 Ashwini Talakayala†1,2
Ashwini Talakayala†1,2 Gopala Krishna Mekala†1
Gopala Krishna Mekala†1 Malireddy K. Reddy3
Malireddy K. Reddy3 Srinivas Ankanagari†2*
Srinivas Ankanagari†2* Mallikarjuna Garladinne†1*
Mallikarjuna Garladinne†1*- 1Plant Molecular Biology Laboratory, Agri Biotech Foundation, Hyderabad, India
- 2Department of Genetics, Osmania University, Hyderabad, India
- 3Crop Improvement Group, International Center for Genetic Engineering and Biotechnology, New Delhi, India
Clustered regularly interspaced short palindromic repeats (CRISPR) associated protein (CRISPR/Cas9) is an adaptive immune system of bacteria to counter the impending viral pathogen attack. With persistent improvements, CRISPR has become a versatile tool for developing molecular immunity against viruses in plants. In the current report, we utilized the Cas9 endonuclease and dual 20 bp-gRNAs targeting two different locations in single-stranded DNA-A of AC1 (rep protein) and AV1 (coat protein) of mungbean yellow mosaic virus for achieving resistance in greengram. The cotyledonary nodal explants were infected with Agrobacterium strain EHA105 harboring pMDC100-Cas9 with AC1 and AV1 gRNA cassettes and generated transgenic plants. The integration of Cas9 and gRNA cassettes in the transformed plants of greengram were confirmed by PCR and dot blot assays. Agroinfiltrated T2 transgenic lines exhibited minimal mosaic symptoms. A drastic reduction in the accumulation of AC1 and AV1 was observed in T2 transformed lines. The T7EI assay indicated that AC1 fragments were edited at a frequency of 46%, 32%, 20%, and AV1 at 38.15%, 40%, and 21.36% in MYMV infected greengram lines T2-6-2-3, T2-6-4-4, and T2-6-4-7, respectively. The manipulation of resistance to MYMV through the editing of the pathogen genome using the CRISPR/Cas9 tool can be a powerful approach to combat viruses and develop resistance in greengram.
Introduction
Greengram (Vigna radiata L) is the third most important legume crop that meets the dietary needs of a large population across the globe. It is being cultivated in about seven million hectares (mha), particularly in the tropical and sub-tropical regions in India (Mishra et al., 2020) with a productivity of 750 kg/ha in India (Nair and Schreinemachers, 2020). The productivity of greengram is low and stagnant due to its susceptibility to various stresses (Mekala et al., 2016), particularly Yellow Mosaic Disease (YMD). YMD is caused by Mungbean Yellow Mosaic Virus (MYMV) and transmitted by whitefly (Bemisia tabaci) in a continuous and circulative manner, where its effectiveness varies with that of the genotype and growth stage (Sudha et al., 2022). MYMV is a bipartite Begomavirus belonging to the family of Geminiviridae (Selvi et al., 2006). It exhibits two genomes, 2.7 kb of DNA-A that code for six genes, including coat protein (AV1), replication-associated protein (AC1), transcriptional activator protein (TrAP), replication enhancer protein (REn) (Fontenelle et al., 2007) and 2.6 kb of DNA-B possessing mobility protein (BC1) and nuclear shuttle protein (BV1) (Hehnle et al., 2004). The symptoms of yellow mosaic disease include yellowing or chlorosis of leaves followed by necrosis, which causes fewer flowers and pods, with immature and abnormal seeds, and reducing the yield of the crop (Karthikeyan et al., 2014). The bipartite genome of this virus makes it amenable for manipulation through genome editing techniques at desired loci in a precise manner (Khan et al., 2017). Mushtaq et al. (2020) reported that CRISPR Cas has become a predominant approach in providing resistance against both DNA and RNA viruses in plants.
The CRISPR system is an antiviral defense mechanism to combat invading pathogens and archea. It relies on the guide RNAs to direct the Cas9 endonuclease to chop the double-stranded DNA in pathogens. Subsequently, these DSBs would be repaired by non-homologous end-joining (NHEJ) or by precise homology-directed repair (HDR). Following this, the unrepaired sequences would be finally degraded (Aouida et al., 2015; Piatek and Mahfouz, 2017). In most cases, mutations generated through NHEJ cannot replicate or move systemically due to the induced frameshift in the viral ORFs leading to untranslated products (Chaparro-Garcia et al., 2015; Hadidi et al., 2016).
Recently, several viral genomes have been edited by CRISPR/Cas9 conferring molecular immunity against viruses in plants. The viral genomes such as beet curly top virus (BCTV) (Ali et al., 2015b), tobacco mosaic virus (TMV) (Cody et al., 2017) beet yellow dwarf virus (BeYDV) (Liang et al., 2016), tomato yellow leaf curl virus (TYLCV) (Tashkandi et al., 2018), Merremia mosaic virus (MeMV) (Ali et al., 2016), tobacco rattle virus (TRV) (Ali et al., 2015a), beet necrotic yellow vein virus (BNYVV), and pea early browning virus (PEBV) (Ali et al., 2018) were edited through CRISPR/Cas9 and achieved durable resistance against viral pathogens, subsequently reduced viral load accumulation in transgenic plants and ceased the pathogenicity in tobacco and Arabidopsis (Talakayala et al., 2022). The transgenic plants expressing dual gRNAs targeting C1 (Replication associated protein) and IR regions of Cotton Leaf Curl Multan virus (CLCuMuV) demonstrated complete resistance to viral diseases in tobacco (Yin et al., 2019). Multiplexed gRNA strategy was applied for targeting different locations of Chilli leaf curl virus in tobacco, which exhibited a high degree of resistance against ChiLCV (Roy et al., 2019). Ji et al. (2015) reported on the induction of resistance to BSCTV (Beet Severe Curly Top Virus) in transgenic Nicotiana benthamiana and Arabidopsis thaliana by targeting a viral intergenic region (IR), CP, and Rep genes using the CRISPR-Cas9 approach.
There are limited experiments performed in crop plants using the CRISPR tool to develop resistance against viral diseases. The Banana streak virus (BSV), a double-stranded DNA Badnavirus belonging to the family of Caulimoviridae, and replicates in Musa spp., was inactivated by expressing sgRNA targeting the coding region of eBSV using the CRISPR/Cas9 tool. The resultant transgenic banana plants showed resistance against eBSV and observed minimal symptoms in comparison to non-edited control plants (Tripathi et al., 2019). CRISPR mutants have failed to exhibit immunity to the African Cassava mosaic virus in Cassava (Mehta et al., 2019). Four gRNAs targeting the MP, CP in the coding region, LIR, WDV, Rep/Rep-A protein in WDV were introduced into wheat. The transgenic plants showed resistance to WDV (wheat dwarf virus) and failed to exhibit any symptoms in wheat (Kis et al., 2019).
