Explore article hub
 Elisa Laiolo1,2,3*†
Elisa Laiolo1,2,3*† Intikhab Alam3†
Intikhab Alam3† Mahmut Uludag3
Mahmut Uludag3 Tahira Jamil1,2,3
Tahira Jamil1,2,3 Susana Agusti1,2
Susana Agusti1,2 Takashi Gojobori3
Takashi Gojobori3 Silvia G. Acinas4
Silvia G. Acinas4 Josep M. Gasol4
Josep M. Gasol4 Carlos M. Duarte1,2,3
Carlos M. Duarte1,2,3- 1Marine Science Program, Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
- 2Red Sea Research Center (RSRC), King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
- 3Computational Bioscience Research Center (CBRC), King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
- 4Department of Marine Biology and Oceanography, Institute of Marine Sciences, Spanish National Research Council (CSIC), Barcelona, Spain
Abstract
The global ocean genome (the pool of genes in marine organisms and the functional information they encode) is a major, untapped resource for science and society with a growing range of biotechnology applications in sectors such as biomedicine, energy, and food. Shotgun sequencing and metagenomics can now be used to catalog the diversity of ocean microbial life and to explore its functional potential, but has been limited by sample coverage, access to suitable sequencing platforms, and computational capacity. Here we provide a novel synthesis of the global ocean genome based on analysis of 2,102 sampled ocean metagenomes, with gene assembly and annotation via the KAUST Metagenome Analysis Platform (KMAP) Global Ocean Gene Catalog 1.0 containing 308.6 million gene clusters. Taxonomically, we report the distribution of marine genes across the tree of life and different ocean basins and depth zone biomes. Functionally, we map its relationship to protein families and biogeochemical processes, including the major microbial metabolic pathways that process three elements that play fundamental roles in biogeochemical cycles and are relevant to climate change. These data extend our understanding of the complex, dynamic nature of the ocean microbiome and its metabolic capabilities. Further research is of critical global importance both to unlock the potential of the ocean genome and to understand and predict the effects of human-induced changes, including pollution and climate change. Further hypothesis-driven research should target under-sampled deep sea and benthic microbial communities using enhanced metagenomic methods, to better understand marine ecosystem functioning. Investment in the necessary computational capacity is essential, as are suitable intellectual property frameworks.
Key points
- With 308.6 million gene clusters, the KAUST Metagenome Analysis Platform (KMAP) Global Ocean Gene Catalog 1.0 is the largest open-source catalog to date matching microbial class with gene function, geographic location, and ecosystem type.
- The catalog offers diverse applications that go beyond advancing our understanding of the ocean microbiome and its metabolic capabilities—it also establishes a baseline for tracking the influence of global warming, pollution, and other changes on ocean health and provides a tool to explore marine genetic resources to discover novel genes with potential uses in medicine, energy, food, and other industries.
- Fungi represented over 50% of the distinct gene clusters identified in the mesopelagic zone. This finding highlights the contribution of fungi to microbial diversity and carries functional consequences for the role of fungi in elemental cycling in the ocean.
- Biomass ratios among domains are not reflective of the corresponding distribution of unique genes: this rough comparison shows that the eukaryotic component of ocean biomass outweighs its contribution to marine genetic diversity, which remains dominated by Bacteria, and that viral genomes contain far more innovation than hitherto realized, even considering that this DNA-based assessment of the global ocean genome omits RNA viruses.
- Analysis of benthic and pelagic samples revealed significant differences in both taxonomic composition and metabolic processes, reflecting the heterogeneous nature of the ocean floor and highlighting the potential for uncovering a vast reservoir of functional genes, as well as the need to further sample and investigate marine benthic metagenomic resources, which remain under-explored compared with the pelagic component.
Introduction
The ocean is the largest habitat in the world, covering 71% of the Earth’s surface and an estimated 99% of the volume of the biosphere, which is approximately 1.335 million km³ (1, 2). The ocean is also the cradle of life: with 3.9 billion years of evolutionary history (3–5), this is reflected in a far more extensive representation of taxa across the tree of life compared with a much narrower representation on land.
Life in the ocean is believed to have started with single-celled organisms operating under anaerobic metabolic processes, such as fermentation (6). Aerobic metabolism occurred much later, facilitated by the advent of photosynthesis approximately 2.4 billion years ago and the oxygenation of Earth’s ocean and the atmosphere (7–9). Hence, anaerobic processes are now limited to sediment, hypoxic waters, or specific marine habitat niches, such as hydrothermal vents. Aerobic processes, meanwhile, dominate in the ocean water column, with aerobes being highly adapted to the presence of molecular oxygen, which is toxic to anaerobic organisms. The origins of the bacterial and archaeal domains in the anoxic Archaean ocean predate the divergence of anaerobic and aerobic metabolisms; aerobic lineages evolved during and after the gradual oxidation of the biosphere. After the advent of multicellularity and the emergence of nuclei as a structure, phylogenetic analyses position a newly evolved branch originating from Archaea (10), leading to the establishment of a distinct domain of life—the eukaryotes.
An analysis of the global genome of the ocean classifying metabolism types and taxonomic domains is thus consistent with the evolution of life in the ocean, acknowledging that the modern water column is mostly oxic and that aerobic microbial life is, therefore, prevalent in the ocean water column. The development of the tree of life in the ocean, dominated by microbial taxa, is also reflected in its functional diversity—represented by the array of metabolic pathways used to source energy and process organic matter.
The global ocean genome, defined as the pool of genes present in marine organisms and the functional information they encode (including protein-coding genes and biosynthetic gene clusters supporting ocean metabolism), is a major resource for science and society, with a growing range of biotechnology applications across many sectors, including health, energy, and food (11). For instance, the discovery and development of the green fluorescence protein (GFP), first isolated from jellyfish and now widely used in medical imaging diagnostics, won Osamu Shimomura, Martin Chalfie, and Roger Y. Tsien the Nobel Prize in Chemistry in 2008 (12). Polymerases sourced from bacteria associated with hydrothermal vents are used in polymerase chain reaction (PCR) tests, which proved critical to global efforts against the SARS-Cov-2 pandemic (13).
The release of the first full genome sequence of a marine organism, that of the marine archaeon Methanococcus jannaschii (14), was followed by the release of the full genome sequence of the first marine photosynthetic organism, the ubiquitous cyanobacterium Prochlorococcus marinus (15), which has the smallest genome of all photosynthetic organisms known to date. These pioneering, descriptive efforts were boosted with the development of functional genomics, enabling the functional annotation of proteins and inference on their role (16, 17), together with shotgun sequencing, pioneered by the Sorcerer II Global Ocean Sampling expedition (2003–2004) in the first metagenomic assessment of a marine plankton community (18, 19).
The term “metagenomics”, first used by Handelsman and collaborators in 1998 (20), is defined as the study of the pooled genetic information contained in an environmental sample (21). Shotgun sequencing, enabling metagenomics, has catalyzed efforts to catalog microbial diversity and explore its functional potential in the ocean, where >99% of microbes—the “unseen majority” (22)—have not been cultured. Metagenomic-based approaches allow multilevel assessments of microbial diversity and functionality across habitats. They are now facilitated by the creation of metagenomic analysis platforms such as the KAUST Metagenomic Analytical Platform (KMAP) (23) and MGnify, the European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI) platform, for the assembly, analysis, and archiving of microbiome data (24).
The rapid reduction in sequencing costs, following the rise of next-generation sequencing technologies, has enabled the generation of gene catalogs and functional characterization of marine microbial communities on a global scale. As a result, the pioneering efforts of the Sorcerer II expedition (18, 19) were greatly expanded, with increased sequencing depth and coverage, by the global Tara Oceans expedition (2009–2013) (25), which focused on microbial communities in the upper ocean, and the Malaspina Expedition (2010–2011), which sampled the tropical and subtropical Atlantic, Indian, and Pacific Oceans (26) and delivered the first global metagenomics assessment of the deep sea (27). In addition, the Ocean Sampling Day program (28) coordinated a metagenomics analysis of coastal sites around the world, and the Hawaii Ocean Time Series developed the most extensive time series of microbial and biogeochemical processes observations available, spanning from October 1988 to the present day (29). In 2015, Tara Oceans released the first global ocean gene catalog of the ocean, including 33.3 million non-redundant genes sourced from 243 samples collected across 68 sampling sites, mostly from the upper ocean (25). Coelho and colleagues released the Global Microbial Gene Catalog (GMGCv1) in 2022, expanding the global gene catalog to a total of 303 million (30), including 88 million 95% non-redundant annotated genes from marine organisms.
The microbial population within the global ocean is highly diverse both in composition—comprising more than 2×106 species (31)—and in roles. Bacteria and Archaea are major players in all biogeochemical cycles and conduct a vast array of metabolic functions (18, 32) involving the uptake, release, and transformation of some of the main climate-active molecules formed from carbon, nitrogen, and sulfur (S1). The carbon cycle is multifaceted and is rooted in carbon dioxide being fixed by autotrophs into organic compounds, mainly using solar energy through photosynthesis. These compounds are transformed and ultimately remineralized through a series of complex cycles supporting all biological processes and their energy requirements. These biogeochemical cycles are all powered by the enzymatic and biosynthetic machinery coded by the genome of the biosphere. Metagenomics allows gene functions to be matched with the corresponding taxonomic identity of the organism to which the gene pertains (32), thereby correlating biodiversity and function in the ecosystem—a major goal in ecology and biogeochemistry. While the importance of bacteria in biogeochemical cycles is well established (33, 34), the role of Archaea in the functioning of marine systems remains poorly understood.
Archaea are now known to be widespread throughout the oceans, where they constitute a relevant fraction of the microbial community (35). Benthic microbes play a major role in oceanic biogeochemical cycles (36–39) and support much of the integrated metabolic activity in the ocean through a broader diversity of processes compared with those supported in the water column, enabled by the sharp biogeochemical and redox gradients within the first centimeters of marine sediments. Yet, efforts to catalog the ocean genome have focused on pelagic microbes, thereby potentially overlooking a wealth of functional diversity.
The term metabolic architecture was introduced recently to refer to the functional structure of marine microbial communities, resolved using functional analyses of metagenomics (27). Metabolic architecture highlights the complex and multilevel interactive nature of the various microbial metabolic pathways present in the community, which creates in the end a metabolic network that contributes to the maintenance of the integrity and functioning of the ecosystem. Metagenomics provides the structure of the network, which provides insights into potential metabolisms, but cannot resolve rate processes.
Here we provide a novel synthesis of the global ocean genome based on the analysis of 2,102 sampled ocean metagenomes, with gene assembly and annotation via the KAUST Metagenome Analysis Platform (KMAP), the KMAP Global Ocean Gene Catalog 1.0, which contains 308.6 million gene clusters. Taxonomically, we report the distribution of marine genes across the tree of life and different ocean basins and depth zone biomes. Functionally, we map its relationship to protein families and biogeochemical processes, including the major microbial metabolic pathways involving climate-active molecules built from carbon, nitrogen, and sulfur.
Results
We analyzed 2,102 metagenomes obtained from the European Nucleotide Archive (see Methods). The metagenomes were dominated by pelagic samples (2016; 95.9%), with only 4.1% (86) of metagenomics samples corresponding to benthic communities (Table 1). Most (78.5%) samples were collected in the upper ocean (depth 0–200 m), which represents only 5.2% of the ocean volume (Table 1, Figure 1); 7.2% samples were collected from the mesopelagic ocean (200–1000 m) and 10.2% from the dark ocean (>1000 m), the largest ocean biome by volume (Table 1, Figure 1).
Table 1
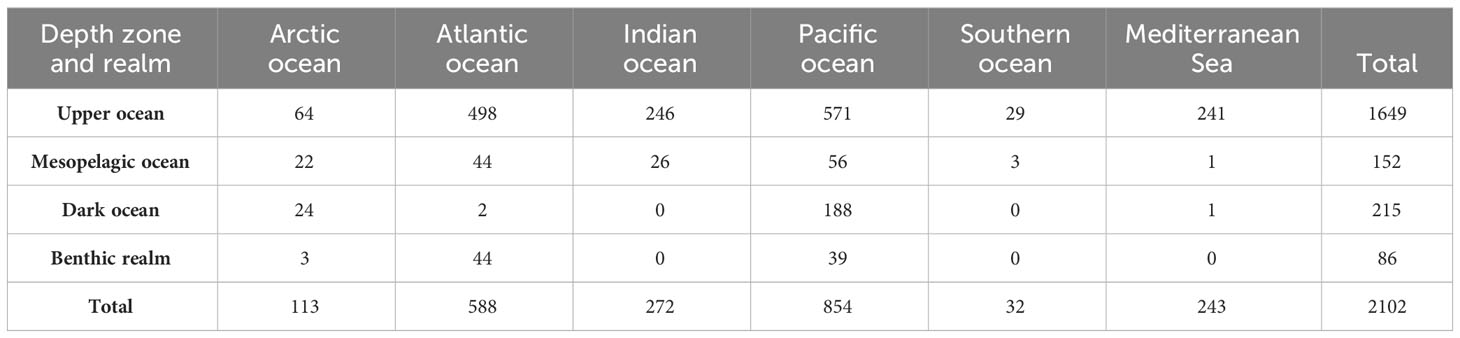
Table 1 Metagenome samples (number) across depth zones and basins.
