
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Sustain. Food Syst. , 13 October 2021
Sec. Agro-Food Safety
Volume 5 - 2021 | https://doi.org/10.3389/fsufs.2021.742353
This article is part of the Research Topic Using Genomics to Inform Food Safety Inspection Systems View all 7 articles
 Phillip Brown1*
Phillip Brown1* Yi Chen2
Yi Chen2 Robin Siletzky3
Robin Siletzky3 Cameron Parsons3
Cameron Parsons3 Lee-Ann Jaykus1,3
Lee-Ann Jaykus1,3 Joseph Eifert4
Joseph Eifert4 Elliot Ryser5
Elliot Ryser5 Catherine M. Logue6
Catherine M. Logue6 Christina Stam3
Christina Stam3 Eric Brown2
Eric Brown2 Sophia Kathariou1,3*
Sophia Kathariou1,3*Listeria monocytogenes is a Gram-positive foodborne pathogen responsible for the severe disease listeriosis and notorious for its ability to persist in food processing plants, leading to contamination of processed, ready-to-eat foods. L. monocytogenes persistence in various food processing environments (FPEs) has been extensively investigated by various subtyping tools, with increasing use of whole genome sequencing (WGS). However, major knowledge gaps remain. There is a need for facility-specific molecular signatures not only for adequate attribution of L. monocytogenes to a specific FPE but also for improved understanding of the ecology and evolution of L. monocytogenes in the food processing ecosystem. Furthermore, multiple strains can be recovered from a single FPE sample, but their diversity can be underestimated with common molecular subtyping tools. In this study we investigated a panel of 54 L. monocytogenes strains from four turkey processing plants in the United States. A combination of WGS and phenotypic assays was employed to assess strain persistence as well as identify facility-specific molecular signatures. Comparative analysis of allelic variation across the whole genome revealed that allelic profiles have the potential to be specific to individual processing plants. Certain allelic profiles remained associated with individual plants even when closely-related strains from other sources were included in the analysis. Furthermore, for certain sequence types (STs) based on the seven-locus multilocus sequence typing scheme, presence and location of premature stop codons in inlA, inlB length, prophage sequences, and the sequence content of a genomic hotspot could serve as plant-specific signatures. Interestingly, the analysis of different isolates from the same environmental sample revealed major differences not only in serotype and ST, but even in the sequence content of strains of the same ST. This study highlights the potential for WGS data to be deployed for identification of facility-specific signatures, thus facilitating the tracking of strain movement through the food chain. Furthermore, deployment of WGS for intra-sample strain analysis allows for a more complete environmental surveillance of L. monocytogenes in food processing facilities, reducing the risk of failing to detect strains that may be clinically relevant and potentially novel.
Listeria monocytogenes is a Gram-positive facultative intracellular pathogen that is ubiquitous in the environment and responsible for the severe foodborne disease listeriosis in humans and other animals. Most cases of human listeriosis involve strains of two major lineages, i.e., lineage I and II, and three serotypes, specifically serotypes 4b and 1/2b (both of lineage I) and serotype 1/2a (lineage II) (Kathariou, 2002; Swaminathan and Gerner-Smidt, 2007; Orsi et al., 2011). Human populations at high risk include immunocompromised individuals, the elderly, pregnant women and their fetuses, and listeriosis can have severe outcomes including septicemia, meningitis, stillbirths, and death (Painter and Slutsker, 2007; Scallan et al., 2011; de Noordhout et al., 2014; Schlech, 2019). L. monocytogenes is responsible for about 1,600 illnesses and an estimated $2.8 billion in adverse economic impacts each year in the United States alone (Scallan et al., 2011; de Noordhout et al., 2014; Hoffman et al., 2015). The severe outcomes of listeriosis and the associated high economic burden render L. monocytogenes a highly problematic agent.
L. monocytogenes can persist in food processing environments (FPEs) for impressively long periods of time, sometimes more than a decade (Kathariou, 2002; Gandhi and Chikindas, 2007; Carpentier and Cerf, 2011). Such persistence is mediated by several adaptations, including the ability to form persistent biofilms, tolerance to sanitizers such as quaternary ammonium compounds, growth at low temperatures, and phage resistance (Kathariou, 2002; Gandhi and Chikindas, 2007; Kim et al., 2008; Carpentier and Cerf, 2011; Kathariou et al., 2017). The increasing employment of whole genome sequencing (WGS) has significantly facilitated the assessment of L. monocytogenes persistence in individual food processing plants, greatly improving the resolution afforded by previous tools such as pulsed-field gel electrophoresis or the seven-locus multilocus sequence typing (MLST) scheme (Fagerlund et al., 2016, 2020; Knudsen et al., 2017; Hurley et al., 2019; Stoller et al., 2019; Harrand et al., 2020; Yang et al., 2020).
Analysis of WGS data from various FPEs has been utilized to identify genetic elements, especially those mediating biocide resistance, that can contribute to persistence (Cherifi et al., 2018; Muhterem-Uyar et al., 2018; Pasquali et al., 2018; Hurley et al., 2019; Cooper et al., 2021). Furthermore, WGS analysis has allowed rigorous, targeted inter-FPE comparisons of strains of the same genotype, e.g., the same MLST-based sequence type (ST). Such strains can sometimes be clearly differentiated based on the facility of their origin (Knudsen et al., 2017; Hurley et al., 2019; Fagerlund et al., 2020; Kwon et al., 2020; Yang et al., 2020). However, they can also exhibit impressive similarity, as suggested by WGS-based detection of virtually identical strains of ST8 isolated 15 years apart from two different salmon processing plants, one in Norway and one in Denmark (Fagerlund et al., 2016). Additional data from such inter-FPE comparisons are critically needed to determine the potential for molecular signatures that may be unique to and specific for particular food processing plants, thus greatly facilitating the tracking of contamination and effectively directing mitigation strategies to the relevant food processing facilities. Furthermore, such comparisons may yield critically-needed insights on specific attributes and adaptations that can mediate persistence of L. monocytogenes in a particular FPE, and can contribute importantly to our understanding of Listeria's ecology and evolution in the food processing ecosystem. Integration of WGS-based data with phenotypic assessments of specific adaptive traits of high relevance to FPE persistence may be especially valuable in this regard.
In previous studies we isolated L. monocytogenes via selective enrichments of environmental samples from selected turkey processing plants in the United States and analyzed the strains for resistance to the quaternary ammonium disinfectant benzalkonium chloride (BC) and to heavy metals (Mullapudi et al., 2008). A high proportion of the serotype 1/2a and 1/2b strains were resistant to BC, with resistance typically accompanied by resistance to the heavy metal cadmium (Mullapudi et al., 2008). Furthermore, the serotype 1/2a and 1/2b strains were also frequently resistant to three wide-host-range lytic phages, including two phages isolated from environmental swabs from the same turkey processing plants (Kim et al., 2008). Recently, phage resistance of some of these turkey processing plant-derived strains of ST321 and ST391 was further characterized via WGS analysis and investigation of phage receptors on the teichoic acid of the cell wall (Brown et al., 2021).
In the current study, a panel of 54 L. monocytogenes strains and two L. welshimeri strains from these turkey processing plants were investigated via an approach that integrated WGS analysis with phenotypic assessments of resistance to BC, cadmium and a panel of four wide-host-range lytic phages. Our objectives were to obtain WGS-based assessments of possible persistence and dissemination among different turkey processing plants, as well as to determine the potential for WGS-based analysis to identify molecular signatures specific to individual facilities. The panel of strains included several that were isolated from the same environmental sample, including different isolates of the same serotype and ST. Therefore, an additional objective was to characterize the effectiveness of WGS-based analysis to reveal genomic diversity among isolates from the same FPE sample.
The L. monocytogenes and L. welshimeri strains used in this study (Supplementary Table 1) were isolated from selective enrichments of environmental samples from four different turkey processing plants (A–D) in the United States from 2003 to 2006 as described previously (Eifert et al., 2005). Multiplex PCR was employed to determine serotype designations as previously described (Doumith et al., 2004), with designations confirmed via analysis of the whole genome sequence data. Unless otherwise indicated, bacterial strains were grown overnight in brain heart infusion broth (BHI; Becton, Dickinson and Co., Sparks, MD) or on BHI agar at 37°C, and were preserved at −80°C in BHI with 20% glycerol.
Listeria strains were grown overnight in BHI at 37°C and genomic DNA was extracted using the DNeasy blood and tissue kit (Qiagen, Valencia, CA). Library preparations were completed using a Nextera XT DNA library preparation kit (Illumina, San Diego, CA, USA) with 1 ng of genomic DNA. Genomic DNA was sequenced using a NextSeq 500 sequencer and the NextSeq 500/500 high-output kit v2.5 (300 cycles, 2 × 150 bp) (Illumina) or a MiSeq desktop sequencer with the MiSeq kit v2 (500 cycles, 2 × 250 bp) (Illumina), according to the manufacturer's instructions. Raw sequence reads were quality-trimmed and assembled de novo using SPAdes v 3.14.1 (Bankevich et al., 2012). The quality of the assemblies was assessed using QUAST v.4.6.4 (Gurevich et al., 2013). Default parameters were used with all software.
Sequence type (ST) designations using the seven-locus MLST scheme and heavy metal/antimicrobial resistance gene allele designations were determined using BIGSdb-Lm hosted by the Institut Pasteur (Moura et al., 2016). Whole-genome MLST (wgMLST) and core-genome MLST (cgMLST) data were analyzed to determine allele differences with the BIGSdb PasteurMLST Genome Comparator (Jolley et al., 2018), with the cgMLST and wgMLST schemes using 1,748 and 3,273 loci, respectively. Incomplete alleles and loci that had identical alleles among all strains of a given sequence type were excluded from analysis. For certain STs (i.e., 3, 5, 6, 9 and 321), strains from additional sources were included in genomic comparisons with the strains from the turkey processing plants. Strains from other sources were chosen based on publicly-available whole genome sequence data and origin from diverse sources, e.g., clinical origin and USDA-FSIS surveillance. For ST321, we also included in the comparisons a panel of six ST321 strains from a cold-smoked salmon facility in the United States (Harrand et al., 2020) while for ST5 we included 12 additional strains of meat origin from Ireland (Hurley et al., 2019. Allelic analysis of selected virulence determinants employed the BIGSdb Pasteur MLST Genome Comparator (Jolley et al., 2018). A phylogenetic tree was created by using 1,748 L. monocytogenes core genes from the PasteurMLST into Ridom SeqSphere+, as previously described (Chen et al., 2016). Minimum spanning trees were constructed with Bionumerics1 version 8.0 (https://www.applied-maths.com) using the seven-locus MLST scheme. Prophage identification employed the proteome comparison function from PAThosystems Resource Integration Center (PATRIC) Bioinformatic Resource Center (PATRICBRC; Davis et al., 2020) and the Phage Search Tool Enhanced Release (PHASTER) (Zhou et al., 2011; Arndt et al., 2016). SNP cluster designations were identified using the NCBI Pathogen Detection Pipeline (https://www.ncbi.nlm.nih.gov/pathogens/) (The NCBI Pathogen Detection Project, 2016). The SNP calls are made in the context of all members of a specific cluster following SNP-calling algorithms developed by NCBI (https://www.ncbi.nlm.nih.gov/pathogens/pathogens_help/#data-processing-clustering).