In the current study, we have identified two gRNAs encoding AC1 (replication-associated protein) and AV1 (coat protein) of MYMV and cloned them in pMDC100 plant expression vector with npt II as plant selection marker gene. Transgenic greengram plants were generated through Agrobacterium using cotyledonary nodes as explants. The putative transformed plants were confirmed by PCR and dot blot detection assays. The T2 generation transgenic plants were Agroinfiltrated for the evaluation of the mosaic symptoms in the leaf region. The transcripts of AC1 and AV1 were checked using quantitative real-time PCR. The viral genome editing was further screened and evaluated by using a T7EI genome editing assay. This is the first report on genome editing in greengram, which would help to curb the viral pathogens in pulse crops.
Materials and methods
Designing 20 bp sgRNAs targeting AC1 and AV1 and construction of plant expression vector for mungbean transformation
The 20 bp gRNAs were designed to target the replication protein (AC1) and coat protein (AV1) of Mungbean yellow mosaic virus genome (Figure 1) using CRISPR Direct software (http:cripsr.dbcls.jp). The developed gRNA cassettes AtUbiquitin6p-AC1 and AtUbiquitin3p-AV1 were cloned in entry vectors EV1 and EV2, respectively, by the gateway cloning approach. The Cas9 nuclease was cloned under the regulation of Double Enhancer 35S CaMV promoter in the entry vector I. Subsequently, all cassettes were sub-cloned sequentially into the final destination vector pMDC100 with npt II as a plant-selectable marker gene (Figure 2A). The pMDC100 vector containing gRNA1 (AtU6p- gRNA AC1 -AtU6t), gRNA2 (AtU3p-gRNA AV1-AtU3t), and Cas9 (DE35S-Cas9-Nos-t) cassettes was mobilized into A. tumefaciens (EHA105) using electroporation following standard protocols. The CRISPR construct was further confirmed by PCR using cassette-specific primers.
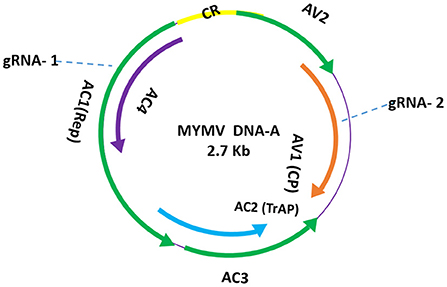
Figure 1. Schematic representation of gRNA locations in MYMV-A genome. The gRNAs designed homologs to AC1- Rep protein and AV1- coat protein (target sites are indicated in dashed arrows).

Figure 2. (A) Cloning of guide RNAs and Cas9 exonuclease in pMDC100 vector: The image depicted shows the single guide locations in MYMV genome and T-DNA region of pMDC100 vector harboring Cas9 nuclease expression cassette, gRNA1, gRNA2, and Kanamycin selectable marker. (B) Agrobacterium EHA105 mediated greengram transformation using CRISPR construct and cotyledonary nodes as explants: (A) seed germination, (B) Co cultivation of EHA105 with cotyledonary node explants, (C) selection on kanamycin medium, (D) resistant plants growing regeneration medium, (E) regenerated plants growing in elongation medium, (F) plant establishment in soil, and (G) Matured plants growing in a glass house.
Agrobacterium-mediated genetic transformation of greengram (var. LGG 460) using cotyledonary nodes as explants
A single colony of Agrobacterium (EHA105) carrying the vector, pMDC100 with CRISPR MYMV construct (Figure 2A) was inoculated in 10 ml of YEM (Yeast Extract Mannitol) broth supplemented with 50 mg/L Kanamycin, 50 mg/L Streptomycin and 20 mg/L Rifampicin. The culture was incubated at 28°C until the OD600 reached 0.8. Following this, the culture was pelleted down by centrifugation at 6,000 rpm for 5 min. The pellet was then re-suspended in 20 ml of MS basal liquid medium and 200 μM of acetosyringone was added to this suspension with OD600 of 0.5–0.6 for the induction of vir genes.
Mature greengram seeds were surface sterilized with 0.1% aqueous (w/v) HgCl2 solution for 5 min, followed by 70 % (v/v) ethanol for 1 min. Subsequently, the sterilized seeds were washed 4–5 times with sterile double distilled water. The seeds were then blot dried on sterile filter paper and inoculated on MSB5 medium supplemented with 2 mg/L 6-benzyl amino purine (BAP). The cotyledonary node explants were excised from 4 d old seedlings, and pricked with a fine needle near the axillary meristematic region. Subsequently, the explants were placed in the Agrobacterium culture and kept in an incubator with agitation for 10 min at 28°C. The infected explants were fortified with a co-cultivation medium (MSB5 + 0.5 mg/l BAP + 200 μM acetosyringone) and incubated at 25 ± 2°C for 2 days under dark conditions. Uninfected control explants were also cultured under similar conditions.
After co-cultivation, the treated explants were washed 5 times with 250 mg/l cefotaxime and dried on sterile filter paper. The explants were then inoculated on shoot induction medium (MSB5 + 0.5 mg/L BAP) supplemented with 50 mg/L Kanamycin and 250 mg/L Cefotaxime and cultured on selection media for 15 days followed by two more selection cycles on the same medium at an interval of 15 days. Regenerated shoots were transferred to sterile wet cotton supplemented with liquid MSB5 + 1 mg/L IBA medium for efficient root induction. After 15 days, the well-developed putative transformed plantlets with well-formed roots were transferred into plastic cups filled with vermiculite and acclimatized under controlled greenhouse conditions. The plants were maintained in a transgenic glasshouse until attaining maturity (Figure 2B).
Molecular confirmation of greengram transformed plants by PCR
The genomic DNA was isolated from the putative transgenics and control plants using the modified CTAB method as described by Vanti et al. (2015). The quality and quantity of DNA samples were determined by a Nano spectrophotometer (GE Healthcare). The PCR master mix was prepared by adding 2 μL 10X Taq buffer, 2.5 mM dNTPs, 10 picomoles of each primer, 5 U of Taq, and 100 ng of genomic DNA from the transformed and control plants. The reaction mixture was made up to 20 μL and kept in a thermo cycler (Thermo Scientifics) using an initial denaturation for 5 min at 94°C, followed by 35 cycles of denaturation for 40 s at 94°C, annealing for 40 s at 52°C for gRNA1 and gRNA2 cassettes, 54°C for Cas9 and 57°C for npt II, extension for 40 s at 72°C and final extension for 5 min. The PCR amplified products were resolved on 1% agarose gel, visualized, and documented in a gel doc system (G-Box Syngene, UK) (Figure 3; Table 1).
