
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.990463
This article is part of the Research Topic Community Series in Manipulating the Immunological Tumor Microenvironment - Volume II View all 20 articles
 Hongshu Sui1†
Hongshu Sui1† Shengyi Dongye2†
Shengyi Dongye2† Xiaocui Liu1†
Xiaocui Liu1† Xinghua Xu1Li Wang3
Xinghua Xu1Li Wang3 Christopher Q. Jin4Minhua Yao1Zhaoqing Gong1
Christopher Q. Jin4Minhua Yao1Zhaoqing Gong1 Daniel Jiang4
Daniel Jiang4 Kexin Zhang4Yaling Liu2,5
Kexin Zhang4Yaling Liu2,5 Hui Liu1*
Hui Liu1* Guomin Jiang4*
Guomin Jiang4* Yanping Su1*
Yanping Su1*Myeloid-derived suppressor cells (MDSCs) are a group of heterogeneous cells which are abnormally accumulated during the differentiation of myeloid cells. Immunosuppression is the main functional feature of MDSCs, which inhibit T cell activity in the tumor microenvironment (TME) and promote tumoral immune escape. The main principle for immunotherapy is to modulate, restore, and remodel the plasticity and potential of immune system to have an effective anti-tumor response. In the TME, MDSCs are major obstacles to cancer immunotherapy through reducing the anti-tumor efficacy and making tumor cells more resistant to immunotherapy. Therefore, targeting MDSCs treatment becomes the priority of relevant studies and provides new immunotherapeutic strategy for cancer treatment. In this review, we mainly discuss the functions and mechanisms of MDSCs as well as their functional changes in the TME. Further, we review therapeutic effects of immunotherapy against MDSCs and potential breakthroughs regarding immunotherapy targeting MDSCs and immune checkpoint blockade (ICB) immunotherapy.
It is known that the incidence of cancer is still rising with poor prognosis and high mortality, despite the continuous improvement of treatment modalities (1). At present, the main treatments for cancer include surgery, radiotherapy, chemotherapy. However, those treatments have severe side effects on healthy cells, and have limitations such as drug resistance. Immunotherapy has increasingly attracted wide attention due to its unique advantages, especially its long-lasting therapeutic effects (2, 3). Recently, it was reported that the immune checkpoint blockade (ICB) immunotherapy, such as those targeting CTLA-4, PD-1, and PD-L1. ICB greatly improved treatment efficacy on tumors with high mutation rates, such as non-small cell lung cancer (NSCLC). In the clinical data statistics of NSCLC, it was found that the composite mutation characteristics are closely related to the pro-inflammatory tumor microenvironment (TME). In addition, the checkpoint proteins are the most commonly used biomarkers for NSCLC patients, especially for high expression of PD-L1 on tumor and PD-1 on T cells (4, 5). However, the response rate for ICB immunotherapy is only 15-20% in various types of solid tumors, which is far from clinical request due to complex TME (6).. Therefore, the impact of cancer on the immune system still needs to be explored.
The immune system consists of many negative feedbacks of inhibitory pathways that are found to suppress the development of excessive immune responses to avoid autoimmune reactions (7, 8). In the process of cancer development, there is a balance complex between cancer and the immune system. The occurrence and development of cancer cause immune escape, promoting cancer progression (9). The long-term inflammatory invasion of cancer cells disrupts the balance of the immune system, making immune cells exhausted, finally leading to continuous tumor growth and metastasis. Therefore, researchers are paying more attention to the anti-tumor immune responses on the TME, where there are plenty of cancer suppressor factors such as tumor-related stromal cells, regulatory T cells (Treg) and immunosuppressive myeloid cells (IMCs) (10). The immunosuppressive cytokines in TME produced by tumor cells, MDSCs and CAFs are crucial factors that mediate T-cell and other immune cell dysfunction. TGF-β signaling pathway, for example, plays an important role in tumor promotion (11, 12). A hallmark of cancer development is persistent inflammatory invasion and immune escape, which induce oncogene protein, reduces tumor suppressor genes, and produces IMCs which include TAMs and MDSCs (13, 14). Plenty of IMCs in TME and their strong inhibitory roles on lymphocytes have become the main obstacle to tumor immunotherapy (15). Here we focus on one type of cell of IMCs, MDSCs, which suppress T cell to promote tumor cell proliferation, metastatic growth and immunotherapy resistance (16).
MDSCs are a class of highly heterogeneous cells derived from immature myeloid progenitors, consisting of myeloid progenitors and precursors of macrophages, granulocytes and DCs, trigger the abnormal state of hematopoietic stem cells in the differentiation process and display immunosuppressive activity on T cells and NK cells during the progression of cancer (17) (Figure 1). MDSCs mainly consist of two cell subsets: granulocytic/polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs) (18). During cancer development, it is stimulated by long-term inflammatory factors and cytokines (e.g., high levels of GM-CSF, VEGF, IL-6, IL-1β, adenosine, HIF1α). This leads to myelogenesis and regulation in the bone marrow, which transforms into pathologically activated cells in tissues; Moderate myelogenesis, changes in cellular metabolism, thereby inhibiting immune function. Immunosuppression roles of MDSCs are mainly regulated through STAT3, STAT1, STAT6, NF-κB, ER stress pathway, Down-regulation of cAMP, COX2, PTGES, CEBPβ, IRF8, RB1 and lipid oxidation (19, 20). MDSCs participate in promoting tumor progression by enhancing tumor angiogenesis and invasion, and facilitating pre-metastatic niche formation (21, 22). Thus, MDSCs play an important role in determining the therapeutic effect of immunotherapy. In this review, we address the regulatory mechanisms of MDSCs as well as their functional changes in the TME, and summarize combined therapeutic effect of targeting tumor MDSCs and ICB.
