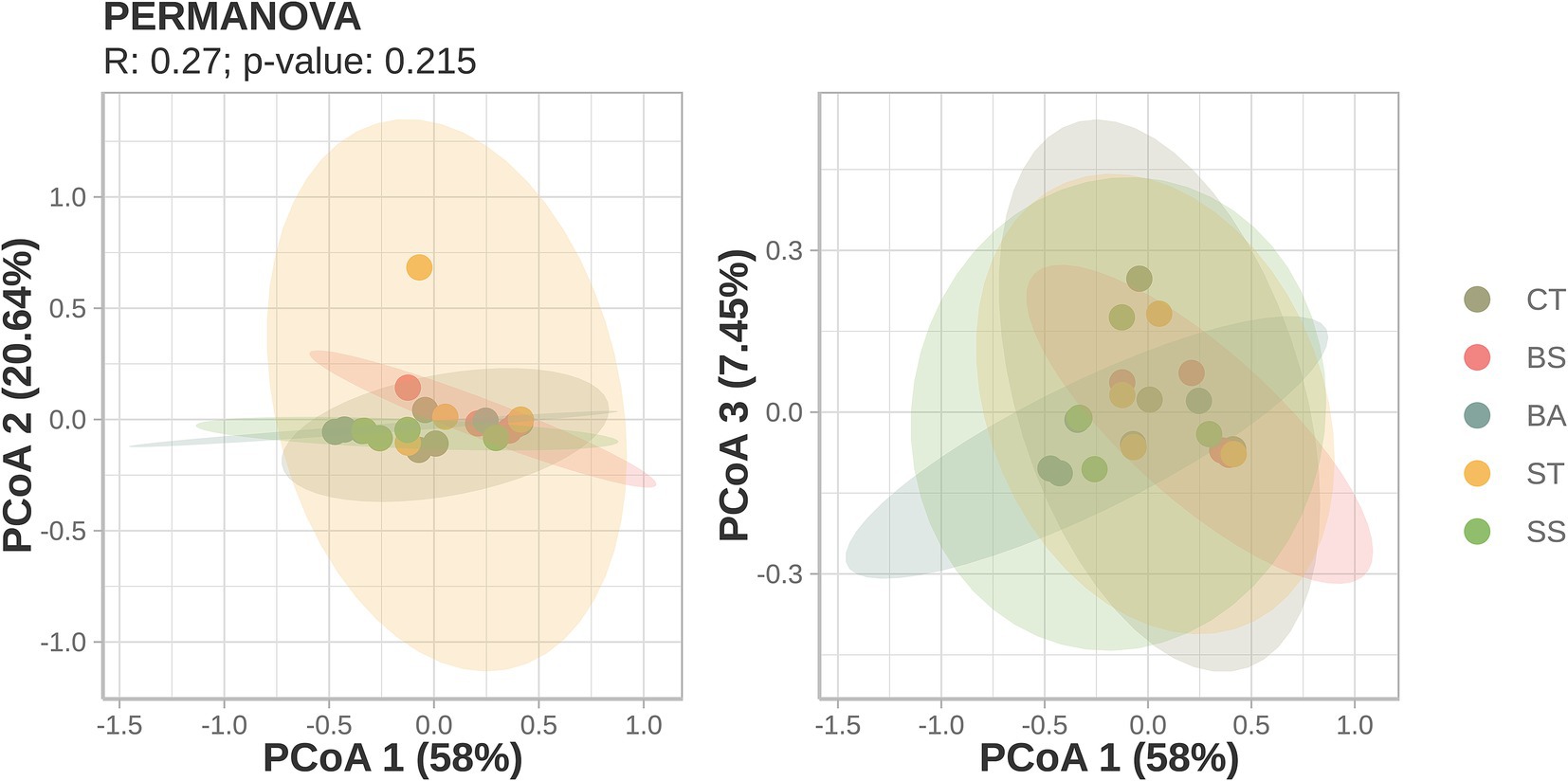
Figure 3. Principal coordinates analysis (PCoA) of Bray–Curtis dissimilarity distances. Axes and variance: the axes represent principal components (PCs) or dimensions that explain variation in the data. Higher percentages on the axes indicate a greater explanation for the variance in community differences. Clustering: points close together indicate similar microbial communities, whereas points farther apart show distinct communities.
Although the prokaryotic communities present in the treatments were narrowly similar, some significant differences were found when prospecting for differentially abundant taxa (DATs). In total, 30 DATs were found, 24 of which were unique. The measures of differences (fold changes), p-values, and relative abundances in the treatments. Differences were concentrated on more specific taxonomic ranks, such as family, genus, and species (Figure 4). Compared with the control treatment (CT), the ST treatment caused the most shifts at 14 DAT (Figure 4C). Of these, 12 were found in greater abundance in the ST samples, while two other species were more prevalent in the CT samples.