Figure 1
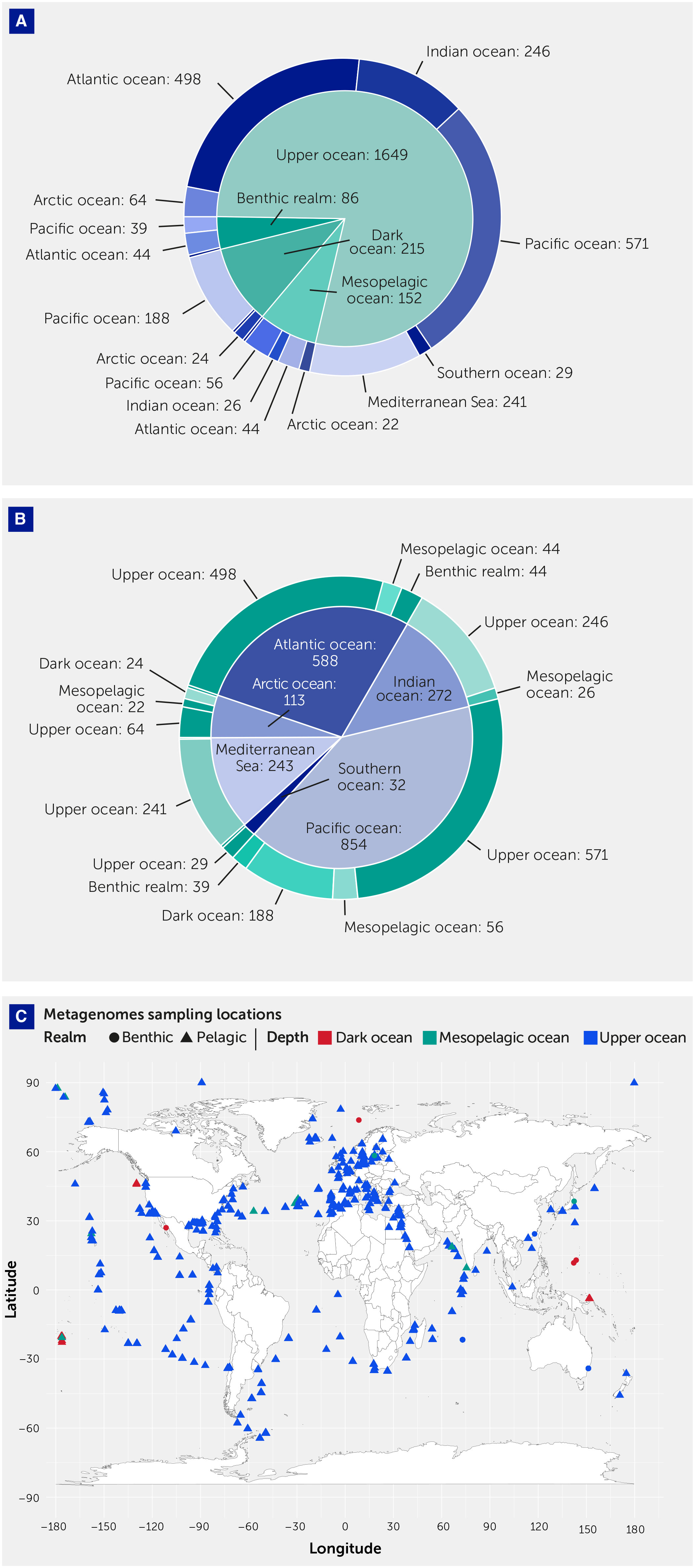
Figure 1 Metagenomes sampling locations. (A) Pie chart showing the sample (number) distribution across basins. (B) Pie chart showing the sample (number) distribution across depth zones. (C) Map summarizing the distribution of the metagenomes across realms, indicated by the polygon shape, and depth zone, indicated by the filling color. The polygon indicates the realm (benthic or pelagic), while the filling color indicates the depth zone: upper ocean, 0–200 m, mesopelagic ocean, 200–1000 m, and dark ocean, >1000 m. The full list of metagenomes analyzed in this study is reported in Supplementary Table S2, including the above-mentioned classification.
Geographically, the Pacific and Atlantic oceans were best represented in the metagenome set, accounting for 40.6% and 28% of samples, respectively. There was a weak representation of polar oceans, with 5.4% and 1.5% of metagenome samples collected in the Arctic and Southern oceans, respectively, and an overrepresentation of the Mediterranean Sea, which accounted for 11.6% of samples while representing only 0.7% of the ocean surface (Table 1, Figure 1).
Future efforts for metagenomic characterization of ocean communities should improve the delivery of essential metadata, including the data (latitude, longitude, depth, temperature, salinity, and nutrient concentration) to apportion the samples to Longhurst biogeochemical provinces (40), which may provide additional insight into selection and adaptation processes, matching communities with functional traits as well as inferring biogeochemical processes from functional metagenomics.
Taxonomic distribution of genes
The KMAP Global Ocean Gene Catalog 1.0 comprises 308.6 million gene clusters, of which 164.8 million (53.4%) were annotated. The gene catalog was dominated by Bacteria (78.26% of the annotated unique gene clusters), followed by Eukaryota (12.21%), Archaea (6.08%), and viruses (3.46%). Since our analysis was based on DNA sequencing, RNA viruses have not been included, and their incorporation is pending the availability of additional data. Each domain had different contributions to the different depth zones and realms: the mesopelagic and dark ocean were relatively rich in unique Archaea genes, whereas the upper ocean was greatly enriched with eukaryotic and viral genes (Figure 2).
Figure 2
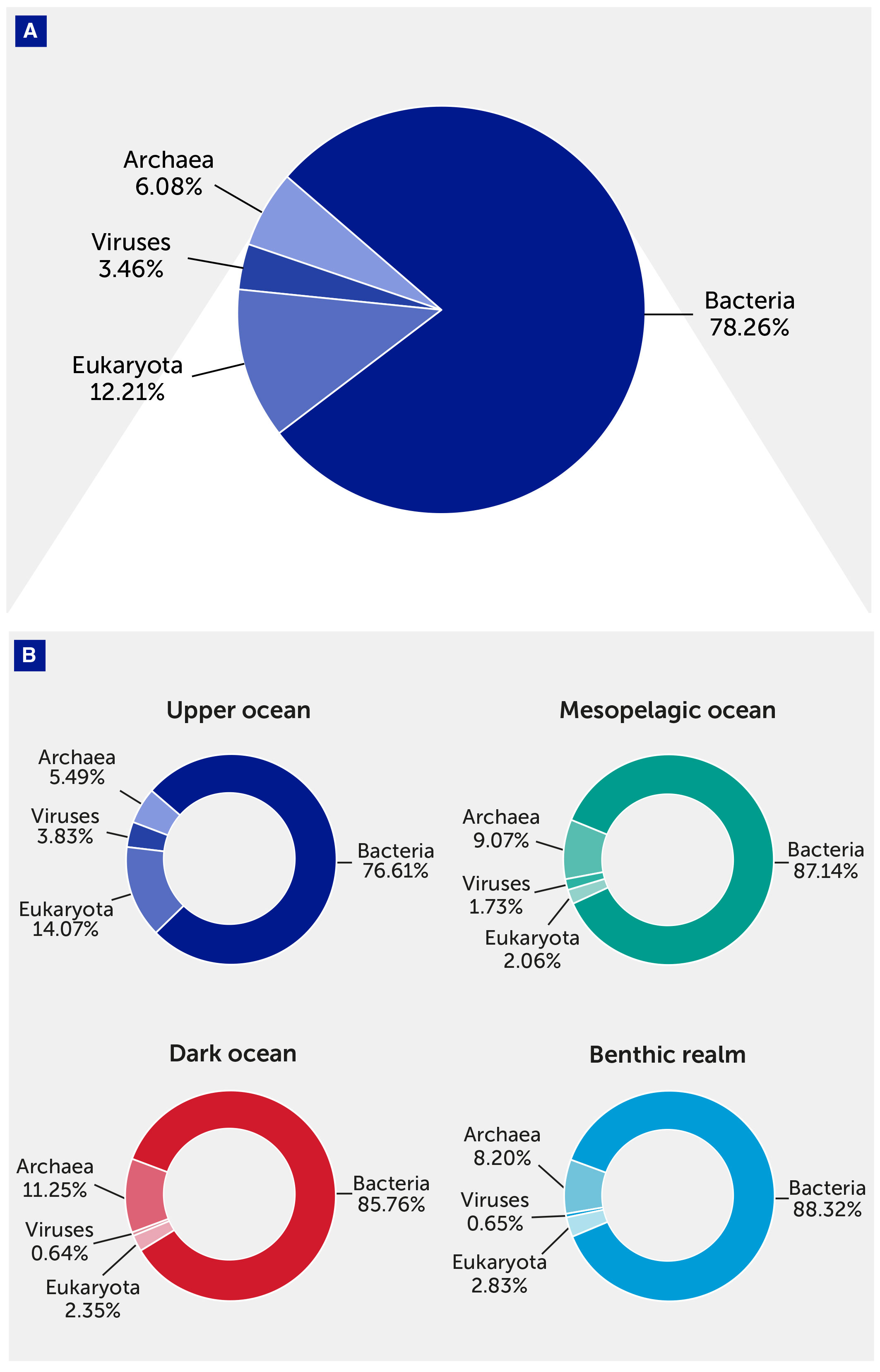
Figure 2 The contribution of taxonomical domains to the gene catalog. (A) Pie chart showing the overall relative contribution of the four taxonomical domains (Archaea, Bacteria, Eukaryota, and viruses) to the gene catalog. (B) Pie charts showing the contribution of the four taxonomical domains to the gene catalog in each depth zone of the pelagic (upper, mesopelagic, and dark ocean) and benthic realms.
Taxonomically, we focused separately on the contribution of Archaea and Bacteria and the eukaryotic groups to the catalog of unique annotated genes.
Regarding Bacteria, the gene clusters related to Pseudomonadota were the bacterial phylum contributing the most to unique annotated genes in the catalog in all realms and depth zones.
Alphaproteobacteria, in particular, dominated the unique gene clusters (Table 2, Figure 3) in all depth zones except the dark ocean, where Gammaproteobacteria contributed the highest number of unique genes. Deltaproteobacteria were highly represented in the gene catalogs of the dark ocean (10.62%) and benthic realm (9.99%). Epsilonproteobacteria were also well-represented in the dark ocean (8.97%), contributing 10-fold more unique gene clusters in this depth zone than in the upper ocean, mesopelagic zone, or benthic realm, where they accounted for less than 1% (Table 2, Figure 3). Bacteroidota showed similar contributions in the upper ocean and the benthic realm (12.69% and 13.84%, respectively) while accounting for around 7% of unique gene clusters in the mesopelagic and dark ocean. Cyanobacteriota showed the highest relative contribution (6.30%) in the upper ocean, while Bacillota reached the highest contribution (1.93%) in the benthic realm. Gene cluster enrichment of other phyla is shown in Figure 3.
Table 2
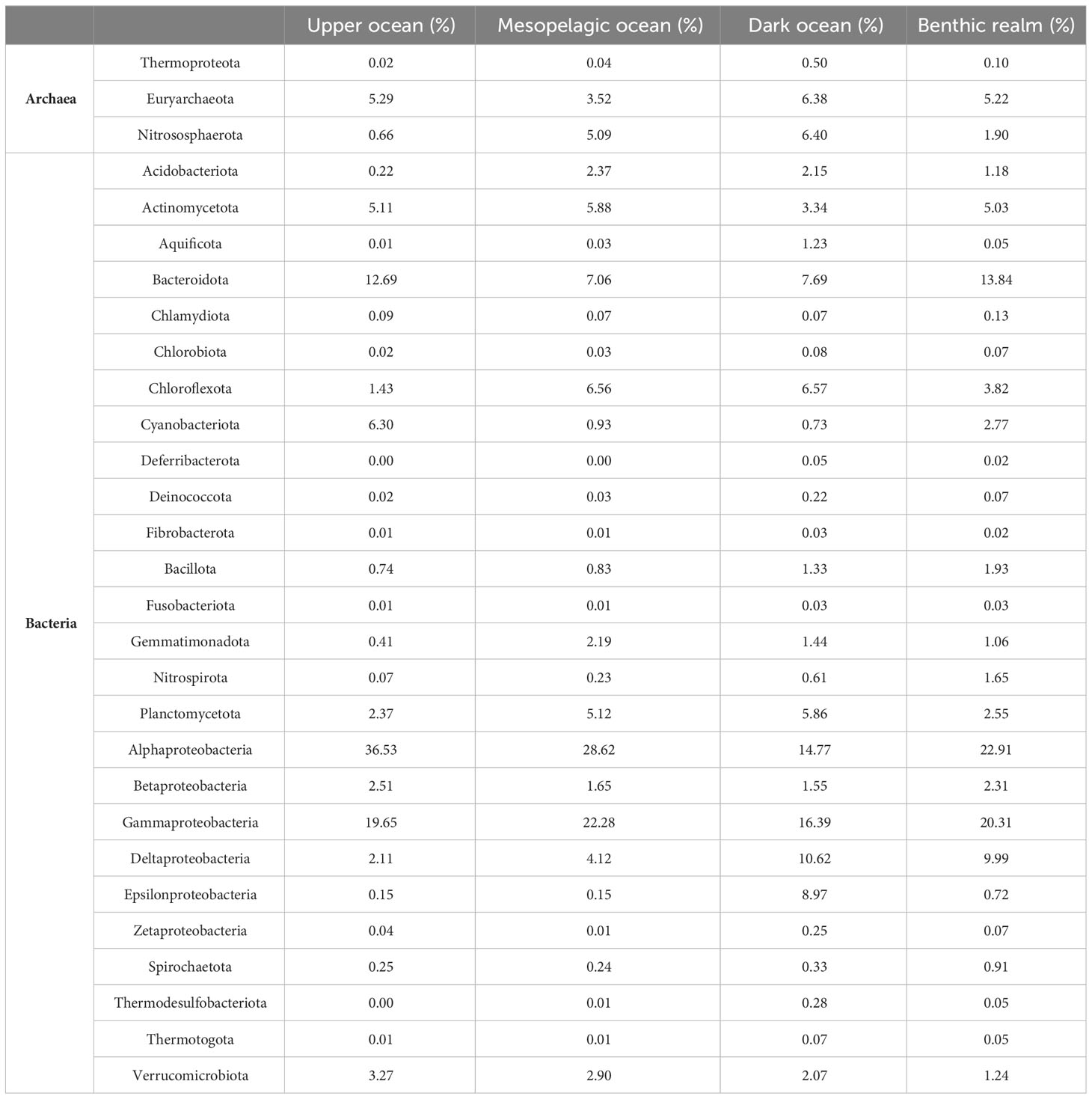
Table 2 Relative (%) contribution of the Archaea and Bacteria taxonomical groups to the gene clusters.