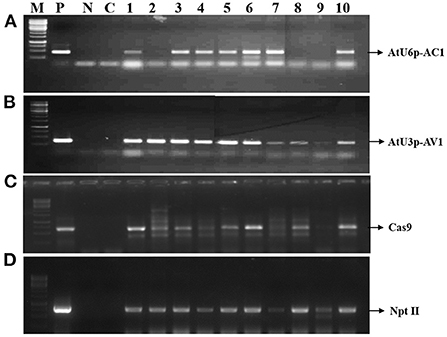
Figure 3. Molecular confirmation of putatively transformed plants (T0): the agarose gel shows the amplification of (A) AtU6p-AC1 of 480 bp, (B) AtU3p-AV1 of 420 bp, (C) Cas9 of 610 bp and, (D) npt II of 730 bp M; 1 Kb ladder, P, positive control; N, negative control; C, control; Lanes 1 to 10; transformed greengram plants.

Table 1. Primers used for detection of transgenes and transcript analysis.
Dot blot analysis
To detect the T-DNA integration in the genome of greengram, a dot blot assay was performed, by using the genomic DNA of PCR-positive T0 transformed plants. The probes AtU6p-gRNA AC1 and AtU3p-gRNA AV1 fragments were labeled with Biotin Deca DNA Labeling Kit (Thermo Fisher Scientific) as per the manufacturer's instructions. Further, 5 μg of genomic DNA from transgenic and control plant samples were spotted on a nylon membrane and cross-linked using a UV cross-linker at 1,200 kJ/cm2 energy level (UVP CL1000). Pre-hybridization, hybridization, washing, and detection of chromogram were performed using Biotin Chromogenic Detection Kit as per the manufacturer's protocols (Figure 4).
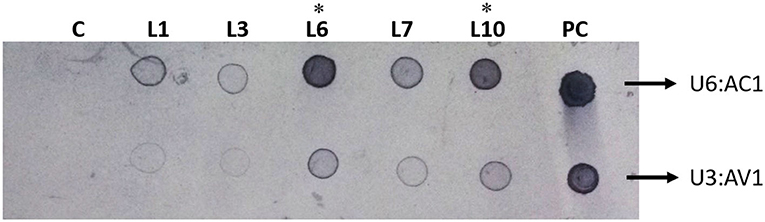
Figure 4. Chromogenic visualization of transformed cassettes in greengram plants by dot blot: DNA (5 μg) from the five PCR-positive T0 plants was loaded on a nylon membrane and probed with U6:AC1 and U3:AV1 gene fragments. The blue color precipitation was observed in Lanes 1, 3, 4, 6, and 10. No band appeared in the control plant (C). *The blue color precipitation of both gRNA cassettes was very high in lanes 6 and 10. Hence plant 6 was used for Sequencing of all fragments and also advanced to the next generation.
Agroinfiltration of CRISPR greengram plants with MYMV genome targeting clone
Initially, the MYMV genome was amplified using Φ29 DNA polymerase following the rolling circle mode of replication. Then the genome was cloned into pCAMBIA2301 vector and mobilized into Agrobacterium LBA4404 by electroporation followed by the selection (Talakayala et al., 2021). The Recombinant Agrobacterium colony was inoculated into YEM medium and grown at 28°C in an orbital shaker. Then, the overnight grown culture (OD600 = 0.3–0.4) was centrifuged and the pellet was suspended in infiltration buffer [10 mM MES (pH 5.7), 10 mM MgCl2, and 100 μM acetosyringone]. Approximately, 4–5-week-old PCR-positive transformed CRISPR plants (T2) and controls were agroinfiltrated using a needleless syringe. The experiment was performed in triplicates. The trifoliate transformed and untransformed leaves were infiltrated and the progress of the symptoms was recorded (Figures 5, 6).
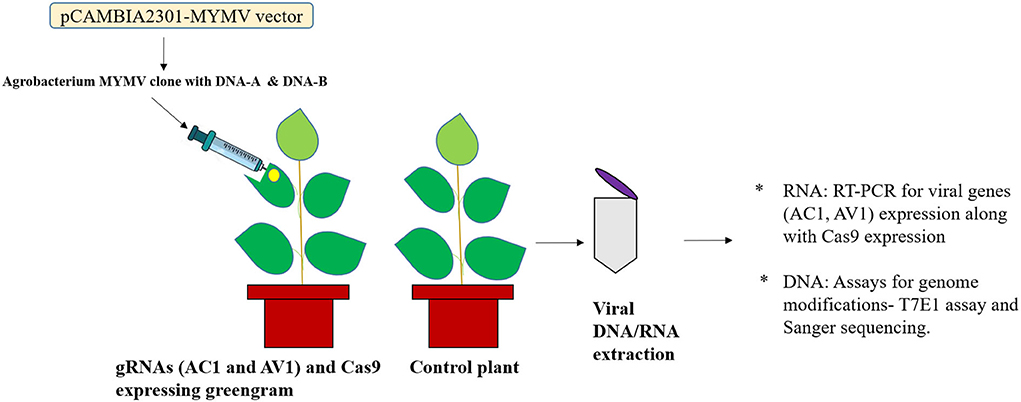
Figure 5. Schematic representation of Agroinfiltration. (A) Agrobacterium containing engineered MYMV genome was infiltrated at abaxial leaf side of control and transgenic greengram plants expressing gRNAs (AC1 and AV1) and Cas9, (B) Samples collected for molecular analysis such as RNA: for analyzing transcripts of RNA and DNA: for detection of viral genome editing using T7EI assay and Sanger sequencing.

Figure 6. Disease Phenotype of Vigna radiata (Greengram) plants of T2 in distal leaves through Agroinfiltration: (A) Infiltrate MYMV clone using a needleless syringe into control and transgenic plants, C- Control greengram leaves showing MYMV disease symptoms within 20 dpi, (B) CRISPR challenged T2 plants 1–7, the Infiltrated area showing symptoms within 20 dpi.
Quantitative real-time PCR for viral transcript analysis
The total RNA was isolated from the Agroinfiltrated leaves of transformed (T2) and control plants and about 5 μg of total RNA was reverse transcribed to cDNA using oligo dT primers according to the manufacturer's instructions (Thermo Scientifics). Quantitative real-time PCR was performed using cDNA as a template for detecting the transcripts of AC1, AV1, and Cas9 genes. The MYMV-AC1, MYMV-AV1, and Cas9 specific primers were used in this study (Table 1).