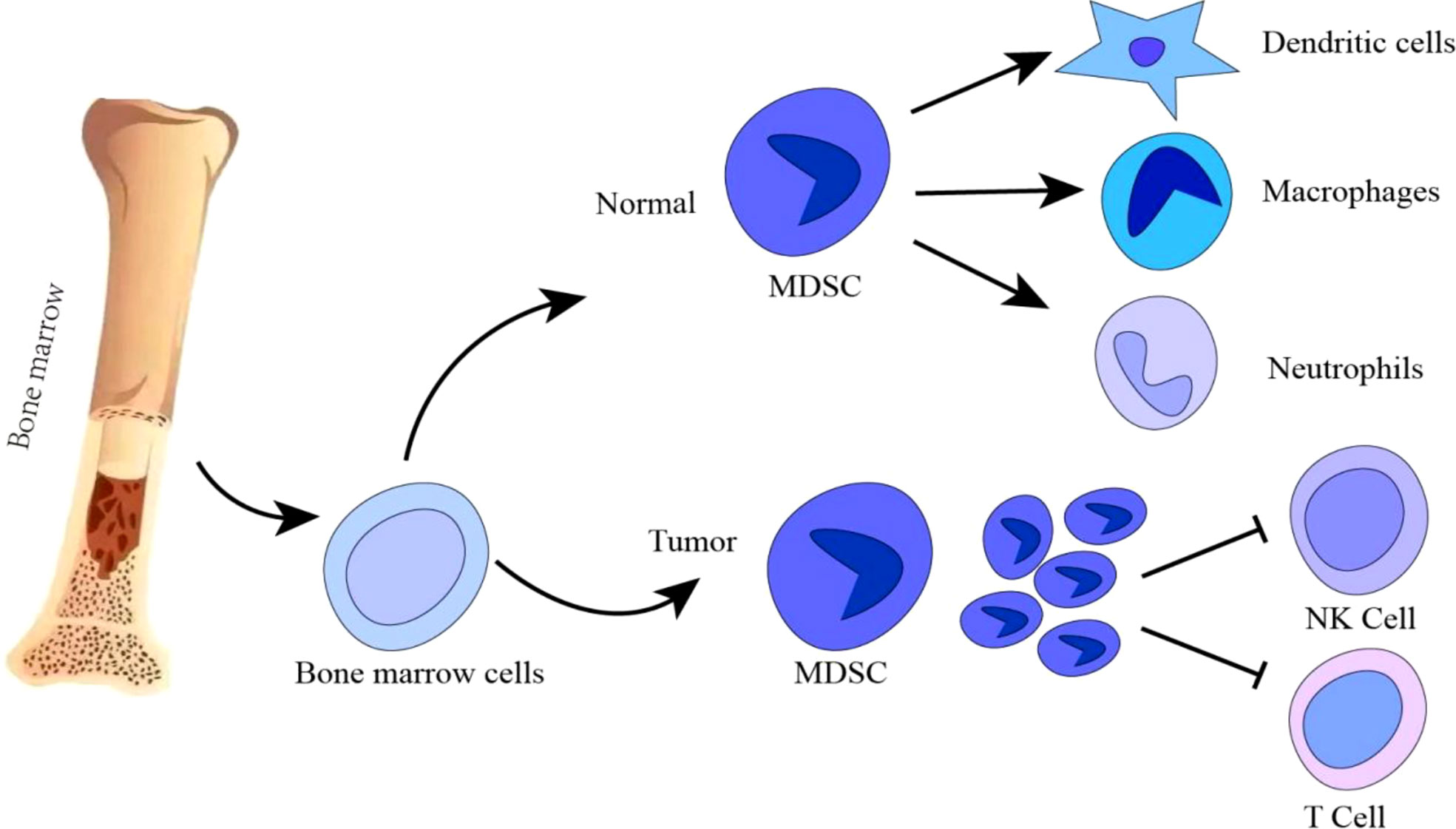
Figure 1 During normal myelogenesis, bone marrow hematopoietic stem cells differentiate into immature cells, which migrate to corresponding peripheral organs and further differentiate into macrophages, dendritic cells or neutrophils. However, in a chronic inflammatory or tumor microenvironment, the differentiation of immature bone marrow cells is blocked, and abnormal accumulation of immature bone marrow cells is induced. Tumor MDSCs are increased to inhibit T/NK cells anti-tumor immune response. Promotion: →; Inhibition: ┤.
During normal myelopoiesis, the major myeloid populations include granulocytes (with the most representative being neutrophils), monocytes, terminally differentiated macrophages (MΦ), and DCs. Those mature myeloid cells are one of the main protective mechanisms against pathogens (23–25) (Figure 1). For instance, classical activation of myeloid cells occurs in response to antipathogenic signals, mainly in the form of toll-like receptor (TLR) ligands, various damage-associated molecular pattern (DAMP) molecules, and pathogen-associated molecular pattern (PAMP) molecules (26). The activation leads to the production of monocytes and neutrophils in the bone marrow, markedly increase phagocytosis, a respiratory burst, the production of pro-inflammatory cytokines and the upregulation of both the major histocompatibility complex (MHC) class II and co-stimulatory molecules. This process usually lasts for only a short time and the reaction disappears with the threat (i.e., pathogen) lost. MDSC formation is different from normal myelogenesis, which is a pathological activation state resulting from continuous stimulation of tumor or chronic inflammation on bone marrow compartment, leading to high levels of ROS, MPO, NO, and most anti-inflammatory cytokines, which prevent the generation of mature bone marrow cells (27, 28). C/EBPα and C/EBPβ play the important role in the formation of bone marrow and affect the formation and development of MDSC. C/EBPα makes the granulocyte lineage specific transition from CMP to granulocyte-monocyte precursors (GMPs), GMP. After the GMP phase, C/EBPα is dispensable, while C/EBPβ plays a major role. C/EBP family members have been found to control the expansion and functional properties of MDSC, and may regulate MDSC differentiation into neutrophils and macrophage. Under pathological conditions, C/EBPα may be down-regulated in MDSCs, but C/EBPβ, C/EBP-δ may be strongly up-regulated (29). MDSC-like cells are continuously activated through signal transduction and activator of transcription 3 (STAT3) or NF-κB pathway (20).However, in some chronic infections and cancers, the signals that activate myeloid cells are quite different (24, 30). These signals are relatively weak and long-lasting in the present of growth factors and inflammatory mediators. Both neutrophils and monocytes under these conditions exhibit immature phenotype and morphology, relatively weak phagocytic activity, increased levels of reactive oxygen species (ROS), nitric oxide (NO) production, Arginase and Prostaglandin E2 (PGE2) (23, 26, 30, 31). Those myeloid cells in the pathological state suppress T cell anti-tumor immune response, are now referred to as MDSCs (32, 33) (Figure 2).
