
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Sustain. Food Syst. , 19 July 2021
Sec. Crop Biology and Sustainability
Volume 5 - 2021 | https://doi.org/10.3389/fsufs.2021.688393
This article is part of the Research Topic Identification and Control of the Geographic Origin of Plant Materials: Investigation of Ambient Influences and Environmental Selection View all 10 articles
 Nancy Bhagat1†
Nancy Bhagat1† Shivali Sharma1†
Shivali Sharma1† Sheetal Ambardar2
Sheetal Ambardar2 Sushmeeta Raj1
Sushmeeta Raj1 Deepika Trakroo1,3
Deepika Trakroo1,3 Micha Horacek4
Micha Horacek4 Rahma Zouagui5
Rahma Zouagui5 Laila Sbabou5
Laila Sbabou5 Jyoti Vakhlu1*
Jyoti Vakhlu1*Host–microbiome interactions are specific and not random, making them defining entities for the host. The hypothesis proposed by various researchers earlier, that both plants and animals harbor specific inheritable core microbiome, is being augmented in the present study. Additionally, a case for using microbial fingerprint as a biomarker, not only for plant identification but also as a geographical indicator, has been investigated, taking Crocus sativus, saffron, as a study material. Crocus sativus, a monogenetic herb, on account of its male sterility and vegetative propagation, is reported to lack genome based molecular markers. Cormosphere microbiome (microbiome associated with corm) has been compared across three geographical locations, in two continents, to identify the core and unique microbiome, during the vegetative phase of its growth. Microbiome analysis done at phylum and genus level, using next generation sequencing technology, revealed that cormosphere at three locations harbored common phyla. At genus level, 24 genera were found common to all three geographical locations, indicating them to be part of the core microbiome of saffron. However, there were some bacterial genera unique to Kashmir, Kishtwar, and Morocco that can be used to develop microbial markers/geographical indicators for saffron grown in these regions. This is a preliminary study, indicating that the location specific bacterial community can be used to develop microbial barcodes but needs further augmentation with high coverage data from other saffron growing geographical regions.
Molecular markers are important tools for plant genome analysis, crop improvement, and development of barcodes for authentic plant identification (Mishra et al., 2016). The development of molecular markers relies on genetic variation in the plants; however, plants lacking genetic variation due to asexual reproduction harbor unidentified or poor resolution molecular markers. Crocus sativus is one amongst such plants that lack genetic variations. Crocus sativus, commonly known as saffron, is an autumn-flowering perennial sterile triploid plant bearing eight chromosomes per set, i.e., 2n = 3x = 24 (Goldblatt et al., 2006; Ahmad et al., 2021). Due to male sterility, it does not produce viable seeds and propagates vegetatively, thereby lacking any variations at the genomic level. In order to propagate vegetatively, clusters of corms, underground bulb-like, starch-storing organs are dug up, divided, and replanted in the soil (Golmohammadi, 2014; Moshtaghi, 2020). The stigmas of saffron flowers are plucked and dried, to be used as a seasoning and coloring agent in food (Mzabri et al., 2017; Jafari et al., 2020). It is the world's highest priced spice with medicinal and aromatic properties, so referred to as ‘golden condiment' (Monika and Neha, 2014; Pandita, 2021; Su et al., 2021). This exotic spice is cultivated worldwide in many countries, among them Iran, Spain, India and Morocco are the major producers (Mykhailenko et al., 2020; Gupta et al., 2021). Saffron is reported to be a monomorphic plant, having no variations at a genomic level across different saffron accessions across the world, as depicted by various molecular markers like RAPD, AFLP, and SSR (Rubio-Moraga et al., 2009; Busconi et al., 2015). However, there are variations in the phenotypic and biochemical characteristics such as the percentage of various metabolites (crocin, picocrocin, and saffranal) reported from different geographical locations (Othman et al., 2020). However, there are no authentic molecular markers that can identify these variations till date (Alavi-Kia et al., 2008; Keify and Beiki, 2012; Mir et al., 2021).
In the last three decades, microbes are established to have a pronounced effect on the biotic or abiotic environments they inhabit (Gupta et al., 2021). No eukaryote lives as an individual but is a meta-organism or holobiont (Guerrero et al., 2013; Vandenkoornhuyse et al., 2015). As established in humans, plants have their own microbiota and studies have shown that the rhizosphere (hotspot for microbial activities) has tremendous microbial density associated with almost 1011 microbial cells/gram of soil, represented by >30,000 prokaryotic species (Berendsen et al., 2012; Chaparro et al., 2014; Ofek-Lalzar et al., 2014). The sum total of the genomes of microbes associated with any environmental niche is referred to as “microbiome”, the term coined by Joshua Lederberg (Lederberg and McCray, 2001).
The rhizosphere of many plants has been reported to contain specific microbiomes that are essential for plant survival and are referred to as the plant's second genome (Berendsen et al., 2012; Ofek-Lalzar et al., 2014). The microbiome and its dynamics have been extensively studied in the rhizosphere of Arabidopsis, Zea mays, Triticum aestivum, Oryza sativa, Vitis vinifera, Crocus sativus, etc. (Ambardar and Vakhlu, 2013; Ambardar et al., 2014, 2016; Mendes and Raaijmakers, 2015; Kandel et al., 2017). Studies also suggest that though the microbiome associated with any plant is organ specific, it may show temporal variation, primarily during developmental stages, geographical location, and cultivation practices (Peiffer et al., 2013; Chaparro et al., 2014; Edwards et al., 2015; Lin et al., 2020).
Despite temporal and spatial variations, the core microbiome is specific for each plant. Core microbiome may be defined as a congregation of micro-organisms within a host's microbiome that remains unaltered across different growth stages, niches, and geographical locations. Core inheritable microbiome, present in all the organisms, is hypothesized to be inherited as a core genome (Walters et al., 2018). The composition of core microbiome is reported to depend on the host plant, and this hypothesis was tested in Arabidopsis by Lundberg et al. (2012) wherein specific microbial communities common to tested plant varieties were established. The presence of core microbiome has been established in rice (Eyre et al., 2019), sponges (Schmitt et al., 2012), corals (Ainsworth et al., 2015; Hernandez-Agreda et al., 2016, 2017), wheat (Kuzniar et al., 2020), human gut (Qin et al., 2010), human hands (Fierer et al., 2008), and sand beaches (Newton et al., 2013).
The present study was initiated to discover the core microbiome of saffron cormosphere, along with unique genera specific to the saffron cultivated at a particular location. Unique genera can be used to develop molecular markers/geographical indicators. In the present study, core microbiome of saffron cormosphere comprises 24 genera that remain unaltered in three locations across two continents, i.e., Africa and Asia. In addition, unique microbes associated with the corm at each location have also been identified and cataloged, which can be used as molecular markers/geographical indicators.
Plant samples were collected from three geographical locations, two in Jammu and Kashmir, India, i.e., Kishtwar (33°19′12.00″ N 75°46′12.00″ E) and Kashmir (34.02°″ N 74.930″ E), and one from Morocco (Taliouine, 30°31′58.00″ N 7°55′32.00″ W) (Figure 1). Plant samples were collected from Kishtwar during three consecutive years (2011, 2012, and 2013; coded as C11, C12, and C13, respectively), whereas from Kashmir in 2013 and Morocco in 2016. In all the cases, sampling was done during the vegetative phase of saffron life cycle. The sample collection was done as per the protocol developed by Ambardar et al. (2014). The temperature and humidity of the sites at the time of sample collection have been tabulated in Table 1.
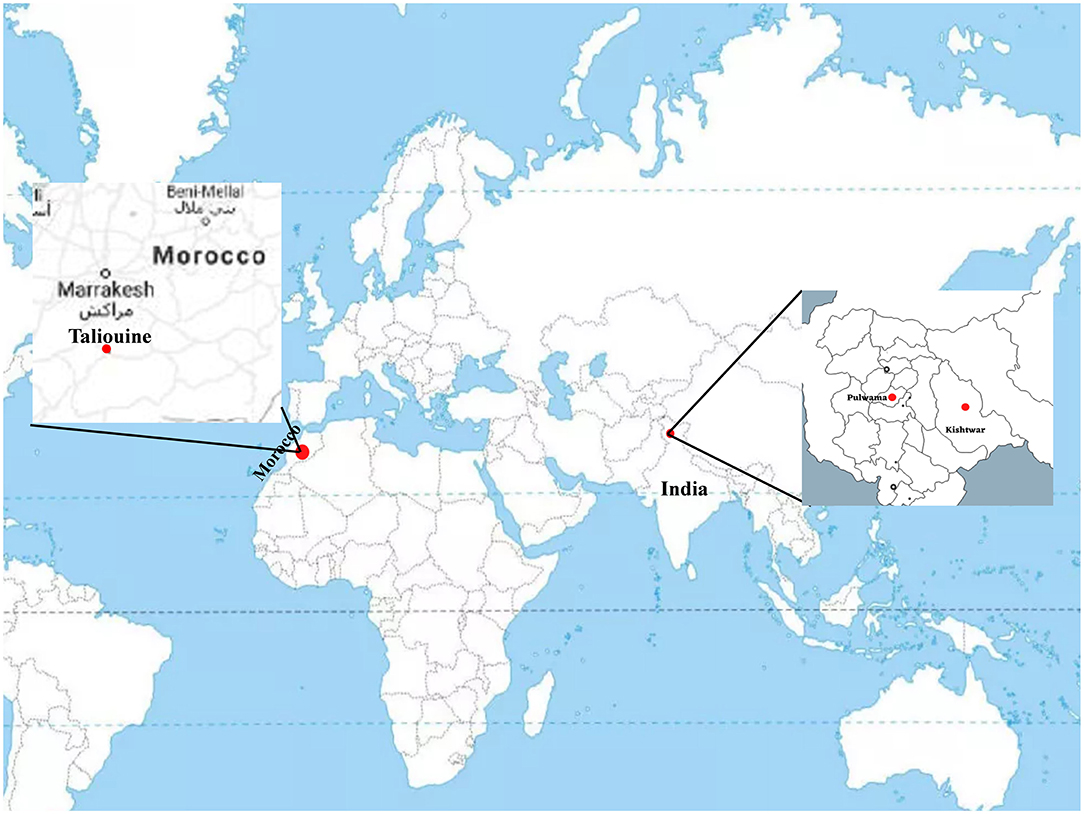
Figure 1. Geographical location of sample collection sites on world map. Kishtwar (33°19′12.00“ N 75°46′12.00″ E), Kashmir (34°1′30″ N 74°.5′80″ E), and Morocco (Taliouine, 30°31′58.00″ N 7°55′32.00″ W).
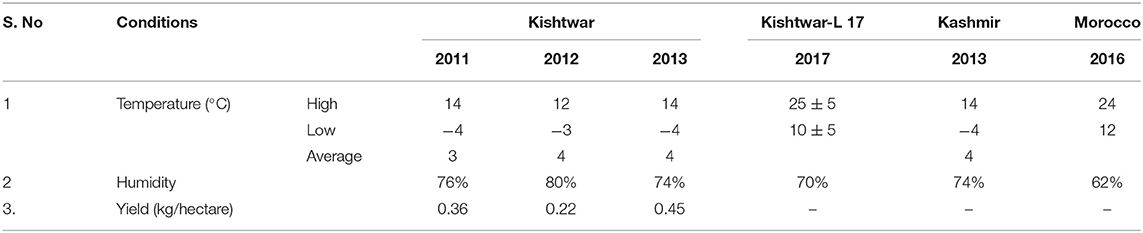
Table 1. The temperature and humidity of the three geographical regions: Kishtwar (during different years), Kishtwar-L 17, Kashmir, and Morocco.
In a separate experiment, corm samples and saffron field soil from Kishtwar region [referred to as Kishtwar-L 17 (L for lab)] were collected during the dormant phase of life cycle (August, 2017) and stored at room temperature (20–25°C) till further use. Corms were sown in the soil (Kishtwar soil) at the start of vegetative phase (October, 2017) and were grown for 2 months. The corms were grown in pots that were incubated in plant growth chamber with day temperature (25 ± 5°C), night temperature (10 ± 5°C), and 70% relative humidity and watered after every 5 days with autoclaved distilled water for a period of 2 months. Cormosphere samples were collected after 2 months in vegetative phase.
The loose soil adhered to corms was removed by shaking corms vigorously. Corm sheath was peeled off from the corms under sterile conditions (inside laminar air flow) and was used for metagenomic DNA extraction. Metagenomic DNA was isolated using three different standardized protocols: Zhou et al. (1996), Wechter et al. (2003), and Pang et al. (2008) to capture maximum microbial diversity. The metagenomic extraction was done in triplicates for each sample with all the three protocols. These triplicate metagenomic DNA extracts were subsequently pooled to get maximum possible diversity for each sample. The quality of metagenomic DNA was accessed on 0.8% agarose gel and quantified using Invitrogen Qubit® 2.0 Fluorometer (Thermofischer, Foster City, CA, USA).
The library preparation of isolated metagenomic DNA from all the samples was done using Truseq Nano DNA Library preparation kit (Catalog No. 20015964, Illumina, CA, USA) as per the instructions of manufacturer. To ensure maximum yield, a high-fidelity amplification step was performed using sparQ HiFi PCR Master mix (Quanta bio, QIAGEN, Beverly Inc. Catalog No. 95192-050). The quality of amplified libraries was analyzed on Bioanalyzer 2100 (Agilent Technologies, catalog number: G2939BA) and was further sequenced using Illumina NextSeq 500 platform with 2 × 150 base paired-end configuration from Xcleries labs limited, Ahmadabad, Gujarat, India. The raw reads were quality checked using FastQC tool kit (Brown et al., 2017). The low quality reads having phred score less than 30 were filtered and trimmed using software Trimmomatic (Bolger et al., 2014). The de novo assembly of high quality paired end reads for all samples was done using CLC Genomics Workbench 6.0 (CLC bio, Aarhus, Denmark) with the following parameters: minimum contig length: 200, automatic word size, perform scaffolding, mismatch cost: 2, insertion cost: 3, deletion cost: 3, length fraction: 0.5, similarity fraction: 0.8. The assembled contigs were then uploaded on MG-RAST (Metagenomic Rapid Annotation using Subsystem Technology) server version 4.0.3 with default parameters for analyzing taxonomic hit distribution and calculation of Shannon-alpha diversity values (Meyer et al., 2008).
