 Qiuxuan Chen
Qiuxuan Chen- 1Cancer Center, Renmin Hospital of Wuhan University, Wuhan, Hubei, China
- 2Institude of Experimental Immunology, University Clinic of Rheinische Friedrich-Wihelms-University, Bonn, Germany
Lung cancer, a common type of malignant neoplasm, has seen significant advancements in the treatment of lung adenocarcinoma (LUAD). However, the management of lung squamous cell carcinoma (LSCC) continues to pose challenges. Traditional treatment methods for LSCC encompass surgical resection, chemotherapy, and radiotherapy. The introduction of targeted therapy and immunotherapy has greatly benefited LSCC patients, but issues such as limited immune response rates and adverse reactions persist. Therefore, gaining a deeper comprehension of the underlying mechanisms holds immense importance. This review provides an in-depth overview of classical signaling pathways and therapeutic targets, including the PI3K signaling pathway, CDK4/6 pathway, FGFR1 pathway and EGFR pathway. Additionally, we delve into alternative signaling pathways and potential targets that could offer new therapeutic avenues for LSCC. Lastly, we summarize the latest advancements in targeted therapy combined with immune checkpoint blockade (ICB) therapy for LSCC and discuss the prospects and challenges in this field.
1 Introduction
Lung cancer, a leading cause of cancer-related deaths worldwide (1), is commonly classified into small-cell and non-small-cell subtypes (2). Non-small-cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancer cases, with lung squamous cell carcinoma (LSCC) making up around 30% of them (3). Smoking history, prolonged exposure to harmful substances, and familial genetic factors significantly contribute to the risk of lung cancer (4–6). Patients diagnosed with lung adenocarcinoma (LUAD) often benefit more from targeted therapy and immunotherapy than those with LSCC (7). Operable mutations are rarely detected in patients with LSCC, resulting in limited treatment options (8). Currently, the primary treatments for LSCC encompass surgical resection, chemotherapy, radiotherapy, targeted therapy, and immunotherapy (6, 9, 10). The standard treatment of early and middle-stage LSCC is primarily surgical treatment, supplemented by radiotherapy and chemotherapy, and the advanced treatment is mainly chemotherapy. However, surgical resection is often unfeasible for advanced LSCC cases (11), and chemotherapy and radiotherapy can frequently lead to toxicity and drug resistance (12, 13). As an emerging approach in LSCC therapy, targeted therapy holds promise in improving patient survival rates and quality of life by inhibiting specific signaling pathways involved in cancer cell growth. Prominent pathway alterations, such as phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT), epidermal growth factor receptor (EGFR), fibroblast growth factor receptor (FGFR), cell cycle protein-dependent kinases 4 and 6 (CDK4/6), and rat sarcoma (RAS) pathways, have been observed. While treatment options for the two subtypes of NSCLC, namely LSCC and LUAD, were previously similar (14), LSCC exhibits a higher prevalence of certain mutated genes, including tumor protein p53, glutamate receptor, metabotropic 8, Erb-B2 receptor tyrosine kinase 4, Kelch-like ECH-associated protein 1 (KEAP1), and mucin 16 (15). However, a single targeted therapeutic approach still presents certain challenges owing to drug resistance and tumor heterogeneity.
Immunotherapy treatment for LSCC shows potential benefits, particularly in histological and mutational states (16, 17). Immune checkpoints, such as programmed death receptors and their ligands, naturally regulate the immune system to prevent overreaction and autoimmune diseases (18). However, tumor cells often exploit programmed death ligands on their surface to bind to programmed death acceptors, impeding the function of T cells, thereby evading immune surveillance (19, 20). Immune checkpoint blockade (ICB) therapy stimulates the immune system to re-recognize tumor cells and increase its aggression by inhibiting the binding of tumor cells to programmed death receptors and their ligands (18, 21). Current ICB first- and second-line therapies for advanced LSCC include programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1) and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) inhibitors (22). Despite these advances, further comprehensive studies are required to address issues such as the search for novel medicines, immune tolerance management, and hazardous side effects (23, 24). ICB therapy can cause unique toxicity by enhancing immune response, consequently inducing immune-related adverse events (25). Additionally, many patients with LSCC develop immune escape owing to the human leukocyte antigen gene mutations (26). Combining immunotherapy with other therapies, including targeted therapy, may offer a reliable solution. Some classical signaling pathways, such as PI3K and CDK4/6, are associated with a tumor mutational burden (TMB) state. Novel targets such as nuclear receptor-binding SET domain protein 3 (NSD3), lysine methyltransferase 2 D (KMT2D), and ubiquitin-specific peptidase 28 (USP28), are related to tumor microenvironment (TME) and immune cells. A recent study involving 1008 patients with LUAD and LSCC suggests that identifying elevated PD-L1 expression levels could effectively guide targeted therapy. This is because PD-L1 is more prevalent in men, smokers, and squamous cell carcinoma tumors with a maximum diameter >3 cm, poorly differentiated, and/or high tumor node metastasis stage (27).
Future LSCC research will continue to focus on individualized treatment plans (28). This review summarizes the current knowledge of potential targetable gene alterations in LSCC, and considers repurposing some targets effective against other cancers, including LUAD. Considering the complex genome of patients with LSCC, we further discuss the possibility of multi-target combination therapy and combining immunotherapy with targeted therapy to improve the efficacy of LSCC treatment. We anticipate more advances in LSCC therapy, which will benefit the clinical outcomes of patients by broadening the pool of possible therapeutic targets, increasing drug selectivity, and implementing treatment approaches like targeted therapy in conjunction with immunotherapy or multi-target combination therapy.
2 Targeting classical targets in LSCC
Previous studies have shown that patients with LSCC exhibit numerous genetic alterations, affecting various signaling pathways. The most commonly mutated genes in lung squamous carcinoma include phosphatase and tensin homolog (PTEN), EGFR, cyclin dependent kinase inhibitor 2A, and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) (29, 30). This section introduces several previously identified classical targets, along with their associated developments, paving the way for new targets and therapeutic strategies (Figure 1).
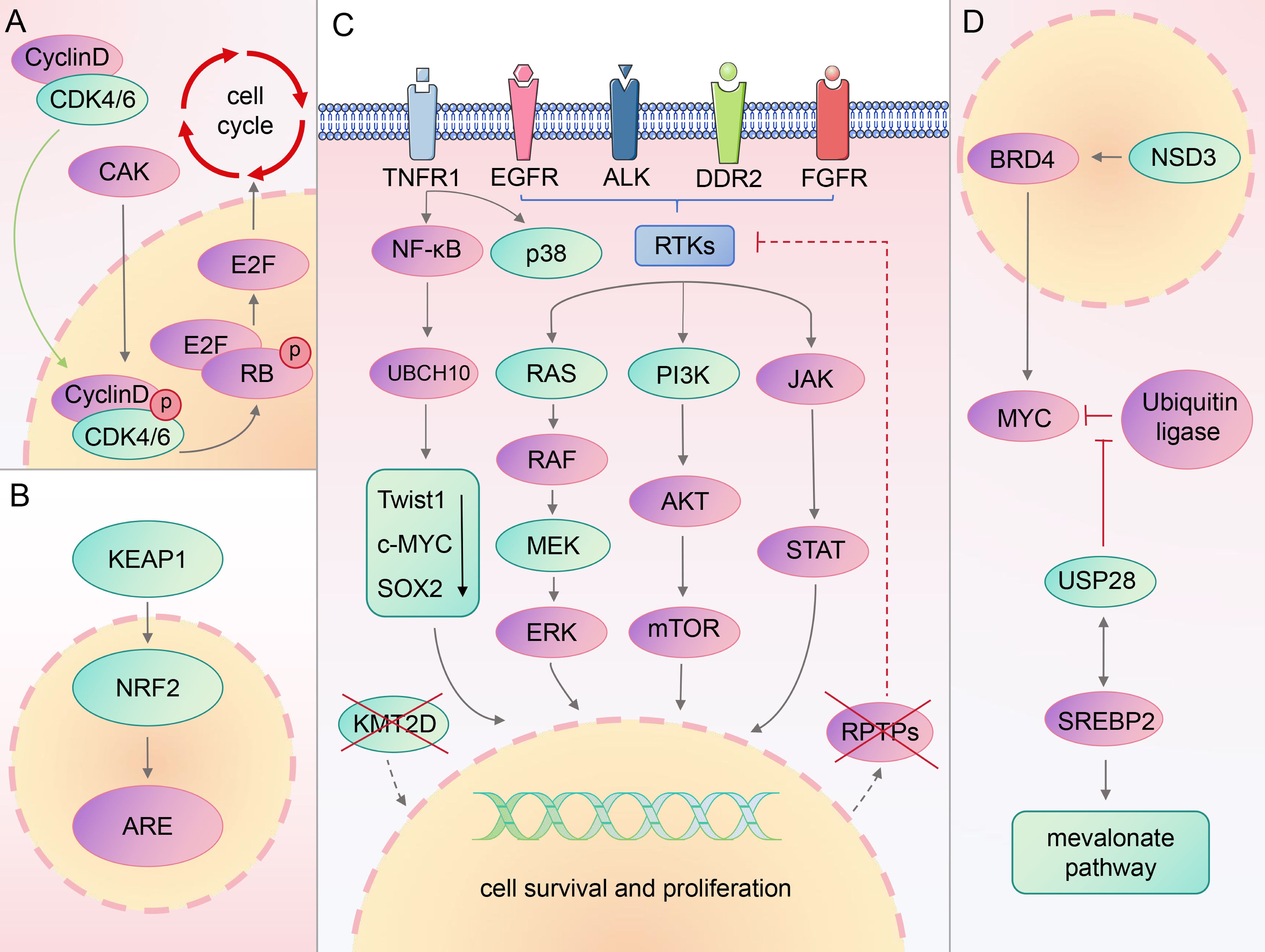
Figure 1. Signaling pathways associated with classical/novel targets.
2.1 PI3K
Research has identified overactivation of the PI3K/AKT signaling pathway as a prevalent etiology among cancers (31). This pathway is disrupted in 68% of LSCC samples (32).
PI3K is divided into three main classes. Class I PI3K, which is strongly associated with cancer (33), is comprised of the catalytic subunit p110 (with four subtypes: α, β, γ, and δ) and regulatory subunit p85 (34, 35). P110α directly binds to the RAS gene or recruits the p85 subunit to the tyrosine phosphorylated receptor and receptor-associated adaptor proteins, signaling downstream plasma membrane-associated tyrosine kinase (36).
The primary manifestations of PI3K signal distortion in LSCC are PIK3CA amplification, PIK3CA mutation, and PTEN loss (37, 38). Tumor suppressor PTEN activation impacts the negative feedback mediating PI3K, significantly inhibiting tumor cell growth and invasion along with the activity of focal adhesion kinase (39, 40). PIK3CA mutations often co-mutate with other oncogenes such as EGFR, RAS, TP53, and so on (41), making it difficult to accurately target them with existing therapies (42). Furthermore, several clinical studies and experimental investigations have yielded negative results. When PI3K is inhibited, adaptive overexpression occurs in several compensatory pathways, leading to drug resistance (43). Furthermore, hyperglycemia has been reported in some patients treated with PI3K/AKT inhibitors (44), possibly due to the promotion of insulin-stimulated glucose uptake and storage by this pathway. Although the PTEN–PI3K axis has been established in LSCC studies, this pathway also plays a role in many normal cellular functions, while mutation-specific inhibitors remain unavailable. Ultimately, we need to deepen our understanding of PI3K’s function in cancer environment and enhance the durability and specificity of relevant treatments.
2.2 EGFR
The erbB-1 gene encodes EGFR, a transmembrane protein crucial for cell functions such as proliferation, growth, apoptosis, repair, and survival (45, 46). Mutations in the EGFR tyrosine kinase region, mainly in exons 18–21, are strongly associated with a subset of patients with LSCC, particularly women and non-smokers (30, 47, 48). Roughly half of patients with lung cancer exhibit EGFR mutations, with a notable increase in EGFR protein expression in LSCC compared with LUAD (49, 50). Furthermore, the mRNA abundance of the five EGFR ligands strongly correlates with EGFR-amplified LSCC cohorts, but no increase in EGFR pathway activity was observed (51). This suggests that EGFR ligand abundance is a more accurate indicator of EGFR inhibitor responsiveness in these patients.
Currently, EGFR-tyrosine kinase inhibitors (TKI) are a key therapeutic option for patients with LSCC and EGFR-activating mutations, significantly improving clinical outcomes (28). However, owing to an altered EGFR signaling pathway and activation of abnormal bypass pathway, acquired resistance of 1st, 2nd, and 3rd generation EGFR-TKI remains inevitable (52, 53). For EGFR-mutated patients with NSCLC who progress after EGFR-TKI treatment, a combination with immune checkpoint inhibitors (ICIs), chemotherapy, and anti-vascularization offers the greatest survival benefit, according to a statistical analysis of 2,085 patients in 17 studies (54).
2.3 FGFR
The FGFR family is key in tissue development and cancer progression, forming complexes with fibroblast growth factor (FGF) which activates several downstream signal transduction pathways (55, 56). Dysregulation of FGFR expression can contribute to tumor cell proliferation, survival, resistance, and immune escape (56), with LSCC showing higher FGFR1 levels than LUAD (37, 57).
