 Bheemapura Shivakumar Harshitha1
Bheemapura Shivakumar Harshitha1 Vikram Jeet Singh2
Vikram Jeet Singh2 Shekharappa Nandakumar1,3
Shekharappa Nandakumar1,3 Sonu Shekhawat1
Sonu Shekhawat1 Brijesh Kumar Dixit1
Brijesh Kumar Dixit1 Shridhar Ragi1
Shridhar Ragi1 Deepak Saran1
Deepak Saran1 Kunnummal Kurungara Vinod1
Kunnummal Kurungara Vinod1 Subbaiyan Gopal Krishnan1
Subbaiyan Gopal Krishnan1 Ranjith Kumar Ellur1Mariappan Nagarajan4
Ranjith Kumar Ellur1Mariappan Nagarajan4 Haritha Bollinedi1
Haritha Bollinedi1 Tapan Kumar Mondal5Ashok Kumar Singh1
Tapan Kumar Mondal5Ashok Kumar Singh1 Prolay Kumar Bhowmick1*
Prolay Kumar Bhowmick1*- 1Division of Genetics, ICAR-Indian Agricultural Research Institute, New Delhi, India
- 2Department of Seed Science and Technology, Acharya Narendra Deva University of Agriculture & Technology, Ayodhya, India
- 3Corteva Agrisciences, Hyderabad Research Centre, Hyderabad, India
- 4Rice Breeding and Genetics Research Centre, Aduthurai, India
- 5ICAR-National Institute for Plant Biotechnology, New Delhi, India
Uppermost internode length (UIL) and panicle exsertion are two important traits that contribute significantly to increased yield in rice. The present study was conducted using recombinant inbred lines (RILs) comprising 201 lines derived from RTN10B and IRG213. These RILs were evaluated at three different locations— Delhi, Karnal, and Aduthurai. We report the mapping of quantitative trait loci (QTLs) for uppermost internode length and the panicle exsertion ratio (PER), as well as QTLs for plant height (PH), tiller number, length of the second internode, length of the third internode, length from the flag leaf to the panicle tip (PFL), and panicle length (PL). Mapping was performed using a custom microsatellite linkage map constructed for the population, having a total span of 2077.1 cM with 104 markers. A total of 22 QTLs were identified for various traits, among which 19 were found distributed in four hotspots. A total of 11 major effect QTLs and 11 minor effect QTLs were identified for various internode length-related traits. Among the four QTLs identified for PFL, three QTLs—qPFL1.1, qPFL1.2, and qPFL2.1—co-localized with previously reported QTLs, while qPFL8.1 was a novel QTL. A major QTL hotspot was identified on chromosome 1, located at the marker interval of RM12055-RM320. The size of the major QTL hotspot on chromosome 1 was 1.4 Mb and contained a total of seven HEs for PER, PFL, PH, PL, UIL, SIL, and TIL. Based on the in-silico analysis of the major QTL hotspot on chromosome 1, we identified 15 putative candidate genes associated with internode length- and panicle-related traits. Furthermore, narrowing the genomic region through the fine mapping of the marker intervals facilitated the identification of candidate genes to enable more precise marker-assisted selection for uppermost internode length and panicle exsertion.
1 Introduction
Rice is one of the major staple foods for the majority of the people in Asia, ensuring food and nutritional security in India, which contributes 23% of global per capita energy and 16% of protein intake (Sonu et al., 2024; Singh et al., 2021). Despite India being the leading country in terms of rice cultivation area, when it comes to production and productivity, China is much ahead of India (Yang et al., 2021; Bi et al., 2024; Zhang et al., 2024). In fact, the large increase in rice productivity in China can largely be ascribed to the development and wide adoption of hybrid rice since the late 1970s. India has an area of 4 mha under hybrid seed production, which is far less than China and is one of the reasons for lower rice productivity in India (Sruthi et al., 2020; Shidenur et al., 2019; Kumar et al., 2017). Hybrid rice has high potential for increasing yield and productivity.
The wild abortive (WA) cytoplasm is the most commonly used male-sterile cytoplasm in rice hybrid seed production because of the availability of many fertility-restorer genes. However, a major hindrance is incomplete panicle exsertion (CPE) (Shidenur et al., 2020; Kumar et al., 2017). It prevents hybrid rice from pollinating normally, which lowers seed yield. IAA (Indole-3-acetic acid) levels in cytoplasmic male sterility (CMS) lines panicles have been shown to be low, which inhibits the gene’s ability to synthesize gibberellic acid (GA). Low amounts of bioactive GA lead to a decrease in the uppermost internode (UI) cell number and cell elongation (Yin et al., 2007). This leads to a reduced rate of outcrossing and reduced seed setting. Panicle exsertion and stem elongation depend on the level of internal GA production by the genotype. A lack of GA production may cause incomplete panicle exsertion. To address this issue, a large amount of external GA3 application is generally preferred to increase panicle exsertion. External application of GA3 increases the cost of cultivation and decreases the seed quality (Honnaiah, 2003). Previous studies have identified many genes for internode elongation, such as sd-1 (Monna et al., 2002), d18 and d35 (Itoh et al., 2004), d1 (Ueguchi-Tanaka et al., 2000), gid2 (Sasaki et al., 2003), gid1 (Ueguchi-Tanaka et al., 2005), and the SNORKEL1and SNORKEL2 genes (Hattori et al., 2009). However, the majority of these genes are targeted for the development of semi-dwarf and submergence-tolerant varieties. Therefore, genotypes with an elongated uppermost internode may offer a possible solution for incomplete panicle exsertion in CMS lines. Uppermost internode elongation is a genetically controlled trait. Several previous studies have investigated the genetics of this trait, and the majority of them reported one recessive gene responsible for uppermost internode elongation.
Indica/japonica-based hybrids are a good source of genetic variation for yield and related traits (Singh et al., 2021; Kallugudi et al., 2022). Many indica and japonica genotypes exhibit good genetic and phenotypic variations for uppermost internode elongation. In 1981, a japonica-based recessive tall spontaneous mutant genotype with an elongated uppermost internode was observed by Rutger and Carnahan. The identified mutant was named the eui mutant. Later on, one locus was mapped on chromosome 5 through trisomic analysis by Librojo and Khush (1986) and fine-mapped to a 98 kb region using a large F2 population (Xu et al., 2004). The dominant Eui gene encodes a GA3-deactivating enzyme, cytochrome P450 monooxygenase, which reduces panicle exsertion (Luo et al., 2006). A mutant recessive gene in this locus leads to complete panicle exsertion. In another study by Yang et al. (2000), two eui mutants were identified in an M2 segregating population, which were treated with gamma irradiation. Of the two mutants, one was allelic to the original eui mutant, while the other was found to be non-allelic and named eui2. Later on, Eui2 was mapped on chromosome number 10 by Yang et al. (2000). Bao-Jian et al. (2008), using the composite interval mapping method, mapped the quantitative trait loci (QTLs) for the uppermost internode length (UIL) in rice under various growing environments utilizing 98 backcross inbred lines (BC1F12 and BC1F13) from backcross of Nipponbare (japonica) / Kasalath (indica) // Nipponbare. Wang et al. (2017) located QTLs in japonica rice that were consistently associated with culm length (CL) and the first to fourth elongated internode lengths (EILs) from the panicle base. Zhao et al. (2021) studied panicle length (PL) and uppermost internode length (UIL) in 120 Cheongcheong/Nagdong doubled haploid populations and reported QTLs associated with internode length. To date, numerous gene(s)/QTLs have been identified for uppermost internode length and panicle exsertion-related traits, but none of them have been used for introgression in crop improvement programs due to the lack of effectiveness of the gene (Yang et al., 1999; Yang et al., 2000).
The success of linkage/QTL mapping depends on the selection of parental lines that exhibit significant phenotypic differences for the trait of interest (Nandakumar et al., 2024). This approach allows for the correlation of the segregating trait with allelic differences between two homozygous parents, which can be easily monitored in their offspring. Therefore, identifying genetic variation for uppermost internode elongation and introgressing it into widely used CMS lines can solve the problem of incomplete panicle exsertion. Developing hybrids based on the eui gene may produce high yield due to improved pollination and increased seed setting. It also reduces the cost of seed production by reducing the external application of GA3 during flowering.
Given this genetic problem, this study was conducted to map the genomic regions controlling uppermost internode elongation and panicle exsertion. This study’s findings may help identify the best genotype with complete panicle exsertion, and the identified QTLs can further be incorporated into elite restorer, maintainer, and CMS lines to enhance exsertion, ultimately boosting hybrid rice yield.
2 Materials and methods
2.1 Plant materials and development of the mapping population
To map the QTLs for uppermost internode length and panicle exsertion, IRG-213 and RTN10B were crossed to create a set of 201 recombinant inbred lines (RILs). The male parent, IRG 213, is a line from the International Rice Research Institute (IRRI) 3 K panel, which is characterized by a longer uppermost internode, complete panicle exsertion, and relatively greater height. In contrast, the female parent, RTN10B, is comparatively dwarf, with a short uppermost internode and incomplete panicle exsertion.
The hybridization of the parents, RTN10B (female) and IRG213 (male), was carried out during the Kharif season of 2018 at the Division of Genetics, ICAR-Indian Agricultural Research Institute (ICAR-IARI), New Delhi. A total of 37 F1 plants were generated and were further grown during the Rabi season of 2018–19 at ICAR-IARI-Rice Breeding & Genetics Research Centre (RBGRC), Aduturai, Tamil Nadu. All 37 F1 plants were phenotyped for uppermost internode length, and hybridity was checked using RM1344. A single true hybrid plant exhibiting phenotypic resemblance of both parents was selected to generate the F2 population. Subsequent filial generations were developed using the single-seed descent method. No selection for any of the superior or inferior traits was made during the process of mapping population development.
By Kharif 2021, a total of 201 RILs in the F6 generation were raised along with the parental lines and evaluated at different locations using an augmented design. The recommended package of practice was adopted for all locations and for all seasons. A Flow chart illustrating the development of the mapping population is presented in Figure 1.