Taxonomic classification up to phylum and genus level was represented in the form of heat maps and principal component analysis (PCA) plots using ClustVis tool (Metsalu and Vilo, 2015). Rarefaction curve was generated using PAST software version 4.03 (Hammer et al., 2001). Circos plot was generated using the tool Circos Version 0.63-9 (Krzywinski et al., 2009).
Whole metagenome sequencing data of the six samples (Kishtwar 2011, 2012, 2013; Kishtwar-L 17, Kashmir 2013; Morocco 2016), collected from three locations during the vegetative period of growth, ranged between 5.3 million reads and 40 million reads. Reads mapped to bacteria and eukaryota ranged from 85.29 to 98.78% and 1 to 14.2% in all the samples, respectively (Table 2). The dynamics of the microbiome of Kishtwar for three consecutive years (2011, 2012, and 2013) were studied, and the average of the 3 years was taken as reference. Subsequently, the Kishtwar average was compared to the microbiome of saffron cormosphere of Kishtwar-L 17, Kashmir (2013), and Morocco (2016) to discover core and location specific unique microbiome.
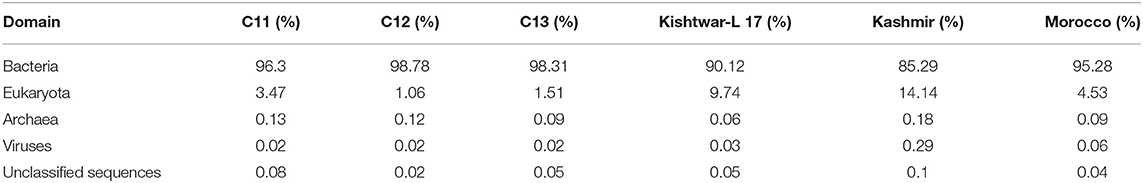
Table 2. Mapping percentage of reads to different domains in all six samples [Kishtwar 2011 (C11), Kishtwar 2012 (C12), Kishtwar 2013 (C13), Kishtwar-L 17, Kashmir (2013), and Morocco (2016)].
At the phylum level, 15 phyla (12 bacterial, 2 fungal, 1 archaeal) were cataloged from the cormosphere of saffron grown in Kishtwar during three consecutive years (2011, 2012, and 2013). The three most dominant phyla were Proteobacteria followed by Actinobacteria and Bacteroidetes. The relative abundance of Proteobacteria, Actinobacteria, and Bacteroidetes varied among the samples, and it was observed that the percentage of Proteobacteria reduced gradually from 2011 to 2013 (54.4 to 48.3%). On the other hand, Actinobacteria and Bacteroidetes increased from 29.5 to 40.9% and 3.6 to 4.36%, respectively (Figure 2).
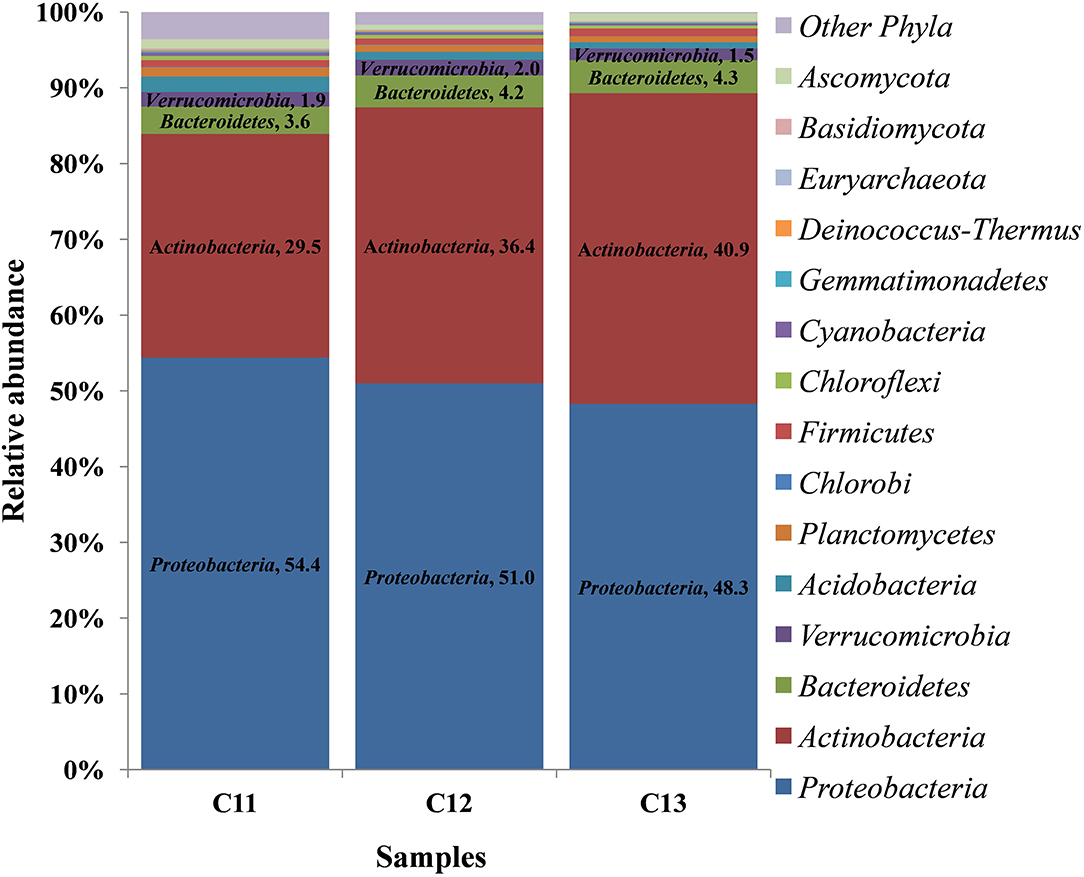
Figure 2. Relative abundance of different phyla in the three consecutive years of Kishtwar (2011, 2012, and 2013). The bar graph depicts 15 phyla (12 bacterial, 2 fungal, 1 archaeal) cataloged from the cormosphere of saffron grown in Kishtwar.
At the genus level, a total 52 genera were cataloged from three Kishtwar samples. Surprisingly, Streptomyces (C11 = 11.5%, C12 = 13.8%, and C13 = 17.7%) belonging to phylum Actinobacteria was the most abundant followed by Mycobacterium, Bradyrhizobium, and Burkholderia all belonging to Proteobacteria (Figure 3). The abundance of three genera Streptomyces, Burkholderia, and Mycobacterium increased from C11 to C13, whereas Bradyrhizobium decreased from 4.58% to 3.22%. Out of total 52 genera cataloged, 42 genera were found common in C11, C12, and C13, which correspond to 81% of total genera. The relative abundance of the 10 genera that were not common (19%) was found to be less than 1% of each genus.
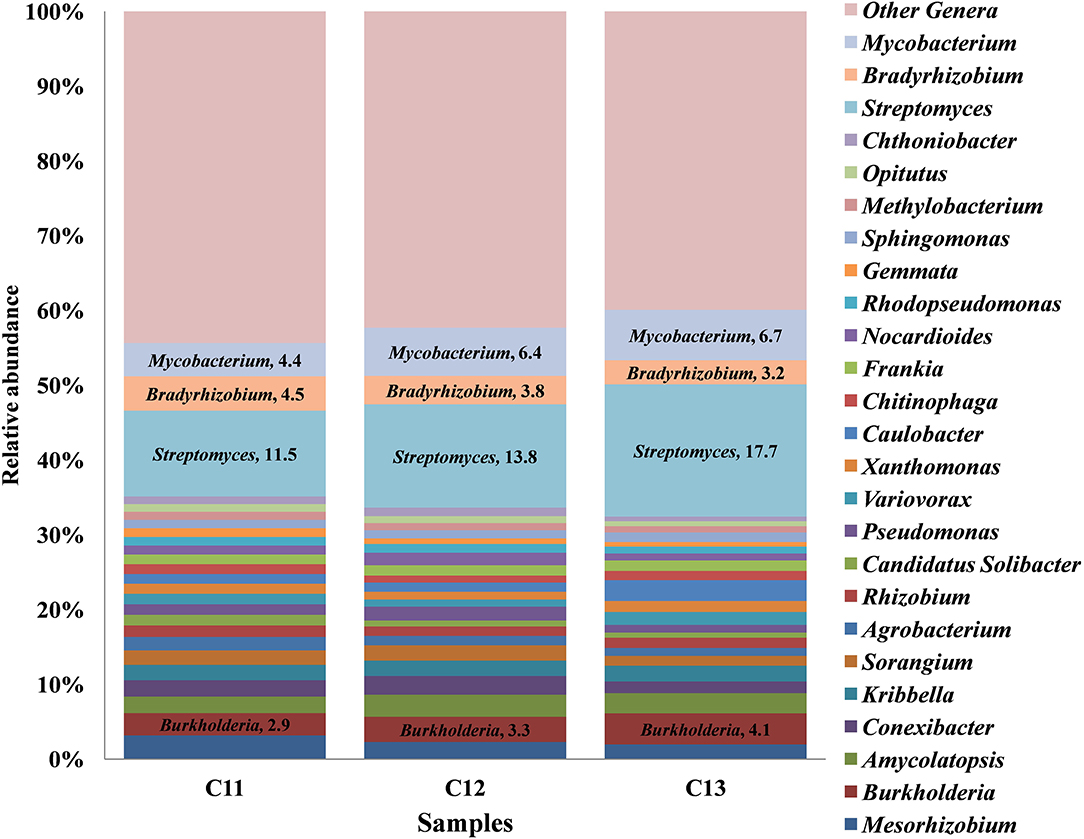
Figure 3. Relative abundance of top 25 genera cataloged from Kishtwar cormosphere in the three consecutive years (2011, 2012, and 2013). The bar graph represents 42 genera common in all the three Kishtwar samples out of 52 genera cataloged in this study.
Core bacteriome of the saffron plant grown in Kishtwar comprised of 42 genera that belongs to five different phyla and corresponds to 81% of the total microbiome (52 genera). Based on diversity/relative representation, 50% (21 genera) belonged to Proteobacteria, 31% (13 genera) to Actinobacteria, 7.2% (3 genera) to Bacteriodetes, 7.2% (3 genera) to Verrumicrobia, and 2.3% (1 genus) to phylum Planctomycetes in the core microbiome of Kishtwar. However, on the basis of abundance, 10 most abundant genera were Streptomyces followed by Mycobacterium, Bradyrhizobium, Burkholderia, Mesorhizobium, Conexibacter, Sorangium, Agrobacterium, Rhizobium, and Pseudomonas. The relative abundance of core genera has been tabulated in Table 3.
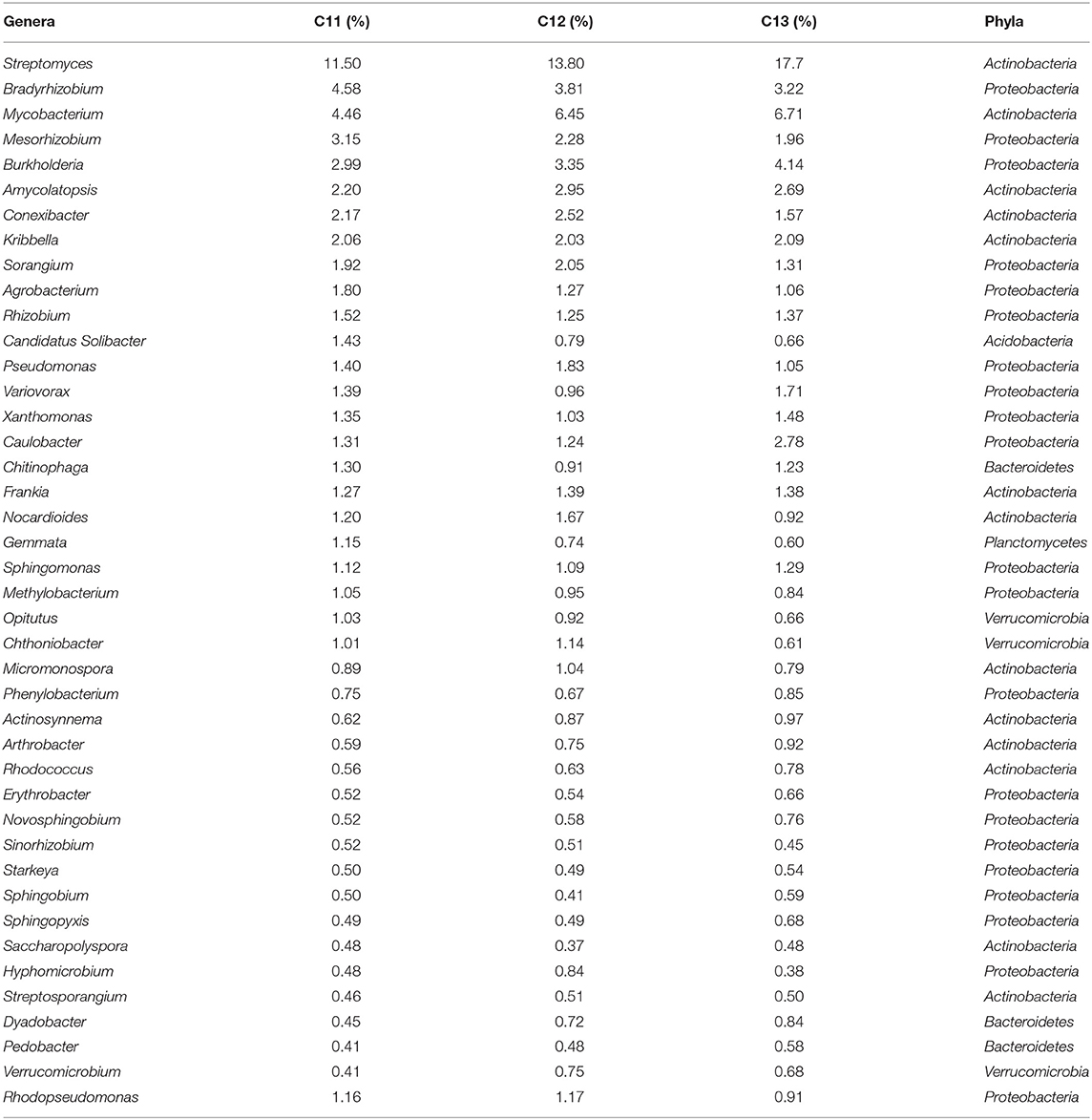
Table 3. Core microbiome of Kishtwar analyzed in 3 years (42 genera) in C11, C12, and C13 with their relative abundance (%).