Phage susceptibility assays were done at 25°C as described previously using the wide-host-range lytic phages A511, P100, 20422-1, and 805405-1 (Brown et al., 2021). Resistance to BC and cadmium was determined as described previously (McLauchlin et al., 1997; Mullapudi et al., 2008). Strains were designated resistant if confluent growth was observed on Iso-Sensitest agar (Oxoid, Hampshire, England) supplemented with 70 μg/mL anhydrous cadmium chloride (Sigma, St. Louis, MO, USA) or on Mueller-Hinton agar (Becton, Dickinson and Co., Sparks, MD, USA) supplemented with 10 μg/mL BC (Acros, Morris Plains, NJ, USA). Plates were examined following incubation for 48 h at 37°C.
Whole genome sequence data for the strains used in this project can be found in Supplementary Table 1 and under BioProject Accession PRJNA215355.
Sampling of four different turkey processing plants (A-D) from 2003 to 2006 resulted in 1,584 samples. The recovery of L. monocytogenes and other Listeria spp. from these samples will be described in a separate presentation. A panel of 54 L. monocytogenes isolates from the four turkey processing plants and two L. welshimeri from Plant A were chosen for whole genome sequencing based on their serotype as well as phenotypic assays including resistance to benzalkonium chloride (BC), cadmium and a panel of four wide-host-range lytic phages (Supplementary Table 1). The L. monocytogenes panel consisted predominantly of strains from Plants A (n = 30) and B (n = 13), with Plants C (n = 8) and D (n = 3) making smaller contributions, and included strains of all three serotypes most commonly encountered in human listeriosis: 1/2a (n = 20), 1/2b (n = 16), and 4b (n = 15) (Table 1). In-silico MLST revealed a total of 19 STs among the 54 L. monocytogenes strains, with several STs encountered in multiple plants (Table 1; Figure 1A). Several STs were also encountered at multiple time points over the three-year sampling period (Table 1; Figure 1B). The two L. welshimeri strains had the same ST, i.e., ST1084 (Supplementary Table 1).
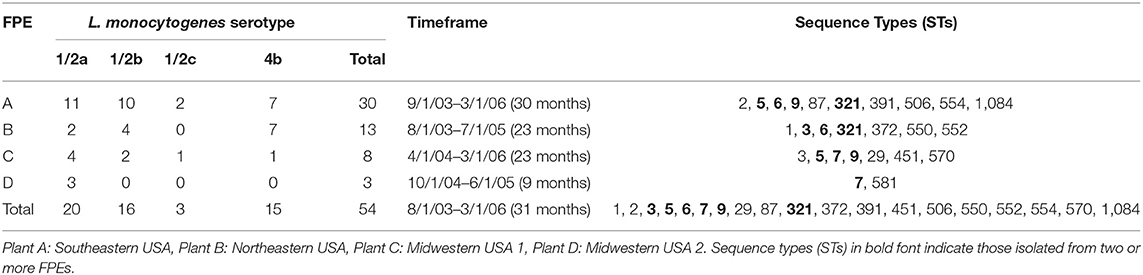
Table 1. Composition of the panel of L. monocytogenes strains investigated in this study.
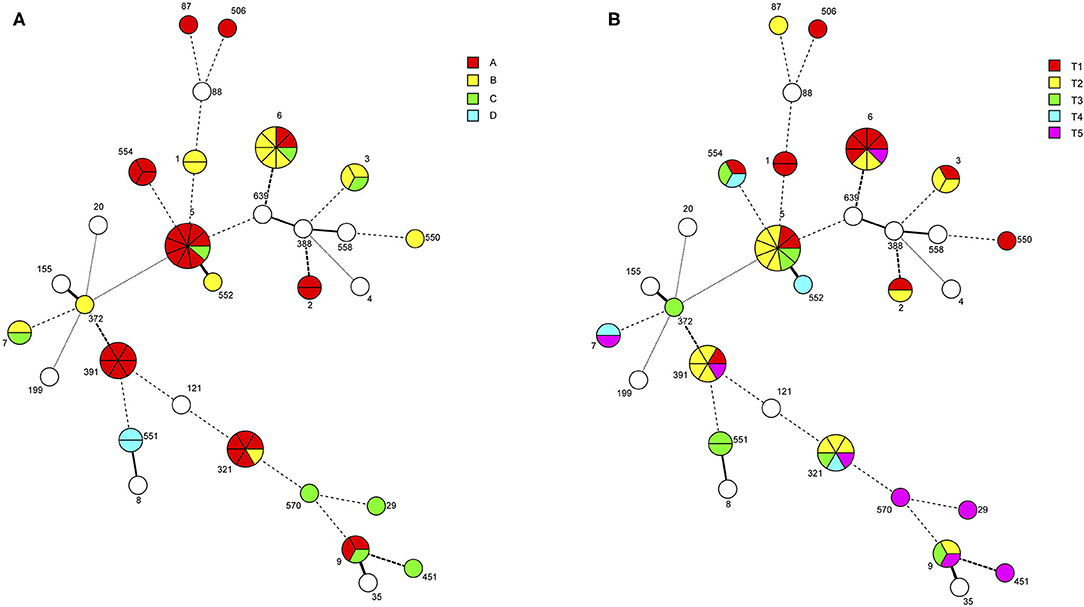
Figure 1. Minimum spanning tree of strains isolated from four turkey processing plants in the United States over a 3-year sampling period. The minimum spanning tree (MST) was constructed using the seven-locus MLST scheme in Bionumerics as described in Materials and Methods. Each circle corresponds to the indicated sequence type (ST). The sections within the circles represent individual strains with a given ST. Circles shown in white are from outside the current study and were included for scaffolding the MST. STs are separated by branches based on seven-locus MLST similarity: one-allele difference (thick solid line), two-allele differences (thin solid line), three-allele differences (thick dashed line), four-allele differences (thin dashed line), more than four-allele differences (thin dotted line). (A) Individual strains are color-coded based on plant of origin: Southeastern USA (Plant A), Northeastern USA (Plant B), Midwestern USA 1 (Plant C), and Midwestern USA 2 (Plant D). (B) Individual strains are color-coded based on isolation date: (T1), September 2003-February 2004; T2, March 2004-August 2004; T3, September 2004-February 2005; T4, March 2005-August 2005; T5, September 2005-March 2006.
BC resistance was found to be common (43%) among these L. monocytogenes strains, with all BC-resistant strains being also resistant to cadmium as previously described (Mullapudi et al., 2008), while several additional strains were resistant to cadmium but susceptible to BC, leading to 65% of the strains exhibiting cadmium resistance (Figure 2; Supplementary Table 1). Phage resistance was also common, with 29/54 (54%) of the strains exhibiting resistance against all four wide-host-range phages in the panel and 39 (69%) exhibiting resistance to at least one of the phages (Supplementary Table 1). Strains of the same ST and from the same plant tended to exhibit the same phage resistance profile, with the notable exceptions of ST5, ST391, and ST554 from Plant A (Supplementary Table 1). While strains of the same ST were found over extended periods of time in the same processing plant, no apparent increase in resistance to BC or phage could be noted, potentially because the selective pressures associated with BC or phage exposure may well have selected for specific resistance traits before sampling began.
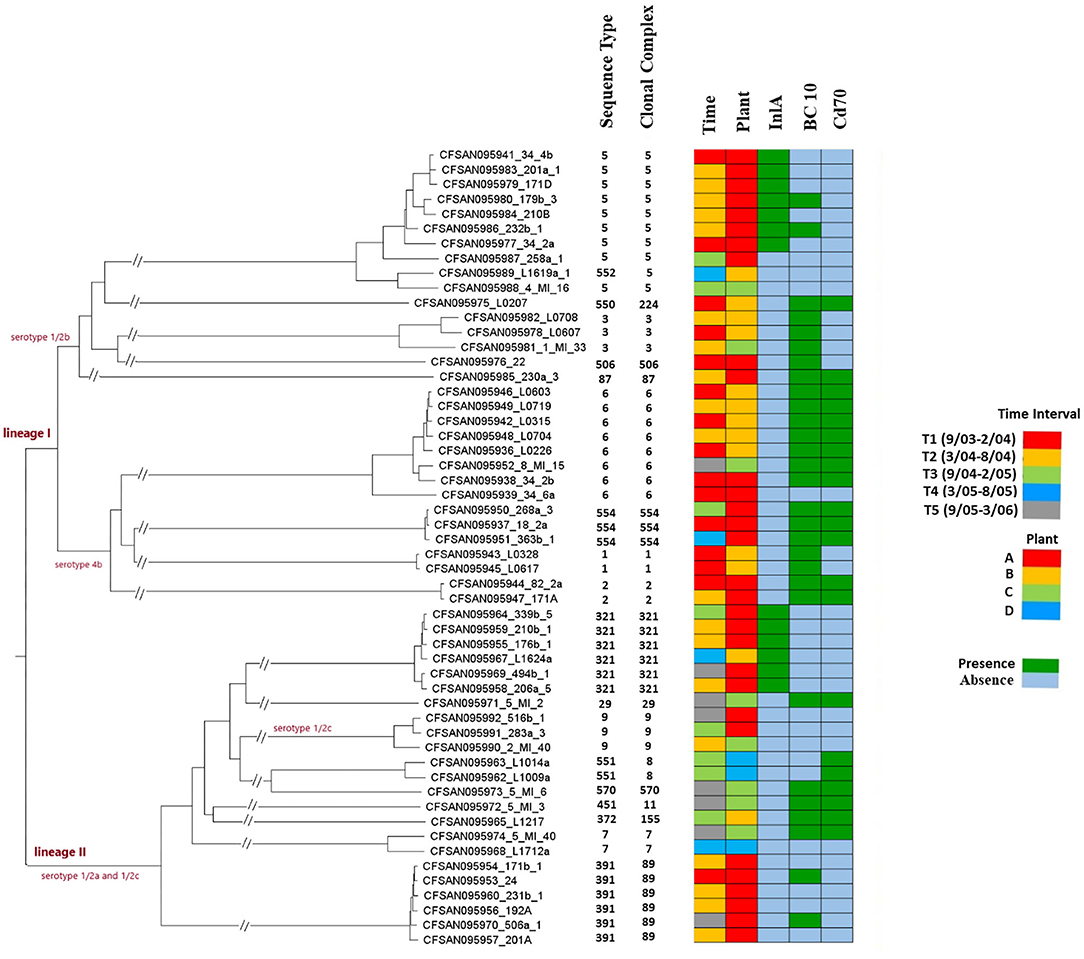
Figure 2. Whole genome MLST-based phylogenetic tree of the 54 L. monocytogenes strains from Plants A-D. Sequence type (ST) and clonal complex (CC) designations are based on the seven-locus MLST scheme. Plant of origin and time period during which the strain was isolated are indicated as shown in the insets. InlA PMSC indicates presence or absence of premature stop codons (PMSCs) in InlA, as shown in the inset. BC10 and CD70 indicate presence or absence of resistance to benzalkonium chloride (BC) and cadmium (CD), respectively, as shown in the inset and resistance was tested as described in Materials and Methods. Two parallel diagonal lines on a branch indicates a break in order to fit greater phylogenetic distances on a single tree.