Figure 3
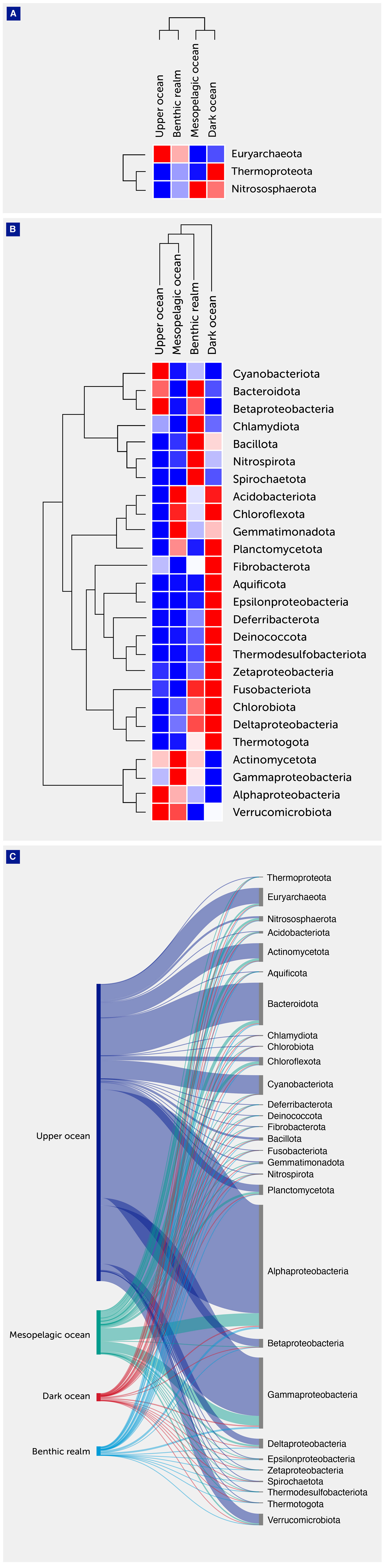
Figure 3 Archaeal and bacterial annotated gene cluster contribution. (A) Heatmap with hierarchical clustering of gene clusters related to the major archaeal phyla. The clades are shown on the right of the graph, while the depth zones of the pelagic (upper, mesopelagic, and dark ocean) and benthic realms are shown on the top. Red color indicates the highest values and blue the lowest. (B) Heatmap with hierarchical clustering of gene clusters related to the major archaeal and bacterial phyla. The clades are shown on the right of the graph, while the depth zones of the pelagic (upper, mesopelagic, and dark ocean) and benthic realms are shown on the top. Red color indicates the highest values and blue the lowest. (C) Sankey plot showing the distribution of the archaeal and bacterial gene clusters among depth zones of the pelagic (upper, mesopelagic, and dark ocean) and benthic realms.
Among Archaea, the phylum Euryarchaeota contributed most to the unique gene clusters across all depth zones and realms with the exception of the dark ocean depth zone where its relative contribution was almost identical to the phylum Nitrososphaerota (6.38% and 6.40%, respectively). This is consistent with previous studies (35, 41) since Nitrososphaerota are mostly chemolithoautotrophs and occur predominantly in the deep ocean, while Euryarchaeota thrive mostly in the photic zone and live heterotrophically, displaying great seasonal and spatial variation and phylogenetic diversity.
For the Eukaryota, gene clusters related to the phylum Metazoa dominated both the upper ocean (43.54%) and dark ocean (78.97%) depth zones, while the clade Fungi dominated the mesopelagic ocean zones, accounting for more than half the number of unique annotated gene clusters (56.7%) (Table 3, Figure 4). The benthic realm showed enrichment of unique annotated gene clusters related to the phylum Chlorophyta (67.75%), followed by the metazoans (11.20%). The enrichment in chlorophyta-related gene clusters of this realm can be attributed to a depositional flux probable of dead photosynthetic organisms from the photic layer, corresponding with the ubiquity of viable photosynthetic organisms in deep sea waters (42) as well as microphytobenthos in the shallow coastal sediments sampled.
Table 3
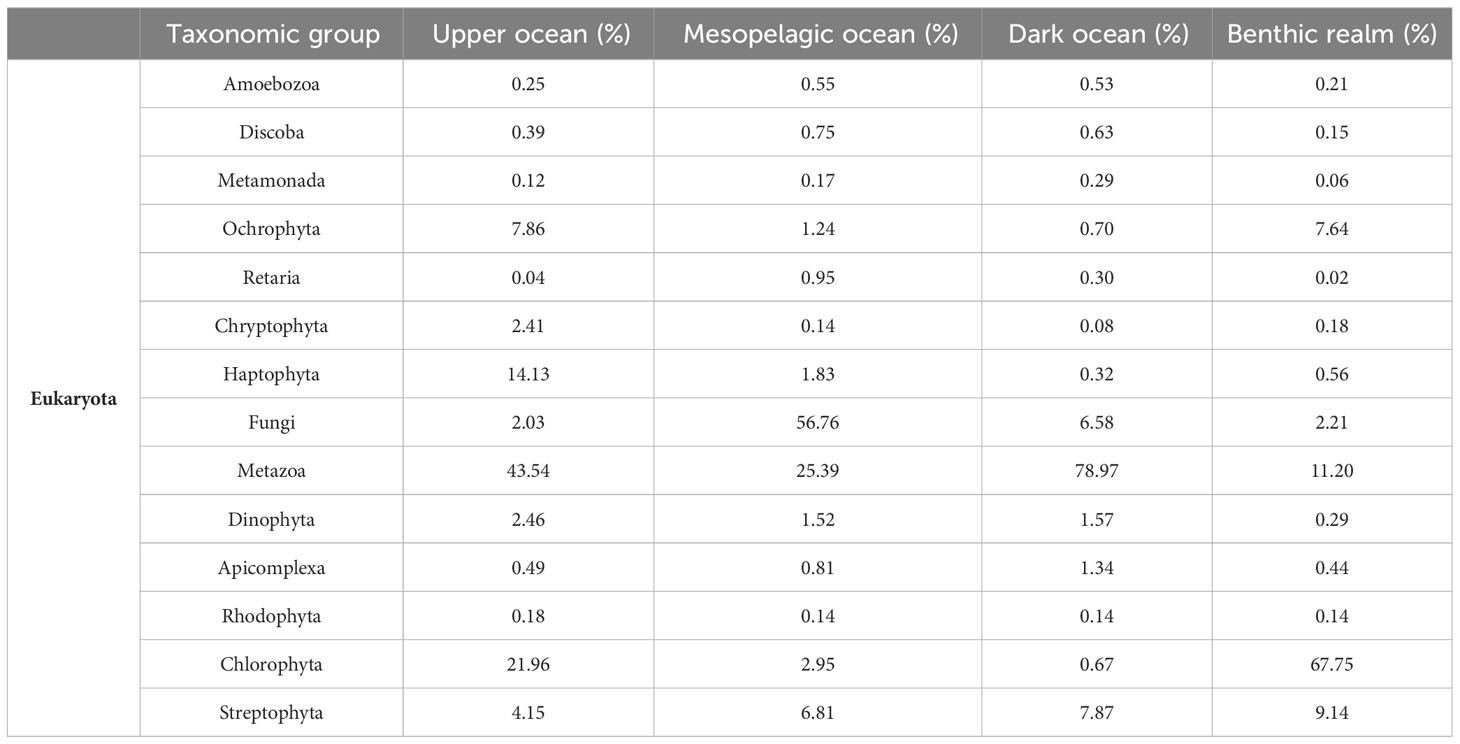
Table 3 Relative (%) contribution of the eukaryotic taxonomical groups to the gene clusters.
Figure 4
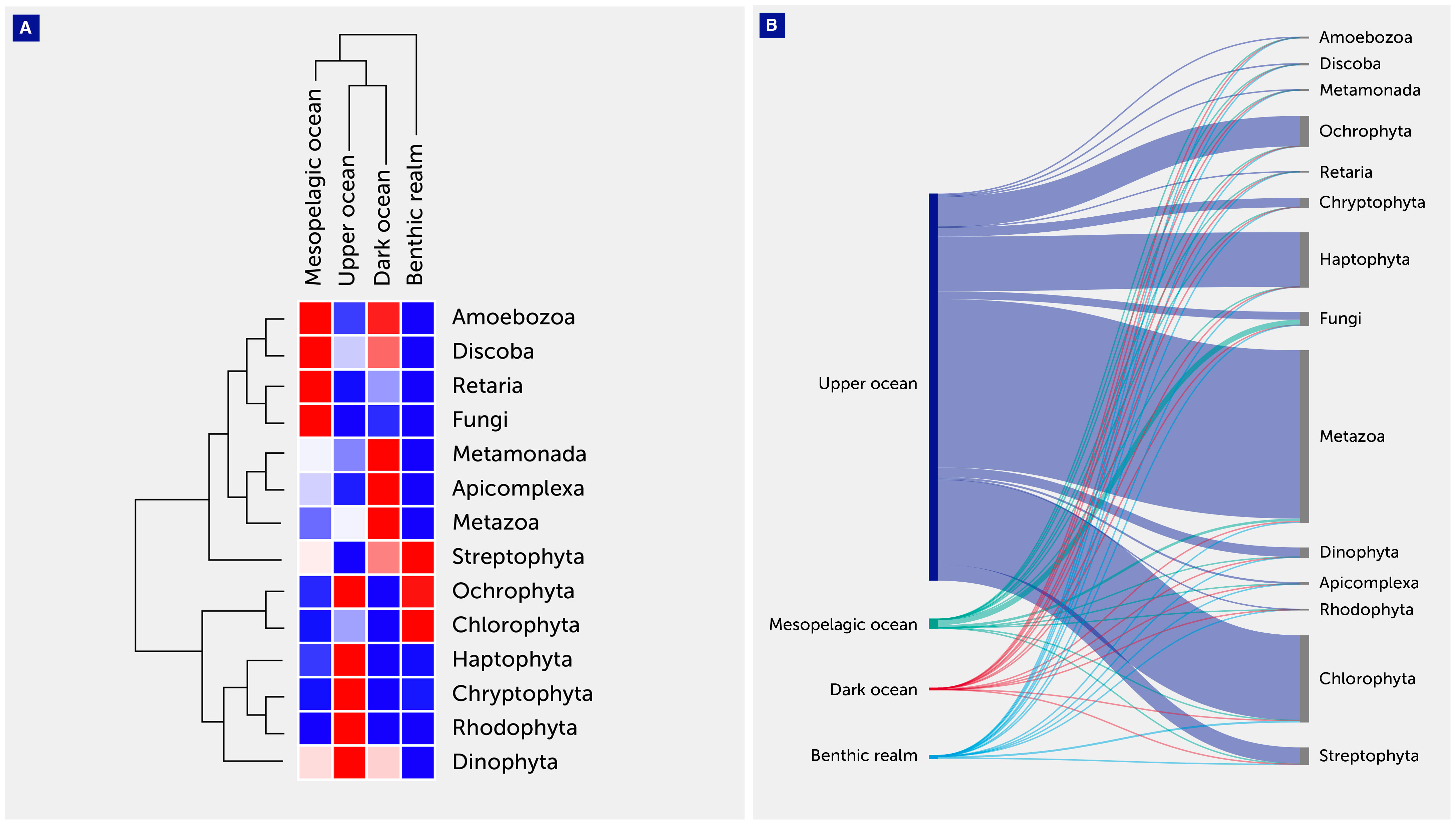
Figure 4 Eukaryotic annotated gene cluster contribution. (A) Heatmap with hierarchical clustering of gene clusters related to the major eukaryotic clades. The clades are shown on the right of the graph, while the depth zone of the pelagic (upper, mesopelagic, and dark ocean) and benthic realms are shown on the top. Red color indicates the highest values and blue the lowest. (B) Sankey plot showing the distribution of the eukaryotic gene clusters among depth zones of the pelagic (upper, mesopelagic, and dark ocean) and benthic realms.