FGF regulates the FGFR signaling pathway, maintaining tissue homeostasis, promoting repair and development, and influencing cell proliferation, differentiation, migration, survival, and angiogenesis (58, 59). FGF19 is crucial in prostate and breast cancer progression as it binds to the FGFR4 and Klotho complexes to initiate downstream signaling, significantly affecting LSCC development and proliferation (60–63). GLI family zinc finger 2 (GLI2), a downstream effector of FGF19, is induced by the TGF-B/SMAD pathway and is significant in promoting cancer metastasis (64). High FGF19 expression is associated with poor outcomes in LSCC, driving cell invasion through GLI2-mediated epithelial-mesenchymal transition (EMT) (65). As a result, therapeutic strategies that focus on the positive feedback loop of FGF19-GLI2 may be effective in treating LSCC.
FGFR emerges as a potential therapeutic target for LSCC (66). Current targeted therapeutics of FGFR include ortho-binding inhibitors, allosteric inhibitors, ligand traps, and small-molecule kinase inhibitors (67). FGFR1 amplification, the most common FGFR mutation affecting approximately 20% of patients with LSCC (51), has not been proven to be a reliable predictor in therapeutic trials (57, 68). Furthermore, the complex action mechanism of NSCLC and the uncertain origins of FGFR1 mutations warrant further research to understand or determine FGFR1 signaling in LSCC pathophysiology.
2.4 ALK
Anaplastic lymphoma kinase (ALK) regulates the function of the frontal cortex and hippocampus of the adult brain as well as influences tumor cell cycle control, thus impacting tumor transformation (69). Although unessential for normal growth and development, ALK's activation of signaling pathways, such as Janus kinase/signal transducer of activation, mitogen-activated protein kinase (MAPK), PI3K/AKT, and mitogen-activated protein kinase kinase (MEK) 5-extracellular signal-regulated kinase (ERK) 5, underscores its essential role in cell proliferation, differentiation, and apoptosis inhibition, making it a potential target for cancer therapy (70).
ALK rearrangements, particularly its fusion with echinodermal microtubule-associated protein-like 4, are common in NSCLC, with ALK mutations present in approximately 5% of cases, mostly patients with LUAD (71–74). Despite this, the large NSCLC patient population keeps ALK as a significant therapeutic target. Targeted therapy with TKIs, including crizotinib (first-generation), alectinib, ceritinib, ansatinib, brigatinib (second-generation), and loratinib (third-generation), has been developed for ALK-rearranged NSCLC (75–77). However, challenges including drug resistance and toxicity require further research and the development of new treatments (78).
2.5 RAS
When the RAS proteins are activated, they initiate several downstream signaling pathways, such as the PI3K/AKT/mTOR network, RAF/MEK/ERK pathway, and RalEGF/Ral route (79, 80). The dysregulation of the RAF/MEK/ERK signaling pathway is linked to tumor growth (81). The PI3K/AKT/mTOR pathway is associated with tumor pathologies (82). Despite initial challenges of “undruggable”, targeted allele-specific inhibitors of RAS, such as Kirsten rat sarcoma virus (KRAS)-G12C targeting drugs, have been developed, potentially changing the treatment landscape for RAS-driven tumors (83).
KRAS mutations are a frequent genetic cause of NSCLC, especially LUAD, but are rare in LSCC, with KRAS mutation rates reported between 1 and 7% in LSCC (84). Controversies exist regarding the presence of KRAS mutations in LSCC; some data suggest that KRAS mutations may be misclassified as LUAD or adenosquamous carcinoma. However, the persistence of KRAS mutations suggests that KRAS mutant LSCC is likely to exist, and future evidence may provide further clarification (85).
2.6 MEK
MEK is an important downstream component of the RAS with two main isoforms: MEK1 and MEK2 (86). It functions by specifically phosphorylating tyrosine and threonine residues in the activation loops of ERK1/2, playing a role in the RAS/RAF/MEK/ERK pathway (87). The MEK mutations are less common in patients (88). However, due to the importance of the RAF/MEK/ERK pathway, MEK is considered a potential target for new cancer therapies. For LSCC patients with V-raf murine sarcoma viral oncogene homolog B (BRAF) or KRAS mutations, monotherapy or combination therapy with MEK inhibitors, including trametinib, binimetinib, selumetinib, and cobimetinib, approved by the United States Food and Drug Administration (FDA), may be a promising treatment strategy. Compared with MEK inhibitor monotherapy, MEK inhibitor combination therapy demonstrates better therapeutic efficacy and less toxic side effects. The clinical efficacy requires further validation (89–91).
2.7 CDK4/6
CDK4/6 are key mediators for cell cycle transition from the G1 to the S phase (92). They contribute to the CDK-activated kinase complex phosphorylation of a CDK4/6 complex and activate cyclin D (93). Then, it phosphorylates the retinoblastoma, preventing it from repressing the E2F family of transcription factors, thus controlling the cell cycle (94). In LSCC, the cell cycle is disrupted owing to a high rate of gene inactivation that regulates CDK4/6 expression, making them potential targets for cancer therapy (95).
CDK4/6 inhibitors have shown promise in treating certain cancers, and their efficacy in treating LSCC is being investigated (96). Regretfully, monotherapy with CDK4/6 inhibitors such as palbociclib and abemaciclib has not been very successful (97, 98). Ongoing research and clinical trials are exploring the potential benefits of CDK4/6 inhibitors, both as monotherapy and in combination with other treatments (99).
2.8 DDR2
Discoidin domain receptor 2 (DDR2) is a member of the RTK family that plays a key role in cell proliferation and survival through EMT (100, 101). Evidence shows that DDR2 signals are closely related to the activation of PI3K/AKT and RAS/MEK/ERK pathways (102). The mutation rate of DDR2 varies among patients with LSCC, ranging from 0 to 4.6%, possibly because of ethnic differences (103–106). A study by Miao et al. demonstrated that DDR2 mRNA levels in LSCC tissue were significantly reduced compared with normal lung tissue (105).
Dasatinib is one of the most effective drugs for inhibiting the proliferation of DDR2-mutated cancer cells and has been approved for leukemia treatment (107). However, its clinical application is limited because of its significant toxicity and complexity of DDR2 signal transduction in lung cancer (102, 108). In addition, the gatekeeper T654I mutation acquired on DDR2 and the deletion of neurofibromin 1 expression through a splice site mutation can result in dasatinib resistance (109). Therefore, it is necessary to fully understand the signal transduction mechanism of DDR2 and develop the second generation of DDR2 inhibitors as soon as possible (110).
3 Other potential treatment targets
In addition to the conventional objectives discussed before, recent novel clinical trials have introduced innovative approaches, generating a treasure trove of valuable data for researchers. The pioneering potential targets might offer a renewed sense of optimism for LSCC treatment. This section delves into a selection of these targets.
3.1 NSD3
The NSD family are selective methyltransferases for histone H3 lysine 36 (H3K36) on nucleosome core particles (111, 112). This family, which operates in an auto-inhibitory state, allows H3K36 dimethylation to be catalyzed by nucleosome-based recognition and modification pathways which is crucial for maintaining chromatin stability and regulating gene expression (113).
Adjacent to FGFR1 is NSD3, which regulates the histone H3 lysine 36 methyltransferase (114). The amplification of NSD3, strongly linked with NSD3 mRNA expression (115), is among the more common genetic alterations in LSCC. Gene set enrichment analysis revealed that MYC targets, E2F targets, G2-M checkpoints, and unfolded protein response (UPR) are highly enriched in NSD3-amplified tumors. Additionally, the non-inflammatory TME condition of NSD3-amplified LSCCs leads to a less-than-optimal clinical response (116). These findings underscore the important carcinogenic function of NSD3 in LSCC and suggest that NSD3 may influence the ability of the immune system to combat tumors.
Considering their substantial role in tumor progression (117), the NSD family is under investigation as a potential therapeutic target for various cancers. However, the development of NSD inhibitors has been slow, largely owing to limitations in bioanalysis methodologies and the unique self-inhibition ring within the SET domain of NSD, making obtaining access to its substrate binding sites challenging (118). Despite these challenges, there has been notable progress in creating small-molecule inhibitors that target specific domains of NSD3 (119, 120), largely attributed to the refinement of the molecular mechanism of histone methylation catalyzed by NSD family proteins. Several inhibitors that target the NSD3 domain or its upstream and downstream signaling targets have been reported, such as BI-9321, a selective antagonist that specifically targets the PWWP1 domain of NSD3, and BRD4, an inhibitor that targets the bromodomain and extraterminal proteins (BET) (120, 121). We look forward to further potential applications of various NSD3 inhibitors.
3.2 KMT2D
The KMT2 family proteins play a pivotal role in chromatin structure regulation through methylation of lysine 3 on histone H4, thereby influencing epigenetic transcriptional activation (75, 122). KMT2D is known for its role as an H3K4 mono-methyltransferase that activates enhancers (123). The KMT2D gene, frequently mutated in LSCC and ranking third in the mutation rate among all cancer-related genes in The Cancer Genome Atlas Pan-Cancer Atlas, is a significant factor in lymphoma and breast cancer (124–127). KMT2D functions as a tumor suppressor in ovarian malignancies and lymphoma; however, it paradoxically promotes tumor growth in gastric and breast cancers (125, 126, 128–130). This dual role highlights the complexity of KMT2D’s function in cancer biology. The role of KMT2D in LSCC remains elusive, but a recent study has shed light on its tumor suppressor function. The study discovered that KMT2D absence leads to the underexpression of receptor-like protein tyrosine phosphatases, resulting in unchecked receptor tyrosine kinases-RAS signaling. The research reveals that inhibiting Src homology-2 protein tyrosine phosphatase and pan-ERBB can mitigate receptor tyrosine kinases-RAS signaling, slow LSCC progression, and extend survival, suggesting that targeting downstream components of KMT2D may be crucial for effective LSCC therapy (131). Furthermore, KMT2D deficiency has been linked to reduced expression of the tumor suppressor gene period circadian regulator 2 in lung cancer, leading to increased glycolysis and tumors proliferating (132).
3.3 KEAP/NRF2
Nuclear factor erythroid 2 related factor 2 (NRF2), a basic leucine zipper (bZIP) transcription factor, is a key regulator related to cell homeostasis (133, 134). Under normal conditions, NRF2 is bound by its repressor, KEAP1, which facilitates the degradation of NRF2 by proteases, maintaining the redox equilibrium (135). The homodimerization of KEAP1 and disruption of the KEAP1-Cullin3 complex under oxidative stress diminishes KEAP1’s ability to target NRF2, allowing NRF2 to enter the nucleus and bind to the regulatory regions of antioxidant genes (136). LUAD frequently exhibits gain-of-function mutations in the NRF2 encoding gene, whereas LSCC is characterized by loss-of-function mutations in KEAP1, affecting ≤21% of cases (137). Furthermore, experimental data indicates that the KEAP1/NRF2 pathway influences cell motility by inhibiting the RAS homolog family member A-Rho-associated protein kinase 1 pathway, affecting tumor cell adhesion and migration (138).
While there is currently no established cure for mutation in this pathway, many innovative and natural electrophilic compounds may serve as potential selective NRF2 activators, exhibiting anti-inflammatory properties (133). This is particularly relevant as NRF2 overactivation in cancer has been linked to chemotherapy and radiation therapy resistance, often resulting in less favorable patient outcomes, such as activating pathways that activate NRF2-mediated cisplatin resistance (137). Additionally, as reported by Sanchez-Ortega et al., reactive oxygen species (ROS) induction in wild-type NRF2/KEAP1 LSCC cells triggers iron death, and local ROS induced generation may be a novel therapeutic strategy for wild-type KEAP1/NRF2 LSCC (139). Surprisingly, Cui et al. found that NRF2 inhibition enhanced cell death and inhibited tumor growth in EGFR-mutated TKI-resistant non-small cell lung cancer, which may provide a new strategy to overcome resistance to EGFR-TKIs (29). Furthermore, the physiological mechanism of the KEAP1/NRF2 pathways is intricate, owing to the dual role of NRF2 as both a proto-oncogene and tumor suppressor in cancer (140). There is an urgent need for pharmaceuticals that specifically target the KEAP1/NRF2 signaling pathway and the adoption of advanced therapeutic approaches such as genetic stratification (141). These approaches could potentially provide more effective treatment options for patients.
3.4 USP28
USP28, a member of the deubiquitinase family, plays a crucial role in ubiquitination by removing ubiquitin tags, thereby regulating protein stability and function (142). Studies have highlighted the importance of USP28 in controlling various cancers, including gliomas, bowel cancer, and breast cancer, suggesting its potential as a novel therapeutic target for LSCC (143–145).
F-box and WD repeat domain containing 7 (FBXW7), a crucial component of the ubiquitin ligase complex, targets several well-known oncoproteins for degradation, including c-MYC, neurogenic locus Notch homolog protein, and c-JUN (145). USP28 disrupts this process by preventing FBXW7-mediated ubiquitination (146). Experimental evidence has demonstrated that inhibiting USP28 significantly curtails tumor growth in LSCC. This therapeutic response to USP28 inhibition occurs regardless of FBXW7 and USP28 mutations, underscoring USP28's promise as a therapeutic target in LSCC treatment (147).