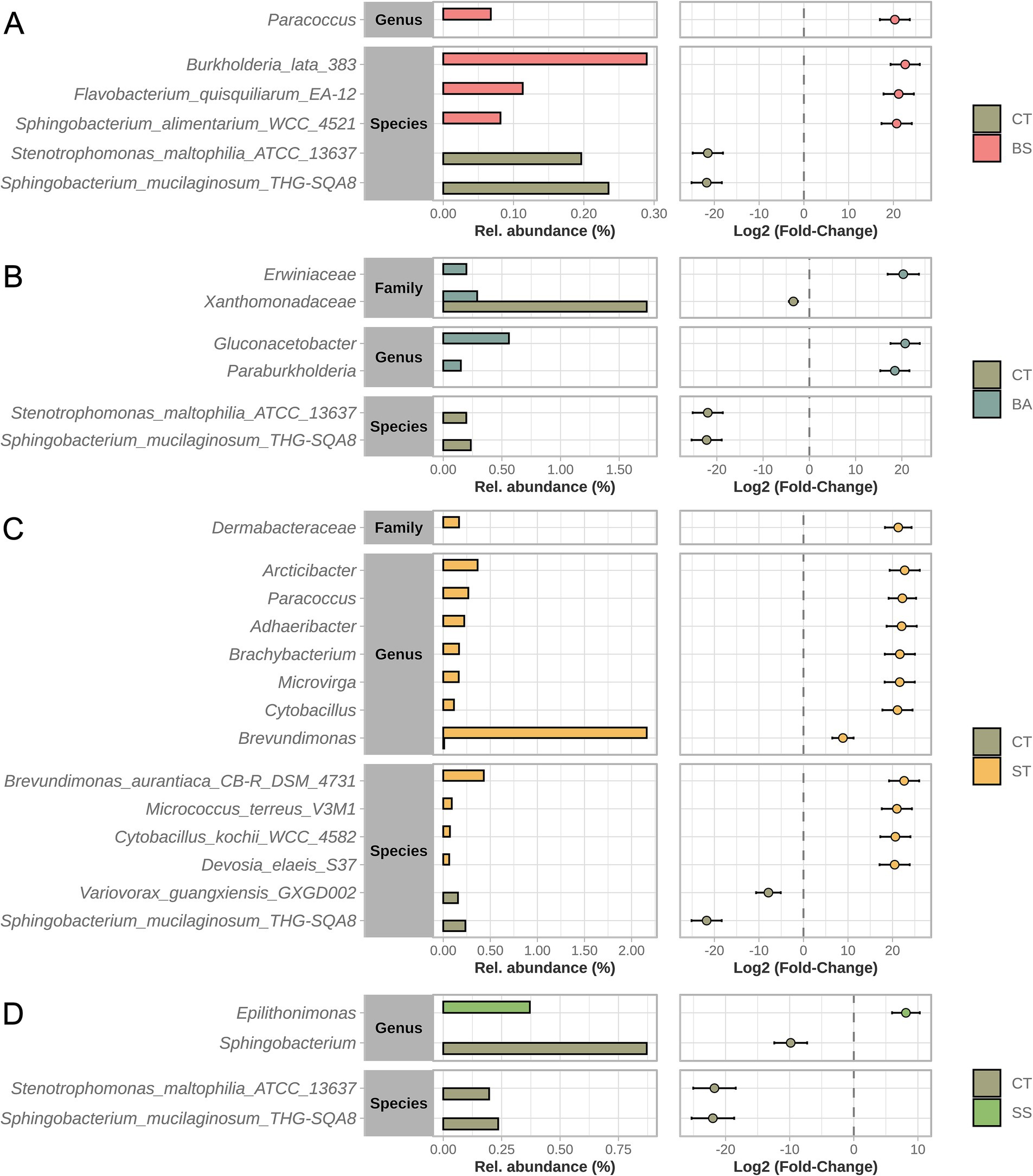
Figure 4. Differentially abundant taxa (DAT). Comparisons were made between each treatment and the control (A–D). All the taxonomic ranks were tested. The bar graph (left) represents the comparison of relative abundances, and the dot-and-whisker graph (right) represents the intensity of the difference (fold-change) and its variation in the samples.
The SS treatment had the least number of shifts (4 DAT), mainly reductions in the abundance of taxa present in the CT treatment (Figure 4D). The abundances of the Sphingobacterium mucilaginosum strain THG-SQA8 and the Stenotrophomonas maltophilia strain ATCC-13637 seemed to be most affected by inoculation since their abundances decreased after 4 and 3 of the treatments, respectively.
