 Guoqi Ya
Guoqi Ya
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 27 September 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.975261
This article is part of the Research TopicReviews in Molecular and Cellular OncologyView all 47 articles
Metastasis is a complex process, which depends on the interaction between tumor cells and host organs. Driven by the primary tumor, the host organ will establish an environment suitable for the growth of tumor cells before their arrival, which is called the pre-metastasis niche. The formation of pre-metastasis niche requires the participation of a variety of cells, in which myeloid-derived suppressor cells play a very important role. They reach the host organ before the tumor cells, and promote the establishment of the pre-metastasis niche by influencing immunosuppression, vascular leakage, extracellular matrix remodeling, angiogenesis and so on. In this article, we introduced the formation of the pre-metastasis niche and discussed the important role of myeloid-derived suppressor cells. In addition, this paper also emphasized the targeting of myeloid-derived suppressor cells as a therapeutic strategy to inhibit the formation of pre-metastasis niche, which provided a research idea for curbing tumor metastasis.
According to statistics, the number of cancer patients in the world has exceeded 19.3 million in 2020, with nearly 10 million cumulative deaths (1). For decades, research of cancer biologist on cancer metastasis has mainly focused on the causes of carcinogenic transformation. In recent years, due to the high mortality related to metastasis, cancer biologists are forced to turn their research to the process of tumor metastasis. After massive research, it is found that the host organ has made a series of preparations for the arrival of tumor cells before tumor metastasis, such as recruiting immune cells, remodeling matrix, generating new blood vessels, etc. These changes create a favorable environment for the colonization and dissemination of tumor cells, that is, the pre-metastasis niche (PMN) (2).
Myeloid-derived suppressor cells (MDSCs) are a group of heterogeneous cells from bone marrow with strong immunosuppressive activity. In the 1970s, MDSCs were first found in a cancer patient. After years of research, it was found that these cells are related to the occurrence and development of inflammation, chronic infection, autoimmune diseases, cancer and other diseases. In order to better describe the origin and function of such cells, researchers suggested that this group of cells be named MDSCs (3). MDSCs play an important role in promoting PMN and maintaining tumor metastasis due to their immunosuppressive function. Increasing evidence that MDSCs contributed to cancer metastasis can bring us the opportunity to develop new therapeutic approaches and enhance patient’s survival. Therefore, understanding how MDSCs can promote and maintain metastasis has laid a foundation for studying some unsolved problems and developing clinical applications. In this article, we introduced the phenotype of MDSCs and the process of PMN formation, and mainly discussed the important contribution of MSDCs in the induction of the PMN and summarized the clinical application of MDSCs targeted therapy.
MDSCs are derived from hematopoietic stem cells (HSCs) (4). Under physiological conditions, HSCs differentiate into common myeloid progenitors (CMPs) and then differentiate into granulocyte-monocyte progenitors (GMPs). Immature myeloid cells (IMCs) such as CMPs and GMPs migrate to peripheral organs and differentiate into mature granulocytes, dendritic cells (DCs) and macrophages, and enter the corresponding tissues and organs to play a normal immune response. Under pathological conditions, such as trauma, infection and cancer, inflammatory factors will inhibit the differentiation of IMCs into mature myeloid cells, make it stay in various differentiation stages, and finally form MDSCs with immunosuppressive function (5).
MDSCs can be divided into polymorphonuclear or granulocytic MDSCs (PMN/G-MDSCs) and monocytic MDSCs (M-MDSCs) according to their phenotypic and morphological characteristics (6). The phenotypes of MDSCs in different species are different. Due to the high expression of myeloid differentiation antigens Gr-1 and CD11b in mice, the phenotype of M-MDSCs in mice is defined as CD11b+Ly6G−Ly6Chigh, while the phenotype of PMN-MDSCs is defined as CD11b+Ly6G+Ly6Clow (7). In 2017, Goldmann (8) and others found a new subgroup of MDSCs in mice infected with staphylococcus aureus and named it Eo-MDSCs. Because Eo-MDSCs resemble eosinophil morphologically and express the eosinophil marker Syglec-F, their phenotype is defined as CD11b+SyglecF+CCR3lowIL-5RαlowSSC-Ahigh.
Compared with mouse MDSCs, human MDSCs lack the Gr-1, and mainly express CD11b, CD33, HLA-DR, Lin (including CD3, CD14, CD15, CD19, AND CD56) (6, 9). Due to the complex classification of human MDSCs, it is widely accepted that M-MDSCs are defined as CD11b+CD14+HLA-DR−CD15− and PMN-MDSCs are defined as CD11b+CD14−CD15+ or CD11b+CD14−CD66b+ (9). In addition to the well-known PMN-MDSCs and M-MDSCs, there is another phenotype of human MDSCs. This class of MDSCs is considered immature due to the absence of granulocyte or monocyte markers and is therefore described as early MDSCs, whose phenotype is defined as Lin−HLA-DR−CD33+ (10). In addition, a subpopulation of MDSCs with a fibrocytic phenotype, known as fibrocytic MDSCs (F-MDSCs), was identified in pediatric metastatic sarcoma patients (11) and healthy neonatal cord blood (12). They defined the F-MDSCs phenotype as CD11b−CD11c−CD33+IL-4Rα+ (11, 13) (Table 1).
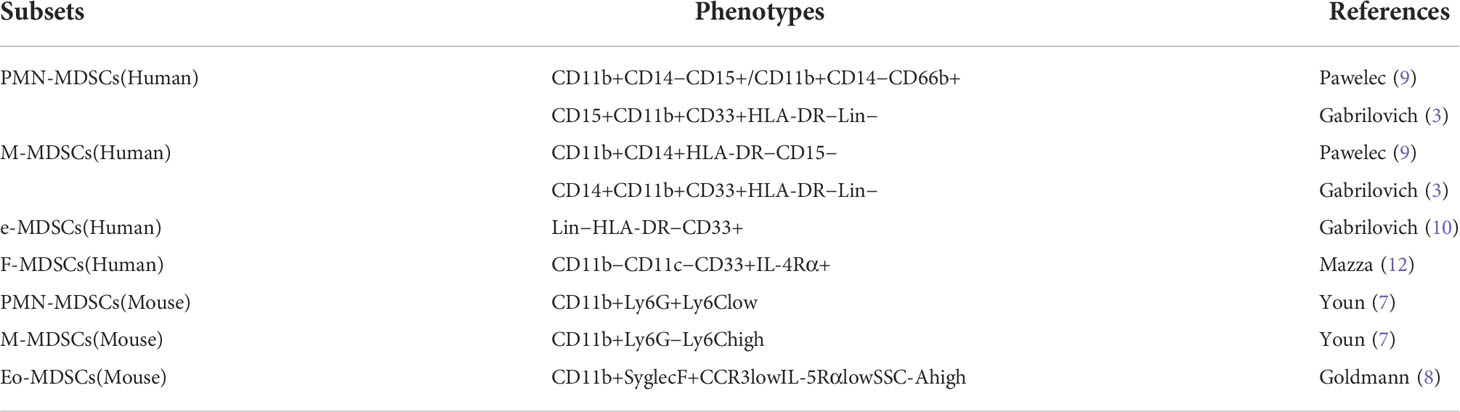
Table 1 Phenotypes of MDSCs.
Tumor metastasis is a multi-step process, including invasion, circulation, exosmosis, colonization and so on (14–16). Christine Chaffer (17) and others summarized this complex cascade reaction into two stages. The first stage is the physical migration of cancer cells from the primary tumor to the host organ, the second stage is when cancer cells colonize the host organ and develop into metastases. However, the host organs that form metastases are not randomly selected, but determined by the organotropism of the primary tumor. The “seed and soil” hypothesis proposed by Steven Paget (18) in 1989 explained organotropism during tumor metastasis, which was also supported by Fidler (19, 20), who showed that the outcome of metastasis depended on the characteristics of tumor cells and host organs. Later researchers also came to light by realizing that breast cancer was found to predispose to bone, liver, brain, and lung metastases, colorectal cancers predominantly develop liver metastases, and tumors originating in the breast, bladder, colon, kidney, head and neck and melanoma have a tendency to cause metastases in the lung (21). In 2005, Kaplan found that myeloid hematopoietic progenitors expressing vascular endothelial growth factor receptor 1 (VEGFR1) arrived at the host organ before tumor cells and changed the microenvironment there (2). Based on these results, they proposed the concept of “premetastatic microenvironment”, in which primary tumors induce host organs to create a microenvironment that supports tumor cell metastasis. The presence of the PMN more strongly explains organ tropism during tumor metastasis and illustrates the importance of the interaction between tumor cells and host organs.
The formation of the PMN is the result of the joint action of multiple cytokines or cellular components including tumor-derived secretory factors (TDSFs), exosomes, bone marrow-derived cells (BMDCs) and extracellular matrix (ECM) (22) (Figure 1). Since PMN formation is a complex dynamic change, this paper mainly summarizes it from the following three aspects.
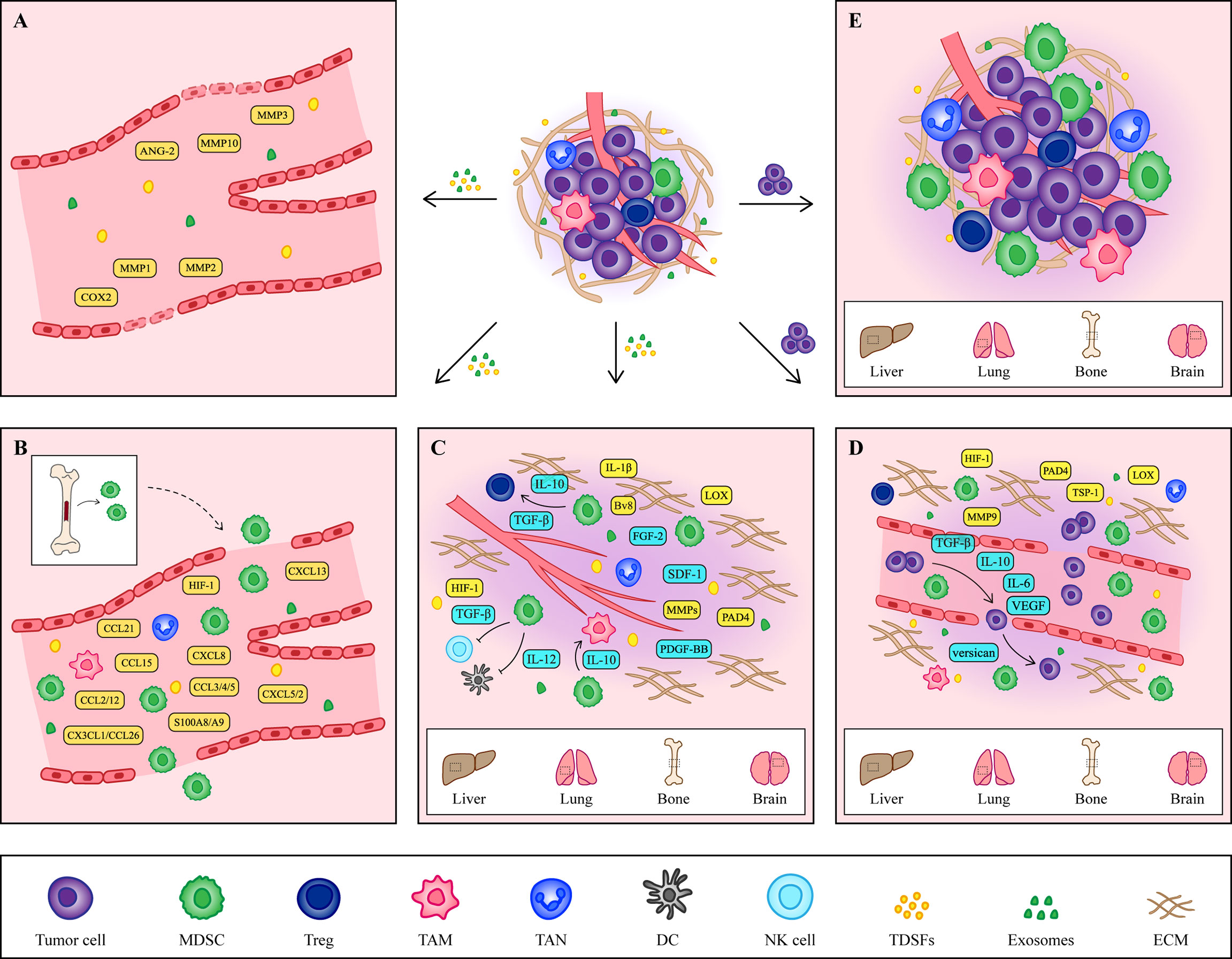
Figure 1 The formation of PMN. (A) Vascular leakage is the initial marker of PMN formation. Cytokines such as COX2, MMP1 and MMP2 secreted by primary tumors increase the permeability of endothelial cells and help the exudation of tumor cells. (B) Under the induction of hypoxic microenvironment and TDSFs, MDSCs and other immune cells are recruited to PMN, creating an excellent immune microenvironment for tumor cells. (C) At the same time, the ECM in the host organ began to change, providing a good environment for the growth of tumor cells. The accumulation of fibronectin and the crosslinking of collagen I provide a platform for the adhesion of MDSCs. At the same time, MMPs promote angiogenesis and contribute to tumor cells intrusion. In addition, some cytokines secreted by MDSCs also contribute to the remodeling of ECM. (D) Tumor cells arriving at the host organ will enter a dormant state, which is regulated by cytokines secreted by tumor cells and MDSCs. When tumor cells wake up, they continue to grow. (E) The formation of the PMN is a comprehensive result of vascular leakage, MDSCs recruitment and ECM remodeling. The tumor cells shed from the primary site and colonized the host organ, eventually forming a distant metastasis.