Overall, the ratio of unique annotated genes from Bacteria to Archaea to Eukaryota to viruses in the KMAP Global Ocean Gene Catalog 1.0 was 22.65: 1.76: 3.53: 1 (Table 4), with the benthic realm and the dark ocean being highly enriched in Bacteria and Archaea gene clusters and the upper ocean being highly enriched both in Eukaryota and Bacteria gene clusters. The upper ocean also presented the highest relative abundance of viral gene clusters among all depth zones and realms. This overall ratio of unique annotated genes implies that Bacteria are the largest contributors to unique genes in the ocean, followed by Eukaryota and Archaea. We note, however, that viral genes are underestimated, as only DNA viruses are included in the catalog. For comparison, the ratio of ocean biomass, derived from total ocean biomass estimates from the work of Bar-On and Milo (2019) (43), is 50: 10: 150: 1 for Bacteria to Archaea to Eukaryota to Viruses in the ocean. The ratio of biomass across Bacteria to viruses does not reflect that of unique genes, showing that the contribution of eukaryotes to ocean biomass is disproportionately higher than that to marine genetic diversity, which is dominated by Bacteria. This rough comparison also shows that viral genomes contain far more innovation than hitherto realized, even considering that this DNA-based assessment of the global ocean genome omits RNA viruses. While it is recognized that DNA viruses modulate microbial diversity, populations, and microbially mediated processes, the diversity and role of RNA viruses are not well studied. Recently reported to be prevalent and likely functionally significant, these represent an important emerging area of exploration in the global ocean genome (44).
Table 4
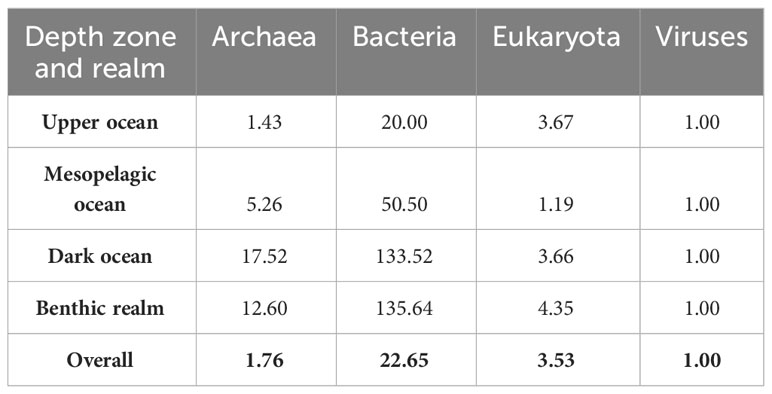
Table 4 Ratios among annotated gene clusters belonging to each taxonomical domain for the benthic realm and each pelagic realm depth zone (upper ocean, mesopelagic ocean, and dark ocean).
Functional distribution of genes
From a functional point of view, gene clusters were grouped into the main relevant Kyoto Encyclopedia of Genes and Genomes (KEGG) categories: Metabolism (KEGG 09100), for gene clusters coding for proteins involved in metabolic processes, Genetic Information Processing (KEGG09120), Environmental Information Processing (KEGG09130), and Signaling and Cellular Processes (KEGG 09183) (Figure 5).
Figure 5
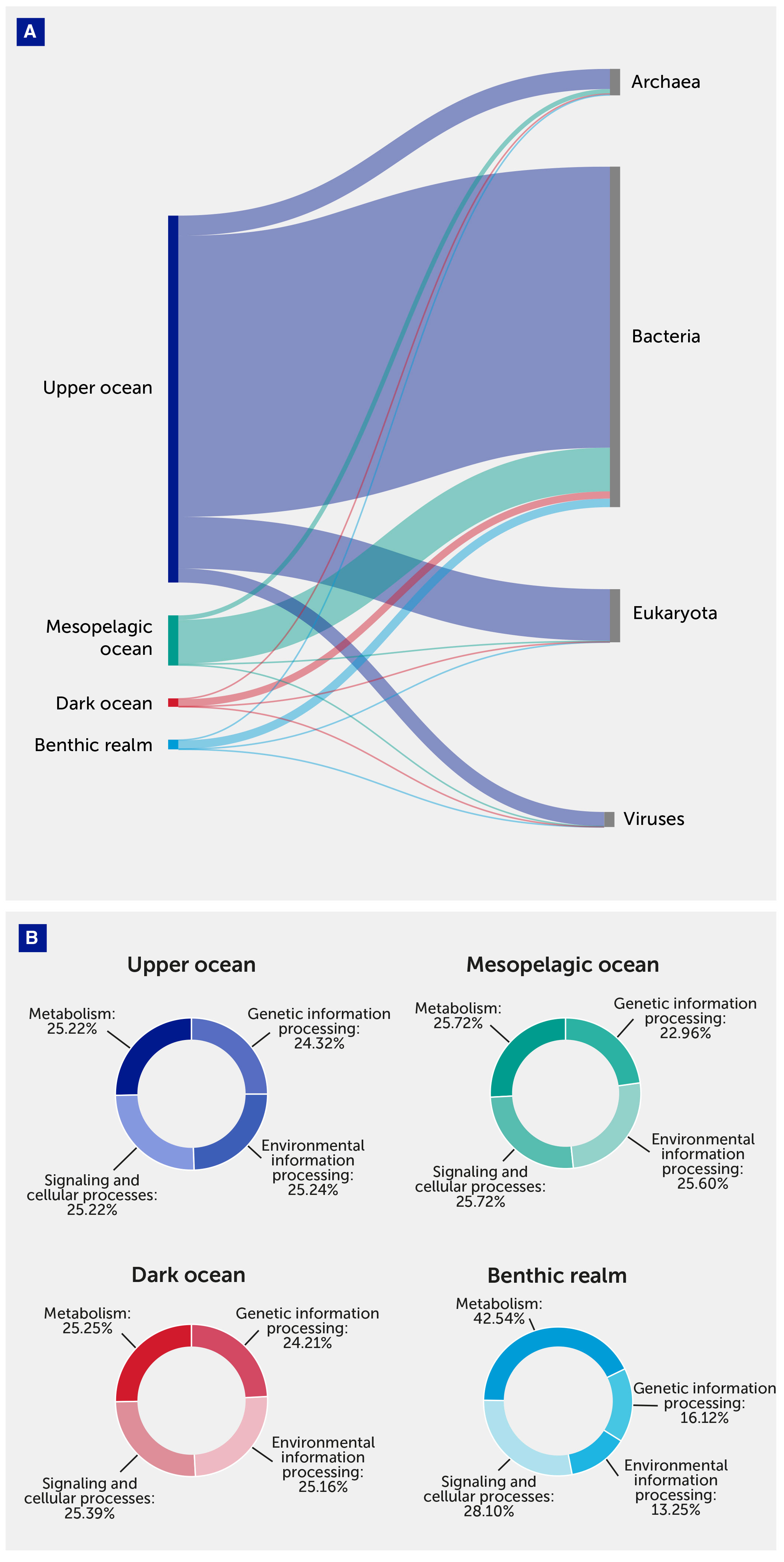
Figure 5 Annotated gene cluster number distribution. (A) Annotated gene cluster distribution between depth zones of the pelagic (upper, mesopelagic, and dark ocean) and benthic realms and taxonomical domains. (B) Annotated gene cluster distribution between depth zones of the pelagic (upper, mesopelagic, and dark ocean) and benthic realms and functional Kyoto Encyclopedia of Genes and Genomes (KEGG) Brite categories.
The benthic realm was enriched by unique gene clusters coding for metabolic processes (KEGG 09100): these represented 42.54% of the total annotated unique gene clusters in this realm compared with about 25% of those in pelagic communities. In contrast, benthic communities were relatively depleted in unique gene clusters coding for Genetic Information Processes (KEGG 09120) and Environmental Information Processing (KEGG 09130) (Figure 5).
Given the importance of microbial metabolic processes in shaping the ocean in every aspect, we assessed the unique gene clusters involved in the most important microbial metabolic pathways of the main elements involved in the formation of climate-active molecules: carbon, nitrogen, and sulfur (Supplementary Table S1). Overall, genes related to acetate utilization and carbon monoxide (CO) oxidation were the predominant unique gene clusters in Bacteria and Archaea (weighted average values, Table 5, Figure 6). In contrast, anaerobic ammonium oxidation and methane oxidation, very important processes for biogeochemical cycles and climate regulation carried out by specific microbial taxonomic groups, were supported by a restricted set of unique gene clusters across the global ocean genome.
Table 5
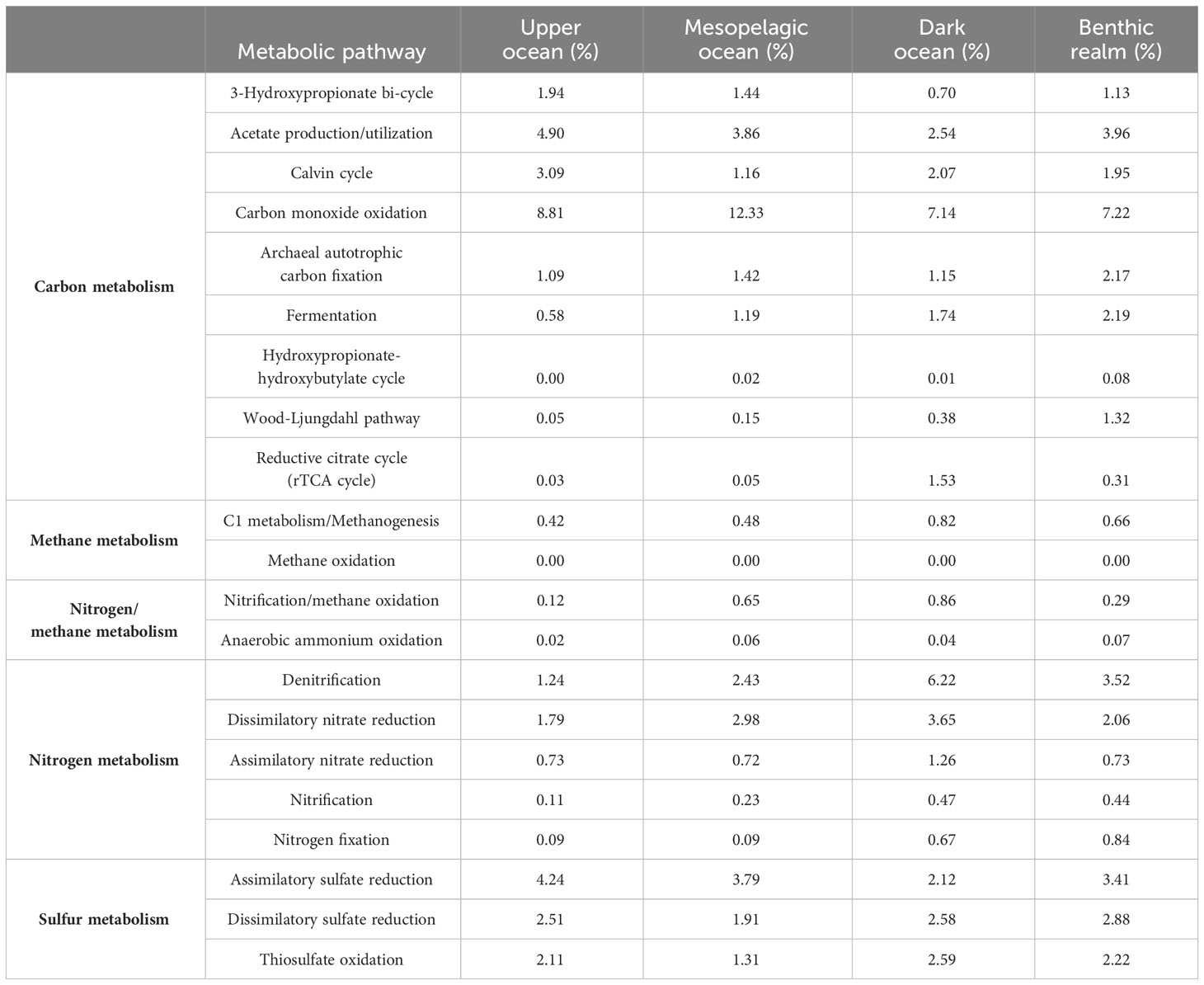
Table 5 Relative abundance (the weighted median proportion of gene clusters relative to important selected metabolic pathways); the KEGG Orthology identifiers related to each metabolic pathway are available in Supplementary Table S1.