Furthermore, studies have delved into the synergistic effects of targeting both USP28 and mevalonate pathways. Abnormalities in the mevalonate pathway are closely linked to tumor growth (148). Within this context, sterol regulatory element-binding protein 2 (SREBP2), a member of the sterol regulatory element-binding proteins, is crucial for cholesterol biosynthesis and uptake (149). Studies have discovered an interaction between SREBP2 and USP28, with both co-localizing in the nucleus of LSCC cells. USP28 can modulate SREBP2 expression, thereby stabilizing it and exerting control over the mevalonate pathway, suppressing tumor growth (150). This insight paves the way for innovative LSCC therapies that target both USP28 and mevalonate pathways, offering promising avenues for treatment.
3.5 p38 MAPK
The p38 MAPK family plays a crucial role in the cellular stress response (151). This kinase group contributes to a stress-response pathway involving three kinase cascades that regulate cell differentiation, survival, and cycle checkpoints (152). Additionally, the dual nature of p38 MAPK has been observed in various cancers, including intestinal, liver, and breast cancer, where it influences both tumor growth and metastasis, making it a potential therapeutic target (153, 154). Despite its promise as a treatment, the clinical application of p38 MAPK inhibitors has been limited owing to systemic adverse effects. Current research is exploring combination therapy and targeting its downstream effectors to enhance its therapeutic efficacy (155). In LSCC context, the role of the p38 pathway in treatment resistance is becoming increasingly clear. Studies have shown that p38 MAPK inhibitors aid in overcoming resistance to gefitinib in NSCLC with EGFR mutations (156). Furthermore, the dual inhibition of FGFR and p38 MAPK has undergone a significant reduction in tumor development and proliferation (157).
3.6 TNFR1
Tumor necrosis factor receptor 1 (TNFR1) is a key mediator of the physiological effects of TNF (158). It activates the nuclear factor-κB (NF-κB) and MAPK pathway, causing necrotic apoptosis (159). The NF-κB pathway, connected to TNFR1, is an important molecular mechanism involved in various cellular processes such as innate immunity, inflammation, cell development, survival, and proliferation (160, 161).
TNFR1, through the NF-κB signaling pathway, stimulates the production of ubiquitin-conjugating Enzyme H10 (UBCH10), an E2 ubiquitin-conjugating enzyme. Activated UBCH10 reduces twist-related protein 1, c-MYC, and SRY-box transcription factor 2 (SOX2) levels, leading to the transformation of differentiated LSCC into dedifferentiated spindle cell carcinoma, contributing to the last stage of LSCC (162). The TNFR1–UBCH10 axis plays a crucial role in LSCC development and metastasis, positioning TNFR1 inhibitors as potential novel treatments for LSCC (Table 1).
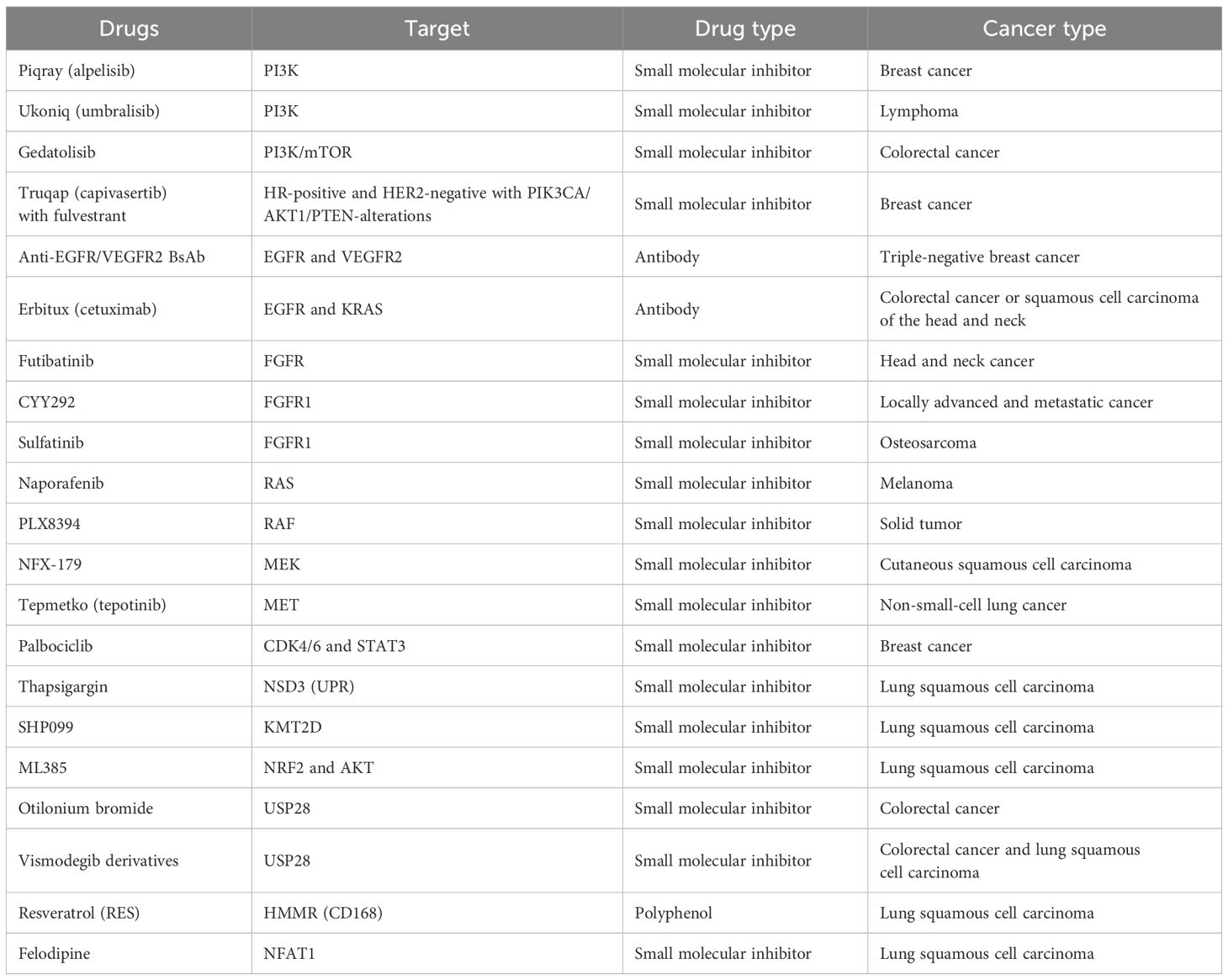
Table 1. Existing drugs that might be used in targeting LSCC.
4 Targeted therapy combined with immunotherapy
At present, immunotherapy stands as an important treatment option for patients with LSCC. Each of these treatments, immunotherapy, chemotherapy, and targeted therapy, faces challenges owing to the diversity of tumor phenotypes and suboptimal clinical response when used independently. Therefore, exploring the potential for combined targeted therapy and immunotherapy could significantly improve patient outcomes.
4.1 Classical pathway with immunotherapy
TMB is a crucial parameter for assessing immunotherapy effectiveness (163). Patients with a high TMB are more likely to benefit from ICI medications (164). High TMB (≥10/Mb) tumors in LSCC demonstrate enhanced immune cell lysis activity and enrichment of CD8+ T cells compared with low TMB cancers. Furthermore, research indicates that seven pathways, including the traditional PI3K/AKT and MAPK, are significantly inversely associated with TMB in LSCC (165).
Recent research suggests that PI3K inhibitors exert specific effects on TME by regulating tumor vasculature, fibroblast activity, and associated protein secretion (166). PI3K-δ and PI3K-γ signaling pathways alter the intrinsic processes of cell populations and influence tumor immune microenvironment (TIME), thereby supporting T-cell inhibition (167). Furthermore, alterations in the PI3K pathway in head and neck squamous cell carcinoma (HNSCC) have been associated with a high density of cytotoxic cells, including central memory CD8+ T and memory CD4+ T cells, which are crucial for immunotherapy and anti-tumor immunity (168).
KRAS mutations, prevalent in NSCLC, have been associated with increased PD-L1 expression, proliferation of tumor-infiltrating lymphocytes (TILs) proliferation, and high TMB (84, 169). Clinical trials suggest that ICIs are effective for patients with specific KRAS mutations, especially the G12C subtype (170). However, not all KRAS mutations are equally responsive to immunotherapy. For instance, the G12D variant is linked to lower TMB and, when co-mutated with TP53, shows reduced PD-L1 expression and immune infiltration (171). Given the prevalence of the G12C subtype in KRAS mutations and the FDA-approval of targeted drugs for this subtype, the synergistic approach of immunotherapy and KRAS targeted therapy holds considerable promise (83).
Additionally, the cell cycle pathway shows a significant positive correlation with TMB in both LUAD and LSCC. Inhibition of CDK4/6 is known to enhance T cell activation, bolster tumor immunological infiltration, reinforce immunological memory, and amplify the anti-tumor immunity elicited by anti-PD-1 antibodies (172, 173). Evidence from ovarian cancer treatment indicates that the synergistic effects of anti-PD-1 and CDK4/6 targeted therapy surpass the benefits of either treatment alone (174). Innovative combinations, such as the CDK4/6 inhibitor palbociclib and the CD73 selective inhibitor AB680, have shown efficacy in colorectal cancer (175). These findings provide hope for the potential of these combinations to increase their efficacy.
4.2 KEAP/NRF2 with immunotherapy
The mutation of the KEAP/NRF2 pathway is associated with a poor prognosis in patients with advanced cancer (176). In addition to being a significant targeted therapeutic option, this pathway mutation influences TIME and the selection of immunotherapy for patients with NSCLC (177). Recent clinical research has highlighted certain tumor metabolic characteristics, such as NRF2-mediated glutamine metabolism, that regulate tumor TIME (178). Glutamate and glutamine metabolism is significantly upregulated in LSCC tissues and plays a significant role in immune and stromal environmental inhibition (179). Immunotherapy for lung cancer benefits from inhibiting the glutamine metabolic pathway in several manners, including the upregulation of PD-L1 expression and reactivation of CD8+ T cells (177, 180). Furthermore, elevated NRF2 levels induce cell senescence. These senescent cells can be eliminated by immune effector cells recruited by NRF2-induced secretory phenotype, establishing the NRF2- NRF2-induced secretory phenotype immune surveillance axis (176).
Considering the multifaceted roles of the KEAP1/NRF2 pathway in both immunotherapy and targeted therapy, combining these two approaches may enhance clinical outcomes for patients with LSCC.
4.3 NSD3 with immunotherapy
Considering the significant variability of tumor immunogenicity and clinical response in patients with LSCC, precise stratification of immunotherapies is crucial. The TME of NSD3-amplified LSCC exhibits a non-inflammatory state with diminished activity in immune-related pathways. High UPR signaling activity may be a major modulator of the non-inflammatory TME phenotype in NSD3-amplified LSCC, as suggested by further molecular characterization (116). This suggests that NSD3 influences TIME and is linked to poorer outcomes in LSCC immunotherapy. Furthermore, NSD3 mutations impact clinical outcomes as they are related to immune cell infiltration and growth signaling in pancreatic cancer (112). While there are currently no therapeutically available NSD3 inhibitors, NSD3 could potentially serve as a biomarker for immunotherapy in combination with targeted therapy.
4.4 KMT2D with immunotherapy
Evidence suggests that patients with mutant KMT2D are likely to respond favorably to immunotherapy, as indicated by the protein's high attachment to major histocompatibility class I (181). Tumors with KMT2D mutations have been observed to exhibit a significantly higher TMB and improved immune infiltration in TIME (125, 182, 183). Furthermore, KMT2D mutations have been found to enhance immunotherapy responses in certain cancers (184). DNA damage, increased mutation burden, intron retention, and activated TE expression contribute to elevated neoantigen synthesis (185). Additionally, KMT2D has been linked to mismatch repair deficiency in prostate cancer (186). KMT2D mutations have been identified as potential predictive markers for melanoma and effective predictors of immune response in colorectal cancer (187, 188). The combination of immunotherapy and targeted therapy for KMT2D holds promising potential for improving treatment outcomes.
4.5 P38 MAPK with immunotherapy
Research indicates that 3-hydroxy-3-methylglutaryl-CoA reductase, an essential enzyme in the mevalonate pathway, regulates the production of PD-1 via p38 MAPK (189). Observations suggest that inhibiting the p38 MAPK pathway stimulates CD8+/CD4+ T cells and fosters the growth of CD8+ T cells (190, 191). A potential treatment approach could involve a temporary boost in immunity by co-blocking PD-1 and p38 MAPK (191). However, p38 MAPK activation in CD8+ T cells compromises the anti-tumor efficacy of some anti-PD-L1 therapies, indicating that a combination of anti-PD-L1 and p38 MAPK-targeted therapies may hold potential (192). Furthermore, a combination of anti-PD-1 therapy and metformin, a drug known to boost the anticancer activity of natural killer cells in a p38 MAPK-dependent manner, may be effective against metastatic melanoma (193).