The microbial community structure was explored through co-occurrence networks (Figure 5 and Table 5). The relationships of taxa at the genus level seemed to be affected by the treatments, as structural shifts were observed (Figure 5). The CT network had intermediate centrality measures in general comparison with other networks.
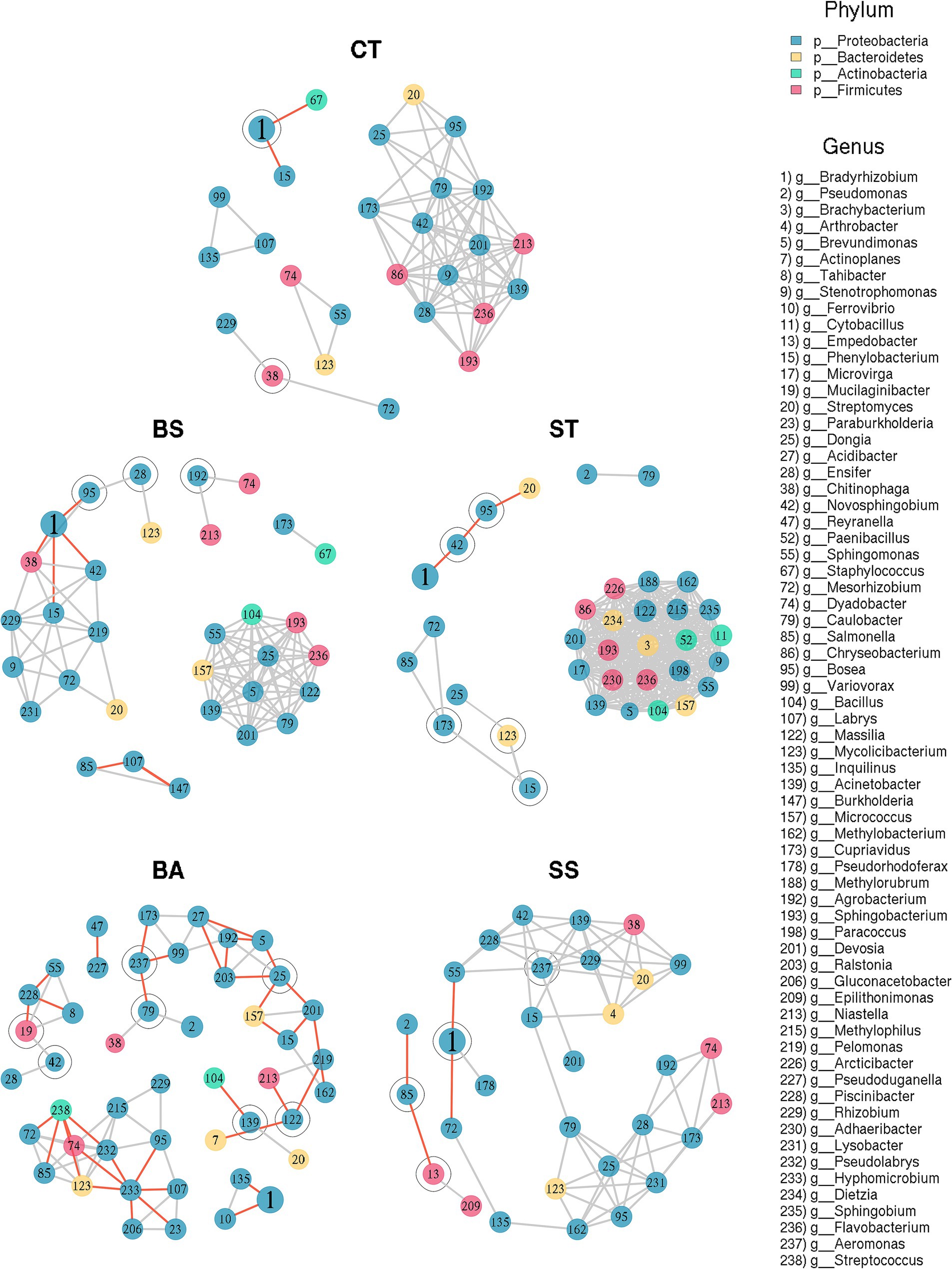
Figure 5. Co-occurrence network graphs of treatments at genus rank. The networks were established using strong Pearson’s correlation coefficients (r = ±0.75) of the relationships at a confidence level of 95% (p < 0.05). Negative relationships are depicted in red, whereas positive relationships are represented in light gray. Articulation points (taxa that generated additional modules in their absence) are denoted by an outer circle.
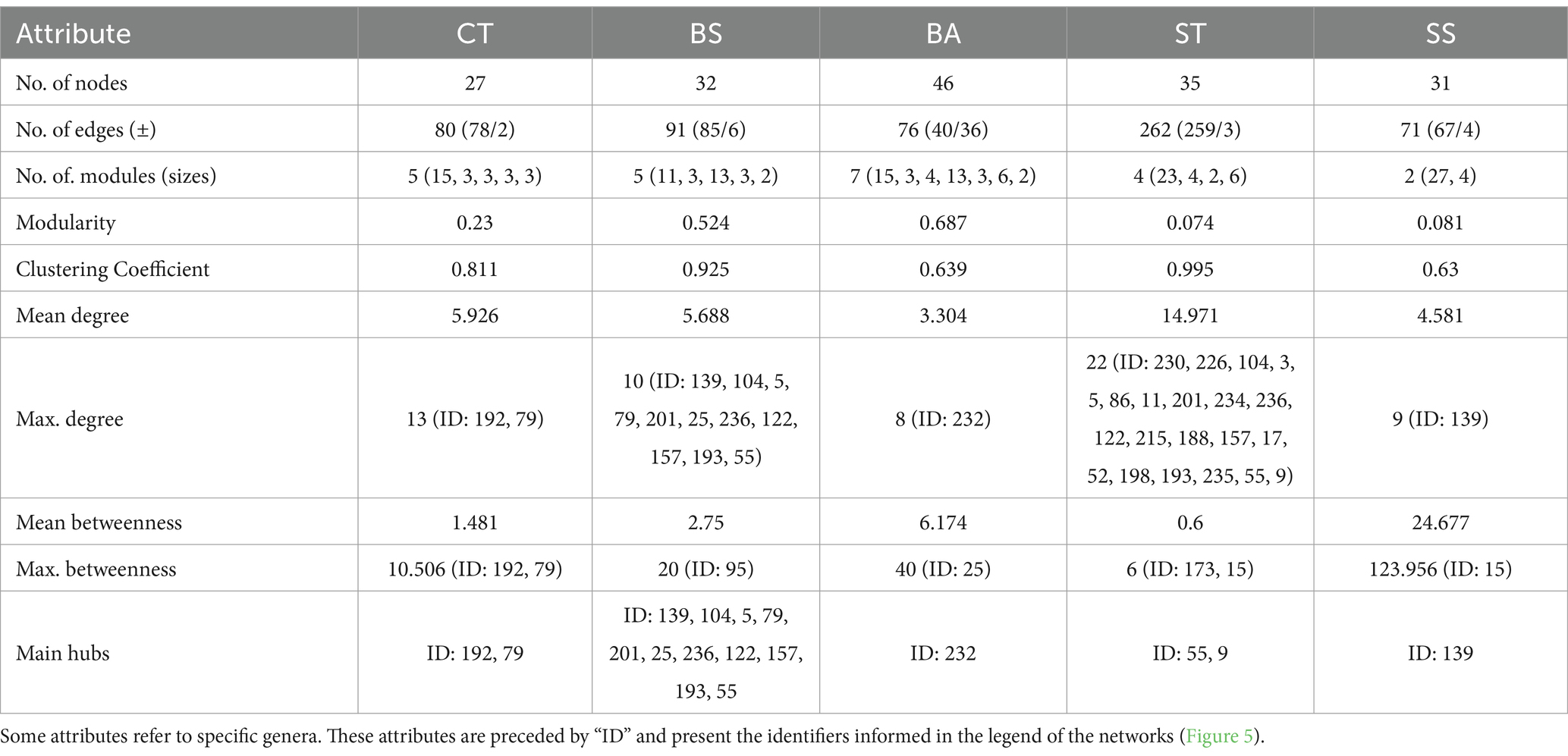
Table 5. Topological features and centrality measures of correlation networks.