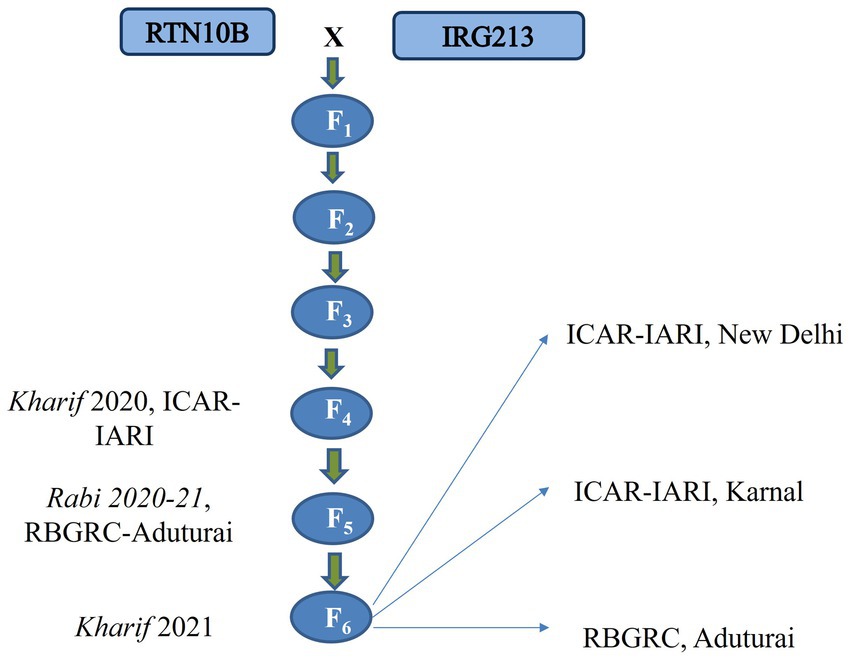
Figure 1. A flow chart depicting the development of the mapping population.
2.2 Multilocation evaluation of the RILs
The RILs were evaluated across three locations in India— New Delhi (DEL), Karnal (KAR), and Aduturai (ADT). The experimental location in DEL was at the Division of Genetics, ICAR-IARI, New Delhi (28 °38’ N; 77 °09′ E; 220 m). In KAR, the experiment was conducted at the research farm of ICAR-IARI Regional Station, Karnal, Haryana (29 °42’ N; 76 °59′ E; 249 m), and in ADT, the experiment was conducted at the research farm of IARI-RBGRC, Aduturai, Tamil Nadu (11 °00’N; 79 °28′ E; 20 m). In DEL and KAR, the soil types were sandy loam, whereas in ADT, it was alluvial clay. At all sites, the experiment was conducted under irrigated transplanted conditions, and the nursery was raised on elevated beds for 21 days before being transplanted with a spacing of 20 × 15 cm into a well-puddled field. The experiments were conducted using an augmented randomized complete block (ARCB) design with four checks: IRG213, RTN10B, EUI, and Pusa 6B. The test genotypes were randomly divided into eight blocks, with four checks replicated in all blocks using the randomization function in PBTools v1.4 (International Rice Research Institute, 2014). The data recorded in the replicated entries (checks) will be used to estimate the blocking effect on the test genotypes. The block effect from all the blocks will be used to generate the adjusted mean of the unreplicated entries.
2.3 Phenotyping
Five plants from each family were randomly selected at physiological maturity, and data were collected on the targeted traits. Plant height (PH), number of tillers (TN), panicle length (PL), length from the uppermost node to the panicle base (UIL), length from the flag leaf collar to the panicle tip (PFL), and length of the top internodes were recorded. The panicle exsertion ratio (PER) was calculated as the ratio of PFL and PL. For subsequent data analysis, the average of the five plants from each family was taken into account. To minimize errors, border plants were excluded.
2.4 Parental polymorphism and linkage map construction
Leaf samples were collected from young leaves, and the CTAB method was used to isolate genomic DNA (Archana et al., 2023). SSR genotyping was performed using PCR after DNA extraction, and the amplicons were resolved by utilizing 3.5% agarose gel electrophoresis. The resolved PCR products were classified as IRG213 type (P1), RTN10B type (P2), and heterozygotes and scored as A, B, and H, respectively. In genotypic data, all monomorphic data and the markers that exhibited segregation distortion were removed. By performing a multipoint analysis using the Kosambi mapping function (Kosambi, 1944; Vinod, 2011) and a logarithm of odds (LOD) value of 3.0 (Lander et al., 1987), a linkage map was created using the QTL IciMapping program V4.2 (Meng et al., 2015). Threshold LOD values for all traits were determined at an alpha value of 0.05 using a permutation test with 1,000 iterations. Centimorgans (cM) were used to measure the map distance.
2.5 Statistical analysis and QTL mapping
The data for plant height (PH), number of tillers (TN), panicle length (PL), length from the uppermost node to the panicle base (UIL), length from the panicle tip to the flag leaf collar (PFL), and length of the top internodes were subjected to analysis of variance (ANOVA) both individually and across different locations using PBTools v.1.4 (International Rice Research Institute, 2014). The software PBTools v1.4 (International Rice Research Institute, 2014) includes the lme4 package, which was used to generate the best linear unbiased predictors (BLUPs) using the restricted maximum likelihood (REML) approach. A histogram was created in the R environment using the rcompanion package. Genetic advance was calculated using the TraitStats package in the R environment. The correlation coefficient among the traits was calculated using the corr function in R and graphically visualized using corrplot. Principal component analysis of the uppermost internode length and panicle exsertion-related traits in rice was performed using an R base, and principal components were selected based on the eigenvalue. All R-based analyses were performed with R Studio v2023.03.1 Build 446 (Posit software). The contribution of the different traits to the principal components was graphically visualized using Circos Table Viewer v0.63–10 (Krzywinski et al., 2009). Using one-dimensional composite interval mapping (Li et al., 2007; Wang, 2009), the QTLs were mapped for all traits and the strategy for additive traits was implemented in QTL IciMapping V4.2. Threshold LOD values for each trait were determined at an alpha value of 0.05 using a permutation test with 1,000 iterations. The walk distance was set to 1 cM, with a 0.01 probability of inclusion for forward stepwise regression, using GGT v2.0 software (Van Berloo, 1999).
2.6 In-silico analysis of the consistent QTLs
The marker interval containing consistent QTLs across locations was selected to look for the presence of putative candidate genes. The Rice Annotation Project Database (RAP-DB) was used to retrieve likely expressed genes located between the marker sites. Based on prior findings, annotated candidate genes already known to function with the target traits were selected.
3 Results
3.1 Phenotypic evaluation of the recombinant inbred lines (RILs)
The site-wise analysis of variance (ANOVA) of the phenotypic data for all traits collected from the RIL population comprising 201 genotypes showed significant variations in all the locations—DEL, KAR, and ADT (Supplementary Table 1). The phenotypic variation for panicle length and panicle exsertion among the RILs, along with the parents, is shown in Figure 2. The pooled ANOVA showed that the GxE interaction (GEI) was highly significant for all traits in all sites (p ≤ 0.01, Supplementary Table 2). The lowest value for PH (69.56 cm) was observed in Karnal, whereas the highest value (191.01 cm) was recorded in Delhi. Similarly, for the trait TN, the lowest value (2.45) was observed in Aduthurai and the highest value (25.35) was observed in Delhi. The lowest UIL1 (22.55 cm) value was recorded in Karnal, whereas the highest value (56.20 cm) was observed in Delhi. For the trait UIL2, the lowest value (9.63 cm) was observed in Karnal, while the highest value (40.30 cm) was recorded in Aduthurai. The value for UIL3 was found to be the lowest (6.32 cm) in Karnal and the highest (36.22 cm) in Delhi, whereas the PFL value was the lowest (13.26 cm) in Delhi and the highest (41.80 cm) in Karnal. The PL values were found to be the lowest (14.34 cm) in Delhi and the highest (32.88 cm) in Karnal, whereas the PER (%) value was the lowest (70.63%) in Delhi and the highest (153.47%) in Karnal (Table 1). The mean performance of the RILs across the locations for all traits is presented in Supplementary Table 3. The frequency distribution histogram and violin plots of the RILs indicated that all traits were normally distributed within the population, thereby confirming the presence of a quantitative inheritance pattern (Figure 3 and Supplementary Figures 1A–C).

Figure 2. Phenotypic variation for panicle length and panicle exsertion among the RILs during Kharif 2021 at ICAR-IARI.
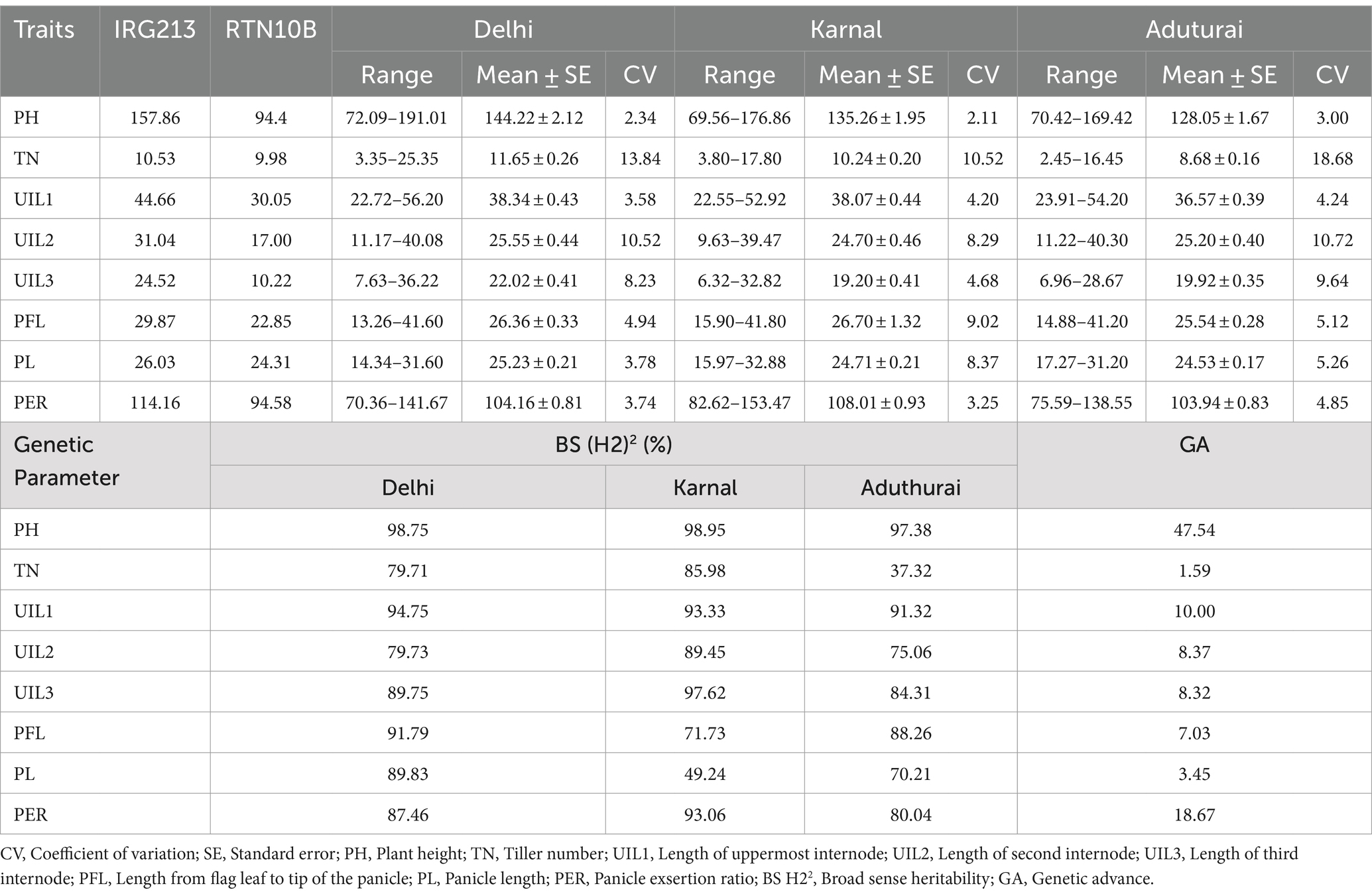
Table 1. Phenotypic evaluation of the recombinant inbred line (RIL) population of ‘IRG213/RTN10B’ across the locations.