In addition to 42 common genera, 10 genera were randomly present in one or two of the three samples. Two genera (Stenotrophomonas and Anaeromyxobacter) belonging to phylum Proteobacteria were present only in C11. Three genera such as Bacillus (Firmicutes), Mucilaginibacte (Bacteriodetes), and Brevundimonas (Proteobacteria) were present only in C13. The Shannon α-diversity of C11, C12, and C13 was found to be 443, 395, and 359, respectively, thereby indicating decrease in microbial diversity from 2011 to 2013. However, decrease in microbial diversity may be due to the depletion of few rare genera having less abundance in consecutive years. To conclude, it was observed that 81% of genera were common in the three consecutive years but their relative abundance varied.
The Kishtwar microbiome (Kishtwar average of C11, C12, and C13, hereafter referred to as Kishtwar-A) was taken as reference microbiome for comparing the diversity and abundance of microbes in Kishtwar-L 17 (plants grown under controlled laboratory conditions) again to confirm stability and inheritability of the corm microbiome. All 15 phyla present in Kishtwar with 3 year average, referred to as Kishtwar-A, were present in Kashmir-L 17. The three major dominant phyla were the same, i.e., Proteobacteria, Actinobacteria, and Bacteroidetes, but their relative abundance varied between Kishtwar-L 17 and Kishtwar-A. Proteobacteria and Actinobacteria were less abundant, whereas Bacteroidetes were more abundant in Kishtwar-L 17 (Figure 4).
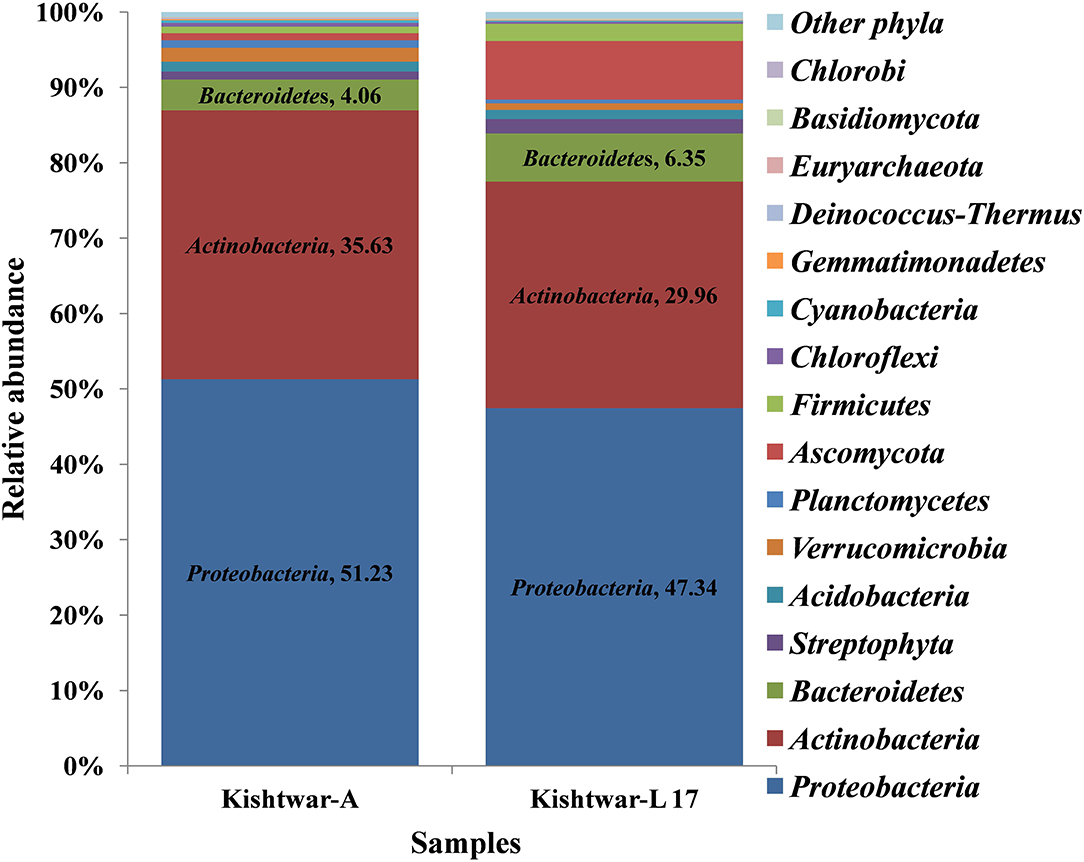
Figure 4. Abundance of different phyla in Kishtwar-A and Kishtwar-L 17. The bar graph depicts 15 phyla (12 bacterial, 2 fungal, 1 archaeal) cataloged from the cormosphere of saffron grown in Kishtwar fields and one laboratory grown sample.
At the genus level, 34 genera were found to be common in Kishtwar-A and Kishtwar-L 17 that constituted the core of Kishtwar region. Similar to Kishtwar-A, the most abundant genera in Kishtwar-L 17 was Streptomyces (17.73%), but the pattern and abundance of other genera varied; Pseudomonas (14.81%) and Burkholderia (8.73%) had higher abundance, and Mycobacterim (3.93%) and Bradyrhizobium (0.6%) were less abundant compared to Kishtwar-A (Figure 5). The alpha diversity index (Shannon indices) was more in Kishtwar-A (399) compared to Kishtwar-L 17 (354). This is further complemented by the rarefaction curve wherein C11 showed the highest species richness and least by Kishtwar-L 17 (Figure 6). In addition, there were six genera unique to Kishtwar-L 17, namely, Achromobacter, Enterobacter, Serratia, Nocardia, Terriglobus, and Sphingobacterium, whereas eight genera (Rhodopseudomonas, Hyphomicrobium, Sphingopyxis, Novosphingobium, Sphingobium, Erythrobacter, Starkeya, and Actinosynnema), present in Kishtwar-A, were completely absent in Kishtwar-L 17.
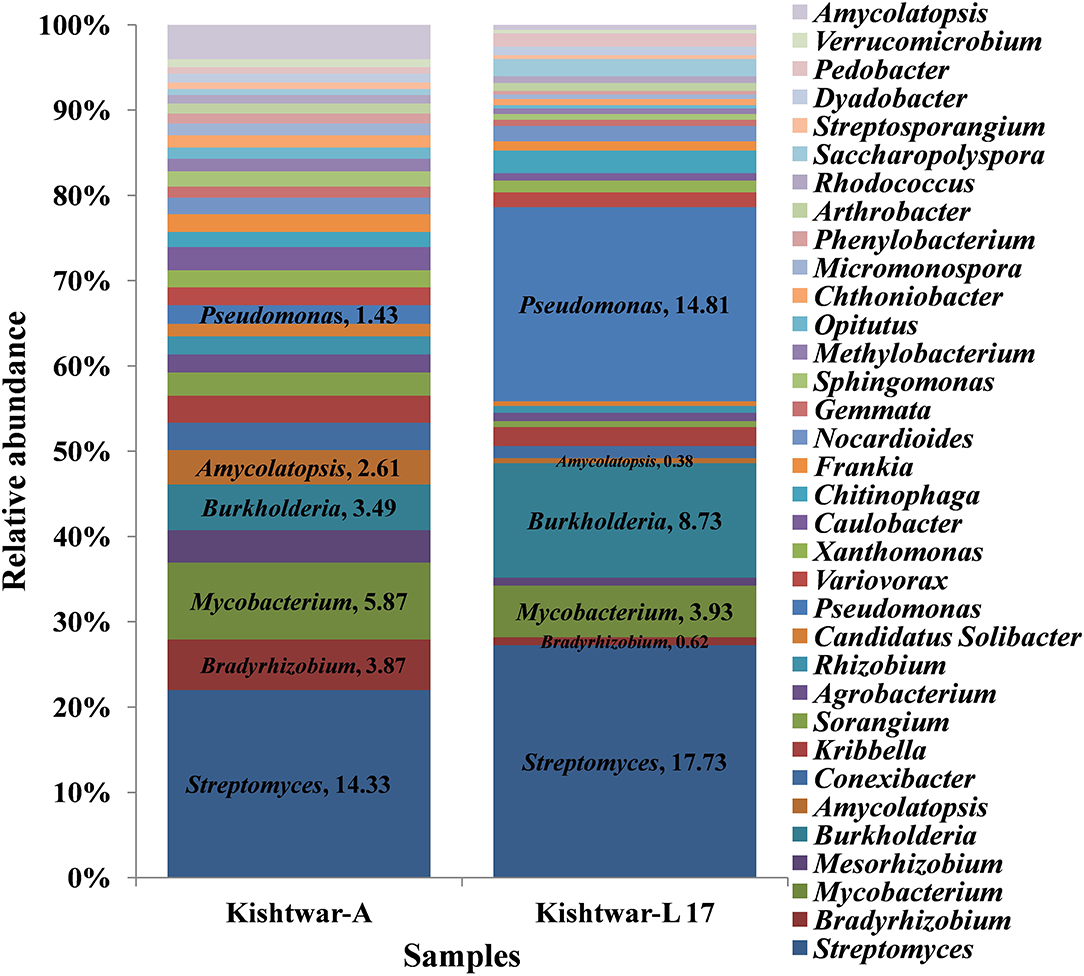
Figure 5. Relative abundance of common 34 genera cataloged from Kishtwar-A and Kishtwar-L 17.
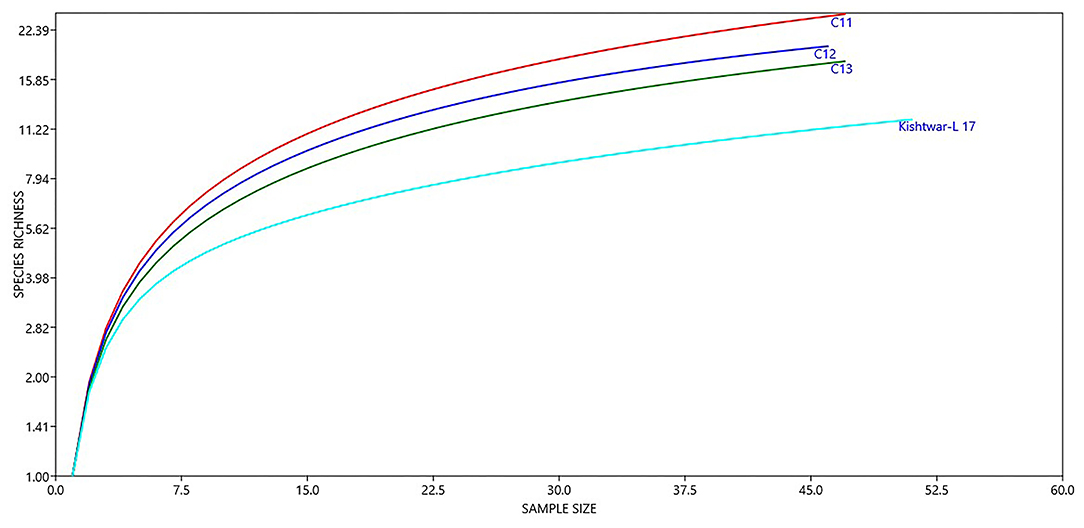
Figure 6. Diversity of different genera in Kishtwar samples (C11, C12, C13, and Kishtwar-L 17). Presence of higher number of species in C11 indicates that it is more enriched than C12, C13, and Kishtwar-L 17.
At the phylum level, 15 phyla (12 bacterial, 2 fungal, 1 archaeal) were cataloged from the cormosphere of saffron corms grown in Kashmir and Morocco. The three dominant bacterial phyla among the samples were Proteobacteria, Bacteroidetes, and Actinobacteria. Proteobacteria was most dominant (Kashmir = 48.46%, Morocco = 55.06%) followed by Bacteroidetes (Kashmir = 12.93% and Morocco = 16.92%) and Actinobacteria (Kashmir = 10.98% and Morocco = 16.34%) Proteobacteria (51.23%), followed by Actinobacteria (35.64%), Bacteroidetes (4.06%), and Verrucomicrobia (1.85%). Though the number of phyla was same in Kishtwar-A, Kashmir, and Morocco, heat maps hierarchical clustering clustered Kashmir and Morocco in one clad and Kishtwar-A in different clad. This represented that the co-relation distance and average linkage of Kishtwar-A were different from Kashmir and Morocco (Figure 7). Relative abundance of different phyla of Kishtwar-A, Kashmir, and Morocco is represented in Figure 8.