Simultaneously, relative gene expression was analyzed using semi-quantitative real-time PCR (BioRad- CFX96, USA). The real-time PCR was performed by preparing the 10 μl reaction mixture containing 5 μl SYBR Green PCR Master mix (Bio-Rad, USA), 30 ng of cDNA pool, and 500 nM of each primer (RT-AC1, RT-AV1, and Cas 9). The amplification was performed with an initial denaturation at 95°C for 5 min followed by 40 cycles of denaturation at 95°C for 10 s, annealing, and extension at 54°C for 30 s. To calculate mean relative expression levels, cDNAs from three independent samples in three replicates were used. The reaction was normalized with VrTubulin as a reference gene. After normalization, the quantity of each mRNA was calculated from the threshold points positioned in the log- linear range. Data from different cDNA samples were compared using the mean Ct values of three technical replicates and normalized with the mean Ct values of the VrTubulin gene. The relative expression ratios were calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001), and the data was analyzed through BioRad CFX Manager Software 3.1 (Figure 7).
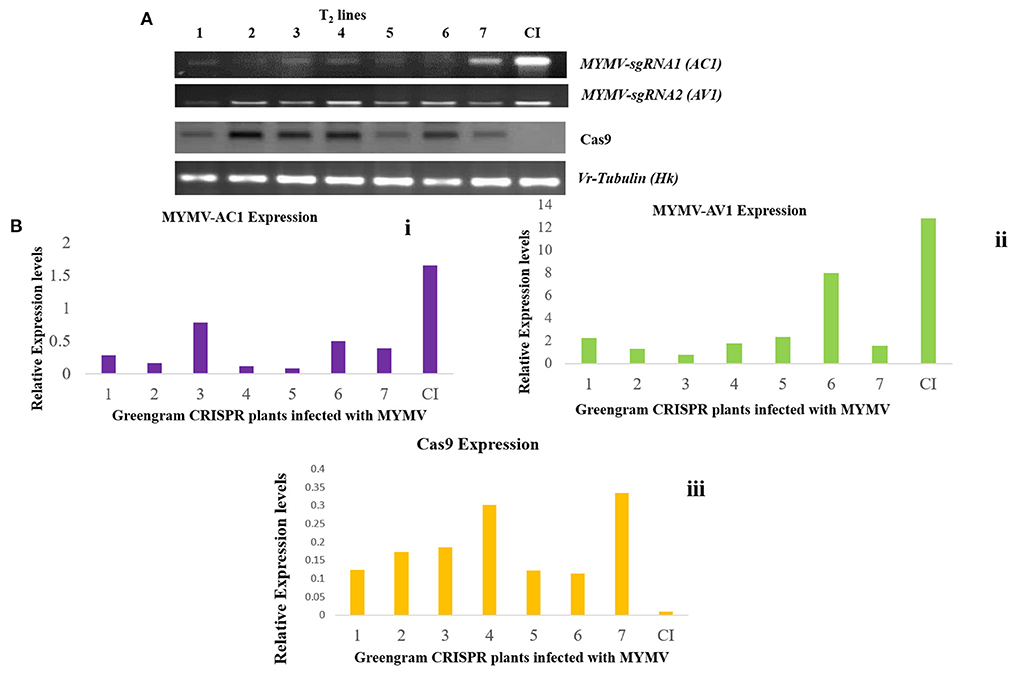
Figure 7. (A) Expression of AC1, AV1, and Cas9 genes in transgenic greengram plants- quantitative PCR showing the transcript expression pattern of MYMV-sgRNA1 (AC1), MYMVsgRNA2 (AV1), Cas9, and Vr Tubulin as reference gene in control and transgenic lines which were infected with MYMV via Agroinfiltration CI: MYMV infected control, 1–7 transgenic greengram plants challenged with MYMV clone. (B) Relative expression of AC1, AV1, and Cas9 genes in transformed and untransformed plants: viral transcript analysis: i. Accumulation of viral transcript was minimal in transformed plants, whereas control showed higher accumulation of AC1 gene, ii. AV1 gene. iii. Cas9 gene expression was normalized by using Vr-Tubulin as a reference standard in viral-challenged plants with MYMV compared with untransformed control plants. Each experiment was carried out in three replicates of treated and untreated plants.
T7EI assay for detecting mutations
To detect the mutation in viral genome, T7 Endonuclease I (T7EI) assay was performed using EnGen® Mutation Detection Kit (NEB) by following the manufacturer's instructions. In the T7EI assay, the samples which were agroinfiltrated with MYMV of T2 lines, from which the DNA fragments containing the target sites i.e., AC1 and AV1 regions covering the sgRNAs of T2 line plants were amplified from the genomic DNA of transformed and control greengram using viral genome-specific primers. The PCR products of T2-6-2-3, T2-6-4-4, and T2-6-4-7 were purified using Quiagen quick column method and their concentration was determined using a Nano spectrophotometer. About 300 ng of PCR products were subjected to denaturation and annealing by the following conditions: 95°C for 5 min, ramp down to 25°C at 0.1 C s−1, and incubated the reaction at 25°C for 30 min. The annealed PCR products were then digested with 5U of T7EI and incubated for 2 h at 37°C. The T7EI digested products were resolved on 3% agarose gel and the images were recorded through gel documentation. The intensity of DNA bands was calculated using Image-J software (http://rsbweb.nih.gov/ij/) (Figure 8A). The samples which were used for this analysis were sent for sequencing to detect the mutation in the viral genome (Figure 8B).