In the process of tumor metastasis, cancer stem cells (CSCs) with self-replication and differentiation ability first lose the cell-cell adhesion ability and detach from the primary site, invade the surrounding tissues and enter lymphatic vessels and blood vessels. In this process, CSCs undergo epithelial mesenchymal transition (EMT) to become circulating tumor cells (CTCs) (14). CTCs entering the blood vessels are surrounded by platelets, clotting factors and fibrin, preventing the killing effect of NK cells. Meanwhile, selectin and integrin secreted by platelets mediate the adhesion and adhesion of CTCs to vascular endothelial cells, preparing for CTCs extravasation (23, 24). Therefore, the changes of vascular endothelial cells are the key markers for the initial establishment of the PMN.
The current study revealed a relationship between vascular stability and TDSFs. Among them, cyclooxygenase 2 (COX2), matrix metalloproteinase 1 (MMP1) and matrix metalloproteinase 2 (MMP2) secreted by primary tumors were found to affect vascular integrity by changing the morphology of endothelial cells and improving vascular permeability in the lung metastasis model of breast cancer mice (25). In a melanoma mouse model of lung metastasis, primary tumors disrupt pulmonary vascular stability by upregulation of angiopoietin-2 (ANG-2), MMP3, and MMP10 in pulmonary blood vessels (26).
In addition to TDSFs, tumor-derived exosomes are also involved in regulating intracellular stability. Human colon cancer cell derived exosomal miR-25-3p increases vascular permeability in mouse lung and liver metastasis models by regulating the expression of VEGF2 and zonula occludens 1 (ZO-1), occludin and Claudin5 in endothelial cells (27). MiR-103, miR-638, miR-663a, miR-3648 and miR-4258 secreted by HCC attenuated the integrity of endothelial cells by down-regulating the expression of VE-cadherin and ZO-1 (28, 29). MiR-3157-3p regulates endothelial cell permeability and promotes angiogenesis by regulating the expression of vascular endothelial growth factor (VEGF), MMP2, MMP9 and occludin in Non-small cell lung carcinoma (30). Changes in the stability and integrity of vascular endothelial cells prepare for CTCs extravasation, so timely prevention of endothelial cell changes in the early stage of cancer can help block the formation of the PMN and prevent tumor metastasis.
In fact, primary tumors shed tens of thousands of cancer cells into the bloodstream each day, but because of oxidative stress, shear forces, and immune system attacks, only a few make it into the host organs to form metastases (31, 32). Therefore, the host organ needs to establish a good immune microenvironment to protect CTCs from elimination by NK cells, CD4+T cells and CD8+T cells. Under the action of TDSFs, BMDCs and immune cells such as regulatory T cells (Tregs), tumor associated macrophages (TAMs), and tumor associated neutrophils (TANs) are recruited to host organs to help establish the immune microenvironment (33, 34). Among them, BMDCs are the main participating cells, which protect CTCs by secreting a large number of cell components, and the MDSCs subgroup that constitutes BMDCs is considered to be the most critical cell group (35).
As an important marker of PMN initiation and evolution, the recruitment of MDSCs is a multi-step process, which is mainly regulated by tumor microenvironment and TDSFs. The hypoxic environment in the tumor microenvironment is considered to be an important factor in inducing the recruitment and activation of MDSCs. Sceneay showed that primary tumor hypoxia provides cytokines and growth factors capable of creating a premetastatic niche through recruitment of MDSCs (36). Then more studies found that hypoxia inducible factor 1 (HIF-1) induced the migration of MDSCs into PMN by up regulating the expression of CCL26 (37), programmed death-ligand 1 (PD-L1) (38) and ectonucleoside triphosphate diphosphohydrolase 2 in tumor cells (39). Among TDSFs, chemokines are the main components regulating the recruitment of MDSCs (Table 2). In addition to chemokines, other TDSFs, such as S100A8/A9, are also involved in the recruitment of MDSCs. S100A8/A9 from breast cancer cells promotes the migration and accumulation of MDSCs through the NF-κB signaling pathway. However, S100A8/A9 not only comes from tumor cells, but also can be synthesized and secreted by MDSCs (60). Therefore, S100A8/A9 may provide an autocrine pathway for PMN to recruit MDSCs.
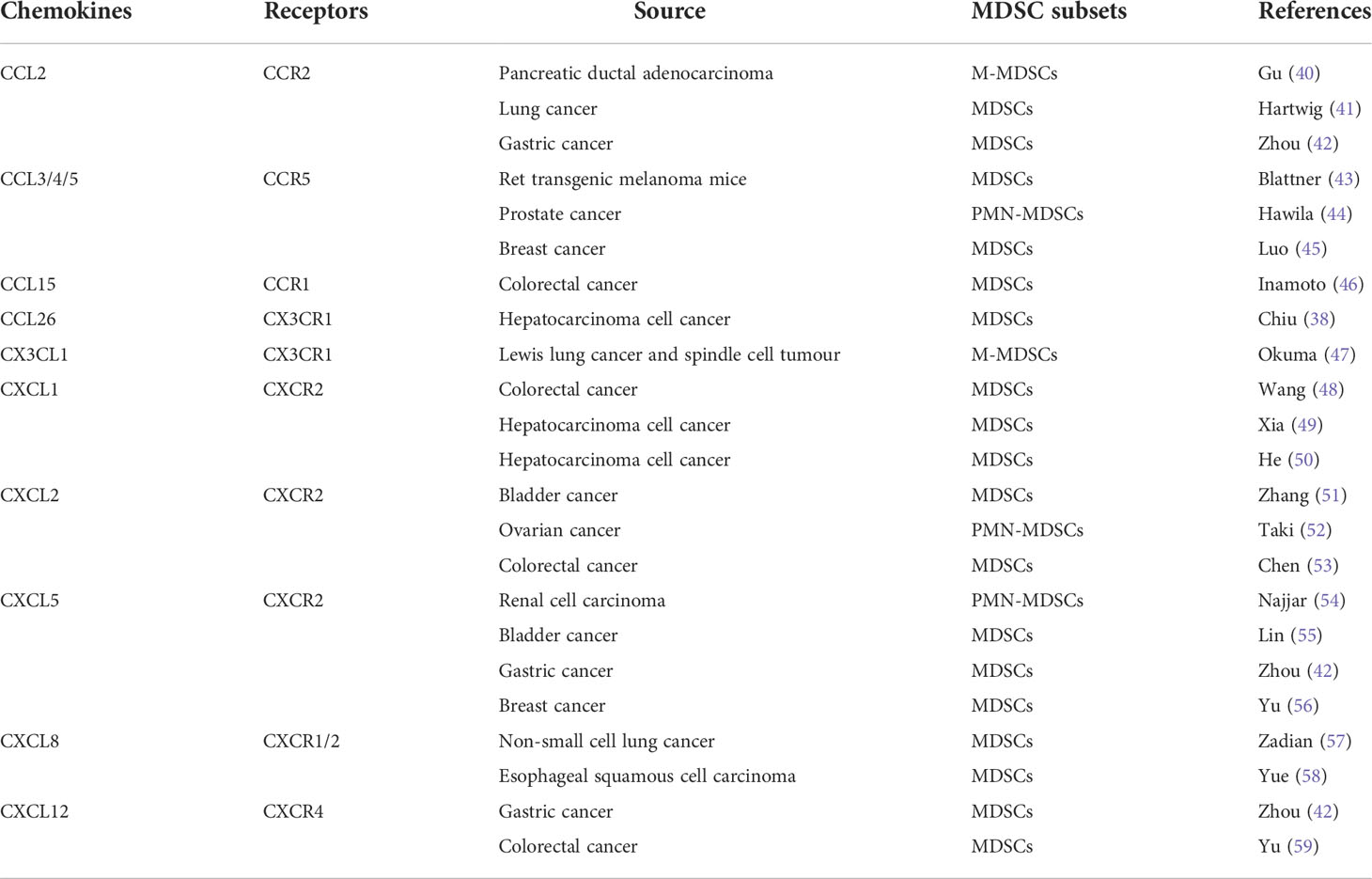
Table 2 Major chemokines involved in regulating MDSCs recruitment.
Since the host organ and the primary site of the tumor are not the same in tissue and structure, in order to establish a PMN suitable for the growth and colonization of CTCs, the ECM structure in the host organ will change, promoting tumor cell migration and invasion into the stromal tissue. During the formation of the PMN, primary tumors remodel ECM by inducing stromal cells to deposit new ECM components or modify old ECM components by TDSFs.
As we all know, Lysyl oxidase (LOX) and MMPs secreted by primary tumors play an important role in ECM remodeling. LOX increases the stiffness of ECM by catalyzing collagen and elastin crosslinking. At the same time, LOX can also promote the recruitment of BMDCs and drive the formation of osteolytic lesions, leading to bone metastasis (61–63). While MMPs play a role in degrading ECM and inducing angiogenesis (64). In addition, recent studies have found that tumor-produced peptidylarginine deiminase 4 (PAD4) also helps to remodel ECM. PAD4 produced by colorectal cancer affects the citrullination of ECM, which promotes EMT and eventually leads to tumor metastasis to the liver. Preventing citrullination of ECM may create an adverse microenvironment for cancer cell growth (65). Therefore, PAD4 and citrullination may be an effective target for the treatment of liver metastases.
CTCs entering the host organ either apoptosis or dormant until all environments become suitable for CTCs to awaken (66). A growing body of research has shown that the composition of ECMs and their effect on adhesion signaling not only provides a favorable environment for the growth of CTCs, but also regulates the dormancy and awakening of tumor cells (67). For example, endothelial-derived thrombospondin-1 (TSP-1) (68) and osteopontin secreted by osteoblasts (69), as well as matrigel, commonly used for cultured cells in vitro (70), can induce tumor cell dormancy. Recently, studies have found that dormant tumor cells assemble a type III collagen-enriched ECM niche. Type III collagen triggers STAT1 activation and nuclear translocation to regulate COL3A1 expression by binding to discoidin domain receptor 1 (DDR1). The increase of COL3A1 expression in turn remodeled the ECM by increasing the curl of the ECM and brought the cells into a dormant state maintained by DDR1 binding (71). Thus, the manipulation of these mechanisms could serve as a barrier to metastasis through disseminated tumor cell dormancy induction. While the awakening of tumor cells is associated with neutrophil elastase and MMP9, which induce the proliferation of dormant CTCs by exposing laminin epitopes associated with CTCs (72). Given the central role of the ECM in tumor metastasis, altering the structure, function, and biological properties of the ECM and modulating adhesion signaling promises to reduce tumor invasion and prevent tumor metastasis, providing new therapeutic strategies for cancer patients.
After a series of changes, the PMN eventually become mature, and subsequently more and more tumor cells migrate to this site to grow, proliferate, and thus form tumor metastases. Although this article divides the process of PMN formation into three main parts, the establishment and eventual progression of PMN into metastases is an integral and dynamic process, and therefore each of these changes is critical and does not act as an independent contributing factor.
Since metastasis is intrinsically an inefficient process, most cancer cells are unable to metastasize to host organs. Primary tumors therefore need to develop strategies that can both suppress immune responses and alter the tissue framework when inducing PMN formation, providing the most effective aid to tumor metastasis. In this strategy, MDSCs with immunosuppressive effect are the best partners for tumor cells, which not only create a favorable growth environment for tumor cells but also provide much help for their metastasis. The important roles played by MDSCs are mainly reflected in two aspects: immunological effects and non-immunological effects. On the one hand, the immunosuppressive effect of MDSCs promotes tumor cell survival. On the other hand, MDSCs promote tumor cell metastasis by promoting EMT and mesenchymal epithelial transition (MET), protecting CTCs and promoting their extravasation, as well as inducing angiogenesis (73, 74).
The immunosuppressive effects exerted by MDSCs include specific immunity and nonspecific immunity, both of which exert corresponding immunosuppressive effects by suppressing T cells. During the formation of the PMN, MDSCs, as the main cell group involved in the establishment of the immune system, play a key role in the survival of CTCs and the establishment and maintenance of an immunosuppressive microenvironment in host organs. The main mechanisms of action include depletion of amino acids required for T cell activation, production of reactive oxygen species (ROS), expression of negative immune checkpoint molecules, expression of relevant enzymes regulating adenosine metabolism and regulation of other immune cells (Figure 2).