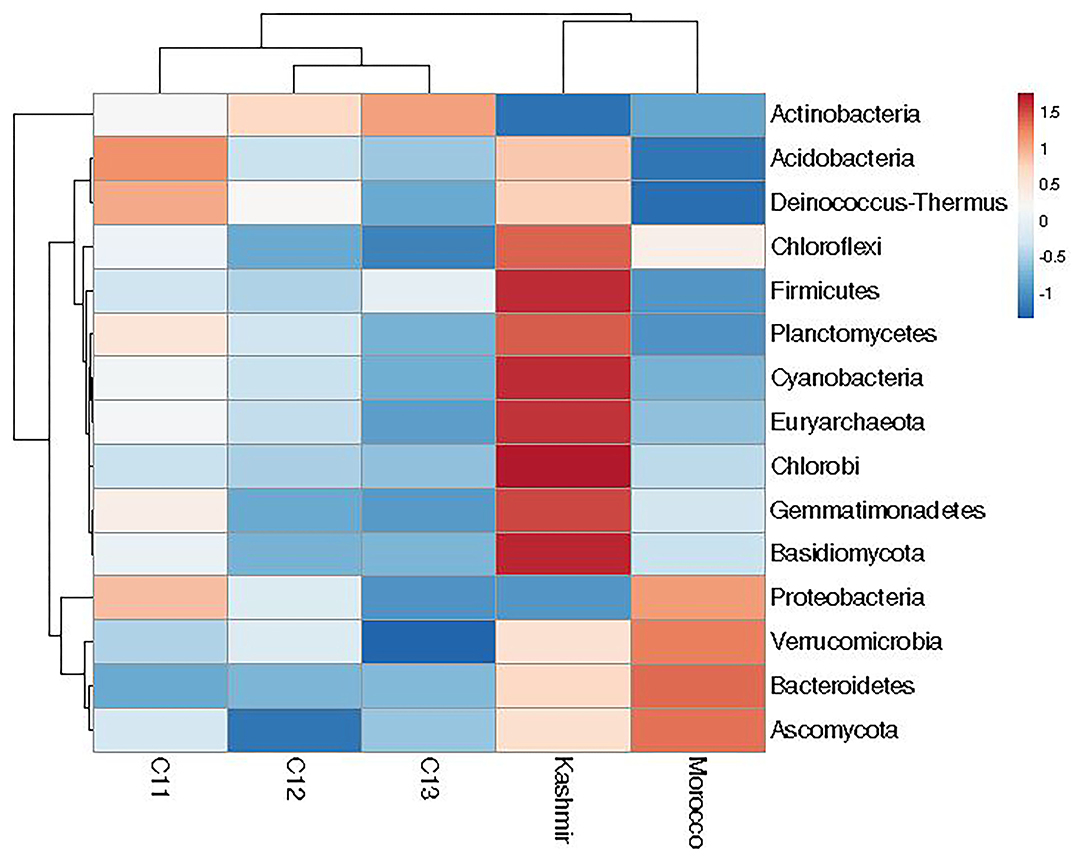
Figure 7. Relative abundance of 15 phyla in the saffron cormosphere from all the three geographical locations (Kashmir, Kishtwar, and Morocco).
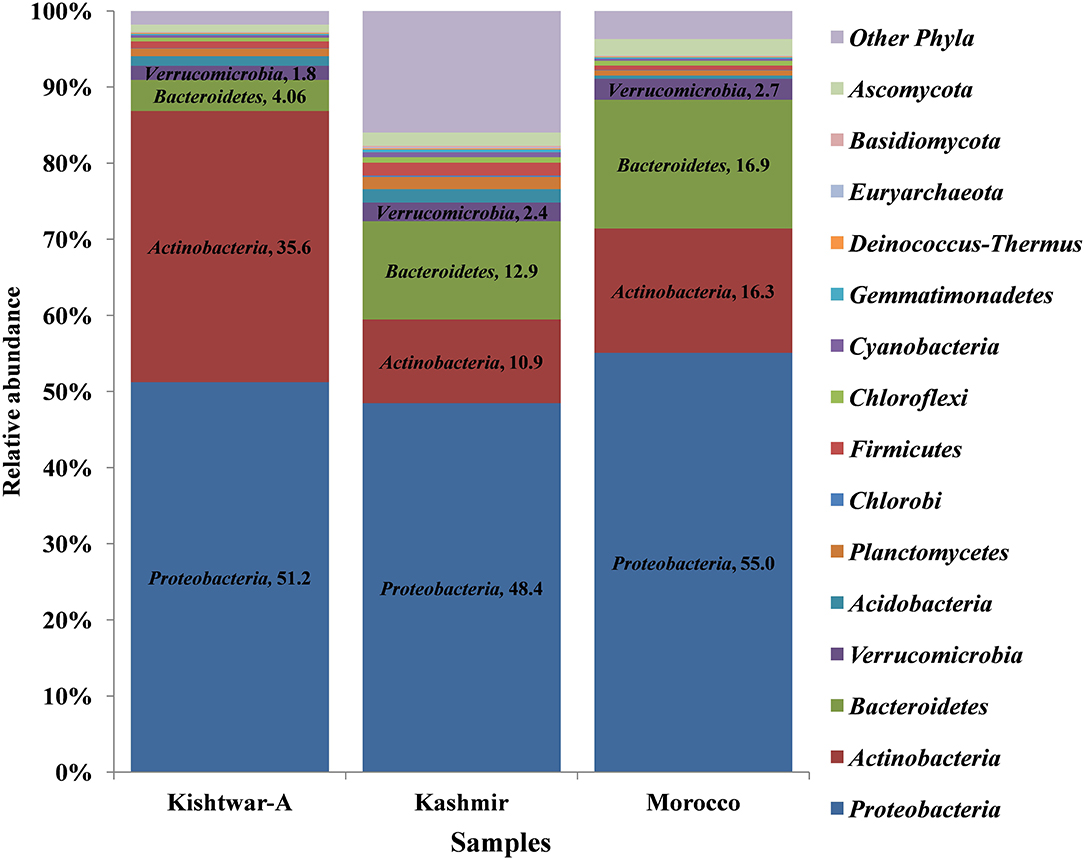
Figure 8. Representation of relative abundance of different phyla in all the three geographical locations (Kishtwar-A, Kashmir, and Morocco). Fifteen major phyla (12 bacterial, 2 fungal, 1 archaeal) were cataloged from the cormosphere of saffron grown in three geographical locations. Proteobacteria was dominant in all the three locations followed by Actinobacteria and Bacteroidetes in Kishtwar-A and Bacteroidetes and Actinobacteria in both Kashmir and Morocco.
At the genus level, abundance dynamics of bacterial genera varied among samples (Figure 9). Kashmir was dominated by Pseudomonas (12.78%), Streptomyces (3.33%), and Chitinophaga (2.65%). Morocco was dominated by Pseudomonas (8.66%), Caulobacter (5.46%), and Pedobacter (4.68%) compared with Kishtwar-A, which was dominated by Streptomyces (14.33%), Mycobacterim (5.87%), and Bradyrhizobium (3.87).
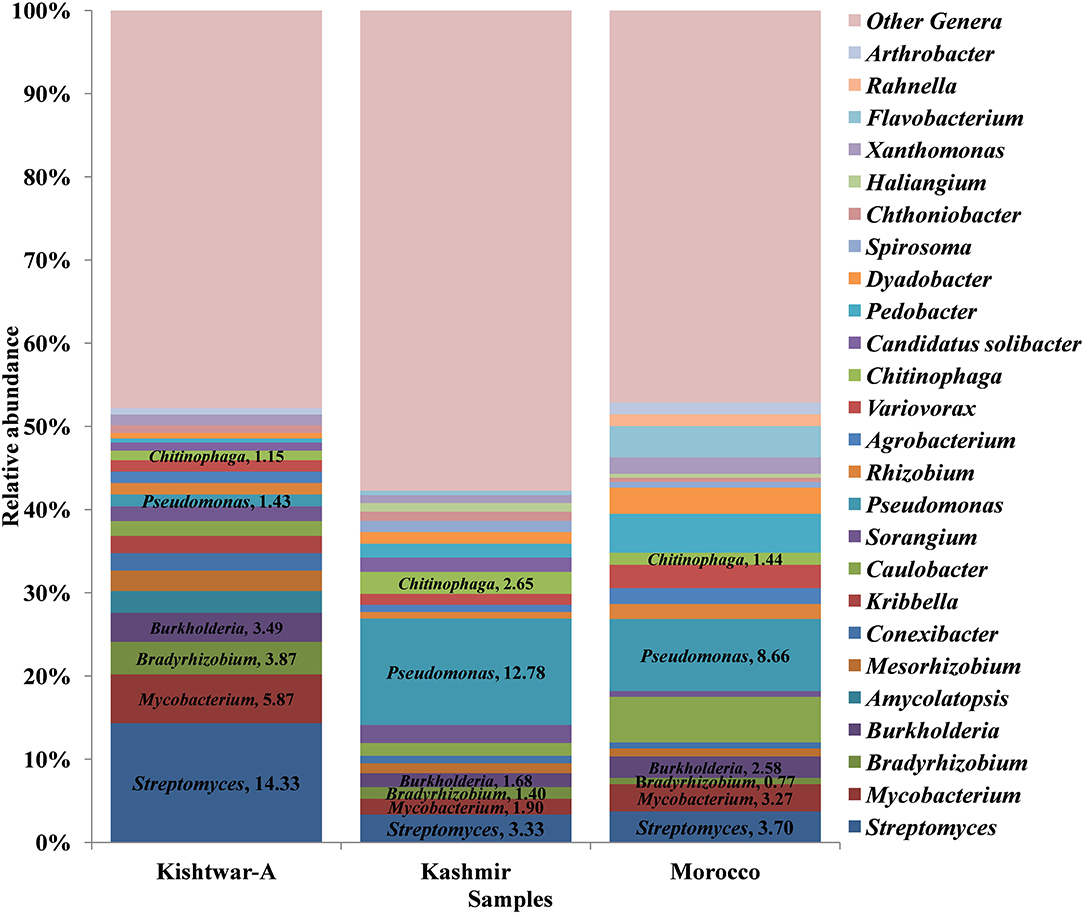
Figure 9. Relative abundance of top 25 genera cataloged from cormosphere of all the three geographical locations (a total of 73 genera were cataloged from three geographical locations: Kishtwar-A, Kashmir, and Morocco). Streptomyces was the dominant genera in Kishtwar-A followed by Mycobacterim, Bradyrhizobium, and Burkholderia. Pseudomonas was the dominant genera in both Kashmir and Morocco followed by Streptomyces, Chitinophaga, and Sorangium in Kashmir and Caulobacter, Pedobacter, and Flavobacterium in Morocco.
Of the total 73 genera cataloged, 24 genera were found common in all the three geographical locations, viz., Kishtwar, Kashmir, and Morocco, corresponding to 32.8% similarity in microbial community across geographical regions in Asian and African sub-continent. Core microbiome of saffron across different geographical location comprised of 24 genera; out of 24 common genera, 13 genera (54.2%) belonged to phylum Proteobacteria, 5 genera (20.8) to Actinobacteria, 3 genera (12.5%) to Bacteriodetes, and 3 genera (12.5%) to Verrumicrobia (Figure 10). The core genera with their relative abundance have been tabulated in Table 4.
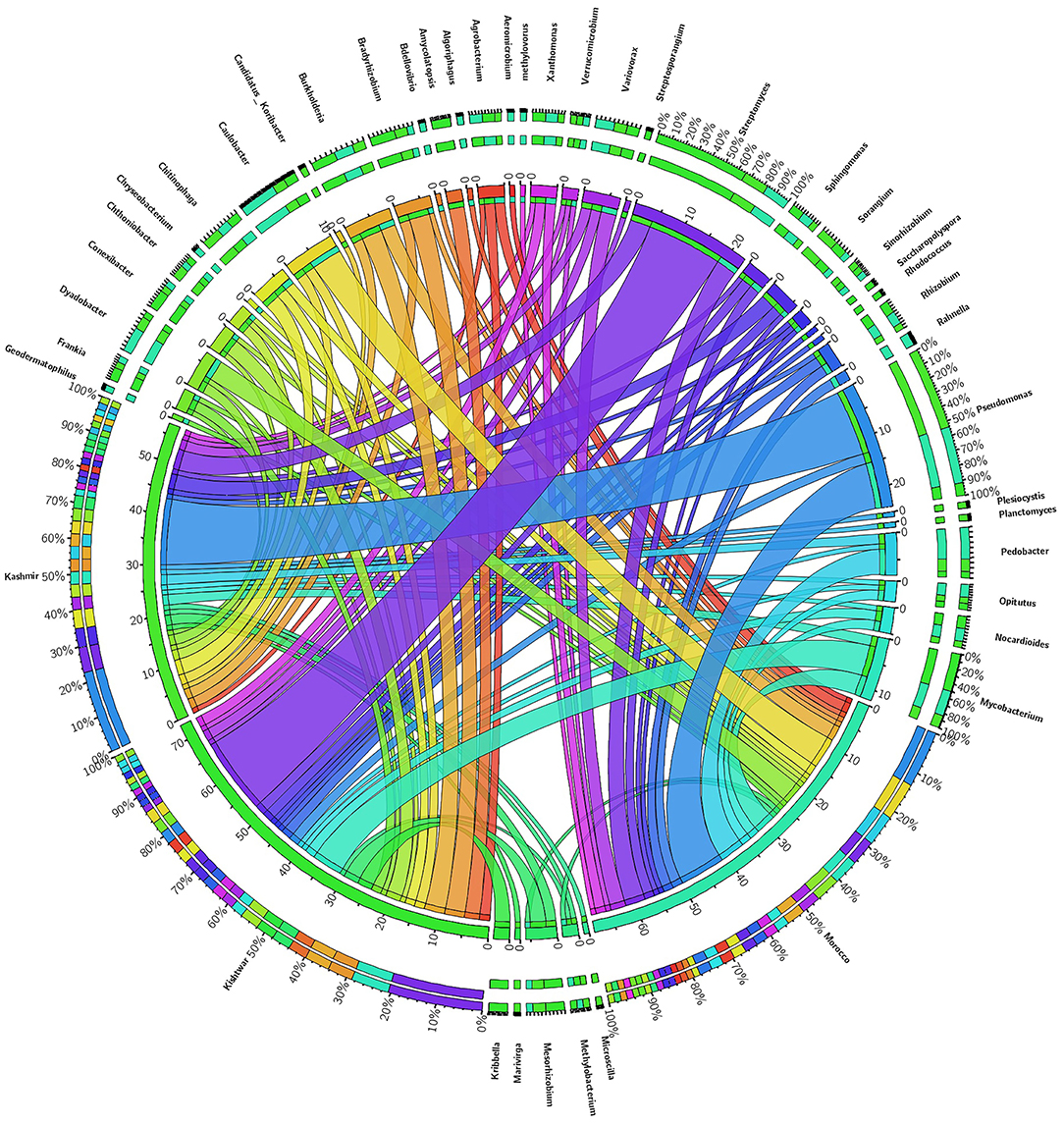
Figure 10. Abundance of core genera and unique genera in three geographical locations. Central line (ribbons) represents the abundance of each genus and connects genera to samples. The outer three rings are the stacked bars representing the relative contribution of particular genera in every sample.