The BS and BA treatments had greater tendencies to form larger modules, with modularity values of 0.524 and 0.687, respectively (Table 5). Additionally, the BA treatment group presented a greater number of modules (7; Table 5). Notably, this treatment had strong negative correlations (Figure 5 and Table 5). The modularity values of the ST and SS treatments were considerably lower than those of the CT network, at 0.074 and 0.081, respectively. The SS had only two modules.
Another aspect noted in the structuring is the density of the modules. The ST treatment presented a module composed of strongly correlated genera (Figure 5), with a high number of edges and a high mean degree of connections per node (Table 5). This resulted in a high clustering coefficient (0.995; Table 5). The same observations apply—to a lesser extent—to the BS treatment. The BA and SS treatments, on the other hand, had opposite trends, as they had a lower number of edges and mean degree by genus. Thus, their clustering coefficients were lower than those of CT (Table 5). Another consequent effect of the greater number of rarefied modules in the BA and SS treatment networks was the greater dependence on intermediate taxa to maintain the internal links of the modules. This can be verified by the highest means and maximums of betweenness of these treatments (Table 5). Finally, changes are observed in the genera considered hubs since the results differed between treatments (except for the BS treatment, whose taxa are present in the densest submodule tied in this measure) (Table 5). However, considering the same exception, all hub taxa belonged to the phylum Proteobacteria (Figure 5 and Table 5).
The data show statistical significance at a significance level of 5% based on the ANOVA. Figure 6 demonstrates that all bacterial treatments exhibited similar or superior performance in productivity and production compared to the control and S. spinosa (SS), which demonstrated the highest mean productivity (4407.32 kg/ha) in contrast to the control (4201.32 kg/ha). For the productivity parameter, the highest value was observed in the SS treatment compared with the control, whereas no statistically significant difference was found between the other treatments and the control. Regarding nitrogen content, only the ST treatment did not exhibit a statistically significant difference from the control, whereas the other treatments demonstrated higher values than the control.
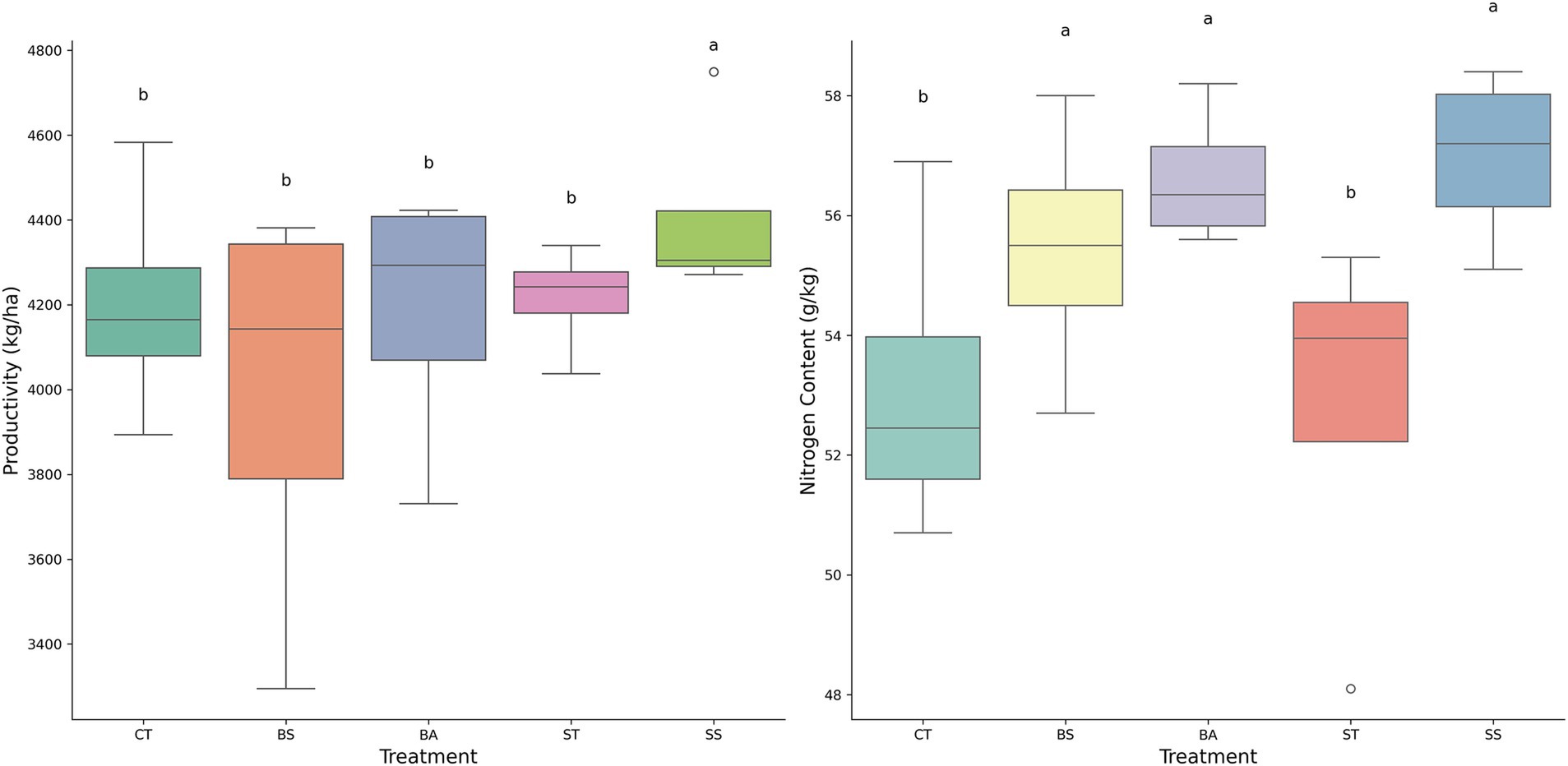
Figure 6. This consisted of two box plots comparing the effects of different treatments (CT, BS, BA, ST, and SS) on two parameters: productivity (kg/ha) on the left and nitrogen content (g/kg) on the right. The SS treatment had the highest value for productivity, whereas the BS, BA, and SS treatments had the highest values for nitrogen content. The same letters indicate no statistical difference between treatments according to Tukey 5%.