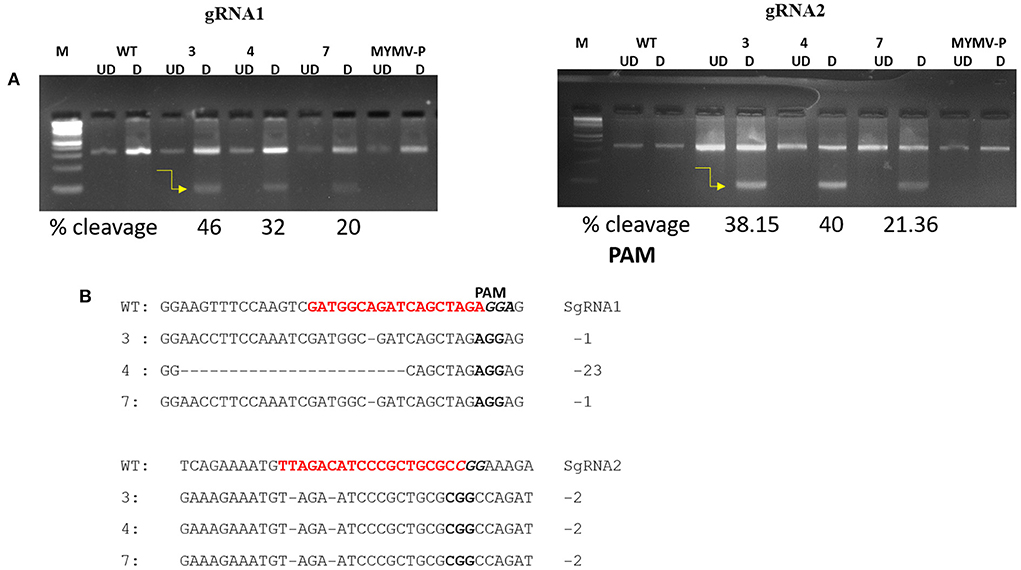
Figure 8. Detection of genome editing by T7EI mutation analysis (A) Detection of mutations and transgenes in three T2 transgenic lines. The PCR products of the MYMV region covering AC1 and AV1 sgRNAs were subjected to T7EI and resolved on 3% agarose gels. The arrow represents the DNA band of the T7EI cleavage products of T2-6-2-3 (46%), T2-6-4-4 (32%), T2-6-4-7 (20%) for gRNA1(AC1), and T2-6-2-3 (38.15%), T2-6-4-4 (40%), T2-6-4-7 (21.36%) for gRNA2 (AV1) ratio of editing. (B) A single base pair/heterozygous mutation was seen in sgRNA1 of T2-6-2-3, T2-6-4-7, and T2-6-4-4 having 23 base pair mutation, sgRNA1 target shown in red, whereas in sgRNA2, two base pair mutation was detected in all T2-6-2-3, T2-6-4-4 and T2-6-4-7, sgRNA2 target shown in red.
Results
Construction of plant expression vector and Agrobacterium-mediated greengram transformation using cotyledonary nodes as explants
The pMDC100 vector harboring Cas9, AtU6p-gRNA AC1, and AtU3p-gRNA AV1 cassettes were developed using the Gateway cloning technology and the vector was confirmed by PCR for all the cloned fragments. Subsequently, the construct was mobilized into Agrobacterium EHA105 strain through the electroporation method following standard protocol for utilization in greengram transformation. Five batches of cotyledonary node explants (~200/ per batch) of greengram (variety LGG-460) were infected with Agrobacterium (EHA105) harboring pMDC100-CRISPR construct. In the preliminary selection, 126 explants were observed to show resistance to Kanamycin (50 mg/L). Out of them, 41 retained resistance to Kanamycin in repeated culture on the selection medium. Finally, 12 plantlets were regenerated on shoot and root induction media. The plantlets were hardened and maintained in a transgenic glass house for further characterization (Figures 2A,B).
Molecular confirmation of greengram plants
The integration of the transferred DNA fragments in the greengram genome was analyzed through PCR using the genomic DNA isolated from 10 putative transformed (T0) and control plants. Out of 10 plants, seven (1, 3, 4, 5, 6, 7, and 10) displayed the amplification of all four fragments including gRNA1 (AtU6p- gRNA AC1), gRNA2 (AtU3p-gRNA AV1), 35s-Cas9 and npt II with expected amplification of 480, 420, 610, and 730 bp fragments, respectively (Figure 3). This indicated that the T-DNA was integrated into the genome of greengram.
Simultaneously, the integration of T-DNA in the genome of greengram was detected using a dot blot detection assay. The genomic DNA was hybridized with Biotin labeled AtUp6-AC1 and AtU3-AV1. Blue color precipitation was observed in Lanes 1, 3, 6, 7, and 10, which was exactly matching with positive control. The intensity of precipitation of both gRNA cassettes was very high in lanes 6 and 10. However, no precipitation was observed in the control (Figure 4). These results also confirmed that the T-DNA harboring gRNA cassettes were integrated into the genome of the greengram.
The amplified products of AtUp6-AC1 and AtU3-AV1 cassettes, Cas9, and npt II from plant no.6 were confirmed with commercial sequencing. The sequences of all four fragments exactly matched with cassette-specific sequences when analyzed through Clustal W. About 42 T1- seeds were collected from T0- 6 plants and germinated in pots in the transgenic greenhouse. Out of 23 plants, 16 samples showed amplification of the AtUp6-AC1 and AtU3-AV1 cassettes using the specific primers. The PCR-positive plants were used for further bioassay and qRT-PCR analysis.
Phenotypic evaluation of CRISPR/Cas9 edited greengram plants to assess their efficacy against MYM virus infection
About 4–5 week old transformed and control plants were grown under controlled conditions and agroinfiltrated with Agrobacterium strain carrying the MYMV clone at 5–6 leaf stage or trifoliate stage. After 10 days post-infection (dpi), minimal mosaic symptoms were exhibited in T2-6-3-1 and T2-6-4-4. No symptoms were observed on the leaves of T2-6-2-2, T2-6-2-3, T2-6-4-5, T2-6-4-6, and T2-6-4-7 plants. However, severe mosaic symptoms were observed in non-transformed control plants (Figure 6). Symptoms were categorized based on the spreading of symptoms with minimal or high mosaic patterns by the following scale: no yellow mosaic symptom- 0; minimal yellow symptoms on leaf area-1; high or severe yellow mosaic symptoms-2. This indicated that the expression of gRNAs and Cas9 led to the editing of the viral genome in transformed plants resulting in the inhibition of virus replication. The leaf tissues were harvested from infested and control plants for further molecular analysis to determine the viral transcript and viral load analysis.
Semi-quantitative real-time PCR for transcript analysis in CRISPR plants
To quantify the expression of the integrated DNA fragments in the infected plants in comparison to the control plants, quantitative PCR was carried out using the cDNA of the transgenic plants as a template. This analysis showed that the transcripts of MYMV-AC1 and AV1 accumulated in transformed plants in comparison to the higher expression level seen in the control sample Agro infiltrated with MYMV. Vr-Tubulin was used as a reference gene and it was expressed equally in all plants. The Cas9 accumulation was also noticed in all the transgenic plants except the control challenged with MYMV. No viral load of AC1 and AV1 were seen in T2-6-2-2, T2-6-2-3, T2-6-4-5, and T2-6-4-7 plants compared with control infected plants with MYMV clone.