Antibiotic resistance determinants identified via the NCBI Pathogen Detection Pipeline were only fosX and lin, both of which are widely conserved across L. monocytogenes, while an additional determinant, abc-F, was identified in ST391 and a small number of other strains (Supplementary Table 1). No other antibiotic resistance determinants were revealed in this panel of strains (Supplementary Table 1).
Strains of the same ST were isolated during multiple sampling months in both Plant A and B. In Plant A, strains of ST 2, 5, 9, 321, 391, and 554 were isolated at least 4 months apart, with six strains of ST391 isolated over a 21-month period (Table 1; Supplementary Table 1; Figure 1). ST391 strains were noteworthy, as they were isolated from Plant A over a 29-month period and belonged to the same SNP cluster (PDS000025393.5), with 19–47 wgMLST allele differences (Supplementary Table 2; Figure 2), in spite of the differences in their phage resistance profiles (Supplementary Table 1). A similar finding was obtained with ST321, which was isolated from Plants A and B and for which strains belonged to the same SNP cluster (PDS000000366.424) with 16–114 wgMLST allele differences (Supplementary Table 2; Figure 2). Even though the ST321 strains from plant A were resistant to all four phages, the strain from Plant B was susceptible (Supplementary Table 1). Several other STs from Plant A had high similarity between strains isolated on different dates, including ST9 (27 wgMLST allele differences, SNP cluster PDS000025433.45) and ST554 (7–12 wgMLST allele differences, SNP cluster PDS000024310.3) (Supplementary Tables 1, 2; Figure 2). In Plant B, strains of ST1 and ST6 were isolated at least 3 months apart, with two highly similar ST1 strains (15 wgMLST allele differences, SNP cluster PDS000083087.1) from different dates and five strains of ST6 in the same SNP cluster (PDS000025190.4) over a 30-month period (Table 1; Supplementary Table 1; Figure 2).
In comparison to these STs, noticeably more divergence was noted among ST5 strains from Plant A, with 27–282 wgMLST allele differences (Supplementary Table 2). The relatively large number of differences in ST5 strains may reflect either multiple introductions of these ST5 strains into Plant A or possibly earlier introductions enabling greater diversification over a longer period of time. However, the findings may also reflect the greater diversification potential of this ST. A previous study reported broad differences in strain diversity among ST5 strains from the same ready-to-eat meat processing plant in China and identified eight clusters of ST5 strains with one such cluster having >70 cgMLST allele differences from the others (Zhang et al., 2021).
While strains of the same ST isolated on different dates from Plant C or D were not identified in the panel, two ST551 strains were isolated on the same day from different samples from Plant D (Supplementary Table 1). These two strains (L1009a, L1014a) differed by 59 wgMLST alleles, indicating that, while they are related and likely diverged from the same introduction into Plant D, they had persisted in this plant long enough to diverge sufficiently to be differentiated by WGS (Supplementary Table 2; Figure 2). However, the possibility of repeated introduction should also be considered.
Both sequenced L. welshimeri strains (266a-1 and 34-3a) were BC and cadmium resistant, harboring the same bcrABC alleles found in the majority of L. monocytogenes strains and also cadA2, which frequently accompanied bcrABC in L. monocytogenes in the current panel (Supplementary Table 1) as well as in larger surveys of L. monocytogenes from the turkey processing plants (Mullapudi et al., 2008). Previous findings showed the potential of bcrABC to be transferred via conjugation from L. welshimeri to L. monocytogenes upon co-selection for cadmium resistance (Katharios-Lanwermeyer et al., 2012). These two L. welshimeri strains were isolated from two different floor drains of the same facility more than 1 year apart, had the same ST (Supplementary Table 1) and were closely related (79 wgMLST allele differences). Even though repeated introduction cannot be excluded, these findings may also suggest that persistence in the FPE is not unique to L. monocytogenes but can also be exhibited by non-pathogenic Listeria spp. FPE persistence in L. welshimeri adds further relevance to the potential transfer of bcrABC-mediated BC resistance from non-pathogenic Listeria spp. to L. monocytogenes within the same FPE.
Genome comparator-based analysis was employed to assess the possibility of strain dissemination among different plants. As discussed above, the five ST321 strains from Plant A belonged to the same SNP cluster (PDS000000366.424) as the single ST321 strain from Plant B (Supplementary Table 1). The phylogenetic tree revealed two clusters, with the Plant B strain being in the same cluster as three ST321 strains from Plant A and (as will be discussed below) harboring the same uncommon premature stop codon in inlA (Figure 2). Even though independent introduction events to Plants A and B from a common source cannot be excluded, such findings constitute suggestive evidence for cross-plant dissemination. Equipment relocation, for instance, was shown to mediate strain transfer between two different food processing plants (Fagerlund et al., 2016).
Evidence of cross-plant dissemination among other STs was uncommon. In the case of ST6, the phylogenetic analysis revealed three groups, one of which consisted of strains from Plant A and C (Figure 2). These two strains differed by 88 wgMLST alleles (Supplementary Table 2) and were both unaffiliated with a SNP cluster (Supplementary Table 1). Thus, if a cross-plant dissemination or independent introduction from a common source event had taken place, significance divergence appears to have ensued, potentially reflecting early introduction events or differences in the ecology of the respective facilities. Cross-plant comparisons with strains of ST3, ST5, and ST9 found a range of 94–357 wgMLST allele differences, with the largest difference found for strains of ST5 (Supplementary Table 2). On the phylogenetic tree the ST5 strains partitioned in several sub-clusters (Figure 2), in agreement with the allelic diversity revealed via genome comparator analysis with cgMLST or wgMLST (Supplementary Table 2). ST5 strains from Plant A partitioned clearly apart from ST5 from Plant C (Figure 2). Interestingly, the ST5 strain from Plant C was closest to the ST552 strain from Plant B (Figure 2). ST5 and ST552 belong to the same clonal complex (CC) in the eight-locus MLST scheme, i.e., CC5 (Figure 2). The observed relatedness but also diversification between the ST5/ST552 strains from Plants B and C (Figure 2) may suggest earlier acquisitions e.g., via cross-plant transfer, and subsequent divergence, as discussed above for ST6 from Plant A and C.
For ST 3 and 9, the phylogenetic tree also reflects plant-specific diversification and does not provide evidence for cross-plant transfer (Figure 2). The ST3 strains from Plant B clustered together and separately from the ST3 strain from Plant C (Figure 2). Similarly, ST9 strains from Plant A grouped separately from ST9 from Plant C (Figure 2). Additional strains from the different plants would need to be analyzed to further assess the potential for cross-plant transfer or other common-source acquisition events for these STs.
While L. monocytogenes is known to persist for extended periods of time in FPEs, less is known about how strains adapt and diversify in a specific FPE as compared to others. Processing facilities can exert certain common selective pressures but no two are identical, with different food commodities, environmental surface types, facility age and complexity, cleaning and sanitation regimens, and FPE microbial community composition, among the possible differences (Carpentier and Cerf, 2011; Ferreira et al., 2014; Mazaheri et al., 2021). These differences can play major roles in genotypic and phenotypic diversification of strains between different processing plants. In addition, silent substitutions are expected to accrue due to genetic drift as populations evolve in physical isolation from each other in different FPEs. This can lead to genetically-related strains harboring plant-specific signatures that may be used to distinguish strains from different plants. Several STs were isolated in multiple processing plants in the current study (Table 1; Figures 1A, 2), giving us a unique opportunity to look for plant-specific genetic traits in these strains. ST7 was not included in this analysis as the panel included only one strain each from two different plants (Figure 2; Supplementary Table 1).
Whole genome MLST (wgMLST) was utilized with FPE-derived (Plants A-D) strains of ST3, 5 (and the closely-related strain of ST552), 6, 9, and 321 to identify putative plant-specific alleles. This analysis identified 387 wgMLST loci with plant-specific alleles, with 219 of these belonging to the core genome (1,748 core genes) (Supplementary Table 3). In order to further assess the potential of allelic variation at these loci to discern among different plants (e.g., Plant A vs. B), the FPE-derived strains were compared not only among themselves but also with several (7–16) other strains of the same ST, including strains from human listeriosis and USDA-FSIS surveillance. This led to a total of 181 wgMLST loci (96 from the core genome) found to have plant-specific alleles within a given sequence type, with 46, 93, 35, 9, and 8 plant-specific alleles being detected in ST3, 5 (and 552), 6, 9 and 321, respectively (Figure 3; Supplementary Table 4). Interestingly, eight of these loci, i.e., lmo0061 (FtsK/SpoIIIE family protein, putative EssC/YukB component of Type VII secretion system), lmo0530 (hypothetical protein), lmo0829 (pyruvate-flavodoxin oxidoreductase), lmo1291 (peptidoglycan O-acetyltransferase YrhL), lmo1499 (murein endolytic transglycosylase MltG), lmo1574 (DNA polymerase III alpha subunit), lmo2370 (aminotransferase), and oatA (O-acetyltransferase), were found to be variable between plants in multiple STs, and lmo1499 was especially noteworthy in exhibiting a plant-specific allele in three of the five STs (Supplementary Tables 3, 4). This suggests that these seven loci may be especially prone to responding to plant-specific selective pressures, potentially leading to adaptive mutations and making them excellent candidates for plant-specific signatures.