Many microorganisms are associated with plants and have a significant effect on various aspects of plant growth and development. These associated microorganisms can enhance nutrient availability in the soil and improve the ability of a plant to absorb nutrients and water. In addition, they can produce phytohormones such as auxins, gibberellins, and cytokinins, which promote root and shoot development, increase a plant’s ability to explore the soil, and enhance its photosynthetic efficiency. Interactions between microorganisms can sometimes be detrimental because they may produce antimicrobial molecules to eliminate one another or compete for the same niche of colonization and nutrient resources. However, these interactions can also be synergistic, where one species of microorganism assists another in colonizing and establishing itself in the soil or host plant (Wallis and Galarneau, 2020; Nakano et al., 2022).
As demonstrated in other studies, the process of inoculating endogenous microorganisms can significantly alter soil and root microbiomes, thereby promoting growth and enhancing the capacity of plants to thrive (Mitter et al., 2016; Nadarajah and Abdul Rahman, 2021).
In this study, the objective was to determine which bacteria would stimulate soybean growth and how the inoculation of these bacteria would impact the root microbiome compared to the control, which did not receive bacterial inoculation. To achieve this goal, several bacteria were inoculated into the soybean crop, including B. subtilis (BS), B. aryabhattai (BA), Streptomyces sp. (ST), and S. spinosa (SS). Interestingly, in terms of promoting plant growth, no significant differences were observed among the treatments, indicating that none of the bacterial strains enhanced soybean productivity. However, according to the PCA analysis, inoculation with S. spinosa and B. aryabhattai showed the potential to improve plant yield. Variables related to nitrogen content in the grains and aboveground parts showed a stronger correlation with S. spinosa inoculation. These results suggest that S. spinosa may play a significant role in increasing nitrogen availability and utilization in plants. Saccharopolyspora spinosa is a bacterium belonging to the Actinobacteria phylum. It is a naturally occurring organism used to produce spinosad, an insect-control agent (Breslin et al., 2000). This organism is involved in a fermentation process that results in spinosad, which has been widely used to control several caterpillars that cause yield losses in many different crops, such as chickpea, corn, cotton, wheat, peanut, soybean, and other valuable agricultural crops (Yang et al., 2019; Mihretie et al., 2020; Qu et al., 2020) because of its low mammalian toxicity, ecotoxicology, and minimal environmental impact.
There is currently no published research indicating that the bacterium S. spinosa exhibits plant growth-promoting properties, nor is there any evidence suggesting that inoculation with this bacterium has any impact on the nitrogen content of soybean grains. However, inoculation with SS may have changed the rhizospheric microbiome, favoring nitrogen absorption by plants.
Phosphorus content in the dry mass was influenced more by B. aryabhattai. This indicated that B. aryabhattai may have a particular impact on phosphorus availability and plant biomass accumulation. Several studies have demonstrated the capacity of bacteria from the genus Bacillus to solubilize phosphorus and increase the phosphorus content in plants (Emami et al., 2020; Pathania et al., 2020; Torres et al., 2024). At present, there is no evidence to suggest that B. arayabhattai can enhance the phosphorus content in soybeans. According to a study by Kang et al. (2023), B. arayabhattai has the capacity to eliminate excess nitrogen from soil through two processes: the conversion of ammonia to other nitrogen forms and the reduction of those forms into harmless nitrogen gas. However, this finding has raised concerns, as the inoculation of this bacterium could potentially harm soybean plants by hindering their ability to absorb nitrogen. Although this effect may occur, Kang et al. (2012) reported that it only occurs under specific circumstances such as cold temperatures and highly alkaline environments, which are typically challenging for other bacteria. Conversely, the control treatment, which received mineral fertilization, did not differ from the treatments. These findings suggest that mineral fertilization can be replaced with the inoculation of these bacteria without any reduction in yield or the need for B. japonicum inoculation in areas where soybeans are consistently produced.
Several studies have demonstrated the capacity of the inoculation of indigenous bacteria to modulate the rhizospheric and root microbiomes to promote plant growth (Dastogeer et al., 2020). However, little information is available on how to manipulate the microbiome to promote plant growth. In the present study, inoculation with four types of bacterial strains influenced the root microbiome in different ways; however, B. japonicum was the most abundant genus in all samples, except for ST (ST treatment), with an abundance of only 12.66%.
The introduction of a bacterium may result in an increase in the population of other bacteria. This increase in population was attributed to the altered environment caused by the introduced bacteria or their influence on the roots of the plant. This phenomenon occurred for all the evaluated strains, except for ST.
Despite the noticeable differences observed in the proportions of the taxonomic profiles (Figure 2), alpha diversity measures were similar between the treatments, as there was no significant difference in pairwise comparisons between the different indices evaluated (Table 4). Some results have shown no differences in alpha diversity between the treatments and have also shown an effect on plant growth (Bueno et al., 2022; dos Santos et al., 2022). When the bacterial inoculant was applied, indigenous bacteria could promote modifications in the microbiome, resulting in both alterations and benefits to the plant. However, these changes were short-lived, and the microbiome returned to its previous state, rendering microbiome evaluation unable to detect any differences. Another study that provided this evidence was conducted by Lobo et al. (2019). In this study, four strains of B. subtilis were inoculated into maize crops under field conditions. Notably, only the treatment that showed a higher yield than the control had fewer B. subtilis-covered roots than the other treatments. This suggests that the set of abilities of bacteria to interact with plants is more important than the number of bacteria in promoting plant growth.