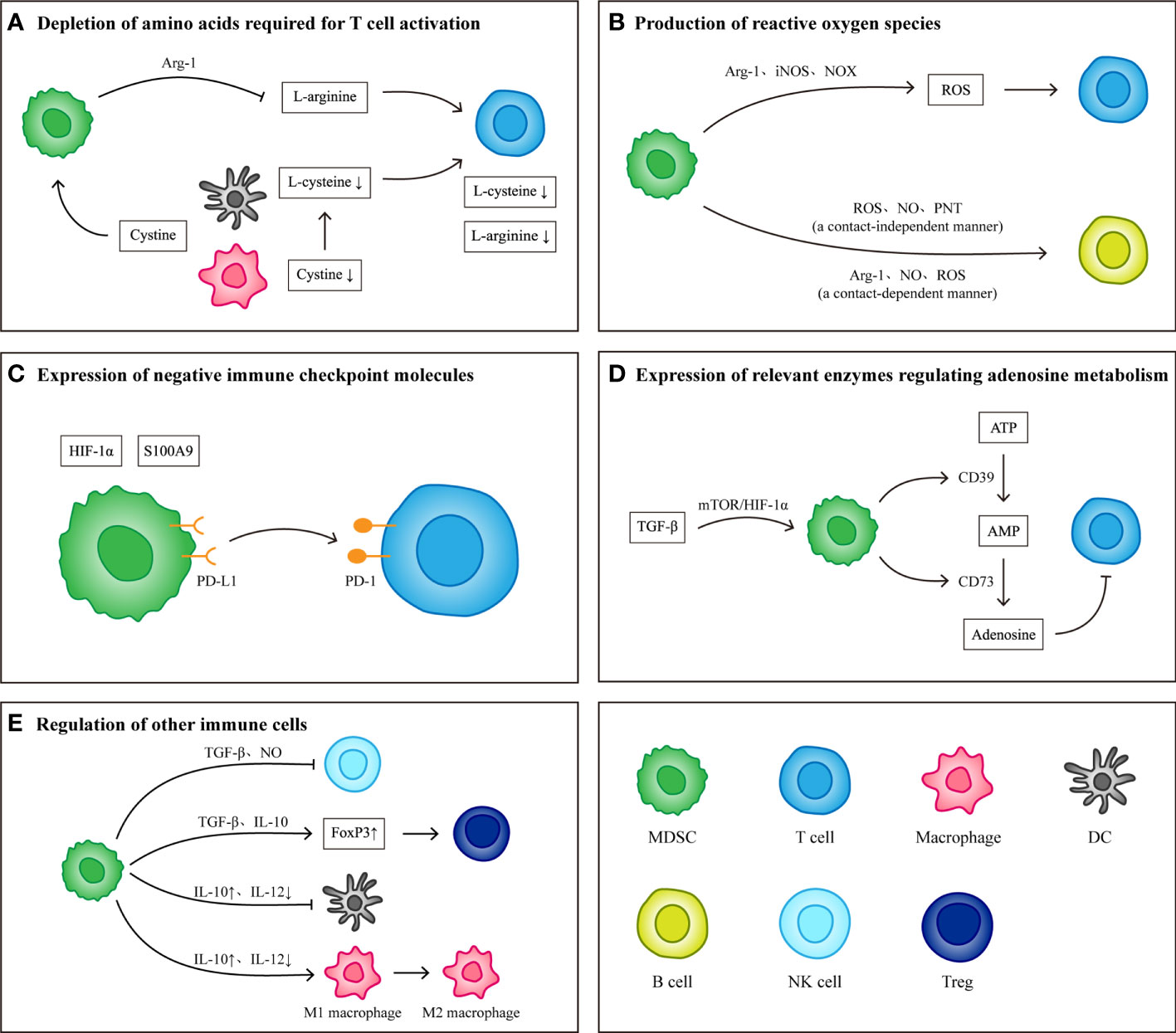
Figure 2 Immunological effects of MDSCs. (A) MDSCs consume L-arginine and L-cysteine required for T cell activation by expressing Arg-1 and ingesting cystine. (B) MDSCs produce reactive oxygen species by expressing Arg-1, iNOS and NOX. Different components of reactive oxygen species have inhibitory effects on T cells and B cells. (C) Under the induction of S100A9 and HIF-1α, the expression levels of PD-L1 on MDSCs increased. High levels of PD-L1 combined with PD-1 on T cells induced T cell apoptosis. (D) TGF-β Induce MDSCs to produce CD39 and CD73, which can induce the conversion of ATP to adenosine. The presence of adenosine inhibits the activation and proliferation of T cells. (E) In addition to inhibiting T cells, MDSCs can also regulate other immune cells to protect tumor cells and establish immune microenvironment. MDSCs inhibit the killing effect of NK cells by producing TGF-β and NO) they weakened the antigen-presenting function of DCs and macrophages by producing IL-10, and transformed M1 macrophages into M2 macrophages) they also secrete TGF-β and IL-10 up regulates the expression of FoxP3 in Tregs and induce the production of FoxP3+ Tregs.
L-arginine and L-cysteine are considered essential amino acids for T cell activation (75, 76). Deficiency in L-arginine leads to a block in the synthesis of CD3ζ in the T cell receptor (TCR), which plays a central role in initiating the signal transduction cascade leading to T cell activation, and therefore downregulation of CD3ζ affects T cell activation and function (77). Concomitant L-arginine deficiency also causes T cell cycle arrest in G0-G1 phase by upregulating cyclin D3 and cyclin-dependent kinase 4 (78). Meanwhile, the participation of L-cysteine is also required during T cell activation. Because T cells lack cystathionase and an intact cystine transporter, it is unable to produce L-cysteine or convert intracellular methionine to L-cysteine. Therefore, the uptake of L-cysteine by T cells depends on the supply of other cells (76).
MDSCs were first shown by Bronte in 2003 to express highly active arginase-1 (Arg-1), and MDSCs inhibit T cell proliferation in an Arg-1-dependent mechanism (79). Subsequently, researchers in multiple studies have confirmed the mechanism by which MDSCs break down L-arginine required by T cells to inhibit T cell proliferation and activation by expressing Arg-1, and showed that Arg-1 can serve as a breakthrough point to block the suppressive effect of MDSCs (80–82). However, it was found that MDSCs mediated T cell suppression does not have to be Arg-1 dependent. In 2018, Bian found that either Arg-1-expressing MDSCs or Arg-1-deficient MDSCs exhibited a strong inhibitory effect on the proliferation of T cells, and found that MDSCs inhibited the proliferation of T cells required direct contact between cells (83). Clearly, further studies are needed to investigate the remaining question regarding the role of arginase-1 MDSC function, if not for mediating the inhibition of T cells.
In addition, MDSCs can competitively take up cystine with macrophages and DCs, which renders macrophages and DCs unable to take up cystine and provide its reduction to L-cysteine to T cells, resulting in limited T cell activation (76).
MDSCs generate ROS, including superoxide anion, H2O2, hydroxyl radical, NO, and others, by expressing Arg-1, inducible nitric oxide synthase (iNOS), and NADPH oxidases (NOX) (84). Although ROS are toxic to most cells, MDSC survive despite their elevated content and release of ROS (85). H2O2 produced by MDSC restricts the activation of T cells by reducing the expression of T cell CD3ζ (86), while NO blocks the proliferation of T cells by inhibiting IL-2 signal (87), and also inhibits NK-cell FcR-mediated functions including antibody-dependent cellular cytotoxicity, cytokine production, and signal transduction in a contact-independent manner (88). In addition, MDSCs can promote peroxynitrite (PNT) production by expressing iNOS, which in turn can inhibit antigen-specific T cell responses by nitrating the TCR so that CD8+ T cells are unable to bind to and respond to peptide major histocompatibility complexs (89, 90).
The production of ROS by MDSCs can inhibit not only T cells and NK cells but also B cells. Rastad have experimentally demonstrated the ability of M-MDSCs to suppress B cells in a cell contact independent manner through the production of ROS, NO, PNT and others in murine AIDS models (91). And then Stiff demonstrated that PMN-MDSCs could inhibit B cell proliferation in a cell contact dependent manner by producing Arg-1, NO, ROS (88). Moreover, multiple studies have reported that the regulation of Tregs by macrophages involves ROS generation (92, 93), and that Tregs are less able to resist oxidative stress in the tumor microenvironment and can undergo apoptosis under the induction of ROS compared with T cells. These apoptotic Tregs mediated immunosuppression via adenosine and A2A pathways (94). Although the interaction of ROS in MDSCs and Tregs is unclear, Tregs mediated suppression has been demonstrated to benefit from oxidative stress conditions. Therefore, it can be speculated that ROS produced by MDSCs may induce the suppressive effects of Tregs.
Immune checkpoint molecules are key molecules that regulate the balance between activation and inhibition of immune responses. In the physiological state, immune checkpoint molecules block inhibitory signals for T cell activation and generate potent antitumor responses, but in tumors, negative immune checkpoint molecules mediate tumor immune escape (95). PD-L1 is a common negative immune checkpoint molecule that induces T cell apoptosis by binding to programmed death-1 (PD-1) receptors on T cells (96). PD-L1 is highly expressed in tumor derived MDSCs, especially in PMN-MDSCs (97, 98). The levels of PD-L1 expression in MDSCs was significantly increased under hypoxia, and blockade of PD-L1 at this time downregulated the immunosuppressive effect of MDSCs on T cells, suggesting that MDSCs could suppress T cells by expressing PD-L1 and that HIF-1α contributes to the induction of PD-L1 expression (37). Later, it was shown that S100A9 secreted by MDSCs also induces PD-L1 expression and that the mechanism is related to aberrant activation of the c-Myc (99). In conclusion, low MDSCs levels before tumor treatment are positively correlated with patient survival, and therefore, MDSCs can be considered as a predictive biomarker when treating with immune checkpoint inhibitors.
Adenosine is an important signaling molecule in regulating the body’s immune response. Ectonucleoside triphosphate diphosphohydrolase 1 (also known as CD39) catalyzes the dephosphorylation of ATP to generate amp, which is then dephosphorylated to generate adenosine under the catalysis of ecto-5’-nucleotidase (also known as CD73). Ultimately, adenosine inhibits the priming activation and proliferation of T cells by reducing the expression of effector molecules on T cells (100, 101). In patients with head and neck squamous cell carcinoma, CD73 expression on PMN-MDSCs was elevated and significantly suppressed T cell proliferation (102). In NSCLC patients, transforming growth factor (TGF-β) induces CD39 and CD73 expression on MDSCs through the mTOR/HIF-1α signaling, which makes CD39+CD73-MDSCs highly enriched in tumor tissue and produces adenosine to inhibit T cell activity (103).
Recent studies, however, have found a dual role for CD73 for proliferation and survival of T cells. On the one hand, CD73 favors the expression of IL-7 receptor α chain on CD8+ T cells and their homeostatic survival; On the other hand, under conditions of antigenic stimulation, CD73 reduces the IL-2 receptor α chain and expression of the antiapoptotic molecule Bcl-2 thereby reducing CD8+ T cell survival (104). It is therefore important to note its dual effects on CD8+ T cells when designing CD73 as a target for antitumor therapy.
Although the immunosuppressive effects of MDSCs are mainly directed against T cells, numerous studies have shown that MDSCs can also regulate immune cells such as NK cells, macrophages, DCs, and Tregs to exert tumor promoting effects.
NK cells have played an important role in antitumor immune responses by directly killing tumor cells. It was found that after co-culture with MDSCs, NK cells had decreased cytolytic capacity, IFN-γ production capacity and natural killer group 2, member D (NKG2D) expression capacity, which was related to TGF-β produced by MDSCs (105). In addition, MDSCs can also affect NK cytotoxicity by producing NO, and this mechanism has been mentioned above.
DCs and macrophages, as antigen-presenting cells, are considered key players in antitumor immune responses. Studies have shown that MDSCs and both can interact to alter the inflammatory environment in the tumor microenvironment. IL-10 produced by MDSCs impairs the antigen-presenting function of DCs and macrophages by downregulating the expression of MHC class II, CD80 and CD86 (106). Since IL-10 is a key factor in regulating IL-12 transcription, an increase in IL-10 leads to a decrease in IL-12, which promotes the transformation of macrophages into M2 phenotype (107).
Tregs, as an important component in tumor associated immunosuppression, are able to inhibit the activation and proliferation of T cells. Under the stimulation of INF-γ, MDSCs secrete TGF-β and IL-10 up regulates the expression of Foxp3, a key transcriptional regulatory protein that determines the differentiation and function of Tregs, and upregulation of Foxp3 enhances the suppressive activity of Tregs (108, 109). In addition, in a study of F-MDSCs, it was found that the suppressive effect of F-MDSCs was not mediated by Arg-1 or iNOS mechanisms but was exerted through the generation of IDO inducing effector T cells to convert into Foxp3+ Tregs and that IDO mediated depletion of tryptophan also downregulated CD3ζ of the TCR leading to blocked T cell activation (110–112).