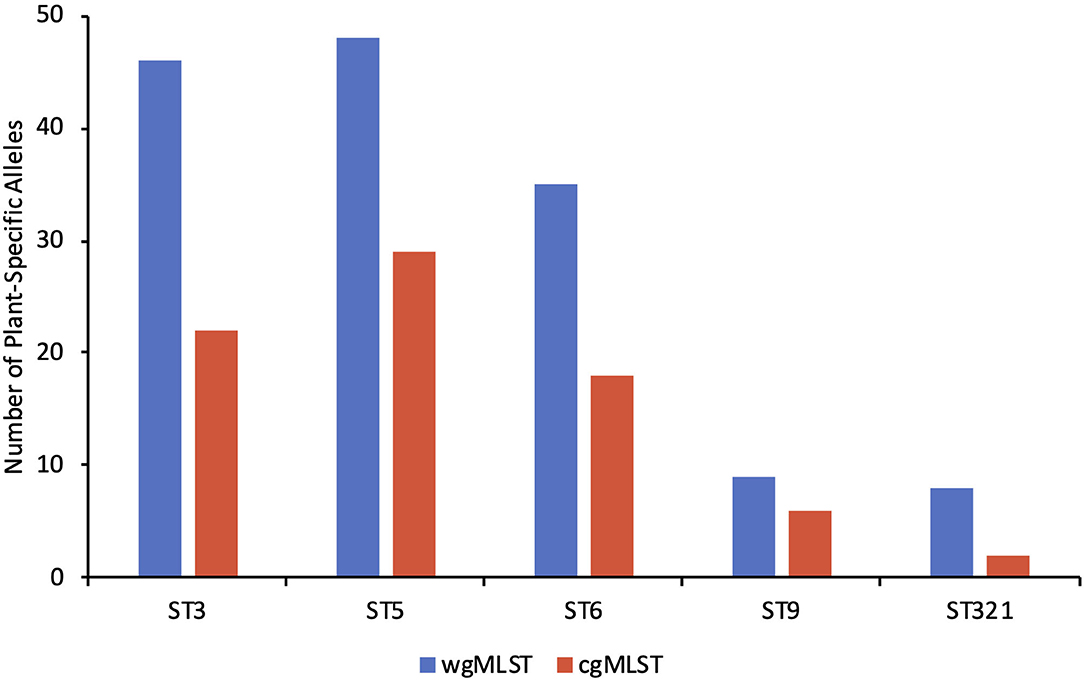
Figure 3. The number of facility-specific alleles based on whole genome MLST (blue) and core genome MLST (red) in sequence types 3, 5, 6, 9, and 321. Lists of the specific plant-specific alleles can be found in Supplementary Table 4.
ST5 (and the related ST552) and ST6 were the most predominant STs in this study that were encountered in multiple plants, and much of our focus was therefore directed to them. For CC5 (ST5 and 552), the ST552 strain from Plant B had more plant-specific alleles (n = 46) than the ST5 strains from Plant A (n = 33) or Plant C (n = 16), i.e., alleles shared among all CC5 strains from the relevant plant but not encountered in CC5 strains from the other plants (Supplementary Table 4). Interestingly, however, of the 16 Plant C-specific alleles, 100% were novel alleles designated as “new” in the genome comparator outputs, having not yet been deposited in the PasteurMLST database, while only 8/33 (24%) of the Plant A-specific alleles and 38/46 (83%) of the Plant B-specific alleles were novel. This suggests that a potentially novel ST5 strain may have been introduced into Plant C, which would be supported by this strain not being affiliated with a SNP cluster (Supplementary Table 1). Allelic content in lmo0883 (transmembrane protein) appears to be a promising plant-specific marker for these strains, as all eight ST5 Plant A strains harbor the same novel allele (new#1) regardless of their SNP cluster designations. On the other hand, the ST5 strains from Plant C harbored a different novel allele (new#2), while a previously-identified allele was harbored by the ST552 (CC5) strain from Plant B and all other (n = 16) ST5 strains in the comparison panel, regardless of source (Supplementary Table 4).
Besides the ST5 and 552 (CC5) strains, ST6 also gave us the opportunity to compare across strains from three different turkey processing plants. In Plant A, the two ST6 strains were isolated from the same sample (sample 34) but were notably different from each other (271 allele differences by wgMLST) as also shown in the phylogenetic tree (Figure 2) and discussed further below, making it difficult to identify any ST6 Plant A-specific alleles (Supplementary Table 2). Nonetheless, we identified 17 and 21 alleles specific for ST6 strains in Plant A and B, respectively, with three loci (lmo2306, lmo2307, and lmo2323) having specific alleles for both plants (Supplementary Table 4). All three loci are annotated as bacteriophage A118-like proteins. The high allelic diversity found among ST6 strains at these loci suggests a high mutation frequency, with each locus exhibiting six to seven novel alleles just among the ST6 strains included in this analysis, and some strains lacking these loci entirely (Supplementary Table 4).
In addition to the six loci (lmo0829, lmo1291, lmo1499, lmo1574, lmo2370, and oatA) that were found to be variable between plants in multiple STs, there were many others where specific alleles were found in all strains from one plant but rarely in other sources (Supplementary Table 3). The occasional detection of these alleles in strains from other sources may potentially help with tracking strain movement. An example of this is ST5, where among our FPE-derived strains allele 33 at lmo2561 appeared specific to Plant C (SK197). This allele was otherwise rare in our comparison panel, found only in one other ST5 strain, of clinical origin (PNUSAL002668) (Supplementary Table 3). Of the ST5 strains in this analysis, SK197 exhibited the least number of wgMLST allele differences (n = 155) from PNUSAL002668, even though the two strains do not belong to the same SNP cluster (Supplementary Table 2 and data not shown).
Similar findings were obtained with ST321, for which strains from Plant A shared the same allele at LMRG_01558 while different alleles at this locus were found in ST321 genomes from Plant B or any of the other sources included in this analysis (Supplementary Table 4). Our panel of ST321 strains from other sources included six from a cold-smoked salmon processing plant in the United States, with three strains from each of the two clusters of ST321 identified in that facility (Harrand et al., 2020). The five ST321 strains from Plant A differed from each other by 16–114 wgMLST alleles while the cold-smoked salmon strains differed from each other by 25–189 wgMLST alleles, indicating somewhat higher diversity (Supplementary Table 2). This could be caused by the longer time period between these strains (17 years) (Harrand et al., 2020) as compared to the Plant A ST321 strains (2 years). Interestingly, some of the Plant A and cold-smoked salmon plant ST321 strains were more closely related to each other than to their ST321 counterparts from the same plant (Supplementary Table 2). The six cold-smoked salmon plant strains had 252 shared alleles that differed from their counterparts in Plants A or B (Supplementary Table 3). However, none of these alleles were unique to the cold-smoked salmon plant, as they were encountered in multiple strains from other sources (Supplementary Table 4).
One unexpected finding was that strains of diverse STs from Plant C had a higher incidence of new alleles based on BIGSdb wgMLST among their plant-specific alleles, with 66/84 (78.6%) of the Plant C-specific alleles being novel as compared to 8/34 (23.5%) and 61/80 (76.3%) for Plants A and B, respectively (Supplementary Table 4). This indicates higher diversification of strains from Plant B and C than from Plant A. This may reflect longer persistence and possibly greater or different selective pressures in Plants B or C, leading to the establishment of a greater number of mutations than in Plant A, or possibly the introduction of novel strains in this particular facility, as speculated earlier for ST5 in Plant C.
Internalin A (inlA) premature stop codon (PMSC) mutations leading to truncations and failure of InlA to anchor to the cell wall have been previously shown to be a frequent feature of serotype 1/2a, 1/2b, and 1/2c strains from processing plants, while remarkably uncommon in serotype 4b (Nightingale et al., 2005; Ward et al., 2010; Kathariou et al., 2017; Hurley et al., 2019). In addition, they are uncommon, regardless of serotype, in L. monocytogenes from natural environments, e.g., water, or from clinical sources (Gorski et al., 2016; Kathariou et al., 2017; Hurley et al., 2019). Truncation of InlA is associated with attenuated virulence in animal models (Nightingale et al., 2005; Kathariou et al., 2017; Disson et al., 2021). We found that inlA PMSCs were ST-specific and not randomly distributed among the sequenced FPE strains. Specifically, they were only encountered in seven of the nine strains of ST5 (serotype 1/2b; T635) and, as previously reported (Brown et al., 2021), in all six ST321 strains (serotype 1/2a; T730) (Supplementary Table 1; Figure 2).
Food-derived ST5 isolates from Poland and China were previously reported to harbor inlA PMSCs (T535, T606, or T753) (Chen et al., 2020a; Kurpas et al., 2020). Our panel of nine ST5 strains consists of eight from Plant A and one from Plant C. The T635 PMSC was noted in seven of the Plant A strains, while the remaining Plant A strain and the ST5 strain from Plant C, as well as the closely-related ST552 (CC5) strain from Plant B, encoded full-length InlA (Supplementary Table 1; Figure 2). This T635 InlA PMSC appears to be novel, not previously reported in a study of 501 L. monocytogenes strains from processing facilities and RTE foods, despite 11 different InlA truncation locations identified among 243 strains (Ward et al., 2010), or in other surveys (Nightingale et al., 2005; Chen et al., 2018; Hurley et al., 2019; Kurpas et al., 2020). Thus, it appears to be a unique and apparently widespread signature in ST5 strains from Plant A, raising the possibility that ST5 strains harboring this particular inlA PMSC may be attributable to this particular FPE.
The ST321 strains from Plant A and Plant B harbored the T730 inlA PMSC, as also reported previously (Brown et al., 2021). ST321 isolates from a variety of sources have been previously reported to harbor inlA PMSCs, but in other locations. Specifically, T699 was found to be a common inlA PMSC in ST321 strains from the cold-smoked fish processing plant in the United States discussed earlier (Harrand et al., 2020), as well as meat and vegetable processing plants in Ireland (Hurley et al., 2019). This T699 inlA PMSC was also found in three USDA-FSIS ST321 strains (Accession Nos. PDT000101443.2, PDT000162336.2, and PDT000208198.2) from ready-to-eat (RTE) meat products. As also noted with the T635 inlA PMSC in ST5, the T730 inlA PMSC in ST321 was not detected in previous surveys (Nightingale et al., 2005; Ward et al., 2010; Chen et al., 2018; Hurley et al., 2019; Kurpas et al., 2020). The T635 and T730 inlA PMSCs identified in our ST5 and ST321 isolates, respectively, may be associated with the specific facility type, i.e., turkey processing plants, potentially conferring fitness advantages to ST321 strains in these FPEs. Other, apparently widespread inlA PMSCs, such as T699, may offer potential fitness benefits in other types of FPEs.