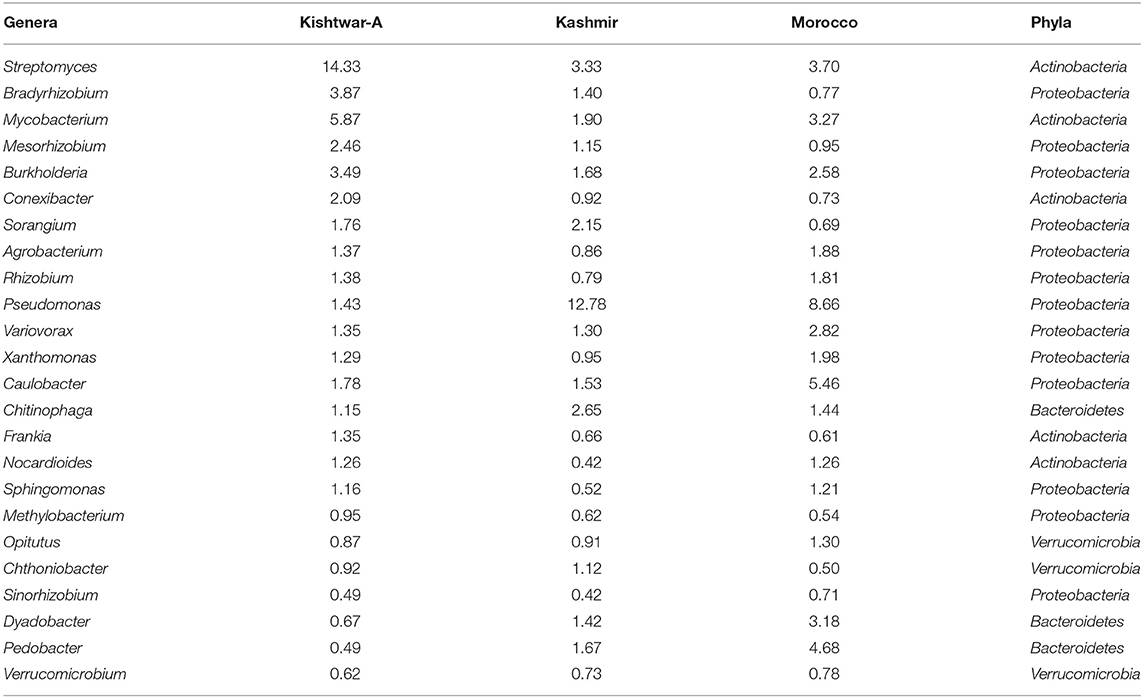
Table 4. Core microbiome of three geographical regions (24 genera) (Kishtwar-A, Kashmir, and Morocco) with their relative abundance (%).
Notable differences were observed in the cormosphere microbiome composition of Kashmir, Kishtwar, and Morocco as depicted by principal component analysis (PCA) that clusters Kishtwar (C11, C12, and C13) samples in close proximity and Kashmir and Morocco samples that clustered away in different coordinate, thereby indicating differences in diversity/abundance pattern (Figure 11). In addition, alpha diversity indices like Shannon indices were highest in case of Kashmir (507) as compared to Morocco (415) and Kishtwar-A (399). This was further complemented by rarefaction curves wherein Kashmir samples were more enriched as depicted by the higher number of species in Kashmir, in comparison to Kishtwar-A and Morocco (Figure 12).
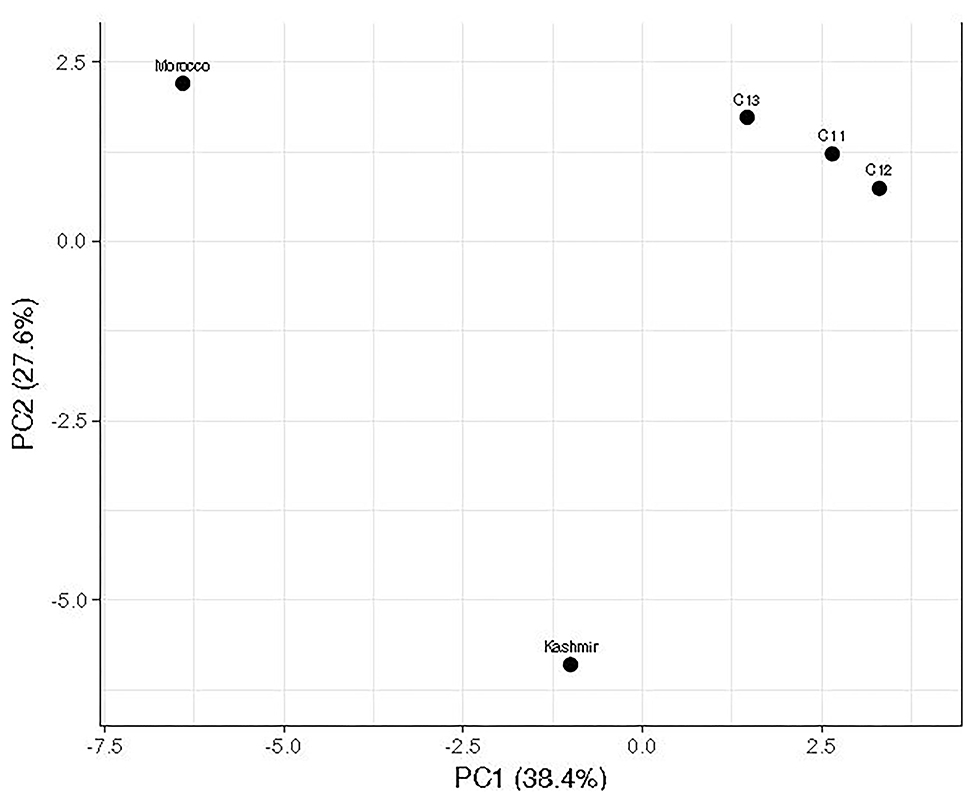
Figure 11. Comparison of microbial diversity of Kishtwar during three consecutive year's sample (C11, C12, and C13) with Kashmir and Morocco. It represents that Kashmir and Morocco are significantly different from the rest of the sample as it does not cluster with Kishtwar sample.
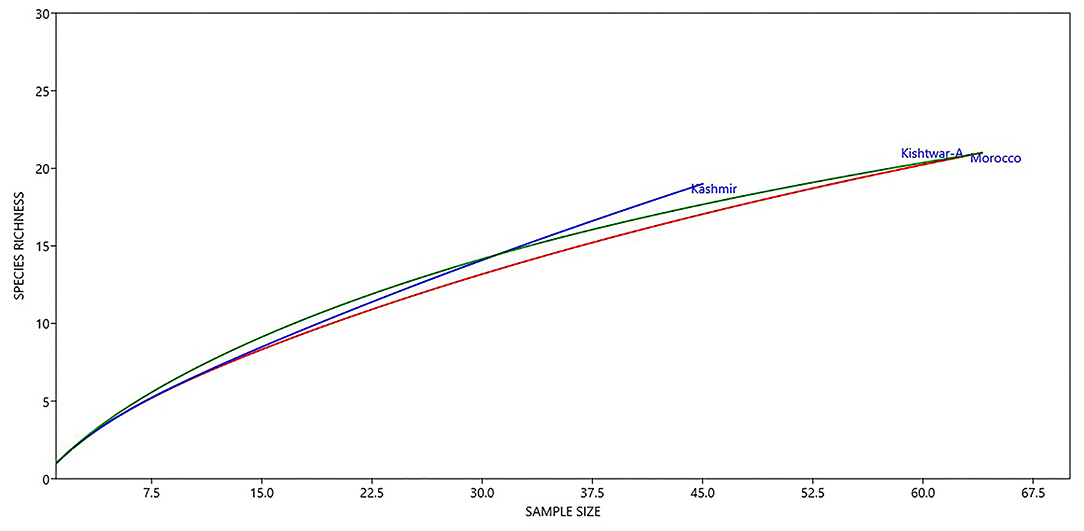
Figure 12. Diversity of different genera across geographical locations (Kishtwar-A, Kashmir, and Morocco). Kashmir samples were more enriched as depicted by higher number of species in Kashmir followed by Kishtwar-A and Morocco.
In addition to the core microbiome, five bacterial genera were unique to Kashmir (Marivirga, Microscilla, Planctomyces, Plesiocystis, Candidatus koribacter), seven genera were unique to Morocco (Rahnella, Bdellovibrio, Methylovorus, Chryseobacterium, and Algoriphagus), and the five unique genera identified specific to Kishtwar were Rhodococcus, Kribella, Streptosporangium, Saccharopolyspora, and Amycolatopsis (Figure 13).
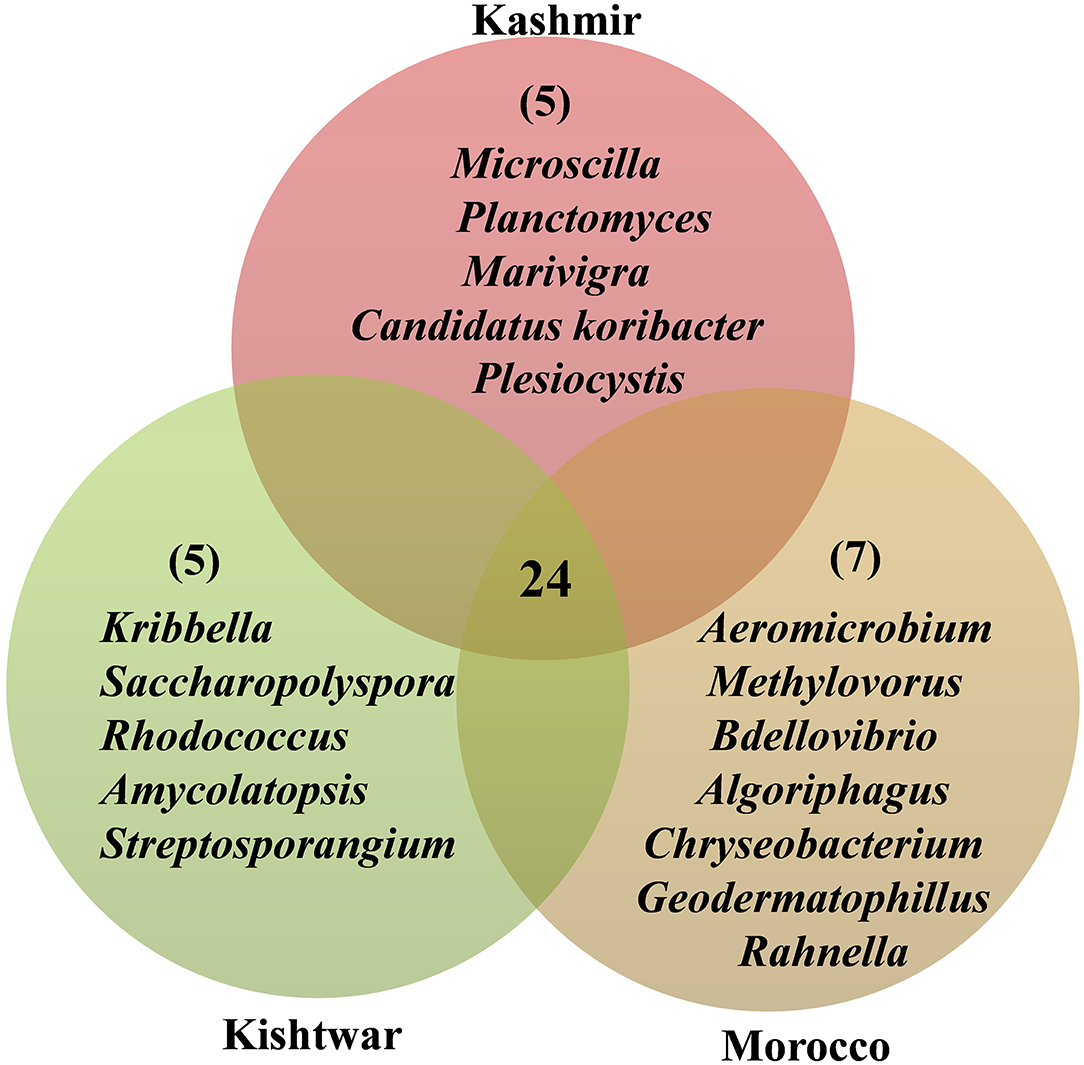
Figure 13. Representing the number of common and unique genera identified in three geographical locations. Twenty-four common genera in all the three geographical locations are referred as the core microbiome of saffron. Five genera were unique to Kishtwar, seven to Morocco, and five to Kashmir.
To conclude, Kishtwar consecutive 3 year samples showed 81% similar genera across the years. The genera that were absent in one or other Kishtwar samples had less than 1% representation in total reads and may be attributed to sequencing bias due to the low relative amount present in whole metagenomic data. This needs to be confirmed by repeated sampling and sequencing across various geographical locations. However, 32.8% of the genera were common in all three locations that can be designated as core microbiome of saffron cormosphere. Bacterial genera that were present in one geographical location and completely absent in the other two locations were observed, thereby representing the specific interactions of these bacterial genera to each geographical location. Unique genera to each place can be developed into biological markers as geographical indicators.
The sequencing data has been submitted to NCBI under the accession number PRJNA705068.