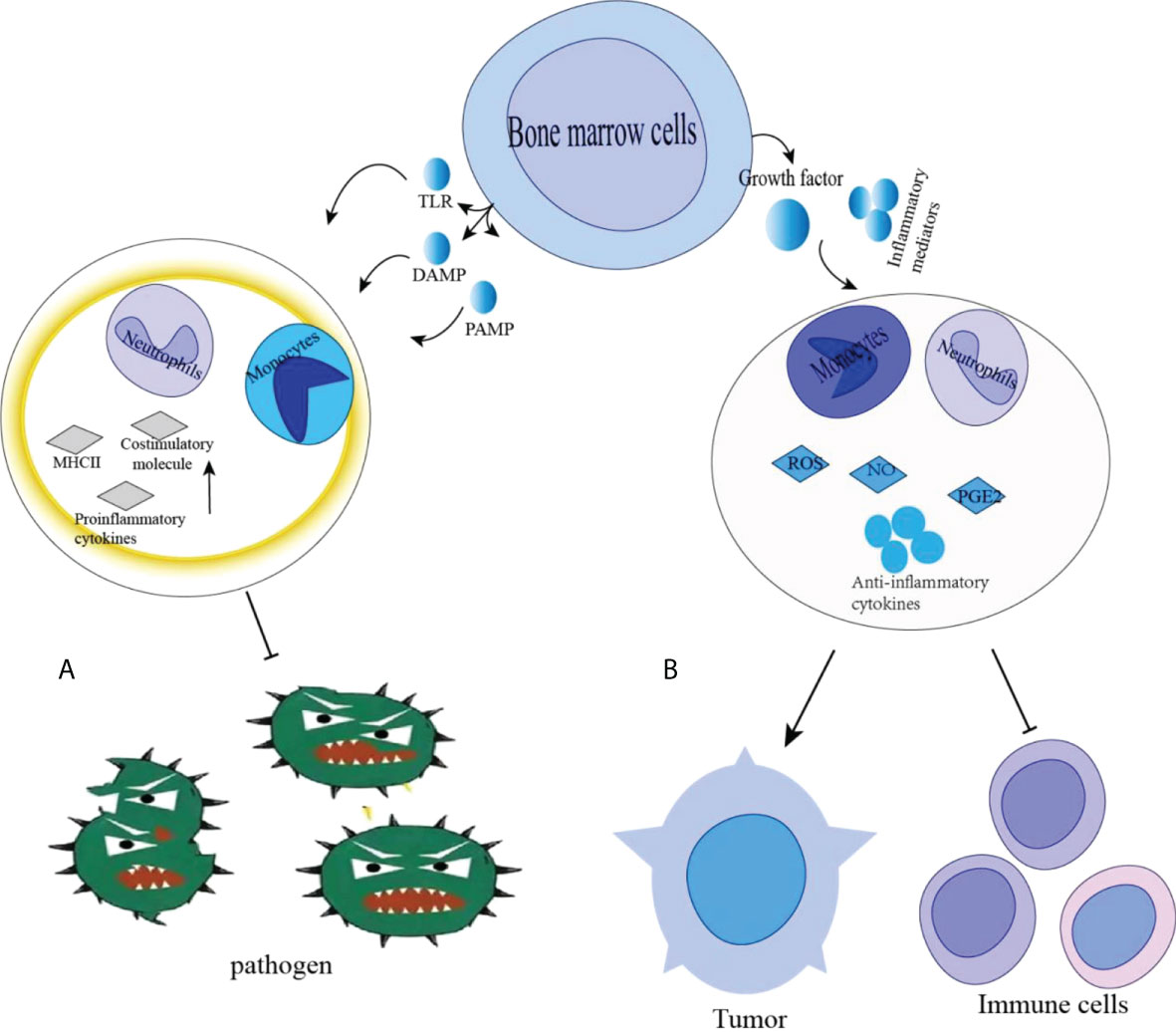
Figure 2 (A) Classical activation of bone marrow cells occurs in response to pathogen signals, mainly in the form of Toll-like receptor (TLR) ligands, various damage-related molecular pattern (DAMP) and pathogen-associated molecular pattern (PAMP) molecules. This results in rapid activation of monocytes and neutrophils in the bone marrow, a significant increase in phagocytosis, respiratory bursts, production of pro-inflammatory cytokines and upregulation of major histocompatibility complex (MHC) class II and costimulatory molecules. (B) In chronic infections and cancer, Immature cell differentiation is blocked. They exhibit relatively weak phagocytic activity, increased levels of reactive oxygen species (ROS) and nitric oxide (NO) production, arginase and PGE2, promoting tumor growth. Promotion: →, Inhibition: ┤.
MDSCs are the main suppressive immune cells with the ability to suppress the adaptive and innate immune responses (34–36). However, the functional mechanisms of MDSCs in immune suppression have been completely uncovered. They can be further divided into two subtypes: PMN-MDSCs and M-MDSCs) (37). PMN-MDSCs are phenotypically and morphologically similar to neutrophils, whereas M-MDSCs are similar to monocytes (38). MDSCs in mice have two distinct subtypes due to different markers on them in mice and humans; In mice, MDSCs are characterized by co-expression of CD11b of α-M integrin, which is considered to be a marker of ubiquitination, and bone marrow differentiation antigen Gr-1 which is a glycosylphosphatidylinositol junction protein, is composed of Ly6C and Ly6G subunits, resulting in two subtypes: CD11b+Ly-6G+Ly-6ChighandCD11b+Ly-6G+Ly-6Clow (39). Since the CD11b+ Ly-6G+ Ly-6 Chigh subtype is morphologically similar to monocytes, it is termed M-MDSCs, and the CD11b+Ly-6G+Ly-6Clow subtype displays a granulocyte-like morphology and is termed granulocytic MDSCs (PMN-MDSCs or G-MDSCs) (34). In human, M-MDSC expression is characterized by CD11b, CD14, HLA-DR and CD15 (40). M-MDSCs can be defined as CD11b+CD15-CD14+HLA-DR-/low MDSCs, while PMN-MDSCs are usually defined as CD11b+CD14-CD15+ (or CD66b+) MDSCs (20, 33). Recently, third subgroup of MDSCs in humans are found to be called early MDSCs, which lacks expression of mature blood cell markers (including CD3, CD14, CD15, CD19 and CD56), thus it is called Lin-HLA-DR-CD33+ (41). consisting mainly of cells with colony-forming potential and other myeloid precursor cells (33, 42).