ST321 strains constitute an especially attractive model system to showcase plant-specific genomic markers, because, in addition to the six strains from the turkey FPEs, ST321 strains have been investigated extensively from an unrelated type of FPE, i.e., a salmon processing plant (Harrand et al., 2020), as discussed above. In addition, ST321 is emerging as a dominant ST among food and FPE-derived isolates of L. monocytogenes (Hurley et al., 2019; Harrand et al., 2020; Matle et al., 2020; Cooper et al., 2021) and is noteworthy for high prevalence of BC resistance (Brown et al., 2021; Cooper et al., 2021). Therefore, ST321 strains from the turkey and salmon FPEs (Harrand et al., 2020) and other sources were investigated further for genomic content in a specific chromosomal hotspot located between lmo301 and lmo305 (Lee et al., 2012). A conserved type IV Mrr restriction-modification (RM) system was detected in this chromosomal hotspot in all five ST321 strains from Plant A and all six from the salmon FPE, as well as in the three USDA-FSIS strains mentioned above. Interestingly, however, the ST321 strain from Plant B lacked this RM system at this genomic location and instead harbored a gene putatively annotated as a lipoprotein (data not shown). Thus, the absence of the type IV Mrr RM system at this genomic hotspot appears to be unique to ST321 from Plant B. Furthermore, we previously showed that the Plant B ST321 strain was susceptible to the panel of wide-host-range lytic phages that we employed, while all five ST321 strains from Plant A were resistant and lacked either N-acetylglucosamine or rhamnose from the teichoic acid of the cell wall, thus preventing phage adsorption (Brown et al., 2021). The extent to which the type IV Mrr RM system may also contribute to phage resistance in the plant A strains of ST321 remains to be elucidated. These phage-resistant strains also harbored the same SNP (nt599, A to T) in lmo1084, a dTDP-4-dehydrorhamnose reductase involved in rhamnose wall teichoic acid glycosylation (Brown et al., 2021). Thus, in addition to the absence of the type IV Mrr RM, this particular SNP in lmo1084 may serve as an excellent plant-specific signature for ST321 originating from Plant A vs. Plant B (Supplementary Table 3).
The comK prophage junction fragments and prophage sequence content have been previously shown to be promising tools to track L. monocytogenes to individual FPEs, potentially reflecting plant-specific adaptations (Verghese et al., 2011; Kwon et al., 2020). STs encountered in multiple processing plants (Table 1) were screened for possible plant-specific prophages. A 43-kb prophage was found in the Plant B ST321 strain L1624a that was absent from the ST321 strains from Plant A or any of other ST321 strains that were examined, including those from the salmon FPE and the USDA-FSIS strains. Analysis via NCBI BLAST revealed no close matches to this prophage, with the best match being Listeriaphage LP-101 (Accession KJ094023.1, query coverage 67%, nucleotide identity 98.23%), classified as a temperate Siphoviridae with a genome size of 43.7 kb. Thus, in addition to the absence of the type IV Mrr RM, the presence of this LP-101-like 43-kb prophage in ST321 may constitute an additional molecular trait that may suggest Plant B origin for ST321 strains. Interestingly, as discussed earlier the ST321 strains from Plants A and B belong to the same SNP cluster (PDS000000366.424) (Supplementary Table 1), in spite of the difference in the plant-associated molecular traits. This, along with their shared and unique inlA PMSC (T730), suggests that these strains likely have a recent common ancestor and then diverged in response to specific FPE-related selective pressures in Plant B.
Plant-specific differences in prophage content were also noted with ST5 strains, with two distinct prophages found only in the ST5 strain from Plant C (SK197), but absent from all eight ST5 strains of Plant A and all 12 strains of meat origin in Ireland (Hurley et al., 2019). These two prophages identified in the ST5 strain from Plant C are 40-kb and 43-kb in size. The 40-kb prophage matches best (Accession DQ003642.1, query coverage 64%, nucleotide identity 95.23%) with A006 (38-kb temperate Siphoviridae phage) while the 43-kb prophage matches best with LP-101 (Accession KJ094023.1, query coverage 70%, nucleotide identity 96.63%), which is 43 kb as discussed above. In contrast to STs 5 and 321, no plant-specific prophages were found among ST6 strains which were also derived from multiple plants (A, B, C) or other STs isolated from more than one plant. In comparison to prophages for which gain or loss in food processing facilities is commonly observed in L. monocytogenes (Muhterem-Uyar et al., 2018; Harrand et al., 2020; Yang et al., 2020), SNPs are stable and likely to be encountered in all genomes, and can therefore better provide permanent plant-specific signatures to distinguish between strains from different plants. Nonetheless, our findings suggest that for certain STs, such as ST321, prophage presence and content, as well as the hotspot-associated type IV Mrr RM system discussed above, are promising genomic traits for plant-specific differentiation.
Phage resistance can be mediated by multiple mechanisms ranging from single-nucleotide polymorphisms (SNPs) in relevant genes to dedicated systems including RM systems, bacteriophage exclusion (BREX) systems, clustered regularly interspaced short palindromic repeats (CRISPR) and prophages (Kim et al., 2012; Di et al., 2014; Goldfarb et al., 2015; Bondy-Denomy et al., 2016; Brauge et al., 2018; Hupfeld et al., 2018; Ofir et al., 2018). SNPs in specific genes have been previously shown to confer phage resistance through loss of wall teichoic acid substituents under laboratory selection for phage resistance as well as in FPE-derived strains (Denes et al., 2015; Brown et al., 2021), and can serve as candidates for plant-specific signatures.
As described earlier, the FPE strains in our panel were analyzed for their resistance profiles to a panel of four wide-host-range lytic phages (A511, P100, 20422-1, and 805405-1) (Kim et al., 2008; Klumpp and Loessner, 2013). Phage resistance was encountered in several STs and the resistance profile tended to be the same for strains from the same plant, with the notable exception of ST5, 391 and 554, where multiple resistance profiles were noted among strains from Plant A (Supplementary Table 1). As described previously, in ST321 phage resistance was encountered in all five strains from Plant A but not in the ST321 strain from Plant B (Brown et al., 2021). The Plant A strains lacked either N-acetylglucosamine or rhamnose from the teichoic acid of the cell wall and did not share a common SNP conferring phage resistance (Brown et al., 2021). Phage resistance profiles of ST3 and ST7 (serotype 1/2b and 1/2a, respectively) also differed between plants (Supplementary Table 1).
Since phage resistance appears to be common among serotype 1/2a, 1/2b, and 1/2c strains from FPEs (Kim et al., 2008), its general usefulness as a facility-specific trait is likely to be limited. This is also reinforced by the fact that phage resistance is rare in serotype 4b, with the notable exception of the temperature-dependent resistance exhibited by ST6 strains (Kim et al., 2008, 2012). Nonetheless, phage resistance data can contribute to our currently limited understanding of the distinct adaptive traits of strains of a certain ST in response to FPE-specific selective regimes which, among other factors, can involve the abundance and composition of the phage community.
Additional phenotypic traits that were investigated included resistance to BC and to the heavy metal cadmium. As discussed above, 43% of the L. monocytogenes strains in our panel were resistant to BC, and BC resistance was always accompanied by resistance to cadmium; the reverse was not true, and we identified several BC-susceptible but cadmium-resistant strains (Figure 2; Supplementary Table 1). All BC-resistant strains in the panel harbored the bcrABC cassette originally identified on plasmid pLM80 (Elhanafi et al., 2010; Dutta et al., 2013). Two cadmium resistance determinants, cadA1 and cadA2, were detected among the cadmium-resistant strains (Supplementary Table 1). When present, bcrABC was accompanied by cadA1 (n = 8), cadA2 (n = 6) or both cadA1 and cadA2 (n = 8) (Supplementary Table 1). All 26 FPE strains harboring cadA1 shared the same novel allele (new#1) (Supplementary Table 1). Two different cadA2 alleles were found among the 24 FPE strains, with most (n = 22) strains harboring the same allele and the remaining two harboring an alternative allele, with only one SNP difference. Both of the latter strains (#22 and 34-6a) were from Plant A but belong to different STs, ST506 and ST6, respectively (Supplementary Table 1), and the shared cadA2 allele that they harbor may thus reflect acquisition of this determinant within this specific facility. Collectively, cadA alleles are extremely conserved and do not have the diversity required to yield plant-specific signatures.
Of the strains harboring bcrABC, all but one had the same allele for both bcrA and bcrB, while 3 alleles were detected among these strains for bcrC (Supplementary Table 1). The bcrC alleles appeared to be ST-specific and not associated with individual processing plants (Supplementary Table 1). Even though these resistance determinants are of limited use as plant-specific markers, information on their presence and variation can enhance our understanding of the FPE ecology and adaptations of L. monocytogenes.
As indicated earlier, the two sequenced L. welshimeri strains (266a-1 and 34-3a) appeared to be persistent in Plant A and were resistant to BC and cadmium, harboring the same bcrABC alleles found in L. monocytogenes strains, as well as cadA2, which frequently accompanied bcrABC in L. monocytogenes in the current study (Supplementary Table 1) as well as in larger surveys of L. monocytogenes isolates from these turkey processing plants (Mullapudi et al., 2010). These L. welshimeri strains also harbored the cadA2 allele encountered in the majority of cadA2-positive L. monocytogenes.
Investigation into potential variation in Listeria pathogenicity island-1 (LIPI-1) genes (Vázquez-Boland et al., 2001) revealed that prfA, plcA, hly, mpl, plcB, and orfX as well as htp, which is tightly controlled by PrfA (Chico-Calero et al., 2002), encoded full-length proteins in all 54 strains (Supplementary Table 1). However, conserved internal deletions (nt 854–959) in actA were found in diverse strains, including those of ST1, 5, 552, and 391 (Supplementary Table 1). This 105-nt internal deletion kept actA in frame but shortened it by 35 amino acids (AA), from 639 to 604 AA. In strains with full-length actA, such as EGD-e, a 35-AA sequence (PPPTDEELRLALPETPMLLGFNAPATSEPSSFEFP) is found twice in tandem but only once in strains containing this internal deletion. This same internal deletion was reported in ST1 isolates from ruminant rhombencephalitis (Dreyer et al., 2016). FPE isolates harboring actA internal deletions were also identified in all 12 meat-derived ST5 isolates from Ireland (Hurley et al., 2019), as well as in ST1 and ST5 isolates from illegally-imported food products in Europe (Rychli et al., 2018). Our analysis of several additional strains of ST1 (n = 9), ST5 (n = 16), and ST391 (n = 4) from diverse sources, including F2365 (ST1) and 2011L-2858 (ST5), indicates that this internal deletion and actA shortening is a clonal feature (Supplementary Table 5). Genome Comparator using PasteurMLST BIGSdb showed ST-specific actA allele conservation among all analyzed strains of ST1 (allele 56), ST5 (allele 120), and ST391 (allele 126) (Supplementary Table 5).