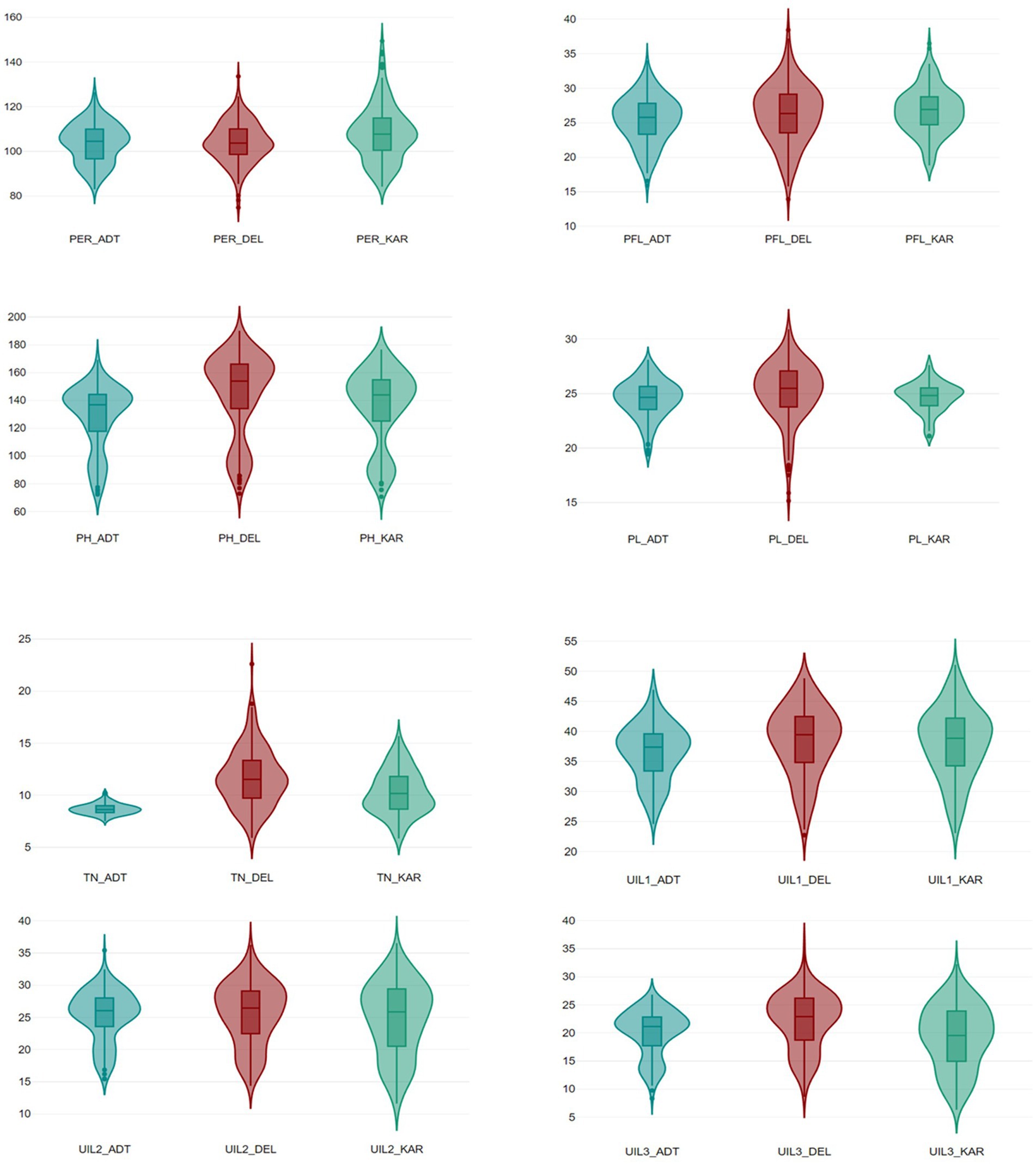
Figure 3. A box plot depicting the phenotypic distribution of PH (Plant height), TN (Tiller number), UIL1 (Length of the uppermost internode), UIL2 (Length of the second internode), UIL3 (Length of the third internode), PFL (Length from the flag leaf base to the panicle tip), PL (Panicle length), and the PER (Panicle exsertion ratio) across the three locations during Kharif 2021.
Among the eight traits studied in Delhi, the highest CV value was observed for TN and the lowest for PH. Similarly, in Karnal, the highest CV value was observed for TN and the lowest for PH. In Aduthurai, the highest CV value was observed for TN and the lowest for PH. High broad-sense heritability was observed for all traits with a range of 37.32–94.75%. The highest heritability was observed for UIL1 in Delhi, whereas the lowest was observed in Aduthurai. The PER showed the highest heritability in Karnal and the lowest in Aduthurai. Higher genetic advance was observed for PH, UIL1, UIL2, UIL3, PFL and PER, FGN, and UFG, whereas the lowest was observed for TN and PL, (Table 1).
3.2 Correlation among the traits
A correlation plot was generated to identify the traits positively contributing to uppermost internode length and panicle exsertion. The correlation coefficients among the traits showed that UIL1, PFL, and UIL2 were positively correlated with the PER (Figure 4). The principal component analysis revealed four major principal components that explain a cumulative variance of 88.41%. The major contributors to the total variance in PC1 was UIL1, followed by PFL, UIL2, PER, and UIL3. Similarly, in PC2, the major contributors were TN, PL, and the PER (Supplementary Table 4). The data visualization using Circos Table Viewer v0.63–10 illustrates the contribution of the various traits on the major principal components (Figure 5). Among the eight traits studied, UIL1 was the most variable trait, followed by UIL3, UIL2, PH, PFL, and the PER (Supplementary Figure 2).

Figure 4. Correlation coefficient among the uppermost internode length and panicle exsertion-related traits using combined BLUPs of Kharif 2021.
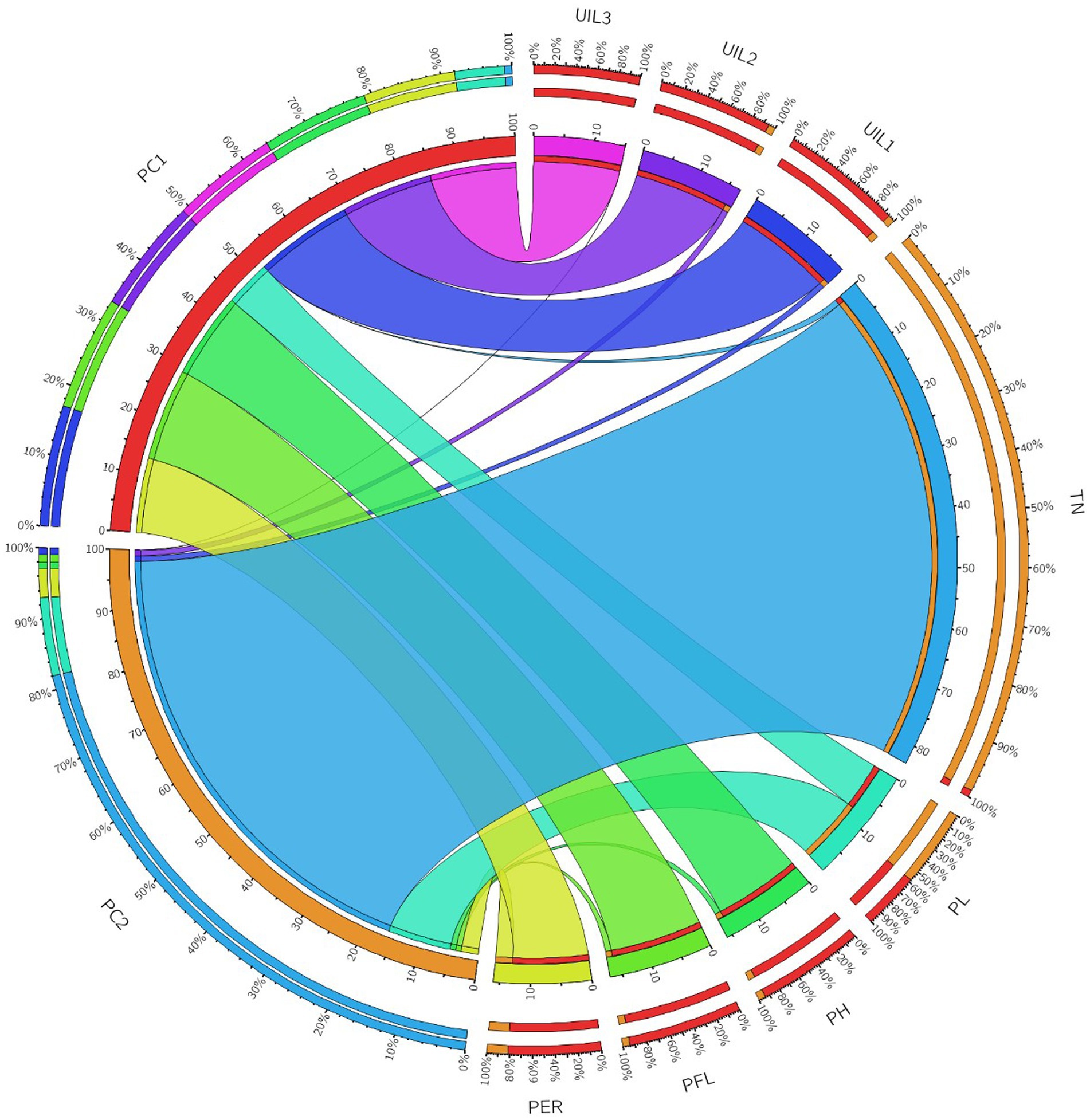
Figure 5. A Circos diagram depicting the contribution of the different traits to the principal components using combined BLUPs during Kharif 2021.