Simultaneously, the relative expression levels of AC1 and AV1, and Cas9 transcripts were analyzed using qRT-PCR in the greengram transgenic plants. The relative expression of AC1 and AV1 in transgenic plants was significantly less than in the infected control (CI) plants. The level of viral cDNA relative to the reference Vr-Tubulin standard was calculated in the virus-challenged plants. The ΔCT value of each RT-PCR was derived by taking the values of the reference standard Vr-Tubulin. The relative expression of AC1 was recorded between 0.07 to 0.6 and AV1 as 0.7 to 1.3 when compared to 1.65 (AC1) and 12.8 (AV1) of control infected plants, respectively. These plants were also used for analyzing the expression of Cas9 in all T2 lines, in which the relative expression was significantly higher in all transgenic plants (T2) compared to the control plants (Figures 7A,B).
Determination of genome editing in greengram using T7EI assay
To validate the efficiency of genome editing, a T7 endonuclease I (T7EI) assay was performed to detect mutations in the two targeted sites of AC1 and AV1 of the MYM viral genome. In this assay, the amplicons (0.35 kb) of AC1 and AV1 from T2 lines-6-2-3, 6-4-4, and 6-4-7 (as T2-6th line targeted sites) were amplified and digested with T7EI endonuclease enzyme. The digested samples of transformed lines were resolved on the 3% agarose gel. However, the wild-type (Control- MYMV infected) plants have failed to show such amplification. The editing efficiencies of AC1 and AV1 observed in these three transformed lines were 46%, 32%, 20%, and 38.15%, 40%, and 21.36%, respectively (Figure 8A). These results indicated that both gRNA locations in AC1 and AV1 were edited efficiently. To sequence the targeted regions in the genome, the MYMV AC1 and AV1 were amplified from the resistant T2 lines and control plants. The purified fragments were sequenced and analyzed for detecting the targeted mutations in the specific locations. There was a heterozygous mutation in all the three T2 lines such as T2-6-2-3, T2-6-4-4, and T2-6-4-7. A single base pair/heterozygous mutation was noticed in gRNA1-AC1, whereas two base pairs were mutated in gRNA2-AV1 when compared with the control MYMV genome after sequencing (Figure 8B).
Discussion
CRISPR/Cas9 is an efficient, adaptive anti-viral defense system that evolved in bacteria and archaea to counter the impending viral pathogen attacks, and was exploited to counter Geminiviruses in plants (Ali et al., 2016). Recent reports on this aspect reveal that the viral genomes of different Geminiviruses infecting plants could be efficiently edited at multiple locations for restricting the viral replication and mobility, thereby restricting the severity of infection in the virus-attacked plants. The CRISPR/Cas9 model is easily adoptable to assemble multiple gRNA cassettes to edit several viral genomes simultaneously at conserved loci of viruses (Baltes et al., 2015; Mehta et al., 2019). In the present investigation, we have cloned two guide RNA cassettes (targeting the genes that code for rep and coat proteins) and codon-optimized Cas9 in a single vector for targeting two different sites in the genome of MYMV in greengram. This is the first report on adopting CRISPR/Cas9 system for editing the viral genome of MYMV in greengram.
Initially, we designed 20 bp spacer sequences from the novel regions in the viral genome for targeting two locations involved in viral replication and coat protein synthesis. Editing of these regions is expected to result in restriction of viral genome replication after the establishment of infection that eventually reduces the virus accumulation in Cas9 expressing plants. Targeting the combination of multiple gRNAs has been proved to be capable of editing effectively in BeYDV, TRV, BSCTV, and ChiLCV rather than single gRNA (Ali et al., 2015b; Baltes et al., 2015; Ji et al., 2015; Roy et al., 2019).
T7EI assay is a novel approach for determining the efficiency of genome editing by using PCR products after subjecting them to digestion with T7 endonuclease for identifying the indel frequencies (Roy et al., 2019). In order to detect the mutations that occur after editing, the PCR products of greengram transgenics after agroinfiltration with MYMV clone with AC1 and AV1 fragments covering the targeted sgRNAs were subjected to digestion with T7 endonuclease on the T7EI assay where the viral genes such as AC1 and AV1 were edited in T2-6-2-3, T2-6-4-4, and T2-6-4-7 (Fig 8A). Baltes et al. (2015) detected the frequency of NHEJ induced mutations in BeYDV genome at predicted cleavage sites as 70, 22.7, 0.03, and 7.85% in gBRBS+, gBM3+, gBM1−and gB9nt+, respectively, in tobacco. The viral load accumulation was drastically reduced in edited transgenic tobacco plants (Baltes et al., 2015).
Multiplexed gRNA strategy was applied for targeting different locations of Chilli leaf curl virus (ChiLCV) genome and the resultant tobacco plants showed a high degree of resistance against ChiLCV with reduced virus accumulation and minimized disease symptoms (Roy et al., 2019). The AC1 and AV1 genes are important for replication and coat proteins of the virus. Editing of these regions has been expected to interfere with viral replication after the viral infection, thereby the reduced viral load and minimizing disease symptoms. A single mutation was observed in the AC1 gene of Agroinfiltrated plants. Baltes et al. (2015) successfully controlled BeYDV, BSCTV (Ji et al., 2015), and TYLCV (Ali et al., 2015a) using genome editing. Up to 97% reduction in viral load of BSCTV was observed in N. benthamiana and A. thaliana by using different constructs (Ji et al., 2015).
The combination of gRNAs homologous to BeYDV reduced the viral load accumulation by 87% in transgenic tobacco (Baltes et al., 2015). In the present investigation, the Cas9 expressing greengram plants exhibited a drastic reduction in viral load. Our qRT-PCR analyses showed that simultaneous targeting of the different sequences of the MYMV resulted in a significant reduction in viral load. These infected plants showed highly reduced MYMV symptoms compared to the control plants. RT-PCR results showed that Cas9 was expressed in all the edited plants except the control and showed a very low viral load compared to the control plant. It indicates that the accumulation of mRNA transcripts of Cas9 is associated with a reduction of viral replication in transgenic plants with low viral expression. Gil-Humanes et al. (2017) reported highly efficient gene targeting of wheat dwarf virus (WDV) by multiplexing, which enabled targeted mutagenesis resulting in mutation frequencies ranging from 12.9 to 20.7%. Similarly, Ali et al. (2016) performed mutation detection analysis in monopartite virus Cotton Leaf Curl Kokhran Virus (CLCuKoV) with a high rate of InDel formation in both ORFs with 18–49% in CP and 35–45% in RCRII. The T7EI enzyme is sensitive to the mismatch sequences of double-stranded DNA and the Cas9 was able to cleave the targeted site to generate DSBs that would be repaired by Non-homologs end joining (NHEJ). Tashkandi et al. (2018) used a T7EI assay to assess the non-homologous end joining after the formation of DSB in N.benthamiana plants, which were challenged with TYLCV within 7 dpi and this has resulted in InDel formation at the Rep sequence in independent T3 homozygous tomato lines with 24 and 29% frequency compared with wild type and with CP sequence of same lines with 4 and 49%. We found that sgRNAs targeting the MYMV regions i.e., AC1 and AV1 sequences were capable of targeting and interfering with replication resulting in viral suppression as revealed by the T7EI assay. The gene editing efficiency as indicated by the percentage of targeted mutations in three transgenic lines of T2 plants was estimated to be T2-6-2-3 (46%), T2-6-4-4 (32%), and T2-6-4-7 (20%) for gRNA1 (AC1) and T2-6-2-3 (38.15%), T2-6-4-4 (40%), and T2-6-4-7 (21.36%) for gRNA2 (AV1) (Figure 8B). Thus, we showed here that the CRISPR/Cas9 system is a highly efficient tool for generating targeted mutation in MYMV-infected greengram plants.