The present study was initiated to investigate the possibility of using microbial fingerprints as molecular marker and/or geographical indicator, particularly in monogenetic crops. Microbial biomarkers have been reported to be used for the determination of biogenic chemical concentration (e.g., phospholipid fatty acids; Lupwayi et al., 2017), evaluation of microbial activity (e.g., photosynthetic activity; Corcoll et al., 2011), and capacity (e.g., adaptation; Pesce et al., 2016). Species specific biomarkers are reported to detect the toxicity in the environment caused by a pathogen bacteria or toxic Cyanobacteria (Bonnineau et al., 2012; Guasch et al., 2017). In plants also, microbiome is reported to have significant variation based on different geographical locations, such as reported in the case of vineyards, Amazonian hardwood, and fragrant orchid. The researchers co-relate the variations in the plant microbiome to various factors such as location/genetics/abiotic and biotic factors (Coller et al., 2019; Skaltsas et al., 2019; Lin et al., 2020). However, reports are emerging on the core microbiome that remains unaltered at various developmental stages of the plants or plants from different locations/niches (Chen et al., 2018; Toju et al., 2018; Lin et al., 2020). The significance of molecular markers/barcodes in selecting/identifying different varieties needs no elaboration.
However, some plants are reported to lack high resolution molecular markers on account of sterility. Crocus sativus, saffron, is reported to lack genetic variation on account of vegetative propagation through corms, as it is male sterile plant (Nemati et al., 2019). The present study was initiated to discover if microbiome based markers can be used as geographical indicators in saffron grown at different geographical locations, i.e., Kashmir and Kishtwar in India and Taliouine in Morocco, also to identify core microbiome in saffron across different locations, if any. The microbiome associated with corm, grown at these three locations, was isolated and analyzed at the vegetative stage to keep the development stage as a constant factor.
The bacteriome and mycobiome of Crocus sativus have been reported earlier by our group, but that study was specific to Kashmir for 1 year only (Ambardar et al., 2014). In the present study, microbiome analysis of cormosphere was initiated in saffron grown in Kishtwar during three consecutive years to check the inheritability of cormosphere microbiome. About 81% of the microbial genera were common in the cormosphere of all 3 years, though their abundance varied (Figure 2). The abundance of the genera belonging to phylum Proteobacteria decreased from C11 (54.4%) to C13 (48.3%), whereas an increase in the abundance of Actinobacteria and Bacteriodetes was observed from C11 (Actinobacteria−29.5%, Bacteriodetes−3.6%) to C13 (Actinobacteria−40.9%, Bacteriodetes−4.36%). Since saffron is a perennial crop, and farming practices were not changed, the climate was more or less the same and yield also did not show any co-relation (Table 1); the exact reason for this variation in abundance is not clear. Eighty-one percent of the genera, common to all the 3 years, are being proposed as the core microbiome of saffron cormosphere grown in Kishtwar. Core microbiome have been earlier reported in various plants across different geographical location, climatic gradients, genotypes, and seasons, namely, Salvia miltiorrhiza (Chen et al., 2018), angiosperm plants (Fitzpatrick et al., 2018), Coffea arabica (Fulthorpe et al., 2020), Orzya sativa (Eyre et al., 2019), Phaseolus vulgaris (Pérez-Jaramillo et al., 2019), Switchgrass (Panicum virgatum L.) and Miscanthus (Miscanthus x giganteus) (Grady et al., 2019), and Panax ginsen, (Nguyen et al., 2016). On the similar lines, phyllosphere microbiome of Switchgrass was compared for two consecutive years (2016 and 2017) and 60% of leaf communities were found common (Grady et al., 2019), which was comparatively less than the present study (81% of cormosphere community was common in saffron).
In the present study, once the inheritability of the core microbiome in field conditions was established, the same was tested under laboratory conditions wherein the saffron corms and soil were collected from Kishtwar in 2017, and grown in pots in the laboratory. Except for the variation of temperature that was around 25°C, the rest of the parameters were kept close to natural conditions, such as soil and light intensity. Out of 81% genera common during three consecutive years from Kishtwar, only 65% were retained under laboratory conditions. Eight genera were missing under lab conditions, but additional six genera were present and were completely absent under field conditions. The aim of laboratory cultivation was to check if the unique genera present in Kishtwar samples are retained if corms are dug and grown somewhere else. It was observed that the unique genera were retained as the five unique genera present in Kishtwar 3 years (C11, C12, and C13) were also present in these samples. This further establishes that the unique genera are inherited and can be used as geographical indicators or markers. In addition, all the microbes, representing the core microbiome of saffron corm characterized from different locations, were also present in laboratory grown samples (discussed in the following sections).
In total, 15 phyla (12 bacterial, 2 fungal, 1 archaeal) cataloged were common among three different locations. The most dominant phylum was Proteobacteria followed by Actinobacteria, Bacteroidetes, Streptophyta, Acidobacteria, and Verrucomicrobia (Figure 8). Proteobacteria have also been reported to be the dominant phylum in the rhizosphere of other plants such as Gossypium hirsutum, Artemisia argyi, Ageratum conyzoides, Erigeron annuus, Bidens biternata, Euphorbia hirta, and Viola japonica (Qiao et al., 2017; Lei et al., 2019). Proteobacteria are the main microbial contributors to plant growth promoting rhizobacteria (PGPR), reported so far. They are fast growing and play a role in diverse metabolic activities that enhance plant growth (Philippot et al., 2013; Rampelotto et al., 2013; Schillaci et al., 2019; Mukhtar et al., 2020). Actinobacteria are the gram positive bacteria that are known to promote plant growth and having biocontrol activity (Viaene et al., 2016). Likewise, Bacteriodetes are the pathogen suppressing members and also have a role in plant growth promotion by phosphorus mobilization (Lidbury et al., 2021). This explains the dominance of Proteobacteria, Actinobacteria, and Bacteroidetes in the cormosphere, which are among the most dominant phyla in the soil and maybe getting transferred from the soil, just like in rhizosphere (Miyashita, 2015; Fierer, 2017; Compant et al., 2019). To our knowledge, cormosphere microbiome has not been studied in any other plant, except for saffron reported previously by our group (Ambardar and Vakhlu, 2013; Ambardar, 2014; Ambardar et al., 2014, 2016; Kour et al., 2018) from Kashmir, India (Chamkhi et al., 2018), and from Taliouine, Morocco. Previous reports by our group have also confirmed the dominance of Proteobacteria in Kashmir saffron rhizosphere and cormosphere, even though the method used was cultivation dependent and 16S targeted metagenome sequencing was different than the methods used in the present study (Ambardar and Vakhlu, 2013; Ambardar et al., 2014) (Supplementary Material 1).
A total of 73 genera were identified after taxonomical characterization of C. sativus cormosphere microbiome from reference samples collected from Kishtwar-A, laboratory grown Kishtwar-L 17, Kashmir, and Morocco. Streptomyces was dominant in Kishtwar-A as well in Kishtwar-L 17, whereas Pseudomonas was dominant in Kashmir and Morocco. Pseudomonas and Streptomyces have been reported in the rhizosphere of various plants like Arabidopsis thaliana, tomato, rice, and maize including saffron (Ambardar and Vakhlu, 2013; Ambardar et al., 2014; Chen et al., 2018; Kour et al., 2018; Chu et al., 2019; Qessaoui et al., 2019). Pseudomonas isolates have been reported to solubilize phosphate, produce siderophores, ammonia, and indole-3-acetic acid, and colonize the roots of tomato plants (Qessaoui et al., 2019) and also have been reported to induce salt tolerance in Arabidopsis thaliana (Chu et al., 2019). Pseudomonas has also been reported to be dominant in saffron rhizosphere grown in Kashmir, India, analyzed using both cultivation dependent and cultivation independent method (Ambardar and Vakhlu, 2013; Ambardar et al., 2014), and Morocco, analyzed using cultivation dependent approaches (Chamkhi et al., 2018). Streptomyces is the largest genus of Actinobacteria phylum comprising 500 species and has been found to promote plant growth (Lehr et al., 2007, 2008; Schrey and Tarkka, 2008; Viaene et al., 2016). It is reported to produce secondary metabolites having antibiotic activities and volatile organic compounds (VOCs) that can exert biocontrol activity (Lucas et al., 2012; Viaene et al., 2016). Streptomyces have been previously reported from the saffron rhizosphere also using cultivation independent approaches (Ambardar et al., 2014). In addition, to Pseudomonas and Streptomyces, 53 have also been reported previously in saffron in Kashmir and Morocco (Ambardar and Vakhlu, 2013; Ambardar et al., 2014, 2016; Chamkhi et al., 2018; Kour et al., 2018) (Supplementary Material 2).
Interestingly, we found that 24 genera were common to all, representing 32.8% of collective bacteriome in the four samples (three field and one laboratory sample). These 24 genera are being proposed to represent the core microbiome of cormosphere of C. sativus across different geographical locations. Core microbiome across different geographical locations has been earlier reported in various plants such as Salvia miltiorrhiza (Chen et al., 2018), Phaseolus vulgaris (Pérez-Jaramillo et al., 2019), Oryza sativa (Eyre et al., 2019), and Gymnadenia conopsea (Lin et al., 2020) and also in Vineyards soil (Coller et al., 2019) and Dalbergia spruceana (Skaltsas et al., 2019). Core microbiome of 21 Salvia miltiorrhiza seeds represented 54% of whole microbiome cataloged from seven different geographic origins, and Pantoea, Pseudomonas, and Sphingomonas were enriched core taxa (Chen et al., 2018). In the case of Phaseolus vulgaris rhizosphere, core microbiome represented 25.9% of total microbiome in native and agricultural soils (Pérez-Jaramillo et al., 2019). Core microbiome of 30 phylogenetically diverse angiosperm plants constitutes 40% of whole microbiome (Fitzpatrick et al., 2018). In rice seeds, the core microbiome was enriched in Rhizobium, Pantoea, Sphingomonas, Methylobacterium, Xanthomonas, Paenibacillus, Alternaria, and Occultifur that have been also reported as PGPB (plant growth promoting bacteria) (Wang et al., 2020). Core microbiome in saffron was 32.8% of the total microbiome, which was comparatively less than Salvia miltiorrhiza. Similar to core microbiome in rice and Salvia miltiorrhiza (seed), saffron's core microbiome was also enriched with Pseudomonas, Rhizobium, Sphingomonas, Xanthomonas, and Methylobacterium. In addition, 22 bacterial genera (out of 24) representing the core microbiome of the C. sativus in the present study were also reported previously in Saffron grown in Kashmir, India, by our group, out of which 19 are PGPB in saffron and other plants (Ambardar and Vakhlu, 2013; Kour et al., 2018; Umadevi et al., 2018; Vergani et al., 2019) (Supplementary Material 3). Agrobacterium, Bacillus, Delftia, Luteibacter, Pantoea, Pseudomonas, Rahnella, Rhizobium, and Variovorax from rhizosphere and Agrobacterium, Pseudomonas, Rhizobium, and Variovorax from cormosphere of saffron grown in Morocco using cultivation dependent methods have been reported earlier as well (Chamkhi et al., 2018). Core microbiome enriched with plant growth promoting bacteria indicates that the plant selects the core microbiome based on their function (Supplementary Material 3). To conclude, the saffron cormosphere core microbiome is conserved across two continents, Asia and Africa, as per this study.
Variation in the microbiome of C. sativus across different geographical locations was confirmed by PCA plots, wherein surprisingly Morocco and Kashmir were clustered away from Kishtwar-A, although Kashmir is geographically closer to Kishtwar (Figure 11). These findings were further complemented by Shannon alpha diversity indices, which were maximum in Kashmir (507) compared to Kishtwar-A (354) and Morocco (415). The Shannon alpha diversity index indicates that microbial communities of each sample have significant differences, though they share core microbiome. There are plenty of reports on the effect of temperature on the diversity and abundance of soil microbes, though those studies have been conducted in reference to climate change (Jansson and Hofmockel, 2020).
The effect of geographical location on the microbiome has been discovered in other plants such as barley (Terrazas et al., 2020) and Gymnadenia conopsea (Lin et al., 2020), and significant variation has been reported. Each location in the present study harbored unique bacteria that could be used as a geographical indicator. The cormosphere of Kashmir had five unique bacteria, namely, Marivirga, Planctomyces, Plesiocystis, Candidatus koribacter, and Microscilla, whereas seven bacteria, namely, Rahnella, Chryseobacterium, Bdellovibrio, Algoriphagus, Methylovorus, Geodermatophilus, and Aeromicrobium, were specific to Morocco and five (Rhodococcus, Kribella, Streptosporangium, Saccharopolyspora, and Amycolatopsis) were specific to Kishtwar (Figure 13). These unique genera have also been reported as PGPB in other plants (Supplementary Material 4).
The bacterial genera (8) that have been reported for the first time from saffron underground parts were Marivirga, Microscilla, Starkeya, Actinosynnema, Saccharopolyspora, Bdellovibrio, Algoriphagus, and Methylovorus, whereas 11 bacterial genera (Planctomyces, Plesiocystis, Candidatus koribacter, Chryseobacterium, Rhodococcus, Rahnella Amycolatopsis, Kribbella, Streptosporangium, Geodermatophilus, and Aeromicrobium) have been reported earlier from saffron rhizosphere in our previous studies from Kashmir and Kishtwar in India and Taliouine in Morocco (Ambardar et al., 2021 unpublished data; Chamkhi et al., 2018). However, the bacteria reported from saffron for the first time have been reported from other plants. Marivirga has been reported from Panax ginseng using 16S rRNA sequencing (Nguyen et al., 2016), Microscilla from Tuber melanosporum (Deveau et al., 2016), Starkeya from root nodules of legume plants (Zakhia et al., 2006), Actinosynnema from beech rhizosphere (Colin et al., 2017), Saccharopolyspora from Colocasia rhizosphere (Intra et al., 2019), Rahnella from Oryza sativa (Sun et al., 2020), Chryseobacterium from sand dune plant rhizosphere (Cho et al., 2010), Algoriphagus from mangrove rhizosphere (Song et al., 2020), and Bdellovibrio and Methylovorus from grassland (Macey et al., 2020). Marivirga, Microscilla, Actinosynnema, and Bdellovibrio have been identified using cultivation independent approach (16S rRNA sequencing), whereas Saccharopolyspora, Chryseobacterium, and Algoriphagus have been identified using cultivation based approaches but their PGP activities have not been estimated yet. However, Starkeya and Rahnella have been reported as PGPRs and isolated using cultivation dependent approaches, whereas Methylovorus is reported to be methylotrophs (Zakhia et al., 2006; Cho et al., 2010; Deveau et al., 2016; Nguyen et al., 2016; Colin et al., 2017; Intra et al., 2019; Macey et al., 2020; Song et al., 2020; Sun et al., 2020).