3.3 Construction of the linkage map
A parental polymorphism survey was conducted between IRG213 and RTN10B using 1,108 SSR markers, with an average density of 92.33 markers per chromosome. A total of 130 markers were found to be polymorphic. Of these, 26 markers were rejected from the study as they exhibited segregation distortion. The remaining 104 polymorphic markers were used to construct the linkage map, and these markers represented a genome diversity of 9.38%. The list of the polymorphic markers, physical positions, and sequence information is presented in Supplementary Table 5. The markers were uniformly distributed across the 12 chromosomes, and a graphical representation of the polymorphic markers is shown in Supplementary Figure 3. On linkage mapping, the markers covered a cumulative map distance of 2077.1 cM, with an average distance of 19.97 cM. The highest number of the polymorphic markers was found on chromosome 1, and the lowest number was found on chromosomes 7 and 9. Marker distribution per chromosome is presented in Supplementary Table 6.
3.4 QTL analysis
QTL analysis using the 104 SSR markers identified a total of 22 major and minor QTLs distributed across the various chromosomes for all traits, based on percentage variance explained (PVE) (%) (Phenotypic variation explained) values. Of the 22 QTLs identified in the current study, 11 were classified as major QTLs and the remaining 11 were classified as minor QTLs. A total of 16 QTLs were identified in Delhi, 11 in Karnal, and 15 in Aduthurai. A few QTLs were consistent across more than one location, including seven QTLs that were present in all locations and six QTLs that were present in two locations. The remaining QTLs were specific to each individual site. The LOD scores for these QTLs ranged from 2.57 to 40.44, whereas the phenotypic variance ranged from 10.20 to 26.68% for the major QTLs and from 1.44 to 5.63% for the minor QTLs. The details of the QTLs, marker interval, LOD, and PVE (%) are shown in Table 2. The QTLs identified for the different traits are graphically represented in Figure 6.
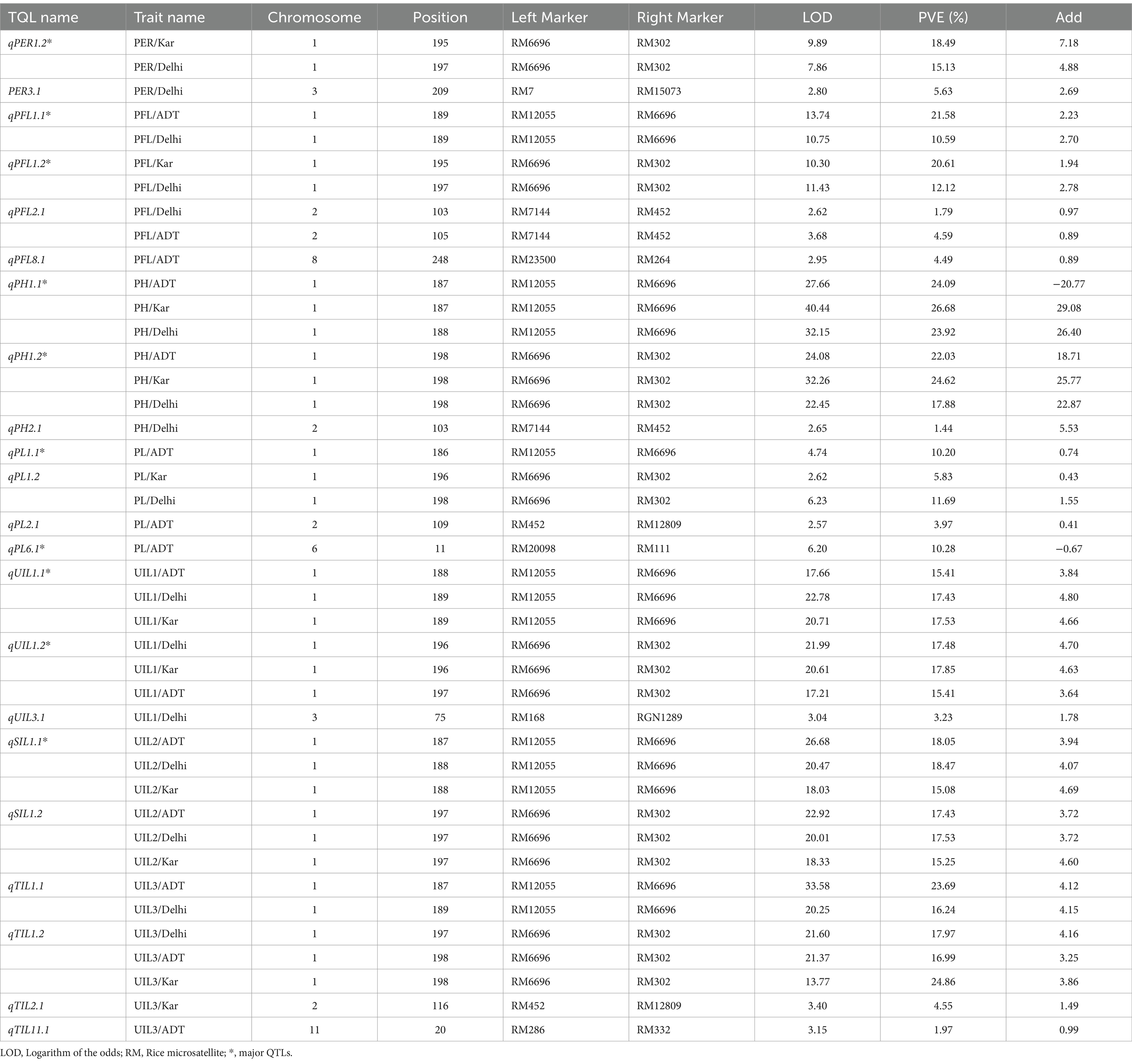
Table 2. QTLs mapped for uppermost internode length and panicle exsertion-related traits in the RIL population of ‘IRG213/RTN10B’.
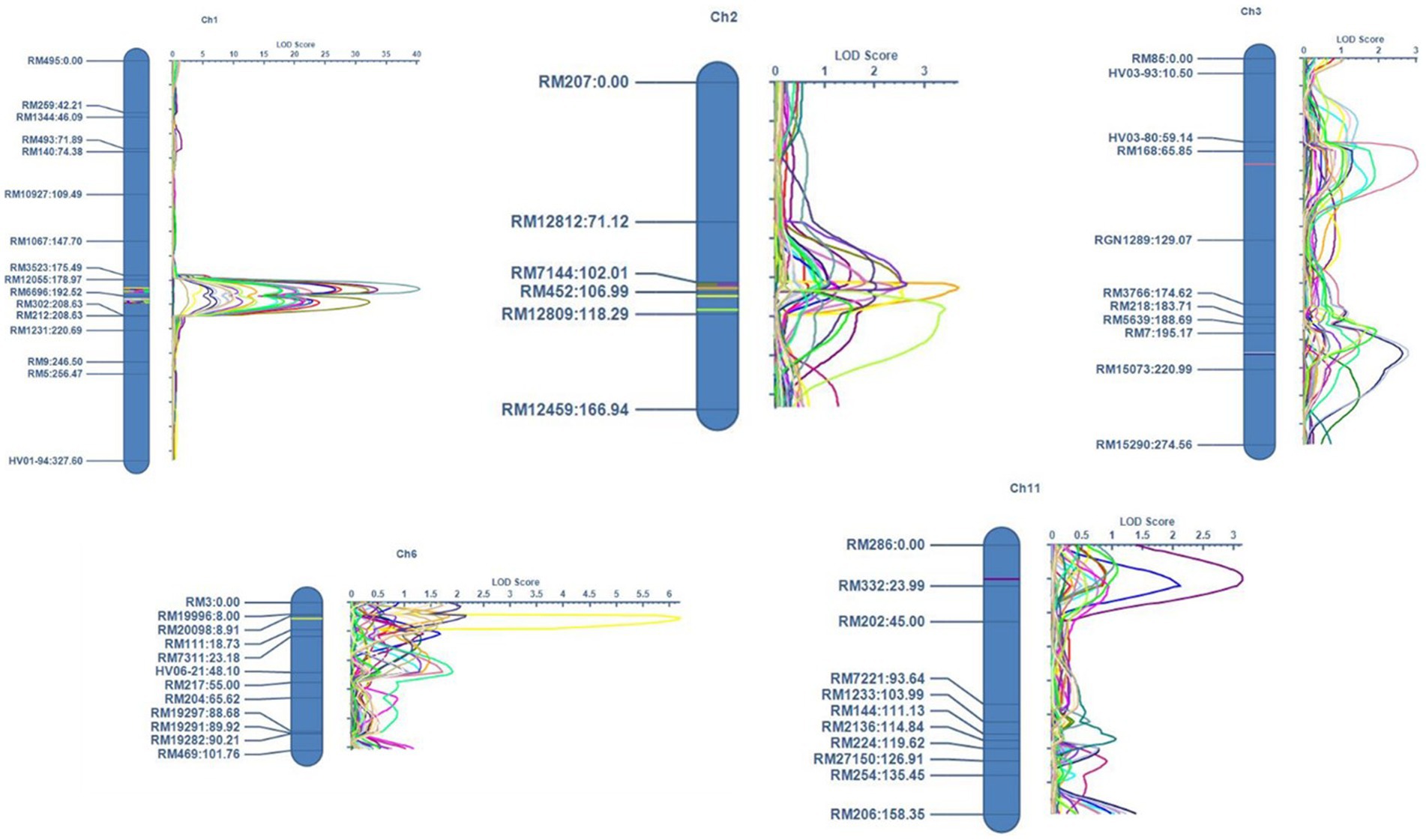
Figure 6. Map positions of the QTLs mapped for PH (Plant height), TN (Tiller number), UIL1 (Length of the uppermost internode), UIL2 (Length of the second internode), UIL3 (Length of the third internode), PFL (Length from the flag leaf to the tip of the panicle), PL (Panicle length), and the PER (Panicle exsertion ratio) in the ‘IRG213’/‘RTN10B’ population across all three location during the Kharif season of 2021.
3.4.1 Length of the uppermost internode (UIL1)
Three quantitative trait loci (QTLs) for uppermost internode length (UIL1) were identified: qUIL1.1, qUIL1.2, and qUIL3.1, located on chromosomes 1 and 3. Among these, qUIL1.1 was a stable major QTL mapped in Delhi, Karnal, and Aduthurai, situated between the markers RM12055 and RM6696, with LOD values ranging from 17.66 to 22.78. The PVE and additive effects were 15.41% and 3.84, respectively, in Aduthurai, 17.43% and 4.80, respectively, in Delhi, and 17.53% and 4.66, respectively, in Karnal. The second major QTL, qUIL1.2, was consistently observed across all locations, located between RM6696 and RM302, with LOD values ranging from 17.21 to 21.99, PVE values of 17.48, 17.85, and 15.41%, and additive effects of 4.70, 4.63, and 3.64 at the respective sites. The minor QTL, qUIL3.1, located between RM168 and RGN1289, had an LOD value of 3.04, a PVE value of 3.23%, and an additive effect of 1.78, all indicating a positive contribution from the allele derived from ‘IRG213’ (Table 2).