Conclusion
In conclusion, we have successfully generated greengram expressing CRISPR components encoding for rep and coat proteins. The Cas9 expressing plants exhibited minimal transcripts of rep and coat proteins. The MYMV genome was efficiently edited by Cas9 endonuclease. The CRISPR/Cas9 tool can be exploited for the transformation of pulse crops for attenuating viral diseases. Our results confirm that the CRISPR/Cas9 tool provides new avenues for viral interference in food crops. It is a proof of concept for editing endogenous genes in greengram and other pulse crops.
Data availability statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author/s.
Author contributions
GM: conceived project, mobilized funds from CSIR-EMRII, designed experiments, and suggestions on conducting experiments. AT: executed all experiments and written MS. MR: provided the CRISPR construct. SA: suggestions and editing and review of the MS. All authors contributed to the article and approved the submitted version.
Acknowledgments
We are sincerely thankful to the Council of Scientific and Industrial Research (CSIR) for financial support –No. 38(1450)/18/EMR II. We thank Executive Director and Agri Biotech Foundation for the support and encouragement.
Conflict of interest
Author MR was employed by International Center for Genetic Engineering and Biotechnology.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ali, Z., Abulfaraj, A., Idris, A., Ali, S., Tashkandi, M., and Mahfouz, M. M. (2015a). CRISPR/Cas9-mediated viral interference in plants. Genome Biol. 16, 1–11. doi: 10.1186/s13059-015-0799-6
Ali, Z., Abul-Faraj, A., Li, L., Ghosh, N., Piatek, M., Mahjoub, A., et al. (2015b). Efficient virus-mediated genome editing in plants using the CRISPR/Cas9 system. Mol. Plant 8, 1288–1291. doi: 10.1016/j.molp.2015.02.011
Ali, Z., Ali, S., Tashkandi, M., Zaidi, S. S. E. A., and Mahfouz, M. M. (2016). CRISPR/Cas9-mediated immunity to geminiviruses: differential interference and evasion. Sci. Rep. 6, 1–13. doi: 10.1038/srep26912
Ali, Z., Eid, A., Ali, S., and Mahfouz, M. M. (2018). Pea early-browning virus-mediated genome editing via the CRISPR/Cas9 system in Nicotiana benthamiana and Arabidopsis. Virus Res. 244, 333–337. doi: 10.1016/j.virusres.2017.10.009
Aouida, M., Eid, A., Ali, Z., Cradick, T., Lee, C., Deshmukh, H., et al. (2015). Efficient fdCas9 synthetic endonuclease with improved specificity for precise genome engineering. PLoS ONE 10, e0133373. doi: 10.1371/journal.pone.0133373
Baltes, N. J., Hummel, A. W., Konecna, E., Cegan, R., Bruns, A. N., Bisaro, D. M., et al. (2015). Conferring resistance to geminiviruses with the CRISPR–Cas prokaryotic immune system. Nat. Plants 1, 1–4. doi: 10.1038/nplants.2015.145
Chaparro-Garcia, A., Kamoun, S., and Nekrasov, V. (2015). Boosting plant immunity with CRISPR/Cas. Genome Biol. 16, 1–4. doi: 10.1186/s13059-015-0829-4
Cody, W. B., Scholthof, H. B., and Mirkov, T. E. (2017). Multiplexed gene editing and protein overexpression using a tobacco mosaic virus viral vector. Plant Physiol. 175, 23–35. doi: 10.1104/pp.17.00411
Fontenelle, M. R., Luz, D. F., Gomes, A. P. S., Florentino, L. H., Zerbini, F. M., and Fontes, E. P. (2007). Functional analysis of the naturally recombinant DNA-A of the bipartite begomovirus Tomato chlorotic mottle virus. Virus Res. 126, 262–267. doi: 10.1016/j.virusres.2007.02.009
Gil-Humanes, J., Wang, Y., Liang, Z., Shan, Q., Ozuna, C. V., Sánchez-León, S., et al. (2017). High-efficiency gene targeting in hexaploid wheat using DNA replicons and CRISPR/Cas9. Plant J. 89, 1251–1262. doi: 10.1111/tpj.13446
Hadidi, A., Flores, R., Candresse, T., and Barba, M. (2016). Next-generation sequencing and genome editing in plant virology. Front. Microbiol. 7, 1325. doi: 10.3389/fmicb.2016.01325
Hehnle, S., Wege, C., and Jeske, H. (2004). Interaction of DNA with the movement proteins of geminiviruses revisited. J. Virol. 78, 7698–7706. doi: 10.1128/JVI.78.14.7698-7706.2004
Ji, X., Zhang, H., Zhang, Y., Wang, Y., and Gao, C. (2015). Establishing a CRISPR–Cas-like immune system conferring DNA virus resistance in plants. Nat. Plants 1, 1–4. doi: 10.1038/nplants.2015.144
Karthikeyan, A., Shobhana, V. G., Sudha, M., Raveendran, M., Senthil, N., Pandiyan, M., et al. (2014). Mungbean yellow mosaic virus (MYMV): a threat to green gram (Vigna radiata) production in Asia. Int. J. Pest. Manage. 60, 314–324. doi: 10.1080/09670874.2014.982230
Khan, Z., Khan, S. H., Mubarik, M. S., Sadia, B., and Ahmad, A. (2017). Use of TALEs and TALEN technology for genetic improvement of plants. Plant Mol. Biol. Rep. 35, 1–19. doi: 10.1007/s11105-016-0997-8
Kis, A., Hamar, E., Tholt, G., Bán, R., and Havelda, Z. (2019). Creating highly efficient resistance against wheat dwarf virus in barley by employing CRISPR/Cas9 system. Plant Biotechnol. J. 17, 1004–1006. doi: 10.1111/pbi.13077
