 Gowthami Sanku1
Gowthami Sanku1 Ravikesavan Rajasekaran1*
Ravikesavan Rajasekaran1* Narayanan Manikanda Boopathi2
Narayanan Manikanda Boopathi2 Iyanar Krishnamoorthy3
Iyanar Krishnamoorthy3 Vichangal Pridiuldi Santhanakrishnan4
Vichangal Pridiuldi Santhanakrishnan4 Vetriventhan Mani5
Vetriventhan Mani5- 1Department of Plant Breeding and Genetics, CPBG, Tamil Nadu Agricultural University, Coimbatore, India
- 2Department of Plant Biotechnology, CPMB&B, Tamil Nadu Agricultural University, Coimbatore, India
- 3Department of Millets, Tamil Nadu Agricultural University, Coimbatore, India
- 4Department of Medicinal and Aromatic Plants, Tamil Nadu Agricultural University, Coimbatore, India
- 5International Crops Research Institute for the Semi-Arid Tropics (ICRISAT), Patancheru, India
Global food and nutritional security are being threatened by abiotic stresses such as drought, salinity, cold, and heat, owing to the rapid and deleterious effects of climate change. Millets represent an incredibly promising agricultural crop in terms of their potential to ensure global food security by virtue of their resiliency against climate change and escalating demand for nutritious food and feed. Considering their durability against climate change and the increasing demand for nutritional food and feed, millets, which refers to the diverse class of small-seeded C4 panicoid grasses, endure remarkable potential for safeguarding the world’s food supply. With a rapid surge in the availability of genomic information through microarray and next-generation sequencing, transcriptomics facilitated the extensive examination and quantification of shifts brought about by abiotic stresses. This stipulates an imperative way of ascertaining the expression of vital genes. This technological advancement targeted toward deciphering the gene expression patterns underpinning the molecular mechanisms/pathways. Other techniques, for instance, genome-wide expression analysis which provides insights into the regulatory networks controlling the cellular processes, genome-wide location analysis which elucidates the control over genes by the transcriptional regulatory proteins, and genomic selection strengthens the reliability of stress tolerance predictions in millet breeding populations. This review emphasizes the impact of transcriptomics on millet improvement by collating the differentially expressed genes (DEGs), and transcription factors (TFs) specific to abiotic stress response in millets which could open advantageous avenues with intriguing opportunities in breeding cultivars for climate resilience.
1 Introduction
Climate change entails several biotic and abiotic stresses on crop plants, seriously threatening agricultural productivity. These abiotic stresses account for a loss of 51–82% of crop output in the context of global agriculture (Oshunsanya et al., 2019). Plants in turn are equipped with various coping mechanisms to evade these stresses; however, during the course of domestication, several crops have lost their ability to withstand the environmental impacts. For instance, wild rice is comparatively more climate resilient than cultivated rice. Cultivated wheat is highly sensitive to temperature, wherein even a 1°C increase in temperature reduces the yield by 3–4% during the grain filling period addressed by accelerated phenological development, a spike in respiration rate and thereby, reduced dry matter production (Fujimura et al., 2012). Semi-domesticated crops like small millets are inherently tolerant to climatic aberrations rendering them the ideal models to uncover the mechanisms underlying abiotic stresses. These small millets include finger millet (Eleusine coracana), foxtail millet (Setaria italica), proso millet (Panicum miliaceum), barnyard millet (Echinochloa crus-galli), kodo millet (Paspalum scrobiculatum), little millet (Panicum sumatrense), browntop millet (Urochloa ramosa), tef (Eragrostis tef), fonio (Digitaria exilis), adlay millet (Coix lacryma-jobi), guinea millet (Brachiaria deflexa), and Taiwan oil millet (Spodiopogon formosanus). The ability of small millets to grow in marginal regions with limited irrigation and poor soil fertility makes these crops a better choice for cultivation in arid and semi-arid areas (Choudhary et al., 2023). Contrary to major cereals, millets pose a number of morpho-physiological, molecular, and biochemical traits that strengthen their resilience to external stressors. The short lifecycle of millets (averaging 12–14 weeks from seed to seed) is of prime relevance to abiotic stress tolerance by avoiding stress (Bandyopadhyay et al., 2017). Furthermore, the C4 mechanism affirms the CO2 content in bundle sheath witnessing an upsurge in the photosynthetic rates with diminishing photorespiration reliant on the temperature and strengthens the in planta catalytic activity of RuBisCO resulting in increased nutrient use efficiency (NUE) and water use efficiency (WUE) in contrast to C3 crops. Millets present a readily navigable system for exploring the features of stress-related responses at the cellular, molecular, and physiological levels owing to their intriguing tolerance against an array of abiotic stresses, such as heat, salt, light, and drought (Sage and Zhu, 2011). The prime attributes concerning abiotic stress in millets are represented in Table 1 and Figure 1.
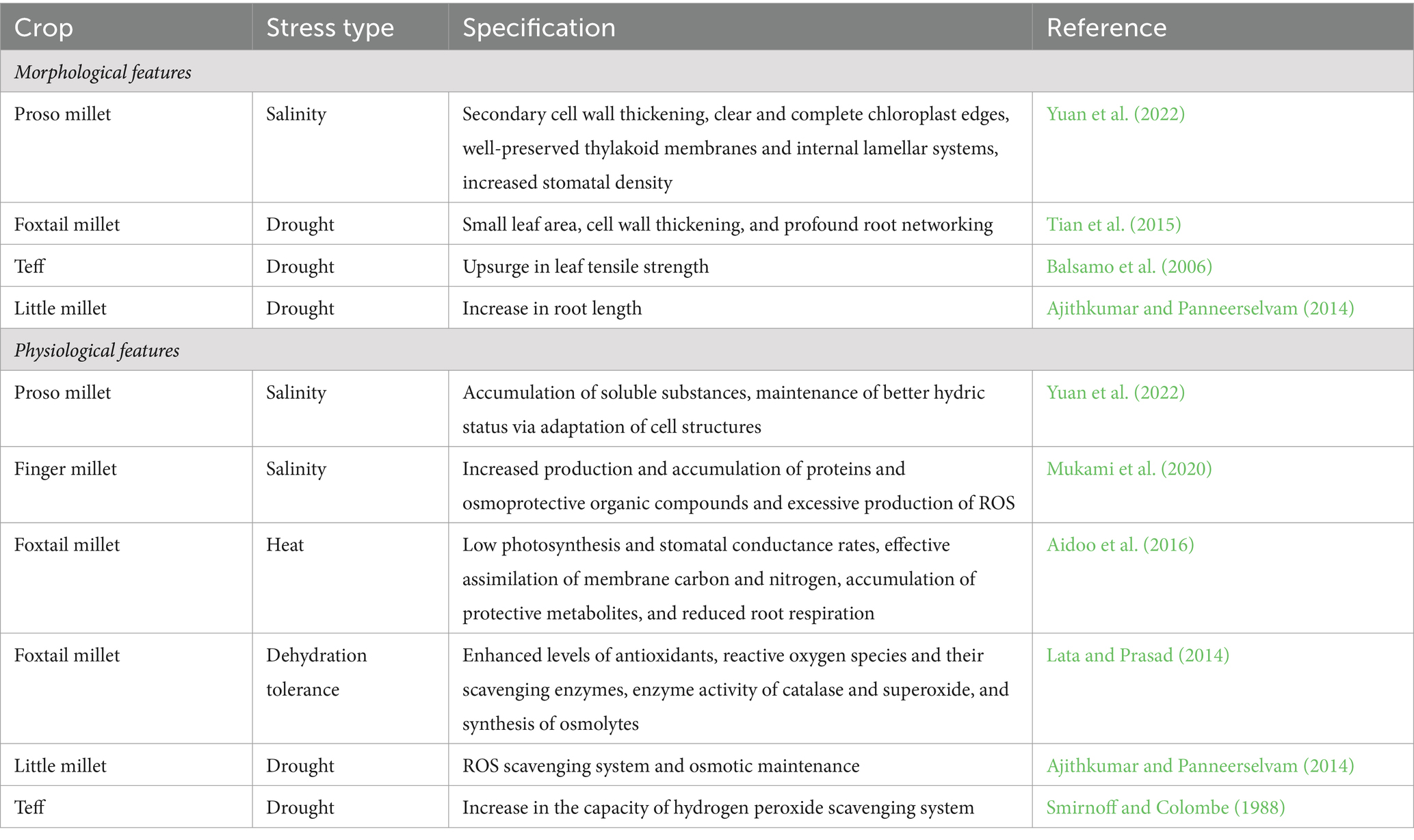
Table 1. Morphological and physiological features of millets in response to abiotic stress tolerance.
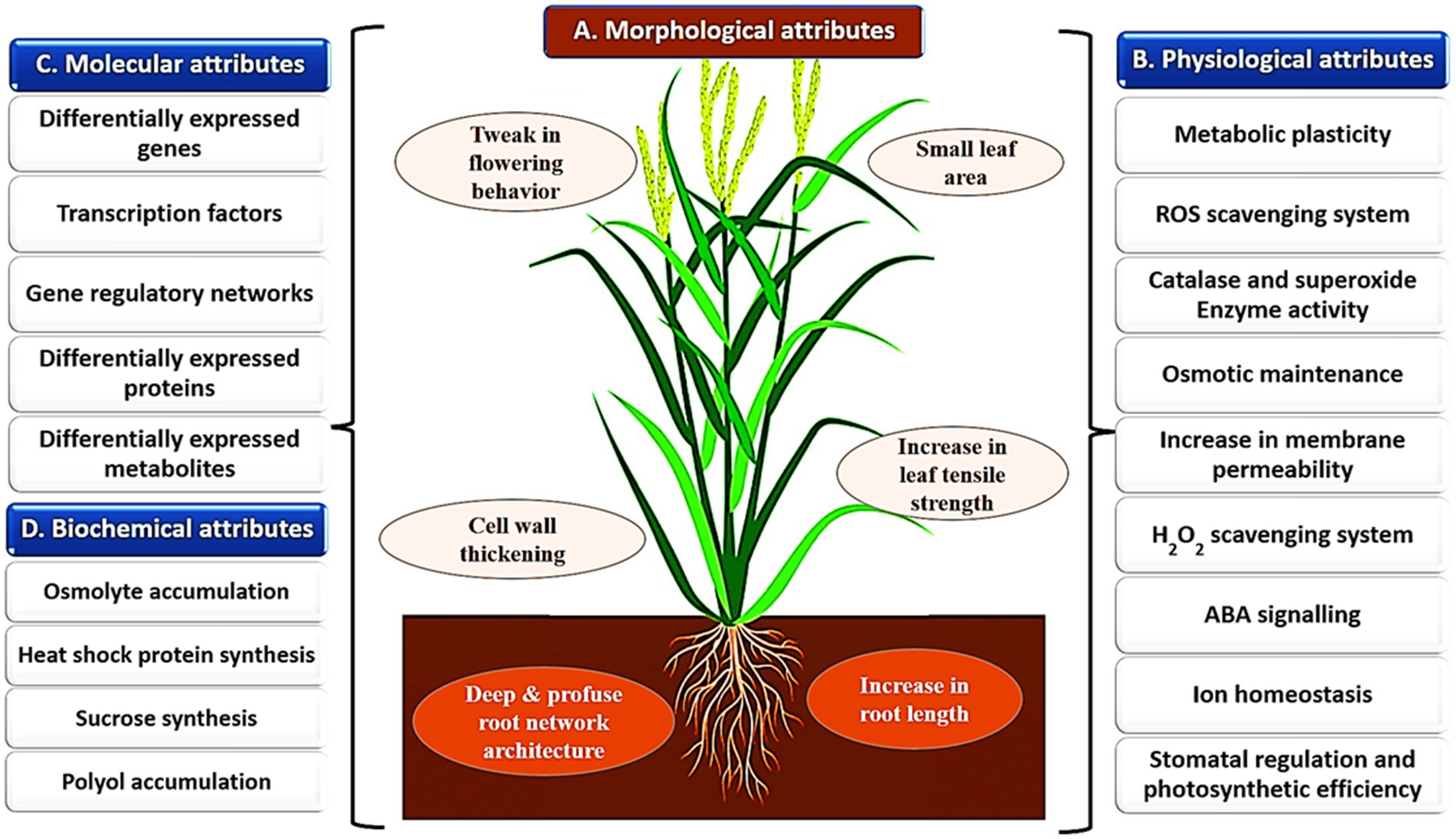
Figure 1. Key attributes of different millets that confer abiotic stress resistance. A. Panel A represents the morphological attributes possessed by the millets such as improved and well-developed root, stem, and leaf characteristics; B. Some of the physiological attributes that enable millets to adapt and survive under adverse environmental conditions are depicted in Panels B and C. The large network of expressed genes, proteins, metabolites, and their regulatory networks constitute molecular attributes in millets concerning abiotic stress resistance; D. A multitude of biochemical attributes involving the metabolic pathways and compounds that help mitigate stress-induced damages and maintain cellular homeostasis are shown in panel D.