In the TME, MDSCs are accumulated to suppress immune function and promote tumor growth through inducing some tumor-derived factors, cytokines and/or chemokines such as interleukin (IL)-6, interferon (IFN)-γ, IL-1β, granulocyte-macrophage colony-stimulating factor (GM-CSF), tumor necrosis factor (TNF)-α and vascular endothelial growth factor (43, 44). In Esophageal Squamous Cell Carcinoma (ESCC), the increase of MDSCs is upregulated through IL-6 or other signaling pathways mediated by aldehyde dehydrogenase (6, 45). The continuous recruitment of MDSCs to tumors is also mediated by interactions between chemokines and chemokine receptors, particularly the interaction between the CC chemokine receptor 5 (CCR5) and its ligand to exaggerate ESCC progression. In melanoma, tumor-infiltrating CCR5+ MDSCs was found to show elevated expression levels of immunosuppressive markers such as PD-L1, Arg1, ROS and NO, exerting stronger immunosuppressive activity compared with its CCR5- counterparts (12). In breast cancer, BCC-Ex induced the increase of myeloid cells by activating the STAT3 signaling pathway, promoting the expansion of MDSCs (27, 46). In the TME, the immunosuppressive function of MDSCs is also mediated through endoplasmic reticulum stress and inhibitor-related enzymes, which include cyclooxygenase 2(COX2), NADPH oxidase 2 (NOX2), Indoleamine 2, 3-dioxygenase (IDO) and arginase 1 (ARG-1) which induces nitrogen Nitric oxide synthase (iNOS or NOS2) (47). MDSCs also regulate the functional activity of other immune cells, such as macrophages, NK cells, Treg, and B cells (48, 49). In a mouse non-T-cell inflammatory oral cancer model (MOC2), peripheral CXCR2+ PMN-MDSCs are pathologically accumulated to enter tumors, inhibiting the functions of NK cells (50, 51). MDSCs also interact with other IMCs such as TAMs to promote their immunosuppressive activities (37, 52) Tables 1, 2.

Table 1 Roles of cytokine and chemokines on tumor MDSCs.

Table 2 Function of target protein on tumor MDSCs.
MDSCs exert its immunosuppressive function through the induction of different cytokines or chemokines. Porta et al. found that inhibition of PGE2/P50/NO axis could prevent the inhibitory function of MDSC and restore the functional effect of anticancer immunotherapy. IFNγ treatment block M-MDSC to produce the tumor-promoting molecule nitric oxide (NO) and PGE2, which promote nuclear accumulation of P50 NF-κB in M-MDSCs, indicating that IFNγ treatment may reverse NO-mediated immunosuppression of MDSCs through PGE2/NO/P50 pathways (53). It has been found in prostate cancer studies that IL-23 produced by MDSCs can promote the development of prostate cancer through activating androgen receptor pathways in castration-resistant prostate cancer (CRPC), promoting cell survival and proliferation in androgen deficiency. Therefore, IL-23 blockage can resist MDSC-mediated castration resistance and produce synergistic effects (54). The expansion of MDSCs is induced in melanoma through NLRP3/IL-1 signaling, suggesting that NLRP3 inhibitors may restore T cell function through reducing the numbers of tumor MDSCs, and the combined treatment with NLRP3 inhibitor and anti-PD-1 is more effective (55). Therefore, Immunotherapy targeting related cytokines or chemokines reverses the suppressive roles of MDSCs on T cells.
In tumor treatment, the durable effects of surgery, radiotherapy, and chemotherapy are relatively low, and immunotherapy has a relatively higher and long-lasting treatment outcome. However, Our data have demonstrated that drug resistance is common in most current immunotherapies due to the immunosuppressive cells in the TME, where MDSC-mediated immunosuppression represents a potential mechanism of resistance to immunotherapy (36). Targeting MDSCs may be an important strategy to overcome immunotherapeutic resistance. In renal cell carcinoma (RCCs), it was found that the recruitment of MDSCs into the tumors induced by a tumor-promoting factor, IL-1β led to immune-suppression on cancer cells. After combined treatment with both anti-IL-1β and anti-PD-L1 antibodies, the number of PD-L1+PMN-MDSCs was reduced in the tumors, blocking tumor progression (56). In an LKB1-deficient NSCLC mouse model, ELR+CXC chemokine promoted NSCLC development and increased levels of ELR+CXC chemokines were positively correlated with the abundance of G-MDSCs in the TME. The depletion of G-MDSCs by anti-Gr-1 antibodies or functional blockade of G-MDSCs by ATRA reverses immunosuppression and made LKB1-deficient tumors sensitive to anti-PD-1 treatment (27, 57).
In head and neck squamous cell carcinoma (HNSCC), the infiltration numbers of CXCR1/2+CD15+ PMN-MDSCs and CD14+ M-MDSCs with immunosuppressive function are significantly increased. Researchers inhibit the infiltration of CXCR2+ PMN-MDSCs into MOC2 tumors through dual inhibitors of CXCR1/2 and SX-682 to enhance the CTL infiltration and increase the therapeutic effects of adoptive transfer of NK cells (12). In neuroblastoma patients, researchers have developed genetically modified NK cells with chimeric receptors NKG2D, which is a cytotoxic receptor activated by non-classical MHC molecules expressed during stressful events such as DNA damage, hypoxia or viral infection. NKG2D is fused to the ζ chain of the cytotoxic T cell receptor (NKG2D.ζ) to become NKG2D.ζ-NK cells (58). Targeting MDSCs with NKG2D.ζ-NK cells inhibits the function of MDSCs and improves the anti-tumor role of tumor-directed chimeric antigen receptor (CAR)-modified T cells (CAR-T) in a xenograft tumor model (59). The liver X nuclear receptor (LXR)/apolipoprotein E (ApoE) axis has been implicated in enhancing anti-tumor activity. LXRβ and LXRα are two members of the nuclear hormone receptor transcription factor family that drive the transcriptional activation of ApoE and other genes involved in cholesterol, fatty acid, and glucose metabolism. Studies have confirmed that LXR and its transcriptional target ApoE can reduce the abundance of MDSCs to inhibit melanoma development (60). Therapeutic LXR agonists reduced the abundance of MDSCs in a mouse tumor model (61). Therefore, Targeting MDSCs treatment may improve the immunotherapy results on the TME.