All inoculated strains demonstrated the ability enhanced the population of B. japonicum in the roots of plants. This phenomenon appears to be primarily influenced by the behavior of B. japonicum rather than the inoculated microorganisms themselves.
A study demonstrated that co-inoculation with Trichoderma harzianum did not increase the number of B. japonicum bacteria but helped soybean plants to form nodules even in the presence of nitrate, which usually stops nodule formation (Iturralde et al., 2020). Co-inoculation of Azospirillum baldaniorum with B. japonicum improves root nodule fixation in lima bean nitrogen (Lopes et al., 2022). Groppa et al. (1998) inoculated B. japonicum with A. brasilense in soybean and verified that nodule number and nodule dry weight were not affected by dual inoculation; however, co-inoculated plants showed a significantly higher proportion of nodules attached to the main root and located in the upper of the root system. The results showed that co-inoculation with B. japonicum and A. brasilense led to an increase in the number of the most active nodules, resulting in greater nitrogen fixation and assimilation.
Compared with the control treatment (CT), the ST treatment caused the greatest shifts at 14 DAT (Figure 4C). Of these, 12 were found to be more abundant in the ST samples, whereas two other species were more prevalent in the CT samples.
Various studies have demonstrated a correlation between high microbial diversity and soil, which is suppressive. The suppressive effect of soil is attributed to the intense competition among microorganisms, which may be a result of high microbial diversity, making it difficult for pathogens to establish and thereby reduce the incidence of diseases (Kotsou et al., 2004; Zhao et al., 2018; dos Santos Souza et al., 2019). Although the effect of high DAT on plant growth remains inconclusive, further research is necessary to determine whether a higher number of DAT is beneficial for promoting plant growth. The results of such studies will be crucial for providing a clearer understanding of this phenomenon.
Co-inoculation of soybean with plant growth-promoting bacteria did not significantly increase the yield compared to mineral fertilization. However, microbial inoculation serves as a viable alternative to mineral fertilization without compromising productivity. Although bacterial inoculation did not enhance the yield, it promoted the growth of B. japonicum in plant roots, with the exception of Streptomyces treatment. This suggests that co-inoculation may be an effective strategy for improving the efficiency of B. japonicum in promoting plant growth. The inoculated strains influenced the root microbiome in different ways, with B. japonicum being the most abundant genus in most of the samples. Further research is required to fully understand the mechanisms underlying these microbial interactions and their implications for soybean productivity. Overall, this study demonstrates the potential of co-inoculation with plant growth-promoting bacteria to modulate the root microbiome and enhance beneficial microbial colonization in soybean.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
MA: Writing – original draft, Writing – review & editing. LC: Writing – original draft, Writing – review & editing. CSa: Writing – original draft, Writing – review & editing. EF: Writing – original draft, Writing – review & editing. CSi: Writing – original draft, Writing – review & editing. DP: Writing – original draft, Writing – review & editing. EZ: Writing – original draft, Writing – review & editing. OB: Writing – original draft, Writing – review & editing. ER: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was financed in part by the Coordination for the Improvement of Higher Education Personnel (CAPES), Brazil, Finance Code 001.
The authors thank FAPESP for financial support Process number 21/10821-8 CNPQ—Process: 302234/2022-5.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
No Generative AI was used in the preparation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Andersen, K., Kirkegaard, R., Karst, S., and Albertsen, M. (2004). ampvis2: an R package to analyse and visualise 16S rRNA amplicon data. bioRxiv.
Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. Babraham Institute, Cambridge, United Kingdom: Babraham Bioinformatics.
Armendariz, A. L., Talano, M. A., Olmos Nicotra, M. F., Escudero, L., Breser, M. L., Porporatto, C., et al. (2019). Impact of double inoculation with Bradyrhizobium japonicum E109 and Azospirillum brasilense Az39 on soybean plants grown under arsenic stress. Plant Physiol. Biochem. 138, 26–35. doi: 10.1016/j.plaphy.2019.02.018
Breslin, W. J., Marty, M. S., Vedula, U., Liberacki, A. B., and Yano, B. L. (2000). Developmental toxicity of spinosad administered by gavage to CD 1 rats and New Zealand white rabbits. Food Chem. Toxicol. 38, 1103–1112. doi: 10.1016/S0278-6915(00)00108-3
Bueno, C. B., dos Santos, R. M., de Souza Buzo, F., de Andrade da Silva, M. S. R., and Rigobelo, E. C. (2022). Effects of chemical fertilization and microbial inoculum on Bacillus subtilis colonization in soybean and maize plants. Front. Microbiol. 13:901157. doi: 10.3389/fmicb.2022.901157
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13:581. doi: 10.1038/nmeth.3869
Cao, L., Qiu, Z., You, J., Tan, H., and Zhou, S. (2005). Isolation and characterization of endophytic streptomycete antagonists of Fusarium wilt pathogen from surface-sterilized banana roots. FEMS Microbiol. Lett. 247, 147–152. doi: 10.1016/j.femsle.2005.05.006
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., McGarrell, D. M., Sun, Y., et al. (2014). Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42. doi: 10.1093/nar/gkt1244
Csardi, G. (2005). The igraph software package for complex network research. Available at: https://www.researchgate.net/publication/221995787 (Accessed February 11, 2024).