3.4.2 Length of the second internode (UIL2)
Two QTLs were identified for UIL2—qSIL1.1 and qSIL1.2—on chromosome 1. qSIL1.1 was flanked by the marker interval of RM12055 and RM6696, with the LOD values ranging from 18.03 to 26.68. The QTL had a PVE value and additive effect of 18.05% and 3.94 in Aduthurai, 18.47% and 4.07 in Delhi, and 15.08% and 4.69 in Karnal. The other QTL, qSIL1.2, was located at the genomic region flanked by the marker interval of RM6696 and RM302. The PVE values of 17.43, 17.53, and 15.25% were recorded in Aduthurai, Delhi, and Karnal, respectively. The QTL had an additive effect of 3.72, 3.72, and 4.6 in Aduthurai, Delhi, and Karnal, respectively. The two reported QTLs for UIL2 showed a positive sign for the additive effect, indicating a contribution from the positive alleles of ‘IRG213’ (Table 2).
3.4.3 Length of the third internode (UIL3)
The QTL analysis for uppermost internode length (UIL3) identified two major QTLs (qTIL1.1 and qTIL1.2) and two minor QTLs (qTIL2.1 and qTIL11.1). The major QTL, qTIL1.1, was located on chromosome 1 between the markers RM12055 and RM6696, and it was mapped at two locations, Aduthurai and Delhi. This QTL exhibited logarithm of odds (LOD) values ranging from 20.25 to 33.58, with percentage variance explained (PVE) of 23.69% in Aduthurai and 16.24% in Delhi, along with additive effects of 4.12 and 4.15, respectively. The second major QTL, qTIL1.2, also located on chromosome 1, demonstrated consistent performance across all three locations, with LOD values ranging from 13.77 to 21.60. Its PVE and additive effects were 17.97% (4.16) in Delhi, 16.99% (3.25) in Aduthurai, and 24.86% (3.86) in Karnal. The minor QTLs qTIL2.1, located on chromosome 2, and qTIL11.1, located on chromosome 11, were located in Karnal and Aduthurai, respectively, showing LOD values of 3.40 (PVE 4.55%, additive effect 1.49) and 3.15 (PVE 1.97%, additive effect 0.99). All four QTLs indicated a positive contribution from the alleles derived from “IRG213” (Table 2).
3.4.4 Plant height (PH)
Three QTLs were reported for plant height, namely qPH1.1, qPH1.2, and qPH2.1, on chromosomes 1 and 2. The QTL qPH1.1 showed consistent performance across all three locations, with LOD values ranging from 27.66 to 40.44, and was mapped in the genomic region flanked by the markers RM12055 and RM6696. The QTL had a PVE value and additive effect of 24.09% and 20.77, respectively, in Aduthurai, 26.68% and 29.08, respectively, in Karnal, and 23.92% and 26.40, respectively, in Delhi. Similarly, the more major QTL qPH1.2 mapped at the genomic region between the markers RM6696 and RM302 showed consistent performance across all three locations. The QTL showed LOD values ranging from 22.45 to 32.26, with a PVE value and additive effect of 22.03% and 18.71, respectively, in Aduthurai, 24.62% and 25.77, respectively, in Karnal, and 17.88% and 22.87, respectively, in Delhi. The minor QTL, qPH2.1, was mapped to the genomic region flanked by RM7144 and RM452 in Delhi, with LOD, PVE, and additive effect values of 2.65, 1.44%, and 5.53, respectively. All QTLs reported for plant height exhibited a positive sign for the additive effect, thereby indicating a contribution from the positive alleles of “IRG213” (Table 2).
3.4.5 Length from the flag leaf collar to the panicle tip (PFL)
Four quantitative trait loci (QTLs) were identified for plant flowering length (PFL), namely qPFL1.1, qPFL1.2, qPFL2.1, and qPFL8.1, located on chromosomes 1, 2, and 8. The major QTL, qPFL1.1, demonstrated consistent performance in Karnal and Delhi, with logarithm of odds (LOD) values ranging from 10.75 to 13.74. It was positioned in the genomic region flanked by the markers RM12055 and RM6696, showing percentage of variance explained (PVE) of 21.58% in Karnal and 10.59% in Delhi, along with additive effects of 2.23 and 2.70, respectively. Similarly, qPFL1.2 was located between RM6696 and RM302, exhibiting LOD values of 10.30 to 11.43 and a PVE value of 20.61% (1.96) in Karnal and 12.12% (2.78) in Delhi. The minor QTL, qPFL2.1, mapped between RM7144 and RM452, showed LOD values ranging from 2.62 to 3.68, with a PVE value of 1.79% (0.97) in Delhi and 4.49% (0.89) in Karnal. The other minor QTL, qPFL8.1, mapped in Aduthurai, showed LOD, PVE, and additive effect values of 2.95, 4.49%, and − 0.89, respectively. Among these, three QTLs exhibited positive additive effects, indicating a contribution from the positive alleles of ‘IRG213’, while qPFL8.1 displayed a negative sign, suggesting contributions from “RTN10B” (Table 2).
3.4.6 Panicle length (PL)
The QTL analysis revealed four QTLs for panicle length, namely qPL1.1, qPL1.2, qPL2.1, and qPL6.1, on chromosomes 1, 2, and 6. qPL1.1 was mapped in the genomic region flanked by the markers RM12055 and RM6696, with LOD, PVE, and additive effect values of 4.74, 10.20%, and 0.74, respectively. The QTL, qPL1.2, showed consistent performance at two locations, namely Karnal and Delhi, and was mapped between the marker intervals of RM6696 and RM302, with LOD values ranging from 2.62 to 6.23. The QTL had a PVE and additive effect value of 5.83% and 0.43, respectively, in Karnal and 11.69% and 1.55, respectively, in Delhi. Similarly, two more QTLs, qPL2.1 and qPL6.1, mapped in Aduthurai, showed LOD values of 2.57 and 6.20, respectively. The genomic region flanked by RM452 and RM12809 contained the minor QTL qPL2.1, which showed a PVE value of 3.97% and an additive effect of 0.41. qPL6.1 showed a PVE value of 10.28%, an additive effect of −0.67, and was located in the marker interval between RM20098 and RM111. Of the four QTLs reported for panicle length, three QTLs showed a positive sign for the additive effect, thus indicating a contribution from the positive alleles of ‘IRG213’ and one QTL qPFL6.1 showed a negative sign, indicating a contribution from “RTN10B” (Table 2).
3.4.7 Panicle exsertion ratio (PER)
Two major and one minor QTLs were reported for PER, namely qPER1.1, qPER1.2, and qPER3.1, on chromosomes 1 and 3. qPER1.1 was mapped in Aduthurai on the genomic region flanked by the markers RM12055 and RM6696 and showed LOD, PVE, and additive effect values of 9.45, 18.10%, and 5.52, respectively. The genomic region flanked by the markers RM6696 and RM302 contained the major QTL qPER1.2, which showed consistent performance at two locations, namely Karnal and Delhi. The QTL had LOD, PVE, and additive effect values of 9.89, 18.10%, and 5.52, respectively, in Karnal and 9.89, 18.49%, and 7.18, respectively, in Delhi. The minor QTL qPER3.1 was mapped in Delhi on the marker intervals between RM7 and RM15073. The QTL showed LOD, PVE, and additive effect values of 2.80, 5.63%, and 2.69, respectively. The QTLs showed a positive sign for the additive effect, thus indicating a contribution from the positive alleles of ‘IRG213’ (Table 2).
3.5 QTL hotspots
Among the 22 QTLs mapped for the different traits, 19 QTLs were located in four QTL hotspots on chromosomes 1, 2, and 3 (Table 3). The first QTL hotspot was found to contain seven QTLs— qPER1.1, qPFL1.1, qPH1.1, qPL1.1, qUIL1.1, qSIL1.1, and qTIL1.1— (Cluster I) located on the long arm of chromosome 1. This QTL was flanked by the markers RM12055 andRM6696 and had an interval length of 1.40 Mb. Similarly, the QTL hotspot on the long arm of chromosome 1 contained seven QTLs—qPER1.2, qPFL1.2, qPH1.2, qPL1.2, qUIL1.2, qSIL1.2, and qTIL1.2—spanning 5.24 Mb and flanked by the markers RM6696 and RM302 (Cluster II). The QTL hotspot on the short arm of chromosome 2 contained three QTLs, namely qPH2.1, qPL2.1, and qTIL2.1 (Cluster III), spanning across a 1.80 Mb region between the markers RM452 and RM12809. The hotspot on the long arm of chromosome 3 contained two QTLs, qPER3.1 and qUIL3.1 (Cluster IV), and was flanked by the markers RM7 and RM15073, spanning 5.48 Mb.
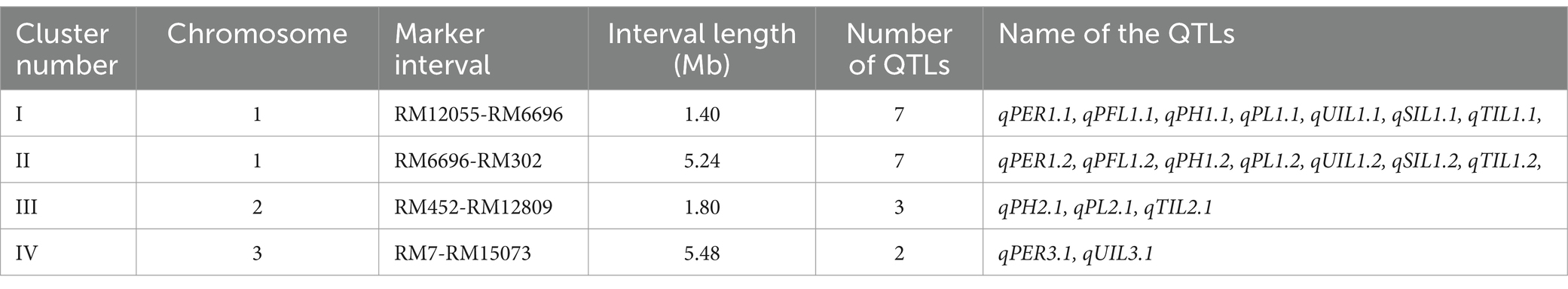
Table 3. QTL hotspots identified in the IRG213 x RTN10B derived RILs during Kharif 2021.
3.6 In-silico analysis
The investigation for the presence of putative candidate genes in the major QTL hotspots (1.40 Mb and 5.24 Mb) identified on chromosome 1 revealed that this region comprised 916 annotated gene models. Among the 916 annotated gene models, 15 genes were putative candidates that may play a key role in the regulation of plant height, internode length, and panicle exsertion. The list of the putative candidate genes for internode length and panicle exsertion is presented in Table 3. Two of these putative genes encoded cytochrome P450, two encoded MYB family transcription factors, two encoded homeobox domain-containing proteins, and one gene each encoded a zinc finger protein, WRKY24, a C3HC4-type domain-containing protein, a ZOS1-16- C2H2 zinc finger protein, gibberellin 20 oxidase 2, cytochrome c, a cyclin-related protein, oligo-peptidase, CXE carboxylesterase, and a chitin-inducible gibberellin-responsive protein (Table 4).