In contrast to major cereals and major millets, small millets have invited less attention pertaining to the production, and research in exploring the mechanisms governing abiotic stress response. The molecular strategies underlying abiotic stress tolerance in plants can be unraveled with the advent of next-generation sequencing to pinpoint the coding and non-coding regions of the genome that regulate stress resilience. Given this, the foxtail millet genome was first sequenced in 2012 by BGI and JGI (Bennetzen et al., 2012; Zhang et al., 2012). In addition to genome sequencing, the researchers also carried out the transcriptome profiling of foxtail millet, particularly of, root, stem, leaf, and spica. This offered a fresh perspective on the tissue-specific gene expression pattern of the genes encoded by the foxtail millet genome. Following this, several transcriptomics studies involving RNA-Seq and macroarray have been performed in other small millet viz., finger millet, little millet, kodo millet, and proso millet. Sequencing the transcriptome of these millet crops exposed to abiotic stresses provides insights into the dynamics of gene expression, and enhancement of genomic resources, thereby providing an efficient approach for the development of markers (Arzani and Ashraf, 2016). In addition, RNA-seq reveals its application in decoding the biological processes and molecular functions governing abiotic stress response and identifies the candidate genes for further functional characterization (Guo et al., 2021; Rajawat, 2018). Given this, the present review focuses on comprehending the transcriptomic approach toward uncovering the gene expression patterns and their regulatory mechanisms toward abiotic stress resilience in minor millets.
2 Transcriptomics
Transcriptomic analysis is one of the most prevalent methods for unraveling of stress-responsive genes through the deployment of advanced technological breakthroughs like RNA-seq which presents an overview of gene expression providing insights into transcriptome as depicted in Figure 2. The entirety of RNA, notably non-coding and mRNA, that is transcribed by a particular cell or tissue in an inevitable state of activity is referred to as the transcriptome. It also covers the exploration of novel transcriptional regions, non-coding region function, structure, and gene transcription level (Rodrigues et al., 2014). The early 1990s witnessed the first endeavors to examine the transcriptome as an ensemble, and since the late 1990s, groundbreaking discoveries in technology have transformed transcriptomics into a sought-after discipline of research (Lowe et al., 2017; Tyagi et al., 2022). Functional genomics researchers are capable of uncovering prospective genes that exercise control over a broad spectrum of biological processes leveraging transcriptome-based gene profiling, which additionally lends comprehensive analyses of functional polymorphisms and patterns of gene expression (Ravikesavan et al., 2023). Deciphering the genes with varying expression patterns by swiftly scanning the transcriptome by means of hybridization-based gene microarray/chip technology, expressed sequence tag-based approach (EST-SAGE), and NGS-based RNA-sequencing (RNA-seq) technology is one of the preliminary objectives of the analysis, in addition to the classification and determination of transcriptional products (McGettigan, 2013; Nejat et al., 2018).
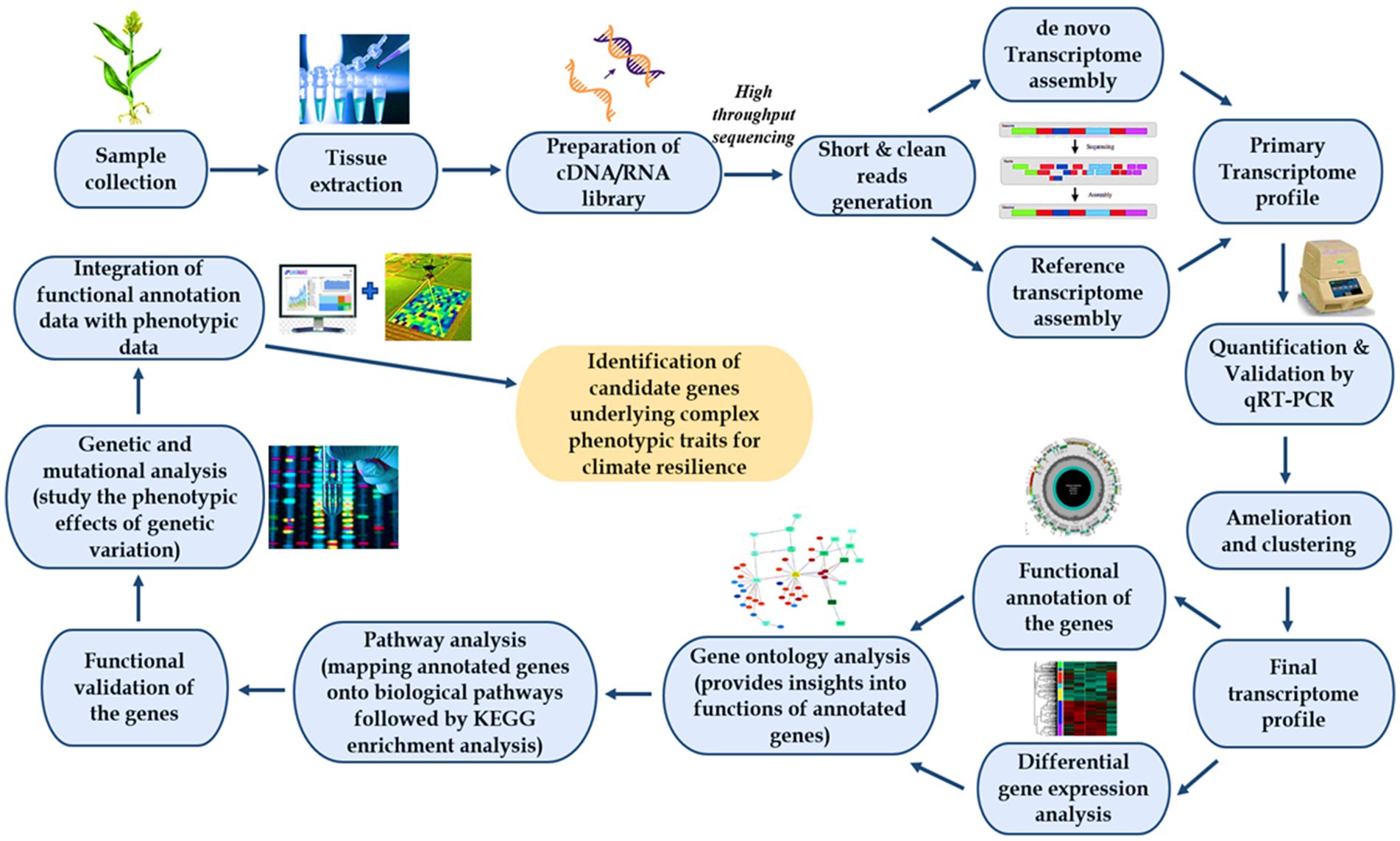
Figure 2. Schematic representation of transcriptomics in the identification of candidate genes for climate resilience. Samples are collected, and tissue is extracted using RNA extraction methods that preserve the integrity and quality of the RNA molecules to ensure accurate downstream analysis followed by mRNA enrichment, fragmentation, and cDNA library preparation for sequencing. High-throughput sequencing is performed generating short and clean reads creating a primary transcriptome profile, which is aided by mapping the processed reads to a reference genome. The final transcriptome profile is developed preceding quantification and validation by deploying qRT-PCR followed by amelioration and clustering of transcripts which can be utilized for annotating genes in addition to elucidation of the differential expression of genes. These genes are then subjected to gene ontology analysis, which assists in comprehending the functional roles of genes within specific biological contexts and pathway analysis to elucidate the interconnectedness of genes and their involvement in various metabolic or signaling pathways. Then, validation is done to reveal the functional roles of annotated genes through overexpression or complementation assays, thereby confirming the biological significance of annotated genes and their contributions to specific phenotypes or biological processes. Genetic and mutational analysis is carried out to uncover the phenotypic expression of genetic variation and the integration of this functional annotation data with phenotyping data generated by means of conventional phenotyping or high throughput phenotyping (imaging platforms and imaging sensors) results in the identification of candidate genes underlying complex molecular mechanisms and phenotypic traits for climate resilience.
This merely reflects that the aforementioned techniques, which were hitherto confined to only a restricted number of species with fully sequenced genomes, have been adopted for the empirical investigation of an expansive spectrum of biological systems. Transcript data from orphan crops cater to exciting prospects for plant breeding in crop improvement, bolstering the copious RNA sequencing (RNA-Seq) data accessible for major crops implying valuable and exacerbated gene pools by unraveling the cellular process underlying the plant stress responses by providing perspective precisely how plants comply with abiotic stresses (Bajay et al., 2023).
3 Transcriptome dynamics of abiotic stress response
Transcriptomics is leveraged to illustrate the intricate regulatory network and expression at the genome-wide level amid stress, filter for novel genes pertinent to tolerance, and quantitatively delve into shifts in plant gene regulation at a given time point and in a specific context (Wang et al., 2020). With an increased likelihood and fostered growth of transcriptomics, millet genomic resources provide genomic data for precision breeding for crop improvement (Banshidhar et al., 2023). The remainder of this article outlines the instances of transcriptomics that focus on abiotic stress resistance in millets.
3.1 Drought stress response
The emergence of drought tolerance entails an accelerated oversight of an array of genes entangled in multiple metabolic pathways linked to the mitigation or restitution of water stress-induced impairment. Owing to the inherent complexity of drought tolerance and the way it interacts with the environment and other genes/characteristics afflict its expression, conventional breeding has encountered fewer positive outcomes than envisaged (Kido et al., 2016). Transcriptomics emphasizes how genetic information is revamped in an organism adhering to an initial stimulation and, thereby assists in defining the molecular avenues for its exploitation in breeding endeavors (Morozova et al., 2009). Guo et al. (2023), while working on three different cultivars of foxtail millet displayed 2,954, 1,531, and 2,344 differentially expressed genes under drought stress and annotated through Gene Ontology (GO) pathway analysis. From this study, it was inferred that the genes were found to be enriched in photosynthesis, chlorophyll metabolism, amino acid metabolism, and carbohydrate metabolism and revealed that trends of transcription changes were consistence with the recurring patterns of drought stress. Transcriptome profiling (RNA-seq) was carried out in little millet by Narayanrao et al. (2023) and disclosed transcriptional stability. He noticed that twenty essential genes were up- or downregulated in the control and treated roots of the OLM-203 genotype in addition to 36 upregulated (root) and 21 downregulated (leaves) serine transcripts from the study. This analysis serves as a guide in providing the fresh insight into the inherent approach of gene regulation in little millet. Cao et al. (2022) performed a transcriptomic analysis in proso millet by deploying PacBio transcriptomic sequencing. The study displayed 245 genes related to energy metabolism; and 219 genes related to plant hormone signal reduction differentially expressed when subjected to drought driven by 495 transcription factors belonging to 10 different families. This study inferred that above said mechanisms were related to the drought resilience of millets and that it tailors to adversity by meticulously altering the magnitude of underlying molecular mechanisms. Suresh et al. (2022) worked on kodo millet by deploying Illumina Hi Seq 4,000 platform for sequencing and identified 9,201, 9,814, and 2,346 differentially expressed genes (DEGs). This study deduced that the gene expression has revealed its relevance to ABA breakdown, detoxification of lipid-related peroxidation products, plant hormones signaling, and metabolism in the aftermath of dehydration stress. Zhang et al. (2012) used the Illumina Hi Seq 2,100 platform for sequencing of foxtail millet genotypes and identified the upregulation of few genes and downregulation of genes (GUS3, scpl20, ATEXPB2). It is inferred from this study that drought tolerance is primarily regulated by genes expressed in transcriptional cascades linked to the cell cycle and DNA replication. A similar study was conducted by Qin et al. (2020) in foxtail millet by deploying RNA-seq technology. He identified the differential expression of a few genes in the wake of drought stress and this study explained that the expressed genes were engaged in metabolic pathways for carbohydrate metabolism, the photosynthesis process, signal transduction, phenylpropanoid production, and osmotic adjustment contributing to resilience to drought stress. Yu et al. (2020) conducted studies on foxtail millet by utilizing the Illumina Hi Seq 4000™ platform and showed that the DREB gene regulatory network, which is triggered by SiCDPK24, enables the millet CDPK (calcium-dependent protein kinases) group to survive drought stress. Studies on finger millet were performed by Li et al. (2021) and revealed the expression of genes BAMY2 and ISA1. This study inferred that these genes were shown to play an imperative part in the response to drought stress because they are strengthened in hydrolase activity, glycosyl bond formation, oxidoreductase activity, sugar binding, and unsaturated fatty acid production. Likewise, Parvathi et al. (2019) revealed the differential expression of genes [serine–threonine protein phosphatase 2A (PP2A), calcineurin B-like interacting protein kinase 31 (CIPK31), farnesyl pyrophosphate synthase (FPS), signal recognition particle receptor α (SRPRα)] while working on finger millet cultivars. This research discovered that these genes were associated with protein targeting, post-translational modification, and signaling networks for increased tolerance. Macroarray-employed sequencing studies were also conducted to reveal the differential expression of genes in finger millet cultivars by Singh et al. (2016). This study revealed that the upregulation of these genes was responsible for intracellular trafficking, rendering boosted plant defense. The studies displaying the differential expression of genes in response to drought stress are tabulated in Table 2.