Major virulence determinants outside of LIPI-1 include inlA and inlB. As discussed earlier, PMSCs in inlA were found to be conserved (T730) in all ST321 strains from Plants A and B, and were also encountered and conserved (T635) in seven of the eight ST5 strains from Plant A (Supplementary Tables 1, 5). Interestingly, shortening of inlB was also found in the same seven Plant A ST5 strains that had PMSCs in inlA (Supplementary Table 5). This inlB shortening was due to internal deletions of nt 1,068–1,209 causing shortening from 630 to 583 AA (Supplementary Table 1). The deletion was conserved in all seven strains, and these strains harbored an identical inlB allele (allele 170) which was also encountered in several other strains of ST5 from other sources (Supplementary Table 5).
The concurrent presence of PMSCs in inlA and internal deletions in inlB in these Plant A strains, together with the internal deletion in actA, suggest selection for FPE adaptations that may promote environmental persistence while also potentially causing attenuated virulence. It was noteworthy that the ST5 strains with concurrent PMSCs in inlA and internal deletions in inlB also tended to be resistant to all four lytic wide-host-range phages that were tested (Supplementary Table 1). These and other aspects of environmental adaptation of these strains remain to be further investigated. In addition to the above-discussed strong association of inlA PMSCs with FPE and food origin in serogroup 1/2 strains (Kathariou et al., 2017), ActA truncation in strains of diverse STs has also been found to be more commonly encountered in FPE and food-derived isolates (190/810, 23.5%) than clinical isolates (10/332, 3.0%) (Painset et al., 2019). Such mutations in major virulence factors in FPE-derived strains highlight the specialization of L. monocytogenes to the FPE at the potential cost of virulence.
FPE strains were also screened for the two pathogenicity islands LIPI-3 and LIPI-4. LIPI-3 was detected in 18/31 lineage I strains, specifically the serotype 4b strains of ST1, 6, and 554 and the serotype 1/2b strains of ST3, 506 and 550, and not among any of the 23 lineage II strains (Supplementary Table 1), in agreement with the previously-reported strong association of LIPI-3 with lineage I (Cotter et al., 2008; Kathariou et al., 2017). Within the Lineage I FPE strains, presence of LIPI-3 was conserved among strains of the same ST (Supplementary Table 1). Despite LIPI-3 being associated with lineage I, it was not detected among any of the ST5/ST552 (CC5) strains from our study (Supplementary Table 1), similarly to findings from other investigations of ST5 (Hurley et al., 2019; Chen et al., 2020a). LIPI-4 was uncommon in our panel with only two of the 54 strains harboring LIPI-4, i.e., the serotype 1/2b strains 230a-3 and #22 of ST87 and 506, respectively (Supplementary Table 1). LIPI-4 rarity is in agreement with the fact that our panel lacked major clones that have been known to harbor it, such as ST4 and ST382 (Maury et al., 2016; Moura et al., 2016; Kathariou et al., 2017; Chen et al., 2020b).
FPEs are commonly contaminated by multiple strains and clones of L. monocytogenes (Hurley et al., 2019; Stoller et al., 2019; Yang et al., 2020), and selective enrichments are typically used to isolate L. monocytogenes from environmental samples (Johnson, 1998). However, this also creates a challenge in ascertaining how many colonies should be isolated from a Listeria-positive selective enrichment. While many colonies are likely to result from identical daughter cells, others may be distinct, resulting from the presence of multiple Listeria strains in the same sample (Ryser et al., 1996).
Among the 54 strains analyzed here, we noted examples where highly diverse strains were obtained from the same environmental swab. Specifically, samples 34, 201, and 210 yielded several distinct strains (Supplementary Table 1; Figure 2). Of special note was sample 34 from Plant A, which yielded four distinct strains of L. monocytogenes, two of ST5 (serotype 1/2b) and two of ST6 (serotype 4b), as well as one strain of L. welshimeri. The pronounced diversity in this sample, including different L. monocytogenes serotypes and two different Listeria species, highlights the importance of isolating multiple colonies from a single enrichment. Furthermore, standard multiplex PCR-based serotyping cannot reveal diversity among strains of the same serotype designation.
Even more noteworthy was the finding that diversity may remain hidden even upon further genotyping such as afforded by the seven-locus MLST scheme. For instance, the two serotype 4b strains from sample 34 (34-2b and 34-6a) were both of ST6 but WGS analysis revealed that they differed by 271 alleles based on wgMLST, with 116 of these belonging to the core genome. One of these strains, 34-6a, was in fact the most distant to all other ST6 strains from Plant A, B, or C combined (Supplementary Table 2, Figure 2). This strain belongs to SNP cluster PDS000025549.3 together with three other strains, all from human listeriosis in the United States in 2017 and 2018, while strain 34-2b was not affiliated with a SNP cluster (Supplementary Table 1). In addition to their noticeable genomic divergence, these two ST6 strains from sample 34 differed in their BC and cadmium resistance profiles (Supplementary Table 1) with 34-6a being resistant to both BC and cadmium while 34-2b was susceptible to both compounds (Supplementary Table 1). Both strains were resistant to all tested wide-host-range lytic phages at 25°C (Supplementary Table 1), reflecting the temperature-regulated RM system unique to ST6 (Kim et al., 2012), but 34-6a was previously found to be resistant to wide-host-range lytic phages when tested at 37°C, while 34-2b was susceptible at this temperature (Kim et al., 2008). Even though a previous analysis of ST6 strains indicated clustering of isolates from the same facility (Kwon et al., 2020) our data clearly show that this is not always the case, with pronounced divergence between ST6 strains even from the same environmental sample.
The two ST5 strains (34-2a and 34-4b) from sample 34 were more closely related, with identical T635 inlA PMSCs, common internal deletions in inlB, shared bcrABC and cadA alleles and also shared phage resistance profiles. However, they still differed by 170 alleles based on wgMLST, with 57 of these in the core genome and did not belong to the same SNP cluster (Supplementary Tables 1, 2). These strains were also placed in clearly distinct branches of the ST5/ST552 (CC5) clade in the phylogenetic tree (Figure 2). While one (34-2a) is not affiliated with a SNP cluster at all, highlighting its genetic novelty, the other (34-4b) belongs to a small three-strain SNP cluster (PDS000083092.1) containing only two additional Plant A strains of ST5 (Supplementary Table 1), and was placed together with these strains in the phylogenetic tree (Figure 2). Even though traditional tests such as multiplex PCR for serotype designations, seven-locus MLST and phage resistance profiles would not be sufficient to differentiate between 34-2a and 34-4b from each other, WGS clearly identified these as distinct strains.
Significantly, as noted above the ST6 strain 34-6a belongs to a SNP cluster which includes three human clinical strains, emphasizing the importance of not missing such a strain, should an epidemiological investigation need to be carried out with a specific FPE. Such findings illustrate the need to isolate and analyze via WGS multiple colonies from a selective enrichment. Not doing so will increase the risk of failing to detect clinically-relevant and potentially novel strains that may not cluster well with other members of the same ST (Supplementary Table 1).
Our study of 54 L. monocytogenes strains from turkey processing plants in the United States reveals the potential of WGS-based analysis of FPE-derived strains to identify facility-specific signatures and to inform aspects of FPE sample analysis. The value of WGS-based analysis was even further enhanced by integration of the WGS data with phenotypic assays of special relevance to the FPE ecosystem such as sanitizer resistance and resistance to lytic phages, with potential for novel insights on the FPE ecology and evolution of L. monocytogenes. We identified plant-specific alleles and other WGS signatures that may prove useful in attributing strains to the FPE of their origin. Additional investigations are needed to assess the potential biological relevance of those plant-specific alleles. We also demonstrated that a single FPE sample may harbor diverse strains whose genetic distinctness would go unnoticed with lower-resolution tools such as serotyping or even the seven-locus MLST scheme. Better understanding of such intra-sample diversity is critically needed not only for our overall understanding of Listeria's FPE ecology but also to help inform the number of L. monocytogenes isolates that may need to be characterized from a single Listeria-positive FPE sample. This would maximize the usefulness of environmental sampling not only for epidemiological investigations but also in monitoring contamination patterns and mitigation effectiveness within a specific FPE.
Strains of the same ST may harbor novel mutations and other genomic traits, including PMSCs in inlA, deletions in inlB, sanitizer resistance genes and prophages that may be used to differentiate among strains from different FPEs. While InlA truncations were found to be not randomly distributed throughout L. monocytogenes and common only in certain STs, the location of the PMSC may be unique to certain plants, though the extent to which such uniqueness may also reflect the specific type of FPE (i.e., turkey slaughter and processing plants) remains to be ascertained. The absence of a specific genetic feature may also serve as a plant-specific marker, as demonstrated by our finding that for certain STs the lack of specific accessory genome components, such as a type IV Mrr system and prophages, were unique to certain processing plants. These plant-specific genetic traits have the potential to be used to track L. monocytogenes movement through the food chain. Future work will be necessary to examine more sequenced strains, from diverse FPEs and regions, in order to further elucidate the distribution of plant-specific genetic traits in L. monocytogenes. The findings from the current study will be useful in designing and informing such future studies.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI [accession: PRJNA215355].
SK and PB: conceptualization. PB: formal analysis and writing—original draft. SK, ER, CL, L-AJ, and JE: funding acquisition. PB, SK, YC, RS, CS, and EB: investigation. SK: project administration. PB, YC, and SK: visualization. PB, SK, YC, RS, CP, L-AJ, JE, ER, CL, and CS: writing—review and editing. All authors have read and agreed to the published version of the manuscript.
This work was partially supported by a National Alliance for Food Safety and Security grant (USDA) and by award 2018-07464 from the USDA National Institute of Food and Agriculture.
Any opinions, findings, conclusions, or recommendations expressed are those of the author and do not necessarily reflect the view of the USDA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to acknowledge the CFSAN GenomeTrackr sequencing team as well as Julie S. Sherwood and Sheri Kernodle for their technical assistance in this project. We thank Sangmi Lee for sharing her insights on the genomic hotspot region.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsufs.2021.742353/full#supplementary-material
1. ^BioNumerics version 8.0 created by Applied Maths NV. Available online at: https://www.applied-maths.com.