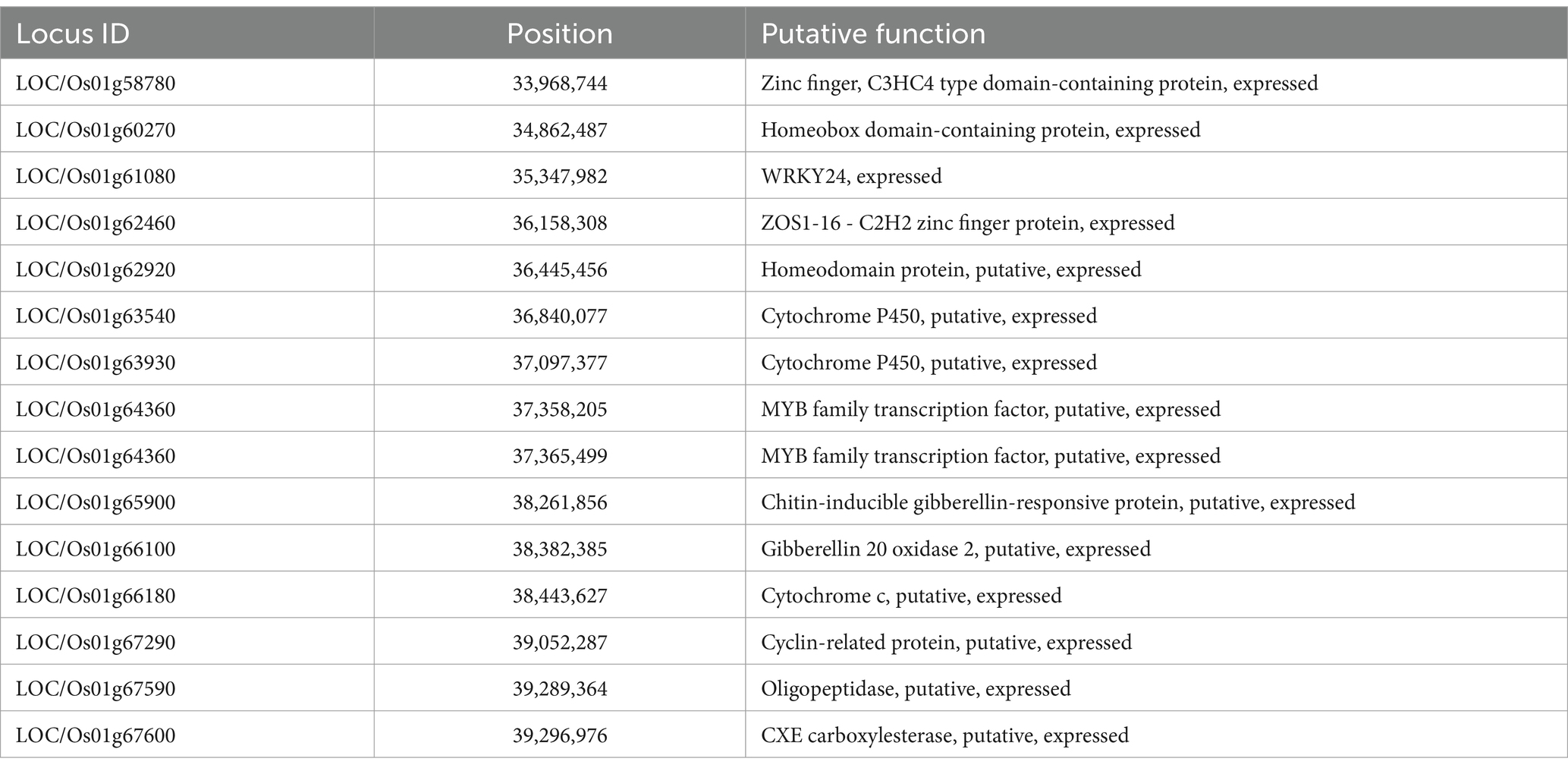
Table 4. Predicted gene models in the QTL hotspot on chromosome 1.
4 Discussion
Incomplete panicle exsertion in wild abortive (WA)-based cytoplasmic male sterility (CMS) lines is one of the major limitations of hybrid rice seed production and seed yield (Priyadarshi et al., 2017; Mohanty and Parida, 2024). Panicle enclosure within the flag leaf sheath leads to a low level of outcrossing and reduced seed setting (Hashim et al., 2021). Although the application of GA3 enhances panicle exsertion by increasing cell elongation, it adds additional cost to seed production (Honnaiah, 2003; Yasir et al., 2022; Karunakaran et al., 2024). To overcome this problem, one can focus on improving panicle exsertion by increasing uppermost internode length. Targeting genes such as AP2/ERF and F-box proteins can enhance panicle exsertion (Ballichatla et al., 2024). Similarly, elongation of uppermost internode length causes the panicle to be exserted from the flag leaf (Dunand and Saichuk, 2014; Gao et al., 2016). Several reports have identified the recessive mutant eui1 type, which controls the tall phenotype in rice and is referred to as the fourth genetic element of hybrid rice. Plants with eui1 have an elongated uppermost internode, which is twice the length of that in normal wild types. It has been reported that it does not influence any other traits (Rutger and Carnahan, 1981; Rutger, 1992). EUI mutants in the segregating M2 population obtained by gamma-ray treatment were identified. One was allelic to the original EUI mutant, while another one was found to be non-allelic and was named eui2 (Yang et al., 1999). Up to this point, neither the CMS line nor the restorer line has been successfully developed with the incorporation of the eui gene, primarily because the gene has shown limited effectiveness (Yang et al., 2002). Therefore, it is necessary to identify effective eui gene(s) or QTLs and understand their mode of action, which can then be used in crop improvement programs to increase the yield of hybrid seed production.
Considering the problem of panicle exertion in CMS lines, the present study revealed significant differences in all eight evaluated traits between the parental lines, IRG213 and RTN10B. IRG 213, a rice line from the IRRI 3 K panel, generally exhibited higher trait values compared to RTN10B, with notable disparities in plant height (PH), total number of tillers (TN), uppermost internode length (UIL1, UIL2, and UIL3), length from the flag leaf to the tip of the panicle (PFL), panicle length (PL), and the panicle exsertion ratio (PER).
Furthermore, we observed that the distribution of the traits in the bi-parental population of IRG213/RTN10B in the F6 generation was stable and showed a continuous and mainly quantitative distribution, providing an ideal condition for QTL mapping (Zhao et al., 2016). The PH distribution showed two peaks, which could be due to genetic variation (e.g., major loci creating distinct height classes), environmental influences, or GEI. The pooled ANOVA showed that the G X E interaction (GEI) was highly significant for all the traits across the locations. However, relatively high broad-sense heritability was observed for the traits such as uppermost internode length, panicle length, length from the flag leaf to the tip of the panicle, and the panicle exsertion ratio, indicating that these traits are more receptive to transmission through breeding (Lingaiah et al., 2015). The site-specific variations are determined to be caused by factors such as soil type, fertility, climate, seasons, and moisture levels (Xu et al., 2016). The site-wise analysis revealed that all the traits assessed in the RILs showed significant differences across the various sites. Among the sites tested in this study, DEL supported the best mean performance of RILs for all the traits, with the lowest performance in ADT.
The spread of polymorphic markers across the genome reflects genetic diversity between the parents and among the RIL population. Notably, chromosome 1 had the highest number of polymorphic markers, highlighting its genetic diversity, while chromosomes 7 and 9 had the lowest number of markers. This distribution of the markers across the chromosomes provided a foundation for the subsequent quantitative trait locus (QTL) analysis. Of the 22 QTLs mapped in this study, 19 were located on chromosomes 1, 2, and 3, while the remaining three were found to be localized at four different hotspots. The clustering of QTLs at various hotspot regions suggests that certain regions are particularly rich in genes significant for the expression of uppermost internode length and panicle exsertion-related traits. Among the hotspots, the highest number of QTLs (seven) were co-mapped on the long arm of chromosome 1 (Cluster I) within a 1.40 Mb region between the markers RM12055 and RM6696. It was interesting to observe that the most desirable trait-contributing QTLs for the PER, PFL, PH, PL, UIL1, UIL2, and UIL3 originated from IRG213. This indicates that the hotspot on chromosome 1 sponsors longer uppermost internode length with complete panicle exsertion. This hotspot region has been reported to contain gene/QTLs for UIL1, UIL2, PL, and the PER in previous reports (Zhu et al., 2011: Zhao et al., 2016; Zhan et al., 2019).
Another cluster on the same chromosome also harbored the desirable QTLs contributed by IRG213. The QTLs identified in this cluster co-localize with those reported in earlier studies, which are associated with PL, UIL1, and PH (Wang et al., 2012; Zhan et al., 2019; Sitoe et al., 2022). The remaining two hotspots contained five QTLs. The hotspot on chromosome 3 (Cluster III) harbored the QTLs for PH, PL,and UIL3, spanning a region of 1.80 M b. The QTL for PL in this hotspot was also reported by Tan et al. (2011). The remaining hotspot on chromosome 2 (Cluster IV) contained the QTLs for the PER and UIL3, spanning a region of 5.48 Mb. The QTLs identified for panicle exsertion co-localize with those reported by Bao-Jian et al. (2008), for PE. Among the four QTLs mapped for PFL, namely qPFL1.1, qPFL1.2, qPFL2.1, and qPFL8.1, only qPFL8.1 was a novel QTL. The QTL, qPFL8.1, was mapped on the long arm of chromosome 8, spanning a region 1.07 Mb. The identification of the novel QTL for PFL opens the possibility for its fine mapping and the identification of candidate genes and their associated functions in PFL. Although the study identified QTLs for the correlated traits such as UIL1, UIL2, UIL3, PFL, PH, and PL on different chromosomes, the genomic regions are quite large. Two hotspots on chromosome 1 spanned 1.40 Mb and 5.24 Mb, while on chromosome 2, the hotspots spanned 1.80 Mb, and on chromosome 3, they spanned 5.48 Mb. The inclusive composite interval was able to map all the QTLs for these traits between 186 cM and 189 cM in cluster 1 and between 195 cM and 198 cM on chromosome 1. Although the linkage distance between the flanking markers is small, further research is needed to identify the physical position of the QTLs and to distinguish the QTLs within the genomic region for the precise transfer of useful QTLs without linkage drag. Similarly, on chromosome 1, two clusters on either side of the marker RM6696 appear to be the same. However, to determine whether both clusters are different or the same, high-density linkage through fine-mapping of the region may be required.