Figure 6
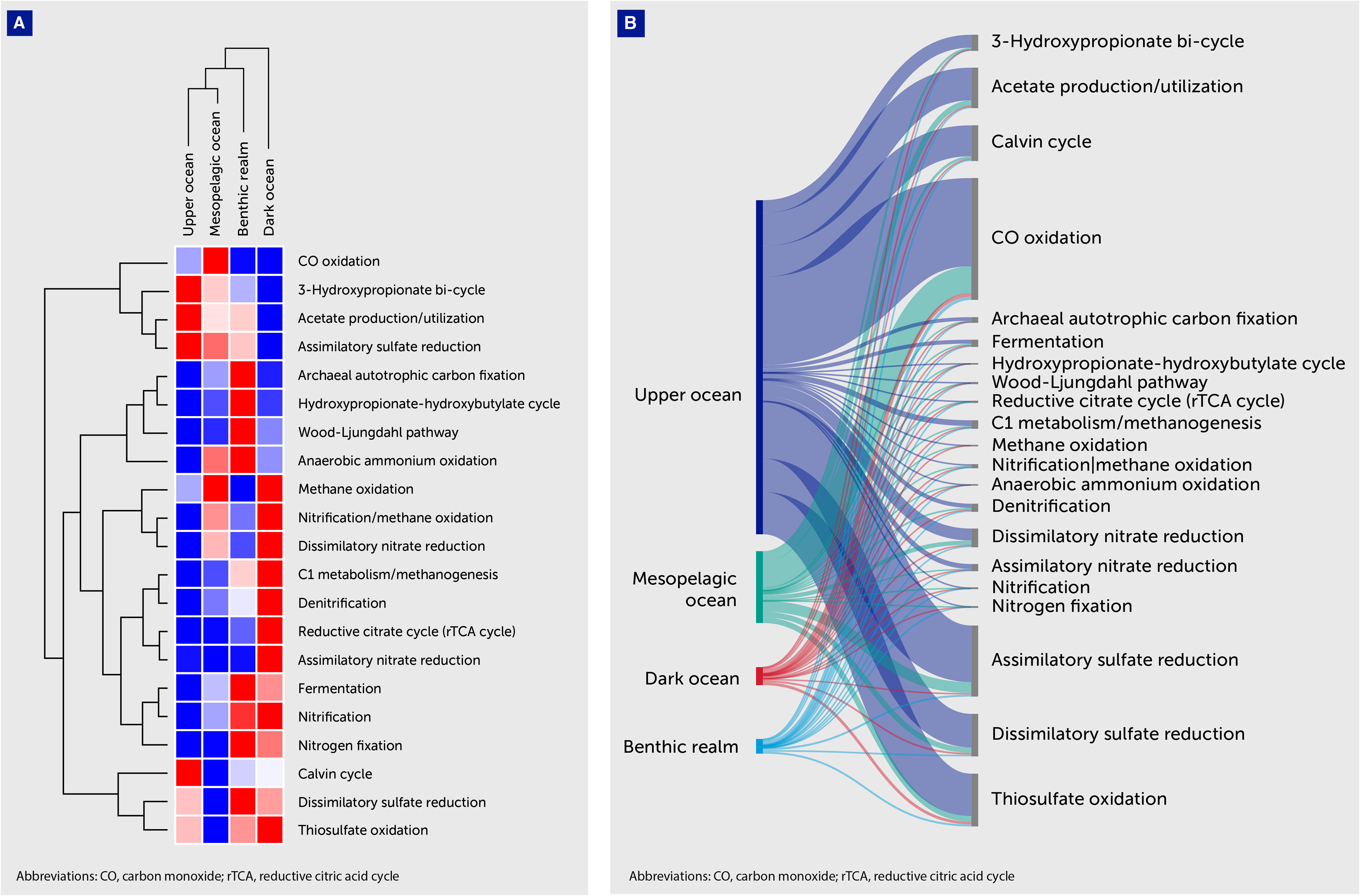
Figure 6 Distribution of average gene abundances related to microbial metabolism. (A) Heatmap with hierarchical clustering of average gene abundances related to microbial metabolic pathways (Table 5). Metabolic pathways are shown on the right of the graph, while the depth zone of the pelagic (upper, mesopelagic, and dark ocean) and benthic realms are shown on the top. Red color indicates the highest values and blue the lowest. (B) Sankey plot showing the distribution of average gene abundances related to prokaryotic metabolic pathways. Gene clusters have been classified according to the depth zone of the pelagic (upper, mesopelagic, and dark ocean) and benthic realms (shown on the left of the graph). The KEGG Orthology identifiers related to each metabolic pathway are available in Supplementary Table S1.
Unique gene clusters related to CO oxidation dominated all depth zones along with acetate utilization ones. Average gene abundances related to CO oxidation ranged from 7.14% in the dark ocean to 12.33% in the mesopelagic ocean, while for acetate utilization the peak abundances were in the upper ocean (4.90%) and in the benthic realm (3.96%), indicating that these processes play an important role in carbon cycling dynamics.
Assimilatory sulfate reduction ranked third in the upper ocean, coupled with a 2.51% relative average abundance of dissimilatory sulfate-reduction-related genes. We observed a notable relative average abundance of genes of the carbon metabolism-related 3-hydroxypropionate bi-cycle, with peak contribution values in the upper ocean (1.94%). The benthic realm showed remarkable contributions to carbon metabolism by the Wood-Ljungdahl pathway (1.32% average gene abundance). Denitrification-related genes showed notable representation in the depths of the ocean, accounting for a 6.22% and a 3.52% average abundance in the dark ocean and the benthic realm, respectively.
Discussion
The KMAP Global Ocean Gene Catalog 1.0 consists of 308.6 million gene clusters, of which 53.4% (164.8 million) have been annotated. This represents remarkable progress compared with the Global Ocean Microbial Reference Gene Catalog (OM-RGC), released in 2015 and based on the Tara Oceans expedition, which included >40 million non-redundant representative genes from viruses, Archaea, Bacteria, and picoeukaryotes (25). Tara Oceans’ sequencing efforts were focused on the upper ocean, providing a global assessment. In 2022, the Global Microbial Gene Catalog (GMGCv1) was released, and it includes 88 million 95% non-redundant annotated genes belonging to marine organisms within the 303 million species-level genes from 14 different habitats (30). Hence, the KMAP Global Ocean Gene Catalog 1.0, which focuses on pelagic and benthic marine habitats, represents an important tool for future comparative studies.
Out of the 2,102 marine metagenomes analyzed, only 86 were sampled from benthic communities, accounting for less than 5% of the total metagenomes, while only 215 out of the 2016 pelagic metagenome samples (10.2%) were sampled from the dark ocean, the largest habitat on Earth.
Marine metagenomics efforts have largely focused on the upper pelagic ocean, the most easily accessible habitat, while benthic and deep-sea environments remain grossly under-sampled in comparison, suggesting that a large pool of genes of marine organisms may be discovered in future efforts targeting benthic and deep-sea environments. However, in contrast to pelagic metagenomics, which has been conducted at global scales by expeditions such as TARA Oceans and Malaspina (26), no systematic attempt to achieve global coverage for benthic metagenomics is available, thereby precluding a deeper classification by major domains of the benthic environment. Recent efforts have targeted and improved the understanding of benthic habitats, some at a regional level and some on a global scale. Dombrowski et al. (45) examined benthic metagenomes from hydrothermal sediments in the Guaymas Basin (Gulf of California), and Langwig et al. (46) reconstructed metagenome-assembled genomes (MAGs) from metagenomes of marine sediments in hydrothermal vents from Guaymas Basin, Gulf of California, and a coastal site in Mesquite Bay, Texas.
Global studies have focused mostly on the recovery of MAGs and provide some insights into benthic diversity. A study by Parks et al. (47), using more than 1,500 public metagenomes, increased the phylogenetic diversity of bacterial and archaeal genome trees by over 30%; this was improved by Nayfach et al. (48), where MAGs were recovered from more than 10,000 publicly available metagenomes from different terrestrial and marine environments and hosts. Publicly available metagenomes were used for these two global scale studies; however, mining metagenomes from public repositories can be a difficult task due to unavailable or incomplete associated metadata. Metadata problems contribute to the underutilization of publicly available metagenomes. Thus, recent efforts tried to offer standardized metadata in public repositories in combination with a user-friendly interface. Planet Microbe (49) is a web-based portal for the open sharing and discovery of past and ongoing oceanographic sequencing efforts. It offers integration of their omics data with their in situ environmental data and information about sampling events, sampling stations, and additional metadata about the cruise tracks, protocols, and instrumentation used. The first release of Planet Microbe included more than 2,000 aquatic samples collected from multiple projects; the majority of samples in this database release are from major global expeditions such as Tara Ocean, Hawaii Ocean Time-series (HOT), and Ocean Sampling Day (OSD). Owing to the sampling strategies of the projects included, most of the samples are pelagic and mostly from surface waters; they did not provide deep insight in the benthic realm. MarineMetagenomeDB is another example of a public repository, created by Nata’ala et al. (50), trying to address the previously mentioned limitations of metagenome-associated metadata. Its objective is to improve the contextualization and ecological interpretation of marine microbial metagenomes, allowing comparison with novel datasets and use in meta-analysis studies. Tools like MarineMetagenomeDB and Planet Microbe can boost efforts focused on better understanding differences among benthic and pelagic realms, leading to works that go beyond the realm or regional scale.
The unique challenges to life in benthic environments imply that both their taxonomic composition (51) and metabolic processes (27) are likely to differ greatly from those in the upper ocean. These differences do not only relate to the depth of the water column but also the variability in microbial communities across the deep ocean (52)—this being a far less homogeneous environment than it was once thought to be. In particular, the benthic compartment, characterized by steep biogeochemical gradients, is likely to contain a large reservoir of undiscovered genes and functions. Moreover, benthic habitats are at risk, with 75–90% of coral reefs predicted to be lost due to climate change even if the most ambitious goals of the Paris Agreement (53) are reached. Despite accounting for less than 0.1% of the total surface (54), coral reefs are biodiversity hotspots; they contribute an estimated US$2.7 trillion per year in ecosystem services (55) and contain a rich microbiome the membership and functions of which are currently the subject of much research (56).
The risks extend even to the deep sea, which has recently been identified as the reservoir holding about 95% of all the synthetic plastics that have ever entered the ocean (57). Along with other pollutants, these plastics may have disrupted deep-sea microbial communities. Likewise, deep-sea mining may also alter benthic habitats, with unpredictable consequences on their functionality and diversity. The abundance of polymetallic nodules in some areas of the deep sea has raised interest in mining these resources to meet the growing needs from electronics and electrification (58). Given the threats to this habitat, developing a baseline understanding of the genome of benthic microbial communities is a matter of urgency.
Whereas biogeochemical cycles in ocean domains have received great attention, this understanding has lagged behind a comparable understanding of its genomic basis for decades. In fact, only the advent of shotgun sequencing two decades ago has allowed this gap in our understanding to be bridged by enabling metagenomics.
The analysis of the architecture of the global ocean genome presented here allows us to match biogeochemical cycling with microbial taxonomy through the taxonomic assignment of the unique genes supporting the various metabolic processes presented in this paper. Hence, resolving the global ocean genome can provide a deeper understanding of the complexity of microbial metabolic capabilities across ocean environments, which should be expanded further in the future. The metagenomic samples corresponding to the data for the ocean gene catalog released here do not discriminate between living or active organisms and inactive or dead ones (59, 60), as it reflects the total pool of genes present in the ocean. Future efforts should strive to distinguish the active or living component from the inactive or dead component, although resolving active versus dead organisms from metabarcoding or metagenomics is not yet straightforward.
The long evolutionary history of the global ocean genome and the diversity of environmental conditions across ocean habitats are reflected in the diversity of present metabolic pathways, such as those involved in carbon metabolism. The range of non-photosynthetic carbon fixation pathways supported by Bacteria and Archaea is impressive and highly specialized: the dicarboxylate/4-hydroxybutyrate cycle, for example, seems to play an important role in the benthic realm, with its combination anaerobic metabolic modules leading to an efficient carbon dioxide (CO2) fixation mechanism.
The upper ocean was rich in genes related to carbon-related metabolic pathways, the Calvin cycle, CO oxidation and acetate utilization, and a sulfur-processing pathway for assimilatory sulfate reduction (Figure 6). In contrast, the benthic realm was rich in genes related to alternative carbon fixation pathways, such as the hydroxypropionate-hydroxybutylate cycle, along with fermentation- and denitrification-related genes. The dark ocean was rich in genes related to denitrification, as in the benthic realm, along with ones related to the reductive citrate cycle and dissimilatory nitrate reduction. The high respiratory activity measured in the dark ocean is difficult to reconcile with the rates of supply of organic carbon produced in the photic layer (39, 61). Recently, it has been suggested that chemolithoautotrophic and mixotrophic organisms, which combine chemolithoautotrophic inorganic carbon fixation and heterotrophy, play a more important role in driving deep-sea metabolism than previously thought (27, 62–64). Indeed, it has been calculated that dark carbon fixation processes contribute a significant fraction of oceanic primary production (65, 66).
Regarding carbon metabolism and in particular CO oxidation, metagenomic analyses of the Sargasso Sea (67, 68) revealed that a significant number of cox genes have proven to be abundant in environmental samples. CO dehydrogenase (CODH) and acetyl-CoA synthase (ACS) serve as pivotal catalysts for growth when utilizing CO. CODH facilitates the oxidation of CO into CO2, yielding low-potential electrons that are essential for cellular processes. Alternatively, CODH can reduce CO2 to CO. When this latter reaction is coordinated with ACS, it creates a system for producing acetyl-CoA from CO2, enabling the synthesis of cellular carbon compounds (69). CO oxidation has been linked to various bacterial groups, including Actinobacterota, Proteobacteria, Bacteroidota, and Chloroflexota phyla. Remarkably, CO oxidation by cox genes is widely distributed in all depth zones, including the dark ocean (27). This high prevalence suggests that CO oxidation serves as a significant energy source for heterotrophic organisms in the deep ocean.
The finding of a relevant presence of genes involved in dissimilatory sulfate reduction in the upper ocean may seem surprising at first, as these are expected to occur in anoxic environments. However, molecular and physiological studies confirm that oxygen minimum zones support pelagic microorganisms capable of utilizing inorganic sulfur compounds to support their energy metabolism, where sulfide products are immediately oxidized and, therefore, do not accumulate in the water column (70–72). The average gene frequency of dissimilatory sulfate-reduction-related genes in the pelagic realm of 1.9–2.88% (Table 5) is conservative based on the estimate that oxygen minimum zones currently constitute 1–7% of the volume of the ocean (73). In contrast, the proportion of assimilatory sulfate-reduction gene clusters showed its peak in the upper ocean; it accounted for an average gene frequency of 4.24%, showing a decreasing contribution towards the deeper depth zones, as expected from the fact that assimilatory sulfate reduction supports sulfur cycling in many organisms, whereas dissimilatory sulfate reduction is expected to be limited to low oxygen environments. A review of metagenomes from pelagic microbial communities from oxygen minimum zones shows that these support an active sulfur cycle with potentially substantial roles in organic carbon input and mineralization and critical links to the nitrogen cycle (70). Moreover, organic aggregates have been shown to provide microenvironments that have demonstrated an ability to support sulfate reduction in the ocean water column (73, 74), which helps explain the prevalence of genes participating in these metabolisms in the water column at different depth zones.