In the TME, some immunoglobulins are co-expressed in immunosuppressive cells, influencing their immunosuppressive roles (62). The tandem action of CD39 and CD73 ectonucleotidases expressed on MDSCs can convert ATP to adenosine on Tregs which is thought to be important mediators of immunosuppression in the TME (63, 64). In NSCLC and melanoma, it was found that high expression levels of CD39 and CD73 on tumor MDSCs are positively correlated with tumor progression (65, 66). TGF-β was found to trigger the phosphorylation of mammalian targets of rapamycin, which subsequently activated hypoxia-inducible factor-1α (HIF-1α) that induced CD39/CD73 expression on MDSC to inhibit T cell function. Therefore, CD39/CD73 on MDSCs may be the novel therapeutic target for tumor treatment (67, 68). Their expression levels on MDSCs are reduced to slow down the tumor progression (69). In the TME, the myeloid cell receptor tyrosine kinases (RTKs) (i.e., TYRO3, AXL, and MERTK) and their ligands (i.e., Gas 6 and protein S) suppress immune responses. In tumor-bearing mice, the expressions of TYRO3, AXL, and MERTK, and their ligands increased >20 folds in M-MDSCs and >15 folds in PMN-MDSCs. Mertk-/-, Axl-/- and Tyro3-/- tumor models were revealed to reduce RTK enzymatic activity, exhibit defective MDSC roles, and display poor tumor migration capacity to tumor-draining lymph nodes (TDLN) (70). The inhibition of TYRO3, AXL, and MERTK reduced the immune suppression function of MDSCs in a STAT3-dependent manner, increased CD8+ T cell infiltration and enhanced treatment efficacy of anti-PD-1 treatment on melanoma (71, 72). These findings suggest that TYRO3, AXL and MERTK, are immunosuppressive and innate immune checkpoint protein, whose inhibitors may improve the TME through downregulating the roles of MDSCs.
CD33 is composed of type 1 membrane proteins and is a transmembrane sialic acid-binding immunoglobulin-like lectin (SIGLEC) possessing two immunoglobin domains that bind to sialic acid and intracellular immunoreceptor tyrosine inhibitory motif (ITIM) (73). CD33 is pathologically overexpressed on MDSCs in blood and tumor tissues from cancer patients to promote tumor growth. Therefore, targeting CD33+ MDSCs can effectively reduce the immunosuppression of MDSCs, slowing down tumor growth (74).
ICB and CAR-T therapies have provided further advantages for cancer immunotherapy (75). Immune checkpoint inhibitors target immune checkpoint molecules, primarily PD-1, PD-L1, and CTLA-4 to restore anti-tumor immune function. Even though some of checkpoint proteins (such as PD-L1) are expressed on the surfaces of both tumor and MDSCs and a few cancer patients show good long-lasting clinical effects after ICB treatment, immunotherapeutic resistance develops in the late stage of most solid tumors.
MDSCs in the TME induced by chemokines or cytokines become major obstacle to compromise the effect of ICB (76, 77). Targeting MDSCs may be a potential breakthrough for ICB (78). It is reported that single immunotherapy treatment on the malignant cholangiocarcinoma (CCA) is not effective due to complex TME. In a mouse model of CAA, PD-L1 is found to be mainly derived from TAMs and MDSCs, promoting tumor progression (79). The response rate for anti-PD-1 treatment is less than 10%. One of the reasons is that the compensatory G-MDSCs mediate immune evasion by impairing the T cell response, and the blockage of TAM alone fails to slow down tumor progression (80). Indeed, the survival time in mice with CCA was longer after combined treatment of G-MDSCs-specific antibodies (i.e., anti-Ly6G antibody, anti-PD-1 antibody and anti-CSF1R antibody), compared with single-antibody treatment (81). In an advanced gastric cancer mouse model, it was found that anti-PD-1 treatment alone was ineffective, since strong infiltration of PMN-MDSCs into tissues inhibited the immune function of CD8+ T cells by increasing the expression of some chemokines or cytokines (i.e., ROS, NO, arginase-1, PGE-2) and interacting with PD-L1/PD-1. Therefore, combination therapy of both blocking PD-1 and targeting MDSCs may overcome the drug resistance in the single immune checkpoint inhibitor treatment (82). In the TME, hypoxia attracts immunosuppressive cells such as MDSCs and TAMs, which are important components of TME, upregulated the expressions levels of both immune checkpoint receptors (such as PD-1 and CTLA-4) and their ligands (such as PD-L1, PD-L2, CD80, and CD86), and induced rapid and selective upregulation of PD-L1 on MDSCs, which is mainly dependent on hypoxia-inducible factor 1α (HIF-1α) rather than HIF-2α. The blockage of PD-L1 under hypoxia reverses the immunosuppressive roles of MDSCs on T cell activation, accompanied by down-regulation of IL-6 and IL-10 in MDSCs. Thus, co-blocking of PD-L1 and HIF-1α reduces the immunosuppressive activity of both MDSCs and TAMs to inhibit tumor development (83). In HNSCC, it was found that the efficacy of anti-CTLA-4 treatment was not significant, mainly due to the recruitment and accumulation of MDSCs. The combined treatment of both G-MDSCs depletion and CTLA-4 inhibitor can achieve an excellent anti-tumor treatment (50). In triple-negative breast cancer (TNBC), aberrant SMAD3 activation promotes metastasis of TNBC through the recruitment of MDSCs. SMAD3 is identified as a non-histone substrate of lysine acetyltransferase 6A (KAT6A). Targeting KAT6A in combination with anti-PD-L1 therapy in TNBC-bearing xenografts models reduced MDSC recruitment, significantly alleviated metastasis potential and increased overall survival (84). For advanced prostate cancer (PCa), the majority of patients are resistant to ICB, due to the accumulation of MDSCs. ICB synergizes with targeting MDSCs therapy with multikinase inhibitors (such as cabozantinib and BEZ235) exhibited stronger anti-tumor activity (85). The inhibition of CXCR4 can promote T-cell infiltration through diminishing the immunosuppressive roles of MDSCs in pancreatic ductal adenocarcinoma (PDAC) (86, 87). Indeed, a clinical study using the combined treatment, including CXCR4 antagonist BL-8040 (motixafortide), Pembrolizumab and chemotherapy has been displayed to increase CD8+ effector T cell infiltration and decrease MDSC infiltration, leading to tumor suppression (88). Therefore, the combination immunotherapy of ICB and targeting MDSCs may provide some evidence for their clinical efficacy Table 3.
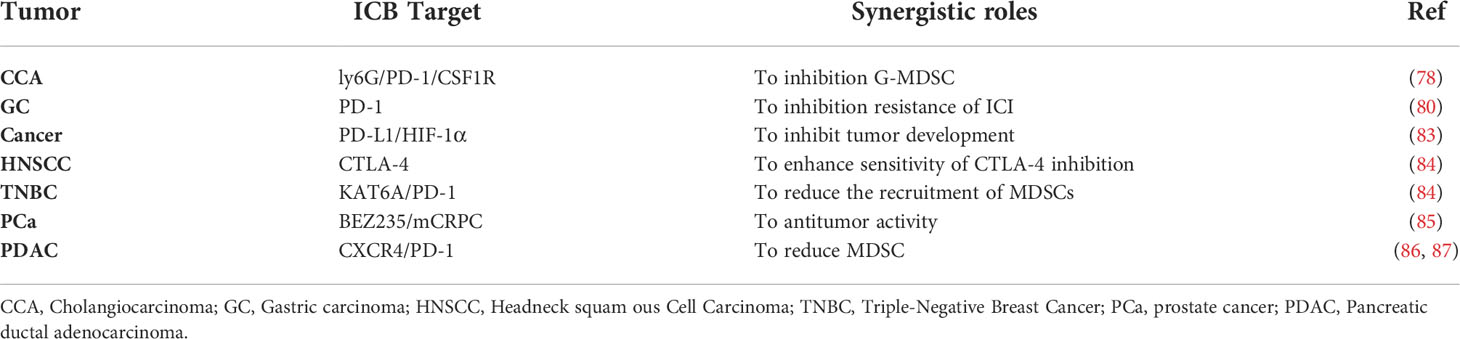
Table 3 Combination immunotherapy of targeting MDSCs and ICB .