Dastogeer, K. M. G., Tumpa, F. H., Sultana, A., Akter, M. A., and Chakraborty, A. (2020). Plant microbiome—an account of the factors that shape community composition and diversity. Curr. Plant Biol. 23:100161. doi: 10.1016/j.cpb.2020.100161
de Souza, R. S. C., Okura, V. K., Armanhi, J. S. L., Jorrín, B., Lozano, N., da Silva, M. J., et al. (2016). Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci. Rep. 6:28744. doi: 10.1038/srep28774
Didion, J. P., Martin, M., and Collins, F. S. (2017). Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ 5:e3720. doi: 10.7717/peerj.3720
dos Santos, R. M., Cueva-Yesquén, L. G., Garboggini, F. F., Desoignies, N., and Rigobelo, E. C. (2022). Inoculum concentration and mineral fertilization: effects on the endophytic microbiome of soybean. Front. Microbiol. 13:900980. doi: 10.3389/fmicb.2022.900980
dos Santos Souza, C. R., de Oliveira Barbosa, A. C., Ferreira, C. F., Souza, F. V. D., de Souza Rocha, L., de Souza, E. H., et al. (2019). Diversity of microorganisms associated to Ananas spp. from natural environment, cultivated and ex situ conservation areas. Sci. Hortic. 243, 544–551. doi: 10.1016/J.SCIENTA.2018.09.015
Emami, S., Alikhani, H. A., Pourbabaee, A. A., Etesami, H., Motasharezadeh, B., and Sarmadian, F. (2020). Consortium of endophyte and rhizosphere phosphate solubilizing bacteria improves phosphorous use efficiency in wheat cultivars in phosphorus deficient soils. Rhizosphere 14:100196. doi: 10.1016/j.rhisph.2020.100196
Groppa, M. D., Zawoznik, M. S., and Tomaro, M. L. (1998). Effect of co-inoculation with Bradyrhizobium japonicum and Azospirillum brasilense on soybean plants. Eur. J. Soil Biol. 34, 75–80. doi: 10.1016/S1164-5563(99)90004-3
Gu, Y., Dong, K., Geisen, S., Yang, W., Yan, Y., Gu, D., et al. (2020). The effect of microbial inoculant origin on the rhizosphere bacterial community composition and plant growth-promotion. Plant Soil 452, 105–117. doi: 10.1007/s11104-020-04545-w
Ikiz, B., Dasgan, H. Y., and Gruda, N. S. (2024). Utilizing the power of plant growth promoting rhizobacteria on reducing mineral fertilizer, improved yield, and nutritional quality of Batavia lettuce in a floating culture. Sci. Rep. 14:1616. doi: 10.1038/s41598-024-51818-w
Iturralde, E. T., Stocco, M. C., Faura, A., Mónaco, C. I., Cordo, C., Pérez-Giménez, J., et al. (2020). Coinoculation of soybean plants with Bradyrhizobium japonicum and Trichoderma harzianum: coexistence of both microbes and relief of nitrate inhibition of nodulation. Biotechnol. Rep. 26:e00461. doi: 10.1016/j.btre.2020.e00461
Kang, S. M., Khan, A. L., Hamayun, M., Hussain, J., Joo, G. J., You, Y. H., et al. (2012). Gibberellin-producing Promicromonospora sp. SE188 improves Solanum lycopersicum plant growth and influences endogenous plant hormones. J. Microbiol. 50, 902–909. doi: 10.1007/s12275-012-2273-4
Kang, X., Zhao, X., Song, X., Wang, D., Shi, G., Duan, X., et al. (2023). Nitrogen removal by a novel strain Priestia aryabhattai KX-3 from East Antarctica under alkaline pH and low-temperature conditions. Process Biochem. 130, 674–684. doi: 10.1016/j.procbio.2023.05.030
Khoso, M. A., Wagan, S., Alam, I., Hussain, A., Ali, Q., Saha, S., et al. (2024). Impact of plant growth-promoting rhizobacteria (PGPR) on plant nutrition and root characteristics: current perspective. Plant Stress 11:100341. doi: 10.1016/j.stress.2023.100341
Khosravi, H., Khoshru, B., Nosratabad, A. F., and Mitra, D. (2024). Exploring the landscape of biofertilizers containing plant growth-promoting rhizobacteria in Iran: progress and research prospects. Curr. Res. Microb. Sci. 7:100268. doi: 10.1016/j.crmicr.2024.100268
Kotsou, M., Mari, I., Lasaridi, K., Chatzipavlidis, I., Balis, C., and Kyriacou, A. (2004). The effect of olive oil mill wastewater (OMW) on soil microbial communities and suppressiveness against Rhizoctonia solani. Appl. Soil Ecol. 26, 113–121. doi: 10.1016/J.APSOIL.2003.12.001
Kumar, V., Chourasia, H. K., Rajani, K., and Kumar, R. R. (2024). Exploration and characterization of high-efficiency phosphate-solubilizing bacteria isolates from chickpea rhizospheric soil. Int. J. Bio-Resour. Stress Manag. 15, 01–09. doi: 10.23910/1.2024.4987a
Lobo, L. L. B., dos Santos, R. M., and Rigobelo, E. C. (2019). Promotion of maize growth using endophytic bacteria under greenhouse and field conditions. Aust. J. Crop. Sci. 13, 2067–2074. doi: 10.21475/ajcs.19.13.12.p2077
Lopes, Á., Setubal, I., Costa, V., Zilli, J., Rodrigues, A., and Bonifacio, A. (2022). Synergism of Bradyrhizobium and Azospirillum baldaniorum improves growth and symbiotic performance in lima bean under salinity by positive modulations in leaf nitrogen compounds. Appl. Soil Ecol. 180:104603. doi: 10.1016/J.APSOIL.2022.104603
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lundberg, D. S., Yourstone, S., Mieczkowski, P., Jones, C. D., and Dangl, J. L. (2013). Practical innovations for high-throughput amplicon sequencing. Nat. Methods 10, 999–1002. doi: 10.1038/nmeth.2634