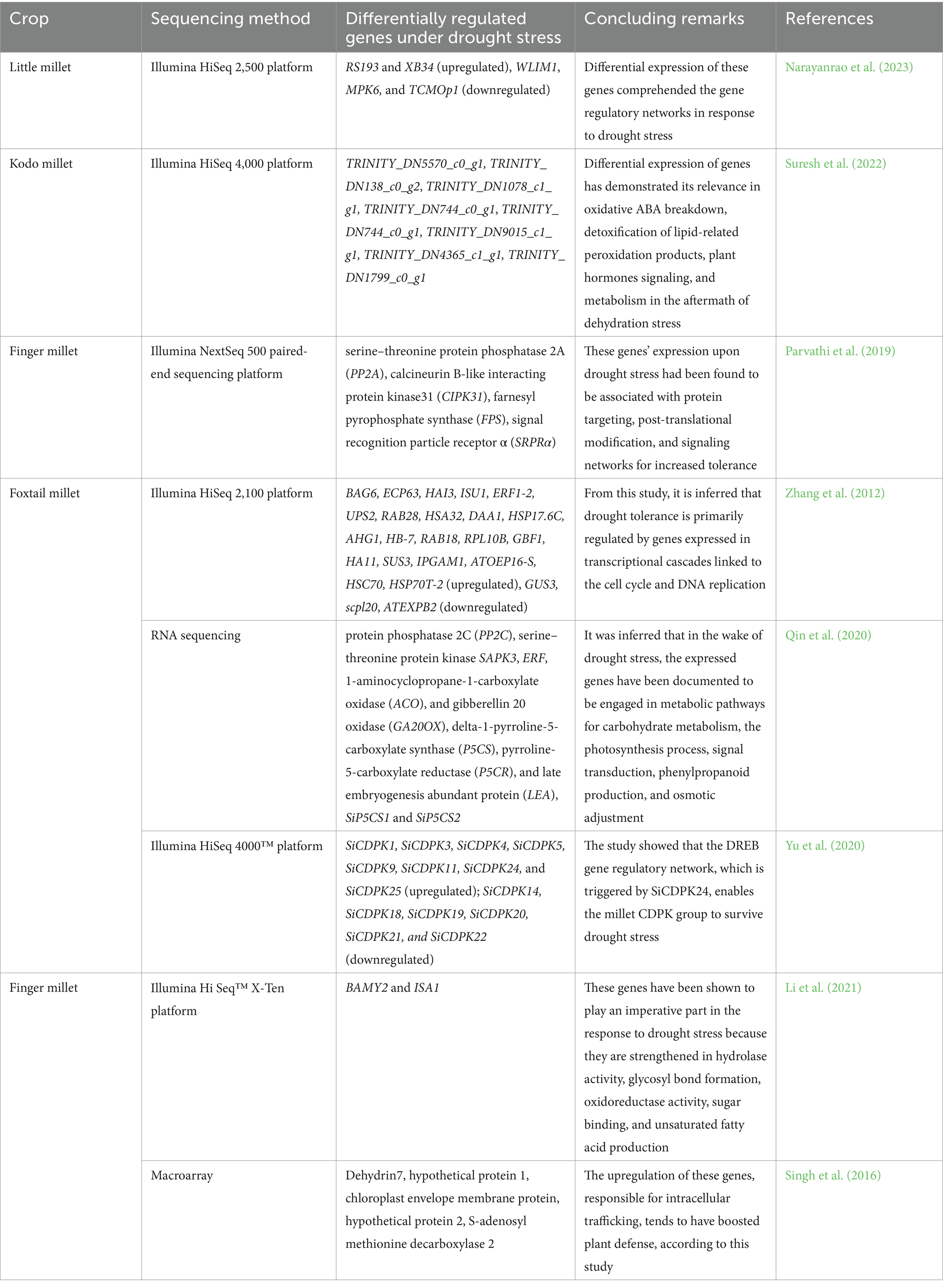
Table 2. Differentially expressed genes in response to drought.
Many transcriptomics studies examining crops under drought stress discern genes, implying a drop in transcripts rooted from photosynthesis and primary energy metabolism and an upsurge in stress signaling and antioxidative and osmoprotective proteins (Kido et al., 2016). From the above-mentioned reports, we can infer that functional analysis of these expressed genes to comprehend their function in regulating drought stress responses in respective millet crops may provide new options for cultivar development that are climate-resilient.
3.2 Salinity stress response
The overall productivity of grain legume crops has been significantly constrained by salt stress, particularly in the aftermath of unpredictable temperatures, injudicious irrigation water consumption, and poor farmland use. Plants react to salt stress by unleashing sophisticated molecular mechanisms explicating the underlying complicated processes which are critical to strengthening our comprehension of plant salinity response (Jha et al., 2019). The increasing demand for salt stress resilience to plants broadened the millet cultivated gene pool by thriving on “adaptive characteristics” in response to salinity stress tolerance. The growing popularity of transcriptomics has rendered it feasible to locate and quantify transcripts which will unveil fresh insights into the molecular drivers by revealing the genetic elements falling behind the salt stress tolerance (Shelden and Roessner, 2013). A large-scale transcriptome data set was derived by Han et al. (2022) to determine the mechanism of salt stress response of salt-tolerant and salt-sensitive accessions deploying the Illumina technology platform. He identified the differential expression of 2019 genes and 736 genes in salt-tolerant and sensitive cultivars, respectively responding to photosynthetic pathways, which will be mostly affected under salt stress. These reports provide important cues for the further characterization and identification of potential candidate genes which fosters the designing of new cultivars with salinity stress resilience. Besides, studies carried out in proso millet cultivars by Yuan et al. (2021) through digital RNA-Seq revealed the differential gene expression patterns of 6,878 and 1940 genes at two time intervals. This study deduced that these genes were reported to regulate phenylpropanoid biosynthesis, carbon metabolism, and auxin and gibberellin signal transduction under conditions of salt stress which facilitates further gene characterization. Das et al. (2020) performed an RNA-seq study to screen the transcriptome of little millet cultivar and revealed differences in regulatory mechanisms governed by differential gene expression patterns in response to drought stress and salinity stress. The results produced by this study could be exploited for further utilization of candidate genes in molecular breeding approaches, thereby, imparting resilience to cultivars. The transcriptomic analysis was conducted by Pan et al. (2019) in fox tail millet cultivars by employing RNA-seq. The study findings displayed the DEGs’ involvement in ion transmembrane transport, redox equilibrium, secondary metabolism, polyamine, and phenylpropanoid biosynthesis in response to salinity. From the study conducted by Puranik et al. (2011) in fox tail millet cultivars, a total of 249 non-redundant ESTs was identified by random EST sequencing and macroarray analysis of these clones. He reported the presence of 159 differentially expressed genes in response to salinity stress, with 115 genes upregulated and 44 downregulated with preferential expression of transcription factors and signaling genes. Sreenivasulu et al. (2004) showed the differential expression of genes governing the cellular metabolism and H2O2 scavenging as a defensive strategy against salt-induced oxidative damage while working on fox tail millet cultivars. The results of transcriptomics revealing their potentiality under salinity stress are furnished in Table 3.
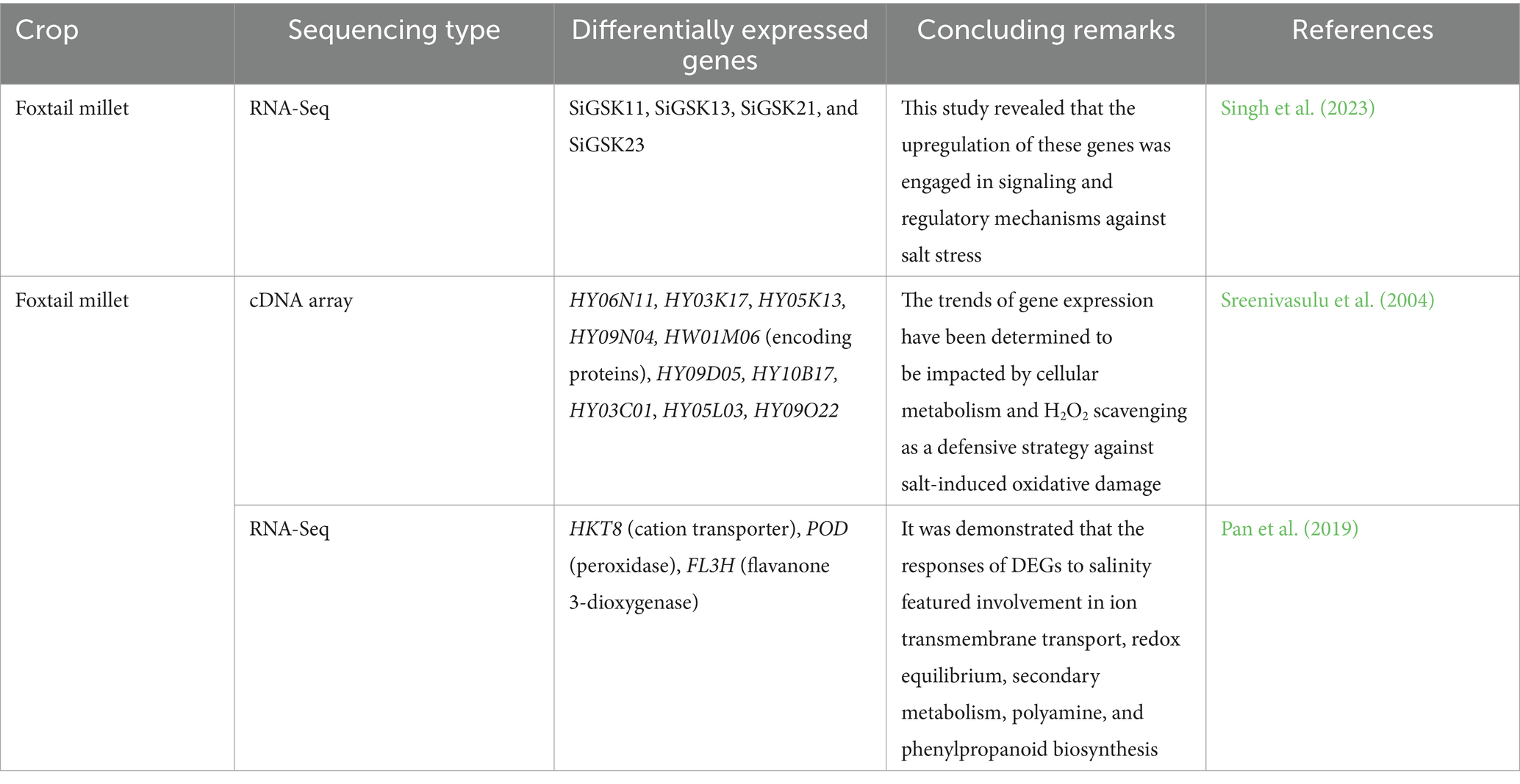
Table 3. Differentially expressed genes in response to salinity.
Genome-wide identification of genes and their transcriptomic profiling driven by transcription factors furnish the groundwork for functional characterization and the unveiling of regulation mechanisms in plant stress responses. This can be exploited for crop enhancement through genome editing and molecular breeding methods, augmenting crop yield and guaranteeing food security in the near future.
3.3 Heat stress response
Heat stress has been viewed as a potential risk to food production in an ever-shifting landscape which seriously impedes nearly all facets of crop growth irrevocably damaging the cellular homeostasis and deteriorating the structural and functional proteins & metabolites. This further leads to the activation of signal molecules like heat shock proteins (HSPs) and molecular chaperones (Sung et al., 2003). The morphological, cellular, physiological, and biochemical changes spurred under heat stress are eventually regulated by the encoding of distinct genes. The transcriptome analysis over heat stress sheds illumination on the regulatory functions of the gene spectrum through the clear comprehension of molecular pathways. This has been rendered more facile by the growing wealth of genome and transcriptomic data (Sailaja et al., 2014). In addition to high dynamics over genomics, the complex nature of transcriptomics is advanced at multiple stages of post-transcriptional regulation, notably, alternate splicing, polyadenylation, RNA editing, and post-transcriptional gene silencing revealing its application in heat stress tolerance. A study was conducted by Hu et al. (2024) for screening foxtail millet cultivars by RNA-seq and revealed the expression of seven SiSUT and 24 SiSWEET genes corresponding to sugar transport and dry matter production. This study serves as a guide explaining the yield condition of the crop when exposed to heat stress. Goyal et al. (2023) reported the differential expression of 684 genes under heat stress treatment with upregulated genes almost thrice that of downregulated genes corresponding to the ERF, WRKY, MYB, NAC, DREB, bZIP, and HSF transcription factor families. This study underpins the candidate genes for their effective utilization in screening the germplasm for heat stress tolerance in finger millet. In the study conducted by Liu et al. (2022) in fox tail millet cultivars by using the RNA-Seq platform, it inferred that shade tolerance is ascribed to genetic regulation of these genes, whose expression has been implicated in photosystem phytochemistry, thiamine functioning, and hormone mechanisms. The research efforts in contemplation with heat stress tolerance are enumerated in Table 4.