Bacteria and Archaea residing within the oxygenated water column of the deep ocean utilize an array of diverse electron donors, including hydrogen, thiosulfate/sulfide, and ammonia, as sources of energy to fuel different metabolic processes, such as nitrification and denitrification. These reduced inorganic compounds play a crucial role in supporting microbial metabolism in the dark ocean (27).
Variations between habitats in the abundance of unique genes contributing to different classes of metabolic processes highlight the existence of distinct metabolic niches in the ocean. Indeed, these probably exist at finer levels of spatial resolution than those presented here. Likely, two key processes involved in nitrogen cycling play fundamental ecological and biogeochemical roles in the ocean (nitrogen fixation and anaerobic ammonium oxidation); they are supported by a very limited set of genes and microbial taxa compared with the diversity of metabolic pathways processing carbon, identifying evolutionary bottlenecks in key metabolic processes.
Although nitrogen fixation is thought to have been restricted to the upper ocean owing to limited reactive nitrogen, our analysis shows an opposing pattern of related gene clusters, with an abundance of nifH genes in deeper depth zones. Indeed, recent analyses identify nitrogen fixation associated with sinking particles in the dark ocean (75) and heterotrophic nitrogen-fixing bacteria in the dark ocean to be associated with organic matter-rich particles (27, 76), which provide substrate as well as microenvironments sheltered from oxygen inhibition of nitrogen fixation (76). However, the significance of nitrogen-fixing bacteria in the dark ocean for water column nitrogen fixation remains to be quantified (77). Indeed, nitrogen fixation is expected to be selected against in the dark ocean, where reactive nitrogen forms are widely available, owing to the prevalence of remineralization of organic matter, while the energy required to fix nitrogen is a limiting resource.
The investigation of the global ocean genome has been a long-standing goal of microbial oceanography, limited by sequencing costs and access to advanced sequencing platforms. These disadvantages are on the verge of being overcome through a recent breakthrough in the sequencing era: the development of miniaturized high-capacity sequencing systems, such as MinION™ (Oxford Nanopore Technologies). These portable systems enable sequencing to be performed on board oceanographic vessels, shortening processing times and increasing reliability in terms of quality and output (78). However, the remaining bottleneck is the need for computational power, as metagenomics datasets keep growing and demand both advanced bioinformatics skills and ongoing access to high-performing supercomputers to align and annotate sequences. For instance, 48.09% of the gene clusters identified here could not be annotated, requiring continuous efforts to blast this large pool of over 150 million gene sequences against newly deposited sequences to functionally characterize them. One factor here is the additional clusters we obtained by applying length overlap (80%) cutoff, as recommended previously (79), to avoid merging shorter genes with bigger ones. This filter was not applied in the OM-RGC derived from the Tara Oceans expedition (30) in which almost 30% of clusters originally remained unannotated despite there being a smaller number of final clusters than in the present analysis (25). Hence, improvements in gene prediction frameworks are needed, together with continuous re-analysis to match these unassigned genes with newly released genes. Efforts to further sequence environments that are underrepresented in the current KMAP Global Ocean Gene Catalog 1.0 must be supported by the development of platforms allowing convenient and open access to computational resources, enabling the exploration of the catalog, as KMAP does (23). Further novel methods are needed to expand our knowledge regarding the non-annotated portion, for example, employing structural aspects given recent developments of AlphaFold2 (80).
Cataloging the genome of the global ocean is a work in progress and will remain so for decades to come. Maintaining the effort, initiated in 2004 (18), to develop a full inventory of marine genomic diversity requires a clear understanding of the benefits to be derived (19). These are many, starting from a basic understanding of the taxonomic structure and diversity of marine communities, ranging from viruses (81) to Archaea and Bacteria (82, 83) and eukaryotes (84, 85), free of the biases introduced by amplicon sequencing (86), as well as accelerating the full genomic description of unculturable marine microbes through the assembly of high-quality metagenome-assembled genomes (87, 88). Functional analyses of annotated genes can help assemble the metabolic architecture of different oceanic environments (27) and elucidate the biogeochemical role of microbial communities (89). Combining taxonomic and functional annotation allows specific roles in biogeochemical processes to be assigned to specific taxa (90)—a fundamental task that has hitherto remained challenging. Functional metagenomics can also probe the functioning of entire microbiomes, as demonstrated by the major effort to resolve the properties of the human gut microbiome (91), therefore describing links between microbial consortia. Biosynthetic gene clusters, cooperating to yield complex molecules, can also be predicted from metagenomics, thereby offering a bridge between genomics and metabolomics, which is currently underexplored in the context of the global ocean genome.
However, future efforts need to be guided by the gap analysis conducted here, targeting under-sampled deep-sea and benthic environments. The exploration of the global ocean genome cannot be based on the simple aggregation of a growing number of samples but instead must be guided by specific hypotheses, targeting specific processes and/or taxa of interest. This study, therefore, does not represent a comprehensive analysis of benthic microbial life owing to the low number of metagenomes and sampling sites included, but it can provide important insights to help formulate hypotheses guiding future efforts.
A question-driven approach, as represented by the KMAP Global Ocean Gene Catalog 1.0, will require broad collaboration since the number of relevant questions is potentially very large and can come from different perspectives, including evolutionary, biogeochemical, ecological, and industrial prospection. Moreover, future efforts must aim to reliably describe the genetic repertoire of a community, as recently shown by a replicated sequencing of a Red Sea community that greatly multiplied the number of sequences retrieved and thereby yielded a far larger gene catalog than those previously retrieved by conventional sequencing (92). Such efforts continue to bring novelty to gene catalogs, including at high levels such as gene families (92). Indeed, next-generation sequencing metagenomics yields 14.7 million unique genes per Tera base-pair sequenced across projects (92), so probing into the genome of the rare ocean biosphere (93) will require enhanced sequencing efforts.
A deeper dive into the genome of the ocean should also include a deeper insight into functional diversity at the ecotype level, examining the functional consequences of relatively minor genomic differences at the genus level, such as those demonstrated for the unicellular cyanobacterium Synechoccocus (94) and the former Prochlorococcus, which is now differentiated into five genera (Prochlorococcus, Eurycolium, Prolificoccus, Thaumococcus, and Riococcus) with distinct genomic and ecological attributes (95). Recent developments are unveiling the role of specific genes in coding for polyfunctional enzymes, which may participate in multiple processes and connect otherwise independent elemental cycles (96). A recent example is the PhoA enzyme; kinetic assays revealed that it exhibits not only the predicted P-monoester activity but also P-diesterase, P-triesterase, and sulfatase activity (97).
Probing deeper into the global ocean genome is vital, because it is the foundation upon which the biodiversity and functionality of all marine ecosystems rest, supporting their resistance and resilience to pressures. Functional redundancy is a foundation of the resilience of ecosystems to disturbance, allowing adaptation to environmental changes and pressures. Indeed, functional redundancy and complementarity are probably the keys to understanding the huge diversity of plankton communities, which has represented a long-standing paradox in ecology. Functional redundancy allows the overall ecosystem functionality to be preserved even in case of stochastic impacts leading to loss of taxonomical groups.
The exploration of the global ocean genome delivers additional findings, such as the dominance of the clade Fungi in the mesopelagic ocean zone, where it accounted for more than half the number of unique annotated gene clusters. This adds to studies that show fungi play an important role in microbial diversity in the mesopelagic ocean (98) and suggests that this diversity may carry important functional consequences. For instance, a recent paper (99) analyzed global metagenomic and metatranscriptomic data covering all major oceanic basins to shed light on fungal peptidase activity, reporting that both total and secretory peptidase genes and transcripts (and the percentage of secretory), as well as the α-diversity of fungal protease genes and transcripts, were higher in the mesopelagic than in the upper water layers. Peptidases are the main enzyme family responsible for cleaving proteins, which constitute a major fraction of marine living and detrital biomass. Based on the expression of carbohydrate-active enzymes (CAZymes) related to this group, Breyer and colleagues hypothesized that fungi are also major contributors to the degradation of proteins in the oceanic water column (99).
Importantly, the genome of the ocean is not a static property but a dynamic one subject to continuing evolution—now forced by human-induced changes in the marine environment, including pollution by plastics and other synthetic pollutants, climate change, and the associated warming, acidification, and deoxygenation. For example, the introduction of synthetic compounds into the ocean may be driving evolutionary processes in microbes to enable their use, as described recently (100, 101). The proliferation of antibiotic resistance genes in marine bacteria (102) also emphasizes the dynamic nature of the ocean genome and its connectivity to changes occurring in other biomes, including the human-dominated environment via sewage and waste (103). Understanding these dynamics is of critical global importance given the foundational importance of the ocean genome to biodiversity and biogeochemical processes.
In conclusion, the taxonomic and metabolic underpinnings of the global ocean genome reported here represent an important step toward integrating marine metagenomic data into an overview of the taxonomic and metabolic distribution of different ocean compartments on a global scale. Understanding the taxonomic basis of metabolic capabilities is a foundation to help predict the functional consequences of ongoing changes in community structure and biodiversity in the ocean. It could also provide a novel perspective to predict the functional consequences of changes driven by climate change, including ocean warming, deoxygenation, and acidification. We also identify gaps, such as benthic metagenomics, where additional effort is required to provide a comprehensive global understanding of the metabolic architecture of the ocean.
Propelling investment into this research will require the resolution of current uncertainties in the ownership of intellectual property derived from the exploration of the ocean genome and the development of a just and equitable framework for benefit sharing (11, 104). On 4 March 2023, the Agreement under the United Nations Convention on the Law of the Sea on the conservation and sustainable use of marine biological diversity of areas beyond national jurisdiction (105) represented a long-awaited milestone decision to share benefits derived from marine genetic resources and delivered a key mechanism of reporting. However, this agreement also poses a new risk: slowing investments in marine genetic resources because of reduced incentives for those making discoveries. This risk comes at an especially crucial time when sequencing technologies are low in cost and efficient, and there is potential for integration with artificial intelligence, potentially prompting unprecedented rates of discovery. Advancing research on the global ocean genome will accelerate the development of blue biotechnology and advance problem-solving across a broad range of sectors, including biomedicine, food, and energy.
Methods
We processed a large number of metagenomic samples available from the European Nucleotide Archive (ENA; www.ebi.ac.uk/ena) as of May 2018, from quality control of reads through to assembly of metagenomes into KMAP, as described in Alam et al. (23). Briefly, using the advanced search function in the ENA, we created a list of FASTQ files for metagenomes, restricting the taxonomy to metagenomes and the shotgun sequencing platform to paired-end Illumina sequencing technology, with a nominal length of >100 base pairs (bp) and a base count higher than 2×108.
The resulting metadata file was filtered for availability of FTP location to download FASTQ files. We downloaded 2,102 metagenomes using wget and GridFTP, also collecting the ENA run, sample, project, and metagenome taxon identifiers. After metadata verification, the 2,102 metagenomes were classified according to their geographical sampling location, depth zone, and realm. The full list of metagenomes analyzed in this study is reported in Supplementary Table S2, including the above-mentioned classification.
Geographically, we categorized the metagenomes into the Atlantic, Pacific, Indian, Arctic, and Southern Oceans; we also included the Mediterranean Sea owing to its peculiar characteristics. We classified the metagenomes from pelagic realm samples into three depth zone categories: the upper ocean, for metagenomes obtained from samples taken between 0 and 200 m, mesopelagic ocean (200–1000 m), and dark ocean (>1000 m). Metagenomes belonging to the benthic realm were not subcategorized owing to their low number.
Upon download, we pre-processed the individual samples for quality control and validation of the pairs using bbduk (http://jgi.doe.gov/data-and-tools/bb-tools/). Assembly and annotation were performed using the KAUST Shaheen II supercomputing infrastructure. Assembly used the MegaHit assembler (106) (final contig size limited to 500 bp) with default options. Complete genes were predicted using Prodigal (107), maintaining a minimum length of 100 bp. Gene sequences were clustered using MMseq2 (108) considering a global sequence identity cutoff of 90% and minimum gene length difference of 80%. Annotation was performed using KMAP (www.cbrc.kaust.edu.sa/kmap). Protein translation of predicted genes was compared using a basic local alignment search tool (BLAST) to Universal Protein Knowledgebase (UniProtKB) to obtain annotations such as taxonomic affiliation, generic functional descriptions, and cross-references, e.g., Cluster of Orthologous Genes (COGs) database.
The taxonomy used in the paper corresponds to that of the National Center for Biotechnology Information (NCBI), confirmed by NCBI’s taxon report checking tool (https://www.ncbi.nlm.nih.gov/Taxonomy/TaxIdentifier/tax_identifier.cgi) (access date: 11 April 2023) and does not capture revisions in some taxa since that date (50, 109). Microbial taxonomy is a very dynamic field and will continue to evolve, requiring an update, in a future release of the Ocean Genome Catalog, when additional data will also be available.
Another BLAST to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database was performed to obtain more specialized functional annotation, given the wide coverage of gene functionalities within this database. All annotations are saved in a Gene Information Table (GIT) containing 19 fields of annotations (for details see: www.cbrc.kaust.edu.sa/aamg/docs/KMAP_Documentation.html).
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsci.2023.1038696/full#supplementary-material
Acknowledgments
We thank the KAUST Supercomputing Laboratory (KSL) for providing access and support to the computer resources required to carry on this work. We thank the late Prof. Vladimir B. Bajic for leading the development of the KMAP used in this study and Allan Anthony Kamau for his contribution to developing it.