In addition to establishing and maintaining an immunosuppressive microenvironment, MDSCs recruited into host organs also play a role in various steps of tumor metastasis, promoting tumor cell growth and colonization. As the best partners of tumor cells, MDSCs play many roles, such as inducing tumor cell invasion and proliferation, aiding tumor cell extravasation, and providing trophic support to tumor cells, and their mechanisms mainly include the following three points (Figure 3).
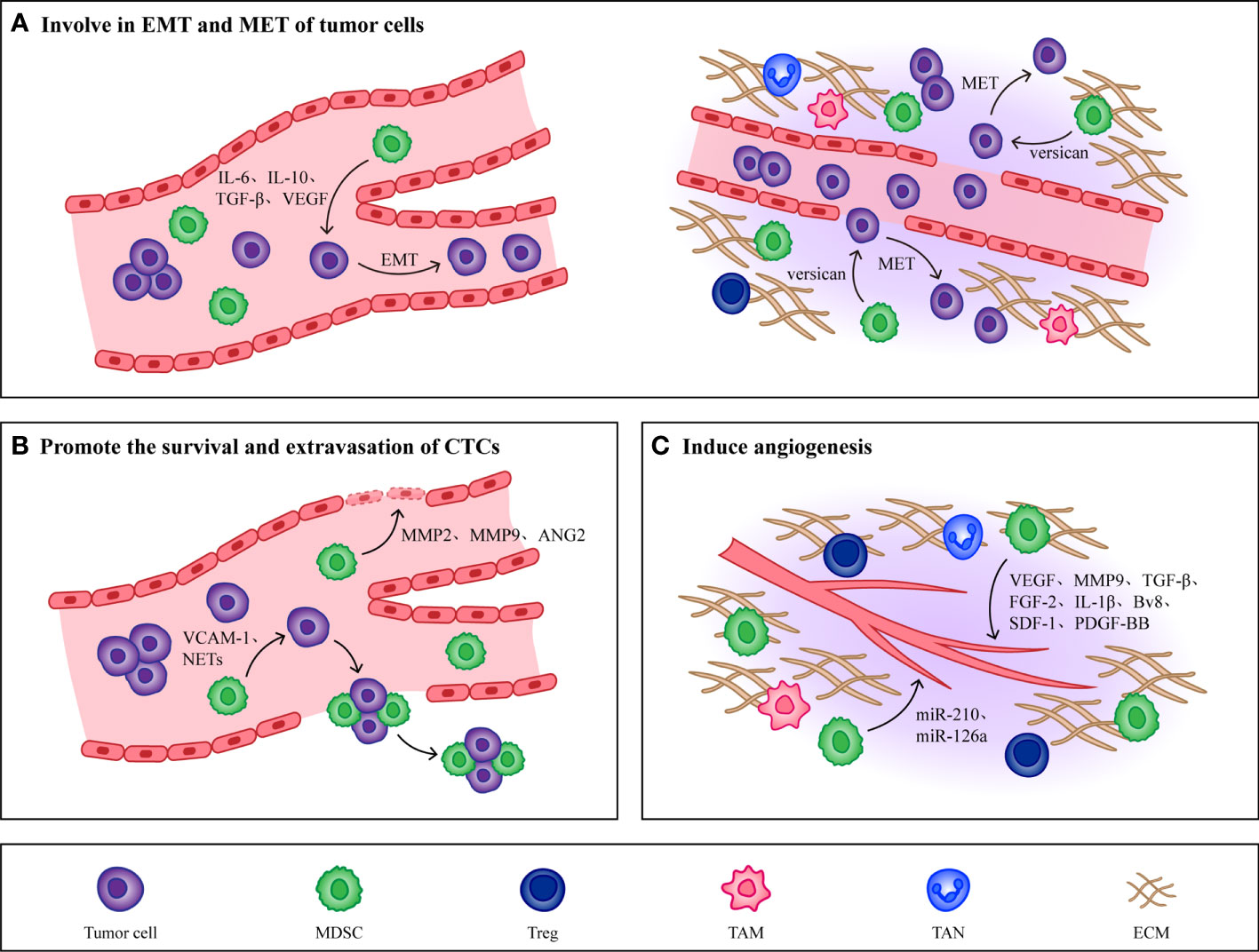
Figure 3 Non immunological effects of MDSCs. (A) MDSCs promote EMT of tumor cells by producing IL-6, IL-10, TGF-β and VEGF, which makes tumor cells obtain high migration and invasion ability. When CTCs reach the host organ, MDSCs will induce CTCs to undergo MET, restore their epithelial phenotype and promote their proliferation. (B) MMP2, MMP9, VEGF and Ang-2 produced by MDSCs effectively increase the permeability of blood vessels and are conducive to the extravasation of CTCs. At the same time, MDSCs combine with CTCs through VCAM-1 and NETs to form CTC/MDSC clusters, which promote the survival and extravasation of CTCs. (C) MDSCs promote angiogenesis in PMN by producing cytokines such as VEGF, MMP9, TGF-β and FGF-2. In addition, MDSCs derived exosomes miR-126a and miR-210 also induce angiogenesis.
The mesenchymal phenotype induced by EMT in primary tumors makes tumor cells lose their intercellular junctions to acquire high migratory and invasive abilities, contributing to the completion of the complex cascade of invasion metastasis (16). Cell transdifferentiation from epithelial to a mesenchymal state is mediated by key transcription factors, such as the basic helix-loop-helix factors TWIST1 and TWIST2 (113), the zinc-finger E-box-binding homeobox factors ZEB1 and ZEB2 (114), and the zinc-finger binding transcription factors SNAI1 and SNAI2 (115), which act as master regulators of cell-cell adhesion, cell polarity and motility. They repress genes associated with an epithelial phenotype and induce the expression of mesenchymal genes, ultimately leading to the cellular hallmarks of EMT.
The most direct evidence for the role of EMT in metastasis comes from the analysis of cancer patients, where it has been reported that immune cells in cancer patients regulate the induction of tumorigenic EMT by secreting cytokines and chemokines, and that tumor cells undergoing EMT also produce immunosuppressive factors and chemokines, which further induce an immunosuppressive state of the tumor microenvironment and thus promote cancer development (116, 117). Various types of cytokines produced by MDSCs, such as VEGF (118), TGF-β (119, 120), IL-6 (121, 122) and IL-10 (123) have been shown to induce EMT in tumor cells. Reported that secreted protein acidic and rich in cysteine (SPARC) from MDSCs was immunosuppressive and tumor promoting in vivo in breast cancer patients, and deletion of SPARC rendered MDSCs with reduced suppressive function and restored EMT, which illustrated that the occurrence of EMT in tumor cells may depend on SPARC secreted by MDSCs (124).
In addition to participating in the induction of EMT, MDSCs also play a role in the met process of tumor cells. Different subpopulations of MDSCs have been shown to play roles at different stages in the tumor invasion metastasis cascade. For example, induction of EMT by M-MDSCs promotes tumor invasion from the primary site to distant site, whereas induction of MET by PMN-MDSCs promotes tumor cell proliferation to support metastatic growth. This illustrates that the regulation of tumor phenotype by MDSCs changes with tumor progression (125). In addition, MDSCs also stimulate MET in tumor cells by secreting versican to attenuate phospho-Smad2 levels (126). In summary, because tumor metastasis is a multistep process and various cancers have different classification criteria, tumor location, tumor type, and tumor stage will all be important to consider in studies on the effects of MDSCs on EMT and MET.
After CTCs detach from the primary tumor into the blood vessels, the mechanical and shear forces present inside the vessels will adversely affect CTCs survival as well as metastasis (127). In addition to the ability of platelets, leukocytes, and macrophages to help CTCs escape from the distress caused by these stresses, MDSCs, because of their potent immunosuppressive properties, can also contribute to the survival of CTCs (128). In pancreatic cancer patients, researchers found a positive correlation between the number of MDSCs and the number of K-RASmut mRNA+ CTCs, which suggested that the presence of MDSCs may promote the proliferation and survival of CTCs (129). Moreover, existing studies have found that MDSCs can interact with CTCs and form CTC/MDSCs clusters to promote CTCs metastasis. CTC/MDSCs cluster promoted the survival and proliferation of CTCS and enhanced their metastatic potency by activating the ROS/Notch/Nodal signaling pathway (130). Recently, it has been reported that vascular cell adhesion molecule-1 (VCAM-1) is required for CTC/MDSCs cluster formation, thus VCAM-1 inhibition could be one of the important factors hindering CTC/MDSCs cluster formation, and developing drugs targeting VCAM-1 may reduce tumor metastasis to some extent (131).
CTCs that survive overcoming the mechanical and shear forces of blood vessels when they move closer and adhere to endothelial cells, MMP2, MMP9, VEGF and Ang-2 produced by MDSCs effectively increase vascular permeability, and upregulate E-selectin, which contributes to adhesion and extravasation of CTCs (26, 132–134). Studies have shown that the formation of neutrophil extracellular traps (NETs) can induce the intravascular coagulation cascade (cancer-related thrombosis), which contributes to the growth of primary tumors, cancer invasiveness, progression and metastasis. Through the use of cecal ligation, Cools Lartigue et al. observed the presence of lung cancer cells encapsulated in NETs in a mouse model of infection. These circulating “packages” were seeded in the liver, forming micrometastases within 48 h and secondary liver cancer 2 weeks after the cancer cell injection (135). Evidence consistent with these observations was provided by Najmeh et al. from the same group, who found a significant association between upregulation of β1-integrin and NET-related entrapment of circulating lung carcinoma cells, further facilitating metastasis formation and cancer spread (136). Theoretically, NETs may play an anti-tumor role by directly killing tumor cells or activating the immune system, but it has been proved that due to partial vascular obstruction and the coagulation microenvironment around NETs, circulating tumor cells can be captured. At the same time, NETs damage endothelial cells and promote the adhesion and extravasation of CTCs.
In recent years, studies have found that PMN-MDSCs are also able to induce the formation of NETs. Alfaro et al. found that IL-8 induced PMN MDSCs to form NETs in the process of studying whether IL-8 secreted by tumors could recruit MDSCs, and observed that tumor cells were captured after NETs were formed (137). In a mouse model of lung metastatic cancer, C5a was shown to induce the expression of high mobility group box 1 (HMGB1) receptors TLR4 and rage in PMN-MDSCs, while the formation of NETs was in turn dependent on HMGB1 produced by cancer cells (138). Thus, CTC/PMN-MDSCs cluster could form NETs under the induction of complement C5a (139). Therefore, hindering the formation of NETs may affect the extravasation of CTCs and thus hinder tumor metastasis, however the presence of MDSCs can directly increase vascular permeability to enhance the extravasation of CTCs. So, the effects of MDSCs and other cytokines on CTCs need to be taken into account when developing therapies that target NETs.
In order for CTCs to colonize the PMN efficiently, PMN would generate new blood vessels to provide nutrients for CTCs to proliferate (24). MDSCs have been shown to induce the PMN angiogenesis through a variety of mechanisms, the most prominent of which is that secretion of VEGFA by MDSCs promotes neovascularization via JAK2/STAT3 signaling (140). In addition to VEGF, other cytokines derived from MDSCs promote tumor angiogenesis, such as MMP9 (141), TGF-β (142), fibroblast growth factor-2 (FGF-2) (143), IL-1β (143), bombina variegata peptide 8 (Bv8) (144), stromal cell-derived factor-1 (SDF-1) (145) and so on. Recently researchers showed that MDSCs expressed high levels of platelet-derived growth factor-BB (PDGF-BB), and they found that this not only promoted tumor angiogenesis but also increased tumor cell metastatic growth (146). In addition, MDSCs derived exosomes also contribute to tumor angiogenesis. MiR-126a released by MDSCs induced tumor angiogenesis and enhanced CTCs adhesion to endothelial cells, which promoted tumor cell metastasis (147). Under hypoxic conditions, HIF-1α upregulates MDSCs derived exosomal miR-210, which not only enhances the function of MDSCs by increasing Arg-1 activity and producing NO, but also regulates endothelial cell activation to induce tumor angiogenesis (148, 149).