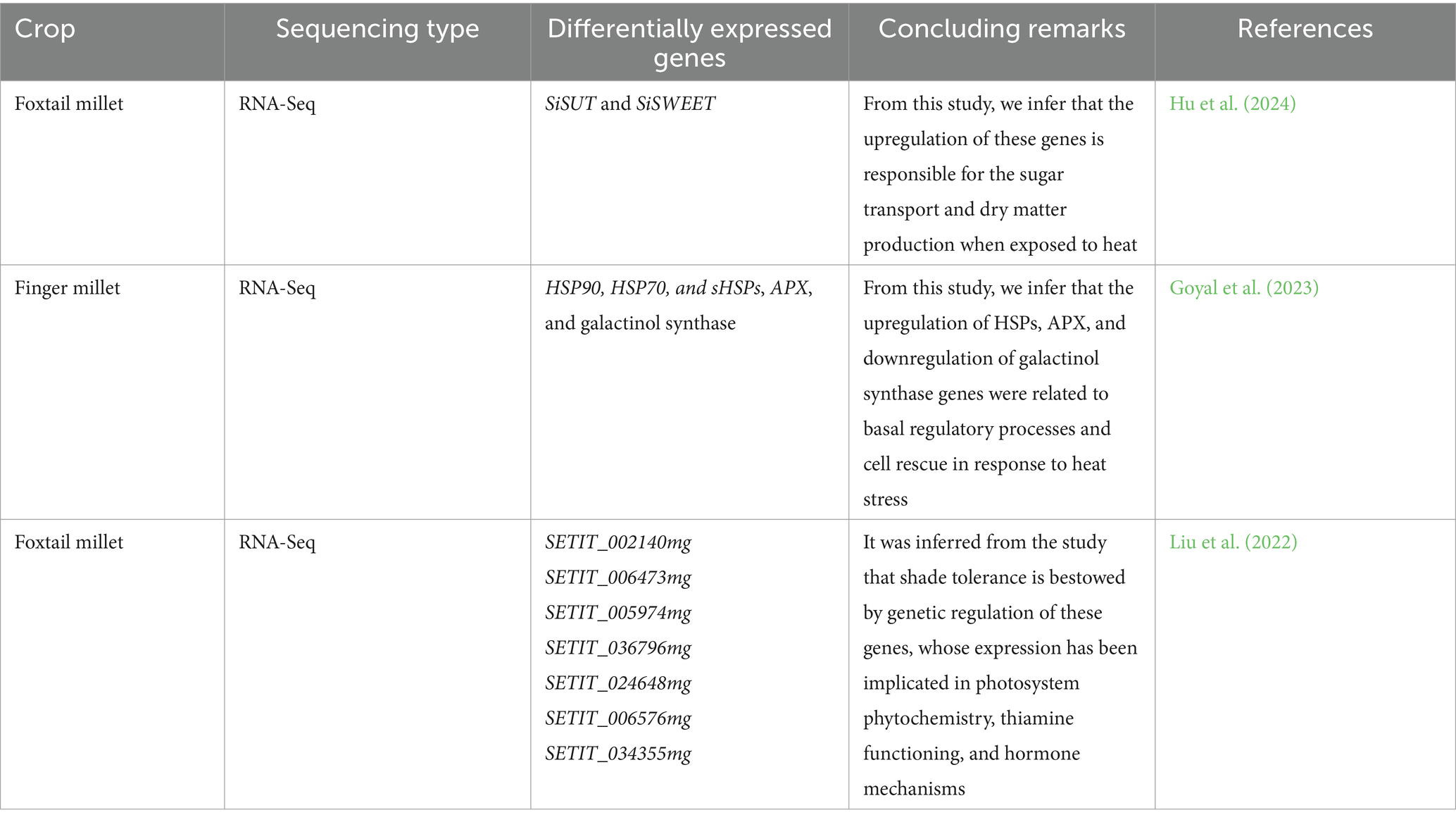
Table 4. Differentially expressed genes in response to heat.
The above-mentioned studies suggest that further investigation into the significance of these genes in controlling thermal stress responses in the corresponding millet crops might lead to the exploring of new avenues for cultivar design by conventional and transgenic approaches.
4 Transcription factors expressed in response to abiotic stress in millets
Transcription factors, known to be significant regulators of gene expression, are indispensable for orchestrating the ideal functioning of several metabolic processes. This process is achieved by adhering itself to cis-regulatory regions located in the promoter region, thereby modifying the target gene’s expression profile (Kajal et al., 2024; Rai et al., 2021). These transcription factors (TFs) constitute the major molecular triggers responsible for regulating how plants develop in response to varying forms of stress by the functional analysis of TFs and their interactions elucidating various signaling cascades (Manna et al., 2021; Khan et al., 2018). Significant advancement has been driven by comprehending the way TFs impact resilience to abiotic stresses like drought with the substantial number of strong candidate genes. Nevertheless, it remains tricky to pin down the transcription factor families and leverage them. Furthermore, academia tends to face pressing difficulties in plants carrying such TF genes that provide the predicted degree of tolerance in field circumstances. For instance, in the reports presented by Das et al. (2020), WRKY transcription factors WRKY1 and WRKY2 were found to be highly expressed in leaves of little millet, shifting by more than 13 and 7-fold, respectively, in the wake of drought stress. Conversely, WRKY3 was almost solely upregulated in root tissue, upgrading by more than 50-fold in response to both salinity and drought. Pan et al. (2020) revealed the mechanism of salinity tolerance in foxtail millet by the functioning of transcription factors PAP1 (MYB75), PAP2 (MYB90), TT2 (MYB123) regulating the activity of anthocyanin gene transcription, thereby elevating the tolerance to salt stress. This study infers that these TFs were found to be potential candidates in the regulation of several responsive upstream and downstream genes, thereby rendering resilience. Xu et al. (2019) demonstrated the role of bHLH, WRKY, NAC, ERF, and MYB transcription factors in response to drought stress regulation and function beside bHLH TF in the control of ROS-mediated signaling thus linking resilience to drought stress. The research efforts in finger millet carried out by Rahman et al. (2014) found the upregulation of transcription factors, i.e., NAC domain, MYB family, bZIP transcription factor, and WRKY29 in regulation to salinity stress in addition to AP2/EREBP family of transcription factors mediating response against abiotic stresses hormone-dependent gene expression. Dehydration-responsive transcription factors viz., bZIP, ERF, NAC, WRKY, bHLH, and MYB were upregulated displaying the differential expression pertaining to dehydration stress in kodo millet by Suresh et al. (2022). These TFs can be considered for further validation based on their contribution to abiotic stress. Some important transcription factors concerning abiotic stress are presented in Table 5.

Table 5. Transcription factors activated in response to abiotic stresses.
5 Omics techniques for abiotic stress resilience in millets
The exploitation of plant species’ genetic resources has sparked owing to the next-generation nucleic acid sequencing technologies, which have opened the door for the advancement of draft genome sequencing, whole genome resequencing, and genotype-by-sequencing platforms (Salgotra and Chauhan, 2023; Ganal et al., 2014). These genomic resources tend to be examined to track down the genomic location or gene(s) that contribute to the desired attribute, which could eventually be introduced via breeding or genetic engineering techniques to the susceptible/elite species.
Abiotic stress tolerance in plants corresponds to a challenging quantitative characteristic monitored by diminutive main-effect quantitative trait loci (QTLs) or the mutually reinforcing effects of several genes (Fan et al., 2015; Hasanuzzaman et al., 2015; Bansal et al., 2014). The majority of the millet species are innately resilient to a variety of abiotic stressors, thereby serving as an intriguing potential source of QTLs or alleles pertinent to environmental hardships that could ultimately be used to further enhance crops. Precise genomic areas linked to the intended phenotype might be geared toward the susceptible crops by the prudent mapping of putative QTLs. Among the minor millets, foxtail millet was found to be extensively studied and genomic information generated, followed by finger millet and proso millet (Dwivedi et al., 2012). Being a naturally stress-resilient millet crop, it is necessary for foxtail millet crop to delineate their contribution to stress tolerance. Singh et al. (2016) conducted an extensive characterization of foxtail millet cultivars contrasting for stress tolerance. He identified 20, 9, 27, 20, and 37 genes belonging to SiHSP100, SiHSP90, SiHSP70, SiHSP60, and SisHSP families, respectively, among which SisHSP-27 revealed the higher expression on exposure to heat stress and salinity stress showing lower levels of methylation. Expression profiling of these genes revealed their potential in conferring abiotic stress tolerance. Comprehensive genome analysis was performed in foxtail millet by Yadav et al. (2016) and they identified 53 SET domain-containing genes. The upregulation of the SiSET14 gene was observed under cold stress and was confirmed to be the candidate gene after its overexpression in yeast. Similar studies by Qie et al. (2014) revealed the presence of 18 QTLs concerning drought and osmotic stress from an interspecific mapping population derived from a cross between the foxtail millet species. A comprehensive genome-wide analysis was performed in foxtail millet by Khan et al. (2014) and he identified 355 mature miRNAs encoding various DNA protein and transcription factors crucial for abiotic stress tolerance. The miRNAs, miR171b were reported to show tolerance to cold stress and miR6248a to dehydration stress. From the aforementioned reports, we infer that the genes identified through genome-wide analysis serve as potential cues in elucidating the response of foxtail millet to abiotic stress tolerance while the studies in other minor millets were still at infancy. However, comparative genomics, which relies on the conservation across closely related plant species, is an intriguing way to acquire information for other millet species with completely unexplored genomes (Akpınar et al., 2013). In addition, species-specific genomic characteristics can also be traversed by utilizing fully annotated reference genome sequences, for instance, homeologous genes with orthologous relationships (Wicker et al., 2011; Glover et al., 2016).
With the advancement in microarray and next-generation sequencing (NGS) technologies, crop genomes and transcriptomes are widely characterized to unravel the molecular response of crops to abiotic stress (Rensink and Robin Buell, 2005; Varshney et al., 2009). Apart from transcription regulation, other significant regulatory mechanisms that govern the potency and precision of plant behaviors to abiotic stress include the translation of mRNA into proteins and their alterations. While transcript data shed light on the protein variant synthesis, sophisticated proteomics techniques are required to identify and measure the post-translational modifications of proteins (Vanderschuren et al., 2013). Heat shock proteins, other chaperones, and enzymes that scavenge reactive oxygen species (ROS) are some of the pathways and proteins that are often modulated by abiotic stressors, despite their complexity and diversity (Kosová et al., 2011). In a proteomic study conducted by Pan et al. (2020) in foxtail millet cultivars using LC–MS/MS analysis, he identified 759 and 990 sites concerning 484 and 633 proteins under salinity in An04 (salt sensitive cultivar) and Yugu2 (salt-tolerant cultivar), respectively, and 1,264 and 1,131 phosphorylation sites corresponding to 789 and 731 proteins were identified between these two varieties before and after salt stress response. This study displayed the differential phosphoprotein regulation in signal transduction, gene expression, and ion transport. From this phosphoproteomic investigation, we infer that this study serves as a gateway to comprehending the regulatory networks involved under stress and further insights into the adaptation mechanisms. Research findings from a study conducted by Pan et al. (2018) in foxtail millet for drought response revealed differential expression of 2,474 proteins among which 321 exhibited expression changes concerning photosynthesis, carbon metabolism, protein synthesis, and ROS scavenging. This study also reported the significance of post-translational modification when subjected to drought based on inconsistent results between protein and related mRNAs in gene expression regulation. Li et al. (2019) carried out proteomic analysis deploying two-dimensional electrophoresis (2-DE) coupled with matrix-assisted laser desorption/ionization-tandem time-of-flight (MALDI-TOF/TOF) analysis in foxtail millet cultivars and identified 104 differentially abundant proteins (DAPs). These DAPs were found to be involved in the regulation of metabolic pathways viz., protein folding, starch and sucrose metabolism, glycolysis/gluconeogenesis, biosynthesis of amino acids, detoxification and defense, protein degradation, tricarboxylic acid cycle, protein synthesis, energy metabolism, pentose phosphate pathway, and signal transduction. We infer from the aforementioned reports that this serves as a valuable inventory of proteins involved in drought stress response. A large-scale transcriptome-proteome data set was derived by Li et al. (2021) in finger millet in response to drought stress. He performed integrated analysis combining proteome and transcriptome and identified 1,305 differentially expressed proteins (DEPs) corresponding to the DEGs (DEPs-DEGs) enriched in hydrolase activity, glycosyl bond formation, oxidoreductase activity, carbohydrate binding, and unsaturated fatty acids metabolism. Two hub genes (BAMY2 and ISA1) were found to be upregulated and reported to be potential candidates for drought tolerance. It is inferred from this study that this coordinated effort of transcriptome and proteome provides valuable insights into enhanced drought tolerance in finger millet.