The preliminary study suggests that saffron across three geographical locations share common core cormosphere microbiome but have certain unique bacteria genera specific for each location. The unique genera specific to a particular location can be used as the geographical indicators and molecular markers in saffron. The study needs to be complemented further by analyzing the microbiome of saffron grown across various geographical locations, as saffron is cultivated now in both the hemispheres. It also needs to be validated by in-depth sequencing and frequent sampling for a couple of consecutive years.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI [accession:PRJNA705068].
JV, MH, and LS gave plan of study. SS, NB, DT, RZ, and JV designed the experiments. SS, NB, SA, and DT conducted the experiments and analyzed the sequencing data. SS, SA, NB, and SR wrote the manuscript and analyzed the taxonomy data. JV, MH, and LS finalized the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Sincere thanks and gratitude to Tahir Ali (Ph.D. Scholar, School of Biotechnology, University of Jammu, Jammu, J&K, India), helping in preparing map showing sample collection sites on the world map and to Mr. Ajmad Malik (Saffron development officer) from Kishtwar (J&K) for providing data on saffron yield. Authors are thankful to UGC for funding the project UGC [42-168/2013 (SR)] and Indo-Austrian project of OeAD GmbH and DST under call number IN2022 to carry out this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsufs.2021.688393/full#supplementary-material
Ahmad, B. R., Shakeel, A. D., Ishrat, B., and Gowhar, R. (2021). “Cytogenetic and bioactive attributes of Crocus sativus (Saffron): a tool to unfold its medicinal mystery,” in Medicinal and Aromatic Plants (New York: Academic Press), 145–167
Ainsworth, T. D., Krause, L., Bridge, T., Torda, G., Raina, J. B., Zakrzewski, M., et al. (2015). The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9, 2261–2274. doi: 10.1038/ismej.2015.39
Alavi-Kia, S. S., Mohammadi, S. A., Aharizad, S., and Moghaddam, M. (2008). Analysis of genetic diversity and phylogenetic relationships in Crocus genus of Iran using inter-retrotransposon amplified polymorphism. Biotechnol. Biotechnol. Equip. 22, 795–800. doi: 10.1080/13102818.2008.10817555
Ambardar, S. (2014). Mining Microbial Diversity of Rhizosphere of Crocus Sativus by Metagenomic Approach. Jammu: University of Jammu.
Ambardar, S., Sangwan, N., Manjula, A., Rajendhran, J., Gunasekaran, P., Lal, R., et al. (2014). Identification of bacteria associated with underground parts of Crocus sativus by 16S rRNA gene targeted metagenomic approach. World J. Microbiol. Biotechnol. 30, 2701–2709. doi: 10.1007/s11274-014-1694-0
Ambardar, S., Singh, H. R., Gowda, M., and Vakhlu, J. (2016). Comparative metagenomics reveal phylum level temporal and spatial changes in mycobiome of belowground parts of Crocus sativus. PLoS ONE 11:e0163300. doi: 10.1371/journal.pone.0163300
Ambardar, S., and Vakhlu, J. (2013). Plant growth promoting bacteria from Crocus sativus rhizosphere. World J. Microbiol. Biotechnol. 29, 2271–2279. doi: 10.1007/s11274-013-1393-2
Berendsen, R. L., Pieterse, C. M., and Bakker, P. A. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bonnineau, C., Moeller, A., Barata, C., Bonet, B., Proia, L., Sans-Pich,é, F., et al. (2012). “Advances in the multibiomarker approach for risk assessment in aquatic ecosystems,” in Emerging and Priority Pollutants in Rivers (Heidelberg: Springer), 147–179
Brown, J., Pirrung, M., and McCue, L. A. (2017). FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139. doi: 10.1093/bioinformatics/btx373
Busconi, M., Colli, L., Sánchez, R. A., Santaella, M., Pascual, M. D. L. M., Santana, O., et al. (2015). AFLP and MS-AFLP analysis of the variation within saffron crocus (Crocus sativus L.) germplasm. PLoS ONE 10:e0123434. doi: 10.1371/journal.pone.0123434
Chamkhi, I., Sbabou, L., and Aurag, J. (2018). Endophytic fungi isolated from Crocus sativus L. (saffron) as a source of bioactive secondary metabolites. Phcog. J. 10, 1143–1148. doi: 10.5530/pj.2018.6.195
Chaparro, J. M., Badri, D. V., and Vivanco, J. M. (2014). Rhizosphere microbiome assemblage is affected by plant development. ISME J. 8, 790–803. doi: 10.1038/ismej.2013.196
Chen, H., Wu, H., Yan, B., Zhao, H., Liu, F., Zhang, H., et al. (2018). Core microbiome of medicinal plant Salvia miltiorrhiza seed: a rich reservoir of beneficial microbes for secondary metabolism? Int. J. Mol. Sci. 19:672. doi: 10.3390/ijms19030672
Cho, S. H., Lee, K. S., Shin, D. S., Han, J. H., Park, K. S., Lee, C. H., et al. (2010). Four new species of Chryseobacterium from the rhizosphere of coastal sand dune plants, Chryseobacterium elymi sp. nov., Chryseobacterium hagamense sp. nov., Chryseobacterium lathyri sp. nov. and Chryseobacterium rhizosphaerae sp. nov. System. Appl. Microbiol. 33, 122–127. doi: 10.1016/j.syapm.2009.12.004
Chu, T. N., Tran, B. T. H., and Hoang, M. T. T. (2019). Plant growth-promoting rhizobacterium Pseudomonas PS01 induces salt tolerance in Arabidopsis thaliana. BMC Res. Notes 12, 1–7. doi: 10.1186/s13104-019-4046-1
Colin, Y., Nicolitch, O., Van Nostrand, J. D., Zhou, J. Z., Turpault, M. P., and Uroz, S. (2017). Taxonomic and functional shifts in the beech rhizosphere microbiome across a natural soil toposequence. Sci. Rep. 7, 1–17. doi: 10.1038/s41598-017-07639-1
Coller, E., Cestaro, A., Zanzotti, R., Bertoldi, D., Pindo, M., Larger, S., et al. (2019). Microbiome of vineyard soils is shaped by geography and management. Microbiome 7, 1–15. doi: 10.1186/s40168-019-0758-7
Compant, S., Samad, A., Faist, H., and Sessitsch, A. (2019). A review on the plant microbiome: ecology, functions, and emerging trends in microbial application. J. Adv. Res. 19, 29–37. doi: 10.1016/j.jare.2019.03.004
Corcoll, N., Bonet, B., Leira, M., and Guasch, H. (2011). Chl-fluorescence parameters as biomarkers of metal toxicity in fluvial biofilms: an experimental study. Hydrobiologia 673, 119–136. doi: 10.1007/s10750-011-0763-8
Deveau, A., Antony-Babu, S., Le Tacon, F., Robin, C., Frey-Klett, P., and Uroz, S. (2016). Temporal changes of bacterial communities in the Tuber melanosporum ecto mycorrhizosphere during ascocarp development. Mycorrhiza 26, 389–399. doi: 10.1007/s00572-015-0679-7
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Nat. Acad. Sci. U.S.A. 112, E911–E920. doi: 10.1073/pnas.1414592112
Eyre, A. W., Wang, M., Oh, Y., and Dean, R. A. (2019). Identification and characterization of the core rice seed microbiome. Phytobiomes J. 3, 148–157. doi: 10.1094/PBIOMES-01-19-0009-R
Fierer, N. (2017). Embracing the unknown: disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 15, 579–590. doi: 10.1038/nrmicro.2017.87
Fierer, N., Hamady, M., Lauber, C. L., and Knight, R. (2008). The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc. Nat. Acad. Sci. 105, 17994–17999. doi: 10.1073/pnas.0807920105
Fitzpatrick, C. R., Copeland, J., Wang, P. W., Guttman, D. S., Kotanen, P. M., and Johnson, M. T. (2018). Assembly and ecological function of the root microbiome across angiosperm plant species. Proc. Nat. Acad. Sci. U.S.A. 115, E1157–E1165. doi: 10.1073/pnas.1717617115
Fulthorpe, R., Martin, A. R., and Isaac, M. E. (2020). Root endophytes of coffee (Coffea arabica): variation across climatic gradients and relationships with functional traits. Phytobiomes J. 4, 27–39. doi: 10.1094/PBIOMES-04-19-0021-R
Goldblatt, P., Davies, T. J., Manning, J. C., van der Bank, M., and Savolainen, V. (2006). Phylogeny of Iridaceae subfamily Crocoideae based on a combined multigene plastid DNA analysis. Aliso 22, 399–411. doi: 10.5642/aliso.20062201.32
Golmohammadi, F. (2014). Saffron and its farming, economic importance, export, medicinal characteristics and various uses in South Khorasan Province-East of Iran. Int. J. Farm. Allied Sci. 3, 566–596.
Grady, K. L., Sorensen, J. W., Stopnisek, N., Guittar, J., and Shade, A. (2019). Assembly and seasonality of core phyllosphere microbiota on perennial biofuel crops. Nat. Commun. 10, 1–10. doi: 10.1038/s41467-019-11974-4
Guasch, H., Bonet, B., Bonnineau, C., and Barral, L. (2017). “Microbial biomarkers,” in Microbial Ecotoxicology (Cham: Springer), pp. 251–281.
Guerrero, R., Margulis, L., and Berlanga, M. (2013). Symbiogenesis: the holobiont as a unit of evolution. Int. Microbiol. 16, 133–143. doi: 10.2436/20.1501.01.188
Gupta, R., Anand, G., Gaur, R., and Yadav, D. (2021). Plant–microbiome interactions for sustainable agriculture: a review. Physiol. Mol. Biol. Plants, 27, 165–179. doi: 10.1007/s12298-021-00927-1
Hammer, Ø., Harper, D. A., and Ryan, P. D. (2001). PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 4:9.
Hernandez-Agreda, A., Gates, R. D., and Ainsworth, T. D. (2017). Defining the core microbiome in corals' microbial soup. Trends Microbiol. 25, 125–140. doi: 10.1016/j.tim.2016.11.003
Hernandez-Agreda, A., Leggat, W., Bongaerts, P., and Ainsworth, T. D. (2016). The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. MBio 7, e00560–16. doi: 10.1128/mBio.00560-16
Intra, B., Euanorasetr, J., Tak,é, A., Inahashi, Y., Mori, M., Panbangred, W., et al. (2019). Saccharopolyspora rhizosphaerae sp. nov., an actinomycete isolated from rhizosphere soil in Thailand. Int. J. Syst. Evol. Microbiol. 69, 1299–1305. doi: 10.1099/ijsem.0.003307
Jafari, S. M., Tsimidou, M. Z., Rajabi, H., and Kyriakoudi, A. (2020). “Bioactive ingredients of saffron: extraction, analysis, applications,” in Saffron (Sawston: Woodhead Publishing), 261–290. doi: 10.1016/B978-0-12-818638-1.00016-2
Jansson, J. K., and Hofmockel, K. S. (2020). Soil microbiomes and climate change. Nat. Rev. Microbiol. 18, 35–46. doi: 10.1038/s41579-019-0265-7
Kandel, S. L., Joubert, P. M., and Doty, S. L. (2017). Bacterial endophyte colonization and distribution within plants. Microorganisms 5:77. doi: 10.3390/microorganisms5040077
Keify, F., and Beiki, A. H. (2012). Exploitation of random amplified polymorphic DNA (RAPD) and sequence-related amplified polymorphism (SRAP) markers for genetic diversity of saffron collection. J. Med. Plants Res. 6, 2761–2768. doi: 10.5897/JMPR11.834
Kour, R., Ambardar, S., and Vakhlu, J. (2018). Plant growth promoting bacteria associated with corm of Crocus sativus during three growth stages. Lett. Appl. Microbiol. 67, 458–464. doi: 10.1111/lam.13042
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kuzniar, A., Włodarczyk, K., Grzadziel, J., Goraj, W., Gałazka, A., and Wolińska, A. (2020). Culture-independent analysis of an endophytic core microbiome in two species of wheat: Triticum aestivum L.(cv.‘Hondia') and the first report of microbiota in Triticum spelta L.(cv.‘Rokosz'). Syst. Appl. Microbiol. 43:126025. doi: 10.1016/j.syapm.2019.126025
Lederberg, J., and McCray, A. T. (2001). Ome SweetOmics—a genealogical treasury of words. The Scientist 15, 8–8.