Breakthroughs in rice genome sequencing and gene annotation have paved the way for identifying and studying putative candidate genes within any marker interval and deciphering their functions (Saito et al., 2004). The significant QTL hotspot in the current study harbored positive QTLs contributed by IRG213. Based on an extensive search of the available literature, annotated databases, and an examination of the functional attributes of the 916 annotated candidate genes, 15 were found to play a direct role in internode length and panicle exsertion. The shortlisted putative candidate genes were directly involved in the regulation of internode elongation and included cytochrome P450, MYB family transcription factors, homeobox domain-containing proteins, zinc finger proteins, WRKY24, C3HC4-type domain-containing proteins, ZOS1-16 - C2H2 zinc finger proteins, gibberellin 20 oxidase 2, cytochrome c, cyclin-related proteins, oligo-peptidase, CXE carboxylesterase, AML1, and chitin-inducible gibberellin-responsive proteins. Cytochrome P450 and gibberellin 20 oxidase-2 are very important genes for regulating uppermost internode length in rice. It has been reported that the tall recessive rice mutant eui contains high levels of bioactive GAs in the uppermost internode and that the Eui gene encodes a cytochrome P450 enzyme that deactivates GAs through GA 16α, 17-epoxidation (Zhu et al., 2006; Zhao et al., 2021). Gibberellin (GA) 20-oxidase (GA20ox2) is a key enzyme that generally catalyzes the penultimate step in GA production. OsGA20ox2 (SD1) is well known as the ‘Green Revolution gene,’ and a loss-of-function mutation in this locus causes semi-dwarfism and plays an important role in plant stature (Oikawa et al., 2004). Several previous studies have indicated that gibberellin-mediated regulation in plants plays an important role in cell elongation of uppermost internode length in rice (Luo et al., 2006; Zhan et al., 2019; Zhao et al., 2021). There are also reports of other genes involved in regulating uppermost internode length and panicle exsertion-related traits, such as cytochrome P450 and gibberellin 20 oxidase2 (Zhu et al., 2006; Kurotani et al., 2015; Mei et al., 2024). A study conducted on complete panicle exsertion (CPE) in the Samba Mahsuri rice variety by Ballichatla et al. (2024) identified significant QTLs on chromosomes 4 and 12, with key SNPs in the genes Os12g0126300 and Os12g0131400 contributing to phenotypic variance. Transcriptomic analysis indicated that the down-regulation of these genes affects the ethylene and gibberellic acid biosynthetic pathways, which are crucial for CPE. Several potential candidate genes identified in the marker interval were found to be highly significant in regulating uppermost internode length and panicle exsertion-related traits. Therefore, further fine mapping of the marker interval will help narrow down the genomic region and can be utilized in marker-assisted introgression into indica-based cytoplasmic sterile lines, which would address the problem of incomplete panicle exertion.
5 Conclusion
In the present study, QTLs for uppermost internode length and panicle exsertion-related traits were mapped, and genomic regions controlling these traits were identified. We identified four QTL hotspots comprising 19 QTLS, with the remaining three QTLs located outside these hotspots. To enable more precise marker-assisted selection for uppermost internode length and panicle exsertion, further fine mapping of the marker intervals can help narrow down genomic regions and facilitate the identification of candidate genes.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
BH: Investigation, Validation, Visualization, Writing – original draft. VS: Writing – review & editing, Data curation, Methodology. SN: Methodology, Formal analysis, Writing – original draft. SS: Software, Writing – original draft. BD: Methodology, Writing – review & editing. SR: Writing – review & editing, Data curation, Methodology. DS: Data curation, Writing – review & editing. KV: Software, Validation, Writing – review & editing. SK: Resources, Software, Writing – review & editing. RE: Software, Validation, Writing – review & editing. MN: Methodology, Writing – review & editing. HB: Software, Validation, Writing – review & editing. TM: Writing – review & editing. AS: Writing – review & editing, Conceptualization. PB: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The first author, BH, is grateful to ICAR for the ICAR-SRF fellowship.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsufs.2024.1470871/full#supplementary-material
References
Archana, R., Vinod, K. K., Gopala Krishnan, S., Vadhana, E. D. C., Bhowmick, P. K., Singh, V. J., et al. (2023). Quantitative trait loci for stay-greenness and agronomic traits provide new insights into chlorophyll homeostasis and nitrogen use in rice. Plant Breed. 142, 312–326. doi: 10.1111/pbr.13086
Ballichatla, S., Gokulan, C. G., Barbadikar, K. M., Hake, A. A., Potupureddi, G., Guha, P. K., et al. (2024). Impairment in a member of AP2/ERF and F-box family protein enhances complete panicle exsertion in rice. J. Exp. Bot. 75, 5611–5626. doi: 10.1093/jxb/erae244
Bao-Jian, Q., Xiao-Biao, Z., Ying-Ying, W., and De-Lin, H. (2008). Mapping of QTL for three panicle exsertion-related traits in rice under different growing environments. Acta Agron. Sin. 34, 389–396. doi: 10.1016/S1875-2780(08)60017-6
Bi, Y., Zhou, B., Ren, P., Chen, X., Zhou, D., Yao, S., et al. (2024). Effects of Bacillus subtilis on cotton physiology and growth under water and salt stress. Agric. Water Manag. 303:109038. doi: 10.1016/j.agwat.2024.109038
Dunand, R., and Saichuk, J. (2014). “Rice growth and development” in Louisiana Rice Production Handbook. ed. J. Saichuk (Baton Rouge, LA: LSU Agricultural Center, Crowley).
Gao, S., Fang, J., Xu, F., Wang, W., and Chu, C. (2016). Rice HOX12 regulates panicle exsertion by directly modulating the expression of Elongated Uppermost Internode1. Plant Cell 28, 680–695. doi: 10.1105/tpc.15.01021
Hashim, S., Ding, P., Ismail, M. F., and Ramli, A. (2021). Floral traits and flowering behaviors of Malaysian rice cytoplasmic male sterile and maintainer lines and its relationship with out-crossing performance. Aust. J. Crop. Sci. 15, 180–186. doi: 10.21475/ajcs.21.15.02.p1732
Hattori, Y., Nagai, K., Furukawa, S., Song, X. J., Kawano, R., Sakakibara, H., et al. (2009). The ethylene response factors SNORKEL1 and SNORKEL2 allow rice to adapt to deep water. Nature 460, 1026–1030. doi: 10.1038/nature08258
Honnaiah, M. (2003). “Economics of hybrid rice cultivation and seed production” in Winter school on advances in hybrid rice technology. eds. B. C. Viraktamath, M. I. Ahmed, and B. Mishra (Hyderabad, India: Directorate of Rice Research), 391–398. Available at: https://knetminer.com (Accessed February 09, 2020).
International Rice Research Institute (2014). Philippines Star for Windows version 2.0.1. Los Baños: International Rice Research Institute.
Itoh, H., Tatsumi, T., Sakamoto, T., Otomo, K., Toyomasu, T., Kitano, H., et al. (2004). A rice semi-dwarf gene, Tan-Ginbozu (D35), encodes the gibberellin biosynthesis enzyme, ent-kaurene oxidase. Plant Mol. Biol. 54, 533–547. doi: 10.1023/B:PLAN.0000038261.21060.47
Kallugudi, J., Singh, V. J., Vinod, K. K., Krishnan, S. G., Nandakumar, S., Dixit, B. K., et al. (2022). Population dynamics of wide compatibility system and evaluation of intersubspecific hybrids by indica-japonica hybridization in rice. Plants 11:1930.
Karunakaran, V., Sivakumar, P., Pandiyan, M., Mathiyazhagan, S., Selvamurugan, M., Baskaran, R., et al. (2024). Effects of plant growth regulators and micronutrients on crop performance, seed germination and seedling vigour in rice (ORYZA SATIVA L.). Applied Ecology & Environmental Research, 22.
Kosambi, D. D. (1944). The estimation of map distance from recombination values. Ann. Eugenics 12, 172–175. doi: 10.1007/978-81-322-3676-4_16
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, A., Bhowmick, P. K., Singh, V. J., Malik, M., Gupta, A. K., Seth, R., et al. (2017). Marker-assisted identification of restorer gene (s) in iso-cytoplasmic restorer lines of WA cytoplasm in rice and assessment of their fertility restoration potential across environments. Physiol. Mol. Biol. Plants 23, 891–909. doi: 10.1007/s12298-017-0464-5
Kurotani, K. I., Hattori, T., and Takeda, S. (2015). Overexpression of a CYP94 family gene CYP94C2b increases internode length and plant height in rice. Plant Signal. Behav. 10:e1046667. doi: 10.1080/15592324.2015.1046667
Lander, E. S., Green, P., Abrahamson, J., Barlow, A., Daly, M. J., Lincoln, S. E., et al. (1987). MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1, 174–181. doi: 10.1016/0888-7543(87)90010-3
Li, H., Guoyou, Y., and Jiankang, W. (2007). A modified algorithm for the improvement of composite interval mapping. Genetics 175, 361–374. doi: 10.1534/genetics.106.066811
Librojo, A. L., and Khush, G. S. (1986). “Chromosomal location of some mutant genes through the use of primary trisomics in rice” in Rice genetics. ed. G. S. Khush (Manila, Philippines: IRRI), 249–255.
Lingaiah, N., Venkanna, V., and Cheralu, C. (2015). Genetic vairability, heritability and genetic advance in rice (Oryza sativa L.). Asian J. Environ. Sci. 46, 917–919.