4.6 TNFR1 with immunotherapy
TNFR1 is crucial for anti-tumor immunity, potentially predicting the immune response (194). In addition, TNFR1 is required for the TNF signaling pathway in melanoma, affecting the efficacy of anti-PD-1 therapy. After anti-PD-1 therapy, TNF insufficiency reduces TIL mortality and increases CD8+ TIL numbers, with similar effects observed in lung cancer (195). Studies suggest that TNFR1 modulates immune cell expression and enhances anti-tumor activity through NF-κB and p38 MAPK signaling cascade, regulating anti-tumor immunity, which is crucial for NSCLC development (196, 197). Furthermore, TNFR1 and interferon-γ signaling cooperate to prevent multistage carcinogenesis as a compromise in either pathway may cause T lymphocytes to promote tumor formation (198).
5 Multi-target combination therapy
5.1 FGFR1 and MAPK
Although several FGFR1 inhibitors have been tested, their clinical effectiveness is limited owing to resistance in various cancers, including lung, breast, colorectal, and melanoma (199, 200). A clinical study suggests that the TAM family of tyrosine kinases, MET, neurotrophic factor receptor pathway, and MAPK pathway are major contributors to FGFR resistance. Specific genes such as KRAS G13D, HRAS, MAP3K8, and BRAF V600E within the MAPK pathway are linked to resistance against the FGFR inhibitor BGJ398 (201). The latter three gene mutations are responsible for PI3K inhibition in breast cancer and RAF/MEK inhibition in BRAF-mutated melanoma (202, 203). BGJ398 inhibits FGFR by deactivating AKT phosphorylation and temporarily suppressing ERK phosphorylation in the NCI-H2077 cell line. However, overexpression of neurotrophic factor receptor pathway 1, MET, and HRAS leads to the resurgence of ERK and AKT activity, indicating an up-regulation trend in ERK activity (201). Increased MAPK signaling gene expression signaling and p38 phosphorylation are common in FGFR inhibitor-sensitive cell lines. Moreover, p38 MAPK overexpression can diminish the effectiveness of FGFR inhibition, inducing resistance (157).
Specifically, in lung cancer cell lines with amplified FGFR1, MET overexpression induces resistance to FGFR inhibitors, indicating a possible inverse correlation between these pathways (204).
5.2 FGFR and EGFR
Commonly used EGFR-TKIs in clinical practice include gefitinib, erlotinib, and afatinib. However, drug resistance remains a challenge (205). KRAS G13D and BRAF V600E, two MAPK family members, increase resistance to erlotinib, gefitinib, and crizotinib in NSCLC and contribute to resistance against FGFR inhibitors (201). Activation of the FGF2/FGFR1 autocrine pathway in EGFR-dependent NSCLC leads to resistance to gefitinib and osimertinib, indicating a potential connection between the FGFR and EGFR pathways (206, 207). In lung cancer cell lines with the FGF2/FGFR1 pathway, targeting this resistance mechanism with EGFR-specific TKI does not significantly increase apoptosis, indicating that FGFR inhibitors may stabilize tumor progression rather than cause tumor regression (208). Therefore, combining FGFR-specific TKIs with EGFR-specific TKIs may serve as an effective targeted therapy for LSCC, potentially delaying the development of acquired resistance in EGFR-driven NSCLC.
5.3 PI3K and KMT2D
Studies have demonstrated that KMT2D modulates SOX2 expression in NSCLC through a PI3K-dependent manner, significantly impacting tumor growth. A KMT2D deficiency leads to reduced phosphoinositide-3-kinase-interacting protein 1 expression and increased phosphorylated AKT, accelerating tumor growth in NSCLC by activating the PI3K/AKT pathway and upregulating SOX2 expression (209). Inhibiting PI3K affects tumor growth in estrogen receptor (ER)-positive breast cancer and activates glucocorticoid-regulated kinase 1 through KMT2D, regulating ER-dependent transcription via a negative feedback loop (126, 210). Patients with ER-positive breast cancer may gain clinical benefit from the combined targeted therapy of KMT2D and PI3K, potentially increasing efficacy and preventing resistance to PI3K inhibition (211, 212). Although promising, these results are preliminary and more research is required to determine the effectiveness of KMT2D and PI3K targeted therapy in LSCC treatment. Considering KMT2D’s role in the PI3K/AKT/SOX2 axis in NSCLC, which affects tumor growth, combined targeted therapy of KMT2D and PI3K may offer new treatment possibilities for LSCC.
6 Discussion
This review explores the current landscape of targeted therapy for LSCC, highlighting both established and emerging targets. Targeted therapies have proven beneficial in LUAD treatment (213, 214). Patients with LSCC exhibit distinct genetic alterations from LUAD; thus, their therapeutic options remain limited (215). The scarcity of therapeutic targets, coupled with modest efficacy and prevalent adverse effects, underscores the need to expand or discover new targets. The NSD family, especially NSD3, is strongly linked to the development of several malignant cancers. The pathogenic mechanism and structure of NSD3 have been largely established. The link between LSCC pathogenesis and KMT2D deficiency has been established, paving the way for the increased development of targeted drugs. NSD3 and KMT2D are promising therapeutic targets, offering hope for the enhancement of treatment strategies for LSCC. The advent of immunotherapy has significantly improved the treatment of patients with LSCC. Nonetheless, limitations such as low immune response rates and drug resistance persist. This study delves into the potential advantages of integrating immunotherapy with targeted therapy and benefits of multi-target combination therapy are discussed.
It is vital to identify appropriate biomarkers to optimize the clinical effectiveness of targeted therapies. KEAP1 has been identified as one of the significantly mutated genes in LSCC (51). In a Japanese retrospective cohort study, approximately 13.4% of patients with LSCC carried the KEAP1 mutation (216). Blocking glutamine metabolism targeting the KEAP/NRF2 pathway is a promising therapeutic strategy. The broad-acting glutamine antagonist sirpiglenastat has been shown to induce anti-tumor efficacy (217), and has therapeutic potential in HNSCC (218). In a recent Lung-MAP next-generation sequencing analysis, David Kozono’s team found that NFE2L2, KEAP1 and PARP4 are a mutually exclusive set of gene mutations (219), indicating KEAP1 as a potential biomarker in LSCC. In addition, recent research suggests that two members of the sequence similarity family, FAM20A and FAM83A, have potential clinical applications in lung squamous cell carcinoma. A recent study based on genomic databases and analysis of clinical samples showed that FAM20A was significantly reduced in LSCC and positively correlated with immune checkpoints such as CTLA-4, leading to reduced survival (220). Moreover, Lu et al. found that FAM83A, which is overexpressed in LSCC, can promote LSCC cell growth by activating the Wnt/β-catenin signaling pathway, and is a potential biomarker (221).
Epigenetic targeted drugs have promise in treating hematological malignancies and solid tumors, with tazemetostat potentially improving outcomes in solid tumors (222, 223). Furthermore, the epigenetic landscape of LSCC has become clear; for example, the demethylation of cancer/testicular antigen is highly associated with lung cancer, with its expression correlating with the immune microenvironment of NSCLC, offering diagnostic and prognostic insights (224, 225). Promoter hypermethylation promotes the transcriptional silencing of tumor suppressor genes, facilitating LSCC diagnosis and prognosis prediction. Studies have proposed various molecular drugs targeting the epigenetic network of LSCC to revert cancer cells to normalcy, including repaglinide, procainamide, and clomipramine (226). DNA methyltransferases, histone methyltransferase enhancer of zeste homolog 2, and so on are potential targets for targeted therapies, indirectly increasing targeted therapies’ efficacy (227). Additionally, NSD3 and KMT2D, as potential new targets, are vital substances in epigenetic regulation, linking targeted and epigenetic therapy. Their roles in histone methyl transfer may pave the way for LSCC innovative treatment through the synergy of epigenetics and targeted therapies. Epigenetic therapy can reverse the resistance mechanisms of gene changes and transcriptional reprogramming (228–231). The potential of combined targeted therapy of DNA methyltransferase inhibitors and venetoclax (antiapoptotic B-cell lymphoma 2 protein inhibitor) in treating hematological malignancies was investigated, and this combination therapy was recognized as a “breakthrough therapy” by the FDA for patients with specific acute myeloid leukemia (228, 232–234). Therefore, epigenetic therapy can also complement targeted therapies by combining, significantly improving efficacy.
Furthermore, emerging antibody-drug conjugates (ADCs) offer a fresh perspective. ADCs comprise a monoclonal antibody that selectively targets specific tumor cell proteins and is linked to a cytotoxic drug known as the payload (235). As of August 2023, fifteen ADCs have been globally approved, targeting molecules such as human epidermal growth factor receptor 2, trophoblast antigen 2, nectin-4, CD22, and CD33. Among these, the ADC cetuximab saratolacan, targeting EGFR, has been approved for HNSCC (236). Early trials in NSCLC suggest potential benefits from ADCs such as trastuzumab emtansine, patritumab deruxtecan, and sacituzumab govitecan (237–239). Moreover, the bispecific delta-like canonical Notch ligand 3/CD3 IgG-like T-cell engager, which binds to both delta-like canonical Notch ligand 3 and immune cells, has inspired therapeutic drugs and antibodies to enhance treatment efficacy (240). The potential of combining targeted therapeutic drugs with antibodies to form conjugates akin to the ADCs is an area of interest for researchers. Furthermore, EGFR inhibition enhances the therapeutic effect of ADC HER3-DXd, suggesting the potential of combining targeted therapy with ADCs for more effective treatment (241).
Considering the landscape of targeted therapy for LSCC, we recommend the following clinical patient selection strategies. Age, smoking status, and overall health can inform eligibility for specific treatment options. Using genomic tests to detect mutated genes and immunohistochemical tests to detect PD-L1 levels facilitates the identification of potential therapeutic targets and development of combination treatment strategies. Finally, with patient knowledge and committee approval, patients may be encouraged to participate in clinically targeted drug trials to improve treatment effectiveness. The integration of various therapeutic options, and updating of clinical practice and research are of great significance to the improvement of therapeutic benefits.
Author contributions
WC: Writing – review & editing. QC: Writing – original draft. XZ: Writing – original draft. JL: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
ADCs: Antibody-drug conjugates
AKT: Protein kinase B
ALK: Anaplastic lymphoma kinase
BRAF: V-raf murine sarcoma viral oncogene homolog B
CDK4/6: Cell cycle protein-dependent kinases 4 and 6
DDR2: Discoidin domain receptor 2
EGFR: Epidermal growth factor receptor
ER: Estrogen receptor
FGFR: Fibroblast growth factor receptor
ICIs: Immune checkpoint inhibitors
KEAP1: Kelch-like ECH-associated protein 1
KMT2D: Lysine methyltransferase 2 D
KRAS: Kirsten rat sarcoma virus
LSCC: Lung squamous cell carcinoma
LUAD: Lung adenocarcinoma
MEK: Mitogen-activated protein kinase kinase
NF-κB: Nuclear factor-κB
NRF2: Nuclear factor erythroid 2 related factor 2
NSCLC: Non-small cell lung cancer
NSD3: Nuclear receptor-binding SET domain protein 3
P38 MAPK: P38 mitogen-activated protein kinases
PD-L1: Programmed Cell Death Ligand 1
PD-1: Programmed Cell Death Protein 1
PI3K: Phosphoinositide 3-kinase
RAS: Rat sarcoma
SOX2: SRY-box transcription factor 2
TILs: Tumor-infiltrating lymphocytes
TIME: Tumor immune microenvironment
TKI: Tyrosine kinase inhibitor
TMB: Tumor mutational burden
TME: Tumor microenvironment
TNFR1: Tumor necrosis factor receptor 1
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. (2015) 65:87–108. doi: 10.3322/caac.21262
2. Liang C, Pan W, Zhou Z, Liu X. Identification of prognostic biomarkers of smoking-related lung cancer. J Thorac Dis. (2024) 16:1438–49. doi: 10.21037/JTD-23-1890/COIF
3. Heist RS, Sequist LV, Engelman JA. Genetic changes in squamous cell lung cancer: a review. J Thorac Oncol. (2012) 7:924–33. doi: 10.1097/JTO.0b013e31824cc334
4. Zhang Q, Cai G, Cui F, Li F, Liang H, Gao L, et al. The relationship of airflow limitation with lung squamous cell carcinoma: evidence from mendelian randomization analysis. J Cancer Res Clin Oncol. (2023) 149:6999–7006. doi: 10.1007/s00432-023-04612-6
5. Khuder SA, Mutgi AB. Effect of smoking cessation on major histologic types of lung cancer. Chest. (2001) 120:1577–83. doi: 10.1378/chest.120.5.1577
6. Cheng Y, Zhang T, Xu Q. Therapeutic advances in non-small cell lung cancer: Focus on clinical development of targeted therapy and immunotherapy. MedComm (Beijing). (2021) 2:692–729. doi: 10.1002/MCO2.105
7. Jeong Y, Hoang NT, Lovejoy A, Stehr H, Newman AM, Gentles AJ, et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discovery. (2017) 7:86–101. doi: 10.1158/2159-8290.CD-16-0127
8. Kim Y, Hammerman PS, Kim J, Yoon JA, Lee Y, Sun JM, et al. Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. J Clin Oncol. (2014) 32:121–8. doi: 10.1200/JCO.2013.50.8556
9. Pan Y, Han H, Labbe KE, Zhang H, Wong K-K. Recent advances in preclinical models for lung squamous cell carcinoma. Oncogene. (2021) 40:2817–29. doi: 10.1038/s41388-021-01723-7
10. Xia W-Y, Feng W, Zhang C-C, Shen Y-J, Zhang Q, Yu W, et al. Radiotherapy for non-small cell lung cancer in the immunotherapy era: the opportunity and challenge-a narrative review. Transl Lung Cancer Res. (2020) 9:2120–36. doi: 10.21037/tlcr-20-827
11. Guo Q, Liu L, Chen Z, Fan Y, Zhou Y, Yuan Z, et al. Current treatments for non-small cell lung cancer. Front Oncol. (2022) 12:945102. doi: 10.3389/fonc.2022.945102
12. Kim ES. Chemotherapy resistance in lung cancer. Adv Exp Med Biol. (2016) 893:189–209. doi: 10.1007/978-3-319-24223-1_10
13. Vojtíšek R. Cardiac toxicity of lung cancer radiotherapy. Rep Pract Oncol Radiother. (2020) 25:13–9. doi: 10.1016/j.rpor.2019.10.007
14. Alberti W, Anderson G, Bartolucci A, Bell D, Villalba JB, Brodin O, et al. Chemotherapy in non-small cell lung cancer: a meta-analysis using updated data on individual patients from 52 randomised clinical trials. Non-small Cell Lung Cancer Collaborative Group. BMJ. (1995) 311:899–909.
15. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. (2010) 466:869–73. doi: 10.1038/nature09208
16. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
17. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
18. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
19. van den Bulk J, Verdegaal EM, de Miranda NF. Cancer immunotherapy: broadening the scope of targetable tumours. Open Biol. (2018) 8. doi: 10.1098/rsob.180037
20. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
21. Pennock GK, Chow LQM. The evolving role of immune checkpoint inhibitors in cancer treatment. Oncologist. (2015) 20:812–22. doi: 10.1634/theoncologist.2014-0422
22. Yuan H, Liu J, Zhang J. The current landscape of immune checkpoint blockade in metastatic lung squamous cell carcinoma. Molecules. (2021) 26. doi: 10.3390/molecules26051392
23. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. (2015) 161:205–14. doi: 10.1016/j.cell.2015.03.030
24. Aldea M, Andre F, Marabelle A, Dogan S, Barlesi F, Soria J-C. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discovery. (2021) 11:874–99. doi: 10.1158/2159-8290.CD-20-1638
25. Yang M, Lin C, Wang Y, Chen K, Zhang H, Li W. Identification of a cytokine-dominated immunosuppressive class in squamous cell lung carcinoma with implications for immunotherapy resistance. Genome Med. (2022) 14. doi: 10.1186/S13073-022-01079-X
26. Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. (2015) 33:1152–8. doi: 10.1038/NBT.3344
27. Liu LU, Xie B, Zhu W, He Q, Zhou J, Liu S, et al. High expression of PD-L1 mainly occurs in non-small cell lung cancer patients with squamous cell carcinoma or poor differentiation. Oncol Res. (2023) 31:275–86. doi: 10.32604/or.2023.028227
28. Kamel-Reid S, Chong G, Ionescu DN, Magliocco AM, Spatz A, Tsao M, et al. EGFR tyrosine kinase mutation testing in the treatment of non-small-cell lung cancer. Curr Oncol. (2012) 19:e67–74. doi: 10.3747/co.19.862
29. Choi J-S, Kang H-M, Na K, Kim J, Kim T-W, Jung J, et al. KEAP1-NRF2 pathway as a novel therapeutic target for EGFR-mutant non-small cell lung cancer. Tuberc Respir Dis (Seoul). (2024). doi: 10.4046/TRD.2024.0087
30. Zhou S, Wang H, Jiang W, Yu Q. Clinicopathological characteristics and EGFR-TKIs efficacies in lung squamous cell carcinoma patients harboring an EGFR sensitizing mutation. Onco Targets Ther. (2019) 12:8863–71. doi: 10.2147/OTT.S225760
31. Arafeh R, Samuels Y. PIK3CA in cancer: The past 30 years. Semin Cancer Biol. (2019) 59:36–49. doi: 10.1016/j.semcancer.2019.02.002
32. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell. (2018) 173:321–337.e10. doi: 10.1016/j.cell.2018.03.035
33. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. (2010) 11:329–41. doi: 10.1038/nrm2882
34. Janku F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat Rev. (2017) 59:93–101. doi: 10.1016/j.ctrv.2017.07.005
35. Peng Y, Wang Y, Zhou C, Mei W, Zeng C. PI3K/akt/mTOR pathway and its role in cancer therapeutics: are we making headway? Front Oncol. (2022) 12:819128. doi: 10.3389/FONC.2022.819128
36. Madsen RR, Vanhaesebroeck B, Semple RK. Cancer-associated PIK3CA mutations in overgrowth disorders. Trends Mol Med. (2018) 24:856–70. doi: 10.1016/j.molmed.2018.08.003
37. Lau SCM, Pan Y, Velcheti V, Wong KK. Squamous cell lung cancer: Current landscape and future therapeutic options. Cancer Cell. (2022) 40:1279–93. doi: 10.1016/j.ccell.2022.09.018
38. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. (2012) 489:519–25. doi: 10.1038/nature11404
39. Mukherjee R, Vanaja KG, Boyer JA, Gadal S, Solomon H, Chandarlapaty S, et al. Regulation of PTEN translation by PI3K signaling maintains pathway homeostasis. Mol Cell. (2021) 81:708–723.e5. doi: 10.1016/j.molcel.2021.01.033
40. Cavazzoni A, La Monica S, Alfieri R, Ravelli A, van der Steen N, Sciarrillo R, et al. Enhanced efficacy of AKT and FAK kinase combined inhibition in squamous cell lung carcinomas with stable reduction in PTEN. Oncotarget. (2017) 8:53068–83. doi: 10.18632/oncotarget.18087
41. Huang Q, Zhou Y, Wang B, Zhao Y, Zhang F, Ding B. Mutational landscape of pan-cancer patients with PIK3CA alterations in Chinese population. BMC Med Genomics. (2022) 15:146. doi: 10.1186/s12920-022-01297-7
42. Liao RG, Watanabe H, Meyerson M, Hammerman PS. Targeted therapy for squamous cell lung cancer. Lung Cancer Manag. (2012) 1:293–300. doi: 10.2217/lmt.12.40
43. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discovery. (2014) 13:140–56. doi: 10.1038/nrd4204
44. Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. (2011) 29:4688–95. doi: 10.1200/JCO.2011.35.5263
45. Arteaga CL. Overview of epidermal growth factor receptor biology and its role as a therapeutic target in human neoplasia. Semin Oncol. (2002) 29:3–9. doi: 10.1053/sonc.2002.35642
46. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. (2004) 64:8919–23. doi: 10.1158/0008-5472.CAN-04-2818
47. Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. (2005) 97:339–46. doi: 10.1093/jnci/dji055
48. Tan AC, Tan DSW. Targeted therapies for lung cancer patients with oncogenic driver molecular alterations. J Clin Oncol. (2022) 40:611–25. doi: 10.1200/JCO.21.01626
49. Gene mutations in squamous cell NSCLC: insignificance of EGFR, KRAS and PIK3CA mutations in prediction of EGFR-TKI treatment efficacy - PubMed . Available online at (Accessed September 17, 2024).
50. Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C, et al. A proteogenomic portrait of lung squamous cell carcinoma. Cell. (2021) 184:4348–4371.e40. doi: 10.1016/J.CELL.2021.07.016
51. Niu Z, Jin R, Zhang Y, Li H. Signaling pathways and targeted therapies in lung squamous cell carcinoma: mechanisms and clinical trials. Signal Transduct Target Ther. (2022) 7. doi: 10.1038/S41392-022-01200-X
52. Ye Z, Huang Y, Ke J, Zhu X, Leng S, Luo H. Breakthrough in targeted therapy for non-small cell lung cancer. BioMed Pharmacother. (2021) 133. doi: 10.1016/J.BIOPHA.2020.111079
53. Chen Z, Vallega KA, Wang D, Quan Z, Fan S, Wang Q, et al. Inhibition of hTERT/telomerase/telomere mediates therapeutic efficacy of osimertinib in EGFR mutant lung cancer. J Exp Med. (2024) 221. doi: 10.1084/JEM.20240435
54. Wang Z, Zhou F, Xu S, Wang K, Ding H. The efficacy and safety of immune checkpoint inhibitors for patients with EGFR-mutated non-small cell lung cancer who progressed on EGFR tyrosine-kinase inhibitor therapy: A systematic review and network meta-analysis. Cancer Med. (2023) 12:18516–30. doi: 10.1002/cam4.6453
55. Dai S, Zhou Z, Chen Z, Xu G, Chen Y. Fibroblast growth factor receptors (FGFRs): structures and small molecule inhibitors. Cells. (2019) 8. doi: 10.3390/cells8060614
56. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. (2010) 10:116–29. doi: 10.1038/nrc2780
57. Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. (2010) 2:62ra93. doi: 10.1126/scitranslmed.3001451
58. Grose R, Dickson C. Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev. (2005) 16:179–86. doi: 10.1016/J.CYTOGFR.2005.01.003
59. McKeehan WL, Wang F, Kan M. The heparan sulfate-fibroblast growth factor family: diversity of structure and function. Prog Nucleic Acid Res Mol Biol. (1998) 59:135–76. doi: 10.1016/S0079-6603(08)61031-4
60. Tiong KH, Tan BS, Choo HL, Chung FFL, Hii LW, Tan SH, et al. Fibroblast growth factor receptor 4 (FGFR4) and fibroblast growth factor 19 (FGF19) autocrine enhance breast cancer cells survival. Oncotarget. (2016) 7:57633–50. doi: 10.18632/ONCOTARGET.9328
61. Feng S, Dakhova O, Creighton CJ, Ittmann M. Endocrine fibroblast growth factor FGF19 promotes prostate cancer progression. Cancer Res. (2013) 73:2551–62. doi: 10.1158/0008-5472.CAN-12-4108
62. Zhao X, Xu F, Dominguez NP, Xiong Y, Xiong Z, Peng H, et al. FGFR4 provides the conduit to facilitate FGF19 signaling in breast cancer progression. Mol Carcinog. (2018) 57:1616–25. doi: 10.1002/MC.22884
63. Li F, Li Z, Han Q, Cheng Y, Ji W, Yang Y, et al. Enhanced autocrine FGF19/FGFR4 signaling drives the progression of lung squamous cell carcinoma, which responds to mTOR inhibitor AZD2104. Oncogene. (2020) 39:3507–21. doi: 10.1038/S41388-020-1227-2
64. Nagao-Kitamoto H, Nagata M, Nagano S, Kitamoto S, Ishidou Y, Yamamoto T, et al. GLI2 is a novel therapeutic target for metastasis of osteosarcoma. Int J Cancer. (2015) 136:1276–84. doi: 10.1002/IJC.29107
65. Zhang Y, Wu T, Wang Y, Chen Z, Chen J, Lu S, et al. Reciprocal FGF19-GLI2 signaling induces epithelial-to-mesenchymal transition to promote lung squamous cell carcinoma metastasis. Cell Oncol (Dordr). (2023) 46:437–50. doi: 10.1007/S13402-022-00760-Y
66. Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. (2019) 16:105–22. doi: 10.1038/s41571-018-0115-y
67. Carter EP, Fearon AE, Grose RP. Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. (2015) 25:221–33. doi: 10.1016/j.tcb.2014.11.003
68. Hong SH, Kim TM. High-resolution genomic configuration of FGFR rearrangements dictates the therapeutic vulnerability of squamous cell lung cancers. Transl Lung Cancer Res. (2024) 13:236–9. doi: 10.21037/TLCR-23-705/COIF
69. Bilsland JG, Wheeldon A, Mead A, Znamenskiy P, Almond S, Waters KA, et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology. (2008) 33:685–700. doi: 10.1038/SJ.NPP.1301446
70. Golding B, Luu A, Jones R, Viloria-Petit AM. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol Cancer. (2018) 17:1–15. doi: 10.1186/S12943-018-0810-4
71. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. (2007) 448:561–6. doi: 10.1038/NATURE05945
72. Ou SHI, Zhu VW, Nagasaka M. Catalog of 5’ Fusion partners in ALK-positive NSCLC circa 2020. JTO Clin Res Rep. (2020) 1. doi: 10.1016/J.JTOCRR.2020.100015
73. Chia PL, Dobrovic A, Dobrovic A, John T. Prevalence and natural history of ALK positive non-small-cell lung cancer and the clinical impact of targeted therapy with ALK inhibitors. Clin Epidemiol. (2014) 6:423–32. doi: 10.2147/CLEP.S69718
74. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. (2009) 27:4247–53. doi: 10.1200/JCO.2009.22.6993
75. Tinsley E, Bredin P, Toomey S, Hennessy BT, Furney SJ. KMT2C and KMT2D aberrations in breast cancer. Trends Cancer. (2024) 10:519–30. doi: 10.1016/J.TRECAN.2024.02.003
76. Wu J, Savooji J, Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol. (2016) 9:1–7. doi: 10.1186/S13045-016-0251-8
77. Testa U, Castelli G, Pelosi E. Alk-rearranged lung adenocarcinoma: From molecular genetics to therapeutic targeting. Tumori. (2024) 110:88–95. doi: 10.1177/03008916231202149
78. Peng L, Zhu L, Sun Y, Stebbing J, Selvaggi G, Zhang Y, et al. Targeting ALK rearrangements in NSCLC: current state of the art. Front Oncol. (2022) 12:863461. doi: 10.3389/FONC.2022.863461
79. Liu F, Yang X, Geng M, Huang M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm Sin B. (2018) 8:552–62. doi: 10.1016/J.APSB.2018.01.008
80. Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. (2001) 294:1299–304. doi: 10.1126/SCIENCE.1062023
81. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EWT, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, Malignant transformation and drug resistance. Biochim Biophys Acta. (2007) 1773:1263–84. doi: 10.1016/J.BBAMCR.2006.10.001
82. Hassan B, Akcakanat A, Holder AM, Meric-Bernstam F. Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surg Oncol Clin N Am. (2013) 22:641–64. doi: 10.1016/J.SOC.2013.06.008
83. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discovery. (2020) 19:533–52. doi: 10.1038/S41573-020-0068-6
84. Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol. (2013) 24:2371–6. doi: 10.1093/ANNONC/MDT205
85. Acker F, Stratmann J, Aspacher L, Nguyen NTT, Wagner S, Serve H, et al. KRAS mutations in squamous cell carcinomas of the lung. Front Oncol. (2021) 11:788084. doi: 10.3389/FONC.2021.788084
86. Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. (2007) 1773:1213–26. doi: 10.1016/J.BBAMCR.2006.10.005
87. Degirmenci U, Wang M, Hu J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells. (2020) 9. doi: 10.3390/CELLS9010198
88. Yaeger R, Corcoran RB. Targeting alterations in the RAF-MEK pathway. Cancer Discovery. (2019) 9:329–41. doi: 10.1158/2159-8290.CD-18-1321
89. Haura EB, Ricart AD, Larson TG, Stella PJ, Bazhenova L, Miller VA, et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. (2010) 16:2450–7. doi: 10.1158/1078-0432.CCR-09-1920
90. Blumenschein GR, Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)†. Ann Oncol. (2015) 26:894–901. doi: 10.1093/ANNONC/MDV072
91. Han J, Liu Y, Yang S, Wu X, Li H, Wang Q. MEK inhibitors for the treatment of non-small cell lung cancer. J Hematol Oncol. (2021) 14:1–12. doi: 10.1186/S13045-020-01025-7
92. Hume S, Dianov GL, Ramadan K. A unified model for the G1/S cell cycle transition. Nucleic Acids Res. (2020) 48:12483–501. doi: 10.1093/NAR/GKAA1002
93. Goel S, DeCristo MJ, McAllister SS, Zhao JJ. CDK4/6 inhibition in cancer: beyond cell cycle arrest. Trends Cell Biol. (2018) 28:911–25. doi: 10.1016/J.TCB.2018.07.002
94. Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. (1999) 98:859–69. doi: 10.1016/S0092-8674(00)81519-6
95. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. (2017) 17:93–115. doi: 10.1038/NRC.2016.138
96. Beaver JA, Amiri-Kordestani L, Charlab R, Chen W, Palmby T, Tilley A, et al. FDA approval: palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res. (2015) 21:4760–6. doi: 10.1158/1078-0432.CCR-15-1185
97. Edelman MJ, Redman MW, Albain KS, McGary EC, Rafique NM, Petro D, et al. SWOG S1400C (NCT02154490)-A phase II study of palbociclib for previously treated cell cycle gene alteration-positive patients with stage IV squamous cell lung cancer (Lung-MAP substudy). J Thorac Oncol. (2019) 14:1853–9. doi: 10.1016/J.JTHO.2019.06.027
98. Goldman JW, Mazieres J, Barlesi F, Dragnev KH, Koczywas M, Göskel T, et al. A randomized phase III study of abemaciclib versus erlotinib in patients with stage IV non-small cell lung cancer with a detectable KRAS mutation who failed prior platinum-based therapy: JUNIPER. Front Oncol. (2020) 10:578756. doi: 10.3389/FONC.2020.578756
99. Zhang J, Xu D, Zhou Y, Zhu Z, Yang X. Mechanisms and implications of CDK4/6 inhibitors for the treatment of NSCLC. Front Oncol. (2021) 11:676041. doi: 10.3389/FONC.2021.676041
100. Elkamhawy A, Lu Q, Nada H, Woo J, Quan G, Lee K. The journey of DDR1 and DDR2 kinase inhibitors as rising stars in the fight against cancer. Int J Mol Sci. (2021) 22. doi: 10.3390/IJMS22126535
101. Walsh LA, Nawshad A, Medici D. Discoidin domain receptor 2 is a critical regulator of epithelial-mesenchymal transition. Matrix Biol. (2011) 30:243–7. doi: 10.1016/J.MATBIO.2011.03.007
102. Fu HL, Valiathan RR, Arkwright R, Sohail A, Mihai C, Kumarasiri M, et al. Discoidin domain receptors: unique receptor tyrosine kinases in collagen-mediated signaling. J Biol Chem. (2013) 288:7430–7. doi: 10.1074/JBC.R112.444158
103. Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt A, Zhou W, et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discovery. (2011) 1:78–89. doi: 10.1158/2159-8274.CD-11-0005
104. Ricordel C, Lespagnol A, Llamas-Gutierrez F, de Tayrac M, Kerjouan M, Fievet A, et al. Mutational landscape of DDR2 gene in lung squamous cell carcinoma using next-generation sequencing. Clin Lung Cancer. (2018) 19:163–169.e4. doi: 10.1016/J.CLLC.2017.10.006
105. Miao L, Wang Y, Zhu S, Shi M, Li Y, Ding J, et al. Identification of novel driver mutations of the discoidin domain receptor 2 (DDR2) gene in squamous cell lung cancer of Chinese patients. BMC Cancer. (2014) 14:1–10. doi: 10.1186/1471-2407-14-369
106. Sasaki H, Shitara M, Yokota K, Okuda K, Hikosaka Y, Moriyama S, et al. DDR2 polymorphisms and mRNA expression in lung cancers of Japanese patients. Oncol Lett. (2012) 4:33–7. doi: 10.3892/OL.2012.684
107. Day E, Waters B, Spiegel K, Alnadaf T, Manley PW, Buchdunger E, et al. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur J Pharmacol. (2008) 599:44–53. doi: 10.1016/J.EJPHAR.2008.10.014
108. Brunner AM, Costa DB, Heist RS, Garcia E, Lindeman NI, Sholl LM, et al. Treatment-related toxicities in a phase II trial of dasatinib in patients with squamous cell carcinoma of the lung. J Thorac Oncol. (2013) 8:1434–7. doi: 10.1097/JTO.0B013E3182A47162
109. Beauchamp EM, Woods BA, Dulak AM, Tan L, Xu C, Gray NS, et al. Acquired resistance to dasatinib in lung cancer cell lines conferred by DDR2 gatekeeper mutation and NF1 loss. Mol Cancer Ther. (2014) 13:475–82. doi: 10.1158/1535-7163.MCT-13-0817
110. Payne LS, Huang PH. Discoidin domain receptor 2 signaling networks and therapy in lung cancer. J Thorac Oncol. (2014) 9:900–4. doi: 10.1097/JTO.0000000000000164
111. Li W, Tian W, Yuan G, Deng P, Sengupta D, Cheng Z, et al. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature. (2021) 590:498–503. doi: 10.1038/s41586-020-03069-8
112. Xiong Q, Zhou Y, Zhang S, Zhang Y, Xu Y, Yang Y, et al. NSD3, a member of nuclear receptor-binding SET domain family, is a potential prognostic biomarker for pancreatic cancer. Cancer Med. (2023) 12:10961–78. doi: 10.1002/cam4.5774
113. Topchu I, Pangeni RP, Bychkov I, Miller SA, Izumchenko E, Yu J, et al. The role of NSD1, NSD2, and NSD3 histone methyltransferases in solid tumors. Cell Mol Life Sci. (2022) 79:285. doi: 10.1007/s00018-022-04321-2
114. Yuan G, Flores NM, Hausmann S, Lofgren SM, Kharchenko V, Angulo-Ibanez M, et al. Elevated NSD3 histone methylation activity drives squamous cell lung cancer. Nature. (2021) 590:504–8. doi: 10.1038/s41586-020-03170-y
115. Voutsadakis IA. Characteristics and prognosis of 8p11.23-amplified squamous lung carcinomas. J Clin Med. (2023) 12. doi: 10.3390/JCM12051711
116. Xu D, Liu S, Wu X, Marti TM, Dorn P, Schmid RA, et al. Dissecting the immunological profiles in NSD3-amplified LUSC through integrative multi-scale analyses. Cancers (Basel). (2022) 14. doi: 10.3390/CANCERS14204997
117. Husmann D, Gozani O. Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol. (2019) 26:880–9. doi: 10.1038/s41594-019-0298-7
118. Shrestha A, Kim N, Lee SJ, Jeon YH, Song JJ, An H, et al. Targeting the nuclear receptor-binding SET domain family of histone lysine methyltransferases for cancer therapy: recent progress and perspectives. J Med Chem. (2021) 64:14913–29. doi: 10.1021/ACS.JMEDCHEM.1C01116
119. Xiu S, Chi X, Jia Z, Shi C, Zhang X, Li Q, et al. NSD3: Advances in cancer therapeutic potential and inhibitors research. Eur J Med Chem. (2023) 256:115440. doi: 10.1016/j.ejmech.2023.115440
120. Böttcher J, Dilworth D, Reiser U, Neumüller RA, Schleicher M, Petronczki M, et al. Fragment-based discovery of a chemical probe for the PWWP1 domain of NSD3. Nat Chem Biol. (2019) 15:822–9. doi: 10.1038/S41589-019-0310-X
121. Nuñez Y, Vera S, Baeza V, Gonzalez-Pecchi V. NSD3 in cancer: unraveling methyltransferase-dependent and isoform-specific functions. Int J Mol Sci. (2024) 25. doi: 10.3390/IJMS25020944
122. Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. (2015) 15:334–46. doi: 10.1038/NRC3929
123. Froimchuk E, Jang Y, Ge K. Histone H3 lysine 4 methyltransferase KMT2D. Gene. (2017) 627:337–42. doi: 10.1016/J.GENE.2017.06.056
124. Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. (2013) 45:1113–20. doi: 10.1038/NG.2764
125. Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. (2015) 21:1190–8. doi: 10.1038/NM.3940
126. Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. (2017) 355:1324–30. doi: 10.1126/SCIENCE.AAH6893
127. Fang Z, Wu X, Xiao L, Wang C, Zhao Y, Zhang Q, et al. Somatic KMT2D loss-of-function mutations in lung squamous cell carcinoma: a single-center cohort study. J Thorac Dis. (2024) 16:3338–49. doi: 10.21037/JTD-24-134/COIF
128. Hillman RT, Celestino J, Terranova C, Beird HC, Gumbs C, Little L, et al. KMT2D/MLL2 inactivation is associated with recurrence in adult-type granulosa cell tumors of the ovary. Nat Commun. (2018) 9. doi: 10.1038/S41467-018-04950-X
129. Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. (2015) 21:1199–208. doi: 10.1038/NM.3943
130. Xiong W, Deng Z, Tang Y, Deng Z, Li M. Downregulation of KMT2D suppresses proliferation and induces apoptosis of gastric cancer. Biochem Biophys Res Commun. (2018) 504:129–36. doi: 10.1016/J.BBRC.2018.08.143
131. Pan Y, Han H, Hu H, Wang H, Song Y, Hao Y, et al. KMT2D deficiency drives lung squamous cell carcinoma and hypersensitivity to RTK-RAS inhibition. Cancer Cell. (2023) 41:88–105.e8. doi: 10.1016/J.CCELL.2022.11.015
132. Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, et al. KMT2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell. (2020) 37:599–617.e7. doi: 10.1016/J.CCELL.2020.03.005
133. Battino M, Giampieri F, Pistollato F, Sureda A, de Oliveira MR, Pittalà V, et al. Nrf2 as regulator of innate immunity: A molecular Swiss army knife! Biotechnol Adv. (2018) 36:358–70. doi: 10.1016/j.bioteChadv.2017.12.012
134. Lin L, Wu Q, Lu F, Lei J, Zhou Y, Liu Y, et al. Nrf2 signaling pathway: current status and potential therapeutic targetable role in human cancers. Front Oncol. (2023) 13:1184079. doi: 10.3389/FONC.2023.1184079
135. Tian J, Wang X, Shi H, Wu H, Wang C, Liu N, et al. Sestrin2/Keap1/Nrf2 pathway regulates mucus hypersecretion in pulmonary epithelium induced by traffic-related PM2.5 and water-soluble extracts. Ecotoxicol Environ Saf. (2023) 264:115455. doi: 10.1016/j.ecoenv.2023.115455
136. Baumel-Alterzon S, Katz LS, Brill G, Jean-Pierre C, Li Y, Tse I, et al. Nrf2 regulates β-cell mass by suppressing β-cell death and promoting β-cell proliferation. Diabetes. (2022) 71:989–1011. doi: 10.2337/db21-0581
137. Pillai R, Hayashi M, Zavitsanou A-M, Papagiannakopoulos T. NRF2: KEAPing tumors protected. Cancer Discovery. (2022) 12:625–43. doi: 10.1158/2159-8290.CD-21-0922