Statements
Author contributions
EL: Writing – original draft, Writing – review & editing, Conceptualization, Formal Analysis, Investigation, Validation, Visualization. IA: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Software, Validation. MU: Writing – original draft, Writing – review & editing, Data curation, Formal Analysis, Investigation, Resources, Software, Validation. TJ: Writing – original draft, Writing – review & editing, Formal Analysis, Visualization. SA: Writing – original draft, Writing – review & editing, Funding acquisition, Investigation. TG: Writing – original draft, Writing – review & editing, Data curation, Investigation, Funding acquisition, Resources, Software. SGA: Writing – original draft, Writing – review & editing, Funding acquisition. JG: Writing – original draft, Writing – review & editing, Formal Analysis, Funding acquisition, Investigation. CD: Writing – original draft, Writing – review & editing, Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization.
Data availability statement
The datasets presented in this study can be found in online repositories. This data can be found here: KMAP Global Ocean Gene Catalog 1.0, https://www.cbrc.kaust.edu.sa/aamg/GOGC.1.0/.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by KAUST through competitive center funding provided to the Computational Bioscience Research Center.
Conflict of interest
The authors declare that the research was conducted in the absence of financial relationships that could be construed as a potential conflict of interest.
The handling editor BB declared a shared consortium IMG/M Data Consortium with the author SGA at the time of review.
The authors IA, SA, TG, CD declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Costanza R. The ecological, economic, and social importance of the oceans. Ecol Econ (1999) 31(2):199–213. doi: 10.1016/S0921-8009(99)00079-8
2. Eakins BW, Sharman GF. Volumes of the World's oceans from ETOPO1 Boulder, CO: NOAA National Geophysical Data Center (2010).
3. Mißbach H, Duda JP, Van Den Kerkhof AM, Lüders V, Pack A, Reitner J, et al. Ingredients for microbial life preserved in 3.5 billion-year-old fluid inclusions. Nat Commun (2021) 12(1):1101. doi: 10.1038/s41467-021-21323-z
4. Pearce BKD, Tupper AS, Pudritz RE, Higgs PG. Constraining the time interval for the origin of life on Earth. Astrobiology (2018) 18(3):343–64. doi: 10.1089/ast.2017.1674
5. Tashiro T, Ishida A, Hori M, Igisu M, Koike M, Méjean P, et al. Early trace of life from 3.95 ga sedimentary rocks in Labrador, Canada. Nature (2017) 549(7673):516–8. doi: 10.1038/nature24019
6. Schönheit P, Buckel W, Martin WF. On the origin of heterotrophy. Trends Microbiol (2016) 24(1):1225. doi: 10.1016/j.tim.2015.10.003
7. Hohmann-Marriott MF, Blankenship RE. Evolution of photosynthesis. Annu Rev Plant Biol (2011) 62:515–48. doi: 10.1146/annurev-arplant-042110-103811
8. Sessions AL, Doughty DM, Welander PV, Summons RE, Newman DK. The continuing puzzle of the great oxidation event. Curr Biol (2009) 19(14):R567–74. doi: 10.1016/j.cub.2009.05.054
9. Rasmussen B, Fletcher IR, Brocks JJ, Kilburn MR. Reassessing the first appearance of eukaryotes and cyanobacteria. Nature (2008) 455(7216):1101–4. doi: 10.1038/nature07381
10. Williams TA, Foster PG, Cox CJ, Embley TM. An archaeal origin of eukaryotes supports only two primary domains of life. Nature (2013) 504(7479):231–6. doi: 10.1038/nature12779
11. Blasiak R, Wynberg R, Grorud-Colvert K, Thambisetty S, Bandarra NM, Canário AVM, et al. The ocean genome and future prospects for conservation and equity. Nat Sustain (2020) 3(8):588–96. doi: 10.1038/s41893-020-0522-9
12. Zimmer M. GFP: from jellyfish to the Nobel Prize and beyond. Chem Soc Rev (2009) 38(10):2823–32. doi: 10.1039/b904023d
13. Tahamtan A, Ardebili A. Real-time RT-PCR in COVID-19 detection: issues affecting the results. Expert Rev Mol Diagn (2020) 20(5):453–4. doi: 10.1080/14737159.2020.1757437
14. Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science (1996) 273(5278):1058–73. doi: 10.1126/science.273.5278.1058
15. Dufresne A, Salanoubat M, Partensky F, Artiguenave F, Axmann IM, Barbe V, et al. Genome sequence of the cyanobacterium Prochlorococcus marinus SS120, a nearly minimal oxyphototrophic genome. Proc Natl Acad Sci USA (2003) 100(17):10020–5. doi: 10.1073/pnas.1733211100
16. Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. MicroBiol Mol Biol Rev (2004) 68(4):669–85. doi: 10.1128/MMBR.68.4.669-685.2004
17. Kennedy J, Marchesi JR, Dobson AD. Metagenomic approaches to exploit the biotechnological potential of the microbial consortia of marine sponges. Appl MicroBiol Biotechnol (2007) 75(1):11–20. doi: 10.1007/s00253-007-0875-2
18. Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science (2004) 304(5667):66–74. doi: 10.1126/science.1093857
19. Nealson KH, Venter JC. Metagenomics and the global ocean survey: what's in it for us, and why should we care? ISME J (2007) 1(3):185–7. doi: 10.1038/ismej.2007.43
20. Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol (1998) 5(10):R245–9. doi: 10.1016/s1074-5521(98)90108-9
21. Kennedy J, Flemer B, Jackson SA, Lejon DP, Morrissey JP, O’gara F, et al. Marine metagenomics: new tools for the study and exploitation of marine microbial metabolism. Mar Drugs (2010) 8(3):608—8. doi: 10.3390/md8030608
22. Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc Natl Acad Sci USA (1998) 95(12):6578–83. doi: 10.1073/pnas.95.12.6578
23. Alam I, Kamau AA, Ngugi DK, Gojobori T, Duarte CM, Bajic VB. KAUST Metagenomic Analysis Platform (KMAP), enabling access to massive analytics of re-annotated metagenomic data. Sci Rep (2021) 11(1):11511. doi: 10.1038/s41598-021-90799-y
24. Mitchell AL, Almeida A, Beracochea M, Boland M, Burgin J, Cochrane G, et al. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res (2020) 48(D1):D570–8. doi: 10.1093/nar/gkz1035
25. Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Ocean plankton. Structure and function of the global ocean microbiome. Science (2015) 348(6237):1261359. doi: 10.1126/science.1261359
26. Duarte CM. Seafaring in the 21st century: the Malaspina 2010 circumnavigation expedition. Limnol Oceanogr Bull (2015) 24(1):11–4. doi: 10.1002/lob.10008
27. Acinas SG, Sánchez P, Salazar G, Cornejo-Castillo FM, Sebastián M, Logares R, et al. Deep ocean metagenomes provide insight into the metabolic architecture of bathypelagic microbial communities. Commun Biol (2021) 4(1):604. doi: 10.1038/s42003-021-02112-2
28. Kopf A, Bicak M, Kottmann R, Schnetzer J, Kostadinov I, Lehmann K, et al. The ocean sampling day consortium. GigaScience (2015) 4(1):1–5. doi: 10.1186/s13742-015-0066-5
29. Karl DM, Church MJ. Microbial oceanography and the Hawaii Ocean Time-series programme. Nat Rev Microbiol (2014) 12(10):699–713. doi: 10.1038/nrmicro3333
30. Coelho LP, Alves R, Del Río ÁR, Myers PN, Cantalapiedra CP, Giner-Lamia J, et al. Towards the biogeography of prokaryotic genes. Nature (2022) 601(7892):252–6. doi: 10.1038/s41586-021-04233-4
31. Curtis TP, Sloan WT, Scannell JW. Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci USA (2002) 99(16):10494–9. doi: 10.1073/pnas.142680199
32. Escobar-Zepeda A, Godoy-Lozano EE, Raggi L, Segovia L, Merino E, Gutiérrez-Rios RM, et al. Analysis of sequencing strategies and tools for taxonomic annotation: defining standards for progressive metagenomics. Sci Rep (2018) 8(1):12034. doi: 10.1038/s41598-018-30515-5
33. Madsen EL. Microorganisms and their roles in fundamental biogeochemical cycles. Curr Opin Biotechnol (2011) 22(3):456–64. doi: 10.1016/j.copbio.2011.01.008
34. Biddanda B, Ogdahl M, Cotner J. Dominance of bacterial metabolism in oligotrophic relative to eutrophic waters. Limnol Oceanogr (2001) 46(3):730–9. doi: 10.4319/lo.2001.46.3.0730
35. Santoro AE, Richter RA, Dupont CL. Planktonic marine archaea. Annu Rev Mar Sci (2019) 11:131–58. doi: 10.1146/annurev-marine-121916-063141
36. Giovannelli D, Molari M, d’Errico G, Baldrighi E, Pala C, Manini E. Large-scale distribution and activity of prokaryotes in deep-sea surface sediments of the Mediterranean Sea and the adjacent Atlantic Ocean. PloS One (2013) 8(8):e72996. doi: 10.1371/journal.pone.0072996
37. Parkes RJ, Cragg BA, Bale SJ, Getlifff JM, Goodman K, Rochelle PA, et al. Deep bacterial biosphere in Pacific Ocean sediments. Nature (1994) 371(6496):410–3. doi: 10.1038/371410a0
38. D'Hondt S, Jørgensen BB, Miller DJ, Batzke A, Blake R, Cragg BA, et al. Distributions of microbial activities in deep subseafloor sediments. Science (2004) 306(5705):2216–21. doi: 10.1126/science.1101155
39. Del Giorgio PA, Duarte CM. Respiration in the open ocean. Nature (2002) 420(6914):379–84. doi: 10.1038/nature01165
41. Zhang CL, Xie W, Martin-Cuadrado AB, Rodriguez-Valera F. Marine Group II Archaea, potentially important players in the global ocean carbon cycle. Front Microbiol (2015) 6:1108. doi: 10.3389/fmicb.2015.01108
42. Agustí S, González-Gordillo JI, Vaqué D, Estrada M, Cerezo MI, Salazar G, et al. Ubiquitous healthy diatoms in the deep sea confirm deep carbon injection by the biological pump. Nat Commun (2015) 6(1):7608. doi: 10.1038/ncomms8608
43. Bar-On YM, Milo R. The biomass composition of the oceans: a blueprint of our blue planet. Cell (2019) 179(7):1451–4. doi: 10.1016/j.cell.2019.11.018
44. Dominguez-Huerta G, Zayed AA, Wainaina JM, Guo J, Tian F, Pratama AA, et al. Diversity and ecological footprint of Global Ocean RNA viruses. Science (2022) 376(6598):1202–8. doi: 10.1126/science.abn6358
45. Dombrowski N, Teske AP, Baker BJ. Expansive microbial metabolic versatility and biodiversity in dynamic Guaymas Basin hydrothermal sediments. Nat Commun (2018) 9(1):4999. doi: 10.1038/s41467-018-07418-0
46. Langwig MV, De Anda V, Dombrowski N, Seitz KW, Rambo IM, Greening C, et al. Large-scale protein level comparison of Deltaproteobacteria reveals cohesive metabolic groups. ISME J (2022) 16(1):307–20. doi: 10.1038/s41396-021-01057-y
47. Parks DH, Rinke C, ChuvoChina M, Chaumeil PA, Woodcroft BJ, Evans PN, et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol (2017) 2(11):1533–42. doi: 10.1038/s41564-017-0012-7
48. Nayfach S, Roux S, Seshadri R, Udwary D, Varghese N, Schulz F, et al. A genomic catalog of Earth’s microbiomes. Nat Biotechnol (2021) 39(4):499–509. doi: 10.1038/s41587-020-0718-6
49. Ponsero AJ, Bomhoff M, Blumberg K, Youens-Clark K, Herz NM, Wood-Charlson EM, et al. Planet Microbe: a platform for marine microbiology to discover and analyze interconnected ‘omics and environmental data. Nucleic Acids Res (2021) 49(D1):D792–802. doi: 10.1093/nar/gkaa637
50. Nata’ala MK, Avila Santos AP, Coelho Kasmanas J, Bartholomäus A, Saraiva JP, Godinho Silva S, et al. MarineMetagenomeDB: a public repository for curated and standardized metadata for marine metagenomes. Environ Microbiome (2022) 17(1):57. doi: 10.1186/s40793-022-00449-7
51. Orcutt BN, Sylvan JB, Knab NJ, Edwards KJ. Microbial ecology of the dark ocean above, at, and below the seafloor. MicroBiol Mol Biol Rev (2011) 75(2):361–422. doi: 10.1128/MMBR.00039-10
52. Pernice MC, Giner CR, Logares R, Perera-Bel J, Acinas SG, Duarte CM, et al. Large variability of bathypelagic microbial eukaryotic communities across the world’s oceans. ISME J (2016) 10(4):945–58. doi: 10.1038/ismej.2015.170
53. Hoegh-Guldberg O, Jacob D, Taylor M, Bindi M, Brown S, Camilloni I, et al. Impacts of 1.5°C global warming on natural and human systems. In: Global Warming of 1.5°C. An IPCC Special Report on the impacts of global warming of 1.5°C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty [Masson-Delmotte V, Zhai P, Pörtner H-O, Roberts D, Skea J, Shukla PR, et al (eds.)]. Cambridge University Press, Cambridge, UK and New York, NY, USA. 175–312. https://doi.org/10.1017/9781009157940.005. Available at: https://www.ipcc.ch/site/assets/uploads/sites/2/2022/06/SR15_Chapter_3_LR.pdf.