In pursuant to most abiotic stress events, plant metabolism is disrupted possibly due to a diminished production of metabolic enzymes, scarcity of substrate, high demand for particular chemicals, or an assortment of these parameters (Obata and Fernie, 2012; Bowne et al., 2011). In order to preserve vital metabolism and adapt by acquiring a new steady state in light of the current stress conditions, the metabolic framework must be reformed. This urges the need for metabolomics in understanding the chemical responses during the growth and development of stress-responsive plants (Sharma et al., 2018; Arbona et al., 2013; Nakabayashi et al., 2015). However, the full potential of metabolomics was less discussed in the minor millets owing to its difficulty to estimate, unlike the case with transcriptome and proteome, as it is not associated with the genome (Carrera et al., 2021; Roychoudhury et al., 2011). A metabolomic assay was performed by Pan et al. (2020) in foxtail millet cultivar in response to salinity in which 28 classes of metabolites were observed, more specifically flavonoids functioning as antioxidants. He also revealed the functional role of metabolic pathways viz., flavonoid biosynthesis, starch, and sucrose metabolism, glutathione metabolism, ascorbate and aldarate metabolism, and glycerophospholipid metabolism while coping with salt stress. Lysophospholipids were found to be functional signal transduction components when subjected to stress. From this study, we infer that the enhanced level of flavonoids and lysophospholipids provides deep insights into understanding the metabolic mechanisms associated with the salt stress. Dhawale (2022) carried out the metabolomic analysis in little millet for drought tolerance by employing gas chromatography–mass spectrometry (GC–MS). He displayed the 60 untargeted metabolites and 31 metabolic pathways, thereby providing valuable information about the metabolomic profile of the little millet cultivar in response to drought. In proso millet, Ma et al. (2023) conducted metabolomic profiling utilizing ultra-high-pressure liquid chromatography and identified five DAMs (l-alanine, l-aspartic acid, biliverdin, l-glutamic acid, and phosphoric acid) concerning to photosynthetic pathway and four DAMs (5-l-glutamine)-l-amino acid (l-gamma-glutamyl-l-amino acid), 5-oxoproline, Cys-Gly (l-cysteinylglycine), and l-glutamic acid (l-glutamate) concerning to antioxidant machinery. This study revealed the compensatory effect of DAMs in response to alkaline stress. This metabolic study was also extended to orphan millet crop, viz., fonio millet for studying the drought stress response by using GC–MS analysis, and he observed 12 significant metabolites, mainly overaccumulation of sugars, fatty acids, and siloxanes. We infer from this study that this could shed light on exploiting the drought-tolerant mechanisms in fonio millet. The above-discussed investigations could provide new insights into the metabolomics of abiotic stress tolerance in millet crop species.
Despite the progress of genomics, transcriptomics, proteomics, and metabolomics, the area of research in phenomics and ionomics is still lagging behind, which is critical for understanding the mechanisms of abiotic stress response. The above-mentioned omics approaches determine to generate large data sets to provide thorough understanding of the plant system. When compared with individual techniques, systems biology and integrating omics approach offer a more comprehensive molecular perspective on the biology of plants. Nevertheless, owing to the inherent data disparities between omics platforms, integrating them remains a challenging task (Macklin, 2019). Thus, rather than performing separate investigation efforts, it is imperative to establish a highly integrated community for plant systems biology research and to create and exchange datasets, software tools, and other resources as depicted in Figure 3. Given the necessity of increased abiotic stress tolerance under unknown climate scenarios, this is exceedingly significant.
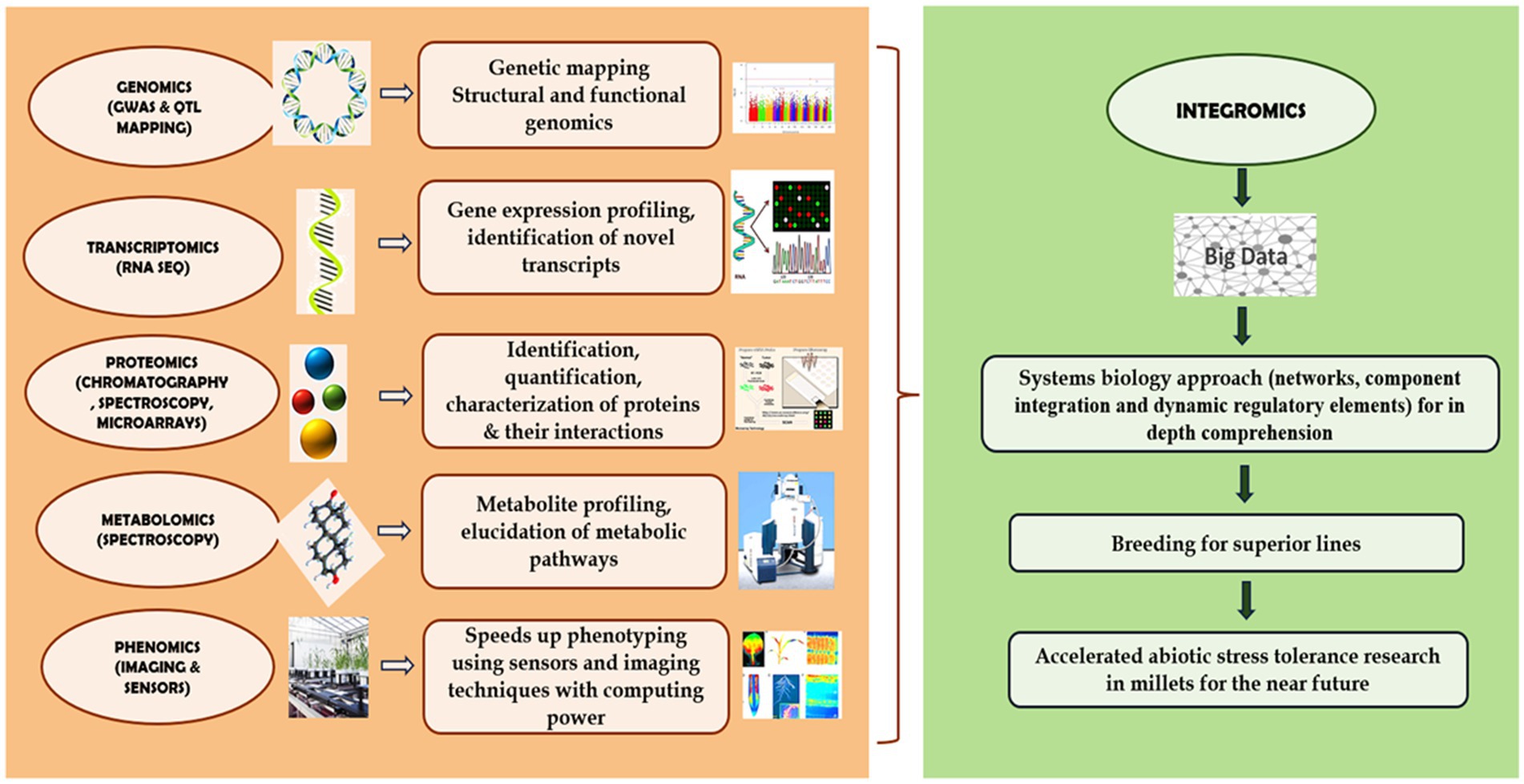
Figure 3. Flow chart of spatial transcriptomics integrated with single-cell transcriptomics for climate-resilient agriculture. Spatial transcriptomics, which enables the simultaneous visualization and quantification of gene expression within intact tissue sections or spatially resolved samples, begins by collecting frozen tissue sections to provide spatial information by in situ capture method, which is performed by coupling of RNA microarray with the spatial barcode. This spatial information generated can also be directed toward library construction and sequencing approaches. Besides, single-cell transcriptomics targeted toward analyzing the gene expression profiles at the level of individual cells within a heterogeneous population, begins with tissue disassociation generating a single-cell barcode, which is currently subjected to sequencing. The sequencing information will be currently tested for data analysis for studying the quality control, read alignment, gene expression quantification, dimensionality reduction, and cell clustering at both the gene and cellular levels for identification and characterization of genes present in the cell/tissue followed by their further validation using qRT-PCR. Genome-wide association studies (GWAS) help in the identification of the regions in the genome associated with the particular trait, and when coupled with bioinformatic tools, they analyze the gene sequences and identify the conserved domains. These validated candidate genes can currently be effectively used in breeding programs through marker-assisted selection (MAS) or genetic engineering approaches for crop improvement concerning climate resilience.
6 Way forward
When the plants are subjected to abiotic stress, the orchestrated activity of single cells in the respective tissues conduces to plant function. A substantial breakthrough arose from the assessment of genetic variation using transcriptomic analysis in expanding our comprehension of plant growth mechanisms and response to stimuli. Nevertheless, this approach loses cell heterogeneity information as it is capable of generating average cell data and is forbidden to delve into an immense number of cells (Bawa et al., 2022). This constraint of transcriptomic analysis opened avenues for the paradigm shift in technological advancement, for instance, cell transcriptomics, which examines the gene expression of individual cells and spatial transcriptomics for positioning cell types, which are identified by mRNA reads in appropriate locations within the histological sections.
6.1 Single-cell transcriptomics
Single-cell transcriptomics assays the concentration of RNA residing in thousands of genes deploying droplet-based methods (encapsulating individual cells in droplets containing barcoded beads for mRNA quantification), laser microdissection (LMD, laser-based isolation of single cells), or fluorescence-activated cell sorting (FACS, the marker gene-associated fluorescence associated with a particular cell type to separate individual cells). This is worked out with the intent to investigate the gene expression level of individual cells within a given population, which facilitates the study of change in cellular developmental pathways, the simulation of transcriptional dynamics, and the deciphering of diverse cell populations (Kanter and Kalisky, 2015). An upsurge in studies on plant single-cell transcriptome studies using high-throughput droplet-based scRNA-seq became apparent in 2019 enabling the formulation of extensive trajectory maps of development and the hunt for new distinctive cell markers (Rich-Griffin et al., 2020). Despite the limitations of single-cell transcriptomics, being strenuous coupled with low throughput, have witnessed an intriguing increase in single-cell transcriptomics in rice, this study revealed the transcriptional regulation of genes in a cell type-specific manner and uncovered the modifications to cell populations in the wake of abiotic stress (Wang Y. et al., 2021). This study in wheat aided in research into genetics by leveraging transcriptional networks, anticipating genetic redundancy, and locating potential genes linked to variables related to agricultural productivity (Wang J. et al., 2021) with the dearth of research findings on millets. Henceforth, concerning millets’ unique characteristics in the context of climate change, single-cell transcriptome research in millets is crucial to feature profound discoveries into the control of gene expression at the cellular level in the aftermath of abiotic stress (Figure 4).