Recent studies found that serine/threonine kinase PIM1 was upregulated in MDSCs in melanoma, which was closely associated with increased FAO and PPARγ-driven lipid metabolism in MDSCs. the increased expression of PIM is negatively related to the therapeutic effect of ICB (89, 90). The inhibition of PIM1 on MDSCs led to the reduced number of MDSCs in the TME, restoring the anti-tumor function of cytotoxic T cells, improving the efficacy of PD-L1 blockade, and overcoming the resistance in ICB-resistant patients (91). In PDAC, CD200 (OX-2; OX-90), a regulator of myeloid cell activity, whose expression is upregulated (92). In preclinical studies, the expression levels of CD200 were elevated in MDSCs in PDAC. The blockade of anti-CD200 antibody reduced the number of intra-tumoral MDSCs, restricting PDAC tumor growth and significantly enhancing the anti-tumor efficacy of ICB and anti-PD-1 antibody (92, 93). Therefore, in the TME, targeting both MDSCs and their surface proteins may reverse the immunotherapy resistance to tumor, providing the theoretical basis and potential breakthrough for clinical treatment of cancer using immunotherapies.
During tumor development, competent metabolic programs promote the proliferation and migration of tumor cells and enhance the immunosuppressive tumor microenvironment (TME) (94, 95). In addition, the exaggerated metabolic activity allows cancer cells to hijack essential nutrients and outcompete neighboring invasive immune cells, thereby weakening anti-tumor immunity (96). As the main heterogeneous population of immunosuppressive cells, MDSCs can be regulated by various mechanisms to affect tumor development, and can also change the metabolic environment of its surrounding environment to exert immunosuppressive function (97, 98). It has been found that the combination of bone marrow (BM) precursors with GM-CSF and IL-6 in vitro has been found to activate L-arginine metabolic enzymes responsible for the immunosuppressive potential of MDSC. The inhibition of L-arginine metabolism enzymes in MSC-1 cells, one cell line derived from primary MDSC, was found to reduce AMPK activity in MSC-1 cells. Subsequently, the inhibition of AMPK activity by specific inhibitor compound C (COMP-C) resulted in the inhibition of L-arginine metabolic enzyme activity to eliminate the immunosuppressive activity of MDSCs (99, 100). The glycolytic metabolite phosphoenolpyruvate (PEP), an important antioxidant, can prevent excessive ROS production, thus reducing the accumulation of MDSCs and inhibiting their immunosuppressive roles. These findings suggest that glycolytic metabolites play an important role in regulating MDSC and may be a potential target (101). In addition, fatty acid metabolism is also proved to be an important part of the development and functional role of MDSC (100, 101). Polyunsaturated fatty acids (PUFA) promote the accumulation of MDSC and enhances the immunosuppressive function of MDSC on T cells through activating the JAK/STAT3 pathway. JAK inhibitor JSI-124 almost completely prevented the effect of PUFA on MDSCs, indicating that fatty acid metabolism may play an important role in the function of MDSC (102, 103). Similar increases in fatty acid uptake and FAo-related enzyme expression are also observed in MDSCs from blood and tumors in Lewis lung cancer (LLC) and McA-38 colorectal adenocarcinoma mouse models. It was also found that MDSCs promoted the increased fatty acid uptake and activated fatty acid oxidation (FAO) (104). The inhibition of FAO alone delays tumor growth and plays an anti-tumor role. FAO inhibition combined with low-dose chemotherapy can completely inhibit the immunosuppressive effect of MDSC, providing better treatment for anti-tumor therapy (105). Therefore, immune-metabolic therapy is now emerging as a major breakthrough direction in the study of MDSCs, which will provide a new strategy for anti-tumor therapy.
The review focuses on immunotherapies against MDSCs and their therapeutic efficacy. In the TME, MDSCs have become the main obstacle to immunotherapy, since they lead to immunotherapeutic resistance. The accumulation of MDSCs can be reduced after targeting chemokines or inflammatory factors, improving the TME and suppressing tumor growth. Moreover, combination treatment with both ICB and targeting MDSCs can effectively inhibit tumor development and progression. These data suggest the effectiveness of targeting MDSCs in TME. Many scientists have conducted in-depth explorations of the immunoglobulins expressed on MDSCs to reveal the regulatory mechanism of these immunoglobulins on the function of MDSCs. However, the functional mechanisms of many immunoglobulins expressed on MDSCs need to be investigated further. In conclusion, immunotherapies targeting MDSCs have significantly increased efficacy and can suppress tumor activity, showing a strong potential to be a new therapeutic strategy for the immunotherapeutic treatment of cancer.
HS, SD, and XL wrote, reviewed, and revised manuscript, figures and tables. XX and LW prepared the figures and tables. MY, ZG, and YL reviewed and revised the manuscript. HL, SP, CJ, DJ, and GJ revised manuscript, figures and tables. All authors contributed to the article and approved the submitted version.
This work was supported by Shandong Medical and Health Science and Technology Development Project (No. 202001010899), Doctor startup fund of Shandong First Medical University (No. 001003053) and Shandong Province graduate education quality curriculum construction project (SDYKC21148, SDYKC18095).