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bondy-Denomy, J., Qian, J., Westra, E. R., Buckling, A., Guttman, D. S., Davidson, A. R., et al. (2016). Prophages mediate defense against phage infection through diverse mechanisms. The ISME Journal, 10, 2854–2866. doi: 10.1038/ismej.2016.79
Brauge, T., Faille, C., Sadovskaya, I., Charbit, A., Benezech, T., Shen, Y., et al. (2018). The absence of N-acetylglucosamine in wall teichoic acids of Listeria monocytogenes modifies biofilm architecture and tolerance to rinsing and cleaning procedures. PloS one, 13:e0190879. doi: 10.1371/journal.pone.0190879
Brown, P., Chen, Y., Parsons, C., Brown, E., Loessner, M. J., Shen, Y., et al. (2021). Whole genome sequence analysis of phage-resistant Listeria monocytogenes serotype 1/2a strains from turkey processing plants. Pathogens 10:199. doi: 10.3390/pathogens10020199
Carpentier, B., and Cerf, O. (2011). Review–Persistence of Listeria monocytogenes in food industry equipment and premises. Int. J. Food Microbiol. 145, 1–8. doi: 10.1016/j.ijfoodmicro.2011.01.005
Chen, M., Cheng, J., Wu, Q., Zhang, J., Chen, Y., Xue, L., et al. (2018). Occurrence, antibiotic resistance, and population diversity of Listeria monocytogenes isolated from fresh aquatic products in China. Front. Microbiol. 9:2215. doi: 10.3389/fmicb.2018.02215
Chen, Y., Chen, M., Wang, J., Wu, Q., Cheng, J., Zhang, J., et al. (2020a). Heterogeneity, characteristics, and public health implications of Listeria monocytogenes in ready-to-eat foods and pasteurized milk in China. Front. Microbiol. 11:642. doi: 10.3389/fmicb.2020.00642
Chen, Y., Chen, Y., Pouillot, R., Dennis, S., Xian, Z., Luchansky, J. B., et al. (2020b). Genetic diversity and profiles of genes associated with virulence and stress resistance among isolates from the 2010-2013 interagency Listeria monocytogenes market basket survey. PLoS ONE 15:e0231393. doi: 10.1371/journal.pone.0231393
Chen, Y., Gonzalez-Escalona, N., Hammack, T. S., Allard, M. W., Strain, E. A., and Brown, E. W. (2016). Core genome multilocus sequence typing for identification of globally distributed clonal groups and differentiation of outbreak strains of Listeria monocytogenes. Appl. Environ. Microbiol. 82, 6258–6272. doi: 10.1128/AEM.01532-16
Cherifi, T., Carrillo, C., Lambert, D., Miniaï, I., Quessy, S., Larivière-Gauthier, G., et al. (2018). Genomic characterization of Listeria monocytogenes isolates reveals that their persistence in a pig slaughterhouse is linked to the presence of benzalkonium chloride resistance genes. BMC Microbiol. 18:220. doi: 10.1186/s12866-018-1363-9
Chico-Calero, I., Suárez, M., González-Zorn, B., Scortti, M., Slaghuis, J., Goebel, W., Vázquez-Boland, J. A., and European Listeria Genome Consortium (2002). Hpt, a bacterial homolog of the microsomal glucose- 6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc. Natl. Acad. Sci. U. S. A. 99, 431–436. doi: 10.1073/pnas.012363899
Cooper, A. L., Carrillo, C. D., Deschênes, M., and Blais, B. W. (2021). Genomic markers for quaternary ammonium compound resistance as a persistence indicator for Listeria monocytogenes contamination in food manufacturing environments. J. Food Prot. 84, 389–398. doi: 10.4315/JFP-20-328
Cotter, P. D., Draper, L. A., Lawton, E. M., Daly, K. M., Groeger, D. S., Casey, P. G., et al. (2008). Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes. PLoS Pathog. 4:e1000144. doi: 10.1371/journal.ppat.1000144
Davis, J. J., Wattam, A. R., Aziz, R. K., Brettin, T., Butler, R., Butler, R. M., et al. (2020). The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res. 48, D606–D612. doi: 10.1093/nar/gkz943
de Noordhout, C. M., Devleesschauwer, B., Angulo, F. J., Verbeke, G., Haagsma, J., Kirk, M., et al. (2014). The global burden of listeriosis: a systematic review and meta-analysis. Lancet Infect. Dis. 14, 1073–1082. doi: 10.1016/S1473-3099(14)70870-9
Denes, T., den Bakker, H. C., Tokman, J. I., Guldimann, C., and Wiedmann, M. (2015). Selection and characterization of phage-resistant mutant strains of Listeria monocytogenes reveal host genes linked to phage adsorption. Appl. Environ. Microbiol. 81, 4295–4305. doi: 10.1128/AEM.00087-15
Di, H., Ye, L., Yan, H., Meng, H., Yamasak, S., and Shi, L. (2014). Comparative analysis of CRISPR loci in different Listeria monocytogenes lineages. Biochem. and Biophys. Res. Commun. 454, 399–403. doi: 10.1016/j.bbrc.2014.10.018
Disson, O., Moura, A., and Lecuit, M. (2021). Making sense of the biodiversity and virulence of Listeria monocytogenes. Trends Microbiol. 29, 811–822. doi: 10.1016/j.tim.2021.01.008
Doumith, M., Buchrieser, C., Glaser, P., Jacquet, C., and Martin, P. (2004). Differentiation of the major Listeria monocytogenes serovars by multiplex PCR. J. Clin. Microbiol. 42, 3819–3822. doi: 10.1128/JCM.42.8.3819-3822.2004
Dreyer, M., Aguilar-Bultet, L., Rupp, S., Guldimann, C., Stephan, R., Schock, A., et al. (2016). Listeria monocytogenes sequence type 1 is predominant in ruminant rhombencephalitis. Sci. Rep. 6:36419. doi: 10.1038/srep36419
Dutta, V., Elhanafi, D., and Kathariou, S. (2013). Conservation and distribution of the benzalkonium chloride resistance cassette bcrABC in Listeria monocytogenes. Appl. Environ. Microbiol. 79, 6067–6074. doi: 10.1128/AEM.01751-13
Eifert, J. D., Curtis, P. A., Bazaco, M. C., Meinersmann, R. J., Berrang, M. E., Kernodle, S., et al. (2005). Molecular characterization of Listeria monocytogenes of the serotype 4b complex (4b, 4d, 4e) from two turkey processing plants. Foodborne Pathog. Dis. 2, 192–200. doi: 10.1089/fpd.2005.2.192
Elhanafi, D., Dutta, V., and Kathariou, S. (2010). Genetic characterization of plasmid-associated benzalkonium chloride resistance determinants in a Listeria monocytogenes strain from the 1998-1999 outbreak. Appl. Environ. Microbiol. 76, 8231–8238. doi: 10.1128/AEM.02056-10
Fagerlund, A., Langsrud, S., and Møretrø, T. (2020). In-depth longitudinal study of Listeria monocytogenes ST9 isolates from the meat processing industry: resolving diversity and transmission patterns using whole-genome sequencing. Appl. Environ. Microbiol. 86:e00579-20. doi: 10.1128/AEM.00579-20
Fagerlund, A., Langsrud, S., Schirmer, B. C., Møretrø, T., and Heir, E. (2016). Genome analysis of Listeria monocytogenes sequence type 8 strains persisting in salmon and poultry processing environments and comparison with related strains. PLoS ONE 11:e0151117. doi: 10.1371/journal.pone.0151117
Ferreira, V., Wiedmann, M., Teixeira, P., and Stasiewicz, M. J. (2014). Listeria monocytogenes persistence in food-associated environments: epidemiology, strain characteristics, and implications for public health. J. Food Prot. 77, 150–170. doi: 10.4315/0362-028X.JFP-13-150
Gandhi, M., and Chikindas, M. L. (2007). Listeria: a foodborne pathogen that knows how to survive. Int. J. Food Microbiol. 113, 1–15. doi: 10.1016/j.ijfoodmicro.2006.07.008
Goldfarb, T., Sberro, H., Weinstock, E., Cohen, O., Doron, S., Charpak-Amikam, Y., et al. (2015). BREX is a novel phage resistance system widespread in microbial genomes. The EMBO J. 34, 169–183. doi: 10.15252/embj.201489455
Gorski, L., Parker, C. T., Liang, A. S., Walker, S., and Romanolo, K. F. (2016). The majority of genotypes of the virulence gene inlA are intact among natural watershed isolates of Listeria monocytogenes from the central California coast. PLoS ONE 11:e0167566. doi: 10.1371/journal.pone.0167566
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics (Oxford, England) 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Harrand, A. S., Jagadeesan, B., Baert, L., Wiedmann, M., and Orsi, R. H. (2020). Evolution of Listeria monocytogenes in a food processing plant involves limited single-nucleotide substitutions but considerable diversification by gain and loss of prophages. Appl. Environ. Microbiol. 86, e02493-19. doi: 10.1128/AEM.02493-19
Hoffman, S., Maculloch, B., and Batz, M. (2015). Economic burden of major foodborne illnesses acquired in the United States. Economic information bulletin 205081, United States Department of Agriculture, Economic Research Service.
Hupfeld, M., Trasanidou, D., Ramazzini, L., Klumpp, J., Loessner, M. J., and Kilcher, S. (2018). A functional type II-A CRISPR-Cas system from Listeria enables efficient genome editing of large non-integrating bacteriophage. Nucleic Acids Res. 46, 6920–6933. doi: 10.1093/nar/gky544
Hurley, D., Luque-Sastre, L., Parker, C. T., Huynh, S., Eshwar, A. K., Nguyen, S. V., et al. (2019). Whole-genome sequencing-based characterization of 100 Listeria monocytogenes isolates collected from food processing environments over a four-year period. mSphere 4:e00252-19. doi: 10.1128/mSphere.00252-19
Johnson, J. L. (1998). Isolation and Identification of Listeria monocytogenes from Meat, Poultry, and Egg Products. U.S. Department of Agriculture Food Safety and Inspection Service Microbiology Laboratory Guidebook, 8-1-8-18, 3rd ed. U.S. Department of Agriculture Food Safety and Inspection Service, Washington, DC.