54. Spalding MD, Ravilious C, Green EP. World atlas of coral reefs. Berkeley, CA: University of California Press (2001).
55. Souter D, Planes S, Wicquart J, Logan M, Obura D, Staub F. Status of coral reefs of the world [2020 report] Global Coral Reef Monitoring Network (GCRMN)/International Coral Reef Initiative (ICRI) (2021). Available at: https://gcrmn.net/2020-report/.
56. van Oppen MJH, Blackall LL. Coral microbiome dynamics, functions and design in a changing world. Nat Rev Microbiol (2019) 17(9):557–67. doi: 10.1038/s41579-019-0223-4
57. Martin C, Young CA, Valluzzi L, Duarte CM. Ocean sediments as the global sink for marine micro-and mesoplastics. Limnol Oceanogr Lett (2022) 7(3):235–43. doi: 10.1002/lol2.10257
58. Miller KA, Thompson KF, Johnston P, Santillo D. An overview of seabed mining including the current state of development, environmental impacts, and knowledge gaps. Front Mar Sci (2018) 4:418. doi: 10.3389/fmars.2017.00418
59. Fricker AM, Podlesny D, Fricke WF. What is new and relevant for sequencing-based microbiome research? A mini-review. J Adv Res (2019) 19:105–12. doi: 10.1016/j.jare.2019.03.006
60. Wisnoski NI, Muscarella ME, Larsen ML, Peralta AL, Lennon JT. Metabolic insight into bacterial community assembly across ecosystem boundaries. Ecology (2020) 101(4):e02968. doi: 10.1002/ecy.2968
61. Duarte CM, Regaudie-de-Gioux A, Arrieta JM, Delgado-Huertas A, Agustí S. The oligotrophic ocean is heterotrophic. Ann Rev Mar Sci (2013) 5:551–69. doi: 10.1146/annurev-marine-121211-172337
62. Swan BK, Martinez-Garcia M, Preston CM, Sczyrba A, Woyke T, Lamy D, et al. Potential for chemolithoautotrophy among ubiquitous bacteria lineages in the dark ocean. Science (2011) 333(6047):1296–300. doi: 10.1126/science.1203690
63. Pachiadaki MG, Sintes E, Bergauer K, Brown JM, Record NR, Swan BK, et al. Major role of nitrite-oxidizing bacteria in dark ocean carbon fixation. Science (2017) 358(6366):1046–51. doi: 10.1126/science.aan8260
64. Baltar F, Herndl GJ. Ideas and perspectives: is dark carbon fixation relevant for oceanic primary production estimates? Biogeosciences (2019) 16(19):3793–9. doi: 10.5194/bg-16-3793-2019
65. Burd BJ, Thomson RE. A review of zooplankton and deep carbon fixation contributions to carbon cycling in the dark ocean. J Mar Syst (2022) 236. doi: 10.1016/j.jmarsys.2022.103800
66. Moran MA, Buchan A, González JM, Heidelberg JF, Whitman WB, Kiene RP, et al. Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature (2004) 432(7019):910–13. doi: 10.1038/nature03170
67. King GM, Weber CF. Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat Rev Microbiol (2007) 5(2):107–18. doi: 10.1038/nrmicro1595
68. Ragsdale SW. Life with carbon monoxide. Crit Rev Biochem Mol Biol (2004) 39(3):165–95. doi: 10.1080/10409230490496577
69. Canfield DE, Stewart FJ, Thamdrup B, De Brabandere L, Dalsgaard T, Delong EF, et al. A cryptic sulfur cycle in oxygen-minimum–zone waters off the Chilean coast. Science (2010) 330(6009):1375–8. doi: 10.1126/science.1196889
70. Stewart FJ. Dissimilatory sulfur cycling in oxygen minimum zones: an emerging metagenomics perspective. Biochem Soc Trans (2011) 39(6):1859–63. doi: 10.1042/BST20110708
71. Callbeck CM, Canfield DE, Kuypers MMM, Yilmaz P, Lavik G, Thamdrup B, et al. Sulfur cycling in oceanic oxygen minimum zones. Limnol Oceanogr (2021) 66(6):2360–92. doi: 10.1002/lno.11759
72. Wright JJ, Konwar KM, Hallam SJ. Microbial ecology of expanding oxygen minimum zones. Nat Rev Microbiol (2012) 10(6):381–94. doi: 10.1038/nrmicro2778
73. Bianchi D, Weber TS, Kiko R, Deutsch C. Global niche of marine anaerobic metabolisms expanded by particle microenvironments. Nat Geosci (2018) 11(4):263–8. doi: 10.1038/s41561-018-0081-0
74. Raven MR, Keil RG, Webb SM. Microbial sulfate reduction and organic sulfur formation in sinking marine particles. Science (2021) 371(6525):178–81. doi: 10.1126/science.abc6035
75. Benavides M, Moisander PH, Berthelot H, Dittmar T, Grosso O, Bonnet S. Mesopelagic N2 fixation related to organic matter composition in the Solomon and B. Seas (southwest Pacific). PloS One (2015) 10(12):e0143775. doi: 10.1371/journal.pone.0143775
76. Farnelid H, Turk-Kubo K, Ploug H, Ossolinski JE, Collins JR, Van Mooy BAS, et al. Diverse diazotrophs are present on sinking particles in the North Pacific Subtropical gyre. ISME J (2019) 13(1):170–82. doi: 10.1038/s41396-018-0259-x
77. Zehr JP, Capone DG. Changing perspectives in marine nitrogen fixation. Science (2020) 368(6492):eaay9514. doi: 10.1126/science.aay9514
78. Tyler AD, Mataseje L, Urfano CJ, Schmidt L, Antonation KS, Mulvey MR, et al. Evaluation of Oxford nanopore’s MinION sequencing device for microbial whole genome sequencing applications. Sci Rep (2018) 8(1):10931. doi: 10.1038/s41598-018-29334-5
79. Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, UniProt Consortium. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics (2015) 31(6):926–32. doi: 10.1093/bioinformatics/btu739
80. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021) 596(7873):583–9. doi: 10.1038/s41586-021-03819-2
81. Roux S, Matthijnssens J, Dutilh BE. Metagenomics in virology. Encyclopedia Virol (2021) 133–40. doi: 10.1016/B978-0-12-809633-8.20957-6
82. Yooseph S, Nealson KH, Rusch DB, McCrow JP, Dupont CL, Kim M, et al. Genomic and functional adaptation in surface ocean planktonic prokaryotes. Nature (2010) 468(7320):60–6. doi: 10.1038/nature09530
83. Ferrera I, Sebastian M, Acinas SG, Gasol JM. Prokaryotic functional gene diversity in the sunlit ocean: stumbling in the dark. Curr Opin Microbiol (2015) 25:33–9. doi: 10.1016/j.mib.2015.03.007
84. Carradec Q, Pelletier E, Da Silva C, Alberti A, Seeleuthner Y, Blanc-Mathieu R, et al. A global ocean atlas of eukaryotic genes. Nat Commun (2018) 9(1):1–3. doi: 10.1038/s41467-017-02342-1
85. Obiol A, Giner CR, Sánchez P, Duarte CM, Acinas SG, Massana R. A metagenomic assessment of microbial eukaryotic diversity in the global ocean. Mol Ecol Resour (2020) 20(3):718–31. doi: 10.1111/1755-0998.13147
86. Schirmer M, Ijaz UZ, D'Amore R, Hall N, Sloan WT, Quince C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res (2015) 43(6):e37. doi: 10.1093/nar/gku1341
87. Tully BJ, Graham ED, Heidelberg JF. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci Data (2018) 5(1):170203. doi: 10.1038/sdata.2017.203
88. Royo-Llonch M, Sánchez P, Ruiz-González C, Salazar G, Pedrós-Alió C, Sebastián M, et al. Compendium of 530 metagenome-assembled bacterial and archaeal genomes from the polar Arctic Ocean. Nat Microbiol (2021) 6(12):1561–74. doi: 10.1038/s41564-021-00979-9
89. Grossart HP, Massana R, McMahon KD, Walsh DA. Linking metagenomics to aquatic microbial ecology and biogeochemical cycles. Limnol Oceanogr (2020) 65(S1):S2–S20. doi: 10.1002/lno.11382
90. Narsing Rao MP, Luo ZH, Dong ZY, Li Q, Liu BB, Guo SX, et al. Metagenomic analysis further extends the role of Chloroflexi in fundamental biogeochemical cycles. Environ Res (2022) 209:112888. doi: 10.1016/j.envres.2022.112888
91. Wang WL, Xu SY, Ren ZG, Tao L, Jiang JW, Zheng SS. Application of metagenomics in the human gut microbiome. World J Gastroenterol (2015) 21(3):803–14. doi: 10.3748/wjg.v21.i3.803
92. Duarte CM, Ngugi DK, Alam I, Pearman J, Kamau A, Eguiluz VM, et al. Sequencing effort dictates gene discovery in marine microbial metagenomes. Environ Microbiol (2020) 22(11):4589–603. doi: 10.1111/1462-2920.15182
93. Lynch MD, Neufeld JD. Ecology and exploration of the rare biosphere. Nat Rev Microbiol (2015) 13(4):217–29. doi: 10.1038/nrmicro3400
94. Ahlgren NA, Belisle BS, Lee MD. Genomic mosaicism underlies the adaptation of marine Synechococcus ecotypes to distinct oceanic iron niches. Environ Microbiol (2020) 22(5):1801–15. doi: 10.1111/1462-2920.14893
95. Tschoeke D, Salazar VW, Vidal L, Campeão M, Swings J, Thompson F, et al. Unlocking the genomic taxonomy of the Prochlorococcus collective. Microb Ecol (2020) 80(3):546–58. doi: 10.1007/s00248-020-01526-5
96. Khersonsky O, Tawfik DS. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu Rev Biochem (2010) 79:471–505. doi: 10.1146/annurev-biochem-030409-143718
97. Srivastava A, Saavedra DEM, Thomson B, García JAL, Zhao Z, Patrick WM, et al. Enzyme promiscuity in natural environments: alkaline phosphatase in the ocean. ISME J (2021) 15(11):3375–83. doi: 10.1038/s41396-021-01013-w
98. Morales SE, Biswas A, Herndl GJ, Baltar F. Global structuring of phylogenetic and functional diversity of pelagic fungi by depth and temperature. Front Mar Sci (2019) 6:131. doi: 10.3389/fmars.2019.00131
99. Breyer E, Zhao Z, Herndl GJ, Baltar F. Global contribution of pelagic fungi to protein degradation in the ocean. Microbiome (2022) 10(1):143. doi: 10.1186/s40168-022-01329-5
100. Meyer-Cifuentes IE, Werner J, Jehmlich N, Will SE, Neumann-Schaal M, Öztürk B. Synergistic biodegradation of aromatic-aliphatic copolyester plastic by a marine microbial consortium. Nat Commun (2020) 11(1):5790. doi: 10.1038/s41467-020-19583-2
101. Muriel-Millán LF, Millán-López S, Pardo-López L. Biotechnological applications of marine bacteria in bioremediation of environments polluted with hydrocarbons and plastics. Appl MicroBiol Biotechnol (2021) 105(19):7171–85. doi: 10.1007/s00253-021-11569-4
102. Hatosy SM, Martiny AC. The ocean as a global reservoir of antibiotic resistance genes. Appl Environ Microbiol (2015) 81(21):7593–9. doi: 10.1128/AEM.00736-15
103. Vijay R, Khobragade PJ, Sohony RA. Water quality simulation of sewage impacts on the west coast of Mumbai, India. Water Sci Technol (2010) 62(2):279–87. doi: 10.2166/wst.2010.237
104. Arnaud-Haond S, Arrieta JM, Duarte CM. Global genetic resources. Marine biodiversity and gene patents. Science (2011) 331(6024):1521–2. doi: 10.1126/science.1200783
105. Agreement under the United Nations Convention on the Law of the Sea on the conservation and sustainable use of marine biological diversity of areas beyond national jurisdiction (2023). Available at: https://www.un.org/bbnj/sites/www.un.org.bbnj/files/draft_agreement_advanced_unedited_for_posting_v1.pdf.
106. Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics (2015) 31(10):1674–6. doi: 10.1093/bioinformatics/btv033
107. Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform (2010) 11(1):119. doi: 10.1186/1471-2105-11-119
108. Steinegger M, Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol (2017) 35(11):1026–8. doi: 10.1038/nbt.3988
Keywords: shotgun metagenomics, microbes, ocean gene catalog, biodiversity, ocean domains, functional genomics, biogeochemical cycling
Citation: Laiolo E, Alam I, Uludag M, Jamil T, Agusti S, Gojobori T, Acinas SG, Gasol JM and Duarte CM. Metagenomic probing toward an atlas of the taxonomic and metabolic foundations of the global ocean genome. Front Sci (2024) 1:1038696. doi: 10.3389/fsci.2023.1038696
Received: 07 September 2022; Accepted: 22 December 2023;
Published: 16 January 2024.
Edited by:
Brett J. Baker, The University of Texas at Austin, United StatesReviewed by:
Gerhard J. Herndl, University of Vienna, AustriaAndreas Teske, University of North Carolina at Chapel Hill, United States
Copyright © 2024 Laiolo, Alam, Uludag, Jamil, Agusti, Gojobori, Acinas, Gasol and Duarte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisa Laiolo, ZWxpc2EubGFpb2xvQGthdXN0LmVkdS5zYQ==
†These authors contributed equally to this work and share first authorship