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meier, M., Liu, Y., Lay-Pruitt, K. S., Takahashi, H., and von Wirén, N. (2020). Auxin-mediated root branching is determined by the form of available nitrogen. Nat. Plants 6, 1136–1145. doi: 10.1038/s41477-020-00756-2
Mihretie, A., Yimer, D., Wudu, E., and Kassaw, A. (2020). Efficacy of insecticides against African bollworm (Helicoverpa armigera Hubner) on chickpea (Cicer arietinum) in the lowlands of Wollo, Northeastern Ethiopia. Cogent Food Agric. 6:1833818. doi: 10.1080/23311932.2020.1833818
Mitter, B., Pfaffenbichler, N., and Sessitsch, A. (2016). Plant-microbe partnerships in 2020. Microb. Biotechnol. 9, 635–640. doi: 10.1111/1751-7915.12382
Mun, B. G., Hussain, A., Park, Y. G., Kang, S. M., Lee, I. J., and Yun, B. W. (2024). The PGPR Bacillus aryabhattai promotes soybean growth via nutrient and chlorophyll maintenance and the production of butanoic acid. Front. Plant Sci. 15:1341993. doi: 10.3389/fpls.2024.1341993
Nadarajah, K., and Abdul Rahman, N. S. N. (2021). Plant-microbe interaction: aboveground to belowground, from the good to the bad. Int. J. Mol. Sci. 22:10388. doi: 10.3390/ijms221910388
Nakano, M., Omae, N., and Tsuda, K. (2022). Inter-organismal phytohormone networks in plant-microbe interactions. Curr. Opin. Plant Biol. 68:102258. doi: 10.1016/j.pbi.2022.102258
Pathania, P., Bhatia, R., and Khatri, M. (2020). Cross-competence and affectivity of maize rhizosphere bacteria Bacillus sp. MT7 in tomato rhizosphere. Sci. Hortic. 272:109480. doi: 10.1016/j.scienta.2020.109480
Paulus, J. M., and Tooy, D. (2024). Effect of plant growth promoting rhizobacteria (PGPR) on growth and yield of soybean. J. Agric. 3, 30–38. doi: 10.47709/joa.v3i01.3613
Qu, Q., Zhang, Z., Peijnenburg, W. J. G. M., Liu, W., Lu, T., Hu, B., et al. (2020). Rhizosphere microbiome assembly and its impact on plant growth. J. Agric. Food Chem. 68, 5024–5038. doi: 10.1021/ACS.JAFC.0C00073
R Core Team (2020). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Available at: https://www.R-project.org/.
R Core Team (2023). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. Available at: https://www.R-project.org/
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Tedersoo, L., Bahram, M., Zinger, L., Nilsson, R. H., Kennedy, P. G., Yang, T., et al. (2022). Best practices in metabarcoding of fungi: from experimental design to results. Mol. Ecol. 31, 2769–2795. doi: 10.1111/mec.16460
Torres, P., Altier, N., Beyhaut, E., Fresia, P., Garaycochea, S., and Abreo, E. (2024). Phenotypic, genomic and in planta characterization of Bacillus sensu lato for their phosphorus biofertilization and plant growth promotion features in soybean. Microbiol. Res. 280:127566. doi: 10.1016/j.micres.2023.127566
Verma, K. K., Joshi, A., Song, X. P., Singh, S., Kumari, A., Arora, J., et al. (2024). Synergistic interactions of nanoparticles and plant growth promoting rhizobacteria enhancing soil-plant systems: a multigenerational perspective. Front. Plant Sci. 15:1376214. doi: 10.3389/fpls.2024.1376214
Wahab, A., Bibi, H., Batool, F., Muhammad, M., Ullah, S., Zaman, W., et al. (2024). Plant growth-promoting rhizobacteria biochemical pathways and their environmental impact: a review of sustainable farming practices. Plant Growth Regul. 104, 637–662. doi: 10.1007/s10725-024-01218-x
Wallis, C. M., and Galarneau, E. R. A. (2020). Phenolic compound induction in plant-microbe and plant-insect interactions: a meta-analysis. Front. Plant Sci. 11:5870753. doi: 10.3389/fpls.2020.580753
Wang, M., Ge, A. H., Ma, X., Wang, X., Xie, Q., Wang, L., et al. (2024). Dynamic root microbiome sustains soybean productivity under unbalanced fertilization. Nat. Commun. 15:1668. doi: 10.1038/s41467-024-45925-5
Wan, W., Qin, Y., Wu, H., Zuo, W., He, H., Tan, J., et al. (2020). Isolation and characterization of phosphorus solubilizing bacteria with multiple phosphorus sources utilizing capability and their potential for lead immobilization in soil. Front. Microbiol. 11:752. doi: 10.3389/fmicb.2020.00752
Yang, X., Chen, X., and Yang, X. (2019). Effect of organic matter on phosphorus adsorption and desorption in a black soil from Northeast China. Soil Tillage Res. 187, 85–91. doi: 10.1016/J.STILL.2018.11.016
Keywords: microbiome, Saccharopolyspora spinosa, Streptomyces, metagenomic, plant growth
Citation: de Andrade da Silva MSR, de Carvalho LAL, Santos CHB, Frezarin ET, da Silva CGN, Pinheiro DG, Zonta E, Babalola OO and Rigobelo EC (2025) Effect of co-inoculation with plant growth-promoting bacteria on the microbiome of soybean roots. Front. Sustain. Food Syst. 9:1505001. doi: 10.3389/fsufs.2025.1505001
Edited by:
Raffaella Balestrini, National Research Council (CNR), ItalyReviewed by:
Huiqiao Pan, Carnegie Institution for Science, United StatesCopyright © 2025 de Andrade da Silva, de Carvalho, Santos, Frezarin, da Silva, Pinheiro, Zonta, Babalola and Rigobelo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Everlon Cid Rigobelo, ZXZlcmxvbi5jaWRAdW5lc3AuYnI=
†ORCID: Everlon Cid Rigobelo, http://orcid.org/0000-0002-9734-3338
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.