Luo, A., Qian, Q., Yin, H., Liu, X., Yin, C., Lan, Y., et al. (2006). EUI1, encoding a putative cytochrome P450 monooxygenase, regulates internode elongation by modulating gibberellin responses in rice. Plant Cell Physiol. 47, 181–191. doi: 10.1093/pcp/pci233
Mei, E., He, M., Xu, M., Tang, J., Liu, J., Liu, Y., et al. (2024). OsWRKY78 regulates panicle exsertion via gibberellin signaling pathway in rice. J. Integr. Plant Biol. 66, 771–786. doi: 10.1111/jipb.13636
Meng, L., Li, H., Zhang, L., and Wang, J. (2015). QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 3, 269–283. doi: 10.1016/j.cj.2015.01.001
Mohanty, J. K., and Parida, S. K. (2024). Stripping off the rice panicle: induced genetic variation awakens the sheathed spikelet for a better yield. J. Exp. Bot. 75, 5459–5462. doi: 10.1093/jxb/erae327
Monna, L., Kitazawa, N., Yoshino, R., Suzuki, J., Masuda, H., Maehara, Y., et al. (2002). Positional cloning of rice semi-dwarfing gene, sd-1: rice “green revolution gene” encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 9, 11–17. doi: 10.1093/dnares/9.1.11
Nandakumar, S., Singh, V. J., Vinod, K. K., Krishnan, S. G., Dixit, B. K., Harshitha, B. S., et al. (2024). Genetic mapping for flag leaf shape in new plant type based recombinant inbred lines in rice (Oryza sativa L.). Indian J. Genet. Plant Breed. 84, 52–62. doi: 10.31742/ISGPB.84.1.4
Oikawa, T., Koshioka, M., Kojima, K., Yoshida, H., and Kawata, M. (2004). A role of OsGA20ox1, encoding an isoform of gibberellin 20-oxidase, for regulation of plant stature in rice. Plant Mol. Biol. 55, 687–700. doi: 10.1007/s11103-004-1692-y
Priyadarshi, R., Arremsetty, H. P. S., Singh, A. K., Khandekar, D., Ulaganathan, K., and Shenoy, V. (2017). Molecular stacking of wide compatibility gene, S5 n and elongated uppermost internode (eui) gene into IR 58025B, an elite maintainer line of rice. J. Plant Biochem. Biotechnol. 26, 425–435. doi: 10.1007/s13562-017-0404-z
Rutger, J. N., and Carnahan, H. L. (1981). A fourth genetic element to facilitate hybrid cereal production—a recessive tall in rice 1. Crop Sci. 21, 373–376. doi: 10.2135/cropsci1981.0011183X002100030005x
Sasaki, A., Itoh, H., Gomi, K., Ueguchi-Tanaka, M., Ishiyama, K., Kobayashi, M., et al. (2003). Accumulation of phosphorylated repressor for gibberellin signaling in an F-box mutant. Science 299, 1896–1898. doi: 10.1126/science.1081077
Saito, K., Hayano-Saito, Y., Maruyama-Funatsuki, W., Sato, Y., and Kato, A. (2004). Physical mapping and putative candidate gene identification of a quantitative trait locus Ctb1 for cold tolerance at the booting stage of rice. Theoretical and Applied Genetics 109, 515–522.
Shidenur, S., Singh, V. J., Vinod, K. K., Gopala Krishnan, S., Ghritlahre, S. K., Bollinedi, H., et al. (2020). Enhanced grain yield in rice hybrids through complementation of fertility restoration by Rf3 and Rf4 genes as revealed by multilocation evaluation of tropical japonica derived rice (Oryza sativa) hybrids. Plant Breed. 139, 743–753. doi: 10.1111/pbr.12818
Shidenur, S., Singh, V. J., Vinod, K. K., Gopala Krishnan, S., Ghritlahre, S. K., Bollinedi, H., et al. (2019). Molecular detection of WA-CMS restorers from tropical japonica-derived lines, their evaluation for fertility resto ration and adaptation. Plant Breed. 138, 553–567. doi: 10.1111/pbr.12701
Singh, V. J., Vinod, K. K., Krishnan, S. G., and Singh, A. K. (2021). “Rice adaptation to climate change: opportunities and priorities in molecular breeding” in Molecular breeding for rice abiotic stress tolerance and nutritional quality. eds. M. A. Hossain, L. Hassan, K. M. Ifterkharuddaula, A. Kumar, and R. Henry (Hoboken: Wiley).
Sitoe, H. M., Zhang, Y., Chen, S., Li, Y., Ali, M., Sowadan, O., et al. (2022). Detection of QTLs for plant height architecture traits in rice (Oryza sativa L.) by association mapping and the RSTEP-LRT method. Plan. Theory 11:999. doi: 10.3390/plants11070999
Sonu, N., Nandakumar, S., Singh, V. J., Pandey, R., Gopala Krishnan, S., Bhowmick, P. K., et al. (2024). Implications of tolerance to iron toxicity on root system architecture changes in rice (Oryza sativa L.). Front. Sustain. Food Syst. 7:1334487. doi: 10.3389/fsufs.2023.1334487
Sruthi, K., Divya, B., Senguttuvel, P., Revathi, P., Kemparaju, K. B., Koteswararao, P., et al. (2020). Evaluation of genetic diversity of parental lines for development of heterotic groups in hybrid rice (Oryza sativa L.). J. Plant Biochem. Biotechnol. 29, 236–252. doi: 10.1007/s13562-019-00529-9
Tan, C. J., Sun, Y. J., Xu, H. S., and Yu, S. B. (2011). Identification of quantitative trait locus and epistatic interaction for degenerated spikelets on the top of panicle in rice. Plant Breed. 130, 177–184. doi: 10.1111/j.1439-0523.2010.01770.x
Ueguchi-Tanaka, M., Ashikari, M., Nakajima, M., Itoh, H., Katoh, E., Kobayashi, M., et al. (2005). Gibberellin Insensitive Dwarf1 encodes a soluble receptor for gibberellin. Nature 437, 693–698. doi: 10.1038/nature04028
Ueguchi-Tanaka, M., Fujisawa, Y., Kobayashi, M., Ashikari, M., Iwasaki, Y., Kitano, H., et al. (2000). Rice dwarf mutant d1, which is defective in the α subunit of the heterotrimeric G protein, affects gibberellin signal transduction. Proc. Natl. Acad. Sci. 97, 11638–11643. doi: 10.1073/pnas.97.21.11638
Van Berloo, R. (1999). Computer note GGT: software for the display of graphical genotypes. J. Hered. 90, 328–329. doi: 10.1093/jhered/90.2.328
Vinod, K. K. (2011). Kosambi and the genetic mapping function. Resonance 16, 540–550. doi: 10.1007/s12045-011-0060-x
Wang, J. (2009). Inclusive composite interval mapping of quantitative trait genes. Acta Agron. Sin. 35, 239–245. doi: 10.3724/SP.J.1006.2009.00239
Wang, J., Gang, S., Yang, L., Zheng, H., Sun, J., Liu, H., et al. (2017). Markers associated with culm length and elongated internode length in japonica rice. Crop Sci. 57, 2329–2344. doi: 10.2135/cropsci2016.06.0533
Wang, P., Zhou, G., Cui, K., Li, Z., and Yu, S. (2012). Clustered QTL for source leaf size and yield traits in rice (Oryza sativa L.). Mol. Breed. 29, 99–113. doi: 10.1007/s11032-010-9529-7
Xu, X., He, P., Zhao, S., Qiu, S., Johnston, A. M., and Zhou, W. (2016). Quantification of yield gap and nutrient use efficiency of irrigated rice in China. Field Crop Res. 186, 58–65. doi: 10.1016/j.fcr.2015.11.011
Xu, Y. H., Zhu, Y. Y., Zhou, H. C., Li, Q., Sun, Z. X., Liu, Y. G., et al. (2004). Identification of a 98-kb DNA segment containing the rice Eui gene controlling uppermost internode elongation, and construction of a TAC transgene sublibrary. Mol. Gen. Genomics. 272, 149–155. doi: 10.1007/s00438-004-1019-3
Yang, R., Huang, R., Zhang, Q., Zhang, S., and Liang, K. (2000). Developing eui-cytoplasmic male sterile lines and applying them in hybrid rice breeding. Int. Rice Res. Notes 25, 11–12.
Yang, X., Wang, J., Xia, X., Zhang, Z., He, J., Nong, B., et al. (2021). OsTTG1, a WD40 repeat gene, regulates anthocyanin biosynthesis in rice. Plant J. 107, 198–214. doi: 10.1111/tpj.15285
Yang, R. C., Yang, S. L., Zhang, Q., and Huang, R. H. (1999). A new elongated uppermost internode gene. Rice Genet. Newsl. 16, 41–43.
Yang, R. C., Zhang, S., Huang, R., Yang, S., and Zhang, Q. (2002). Breeding technology of eui-hybrids of rice. Agril. Sci. China 1, 359–363.
Yasir, T. A., Wasaya, A., Azhar, W., Kanwal, S., Sarwar, N., Rehmani, M. I. A., et al. (2022). “Plant growth regulators for Rice production in changing environment” in Modern techniques of Rice crop production. ed. K. Saima (Singapore: Springer), 257–274.
Yin, C., Gan, L., Ng, D., Zhou, X., and Xia, K. (2007). Decreased panicle-derived indole-3-acetic acid reduces gibberellin A1 level in the uppermost internode, causing panicle enclosure in male sterile rice Zhenshan 97A. J. Exp. Bot. 58, 2441–2449. doi: 10.1093/jxb/erm077
Zhan, C., Hu, J., Pang, Q., Yang, B., Cheng, Y., Xu, E., et al. (2019). Genome-wide association analysis of panicle exsertion and uppermost internode in rice (Oryza sativa L.). Rice 12, 72–13. doi: 10.1186/s12284-019-0330-x
Zhang, L., Chen, C., Li, Y., Suo, C., Zhou, W., Liu, X., et al. (2024). Enhancing aphid resistance in horticultural crops: a breeding prospective. Hortic. Res. 2024:uhae275. doi: 10.1093/hr/uhae275
Zhao, C. F., Chen, T., Zhao, Q. Y., Zhou, L. H., Zhao, L., Zhang, Y. D., et al. (2016). Analysis of QTLs for panicle exsertion and its relationship with yield and yield-related traits in rice (Oryza sativa L.). Genet. Mol. Res. 15:gmr.15027423. doi: 10.4238/gmr.15027423
Zhao, D. D., Son, J. H., Farooq, M., and Kim, K. M. (2021). Identification of candidate gene for internode length in rice to enhance resistance to lodging using QTL analysis. Plan. Theory 10:1369. doi: 10.3390/plants10071369
Zhu, L., Hu, J., Zhu, K., Fang, Y., Gao, Z., He, Y., et al. (2011). Identification and characterization of SHORTENED UPPERMOST INTERNODE 1, a gene negatively regulating uppermost internode elongation in rice. Plant Mol. Biol. 77, 475–487. doi: 10.1007/s11103-011-9825-6
Keywords: rice, QTL mapping, RIL, uppermost internode length, panicle exsertion
Citation: Harshitha BS, Singh VJ, Nandakumar S, Shekhawat S, Dixit BK, Ragi S, Saran D, Vinod KK, Krishnan SG, Ellur RK, Nagarajan M, Bollinedi H, Mondal TK, Singh AK and Bhowmick PK (2024) Uncovering QTLs for uppermost internode length and panicle exsertion in rice (Oryza sativa L.). Front. Sustain. Food Syst. 8:1470871. doi: 10.3389/fsufs.2024.1470871
Edited by:
Autar Krishen Mattoo, Agricultural Research Service (USDA), United StatesReviewed by:
Mohamed El-Soda, Cairo University, EgyptYuzhou Xu, Kansas State University, United States
Copyright © 2024 Harshitha, Singh, Nandakumar, Shekhawat, Dixit, Ragi, Saran, Vinod, Krishnan, Ellur, Nagarajan, Bollinedi, Mondal, Singh and Bhowmick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prolay Kumar Bhowmick, cHJvbGF5Ymhvd21pY2tAZ21haWwuY29t