Early detection of the PMN formation during the course of cancer treatment helps to optimize treatment regimens and improve patient prognosis. Although there is no clinical technique at this stage available to directly visualize the dynamic process of tumor metastasis, the detection of certain specific immune cells or associated molecules in the PMN by radiological imaging offers a viable option to detect the PMN formation. In 2012, Shokeen and others used PET/CT to image very late antigen-4 (VLA-4, also known as α4β1 integrin) expressing MDSCs in PMN (150). Li and others evaluated the effectiveness of CT images in quantifying the microenvironment changes in the premetastatic lung. Their results suggested that changes in the lung microenvironment can be identified by CT before primary breast cancer lung metastasis (151). Based on the role of S100A8/A9 in promoting MDSCs accumulation and inducing the PMN, researchers reported the application of antibody-based SPECT for detection of S100A8/A9 in vivo as an imaging marker for pre-metastatic tissue priming (152). In addition to S100A8/A9, other MDSCs derived exosomes, such as miR-126a and miR-210 mentioned earlier, have also been implicated in PMN formation. But there are no effective tracers for these molecules, and their distribution profile in the pre metastatic microenvironment is unclear. Therefore, further studies are needed to test MDSCs derived exosomes as an indicator of the PMN.
With the development of cancer treatment technology, the rise of immunotherapy exemplifies the shift in cancer treatment from predominantly tumor suppressor to predominantly PMN modulating. Although targeted therapies to PMN can potentially inhibit tumor metastasis, it is difficult to target drugs to PMN owing to the aberrant tumor microenvironment generation that results from increased vascular permeability in PMN (33). Currently, clinical therapeutic strategies targeting PMN have shifted to specifically targeting PMN composition to suppress tumor metastasis. As one of the most important cells driving PMN formation, MDSCs can serve as an important target for inhibiting tumor metastasis. Currently, therapeutic strategies targeting MDSCs both inhibit MDSCs differentiation and accumulation as well as their function (Figure 4).
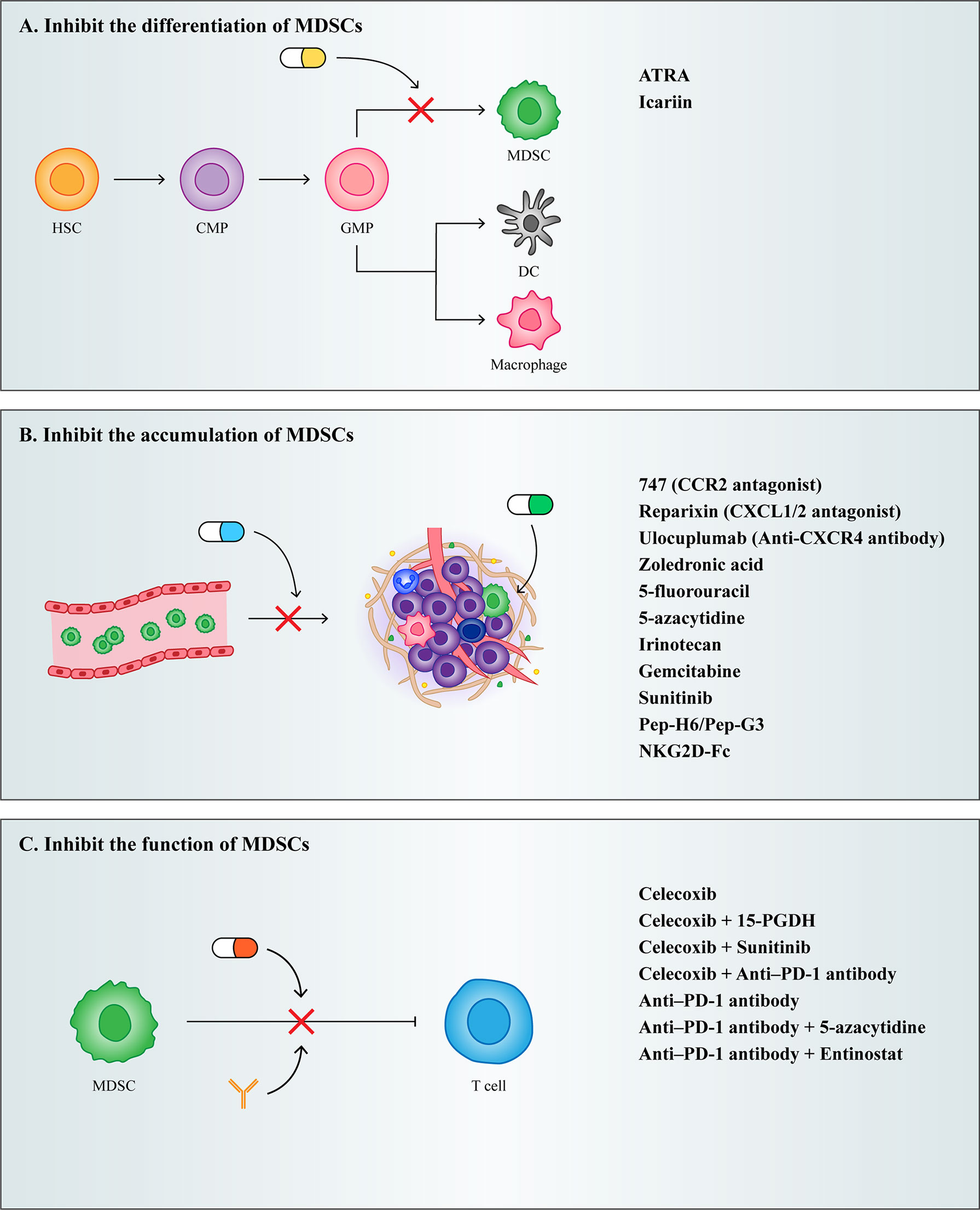
Figure 4 Treatment strategies for MDSCs. (A) Inhibit the differentiation of MDSCs. (B) Inhibit the accumulation of MDSCs. (C) Inhibit the function of MDSCs.
As described previously, IMC differentiates into mature granulocytes, macrophages, and DCs under normal circumstances, and when this process is blocked by certain cytokines, it results in the accumulation of MDSCs. Therefore, the induction of MDSCs into mature myeloid cells may be a potential strategy for the treatment of cancer.
Vitamin A has been shown to induce MDSCs differentiation. Studies have shown that all trans retinoic acid (ATRA), a metabolite intermediate of vitamin A, induces the rapid differentiation of MDSCs into macrophages and DCs by activating the ERK1/2 pathway, which reduces the suppressive function of MDSCs (153). However, it has been shown that ATRA also promotes Foxp3 expression and enhances the function of CD4+ Tregs. Therefore, while using ATRA to target MDSCs for therapy, it is important to note that ATRA may induce the generation of Tregs and thereby promote tumor development (154).
Furthermore, Chinese scholars have proved that Icariin, the main active component of Herba Epimedii, can mediate anti-inflammatory function. Here, they showed that treatment of tumor bearing mice using icariin derivatives could significantly decrease the percentage of MDSCs and their differentiation into macrophages and DCs (155).
Because MDSCs express high levels of chemokine receptors and are recruited to PMN by tumor derived chemokines, preventing the recruitment of MDSCs could be achieved by targeting chemokine receptors on MDSCs. Chemokine receptor antagonists in multiple clinical trials, such as the CCR2 antagonist 747 (156), the CXCR1/2 antagonist Reparixin (157), and the CXCR4 antibody Ulocuplumab (158), have all been shown to reduce the infiltration of MDSCs in tumors, ultimately reducing their metastatic potential.
In addition to chemokine antagonists, aminobisphosphonates have also been shown to inhibit the recruitment of MDSCs. Aminobisphosphonates reduce MMP9 expression and the number of macrophages in the tumor stroma and decrease MDSCs infiltration in bone marrow and peripheral blood by decreasing serum pro-MMP9 and VEGF (159). In a mouse model of pancreatic cancer, the use of zoledronic acid reduced MDSCs accumulation and slowed tumor growth (160).
Depleting MDSCs recruited into PMN is also an effective therapeutic strategy. Low dose chemotherapeutic drugs have been shown to effectively eliminate MDSCs in tumor bearing mice. Given to tumor bearing mice, a significant decrease in the number of MDSCs can be observed after treatment with various chemotherapeutic agents, such as 5-fluorouracil (161), irinotecan (162), gemcitabine (163), and sunitinib (164). In addition to chemotherapeutic agents, researchers have developed fusion proteins capable of targeting MDSCs. Qin developed a novel therapeutic peptide-Fc fusion protein that specifically depletes MDSCs without affecting other pro-inflammatory classes of immune cells (165). Subsequently, Feng synthesized an NKG2D-Fc fusion protein, which reduced infiltrating MDSCs in tumors by binding to NKG2D expressing MDSCs (166).
Because MDSCs play an important role in the tumor microenvironment and PMN due to their powerful immunosuppressive function, alleviating the immunosuppressive mechanisms of MDSCs has become a major therapeutic target to re-establish T-cell activity.
COX2 inhibitors have been reported to act as immunosuppressive agents to improve tumor patient survival. COX2 induces ROS production in MDSCs through the production of prostaglandin E2 (PGE2), and disruption of COX2/PGE2 signaling contributes to the attenuation of the suppressive function of MDSCs. In a murine mesothelioma model, the use of a single COX2 inhibitor, celecoxib, was able to reduce MDSCs levels and reverse T cell function (167). It was subsequently shown that treatment with celecoxib in combination with 15-hydroxyprostaglandin dehydrogenase (15-PGDH) (168), sunitinib (169), and PD-1 (170) in a murine metastatic tumor model more effectively reduced MDSCs levels as well as increased T cell numbers. Therefore, multiple drug combination therapy may be used selectively on therapeutic strategies targeting MDSCs for the patient’s own condition.
In the last two years, researchers found that triggering receptor expressed on myeloid cells (TREMs) has emerged as a potent regulator of innate immune responses, and observed that TREM1 was expressed in M-MDSCs and PMN-MDSCs, but TREM2 was expressed only in the M-MDSCs. They performed contrasting experiments by overexpressing and underexpressing TREM1 and observed worse prognosis in cancer patients with increased levels of TREM1 (171). Martina’s experiments suggest that TREM2 deficiency may promote increased T cell activation and may enhance responsiveness to anti-PD-1 checkpoint blockade, and treatment with anti-TREM2 mAb curbed tumor growth and fostered regression when combined with anti-PD-1 (172). Therefore, TREMs may be a useful biomarker of tumor progression, and developing TREMs inhibitors might be able to effectively combat tumor development.
Nowadays, combining MDSCs targeted therapy and immunotherapy has emerged as a new therapeutic strategy in improving the therapeutic efficacy of targeted MDSCs. The investigators found that mice still had large primary tumors and metastases that were not eliminated after treatment with a single anti-PD-1, whereas when anti-PD-1 was combined with 5-azacytidine and entinostat, both primary tumors in mice were eliminated and no metastases appeared (173). This indicates that immunotherapy combined with MDSCs targeted therapy is more effective in improving tumor progression and improving patient survival.
PMN formation reflects the dynamic relationship between tumor cells and the microenvironment of metastatic sites. Exploring PMN formation as well as the understanding of PMN biology has largely relied on mouse models of lung metastases. Thus, more clinical studies are needed, and more direct assays should be developed on the basis of existing techniques for PMN detection in order to observe the dynamic course of tumor metastasis.
In PMN formation, MDSCs play an important immunosuppressive role because of their specific heterogeneity. PMN-MDSCs suppress T cell function mainly through the production of ROS, and because ROS are very labile, PMN-MDSCs need to be in close contact with T cells to exert suppressive effects. In contrast, M-MDSCs suppress T cell activation by producing large amounts of iNOS, Arg-1, and some immunosuppressive factors. The half-life of iNOS, Arg-1, is much longer compared to ROS, so M-MDSCs need not come into close contact with T cells only to exert their suppressive effect. This illustrates the higher inhibitory activity of M-MDSCs than PMN-MDSCs (84, 174–177). Compared with the first two subsets of MDSCs, aspects such as the origin and function of e-MDSCs are generally less well recognized. It has been suggested that e-MDSCs do not have an inhibitory effect on the proliferation of T cells (178), and conversely, it has been shown that e-MDSCs have a very low inhibitory effect (179). Therefore, whether e-MDSCs can be considered as M-MDSCs, PMN-MDSCs such functional MDSCs is still controversial and needs further exploration. In contrast, F-MDSCs exert immunosuppressive effects by inducing the conversion of effector T cells into Tregs through the production of IDO, as described previously.
In addition to their own immunosuppressive functions, MDSCs have been shown to play important roles in vascular leakage, ECM remodeling, angiogenesis, and so on. Based on the role of MDSCs at various stages in PMN formation, researchers have developed therapeutic approaches to target MDSCs to detect the tumor metastasis situation at early stages of cancer. Although numerous studies have demonstrated the pro metastatic role of MDSCs, additional studies have shown that MDSCs can suppress tumor metastasis in some contexts. High amounts of anti-angiogenic TSP-1 are produced by MDSCs in tumors that lack metastatic potential, and knockdown of TSP-1 restores the pro tumor effects of MDSCs (180). Attention needs therefore to be paid to the effects of TSP-1 in therapies that block tumor metastasis by targeting MDSCs. Nowadays, for targeted therapy of MDSCs, it is important to focus not only on MDSCs regulatory network but also on the mutual communication between MDSCs and other cells or components, which has the potential to provide new options for cancer therapy.