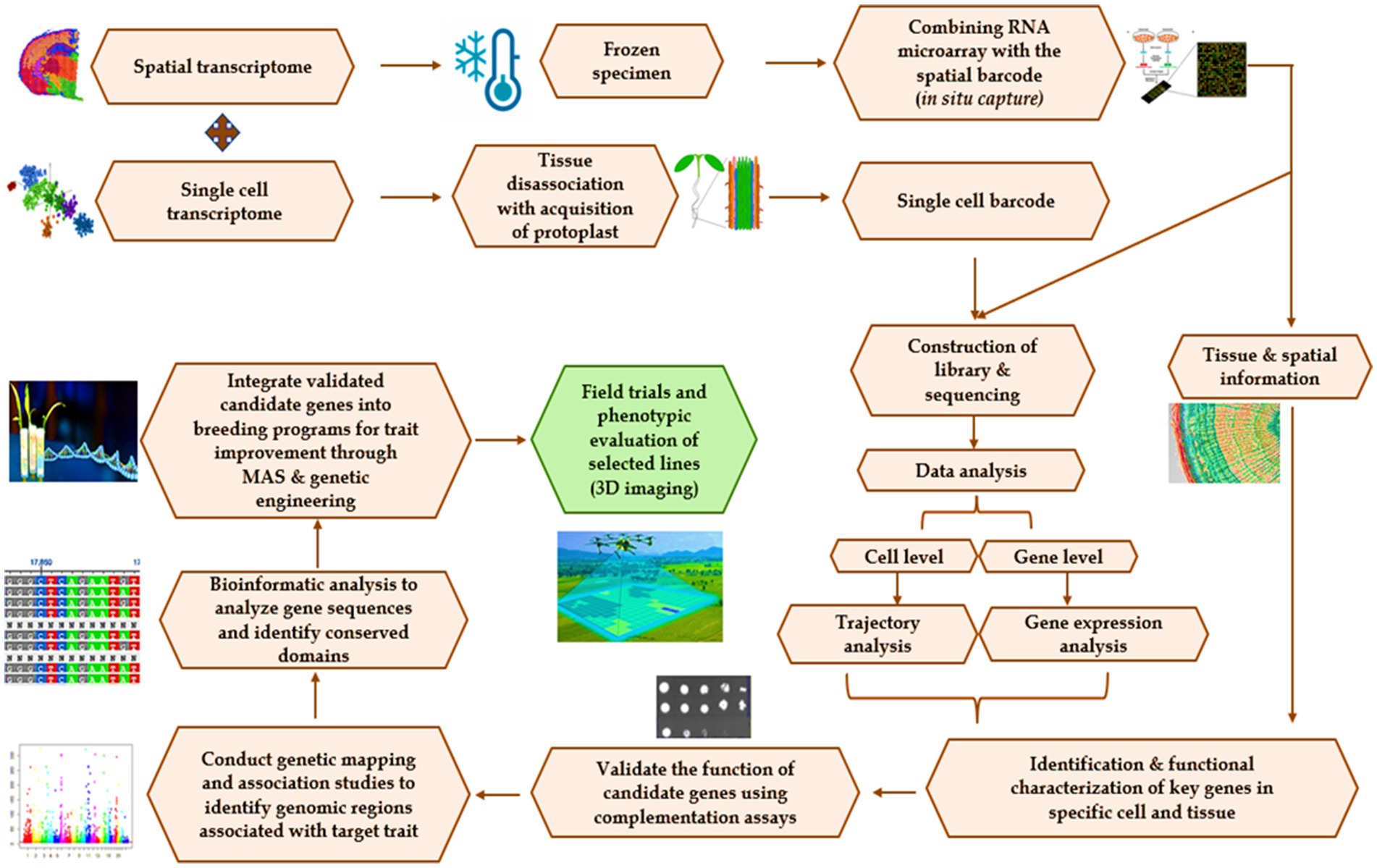
Figure 4. Process chart integrating the multi-omics approaches with systems biology for accelerated abiotic stress tolerance.
6.2 Spatial transcriptomics
The growing interest in spatial transcriptomics has been driven by the critical capacity to correlate gene expression and cell location knowing that the spatial position of cells is a prerequisite for figuring out their physical interactions in the development of tissues (Moses and Pachter, 2022). This approach signified the continued retention of the spatial information of individual cells simultaneously distinguishing cell kinds and assessing their heterogeneity (Larsson et al., 2022). Improvements in recent years in ST have made it achievable to examine single cells of plant tissues from a spatial standpoint utilizing laser capture microscopy methods (sorts the target tissue areas and isolated tissues are subjected to gene expression analysis); imaging-based methods (high-resolution microscopy imaging to identify the spatial location of target RNAs in the cells); and in situ capture sequencing methods (leveraging barcodes to retrieve the associated cells’ mRNA for reverse transcription, imprinting spatial data onto the RNA transcripts followed by sequencing). These techniques bring about significant results in distinguishing different cell types, reestablishing cell-fate lineages, and elucidating how cells interact (Yin et al., 2023). From the findings of Moreno-Villena et al. (2022) in Portulaca, the differential spatial gene expression of the C4 and CAM pathway was demonstrated in mesophyll and bundle sheath cells, when subjected to drought. This research finding revealed the intriguing viewpoint in the C4 photosynthetic pathway through a reliable examination of plant tissue architecture. Consequently, by virtue of millets being C4 plants, spatial transcriptomics study can also be tapped into millet research for improved resilience to climate change by examining the spatial expression of genes when exposed to stress conditions. The integration of spatial transcriptomics into single-cell transcriptomics makes it feasible for plant researchers to uncover the key genes present in specific cells or tissues against the backdrop of abiotic stress as depicted in Figure 2.
6.3 Integrating spatial transcriptomics with 3D imaging in plants
In the course of the intervening years, the field of spatial transcriptomics has swiftly emerged, predominantly because of its practical applicability in spatially resolving, capturing, and capturing RNA from tissue slices at or near single-cell resolution. In addition, it involves subcellular mapping to provide a two-dimensional spatial analysis of gene expression (Cho et al., 2021). The enticing likelihood of acquiring these data in three dimensions currently exists, but it will entail reassembling the tissue from a serial array of two-dimensional optical or tactile tissue slices. This is made possible by the application of optical techniques such as light sheet fluorescence microscopy (LSFM), expansion microscopy (ExM), X-ray microscopy (XRM), and X-ray computed tomography (XCT) (Gurazada et al., 2021). Their unparalleled 3D visual information may enhance spatial transcriptomic techniques by functioning as a model volumetric framework of individual plant cells within particular plant tissues and organs (Cox et al., 2022). When superimposed by the expression data over this framework, it will provide a real-world spatial context, which is achieved through building an enclosure by piling several serial sections; then followed by merging their imaging and biochemical data into a computationally generated 3D framework utilizing computational tools like ST spot detector. This detector is customized to assess ST imaging data interspersed over a high-quality tissue picture, thereby revealing gene expression information specified by 2D spatial coordinates (Wong et al., 2018). This approach can be used in practical research of abiotic stress resilience with the molecular information provided by spatial transcriptomics. When ST merges with the phenotyping deploying the optical imagery, it provides deep insights into plant system architecture adaptability to the stress conditions.
6.4 Transcriptomics and plant stress memory
The tendency of plants to “remember” previous instances of stress and react differentially to successive stress exposures by means of imprinting, training, priming, hardening, conditioning, or acclimation is referred to as memory response (Avramova, 2015). Berry and Dean (2015) delineated between cis molecular memory, which necessitates physical signatures embarked on the chromatin, such as DNA methylation or histone changes, and trans memory, which thrives by feedback loops and the intensity of a diffusible signal that traverses via cytosol segmentation. Scientific inquiry into the accrual of pivotal signaling molecules, transcription factors (TFs), and chromatin alterations have contributed to broadening our awareness about the processes engaged in the stress-induced memory response (Crisp et al., 2016). Memory patterns were readily apparent intuitively as an outcome of inquiries into plant transcriptional memory, which is in dire need of rigorous characterization to get biologically significant revelations, particularly in the realm of the RNA-Seq research. This allows for concurrent assessment of a multitude of memory genes and hyperlinking of these patterns to biological pathways associated with stress (Ding et al., 2013). In a study conducted by Ding et al. (2012) in Arabidopsis, he reported the spike in transcript levels of RD29B and RAB18 genes on multiple exposures to dehydration stress and explained that the tenacity of RNA pol II and methylation marks correlated with the duration of transcriptional memory, corroborating the contention that chromatin remodeling processes play a part in the memory response. This relevance of transcriptomics in plant stress memory driven by stress conditions is still in its infancy in millets, which serves as a potent mechanism for probing the basic facets of the transcriptional memory response in plants.
7 Conclusion
Abiotic stresses weigh in almost 50% of the average returns for cereal crops, making them the root cause of agricultural catastrophe on a global scale. Consequently, broadening the cultivation is necessary to bridge the widening disparity among agricultural supplies and the growing populace via fostering resilience to stress by implementing changes at cellular, physiological, and molecular levels. Taking advantage of their suitability for cultivation in arid and semi-arid areas, millets are characterized and uniquely defined by their propensity to adapt to detrimental abiotic stresses. However, their effective use in agricultural development is endangered by an acute shortage of germplasm and a dearth of genetic diversity information, which could be accomplished by conventional and molecular approaches. In contrast to conventional breeding with its limitations, leveraging biotechnological strategies to circumvent abiotic challenges productively calls for an in-depth consciousness of the intended crop species. This can be achieved through the assessment from the molecular level down to the whole plant, which is conceivable by spurring advancement of transcriptomics. The empirical investigation has been upgraded by next-generation sequencing tools, which enable the decoding of cell transcript sequences/differentially expressed genes avoiding the complications triggered by huge genomes, reminiscently millets. Genome-scale data collected via various simulations pursuant to distinct ecological circumstances are readily analyzed by adopting advanced bioinformatics algorithms to discern and deduce significant patterns in tandem with specific cis-elements and regulatory genes, such as TFs aiding in examining the metabolic and regulatory networks. Recent advancements in transcriptomics, viz., single-cell and spatial transcriptomics, are found to decipher gene expression at the cellular level from a spatial standpoint. This consolidated effort of transcriptomics which assists in uncovering and scoring molecular markers, precisely SNPs, ascertaining the discrepancies in gene sequences and gene expression patterns, may further be utilized in MAS or genetic transformations to breed for varieties with climate resilience, thereby contributing to millet improvement.
Author contributions
GS: Writing – original draft, Writing – review & editing. RR: Conceptualization, Supervision, Writing – review & editing, Validation. NB: Conceptualization, Supervision, Validation, Writing – review & editing. IK: Writing – review & editing, Supervision. VS: Writing – review & editing, Supervision. VM: Writing – review & editing, Supervision.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aidoo, M. K., Bdolach, E., Fait, A., Lazarovitch, N., and Rachmilevitch, S. (2016). Tolerance to high soil temperature in foxtail millet (Setaria italica L.) is related to shoot and root growth and metabolism. Plant Physiol. Biochem. 106, 73–81. doi: 10.1016/j.plaphy.2016.04.038
Ajithkumar, I. P., and Panneerselvam, R. (2014). ROS scavenging system, osmotic maintenance, pigment and growth status of Panicum sumatrense Roth. Under drought stress. Cell Biochem. Biophys. 68, 587–595. doi: 10.1007/s12013-013-9746-x
Akpınar, B. A., Lucas, S. J., and Budak, H. (2013). Genomics approaches for crop improvement against abiotic stress. Sci. World J. 2013:361921. doi: 10.1155/2013/361921
Arbona, V., Matías, M., Carlos de, O., and Aurelio, G-C. (2013). Metabolomics as a tool to investigate abiotic stress tolerance in plants. Int J Mol Sci. 68, 4885–4911.
Arzani, A., and Ashraf, M. (2016). Smart engineering of genetic resources for enhanced salinity tolerance in crop plants. Crit. Rev. Plant Sci. 35, 146–189. doi: 10.1080/07352689.2016.1245056
Avramova, Z. (2015). Transcriptional ‘memory’of a stress: transient chromatin and memory (epigenetic) marks at stress-response genes. Plant J. 83, 149–159. doi: 10.1111/tpj.12832
Bajay, S. K., Ferreira, R. C. U., Pimenta, R. J. G., Mancini, M., Aono, A. H., Niederauer, G. F., et al. (2023). “Transcriptomics in agricultural sciences: capturing changes in gene regulation during abiotic or biotic stress” in Transcriptome profiling (Cambridge, MA: Academic Press), 257–283.
Balsamo, R. A., Vander Willigen, C., Bauer, A. M., and Farrant, J. (2006). Drought tolerance of selected Eragrostis species correlates with leaf tensile properties. Ann. Bot. 97, 985–991. doi: 10.1093/aob/mcl068
Bandyopadhyay, T., Muthamilarasan, M., and Prasad, M. (2017). Millets for next generation climate-smart agriculture. Front. Plant Sci. 8:1266. doi: 10.3389/fpls.2017.01266
Bansal, K. C., Lenka, S. K., and Mondal, T. K. (2014). Genomic resources for breeding crops with enhanced abiotic stress tolerance. Plant Breed. 133, 1–11. doi: 10.1111/pbr.12117
Banshidhar, S. P., Singh, A., Jaiswal, P., Singh, M. K., Meena, K. R., and Singh, S. K. (2023). The potentialities of omics resources for millet improvement. Funct. Integr. Genomics 23:210. doi: 10.1007/s10142-023-01149-2
Bawa, G., Liu, Z., Xiaole, Y., Qin, A., and Sun, X. (2022). Single-cell RNA sequencing for plant research: insights and possible benefits. Int. J. Mol. Sci. 23:4497. doi: 10.3390/ijms23094497
Bennetzen, J. L., Schmutz, J., Wang, H., Percifield, R., Hawkins, J., Pontaroli, A. C., et al. (2012). Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 30, 555–561. doi: 10.1038/nbt.2196
Berry, S., and Dean, C. (2015). Environmental perception and epigenetic memory: mechanistic insight through FLC. Plant J. 83, 133–148. doi: 10.1111/tpj.12869
Bowne, J., Bacic, A., Tester, M., and Roessner, U. (2011). “Abiotic stress and metabolomics” in Annual plant reviews volume 43: biology of plant metabolomics. ed. R. D. Hall (Hoboken, NJ: Blackwell Publishing Ltd.).