We thank Mr. Steven Qu from MIT who helped to revise and polish the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends–an update. Cancer Epidemiol Biomarkers Prev (2016) 25(1):16–27. doi: 10.1158/1055-9965.EPI-15-0578
2. Zaimy MA, Saffarzadeh N, Mohammadi A, Pourghadamyari H, Izadi P, Sarli A, et al. New methods in the diagnosis of cancer and gene therapy of cancer based on nanoparticles. Cancer Gene Ther (2017) 24(6):233–43. doi: 10.1038/cgt.2017.16
4. Pan D, Hu AY, Antonia SJ, Li C-Y. A gene mutation signature predicting immunotherapy benefits in patients with nsclc. J Thorac Oncol (2021) 16(3):419–27. doi: 10.1016/j.jtho.2020.11.021
5. Anichini A, Perotti VE, Sgambelluri F, Mortarini R. Immune escape mechanisms in non small cell lung cancer. Cancers (Basel) (2020) 12(12):3605. doi: 10.3390/cancers12123605
6. Baba Y, Nomoto D, Okadome K, Ishimoto T, Iwatsuki M, Miyamoto Y, et al. Tumor immune microenvironment and immune checkpoint inhibitors in esophageal squamous cell carcinoma. Cancer Sci (2020) 111(9):3132–41. doi: 10.1111/cas.14541
7. Theivanthiran B, Evans KS, DeVito NC, Plebanek M, Sturdivant M, Wachsmuth LP, et al. A tumor-intrinsic pd-L1/Nlrp3 inflammasome signaling pathway drives resistance to anti-Pd-1 immunotherapy. J Clin Invest (2020) 130(5):2570–86. doi: 10.1172/JCI133055
8. Bruni D, Angell HK, Galon J. The immune contexture and immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer (2020) 20(11):662–80. doi: 10.1038/s41568-020-0285-7
9. Muenst S, Läubli H, Soysal SD, Zippelius A, Tzankov A, Hoeller S. The immune system and cancer evasion strategies: Therapeutic concepts. J Intern Med (2016) 279(6):541–62. doi: 10.1111/joim.12470
10. Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells (2020) 9(3):561. doi: 10.3390/cells9030561
11. Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, et al. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci (2015) 106(2):134–42. doi: 10.1111/cas.12584
12. Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of mdsc trafficking with sx-682, a Cxcr1/2 inhibitor, enhances nk-cell immunotherapy in head and neck cancer models. Clin Cancer Res (2020) 26(6):1420–31. doi: 10.1158/1078-0432.CCR-19-2625
13. Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P, et al. Aging and cancer: The role of macrophages and neutrophils. Ageing Res Rev (2017) 36:105–16. doi: 10.1016/j.arr.2017.03.008
14. Kim J, Bae J-S. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflammation (2016) 2016:6058147. doi: 10.1155/2016/6058147
15. Marzagalli M, Ebelt ND, Manuel ER. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin Cancer Biol (2019) 59:236–50. doi: 10.1016/j.semcancer.2019.08.002
16. Osipov A, Saung MT, Zheng L, Murphy AG. Small molecule immunomodulation: The tumor microenvironment and overcoming immune escape. J Immunother Cancer (2019) 7(1):224. doi: 10.1186/s40425-019-0667-0
17. Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol (2020) 17(1):1–12. doi: 10.1038/s41423-019-0306-1
18. Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and pmn-mdsc: Their biological role and interaction with stromal cells. Semin Immunol (2018) 35:19–28. doi: 10.1016/j.smim.2017.12.004
19. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21(8):485–98. doi: 10.1038/s41577-020-00490-y
20. Hegde S, Leader AM, Merad M. Mdsc: Markers, development, states, and unaddressed complexity. Immunity (2021) 54(5):875–84. doi: 10.1016/j.immuni.2021.04.004
21. Li MO, Wolf N, Raulet DH, Akkari L, Pittet MJ, Rodriguez PC, et al. Innate immune cells in the tumor microenvironment. Cancer Cell (2021) 39(6):725–9. doi: 10.1016/j.ccell.2021.05.016
22. Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA, et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci Immunol (2020) 5(44):eaay6017. doi: 10.1126/sciimmunol.aay6017
23. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science (2010) 327(5966):656–61. doi: 10.1126/science.1178331
24. Veglia F, Gabrilovich DI. Dendritic cells in cancer: The role revisited. Curr Opin Immunol (2017) 45:43–51. doi: 10.1016/j.coi.2017.01.002
25. Strauss L, Guarneri V, Gennari A, Sica A. Implications of metabolism-driven myeloid dysfunctions in cancer therapy. Cell Mol Immunol (2021) 18(4):829–41. doi: 10.1038/s41423-020-00556-w
26. Kruger P, Saffarzadeh M, Weber ANR, Rieber N, Radsak M, von Bernuth H, et al. Neutrophils: Between host defence, immune modulation, and tissue injury. PloS Pathog (2015) 11(3):e1004651. doi: 10.1371/journal.ppat.1004651
27. Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol (2015) 98(6):913–22. doi: 10.1189/jlb.4RI0515-204R
28. Ostrand-Rosenberg S, Beury DW, Parker KH, Horn LA. Survival of the fittest: How myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol Immunother (2020) 69(2):215–21. doi: 10.1007/s00262-019-02388-8
29. Wang W, Xia X, Mao L, Wang S. The Ccaat/Enhancer-binding protein family: Its roles in mdsc expansion and function. Front Immunol (2019) 10:1804. doi: 10.3389/fimmu.2019.01804
30. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis (2009) 30(7):1073–81. doi: 10.1093/carcin/bgp127
31. Gomez S, Tabernacki T, Kobyra J, Roberts P, Chiappinelli KB. Combining epigenetic and immune therapy to overcome cancer resistance. Semin Cancer Biol (2020) 65:99–113. doi: 10.1016/j.semcancer.2019.12.019
32. Umansky V, Blattner C, Gebhardt C, Utikal J. The role of myeloid-derived suppressor cells (Mdsc) in cancer progression. Vaccines (Basel) (2016) 4(4):36. doi: 10.3390/vaccines4040036
33. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19(2):108–19. doi: 10.1038/s41590-017-0022-x
34. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
35. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res (2017) 5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297
36. Gao X, Sui H, Zhao S, Gao X, Su Y, Qu P. Immunotherapy targeting myeloid-derived suppressor cells (Mdscs) in tumor microenvironment. Front Immunol (2020) 11:585214. doi: 10.3389/fimmu.2020.585214