GY drafted the article and did literature search. WR revised the article, provided ideas on article structure, and added some content. RQ, JH and SZ modified the arrangement of the article and helped to improve the accuracy of the language. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (82174146) and grants from Henan Provincial Science and Technology Research Project (grant no. 212102310639), Key Scientific Research Project of Colleges and Universities in Henan Province (grant no. 19zx009).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Arg-1, arginase-1; BMDCs, bone marrow-derived cells; COX2, cyclooxygenase 2; CSCs, cancer stem cells; CTCs, circulating tumor cells; DCs, dendritic cells; DDR1, discoidin domain receptor 1, DDR1; ECM, extracellular matrix; e-MDSCs, early myeloid derived suppressor cells; EMT, epithelial mesenchymal transition; F-MDSCs, fibrocytic myeloid derived suppressor cells; IDO, indolamine 2, 3-dioxygenase; iNOS, inducible nitric oxide synthase; MET, mesenchymal epithelial transition; MDSCs, myeloid derived suppressor cells; M-MDSCs: monocytic myeloid derived suppressor cells; NETs, neutrophil extracellular traps; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; PMN, pre-metastatic niche; PMN-MDSCs, polymorphonuclear myeloid derived suppressor cells; ROS, reactive oxygen species; TDSFs, tumor-derived secretory factors; TGF-β, transforming growth factor; Tregs, regulatory T cells; TREMs, triggering receptor expressed on myeloid cells; VEGF, vascular endothelial growth factor; ZO-1, zonula occludens 1.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature (2005) 438(7069):820–7. doi: 10.1038/nature04186
3. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res (2007) 67(1):425–6. doi: 10.1158/0008-5472.CAN-06-3037
4. Talmadge JE, Gabrilovich DI. History of myeloid derived suppressor cells. Nat Rev Cancer (2013) 13(10):739–52. doi: 10.1038/nrc3581
5. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
6. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
7. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol (2008) 181(8):5791–802. doi: 10.4049/jimmunol.181.8.5791
8. Goldmann O, Beineke A, Medina E. Identification of a novel subset of myeloid-derived suppressor cells during chronic staphylococcal infection that resembles immature eosinophils. J Infect Dis (2017) 216(11):1444–51. doi: 10.1093/infdis/jix494
9. Pawelec G, Verschoor CP, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: Not only in tumor immunity. Front Immunol (2019) 10:1099. doi: 10.3389/fimmu.2019.01099
10. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
11. Zhang H, Maric I, DiPrima MJ, Khan J, Orentas RJ, Kaplan RN, et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood (2013) 122(7):1105–13. doi: 10.1182/blood-2012-08-449413
12. Mazza EM, Zoso A, Mandruzzato S, Bronte V, Serafini P, Inverardi L, et al. Gene expression profiling of human fibrocytic myeloid-derived suppressor cells (f-MDSCs). Genomics Data (2014) 2:389–92. doi: 10.1016/j.gdata.2014.10.018
13. Gunaydin G, Kesikli SA, Guc D. Cancer associated fibroblasts have phenotypic and functional characteristics similar to the fibrocytes that represent a novel MDSC subset. Oncoimmunology (2015) 4(9):e1034918. doi: 10.1080/2162402X.2015
14. Cole K, Pravoverov K, Talmadge JE. Role of myeloid-derived suppressor cells in metastasis. Cancer Metastasis Rev (2021) 40(2):391–411. doi: 10.1007/s10555-020-09947-x
15. Nguyen DX, Bos PD, Massagué. J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer (2009) 9(4):274–84. doi: 10.1038/nrc2622
16. Zhang Y, Weinberg RA. Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front Med (2018) 12(4):361–73. doi: 10.1007/s11684-018-0656-6
17. Chaffer CL. Weinberg RA. A Perspective Cancer Cell Metastasis. Sci (2011) 331(6024):1559–64. doi: 10.1126/science.1203543
18. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev (1989) 8(2):98–101.
19. Poste G, Fidler IJ. The pathogenesis of cancer metastasis. Nature (1980) 283(5743):139–46. doi: 10.1038/283139a0
20. Hart IR, Fidler IJ. Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer Res (1980) 40(7):2281–7.
21. Chu JE, Allan AL. The role of cancer stem cells in the organ tropism of breast cancer metastasis: A mechanistic balance between the "Seed" and the "Soil"? Int J Breast Cancer (2012) 2012:209748. doi: 10.1155/2012/209748
22. Zhou Y, Han M, Gao J. Prognosis and targeting of pre-metastatic niche. J Controlled release Off J Controlled Release Soc (2020) 325:223–34. doi: 10.1016/j.jconrel.2020.06.037
23. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer (2011) 11(2):123–34. doi: 10.1038/nrc3004
24. Wang H, Pan J, Barsky L, Jacob JC, Zheng Y, Gao C, et al. Characteristics of pre-metastatic niche: the landscape of molecular and cellular pathways. Mol Biomedicine (2021) 2(1):3. doi: 10.1186/s43556-020-00022-z
25. Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature (2007) 446(7137):765–70. doi: 10.1038/nature05760
26. Huang Y, Song N, Ding Y, Yuan S, Li X, Cai H, et al. Pulmonary vascular destabilization in the premetastatic phase facilitates lung metastasis. Cancer Res (2009) 69(19):7529–37. doi: 10.1158/0008-5472
27. Zeng Z, Li Y, Pan Y, Lan X, Song F, Sun J, et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat Commun (2018) 9(1):5395. doi: 10.1038/s41467-018-07810-w
28. Fang JH, Zhang ZJ, Shang LR, Luo YW, Lin YF, Yuan Y, et al. Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology (2018) 68(4):1459–75. doi: 10.1002/hep.29920
29. Yokota Y, Noda T, Okumura Y, Kobayashi S, Iwagami Y, Yamada D, et al. Serum exosomal miR-638 is a prognostic marker of HCC via downregulation of VE-cadherin and ZO-1 of endothelial cells. Cancer Sci (2021) 112(3):1275–88. doi: 10.1111/cas.14807
30. Ma Z, Wei K, Yang F, Guo Z, Pan C, He Y, et al. Tumor-derived exosomal miR-3157-3p promotes angiogenesis, vascular permeability and metastasis by targeting TIMP/KLF2 in non-small cell lung cancer. Cell Death Dis (2021) 12(9):840. doi: 10.1038/s41419-021-04037-4
31. Cheung KJ, Ewald AJ. A collective route to metastasis: Seeding by tumor cell clusters. Science (2016) 352(6282):167–9. doi: 10.1126/science.aaf6546
32. Lo HC, Xu Z, Kim IS, Pingel B, Aguirre S, Kodali S, et al. Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat Cancer (2020) 1(7):709–22. doi: 10.1038/s43018-020-0068-9
33. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer (2017) 17(5):302–17. doi: 10.1038/nrc.2017.6
34. Doglioni G, Parik S, Fendt SM. Interactions in the (Pre)metastatic niche support metastasis formation. Front Oncol (2019) 9:219. doi: 10.3389/fonc.2019.00219
35. Wang Y, Ding Y, Guo N, Wang S. MDSCs: Key criminals of tumor pre-metastatic niche formation. Front Immunol (2019) 10:172. doi: 10.3389/fimmu.2019.00172
36. Sceneay J, Chow MT, Chen A, Halse HM, Wong CS, Andrews DM, et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res (2012) 72(16):3906–11. doi: 10.1158/0008-5472.CAN-11-3873
37. Chiu DK, Xu IM, Lai RK, Tse AP, Wei LL, Koh HY, et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-c motif) ligand 26. Hepatology (2016) 64(3):797–813. doi: 10.1002/hep.28655
38. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
39. Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun (2017) 8(1):517. doi: 10.1038/s41467-017-00530-7
40. Gu H, Deng W, Zheng Z, Wu K, Sun F. CCL2 produced by pancreatic ductal adenocarcinoma is essential for the accumulation and activation of monocytic myeloid-derived suppressor cells. Immun Inflamm Dis (2021) 9(4):1686–95. doi: 10.1002/iid3.523
41. Hartwig T, Montinaro A, von Karstedt S, Sevko A, Surinova S, Chakravarthy A, et al. The TRAIL-induced cancer secretome promotes a tumor-supportive immune microenvironment via CCR2. Mol Cell (2017) 65(4):730–742.e5. doi: 10.1016/j.molcel.2017.01.021
42. Zhou Y, Guo F. A selective sphingosine-1-phosphate receptor 1 agonist SEW-2871 aggravates gastric cancer by recruiting myeloid-derived suppressor cells. J Biochem (2018) 163(1):77–83. doi: 10.1093/jb/mvx064
43. Blattner C, Fleming V, Weber R, Himmelhan B, Altevogt P, Gebhardt C, et al. CCR5+ myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res (2018) 78(1):157–67. doi: 10.1158/0008-5472.CAN-17-0348
44. Hawila E, Razon H, Wildbaum G, Blattner C, Sapir Y, Shaked Y, et al. CCR5 directs the mobilization of CD11b+Gr1+Ly6Clow polymorphonuclear myeloid cells from the bone marrow to the blood to support tumor development. Cell Rep (2017) 21(8):2212–22. doi: 10.1016/j.celrep.2017.10.104
45. Luo A, Meng M, Wang G, Han R, Zhang Y, Jing X, et al. Myeloid-derived suppressor cells recruited by chemokine (C-c motif) ligand 3 promote the progression of breast cancer via phosphoinositide 3-Kinase-Protein kinase b-mammalian target of rapamycin signaling. J Breast Cancer (2020) 23(2):141–61. doi: 10.4048/jbc.2020.23.e26
46. Inamoto S, Itatani Y, Yamamoto T, Minamiguchi S, Hirai H, Iwamoto M, et al. Loss of SMAD4 promotes colorectal cancer progression by accumulation of myeloid-derived suppressor cells through the CCL15-CCR1 chemokine axis. Clin Cancer Res an Off J Am Assoc Cancer Res (2016) 22(2):492–501. doi: 10.1158/1078-0432.CCR-15-0726
47. Okuma A, Hanyu A, Watanabe S, Hara E. p16Ink4a and p21Cip1/Waf1 promote tumour growth by enhancing myeloid-derived suppressor cells chemotaxis. Nat Commun (2017) 8(1):2050. doi: 10.1038/s41467-017-02281-x
48. Wang D, Sun H, Wei J, Cen B, DuBois RN. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res (2017) 77(13):3655–65. doi: 10.1158/0008-5472.CAN-16-3199
49. Xia S, Wu J, Zhou W, Zhang M, Zhao K, Liu J, et al. SLC7A2 deficiency promotes hepatocellular carcinoma progression by enhancing recruitment of myeloid-derived suppressors cells. Cell Death Dis (2021) 12(6):570. doi: 10.1038/s41419-021-03853-y
50. He Q, Huang W, Liu D, Zhang T, Wang Y, Ji X, et al. Homeobox B5 promotes metastasis and poor prognosis in hepatocellular carcinoma, via FGFR4 and CXCL1 upregulation. Theranostics (2021) 11(12):5759–77. doi: 10.7150/thno.57659
51. Zhang H, Ye YL, Li MX, Ye SB, Huang WR, Cai TT, et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene (2017) 36(15):2095–104. doi: 10.1038/onc.2016.367
52. Taki M, Abiko K, Baba T, Hamanishi J, Yamaguchi K, Murakami R, et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat Commun (2018) 9(1):1685. doi: 10.1038/s41467-018-03966-7
53. Chen L, Jin XH, Luo J, Duan JL, Cai MY, Chen JW, et al. ITLN1 inhibits tumor neovascularization and myeloid derived suppressor cells accumulation in colorectal carcinoma. Oncogene (2021) 40(40):5925–37. doi: 10.1038/s41388-021-01965-5
54. Najjar YG, Rayman P, Jia X, Pavicic PG Jr, Rini BI, Tannenbaum C, et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1β, IL8, CXCL5, and mip-1α. Clin Cancer Res an Off J Am Assoc Cancer Res (2017) 23(9):2346–55. doi: 10.1158/1078-0432.CCR-15-1823
55. Lin N, Chen L, Zhang Y, Yang Y, Zhang L, Chen L, et al. KIF4A promotes tumor progression of bladder cancer via CXCL5 dependent myeloid-derived suppressor cells recruitment. Sci Rep (2022) 12(1):6015. doi: 10.1038/s41598-022-10029-x
56. Yu F, Shi Y, Wang J, Li J, Fan D, Ai W. Deficiency of kruppel-like factor KLF4 in mammary tumor cells inhibits tumor growth and pulmonary metastasis and is accompanied by compromised recruitment of myeloid-derived suppressor cells. Int J Cancer (2013) 133(12):2872–83. doi: 10.1002/ijc.28302
57. Zadian SS, Adcock IM, Salimi B, Mortaz E. Circulating levels of monocytic myeloid-derived suppressor cells (M-MDSC) and CXCL-8 in non-small cell lung cancer (NSCLC). Tanaffos (2021) 20(1):15–21.