138. Ko E, Kim D, Min DW, Kwon S-H, Lee J-Y. Nrf2 regulates cell motility through RhoA-ROCK1 signalling in non-small-cell lung cancer cells. Sci Rep. (2021) 11:1247. doi: 10.1038/s41598-021-81021-0
139. Sánchez-Ortega M, Garrido A, Cirauqui C, Sanz-Gonzalez L, Hernández MC, González-García A, et al. A potential therapeutic strategy based on acute oxidative stress induction for wild-type NRF2/KEAP1 lung squamous cell carcinoma. Redox Biol. (2024) 75. doi: 10.1016/J.REDOX.2024.103305
140. Zhang B, Ma Z, Tan B, Lin N. Targeting the cell signaling pathway Keap1-Nrf2 as a therapeutic strategy for adenocarcinomas of the lung. Expert Opin Ther Targets. (2019) 23:241–50. doi: 10.1080/14728222.2019.1559824
141. Dempke WCM, Reck M. KEAP1/NRF2 (NFE2L2) mutations in NSCLC - Fuel for a superresistant phenotype? Lung Cancer. (2021) 159:10–7. doi: 10.1016/j.lungcan.2021.07.006
142. Wang X, Liu Z, Zhang L, Yang Z, Chen X, Luo J, et al. Targeting deubiquitinase USP28 for cancer therapy. Cell Death Dis. (2018) 9. doi: 10.1038/S41419-017-0208-Z
143. Cao C, Vasilatos SN, Bhargava R, Fine JL, Oesterreich S, Davidson NE, et al. Functional interaction of histone deacetylase 5 (HDAC5) and lysine-specific demethylase 1 (LSD1) promotes breast cancer progression. Oncogene. (2017) 36:133–45. doi: 10.1038/ONC.2016.186
144. Wang Z, Song Q, Xue J, Zhao Y, Qin S. Ubiquitin-specific protease 28 is overexpressed in human glioblastomas and contributes to glioma tumorigenicity by regulating MYC expression. Exp Biol Med (Maywood). (2016) 241:255–64. doi: 10.1177/1535370215595468
145. Diefenbacher ME, Popov N, Blake SM, Schülein-Völk C, Nye E, Spencer-Dene B, et al. The deubiquitinase USP28 controls intestinal homeostasis and promotes colorectal cancer. J Clin Invest. (2014) 124:3407–18. doi: 10.1172/JCI73733
146. Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, et al. The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol. (2007) 9:765–74. doi: 10.1038/NCB1601
147. Josue Ruiz E, Pinto-Fernandez A, Turnbull AP, Lan L, Charlton TM, Scott HC, et al. USP28 deletion and small-molecule inhibition destabilizes c-MYC and elicits regression of squamous cell lung carcinoma. Elife. (2021) 10. doi: 10.7554/ELIFE.71596
148. Guerra B, Recio C, Aranda-Tavío H, Guerra-Rodríguez M, García-Castellano JM, Fernández-Pérez L. The mevalonate pathway, a metabolic target in cancer therapy. Front Oncol. (2021) 11:626971. doi: 10.3389/FONC.2021.626971
149. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). (2018) 38:1–14. doi: 10.1186/S40880-018-0301-4
150. Maier CR, Hartmann O, Prieto-Garcia C, Al-Shami KM, Schlicker L, Vogel FCE, et al. USP28 controls SREBP2 and the mevalonate pathway to drive tumour growth in squamous cancer. Cell Death Differ. (2023) 30:1710–25. doi: 10.1038/S41418-023-01173-6
151. Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med. (2009) 15:369–79. doi: 10.1016/J.MOLMED.2009.06.005
152. Lee S, Rauch J, Kolch W. Targeting MAPK signaling in cancer: mechanisms of drug resistance and sensitivity. Int J Mol Sci. (2020) 21. doi: 10.3390/IJMS21031102
153. Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. (2007) 11:191–205. doi: 10.1016/J.CCR.2006.12.013
154. Igea A, Nebreda AR. The stress kinase p38α as a target for cancer therapy. Cancer Res. (2015) 75:3997–4002. doi: 10.1158/0008-5472.CAN-15-0173
155. Martínez-Limón A, Joaquin M, Caballero M, Posas F, de Nadal E. The p38 pathway: from biology to cancer therapy. Int J Mol Sci. (2020) 21. doi: 10.3390/IJMS21061913
156. Yeung YT, Yin S, Lu B, Fan S, Yang R, Bai R, et al. Losmapimod overcomes gefitinib resistance in non-small cell lung cancer by preventing tetraploidization. EBioMedicine. (2018) 28:51–61. doi: 10.1016/J.EBIOM.2018.01.017
157. Zarczynska I, Gorska-Arcisz M, Cortez AJ, Kujawa KA, Wilk AM, Skladanowski AC, et al. p38 mediates resistance to FGFR inhibition in non-small cell lung cancer. Cells. (2021) 10. doi: 10.3390/CELLS10123363
158. Li R, Zeng L, Zhao H, Deng J, Pan L, Zhang S, et al. ATXN2-mediated translation of TNFR1 promotes esophageal squamous cell carcinoma via m6A-dependent manner. Mol Ther. (2022) 30:1089–103. doi: 10.1016/J.YMTHE.2022.01.006
159. Dondelinger Y, Darding M, Bertrand MJM, Walczak H. Poly-ubiquitination in TNFR1-mediated necroptosis. Cell Mol Life Sci. (2016) 73:2165–76. doi: 10.1007/S00018-016-2191-4
160. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. (2011) 21:103–15. doi: 10.1038/CR.2010.178
161. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. (2008) 132:344–62. doi: 10.1016/J.CELL.2008.01.020
162. Xiao Z, Shi G, Xi S, Singh AK, Willette-Brown J, Li X, et al. A TNFR1-UBCH10 axis drives lung squamous cell carcinoma dedifferentiation and metastasis through a cell-autonomous signaling loop. Cell Death Dis. (2022) 13. doi: 10.1038/S41419-022-05308-4
163. Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. (2019) 30:44–56. doi: 10.1093/annonc/mdy495
164. Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. (2019) 51:202–6. doi: 10.1038/s41588-018-0312-8
165. Li M, Gao X, Wang X. Identification of tumor mutation burden-associated molecular and clinical features in cancer by analyzing multi-omics data. Front Immunol. (2023) 14:1090838. doi: 10.3389/fimmu.2023.1090838
166. Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discovery. (2016) 6:1090–105. doi: 10.1158/2159-8290.CD-16-0716
167. Chandrasekaran S, Funk CR, Kleber T, Paulos CM, Shanmugam M, Waller EK. Strategies to overcome failures in T-cell immunotherapies by targeting PI3K-δ and -γ. Front Immunol. (2021) 12:718621. doi: 10.3389/fimmu.2021.718621
168. Wang L, Chen K, Weng S, Xu H, Ren Y, Cheng Q, et al. PI3K pathway mutation predicts an activated immune microenvironment and better immunotherapeutic efficacy in head and neck squamous cell carcinoma. World J Surg Oncol. (2023) 21:72. doi: 10.1186/s12957-023-02938-6
169. Liu C, Zheng S, Jin R, Wang X, Wang F, Zang R, et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. (2020) 470:95–105. doi: 10.1016/J.CANLET.2019.10.027
170. Seegobin K, Majeed U, Wiest N, Manochakian R, Lou Y, Zhao Y. Immunotherapy in non-small cell lung cancer with actionable mutations other than EGFR. Front Oncol. (2021) 11:750657. doi: 10.3389/FONC.2021.750657
171. Sun L, Hsu M, Cohen RB, Langer CJ, Mamtani R, Aggarwal C. Association between KRAS variant status and outcomes with first-line immune checkpoint inhibitor-based therapy in patients with advanced non-small-cell lung cancer. JAMA Oncol. (2021) 7:937–9. doi: 10.1001/JAMAONCOL.2021.0546
172. Heckler M, Ali LR, Clancy-Thompson E, Qiang L, Ventre KS, Lenehan P, et al. Inhibition of CDK4/6 promotes CD8 T-cell memory formation. Cancer Discovery. (2021) 11:2564–81. doi: 10.1158/2159-8290.CD-20-1540
173. Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discovery. (2018) 8:216–33. doi: 10.1158/2159-8290.CD-17-0915
174. Zhang QF, Li J, Jiang K, Wang R, Ge JL, Yang H, et al. CDK4/6 inhibition promotes immune infiltration in ovarian cancer and synergizes with PD-1 blockade in a B cell-dependent manner. Theranostics. (2020) 10:10619–33. doi: 10.7150/THNO.44871
175. Noh JY, Lee IP, Han NR, Kim M, Min YK, Lee SY, et al. Additive effect of CD73 inhibitor in colorectal cancer treatment with CDK4/6 inhibitor through regulation of PD-L1. Cell Mol Gastroenterol Hepatol. (2022) 14:769–88. doi: 10.1016/J.JCMGH.2022.07.005
176. Baird L, Taguchi K, Zhang A, Takahashi Y, Suzuki T, Kensler TW, et al. A NRF2-induced secretory phenotype activates immune surveillance to remove irreparably damaged cells. Redox Biol. (2023) 66:102845. doi: 10.1016/j.redox.2023.102845
177. Xu K, Ma J, Hall SRR, Peng R-W, Yang H, Yao F. Battles against aberrant KEAP1-NRF2 signaling in lung cancer: intertwined metabolic and immune networks. Theranostics. (2023) 13:704–23. doi: 10.7150/thno.80184
178. Leone RD, Zhao L, Englert JM, Sun I-M, Oh M-H, Sun I-H, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. (2019) 366:1013–21. doi: 10.1126/science.aav2588
179. Xue W, Wu K, Guo X, Chen C, Huang T, Li L, et al. The pan-cancer landscape of glutamate and glutamine metabolism: A comprehensive bioinformatic analysis across 32 solid cancer types. Biochim Biophys Acta Mol Basis Dis. (2024) 1870. doi: 10.1016/J.BBADIS.2023.166982
180. Byun J-K, Park M, Lee S, Yun JW, Lee J, Kim JS, et al. Inhibition of glutamine utilization synergizes with immune checkpoint inhibitor to promote antitumor immunity. Mol Cell. (2020) 80:592–606.e8. doi: 10.1016/j.molcel.2020.10.015
181. Li X. Emerging role of mutations in epigenetic regulators including MLL2 derived from The Cancer Genome Atlas for cervical cancer. BMC Cancer. (2017) 17:1–11. doi: 10.1186/S12885-017-3257-X
182. Shi Y, Lei Y, Liu L, Zhang S, Wang W, Zhao J, et al. Integration of comprehensive genomic profiling, tumor mutational burden, and PD-L1 expression to identify novel biomarkers of immunotherapy in non-small cell lung cancer. Cancer Med. (2021) 10:2216–31. doi: 10.1002/CAM4.3649
183. Amin R, Braza MS. The follicular lymphoma epigenome regulates its microenvironment. J Exp Clin Cancer Res. (2022) 41. doi: 10.1186/S13046-021-02234-9
184. Bao X, Li Q, Chen J, Chen D, Ye C, Dai X, et al. Molecular subgroups of intrahepatic cholangiocarcinoma discovered by single-cell RNA sequencing-assisted multiomics analysis. Cancer Immunol Res. (2022) 10:811–28. doi: 10.1158/2326-6066.CIR-21-1101
185. Wang G, Chow RD, Zhu L, Bai Z, Ye L, Zhang F, et al. CRISPR-GEMM pooled mutagenic screening identifies KMT2D as a major modulator of immune checkpoint blockade. Cancer Discovery. (2020) 10:1912–33. doi: 10.1158/2159-8290.CD-19-1448
186. Fang B, Wei Y, Pan J, Zhang T, Ye D, Zhu Y. The somatic mutational landscape of mismatch repair deficient prostate cancer. J Clin Med. (2023) 12. doi: 10.3390/JCM12020623
187. Liu R, Niu Y, Liu C, Zhang X, Zhang J, Shi M, et al. Association of KMT2C/D loss-of-function variants with response to immune checkpoint blockades in colorectal cancer. Cancer Sci. (2023) 114:1229–39. doi: 10.1111/CAS.15716
188. Zhao L, Luo T, Jiang J, Wu J, Zhang X. Eight gene mutation-based polygenic hazard score as a potential predictor for immune checkpoint inhibitor therapy outcome in metastatic melanoma. Front Mol Biosci. (2022) 9:1001792. doi: 10.3389/FMOLB.2022.1001792
189. Pokhrel RH, Acharya S, Ahn JH, Gu Y, Pandit M, Kim JO, et al. AMPK promotes antitumor immunity by downregulating PD-1 in regulatory T cells via the HMGCR/p38 signaling pathway. Mol Cancer. (2021) 20:1–15. doi: 10.1186/S12943-021-01420-9
190. Lu Y, Zhang M, Wang S, Hong B, Wang Z, Li H, et al. p38 MAPK-inhibited dendritic cells induce superior antitumour immune responses and overcome regulatory T-cell-mediated immunosuppression. Nat Commun. (2014) 5. doi: 10.1038/NCOMMS5229
191. Henson SM, Macaulay R, Riddell NE, Nunn CJ, Akbar AN. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8(+) T-cell proliferation by distinct pathways. Eur J Immunol. (2015) 45:1441–51. doi: 10.1002/EJI.201445312