Lehr, N. A., Schrey, S. D., Bauer, R., Hampp, R., and Tarkka, M. T. (2007). Suppression of plant defence response by a mycorrhiza helper bacterium. New Phytol. 174, 892–903. doi: 10.1111/j.1469-8137.2007.02021.x
Lehr, N. A., Schrey, S. D., Hampp, R., and Tarkka, M. T. (2008). Root inoculation with a forest soil Streptomycete leads to locally and systemically increased resistance against phytopathogens in Norway spruce. New Phytol. 177, 965–976. doi: 10.1111/j.1469-8137.2007.02322.x
Lei, S., Xu, X., Cheng, Z., Xiong, J., Ma, R., Zhang, L., et al. (2019). Analysis of the community composition and bacterial diversity of the rhizosphere microbiome across different plant taxa. Microbiology Open 8:e00762. doi: 10.1002/mbo3.762
Lidbury, I. D., Borsetto, C., Murphy, A. R., Bottrill, A., Jones, A. M., Bending, G. D., et al. (2021). Niche-adaptation in plant-associated Bacteroidetes favours specialisation in organic phosphorus mineralisation. ISME J. 15, 1040–1055. doi: 10.1038/s41396-020-00829-2
Lin, M., Xiong, H., Xiang, X., Zhou, Z., Liang, L., and Mei, Z. (2020). The effect of plant geographical location and developmental stage on root-associated microbiomes of Gymnadenia conopsea. Front. Microbiol. 11:1257. doi: 10.3389/fmicb.2020.01257
Lucas, X., Senger, C., Erxleben, A., Grüning, B. A., Döring, K., Mosch, J., et al. (2012). StreptomeDB: a resource for natural compounds isolated from Streptomyces species. Nucleic Acids Res. 41, D1130–D1136. doi: 10.1093/nar/gks1253
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatti, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488, 86–90. doi: 10.1038/nature11237
Lupwayi, N. Z., Larney, F. J., Blackshaw, R. E., Kanashiro, D. A., and Pearson, D. C. (2017). Phospholipid fatty acid biomarkers show positive soil microbial community responses to conservation soil management of irrigated crop rotations. Soil Till. Res. 168, 1–10. doi: 10.1016/j.still.2016.12.003
Macey, M. C., Pratscher, J., Crombie, A. T., and Murrell, J. C. (2020). Impact of plants on the diversity and activity of methylotrophs in soil. Microbiome 8, 1–17. doi: 10.1186/s40168-020-00801-4
Mendes, R., and Raaijmakers, J. M. (2015). Cross-kingdom similarities in microbiome functions. ISME J. 9, 1905–1907. doi: 10.1038/ismej.2015.7
Metsalu, T., and Vilo, J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566–W570. doi: 10.1093/nar/gkv468
Meyer, F., Paarmann, D., D'Souza, M., Olson, R., and Glass, E. M. (2008). The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. bmcbioinformatics. 9:386. doi: 10.1186/1471-2105-9-386
Mir, M. A., Mansoor, S., Sugapriya, M., Alyemeni, M. N., Wijaya, L., and Ahmad, P. (2021). Deciphering genetic diversity analysis of saffron (Crocus sativus L.) using RAPD and ISSR markers. Saudi J. Biol. Sci. 28, 1308–1317. doi: 10.1016/j.sjbs.2020.11.063
Mishra, P., Kumar, A., Nagireddy, A., Mani, D. N., Shukla, A. K., Tiwari, R., et al. (2016). DNA barcoding: an efficient tool to overcome authentication challenges in the herbal market. Plant Biotechnol. J. 14, 8–21. doi: 10.1111/pbi.12419
Miyashita, N. T. (2015). Contrasting soil bacterial community structure between the phyla Acidobacteria and Proteobacteria in tropical Southeast Asian and temperate Japanese forests. Genes Genet. Syst. 90, 61–77. doi: 10.1266/ggs.90.61
Monika, T., and Neha, S. (2014). Saffron: a golden condiment and a repository of nutraceutical potential. Food Sci. Res. J. 5, 59–67.
Moshtaghi, N. (2020). “Tissue and cell culture of saffron,” in Saffron (Sawston: Woodhead Publishing), 229–246. doi: 10.1016/B978-0-12-818638-1.00014-9
Mukhtar, S., Hirsch, A. M., Khan, N., Malik, K. A., Humm, E. A., Pellegrini, M., and Mehnaz, S. (2020). Impact of soil salinity on the cowpea nodule-microbiome and the isolation of halotolerant PGPR strains to promote plant growth under salinity stress. Phytobiomes J. 4, 364–374. doi: 10.1094/PBIOMES-09-19-0057-R
Mykhailenko, O., Desenko, V., Ivanauskas, L., and Georgiyants, V. (2020). Standard operating procedure of Ukrainian Saffron Cultivation According with Good Agricultural and Collection Practices to assure quality and traceability. Ind. Crops Prod. 151:112376. doi: 10.1016/j.indcrop.2020.112376
Mzabri, I., Legsayer, M., Chetouani, M., Aamar, A., Kouddane, N., Boukroute, A., et al. (2017). Saffron (Crocus sativus L.) yield parameter assessment of abiotic stressed corms stored in low temperature. J. Mater. Environ. Sci. 8, 3588–3597.
Nemati, Z., Harpke, D., Gemicioglu, A., Kerndorff, H., and Blattner, F. R. (2019). Saffron (Crocus sativus) is an autotriploid that evolved in Attica (Greece) from wild Crocus cartwrightianus. Mol. Phylogenet. Evol. 136:14–20. doi: 10.1016/j.ympev.2019.03.022
Newton, R. J., Huse, S. M., Morrison, H. G., Peake, C. S., Sogin, M. L., and McLellan, S. L. (2013). Shifts in the microbial community composition of Gulf Coast beaches following beach oiling. PLoS ONE 8:74265. doi: 10.1371/journal.pone.0074265
Nguyen, N. L., Kim, Y. J., Hoang, V. A., Subramaniyam, S., Kang, J. P., Kang, C. H., et al. (2016). Bacterial diversity and community structure in Korean ginseng field soil are shifted by cultivation time. PLoS ONE 11:e0155055. doi: 10.1371/journal.pone.0155055
Ofek-Lalzar, M., Sela, N., Goldman-Voronov, M., Green, S. J., Hadar, Y., and Minz, D. (2014). Niche and host-associated functional signatures of the root surface microbiome. Nat. Commun. 5, 1–9. doi: 10.1038/ncomms5950
Othman, R., Hatta, F. A. M., Hassan, N. M., and Kamoona, S. (2020). Characterization of carotenoids content and composition of saffron from different localities. J. Pharm. Nutr. Sci. 10, 34–40. doi: 10.29169/1927-5951.2020.10.01.6
Pandita, D. (2021). “Saffron (Crocus sativus L.): Phytochemistry, therapeutic significance and omics-based biology,” in Medicinal and Aromatic Plants. (New York: Academic Press), 325–396. doi: 10.1016/B978-0-12-819590-1.00014-8
Pang, M. F., Abdullah, N., Lee, C. W., and Ng, C. C. (2008). Isolation of high molecular weight DNA from forest topsoil for metagenomic analysis. Asia Pacific J. Mol. Biol. Biotechnol. 16, 35–41.
Peiffer, J. A., Spor, A., Koren, O., Jin, Z., Tringe, S. G., Dangl, J. L., et al. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Nat. Acad. Sci. 110, 6548–6553. doi: 10.1073/pnas.1302837110
Pérez-Jaramillo, J. E., de Hollander, M., Ramírez, C. A., Mendes, R., Raaijmakers, J. M., and Carrión, V. J. (2019). Deciphering rhizosphere microbiome assembly of wild and modern common bean (Phaseolus vulgaris) in native and agricultural soils from Colombia. Microbiome 7, 1–16. doi: 10.1186/s40168-019-0727-1
Pesce, S., Margoum, C., and Foulquier, A. (2016). Pollution-induced community tolerance for in situ assessment of recovery in river microbial communities following the ban of the herbicide diuron. Agric. Ecosyst. Environ. 221, 79–86. doi: 10.1016/j.agee.2016.01.009
Philippot, L., Raaijmakers, J. M., Lemanceau, P., and Van Der Putten, W. H. (2013). Going back to the roots: the microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 11, 789–799. doi: 10.1038/nrmicro3109
Qessaoui, R., Bouharroud, R., Furze, J. N., El Aalaoui, M., Akroud, H., Amarraque, A., et al. (2019). Applications of new rhizobacteria Pseudomonas isolates in agroecology via fundamental processes complementing plant growth. Sci. Rep. 9, 1–10. doi: 10.1038/s41598-019-49216-8
Qiao, Q., Wang, F., Zhang, J., Chen, Y., Zhang, C., Liu, G., et al. (2017). The variation in the rhizosphere microbiome of cotton with soil type, genotype and developmental stage. Sci. Rep. 7, 1–10. doi: 10.1038/s41598-017-04213-7
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. nature 464, 59–65. doi: 10.1038/nature08821
Rampelotto, P. H., de Siqueira Ferreira, A., Barboza, A. D. M., and Roesch, L. F. W. (2013). Changes in diversity, abundance, and structure of soil bacterial communities in Brazilian Savanna under different land use systems. Microb. Ecol. 66, 593–607. doi: 10.1007/s00248-013-0235-y
Rubio-Moraga, A., Castillo-López, R., Gómez-Gómez, L., and Ahrazem, O. (2009). Saffron is a monomorphic species as revealed by RAPD, ISSR and microsatellite analyses. BMC Res. Notes 2, 1–5. doi: 10.1186/1756-0500-2-189
Schillaci, M., Gupta, S., Walker, R., and Roessner, U. (2019). “The role of plant growth-promoting bacteria in the growth of cereals under abiotic stresses,” in Root Biology-Growth, Physiology, and Functions (London: Intechopen), 1–21. doi: 10.5772/intechopen.87083
Schmitt, S., Tsai, P., Bell, J., Fromont, J., Ilan, M., Lindquist, N., et al. (2012). Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 6, 564–576. doi: 10.1038/ismej.2011.116
Schrey, S. D., and Tarkka, M. T. (2008). Friends and foes: Streptomyces as modulators of plant disease and symbiosis. Antonie Van Leeuwenhock 94, 11–19. doi: 10.1007/s10482-008-9241-3
Skaltsas, D. N., Badotti, F., Vaz, A. B. M., da Silva, F. F., Gazis, R., Wurdack, K., et al. (2019). Exploration of stem endophytic communities revealed developmental stage as one of the drivers of fungal endophytic community assemblages in two Amazonian hardwood genera. Sci. Rep. 9, 1–14. doi: 10.1038/s41598-019-48943-2
Song, Z. M., Wang, K. L., Yin, Q., Chen, C. C., and Xu, Y. (2020). Algoriphagus kandeliae sp. nov., isolated from mangrove rhizosphere soil. Int. J. Syst. Evol. Microbiol. 70, 1672–1677. doi: 10.1099/ijsem.0.003954
Su, X., Yuan, C., Wang, L., Chen, R., Li, X., Zhang, Y., et al. (2021). The beneficial effects of saffron extract on potential oxidative stress in cardiovascular diseases. Oxid. Med. Cell. Longev. 1–14. doi: 10.1155/2021/6699821
Sun, X., Ma, W., Xu, Y., Jin, X., and Ni, H. (2020). Complete genome sequence of Rahnella aquatilis MEM40, a plant growth-promoting rhizobacterium isolated from rice rhizosphere soil, with antagonism against Magnaporthe oryzae and Fusarium graminearum. Microbiol. Resour. Announc. 9, e00651–20. doi: 10.1128/MRA.00651-20
Terrazas, R. A., Balbirnie-Cumming, K., Morris, J., Hedley, P. E., Russell, J., Paterson, E., et al. (2020). A footprint of plant eco-geographic adaptation on the composition of the barley rhizosphere bacterial microbiota. Sci. Rep. 10, 1–13. doi: 10.1038/s41598-020-69672-x
Toju, H., Peay, K. G., Yamamichi, M., Narisawa, K., Hiruma, K., Naito, K., et al. (2018). Core microbiomes for sustainable agroecosystems. Nat. Plants 4, 247–257. doi: 10.1038/s41477-018-0139-4
Umadevi, P., Anandaraj, M., Srivastav, V., and Benjamin, S. (2018). Trichoderma harzianum MTCC 5179 impacts the population and functional dynamics of microbial community in the rhizosphere of black pepper (Piper nigrum L.). Braz. J. Microbiol. 49, 463–470. doi: 10.1016/j.bjm.2017.05.011
Vandenkoornhuyse, P., Quaiser, A., Duhamel, M., Le Van, A., and Dufresne, A. (2015). The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206. doi: 10.1111/nph.13312
Vergani, L., Mapelli, F., Suman, J., Cajthaml, T., Uhlik, O., and Borin, S. (2019). Novel PCB-degrading Rhodococcus strains able to promote plant growth for assisted rhizoremediation of historically polluted soils. PLoS ONE 14:e221253. doi: 10.1371/journal.pone.0221253
Viaene, T., Langendries, S., Beirinckx, S., Maes, M., and Goormachtig, S. (2016). Streptomyces as a plant's best friend? FEMS Microbiol. Ecol. 92. doi: 10.1093/femsec/fiw119
Walters, W. A., Jin, Z., Youngblut, N., Wallace, J. G., Sutter, J., Zhang, W., et al. (2018). Large-scale replicated field study of maize rhizosphere identifies heritable microbes. Proc. Natl. Acad. Sci. U.S.A. 115, 7368–7373. doi: 10.1073/pnas.1800918115
Wang, M., Eyre, A. W., Thon, M. R., Oh, Y., and Dean, R. A. (2020). Dynamic changes in the microbiome of rice during shoot and root growth derived from seeds. Front. Microbiol. 11:2183. doi: 10.3389/fmicb.2020.559728
Wechter, P., Williamson, J., Robertson, A., and Kluepfel, D. (2003). A rapid, cost-effective procedure for the extraction of microbial DNA from soil. World J. Microbiol. Biotechnol. 19, 85–91. doi: 10.1023/A:1022587806945
Zakhia, F., Jeder, H., Willems, A., Gillis, M., Dreyfus, B., and De Lajudie, P. (2006). Diverse bacteria associated with root nodules of spontaneous legumes in Tunisia and first report for nifH-like gene within the genera Microbacterium and Starkeya. Microb. Ecol. 51, 375–393. doi: 10.1007/s00248-006-9025-0
Keywords: barcodes, microbiome, microbial fingerprint, biomarker, cormosphere, saffron
Citation: Bhagat N, Sharma S, Ambardar S, Raj S, Trakroo D, Horacek M, Zouagui R, Sbabou L and Vakhlu J (2021) Microbiome Fingerprint as Biomarker for Geographical Origin and Heredity in Crocus sativus: A Feasibility Study. Front. Sustain. Food Syst. 5:688393. doi: 10.3389/fsufs.2021.688393
Received: 30 March 2021; Accepted: 14 June 2021;
Published: 19 July 2021.
Edited by:
Everlon Cid Rigobelo, São Paulo State University, BrazilReviewed by:
Aejaz Ahmad Dar, Sher-e-Kashmir University of Agricultural Sciences and Technology of Jammu, IndiaCopyright © 2021 Bhagat, Sharma, Ambardar, Raj, Trakroo, Horacek, Zouagui, Sbabou and Vakhlu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jyoti Vakhlu, anlvdGltZXRhZ2Vub21pY0BnbWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.