58. Yue D, Liu S, Zhang T, Wang Y, Qin G, Chen X, et al. NEDD9 promotes cancer stemness by recruiting myeloid-derived suppressor cells via CXCL8 in esophageal squamous cell carcinoma. Cancer Biol Med (2021) 18(3):705–20. doi: 10.20892/j.issn.2095-3941.2020.0290
59. Yu X, Wang D, Wang X, Sun S, Zhang Y, Wang S, et al. CXCL12/CXCR4 promotes inflammation-driven colorectal cancer progression through activation of RhoA signaling by sponging miR-133a-3p. J Exp Clin Cancer Res (2019) 38(1):32. doi: 10.1186/s13046-018-1014-x
60. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol (2008) 181(7):4666–75. doi: 10.4049/jimmunol.181.7.4666
61. Dinca SC, Greiner D, Weidenfeld K, Bond L, Barkan D, Jorcyk CL. Novel mechanism for OSM-promoted extracellular matrix remodeling in breast cancer: LOXL2 upregulation and subsequent ECM alignment. Breast Cancer Res (2021) 23(1):56. doi: 10.1186/s13058-021-01430-x
62. Wu S, Zheng Q, Xing X, Dong Y, Wang Y, You Y, et al. Matrix stiffness-upregulated LOXL2 promotes fibronectin production, MMP9 and CXCL12 expression and BMDCs recruitment to assist pre-metastatic niche formation. J Exp Clin Cancer Res (2018) 37(1):99. doi: 10.1186/s13046-018-0761-z
63. Cox TR, Rumney RMH, Schoof EM, Perryman L, Høye AM, Agrawal A, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature (2015) 522(7554):106–10. doi: 10.1038/nature14492
64. Paolillo M, Schinelli S. Extracellular matrix alterations in metastatic processes. Int J Mol Sci (2019) 20(19):4947. doi: 10.3390/ijms20194947
65. Yuzhalin AE, Gordon-Weeks AN, Tognoli ML, Jones K, Markelc B, Konietzny R, et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat Commun (2018) 9(1):4783. doi: 10.1038/s41467-018-07306-7
66. Khadge S, Cole K, Talmadge JE. Myeloid derived suppressor cells and the release of micro-metastases from dormancy. Clin Exp Metastasis (2021) 38(3):279–93. doi: 10.1007/s10585-021-10098-8
67. Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer (2010) 46(7):1181–8. doi: 10.1016/j.ejca.2010.02.027
68. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol (2013) 15(7):807–17. doi: 10.1038/ncb2767
69. Boyerinas B, Zafrir M, Yesilkanal AE, Price TT, Hyjek EM, Sipkins DA. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood (2013) 121(24):4821–31. doi: 10.1182/blood-2012-12-475483
70. Keeratichamroen S, Lirdprapamongkol K, Svasti J. Mechanism of ECM-induced dormancy and chemoresistance in A549 human lung carcinoma cells. Oncol Rep (2018) 39(4):1765–74. doi: 10.3892/or.2018.6258
71. Di Martino JS, Nobre AR, Mondal C, Taha I, Farias EF, Fertig EJ, et al. A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancy. Nat Cancer (2022) 3(1):90–107. doi: 10.1038/s43018-021-00291-9
72. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science (2018) 361(6409):eaao4227. doi: 10.1126/science.aao4227
73. Safarzadeh E, Orangi M, Mohammadi H, Babaie F, Baradaran B. Myeloid-derived suppressor cells: Important contributors to tumor progression and metastasis. J Cell Physiol (2018) 233(4):3024–36. doi: 10.1002/jcp.26075
74. Cheng JN, Yuan YX, Zhu B, Jia Q. Myeloid-derived suppressor cells: A multifaceted accomplice in tumor progression. Front Cell Dev Biol (2021) 9:740827. doi: 10.3389/fcell.2021.740827
75. Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev (2008) 222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x
76. Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res (2010) 70(1):68–77. doi: 10.1158/0008-5472.CAN-09-2587
77. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by l-arginine. J Biol Chem (2002) 277(24):21123–9. doi: 10.1074/jbc.M110675200
78. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109(4):1568–73. doi: 10.1182/blood-2006-06-031856
79. Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol (2003) 170(1):270–8. doi: 10.4049/jimmunol.170.1.270
80. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res (2005) 65(8):3044–8. doi: 10.1158/0008-5472.CAN-04-4505
81. Heuvers ME, Muskens F, Bezemer K, Lambers M, Dingemans AC, Groen HJM, et al. Arginase-1 mRNA expression correlates with myeloid-derived suppressor cell levels in peripheral blood of NSCLC patients. Lung Cancer (2013) 81(3):468–74. doi: 10.1016/j.lungcan.2013.06.005
82. Cao Y, Feng Y, Zhang Y, Zhu X, Jin F. L-arginine supplementation inhibits the growth of breast cancer by enhancing innate and adaptive immune responses mediated by suppression of MDSCs in vivo. . BMC Cancer (2016) 16:343. doi: 10.1186/s12885-016-2376-0
83. Bian Z, Abdelaal AM, Shi L, Liang H, Xiong L, Kidder K, et al. Arginase-1 is neither constitutively expressed in nor required for myeloid-derived suppressor cell-mediated inhibition of T-cell proliferation. Eur J Immunol (2018) 48(6):1046–58. doi: 10.1002/eji.201747355
84. Ohl K, Tenbrock K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front Immunol (2018) 9:2499. doi: 10.3389/fimmu.2018.02499
85. Beury DW, Carter KA, Nelson C, Sinha P, Hanson E, Nyandjo M, et al. Myeloid-derived suppressor cell survival and function are regulated by the transcription factor Nrf2. J Immunol (2016) 196(8):3470–8. doi: 10.4049/jimmunol.1501785
86. Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res (2001) 61(12):4756–60.
87. Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol (2002) 168(2):689–95. doi: 10.4049/jimmunol.168.2.689
88. Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing fc receptor-mediated natural killer cell function. Clin Cancer Res an Off J Am Assoc Cancer Res (2018) 24(8):1891–904. doi: 10.1158/1078-0432.CCR-17-0691
89. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13(7):828–35. doi: 10.1038/nm1609
90. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer (2014) 134(12):2853–64. doi: 10.1002/ijc.28622
91. Rastad JL, Green WR. Myeloid-derived suppressor cells in murine AIDS inhibit b-cell responses in part via soluble mediators including reactive oxygen and nitrogen species, and TGF-β. Virology (2016) 499:9–22. doi: 10.1016/j.virol.2016.08.031
92. Kraaij MD, Savage ND, van der Kooij SW, Koekkoek K, Wang J, van den Berg JM, et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc Natl Acad Sci United States America (2010) 107(41):17686–91. doi: 10.1073/pnas.1012016107
93. Kraaij MD, van der Kooij SW, Reinders ME, Koekkoek K, Rabelink TJ, van Kooten C, et al. Dexamethasone increases ROS production and T cell suppressive capacity by anti-inflammatory macrophages. Mol Immunol (2011) 49(3):549–57. doi: 10.1016/j.molimm.2011.10.002
94. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol (2017) 18(12):1332–41. doi: 10.1038/ni.3868
95. Li B, Chan HL, Chen P. Immune checkpoint inhibitors: Basics and challenges. Curr medicinal Chem (2019) 26(17):3009–25. doi: 10.2174/0929867324666170804143706
96. Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med (2017) 214(4):895–904. doi: 10.1084/jem.20160801
97. Yamauchi Y, Safi S, Blattner C, Rathinasamy A, Umansky L, Juenger S, et al. Circulating and tumor myeloid-derived suppressor cells in resectable non-small cell lung cancer. Am J Respir Crit Care Med (2018) 198(6):777–87. doi: 10.1164/rccm.201708-1707OC
98. Gebhardt C, Sevko A, Jiang H, Lichtenberger R, Reith M, Tarnanidis K, et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin Cancer Res an Off J Am Assoc Cancer Res (2015) 21(24):5453–9. doi: 10.1158/1078-0432.CCR-15-0676
99. Cheng P, Eksioglu EA, Chen X, Kandell W, Le Trinh T, Cen L, et al. S100A9-induced overexpression of PD-1/PD-L1 contributes to ineffective hematopoiesis in myelodysplastic syndromes. Leukemia (2019) 33(8):2034–46. doi: 10.1038/s41375-019-0397-9
100. Linnemann C, Schildberg FA, Schurich A, Diehl L, Hegenbarth SI, Endl E, et al. Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology (2009) 128(1 Suppl):e728–37. doi: 10.1111/j.1365-2567.2009.03075.x
101. Mastelic-Gavillet B, Navarro Rodrigo B, Décombaz L, Wang H, Ercolano G, Ahmed R, et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T cells. J Immunotherapy Cancer (2019) 7(1):257. doi: 10.1186/s40425-019-0719-5
102. Zheng W, Zhu Y, Chen X, Zhao J. CD73 expression in myeloid-derived suppressor cells is correlated with clinical stages in head and neck squamous cell carcinomas. Ann Trans Med (2021) 9(14):1148. doi: 10.21037/atm-21-2589
103. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology (2017) 6(6):e1320011. doi: 10.1080/2162402X.2017.1320011
104. Rosemblatt MV, Parra-Tello B, Briceño P, Rivas-Yáñez E, Tucer S, Saavedra-Almarza J, et al. Ecto-5'-Nucleotidase (CD73) regulates the survival of CD8+ T cells. Front Cell Dev Biol (2021) 9:647058. doi: 10.3389/fcell.2021.647058
105. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol (2009) 182(1):240–9. doi: 10.4049/jimmunol.182.1.240
106. Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, Ostrand-Rosenberg S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukocyte Biol (2014) 96(6):1109–18. doi: 10.1189/jlb.3A0414-210R
107. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol (2007) 179(2):977–83. doi: 10.4049/jimmunol.179.2.977
108. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi: 10.1158/0008-5472.CAN-05-1299
109. Saleh R, Elkord E. FoxP3+ T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett (2020) 490:174–85. doi: 10.1016/j.canlet.2020.07.022
110. Zoso A, Mazza EM, Bicciato S, Mandruzzato S, Bronte V, Serafini P, et al. Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via treg-cell expansion. Eur J Immunol (2014) 44(11):3307–19. doi: 10.1002/eji.201444522
111. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity (2005) 22(5):633–42. doi: 10.1016/j.immuni.2005.03.013
112. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol (2006) 176(11):6752–61. doi: 10.4049/jimmunol.176.11.6752
113. Lee TK, Poon RT, Yuen AP, Ling MT, Kwok WK, Wang XH, et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin Cancer Res an Off J Am Assoc Cancer Res (2006) 12(18):5369–76. doi: 10.1158/1078-0432.CCR-05-2722
114. Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res (2008) 68(2):537–44. doi: 10.1158/0008-5472.CAN-07-5682
115. Hsu DS, Wang HJ, Tai SK, Chou CH, Hsieh CH, Chiu PH, et al. Acetylation of snail modulates the cytokinome of cancer cells to enhance the recruitment of macrophages. Cancer Cell (2014) 26(4):534–48. doi: 10.1016/j.ccell.2014.09.002
116. Singh M, Yelle N, Venugopal C, Singh SK. EMT: Mechanisms and therapeutic implications. Pharmacol Ther (2018) 182:80–94. doi: 10.1016/j.pharmthera.2017.08.009
117. Taki M, Abiko K, Ukita M, Murakami R, Yamanoi K, Yamaguchi K, et al. Tumor immune microenvironment during epithelial-mesenchymal transition. Clin Cancer Res an Off J Am Assoc Cancer Res (2021) 27(17):4669–79. doi: 10.1158/1078-0432.CCR-20-4459
118. Oka N, Soeda A, Inagaki A, Onodera M, Maruyama H, Hara A, et al. VEGF promotes tumorigenesis and angiogenesis of human glioblastoma stem cells. Biochem Biophys Res Commun (2007) 360(3):553–9. doi: 10.1016/j.bbrc.2007.06.094
119. Fu XG, Deng J, Xu WJ, Chen JY, Sun J, Deng H. Histidine decarboxylase-expressing PMN-MDSC-derived TGF-β1 promotes the epithelial-mesenchymal transition of metastatic lung adenocarcinoma. Int J Clin Exp Pathol (2020) 13(6):1361–71.