192. Liu X, Wu X, Cao S, Harrington SM, Yin P, Mansfield AS, et al. B7-H1 antibodies lose antitumor activity due to activation of p38 MAPK that leads to apoptosis of tumor-reactive CD8+ T cells. Sci Rep. (2016) 6. doi: 10.1038/SREP36722
193. Xia W, Qi X, Li M, Wu Y, Sun L, Fan X, et al. Metformin promotes anticancer activity of NK cells in a p38 MAPK dependent manner. Oncoimmunology. (2021) 10. doi: 10.1080/2162402X.2021.1995999
194. Zhang C, Zhang G, Sun N, Zhang Z, Zhang Z, Luo Y, et al. Comprehensive molecular analyses of a TNF family-based signature with regard to prognosis, immune features, and biomarkers for immunotherapy in lung adenocarcinoma. EBioMedicine. (2020) 59. doi: 10.1016/J.EBIOM.2020.102959
195. Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun. (2017) 8. doi: 10.1038/S41467-017-02358-7
196. Yang Y, Sun M, Yao W, Wang F, Li X, Wang W, et al. Compound kushen injection relieves tumor-associated macrophage-mediated immunosuppression through TNFR1 and sensitizes hepatocellular carcinoma to sorafenib. J Immunother Cancer. (2020) 8. doi: 10.1136/JITC-2019-000317
197. Benoot T, Piccioni E, De Ridder K, Goyvaerts C. TNFα and immune checkpoint inhibition: friend or foe for lung cancer? Int J Mol Sci. (2021) 22. doi: 10.3390/IJMS22168691
198. Müller-Hermelink N, Braumüller H, Pichler B, Wieder T, Mailhammer R, Schaak K, et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell. (2008) 13:507–18. doi: 10.1016/J.CCR.2008.04.001
199. Yue S, Li Y, Chen X, Wang J, Li M, Chen Y, et al. FGFR-TKI resistance in cancer: current status and perspectives. J Hematol Oncol. (2021) 14:23. doi: 10.1186/s13045-021-01040-2
200. Zhou Y, Wu C, Lu G, Hu Z, Chen Q, Du X. FGF/FGFR signaling pathway involved resistance in various cancer types. J Cancer. (2020) 11:2000–7. doi: 10.7150/JCA.40531
201. Bockorny B, Rusan M, Chen W, Liao RG, Li Y, Piccioni F, et al. RAS-MAPK reactivation facilitates acquired resistance in FGFR1-amplified lung cancer and underlies a rationale for upfront FGFR-MEK blockade. Mol Cancer Ther. (2018) 17:1526–39. doi: 10.1158/1535-7163.MCT-17-0464
202. Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. (2010) 468:968–72. doi: 10.1038/nature09627
203. Le X, Antony R, Razavi P, Treacy DJ, Luo F, Ghandi M, et al. Systematic functional characterization of resistance to PI3K inhibition in breast cancer. Cancer Discovery. (2016) 6:1134–47. doi: 10.1158/2159-8290.CD-16-0305
204. Kim S-M, Kim H, Yun MR, Kang HN, Pyo K-H, Park HJ, et al. Activation of the Met kinase confers acquired drug resistance in FGFR-targeted lung cancer therapy. Oncogenesis. (2016) 5:e241. doi: 10.1038/oncsis.2016.48
205. He J, Huang Z, Han L, Gong Y, Xie C. Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer (Review). Int J Oncol. (2021) 59:1–20. doi: 10.3892/ijo.2021.5270
206. Terai H, Soejima K, Yasuda H, Nakayama S, Hamamoto J, Arai D, et al. Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res. (2013) 11:759–67. doi: 10.1158/1541-7786.MCR-12-0652
207. Kim TM, Song A, Kim DW, Kim S, Ahn YO, Keam B, et al. Mechanisms of acquired resistance to AZD9291: A mutation-selective, irreversible EGFR inhibitor. J Thorac Oncol. (2015) 10:1736–44. doi: 10.1097/JTO.0000000000000688
208. Ware KE, Hinz TK, Kleczko E, Singleton KR, Marek LA, Helfrich BA, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis. (2013) 2:e39. doi: 10.1038/oncsis.2013.4
209. Yang Y, Qiu R, Weng Q, Xu Z, Song J, Zhao S, et al. MLL4 regulates the progression of non-small-cell lung cancer by regulating the PI3K/AKT/SOX2 axis. Cancer Res Treat. (2023) 55:778–803. doi: 10.4143/CRT.2022.1042
210. Toska E, Castel P, Chhangawala S, Arruabarrena-Aristorena A, Chan C, Hristidis VC, et al. PI3K inhibition activates SGK1 via a feedback loop to promote chromatin-based regulation of ER-dependent gene expression. Cell Rep. (2019) 27:294–306.e5. doi: 10.1016/J.CELREP.2019.02.111
211. Jones RB, Farhi J, Adams M, Parwani KK, Cooper GW, Zecevic M, et al. Targeting MLL methyltransferases enhances the antitumor effects of PI3K inhibition in hormone receptor-positive breast cancer. Cancer Res Commun. (2022) 2:1569–78. doi: 10.1158/2767-9764.CRC-22-0158
212. Koren S, Bentires-Alj M. Tackling resistance to PI3K inhibition by targeting the epigenome. Cancer Cell. (2017) 31:616–8. doi: 10.1016/J.CCELL.2017.04.010
213. Denisenko TV, Budkevich IN, Zhivotovsky B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. (2018) 9. doi: 10.1038/S41419-017-0063-Y
214. Zhang C, Mei W, Zeng C. Oncogenic Neuregulin 1 gene (NRG1) fusions in cancer: A potential new therapeutic opportunities. Biochim Biophys Acta Rev Cancer. (2022) 1877. doi: 10.1016/J.BBCAN.2022.188707
215. Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. (2016) 48:607–16. doi: 10.1038/NG.3564
216. Iwasaki T, Shirota H, Sasaki K, Ouchi K, Nakayama Y, Oshikiri H, et al. Specific cancer types and prognosis in patients with variations in the KEAP1-NRF2 system: A retrospective cohort study. Cancer Sci. (2024). doi: 10.1111/CAS.16355
217. Yokoyama Y, Estok TM, Wild R. Sirpiglenastat (DRP-104) induces antitumor efficacy through direct, broad antagonism of glutamine metabolism and stimulation of the innate and adaptive immune systems. Mol Cancer Ther. (2022) 21:1561–72. doi: 10.1158/1535-7163.MCT-22-0282
218. Allevato MM, Trinh S, Koshizuka K, Nachmanson D, Nguyen TTC, Yokoyama Y, et al. A genome-wide CRISPR screen reveals that antagonism of glutamine metabolism sensitizes head and neck squamous cell carcinoma to ferroptotic cell death. Cancer Lett. (2024) 598. doi: 10.1016/J.CANLET.2024.217089
219. Kozono D, Hua X, Wu MC, Tolba KA, Waqar SN, Dragnev KH, et al. Lung-MAP next-generation sequencing analysis of advanced squamous cell lung cancers (SWOG S1400). J Thorac Oncol. (2024). doi: 10.1016/J.JTHO.2024.07.024
220. Zhang Y, Sun Q, Liang Y, Yang X, Wang H, Song S, et al. FAM20A: a potential diagnostic biomarker for lung squamous cell carcinoma. Front Immunol. (2024) 15:1424197. doi: 10.3389/FIMMU.2024.1424197
221. Wang C, Zhang J, Wang H, Chen R, Lu M. Family with sequence similarity 83, member A (FAM83A) inhibits ferroptosis via the Wnt/β-catenin pathway in lung squamous cell cancer. Cell Death Discovery. (2024) 10. doi: 10.1038/S41420-024-02101-4
222. Jin N, George TL, Otterson GA, Verschraegen C, Wen H, Carbone D, et al. Advances in epigenetic therapeutics with focus on solid tumors. Clin Epigenet. (2021) 13:1–27. doi: 10.1186/S13148-021-01069-7
223. Straining R, Eighmy W. Tazemetostat: EZH2 inhibitor. J Adv Pract Oncol. (2022) 13:158–63. doi: 10.6004/JADPRO.2022.13.2.7
224. Yang P, Qiao Y, Meng M, Zhou Q. Cancer/testis antigens as biomarker and target for the diagnosis, prognosis, and therapy of lung cancer. Front Oncol. (2022) 12:864159. doi: 10.3389/FONC.2022.864159
225. Hikmet F, Rassy M, Backman M, Méar L, Mattsson JSM, Djureinovic D, et al. Expression of cancer-testis antigens in the immune microenvironment of non-small cell lung cancer. Mol Oncol. (2023) 17:2603–17. doi: 10.1002/1878-0261.13474
226. Yeh SJ, Chang CA, Li CW, Wang LHC, Chen BS. Comparing progression molecular mechanisms between lung adenocarcinoma and lung squamous cell carcinoma based on genetic and epigenetic networks: big data mining and genome-wide systems identification. Oncotarget. (2019) 10:3760–806. doi: 10.18632/ONCOTARGET.26940
227. Wang J, Elkrief A, Guo W, Shukla N, Gounder M, Ladanyi M. Prospects for epigenetic targeted therapies of bone and soft-tissue sarcomas. Sarcoma. (2021) 2021. doi: 10.1155/2021/5575444
228. Babar Q, Saeed A, Tabish TA, Pricl S, Townley H, Thorat N. Novel epigenetic therapeutic strategies and targets in cancer. Biochim Biophys Acta Mol Basis Dis. (2022) 1868. doi: 10.1016/J.BBADIS.2022.166552
229. Morel D, Jeffery D, Aspeslagh S, Almouzni G, Postel-Vinay S. Combining epigenetic drugs with other therapies for solid tumours - past lessons and future promise. Nat Rev Clin Oncol. (2020) 17:91–107. doi: 10.1038/S41571-019-0267-4
230. Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther. (2019) 4. doi: 10.1038/S41392-019-0095-0
231. Roberti A, Valdes AF, Torrecillas R, Fraga MF, Fernandez AF. Epigenetics in cancer therapy and nanomedicine. Clin Epigenet. (2019) 11:1–18. doi: 10.1186/S13148-019-0675-4
232. Bose P, Gandhi V, Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma. (2017) 58:2026–39. doi: 10.1080/10428194.2017.1283032
233. Tsao T, Shi Y, Kornblau S, Lu H, Konoplev S, Antony A, et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann Hematol. (2012) 91:1861–70. doi: 10.1007/S00277-012-1537-8
234. DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. (2019) 133:7–17. doi: 10.1182/BLOOD-2018-08-868752
235. Merle G, Friedlaender A, Desai A, Addeo A. Antibody drug conjugates in lung cancer. Cancer J. (2022) 28:429–35. doi: 10.1097/PPO.0000000000000630
236. Gomes-da-Silva LC, Kepp O, Kroemer G. Regulatory approval of photoimmunotherapy: photodynamic therapy that induces immunogenic cell death. Oncoimmunology. (2020) 9. doi: 10.1080/2162402X.2020.1841393
237. Li BT, Shen R, Buonocore D, Olah ZT, Ni A, Ginsberg MS, et al. Ado-trastuzumab emtansine for patients with HER2-mutant lung cancers: results from a phase II basket trial. J Clin Oncol. (2018) 36:2532–7. doi: 10.1200/JCO.2018.77.9777
238. Jänne PA, Baik C, Su WC, Johnson ML, Hayashi H, Nishio M, et al. Efficacy and safety of patritumab deruxtecan (HER3-DXd) in EGFR inhibitor-resistant, EGFR-mutated non-small cell lung cancer. Cancer Discovery. (2022) 12:74–89. doi: 10.1158/2159-8290.CD-21-0715
239. Heist RS, Guarino MJ, Masters G, Purcell WT, Starodub AN, Horn L, et al. Therapy of advanced non-small-cell lung cancer with an SN-38-anti-trop-2 drug conjugate, sacituzumab govitecan. J Clin Oncol. (2017) 35:2790–7. doi: 10.1200/JCO.2016.72.1894
240. Wermke M, Felip E, Gambardella V, Kuboki Y, Morgensztern D, Hamed ZO, et al. Phase I trial of the DLL3/CD3 bispecific T-cell engager BI 764532 in DLL3-positive small-cell lung cancer and neuroendocrine carcinomas. Future Oncol. (2022) 18:2639–49. doi: 10.2217/FON-2022-0196
Keywords: lung squamous cell carcinoma, non-small-cell lung cancer, targeted therapies, immune microenvironment, multi-target combination
Citation: Chen Q, Zheng X, Cheng W and Li J (2024) Landscape of targeted therapies for lung squamous cell carcinoma. Front. Oncol. 14:1467898. doi: 10.3389/fonc.2024.1467898
Received: 24 July 2024; Accepted: 08 October 2024;
Published: 31 October 2024.
Edited by:
Chun Wai Mai, UCSI University, MalaysiaReviewed by:
Savvas Lampridis, Imperial College London, United KingdomQi Zhang, Houston Methodist Research Institute, United States
Copyright © 2024 Chen, Zheng, Cheng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiting Cheng, joycvt@126.com
†These authors have contributed equally to this work and share first authorship