Liang, G., Zhang, H., Lou, D., and Yu, D. (2016). Selection of highly efficient sgRNAs for CRISPR/Cas9-based plant genome editing. Sci. Rep. 6, 1–8. doi: 10.1038/srep21451
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Mehta, D., Stürchler, A., Anjanappa, R. B., Zaidi, S. S. E. A., Hirsch-Hoffmann, M., Gruissem, W., et al. (2019). Linking CRISPR-Cas9 interference in cassava to the evolution of editing-resistant geminiviruses. Genome Biol. 20, 1–10. doi: 10.1186/s13059-019-1678-3
Mekala, G. K., Juturu, V. N., Mallikarjuna, G., Kirti, P. B., and Yadav, S. K. (2016). Optimization of Agrobacterium-mediated genetic transformation of shoot tip explants of green gram (Vigna radiata (L.) Wilczek). Plant Cell Tiss. Organ. Cult. 127, 651–663. doi: 10.1007/s11240-016-1085-3
Mishra, G. P., Dikshit, H. K., Sv, R., Tripathi, K., Kumar, R. R., Aski, M., et al. (2020). Yellow mosaic disease (YMD) of mungbean (Vigna radiata (L.) Wilczek): current status and management opportunities. Front. Plant Sci. 11, 918. doi: 10.3389/fpls.2020.00918
Mushtaq, M., Mukhtar, S., Sakina, A., Dar, A. A., Bhat, R., Deshmukh, R., et al. (2020). Tweaking genome-editing approaches for virus interference in crop plants. Plant Physiol. Biochem. 147, 242–250. doi: 10.1016/j.plaphy.2019.12.022
Nair, R., and Schreinemachers, P. (2020). “Global status and economic importance of mungbean,” in The Mungbean Genome. Compendium of Plant Genomes, eds R. Nair, R. Schafleitner, and S. H. Lee (Cham: Springer).
Piatek, A., and Mahfouz, M. M. (2017). Targeted genome regulation via synthetic programmable transcriptional regulators. Crit. Rev. Biotechnol. 37, 429–440. doi: 10.3109/07388551.2016.1165180
Roy, A., Zhai, Y., Ortiz, J., Neff, M., Mandal, B., Mukherjee, S. K., et al. (2019). Multiplexed editing of a begomovirus genome restricts escape mutant formation and disease development. PLoS ONE 14, e0223765. doi: 10.1371/journal.pone.0223765
Selvi, R., Muthiah, A. R., Manivannan, N., Raveendran, T. S., Manickam, A., and Samiyappan, R. (2006). Tagging of RAPD marker for MYMV resistance in mungbean (Vigna radiata (L.) Wilczek). Asian J. Plant Sci. 5, 277–280. doi: 10.3923/ajps.2006.277.280
Sudha, M., Karthikeyan, A., Madhumitha, B., Veera Ranjani, R., Kanimoli Mathivathana, M., Dhasarathan, M., et al. (2022). Dynamic transcriptome profiling of mungbean genotypes unveil the genes respond to the infection of mungbean yellow mosaic virus. Pathogens 11, 190. doi: 10.3390/pathogens11020190
Talakayala, A., Ankanagari, S., and Garladinne, M. (2022). CRISPR-Cas Genome editing system: a versatile tool for developing disease resistant crops. Plant Stress 3, 100056. doi: 10.1016/j.stress.2022.100056
Talakayala, A., Prathyusha, V. B., Divya, D., Ankanagari, S., and Garladinne, M. (2021). Molecular cloning of MYMV genome and infectivity of yellow mosaic virus in green gram using different viral transmission tools. Biosci. Biotechnol. Res. Asia 18, 467. doi: 10.13005/bbra/2932
Tashkandi, M., Ali, Z., Aljedaani, F. R., Shami, A., and Mahfouz, M. M. (2018). Engineering resistance against Tomato yellow leaf curl virus via the CRISPR/Cas9 system in tomato. Plant Signal. Behav. 13, e1525996. doi: 10.1080/15592324.2018.1525996
Tripathi, J. N., Ntui, V. O., Ron, M., Muiruri, S. K., Britt, A., and Tripathi, L. (2019). CRISPR/Cas9 editing of endogenous banana streak virus in the B genome of Musa spp. overcomes a major challenge in banana breeding. Commun. Biol. 2, 1–11. doi: 10.1038/s42003-019-0288-7
Vanti, G. L., Katageri, I. S., Swamy, B. M., Sangannavar, P. A., Methre, R., and Hirematha, V. (2015). Simple, rapid, economical and high yielding method for extracting genomic DNA from cotton (Gossypium spp.). Electron J. Plant Breeding 6, 1164–1168.
Keywords: clustered regularly interspaced short palindromic repeats (CRISPR), mungbean yellow mosaic (MYMV) virus, guide RNA (gRNA), replication protein (AC1), coat protein (AV1), agroinfiltration, T7E1-T7 endonuclease I assay
Citation: Talakayala A, Mekala GK, Reddy MK, Ankanagari S and Garladinne M (2022) Manipulating resistance to mungbean yellow mosaic virus in greengram (Vigna radiata L): Through CRISPR/Cas9 mediated editing of the viral genome. Front. Sustain. Food Syst. 6:911574. doi: 10.3389/fsufs.2022.911574
Received: 02 April 2022; Accepted: 15 June 2022;
Published: 12 August 2022.
Edited by:
Pranjal Yadava, Indian Agricultural Research Institute (ICAR), IndiaReviewed by:
Uday Chand Jha, Indian Institute of Pulses Research (ICAR), IndiaPradeep Kumar, University of Lucknow, India
Copyright © 2022 Talakayala, Mekala, Reddy, Ankanagari and Garladinne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mallikarjuna Garladinne, Z2FscmFkaW5uZW1hcmp1bkBnbWFpbC5jb20=; Srinivas Ankanagari, YXNyaW5pdmFzQG9zbWFuaWEuYWMuaW4=
†ORCID: Ashwini Talakayala orcid.org/0000-0003-4528-4871
Gopala Krishna Mekala orcid.org/0000-0001-5287-1201
Srinivas Ankanagari orcid.org/0000-0001-7631-6536
Mallikarjuna Garladinne orcid.org/0000-0002-8877-7118