Jolley, K. A., Bray, J. E., and Maiden, M. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Katharios-Lanwermeyer, S., Rakic-Martinez, M., Elhanafi, D., Ratani, S., Tiedje, J. M., and Kathariou, S. (2012). Co-selection of cadmium and benzalkonium chloride resistance in conjugative transfers from nonpathogenic Listeria spp. to other Listeriae. Appl. Environ. Microbiol. 78, 7549–7556. doi: 10.1128/AEM.02245-12
Kathariou, S. (2002). Listeria monocytogenes virulence and pathogenicity, a food safety perspective. J. Food Prot. 65, 1811–1829. doi: 10.4315/0362-028X-65.11.1811
Kathariou, S., Evans, P., and Dutta, V. (2017). “Strain-specific virulence differences in Listeria monocytogenes: current perspectives in addressing an old and vexing issue,” in Foodborne Pathogens. Food Microbiology and Food Safety, eds J. Gurtler, M. Doyle, and J. Kornacki (Cham: Springer), 61–92. doi: 10.1007/978-3-319-56836-2_3
Kim, J. W., Dutta, V., Elhanafi, D., Lee, S., Osborne, J. A., and Kathariou, S. (2012). A novel restriction-modification system is responsible for temperature-dependent phage resistance in Listeria monocytogenes ECII. Appl. Environ. Microbiol. 78, 1995–2004. doi: 10.1128/AEM.07086-11
Kim, J. W., Siletzky, R. M., and Kathariou, S. (2008). Host ranges of Listeria-specific bacteriophages from the turkey processing plant environment in the United States. Appl. Environ. Microbiol. 74, 6623–6630. doi: 10.1128/AEM.01282-08
Klumpp, J., and Loessner, M. J. (2013). Listeria phages: genomes, evolution, and application. Bacteriophage 3:e26861. doi: 10.4161/bact.26861
Knudsen, G. M., Nielsen, J. B., Marvig, R. L., Ng, Y., Worning, P., Westh, H., et al. (2017). Genome-wide-analyses of Listeria monocytogenes from food-processing plants reveal clonal diversity and date the emergence of persisting sequence types. Environ. Microbiol. Rep. 9, 428–440. doi: 10.1111/1758-2229.12552
Kurpas, M., Osek, J., Moura, A., Leclercq, A., Lecuit, M., and Wieczorek, K. (2020). Genomic characterization of Listeria monocytogenes isolated from ready-to-eat meat and meat processing environments in Poland. Front. Microbiol. 11:1412. doi: 10.3389/fmicb.2020.01412
Kwon, H. J., Chen, Z., Evans, P., Meng, J., and Chen, Y. (2020). Characterization of mobile genetic elements using long-read sequencing for tracking Listeria monocytogenes from Food Processing Environments. Pathogens 9:822. doi: 10.3390/pathogens9100822
Lee, S., Ward, T. J., Siletzky, R. M., and Kathariou, S. (2012). Two novel type II restriction-modification systems occupying genomically equivalent locations on the chromosomes of Listeria monocytogenes strains. Appl. Environ. Microbiol. 78, 2623–2630. doi: 10.1128/AEM.07203-11
Matle, I., Mafuna, T., Madoroba, E., Mbatha, K. R., Magwedere, K., and Pierneef, R. (2020). Population structure of non-ST6 Listeria monocytogenes isolated in the red meat and poultry value chain in South Africa. Microorganisms 8:1152. doi: 10.3390/microorganisms8081152
Maury, M. M., Tsai, Y. H., Charlier, C., Touchon, M., Chenal-Francisque, V., Leclercq, A., et al. (2016). Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 48, 308–313. doi: 10.1038/ng.3501
Mazaheri, T., Cervantes-Huamán, B., Bermúdez-Capdevila, M., Ripolles-Avila, C., and Rodríguez-Jerez, J. J. (2021). Listeria monocytogenes biofilms in the food industry: is the current hygiene program sufficient to combat the persistence of the pathogen?. Microorganisms 9:181. doi: 10.3390/microorganisms9010181
McLauchlin, J., Hampton, M. D., Shah, S., Threlfall, E. J., Wieneke, A. A., and Curtis, G. D. (1997). Subtyping of Listeria monocytogenes on the basis of plasmid profiles and arsenic and cadmium susceptibility. J. Appl. Microbiol. 83, 381–388. doi: 10.1046/j.1365-2672.1997.00238.x
Moura, A., Criscuolo, A., Pouseele, H., Maury, M. M., Leclercq, A., Tarr, C., et al. (2016). Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol. 2:16185. doi: 10.1038/nmicrobiol.2016.185
Muhterem-Uyar, M., Ciolacu, L., Wagner, K. H., Wagner, M., Schmitz-Esser, S., and Stessl, B. (2018). New aspects on Listeria monocytogenes ST5-ECVI predominance in a heavily contaminated cheese processing environment. Front. Microbiol. 9:64. doi: 10.3389/fmicb.2018.00064
Mullapudi, S., Siletzky, R. M., and Kathariou, S. (2008). Heavy-metal and benzalkonium chloride resistance of Listeria monocytogenes isolates from the environment of turkey-processing plants. Appl. Environ. Microbiol. 74, 1464–1468. doi: 10.1128/AEM.02426-07
Mullapudi, S., Siletzky, R. M., and Kathariou, S. (2010). Diverse cadmium resistance determinants in Listeria monocytogenes isolates from the turkey processing plant environment. Appl. Environ. Microbiol. 76, 627–630. doi: 10.1128/AEM.01751-09
Nightingale, K. K., Windham, K., Martin, K. E., Yeung, M., and Wiedmann, M. (2005). Select Listeria monocytogenes subtypes commonly found in foods carry distinct nonsense mutations in inlA, leading to expression of truncated and secreted internalin A, and are associated with a reduced invasion phenotype for human intestinal epithelial cells. Appl. Environ. Microbiol. 71, 8764–8772. doi: 10.1128/AEM.71.12.8764-8772.2005
Ofir, G., Melamed, S., Sberro, H., Mukamel, Z., Silverman, S., Yaakov, G., et al. (2018). DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat. Microbiol. 3, 90–98. doi: 10.1038/s41564-017-0051-0
Orsi, R. H., den Bakker, H. C., and Wiedmann, M. (2011). Listeria monocytogenes lineages: genomics, evolution, ecology, and phenotypic characteristics. Int. J. Med. Microbiol. 301, 79–96. doi: 10.1016/j.ijmm.2010.05.002
Painset, A., Björkman, J. T., Kiil, K., Guillier, L., Mariet, J. F., Félix, B., et al. (2019). LiSEQ - whole-genome sequencing of a cross-sectional survey of Listeria monocytogenes in ready-to-eat foods and human clinical cases in Europe. Microb. Genom. 5:e000257. doi: 10.1099/mgen.0.000257
Painter, J., and Slutsker, L. (2007). “Listeriosis in humans,” in Listeria, listeriosis and Food Safety, 3rd Edn, eds E. T. Ryser and E. H. Marth (Boca Raton: CRC Press Taylor and Francis Group), 85–109. doi: 10.1201/9781420015188.ch4
Pasquali, F., Palma, F., Guillier, L., Lucchi, A., De Cesare, A., and Manfreda, G. (2018). Listeria monocytogenes sequence types 121 and 14 repeatedly isolated within one year of sampling in a rabbit meat processing plant: persistence and ecophysiology. Front. Microbiol. 9:596. doi: 10.3389/fmicb.2018.00596
Rychli, K., Stessl, B., Szakmary-Brändle, K., Strauß, A., Wagner, M., and Schoder, D. (2018). Listeria monocytogenes isolated from illegally imported food products into the European Union harbor different virulence factor variants. Genes 9:428. doi: 10.3390/genes9090428
Ryser, E. T., Arimi, S. M., Bunduki, M. M., and Donnelly, C. W. (1996). Recovery of different Listeria ribotypes from naturally contaminated, raw refrigerated meat and poultry products with two primary enrichment media. Appl. Environ. Microbiol. 62, 1781–1787. doi: 10.1128/aem.62.5.1781-1787.1996
Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M. A., Roy, S. L., et al. (2011). Foodborne illness acquired in the United States–major pathogens. Emerging Infect. Dis. 17, 7–15. doi: 10.3201/eid1701.P11101
Schlech, W. F. (2019). Epidemiology and clinical manifestations of Listeria monocytogenes infection. Microbiol. Spectrum 7:GPP3-0014-2018. doi: 10.1128/microbiolspec.GPP3-0014-2018
Stoller, A., Stevens, M., Stephan, R., and Guldimann, C. (2019). Characteristics of Listeria monocytogenes strains persisting in a meat processing facility over a 4-Year Period. Pathogens 8:32. doi: 10.3390/pathogens8010032
Swaminathan, B., and Gerner-Smidt, P. (2007). The epidemiology of human listeriosis. Microb. Infect. 9, 1236–1243. doi: 10.1016/j.micinf.2007.05.011
The NCBI Pathogen Detection Project (2016). Bethesda, MD: National Library of Medicine (US), National Center for Biotechnology Information. Available online at: https://www.ncbi.nlm.nih.gov/pathogens/ (accessed September 09, 2021).
Vázquez-Boland, J. A., Kuhn, M., Berche, P., Chakraborty, T., Domínguez-Bernal, G., Goebel, W., et al. (2001). Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14, 584–640. doi: 10.1128/CMR.14.3.584-640.2001
Verghese, B., Lok, M., Wen, J., Alessandria, V., Chen, Y., Kathariou, S., et al. (2011). comK prophage junction fragments as markers for Listeria monocytogenes genotypes unique to individual meat and poultry processing plants and a model for rapid niche-specific adaptation, biofilm formation, and persistence. Appl. Environ. Microbiol. 77, 3279–3292. doi: 10.1128/AEM.00546-11
Ward, T. J., Evans, P., Wiedmann, M., Usgaard, T., Roof, S. E., Stroika, S. G., et al. (2010). Molecular and phenotypic characterization of Listeria monocytogenes from U.S. Department of Agriculture Food Safety and Inspection Service surveillance of ready-to-eat foods and processing facilities. J. Food Protect. 73, 861–869. doi: 10.4315/0362-028X-73.5.861
Yang, H., Hoffmann, M., Allard, M. W., Brown, E. W., and Chen, Y. (2020). Microevolution and gain or loss of mobile genetic elements of outbreak-related Listeria monocytogenes in food processing environments identified by whole genome sequencing analysis. Front. Microbiol. 11:866. doi: 10.3389/fmicb.2020.00866
Zhang, H., Que, F., Xu, B., Sun, L., Zhu, Y., Chen, W., et al. (2021). Identification of Listeria monocytogenes contamination in a ready-to-eat meat processing plant in China. Front. Microbiol. 12:628204. doi: 10.3389/fmicb.2021.628204
Keywords: Listeria monocytogenes, inlA, whole genome sequencing, turkey processing plant, persistence, plant-specific signatures, cgMLST, wgMLST
Citation: Brown P, Chen Y, Siletzky R, Parsons C, Jaykus L-A, Eifert J, Ryser E, Logue CM, Stam C, Brown E and Kathariou S (2021) Harnessing Whole Genome Sequence Data for Facility-Specific Signatures for Listeria monocytogenes: A Case Study With Turkey Processing Plants in the United States. Front. Sustain. Food Syst. 5:742353. doi: 10.3389/fsufs.2021.742353
Received: 16 July 2021; Accepted: 10 September 2021;
Published: 13 October 2021.
Edited by:
Jasna Kovac, The Pennsylvania State University (PSU), United StatesReviewed by:
M. Leonor Faleiro, University of Algarve, PortugalCopyright © 2021 Brown, Chen, Siletzky, Parsons, Jaykus, Eifert, Ryser, Logue, Stam, Brown and Kathariou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophia Kathariou, c2thdGhhckBuY3N1LmVkdQ==; Phillip Brown, cGVicm93bjRAbmNzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.