Cao, X., Yulu, H., Song, J., Feng, H., Wang, J., Chen, L., et al. (2022). Transcriptome sequencing and metabolome analysis reveals the molecular mechanism of drought stress in millet. Int. J. Mol. Sci. 23:10792. doi: 10.3390/ijms231810792
Carrera, F. P., Noceda, C., Maridueña-Zavala, M. G., and Cevallos-Cevallos, J. M. (2021). Metabolomics, a powerful tool for understanding plant abiotic stress. Agronomy 11:824. doi: 10.3390/agronomy11050824
Cho, C.-S., Xi, J., Si, Y., Park, S.-R., Hsu, J.-E., Kim, M., et al. (2021). Microscopic examination of spatial transcriptome using Seq-scope. Cell 184, 3559–3572. doi: 10.1016/j.cell.2021.05.010
Choudhary, P., Shukla, P., and Muthamilarasan, M. (2023). Genetic enhancement of climate-resilient traits in small millets: a review. Heliyon 9:e14502. doi: 10.1016/j.heliyon.2023.e14502
Cox, K. L., Gurazada, S. G. R., Duncan, K. E., Czymmek, K. J., Topp, C. N., and Meyers, B. C. (2022). Organizing your space: the potential for integrating spatial transcriptomics and 3D imaging data in plants. Plant Physiol. 188, 703–712. doi: 10.1093/plphys/kiab508
Crisp, P. A., Ganguly, D., Eichten, S. R., Borevitz, J. O., and Pogson, B. J. (2016). Reconsidering plant memory: intersections between stress recovery, RNA turnover, and epigenetics. Sci. Adv. 2:e1501340. doi: 10.1126/sciadv.1501340
Das, R. R., Pradhan, S., and Parida, A. (2020). De-novo transcriptome analysis unveils differentially expressed genes regulating drought and salt stress response in Panicum sumatrense. Sci. Rep. 10:21251. doi: 10.1038/s41598-020-78118-3
Dhawale, R. N. (2022). Metabolomic profiling of drought-tolerant little millet (Panicum sumatrense L.) genotype in response to drought stress. Pharm. Innov. 11, 1754–1762.
Ding, Y., Fromm, M., and Avramova, Z. (2012). Multiple exposures to drought'train'transcriptional responses in Arabidopsis. Nat. Commun. 3:740. doi: 10.1038/ncomms1732
Ding, Y., Liu, N., Virlouvet, L., Riethoven, J.-J., Fromm, M., and Avramova, Z. (2013). Four distinct types of dehydration stress memory genes in Arabidopsis thaliana. BMC Plant Biol. 13, 1–11. doi: 10.1186/1471-2229-13-229
Dwivedi, S. L., Upadhyaya, H. D., Senthilvel, S., Hash, C. T., Fukunaga, K., Diao, X., et al. (2012). Millets: Genetic and genomic resources. 247–375.
Fan, Y., Shabala, S., Ma, Y., Rugen, X., and Zhou, M. (2015). Using QTL mapping to investigate the relationships between abiotic stress tolerance (drought and salinity) and agronomic and physiological traits. BMC Genomics 16, 1–11. doi: 10.1186/s12864-015-1243-8
Fujimura, S., Shi, P., Iwama, K., Zhang, X., Gopal, J., and Jitsuyama, Y. (2012). Effects of CO2 increase on wheat growth and yield under different atmospheric pressures and their interaction with temperature. Plant Product. Sci. 15, 118–124. doi: 10.1626/pps.15.118
Ganal, M. W., Wieseke, R., Luerssen, H., Durstewitz, G., Graner, E.-M., Plieske, J., et al. (2014). “High-throughput SNP profiling of genetic resources in crop plants using genotyping arrays” in Genomics of Plant Genetic Resources. eds. R. Tuberosa, A. Graner, and E. Frison (Dordrecht: Springer).
Glover, N. M., Redestig, H., and Dessimoz, C. (2016). Homoeologs: what are they and how do we infer them? Trends Plant Sci. 21, 609–621. doi: 10.1016/j.tplants.2016.02.005
Goyal, E., Singh, A. K., Mahajan, M. M., and Kumar, K. (2023). Comparative transcriptome analysis in contrasting finger millet (Eleusine coracana (L.) Gaertn) genotypes for heat stress. Mol. Biol. Rep. 51:283. doi: 10.1007/s11033-024-09233-x
Guo, Y., Hao, D., Wang, X., Wang, H., Zehao, W., Yang, P., et al. (2023). Comparative transcriptomics reveals key genes contributing to the differences in drought tolerance among three cultivars of foxtail millet (Setaria italica). Plant Growth Regul. 99, 45–64. doi: 10.1007/s10725-022-00875-0
Guo, J., Huang, Z., Sun, J., Cui, X., and Liu, Y. (2021). Research progress and future development trends in medicinal plant transcriptomics. Front. Plant Sci. 12:691838. doi: 10.3389/fpls.2021.691838
Gurazada, S. G., Ranjan, K. L., Jr, C., Czymmek, K. J., and Meyers, B. C. (2021). Space: the final frontier—achieving single-cell, spatially resolved transcriptomics in plants. Emerg. Topics Life Sci. 5, 179–188. doi: 10.1042/ETLS20200274
Han, F., Sun, M., He, W., Guo, S., Feng, J., Wang, H., et al. (2022). Transcriptome analysis reveals molecular mechanisms under salt stress in leaves of foxtail millet (Setaria italica l.). Plan. Theory 11:1864. doi: 10.3390/plants11141864
Hasanuzzaman, M., Rajib Roychowdhury, J. O. Y. D. I. P., Karmakar, N., Dey, K. N., and Fujita, M. (2015). “Recent advances in biotechnology and genomic approaches for abiotic stress tolerance in crop plants” in Genomics and proteomics: Concepts, technologies and applications. ed. K. F. Au (Burlington: Apple Academic Press).
Hittalmani, S., Mahesh, H. B., Shirke, M. D., Biradar, H., Govindareddy Uday, Y. R., Aruna, H. C. L., et al. (2017). Genome and transcriptome sequence of finger millet (Eleusine coracana (L.) Gaertn.) provides insights into drought tolerance and nutraceutical properties. BMC Genomics 18, 1–16. doi: 10.1186/s12864-017-3850-z
Hu, M., Yang, M., Liu, J., Huang, H., Luan, R., Yue, H., et al. (2024). Physiological investigation and transcriptome analysis reveals the mechanisms of Setaria italica’s yield formation under heat stress. Int. J. Mol. Sci. 25:3171. doi: 10.3390/ijms25063171
Jha, U. C., Bohra, A., Jha, R., and Parida, S. K. (2019). Salinity stress response and ‘omics’ approaches for improving salinity stress tolerance in major grain legumes. Plant Cell Rep. 38, 255–277. doi: 10.1007/s00299-019-02374-5
Kajal, R. O., Lohani, P., Deshmukh, R., and Salvi, P. (2024). Engineering the transcriptional regulatory network to improve abiotic stress tolerance in crop plants: taming the tough time. J. Plant Growth Regul. 43, 25–37. doi: 10.1007/s00344-023-11057-1
Kanter, I., and Kalisky, T. (2015). Single cell transcriptomics: methods and applications. Front. Oncol. 5:53. doi: 10.3389/fonc.2015.00053
Khan, S.-A., Li, M.-Z., Wang, S.-M., and Yin, H.-J. (2018). Revisiting the role of plant transcription factors in the battle against abiotic stress. Int. J. Mol. Sci. 19:1634. doi: 10.3390/ijms19061634
Khan, Y., Yadav, A., Bonthala, V. S., Muthamilarasan, M., Yadav, C. B., and Prasad, M. (2014). Comprehensive genome-wide identification and expression profiling of foxtail millet [Setaria italica (L.)] miRNAs in response to abiotic stress and development of miRNA database. Plant Cell Tissue Organ. Cult. 118, 279–292. doi: 10.1007/s11240-014-0480-x
Kido, É. A., Ferreira-Neto, J. R. C., Pandolfi, V., de Melo Souza, A. C., and Benko-Iseppon, A. M. (2016). “Drought stress tolerance in plants: insights from transcriptomic studies” in Drought Stress Tolerance in Plants. eds. M. Hossain, S. Wani, S. Bhattacharjee, D. Burritt, and L. S. Tran (Cham: Springer).
Kosová, K., Vítámvás, P., Prášil, I. T., and Renaut, J. (2011). Plant proteome changes under abiotic stress—contribution of proteomics studies to understanding plant stress response. J. Proteome 74, 1301–1322. doi: 10.1016/j.jprot.2011.02.006
Larsson, L., Bergenstråhle, L., He, M., Andrusivova, Z., and Lundeberg, J. (2022). Snapshot: spatial transcriptomics. Cell 185:2840. doi: 10.1016/j.cell.2022.06.002
Lata, C., and Prasad, M. (2014). Association of an allele-specific marker with dehydration stress tolerance in foxtail millet suggests SiDREB2 to be an important QTL. J. Plant Biochem. Biotechnol. 23, 119–122. doi: 10.1007/s13562-013-0193-y
Li, J., Li, X., Yang, Q., Luo, Y., Gong, X., Zhang, W., et al. (2019). Proteomic changes in the grains of foxtail millet (Setaria italica (L.) beau) under drought stress. Span. J. Agric. Res. 17:e0802. doi: 10.5424/sjar/2019172-14300
Li, J., Wang, Y., Wang, L., Zhu, J., Deng, J., Tang, R., et al. (2021). Integration of transcriptomic and proteomic analyses for finger millet [Eleusine coracana (L.) Gaertn.] in response to drought stress. PLoS One 16:e0247181. doi: 10.1371/journal.pone.0247181
Liu, D., Yanjiao, C., Zilong, Z., Jing, Z., Suying, L., and Zhengli, L. (2022). Transcriptome analysis and mining of genes related to shade tolerance in foxtail millet. Setaria italica (L.) P. Beauv. Royal Soc. Open Sci. 9:220953.
Lowe, R., Shirley, N., Bleackley, M., Dolan, S., and Shafee, T. (2017). Transcriptomics technologies. PLoS Comput. Biol. 13:e1005457. doi: 10.1371/journal.pcbi.1005457
Ma, Q., Wang, H., Enguo, W., Yuhao Yuan, Y., Feng, L. Z., and Feng, B. (2023). Comprehensive physiological, transcriptomic, and metabolomic analysis of the response of Panicum miliaceum L. roots to alkaline stress. Land Degrad. Dev. 34, 2912–2930. doi: 10.1002/ldr.4656
Macklin, P. (2019). Key challenges facing data-driven multicellular systems biology. Gigascience 8:giz127. doi: 10.1093/gigascience/giz127
Manna, M., Thakur, T., Chirom, O., Mandlik, R., Deshmukh, R., and Salvi, P. (2021). Transcription factors as key molecular target to strengthen the drought stress tolerance in plants. Physiol. Plant. 172, 847–868. doi: 10.1111/ppl.13268
McGettigan, P. A. (2013). Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 17, 4–11. doi: 10.1016/j.cbpa.2012.12.008
Moreno-Villena, J. J., Zhou, H., Gilman, I. S., Lori Tausta, S., Maurice Cheung, C. Y., and Edwards, E. J. (2022). "spatial resolution of an integrated C4+ CAM photosynthetic metabolism." science. Advances 8:eabn2349. doi: 10.1126/sciadv.abn2349
Morozova, O., Hirst, M., and Marra, M. A. (2009). Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genomics Hum. Genet. 10, 135–151. doi: 10.1146/annurev-genom-082908-145957
Moses, L., and Pachter, L. (2022). Museum of spatial transcriptomics. Nat. Methods 19, 534–546. doi: 10.1038/s41592-022-01409-2
Mukami, A., Ng’etich, A., Syombua, E., Oduor, R., and Mbinda, W. (2020). Varietal differences in physiological and biochemical responses to salinity stress in six finger millet plants. Physiol. Mol. Biol. Plants 26, 1569–1582. doi: 10.1007/s12298-020-00853-8
Nakabayashi, R., and Saito, K. (2015). Integrated metabolomics for abiotic stress responses in plants. Curr. Opin. Plant Biol. 24, 10–16. doi: 10.1016/j.pbi.2015.01.003
Narayanrao, D. R., Tomar, R. S., Sm, P., Jasminkumar, K., Ashish, G., Chauhan, N. M., et al. (2023). De novo transcriptome sequencing of drought tolerance–associated genes in little millet (Panicum sumatrense L.). Funct. Integr. Genomics 23:303. doi: 10.1007/s10142-023-01221-x
Nejat, N., Ramalingam, A., and Mantri, N. (2018). Advances in transcriptomics of plants. Adv. Biochem. Eng. Biotechnol. 164, 161–185. doi: 10.1007/10_2017_52
Obata, T., and Fernie, A. R. (2012). The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 69, 3225–3243. doi: 10.1007/s00018-012-1091-5
Oshunsanya, S. O., Nwosu, N. J., and Li, Y. (2019). "Abiotic stress in agricultural crops under climatic conditions." M. Jhariya, A. Banerjee, R. Meena, and D. Yadav (eds) Sustainable agriculture, forest and environmental management. Springer, Singapore.