120. Schlegel NC, von Planta A, Widmer DS, Dummer R, Christofori G. PI3K signalling is required for a TGFβ-induced epithelial-mesenchymal-like transition (EMT-like) in human melanoma cells. Exp Dermatol (2015) 24(1):22–8. doi: 10.1111/exd.12580
121. Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res (2011) 9(12):1658–67. doi: 10.1158/1541-7786.MCR-11-0271
122. Kim SY, Kang JW, Song X, Kim BK, Yoo YD, Kwon YT, et al. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signalling (2013) 25(4):961–9. doi: 10.1016/j.cellsig.2013.01.007
123. Yang L, Dong Y, Li Y, Wang D, Liu S, Wang D, et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-κB/Notch1 pathway in non-small cell lung cancer. Int J Cancer (2019) 145(4):1099–110. doi: 10.1002/ijc.32151
124. Sangaletti S, Talarico G, Chiodoni C, Cappetti B, Botti L, Portararo P, et al. SPARC Is a new myeloid-derived suppressor cell marker licensing suppressive activities. Front Immunol (2019) 10:1369. doi: 10.3389/fimmu.2019.01369
125. Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun (2017) 8:14979. doi: 10.1038/ncomms14979
126. Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res (2012) 72(6):1384–94. doi: 10.1158/0008-5472.CAN-11-2905
127. Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer (2011) 11(7):512–22. doi: 10.1038/nrc3080
128. Heeke S, Mograbi B, Alix-Panabières C, Hofman P. Never travel alone: The crosstalk of circulating tumor cells and the blood microenvironment. Cells (2019) 8(7):714. doi: 10.3390/cells8070714
129. Arnoletti JP, Zhu X, Almodovar AJ, Veldhuis PP, Sause R, Griffith E, et al. Portal venous blood circulation supports immunosuppressive environment and pancreatic cancer circulating tumor cell activation. Pancreas (2017) 46(1):116–23. doi: 10.1097/MPA.0000000000000667
130. Sprouse ML, Welte T, Boral D, Liu HN, Yin W, Vishnoi M, et al. PMN-MDSCs enhance CTC metastatic properties through reciprocal interactions via ROS/Notch/Nodal signaling. Int J Mol Sci (2019) 20(8):1916. doi: 10.3390/ijms20081916
131. Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature (2019) 566(7745):553–7. doi: 10.1038/s41586-019-0915-y
132. Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, et al. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res (2010) 70(15):6139–49. doi: 10.1158/0008-5472.CAN-10-0706
133. Hiratsuka S, Goel S, Kamoun WS, Maru Y, Fukumura D, Duda DG, et al. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via e-selectin up-regulation. Proc Natl Acad Sci United States America (2011) 108(9):3725–30. doi: 10.1073/pnas.1100446108
134. Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci United States America (2009) 106(16):6742–7. doi: 10.1073/pnas.0902280106
135. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest (2013) 123(8):3446–58. doi: 10.1172/JCI67484
136. Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F, et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int J Cancer (2017) 140(10):2321–30. doi: 10.1002/ijc.30635
137. Alfaro C, Teijeira A, Oñate C, Pérez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res an Off J Am Assoc Cancer Res (2016) 22(15):3924–36. doi: 10.1158/1078-0432.CCR-15-2463
138. Kim SW, Lee JK. Role of HMGB1 in the interplay between NETosis and thrombosis in ischemic stroke: A review. Cells (2020) 9(8):1794. doi: 10.3390/cells9081794
139. Ortiz-Espinosa S, Morales X, Senent Y, Alignani D, Tavira B, Macaya I, et al. Complement C5a induces the formation of neutrophil extracellular traps by myeloid-derived suppressor cells to promote metastasis. Cancer Lett (2022) 529:70–84. doi: 10.1016/j.canlet.2021.12.027
140. Liu JF, Deng WW, Chen L, Li YC, Wu L, Ma SR, et al. Inhibition of JAK2/STAT3 reduces tumor-induced angiogenesis and myeloid-derived suppressor cells in head and neck cancer. Mol Carcinogenesis (2018) 57(3):429–39. doi: 10.1002/mc.22767
141. Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell (2008) 13(3):193–205. doi: 10.1016/j.ccr.2007.11.032
142. Aguilera KY, Rivera LB, Hur H, Carbon JG, Toombs JE, Goldstein CD, et al. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res (2014) 74(4):1032–44. doi: 10.1158/0008-5472.CAN-13-2800
143. Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest (2008) 118(10):3367–77. doi: 10.1172/JCI35213
144. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature (2007) 450(7171):825–31. doi: 10.1038/nature06348
145. Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med (2006) 12(5):557–67. doi: 10.1038/nm1400
146. Hsu YL, Yen MC, Chang WA, Tsai PH, Pan YC, Liao SH, et al. CXCL17-derived CD11b+Gr-1+ myeloid-derived suppressor cells contribute to lung metastasis of breast cancer through platelet-derived growth factor-BB. Breast Cancer Res (2019) 21(1):23. doi: 10.1186/s13058-019-1114-3
147. Deng Z, Rong Y, Teng Y, Zhuang X, Samykutty A, Mu J, et al. Exosomes miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene (2017) 36(5):639–51. doi: 10.1038/onc.2016.229
148. Noman MZ, Janji B, Hu S, Wu JC, Martelli F, Bronte V, et al. Tumor-promoting effects of myeloid-derived suppressor cells are potentiated by hypoxia-induced expression of miR-210. Cancer Res (2015) 75(18):3771–87. doi: 10.1158/0008-5472.CAN-15-0405
149. Kosaka N, Iguchi H, Hagiwara K, Yoshioka Y, Takeshita F, Ochiya T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J Biol Chem (2013) 288(15):10849–59. doi: 10.1074/jbc.M112.446831
150. Shokeen M, Zheleznyak A, Wilson JM, Jiang M, Liu R, Ferdani R, et al. Molecular imaging of very late antigen-4 (α4β1 integrin) in the premetastatic niche. J Nucl Med Off publication Soc Nucl Med (2012) 53(5):779–86. doi: 10.2967/jnumed.111.100073
151. Li R, Qi Y, Han M, Geng B, Wang G, Han M. Computed tomography reveals microenvironment changes in premetastatic lung. Eur Radiol (2021) 31(6):4340–9. doi: 10.1007/s00330-020-07500-6
152. Eisenblaetter M, Flores-Borja F, Lee JJ, Wefers C, Smith H, Hueting R, et al. Visualization of tumor-immune interaction-Target-Specific imaging of S100A8/A9 reveals pre-metastatic niche establishment. Theranostics (2017) 7(9):2392–401. doi: 10.7150/thno.17138
153. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res (2007) 67(22):11021–8. doi: 10.1158/0008-5472.CAN-07-2593
154. Ma J, Liu Y, Li Y, Gu J, Liu J, Tang J, et al. Differential role of all-trans retinoic acid in promoting the development of CD4+ and CD8+ regulatory T cells. J Leukocyte Biol (2014) 95(2):275–83. doi: 10.1189/jlb.0513297
155. Zhou J, Wu J, Chen X, Fortenbery N, Eksioglu E, Kodumudi KN, et al. Icariin and its derivative, ICT, exert anti-inflammatory, anti-tumor effects, and modulate myeloid derived suppressive cells (MDSCs) functions. Int Immunopharmacol (2011) 11(7):890–8. doi: 10.1016/j.intimp.2011.01.007
156. Yao W, Ba Q, Li X, Li H, Zhang S, Yuan Y, et al. A natural CCR2 antagonist relieves tumor-associated macrophage-mediated immunosuppression to produce a therapeutic effect for liver cancer. EBioMedicine (2017) 22:58–67. doi: 10.1016/j.ebiom.2017.07.014
157. Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, et al. Phase ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2-Negative metastatic breast cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2017) 23(18):5358–65. doi: 10.1158/1078-0432.CCR-16-2748
158. Ghobrial IM, Liu CJ, Redd RA, Perez RP, Baz R, Zavidij O, et al. A phase Ib/II trial of the first-in-Class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma. Clin Cancer Res an Off J Am Assoc Cancer Res (2020) 26(2):344–53. doi: 10.1158/1078-0432.CCR-19-0647
159. Melani C, Sangaletti S, Barazzetta FM, Werb Z, Colombo MP. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res (2007) 67(23):11438–46. doi: 10.1158/0008-5472.CAN-07-1882
160. Porembka MR, Mitchem JB, Belt BA, Hsieh CS, Lee HM, Herndon J, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunotherapy (2012) 61(9):1373–85. doi: 10.1007/s00262-011-1178-0
161. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res (2010) 70(8):3052–61. doi: 10.1158/0008-5472.CAN-09-3690
162. Kanterman J, Sade-Feldman M, Biton M, Ish-Shalom E, Lasry A, Goldshtein A, et al. Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res (2014) 74(21):6022–35. doi: 10.1158/0008-5472.CAN-14-0657
163. Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Trans Med (2016) 14(1):282. doi: 10.1186/s12967-016-1037-z
164. Zahoor H, Mir MC, Barata PC, Stephenson AJ, Campbell SC, Fergany A, et al. Phase II trial of continuous treatment with sunitinib in patients with high-risk (BCG-refractory) non-muscle invasive bladder cancer. Investigational New Drugs (2019) 37(6):1231–8. doi: 10.1007/s10637-018-00716-w
165. Qin H, Lerman B, Sakamaki I, Wei G, Cha SC, Rao SS, et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med (2014) 20(6):676–81. doi: 10.1038/nm.3560
166. Feng PH, Lam B, Tseng SH, Kung YJ, Farmer E, Cheng MA, et al. NKG2D-fc fusion protein promotes antitumor immunity through the depletion of immunosuppressive cells. Cancer Immunol Immunotherapy (2020) 69(10):2147–55. doi: 10.1007/s00262-020-02615-7
167. Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JG, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC Funct BMC Cancer (2010) 10:464. doi: 10.1186/1471-2407-10-464
168. Zhang B, Ma X, Li Z, Gao X, Wang F, Liu L, et al. Celecoxib enhances the efficacy of 15-hydroxyprostaglandin dehydrogenase gene therapy in treating murine breast cancer. J Cancer Res Clin Oncol (2013) 139(5):797–807. doi: 10.1007/s00432-013-1381-9
169. Zhao Q, Guo J, Wang G, Chu Y, Hu X. Suppression of immune regulatory cells with combined therapy of celecoxib and sunitinib in renal cell carcinoma. Oncotarget (2017) 8(1):1668–77. doi: 10.18632/oncotarget.13774
170. Chen JS, Chou CH, Wu YH, Yang MH, Chu SH, Chao YS, et al. CC-01 (chidamide plus celecoxib) modifies the tumor immune microenvironment and reduces tumor progression combined with immune checkpoint inhibitor. Sci Rep (2022) 12(1):1100. doi: 10.1038/s41598-022-05055-8
171. Ford JW, Gonzalez-Cotto M, MacFarlane AW4, Peri S, Howard OMZ, Subleski JJ, et al. Tumor-infiltrating myeloid cells Co-express TREM1 and TREM2 and elevated TREM-1 associates with disease progression in renal cell carcinoma. Front Oncol (2022) 11:662723. doi: 10.3389/fonc.2021.662723
172. Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell (2020) 182(4):886–900.e17. doi: 10.1016/j.cell.2020.07.013
173. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci United States America (2014) 111(32):11774–9. doi: 10.1073/pnas.1410626111
174. Liu Y, Wei G, Cheng WA, Dong Z, Sun H, Lee VY, et al. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol Immunotherapy (2018) 67(8):1181–95. doi: 10.1007/s00262-018-2175-3
175. Navasardyan I, Bonavida B. Regulation of T cells in cancer by nitric oxide. Cells (2021) 10(10):2655. doi: 10.3390/cells10102655
176. Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, et al. L-arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res (2015) 75(2):275–83. doi: 10.1158/0008-5472.CAN-14-1491
177. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
178. Nagatani Y, Funakoshi Y, Suto H, Imamura Y, Toyoda M, Kiyota N, et al. Immunosuppressive effects and mechanisms of three myeloid-derived suppressor cells subsets including monocytic-myeloid-derived suppressor cells, granulocytic-myeloid-derived suppressor cells, and immature-myeloid-derived suppressor cells. J Cancer Res Ther (2021) 17(4):1093–100. doi: 10.4103/jcrt.JCRT_1222_20
179. Lang S, Bruderek K, Kaspar C, Höing B, Kanaan O, Dominas N, et al. Clinical relevance and suppressive capacity of human myeloid-derived suppressor cell subsets. Clin Cancer Res an Off J Am Assoc Cancer Res (2018) 24(19):4834–44. doi: 10.1158/1078-0432.CCR-17-3726
Keywords: myeloid-derived suppressor cells, pre-metastatic niche, circulating tumor cells, immunosuppression, targeted therapy
Citation: Ya G, Ren W, Qin R, He J and Zhao S (2022) Role of myeloid-derived suppressor cells in the formation of pre-metastatic niche. Front. Oncol. 12:975261. doi: 10.3389/fonc.2022.975261
Received: 22 June 2022; Accepted: 12 September 2022;
Published: 27 September 2022.
Edited by:
Ramesh k. Ganju, The Ohio State University, United StatesReviewed by:
Stefano Ugel, University of Verona, ItalyCopyright © 2022 Ya, Ren, Qin, He and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weihong Ren, ren_weihong@163.com
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.