Pan, J., Li, Z., Dai, S., Ding, H., Wang, Q., Li, X., et al. (2020). Integrative analyses of transcriptomics and metabolomics upon seed germination of foxtail millet in response to salinity. Sci. Rep. 10:13660. doi: 10.1038/s41598-020-70520-1
Pan, J., Li, Z., Wang, Q., Garrell, A. K., Liu, M., Guan, Y., et al. (2018). Comparative proteomic investigation of drought responses in foxtail millet. BMC Plant Biol. 18, 1–19. doi: 10.1186/s12870-018-1533-9
Pan, J. W., Li, Z., Wang, Q. G., Guan, Y. A., Li, X. B., Ding, G. H., et al. (2019). Transcriptomics analysis of NaCl response in foxtail millet (Setaria italica L.) seeds at germination stage. Sci. Agric. Sin. 52, 3964–3975. doi: 10.3864/j.issn.0578-1752.2019.22.003
Parvathi, M. S., Karaba, N. N., Nanja Reddy, Y. A., Naika, M. B. N., and Channabyre, M. V. G. (2019). Transcriptome analysis of finger millet (Eleusine coracana (L.) Gaertn.) reveals unique drought responsive genes. J. Genet. 98:46.
Puranik, S., Jha, S., Srivastava, P. S., Sreenivasulu, N., and Prasad, M. (2011). Comparative transcriptome analysis of contrasting foxtail millet cultivars in response to short-term salinity stress. J. Plant Physiol. 168, 280–287. doi: 10.1016/j.jplph.2010.07.005
Qie, L., Jia, G., Zhang, W., Schnable, J., Shang, Z., Li, W., et al. (2014). Mapping of quantitative trait locus (QTLs) that contribute to germination and early seedling drought tolerance in the interspecific cross Setaria italica× Setaria viridis. PloS one 9:e101868. doi: 10.1371/journal.pone.0101868
Qin, L., Erying, C., Feifei, L., Xiao, Y., Zhenyu, L., Yanbing, Y., et al. (2020). Genome-wide gene expression profiles analysis reveal novel insights into drought stress in foxtail millet (Setaria italica L.). Int J Mol Sci. 21:8520.
Rahman, H. N., Jagadeeshselvam, R., Valarmathi, B., Sachin, R., Sasikala, N., Senthil, D., et al. (2014). Transcriptome analysis of salinity responsiveness in contrasting genotypes of finger millet (Eleusine coracana L.) through RNA-sequencing. Plant Mol. Biol. 21, 485–503.
Rai, G. K., Jamwal, G., Rai, G., and Singh, M. (2021). Understanding transcription factors in plant response to drought stress. Indian J. Agric. Sci. 34, 116–125. doi: 10.5958/0974-4479.2021.00016.2
Rajawat, J. (2018). "Transcriptomics." Omics approaches, technologies and applications: integrative approaches for understanding OMICS data. Dorval: Dokumen.pub.
Ravikesavan, R., Jeeva, G., Poornima Jency, J., Muthamilarasan, M., and Francis, N. (2023). “Kodo millet (Paspalum scorbiculatum L.)” in Neglected and underutilized crops. eds. M. Farooq and K. H. M. Siddique (Cambridge, MA: Academic Press).
Rensink, W. A., and Robin Buell, C. (2005). Microarray expression profiling resources for plant genomics. Trends Plant Sci. 10, 603–609. doi: 10.1016/j.tplants.2005.10.003
Rich-Griffin, C., Stechemesser, A., Finch, J., Lucas, E., Ott, S., and Schäfer, P. (2020). Single-cell transcriptomics: a high-resolution avenue for plant functional genomics. Trends Plant Sci. 25, 186–197. doi: 10.1016/j.tplants.2019.10.008
Rodrigues, C. M., Mafra, V. S., and Machado, M. A. (2014) "Transcriptomics." A. Borém and R. Fritsche-Neto Omics in plant breeding : 33–57. Hoboken, NJ: Wiley.
Roychoudhury, A., Datta, K., and Datta, S. K. (2011). “Abiotic stress in plants: from genomics to metabolomics” in Omics and plant abiotic stress tolerance. ed. S. G. Sarvajeet (Potomac, MD: Bentham Science).
Sage, R. F., and Zhu, X.-G. (2011). Exploiting the engine of C4 photosynthesis. J. Exp. Bot. 62, 2989–3000. doi: 10.1093/jxb/err179
Sailaja, B., Mangrauthia, S. K., Sarla, N., and Voleti, S. R. (2014). "Transcriptomics of heat stress in plants." P. Ahmad, M. Wani, M. Azooz, and L. S. Phan Tran Improvement of Crops in the Era of Climatic Changes : Springer, New York, NY.
Salgotra, R. K., and Chauhan, B. S. (2023). Genetic diversity, conservation, and utilization of plant genetic resources. Genes 14:174. doi: 10.3390/genes14010174
Sharma, K., Sarma, S., Bohra, A., Mitra, A., Sharma, N. K., and Kumar, A. (2018). Plant metabolomics: An emerging technology for crop improvement. New visions in plant science. London: Intech open.
Shelden, M. C., and Roessner, U. (2013). Advances in functional genomics for investigating salinity stress tolerance mechanisms in cereals. Front. Plant Sci. 4:123. doi: 10.3389/fpls.2013.00123
Singh, R. K., Jaishankar, J., Muthamilarasan, M., Shweta, S., Dangi, A., and Prasad, M. (2016). Genome-wide analysis of heat shock proteins in C4 model, foxtail millet identifies potential candidates for crop improvement under abiotic stress. Sci. Rep. 6:32641. doi: 10.1038/srep32641
Singh, J., Varshney, V., Tak, N., and Jha, S. (2023). Genome-wide identification and expression analysis of glycogen synthase kinase encoding genes in foxtail millet (Setaria italica L.) under salinity, dehydration, and oxidative stress. Plant Stress 8:100165. doi: 10.1016/j.stress.2023.100165
Smirnoff, N., and Colombe, S. V. (1988). Drought influences the activity of enzymes of the chloroplast hydrogen peroxide scavenging system. J. Exp. Bot. 39, 1097–1108. doi: 10.1093/jxb/39.8.1097
Sreenivasulu, N., Manoela, M., Harischandra, S. P., Ulrich, W., and Winfriede, W. (2004). Transcriptome changes in foxtail millet genotypes at high salinity: Identification and characterization of a PHGPX gene specifically up-regulated by NaCl in a salt-tolerant line. J. Plant Physiol. 161, 467–477.
Sung, D.-Y., Kaplan, F., Lee, K.-J., and Guy, C. L. (2003). Acquired tolerance to temperature extremes. Trends Plant Sci. 8, 179–187. doi: 10.1016/S1360-1385(03)00047-5
Suresh, B. V., Choudhary, P., Aggarwal, P. R., Rana, S., Singh, R. K., Ravikesavan, R., et al. (2022). De novo transcriptome analysis identifies key genes involved in dehydration stress response in kodo millet (Paspalum scrobiculatum L.). Genomics 114:110347. doi: 10.1016/j.ygeno.2022.110347
Tian, B. H., Liu, L. Y., Zhang, L. X., Song, S. X., Wang, J. G., Wu, L. F., et al. (2015). Characterization of culm morphology, anatomy and chemical composition of foxtail millet cultivars differing in lodging resistance. J. Agric. Sci. 153, 1437–1448. doi: 10.1017/S0021859614001105
Tyagi, P., Singh, D., Mathur, S., Singh, A., and Ranjan, R. (2022). Upcoming progress of transcriptomics studies on plants: an overview. Front. Plant Sci. 13:1030890. doi: 10.3389/fpls.2022.1030890
Vanderschuren, H., Lentz, E., Zainuddin, I., and Gruissem, W. (2013). Proteomics of model and crop plant species: status, current limitations and strategic advances for crop improvement. J. Proteome 93, 5–19. doi: 10.1016/j.jprot.2013.05.036
Varshney, R. K., Nayak, S. N., May, G. D., and Jackson, S. A. (2009). Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 27, 522–530. doi: 10.1016/j.tibtech.2009.05.006
Wang, Y., Huan, Q., Li, K., and Qian, W. (2021). Single-cell transcriptome atlas of the leaf and root of rice seedlings. J. Genet. Genomics 48, 881–898. doi: 10.1016/j.jgg.2021.06.001
Wang, X., Li, N., Li, W., Gao, X., Cha, M., Qin, L., et al. (2020). Advances in Transcriptomics in the response to stress in plants. Global Med. Gen. 7, 030–034. doi: 10.1055/s-0040-1714414
Wang, J., Li, Y., Tianwen, W., Miao, C., Xie, M., Ding, B., et al. (2021). Single-cell-type transcriptomic analysis reveals distinct gene expression profiles in wheat guard cells in response to abscisic acid. Funct. Plant Biol. 48, 1087–1099. doi: 10.1071/FP20368
Wicker, T., Mayer, K. F. X., Gundlach, H., Martis, M., Steuernagel, B., Scholz, U., et al. (2011). Frequent gene movement and pseudogene evolution is common to the large and complex genomes of wheat, barley, and their relatives. Plant Cell 23, 1706–1718. doi: 10.1105/tpc.111.086629
Wong, K., Navarro, J. F., Bergenstråhle, L., Ståhl, P. L., and Lundeberg, J. (2018). ST spot detector: a web-based application for automatic spot and tissue detection for spatial Transcriptomics image datasets. Bioinformatics 34, 1966–1968. doi: 10.1093/bioinformatics/bty030
Xu, B.-Q., Gao, X.-L., Gao, J.-F., Jing, L. I., Pu, Y. A. N. G., and Feng, B.-L. (2019). Transcriptome profiling using RNA-seq to provide insights into foxtail millet seedling tolerance to short-term water deficit stress induced by PEG-6000. J. Integr. Agric. 18, 2457–2471. doi: 10.1016/S2095-3119(19)62576-1
Yadav, C. B., Muthamilarasan, M., Dangi, A., Shweta, S., and Prasad, M. (2016). Comprehensive analysis of SET domain gene family in foxtail millet identifies the putative role of SiSET14 in abiotic stress tolerance. Sci. Rep. 6:32621. doi: 10.1038/srep32621
Yin, R., Xia, K., and Xun, X. (2023). Spatial transcriptomics drives a new era in plant research. Plant J. 116, 1571–1581. doi: 10.1111/tpj.16437
Yu, A., Zhao, J., Wang, Z., Cheng, K., Zhang, P., Tian, G., et al. (2020). Transcriptome and metabolite analysis reveal the drought tolerance of foxtail millet significantly correlated with phenylpropanoids-related pathways during germination process under PEG stress. BMC Plant Biol. 20, 1–17. doi: 10.1186/s12870-020-02483-4
Yuan, Y., Caoyang, W., Liu, L., Ma, Q., Yang, Q., and Feng, B. (2022). Unravelling the distinctive growth mechanism of proso millet (Panicum miliaceum L.) under salt stress: from root-to-leaf adaptations to molecular response. GCB Bioenergy 14, 192–214. doi: 10.1111/gcbb.12910
Yuan, Y. H., Li, J., Ma, H. C., Yang, Q. H., Liu, C. J., and Feng, B. L. (2021). Salt-tolerant broomcorn millet (Panicum miliaceum L.) resists salt stress via modulation of cell wall biosynthesis and Na+ balance. Land Degrad. Dev. 32, 518–532. doi: 10.1002/ldr.3717
Keywords: abiotic stress, millets, transcriptomics, differentially expressed genes, transcription factors
Citation: Sanku G, Rajasekaran R, Boopathi NM, Krishnamoorthy I, Santhanakrishnan VP and Mani V (2024) Transcriptomic response of minor millets to abiotic stresses. Front. Sustain. Food Syst. 8:1435437. doi: 10.3389/fsufs.2024.1435437
Edited by:
Rubab Zahra Naqvi, National Institute for Biotechnology and Genetic Engineering, PakistanReviewed by:
Mumtaz Ali Saand, Zhejiang University, ChinaMuhammad Jawad Akbar Awan, National Institute for Biotechnology and Genetic Engineering, Pakistan
Copyright © 2024 Sanku, Rajasekaran, Boopathi, Krishnamoorthy, Santhanakrishnan and Mani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ravikesavan Rajasekaran, Y2hpdGh1cmFndWxAZ21haWwuY29t