37. Yang Z, Guo J, Weng L, Tang W, Jin S, Ma W. Myeloid-derived suppressor cells-new and exciting players in lung cancer. J Hematol Oncol (2020) 13(1):10. doi: 10.1186/s13045-020-0843-1
38. Zhang Md J, Zhang Md L, Yang Md Y, Liu Md Q, Ma Md H, Huang Md A, et al. Polymorphonuclear-mdscs facilitate tumor regrowth after radiation by suppressing Cd8 T cells. Int J Radiat Oncol Biol Phys (2021) 109(5):1533–46. doi: 10.1016/j.ijrobp.2020.11.038
39. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111(8):4233–44. doi: 10.1182/blood-2007-07-099226
40. Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
41. Condamine T, Dominguez GA, Youn J-I, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized ldl receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol (2016) 1(2):aaf8943. doi: 10.1126/sciimmunol.aaf8943
42. Okła K, Czerwonka A, Wawruszak A, Bobiński M, Bilska M, Tarkowski R, et al. Clinical relevance and immunosuppressive pattern of circulating and infiltrating subsets of myeloid-derived suppressor cells (Mdscs) in epithelial ovarian cancer. Front Immunol (2019) 10:691. doi: 10.3389/fimmu.2019.00691
43. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (Mdscs) during tumour progression. Br J Cancer (2019) 120(1):16–25. doi: 10.1038/s41416-018-0333-1
44. Weber R, Riester Z, Hüser L, Sticht C, Siebenmorgen A, Groth C, et al. Il-6 regulates Ccr5 expression and immunosuppressive capacity of mdsc in murine melanoma. J Immunother Cancer (2020) 8(2):e000949. doi: 10.1136/jitc-2020-000949
45. Tcyganov EN, Hanabuchi S, Hashimoto A, Campbell D, Kar G, Slidel TW, et al. Distinct mechanisms govern populations of myeloid-derived suppressor cells in chronic viral infection and cancer. J Clin Invest (2021) 131(16):e145971. doi: 10.1172/JCI145971
46. Liu Q-W, Chen Y, Li J-Y, Xiao L, Zhang W-J, Zhao J-L, et al. Bone marrow cells are differentiated into mdscs by bcc-ex through down-regulating the expression of Cxcr4 and activating Stat3 signalling pathway. J Cell Mol Med (2021) 25(12):5497–510. doi: 10.1111/jcmm.16559
47. Zhang Y, Wang X, Zhang R, Wang X, Fu H, Yang W. Mdscs interactions with other immune cells and their role in maternal-fetal tolerance. Int Rev Immunol (2021) 15:1–18. doi: 10.1080/08830185.2021.1938566
48. Shu C-C, Pan S-W, Feng J-Y, Wang J-Y, Chan Y-J, Yu C-J, et al. The clinical significance of programmed death-1, regulatory T cells and myeloid derived suppressor cells in patients with nontuberculous mycobacteria-lung disease. J Clin Med (2019) 8(5):736. doi: 10.3390/jcm8050736
49. Wang Y, Schafer CC, Hough KP, Tousif S, Duncan SR, Kearney JF, et al. Myeloid-derived suppressor cells impair b cell responses in lung cancer through il-7 and Stat5. J Immunol (2018) 201(1):278–95. doi: 10.4049/jimmunol.1701069
50. Clavijo PE, Moore EC, Chen J, Davis RJ, Friedman J, Kim Y, et al. Resistance to ctla-4 checkpoint inhibition reversed through selective elimination of granulocytic myeloid cells. Oncotarget (2017) 8(34):55804–20. doi: 10.18632/oncotarget.18437
51. Moore E, Clavijo PE, Davis R, Cash H, Van Waes C, Kim Y, et al. Established T cell-inflamed tumors rejected after adaptive resistance was reversed by combination sting activation and pd-1 pathway blockade. Cancer Immunol Res (2016) 4(12):1061–71. doi: 10.1158/2326-6066.CIR-16-0104
52. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (Mdsc), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol (2012) 22(4):275–81. doi: 10.1016/j.semcancer.2012.01.011
53. Porta C, Consonni FM, Morlacchi S, Sangaletti S, Bleve A, Totaro MG, et al. Tumor-derived prostaglandin E2 promotes P50 nf-κb-Dependent differentiation of monocytic mdscs. Cancer Res (2020) 80(13):2874–88. doi: 10.1158/0008-5472.CAN-19-2843
54. Calcinotto A, Spataro C, Zagato E, Di Mitri D, Gil V, Crespo M, et al. Il-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature (2018) 559(7714):363–9. doi: 10.1038/s41586-018-0266-0
55. Tengesdal IW, Menon DR, Osborne DG, Neff CP, Powers NE, Gamboni F, et al. Targeting tumor-derived Nlrp3 reduces melanoma progression by limiting mdscs expansion. Proc Natl Acad Sci U.S.A. (2021) 118(10):e2000915118. doi: 10.1073/pnas.2000915118
56. Najjar YG, Rayman P, Jia X, Pavicic PG, Rini BI, Tannenbaum C, et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of Il1β, Il8, Cxcl5, and mip-1α. Clin Cancer Res (2017) 23(9):2346–55. doi: 10.1158/1078-0432.CCR-15-1823
57. Li R, Salehi-Rad R, Crosson W, Momcilovic M, Lim RJ, Ong SL, et al. Inhibition of granulocytic myeloid-derived suppressor cells overcomes resistance to immune checkpoint inhibition in Lkb1-deficient non-small cell lung cancer. Cancer Res (2021) 81(12):3295–308. doi: 10.1158/0008-5472.CAN-20-3564
58. González S, López-Soto A, Suarez-Alvarez B, López-Vázquez A, López-Larrea C. Nkg2d ligands: Key targets of the immune response. Trends Immunol (2008) 29(8):397–403. doi: 10.1016/j.it.2008.04.007
59. Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. Nk cells expressing a chimeric activating receptor eliminate mdscs and rescue impaired car-T cell activity against solid tumors. Cancer Immunol Res (2019) 7(3):363–75. doi: 10.1158/2326-6066.CIR-18-0572
60. Lin C-Y, Gustafsson J-Å. Targeting liver X receptors in cancer therapeutics. Nat Rev Cancer (2015) 15(4):216–24. doi: 10.1038/nrc3912
61. Pencheva N, Buss CG, Posada J, Merghoub T, Tavazoie SF. Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell (2014) 156(5):986–1001. doi: 10.1016/j.cell.2014.01.038
62. Bastid J, Regairaz A, Bonnefoy N, Déjou C, Giustiniani J, Laheurte C, et al. Inhibition of Cd39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res (2015) 3(3):254–65. doi: 10.1158/2326-6066.CIR-14-0018
63. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases Cd39 and Cd73: Novel checkpoint inhibitor targets. Immunol Rev (2017) 276(1):121–44. doi: 10.1111/imr.12528
64. Häusler SFM, Montalbán del Barrio I, Strohschein J, Chandran PA, Engel JB, Hönig A, et al. Ectonucleotidases Cd39 and Cd73 on ovca cells are potent adenosine-generating enzymes responsible for adenosine receptor 2a-dependent suppression of T cell function and nk cell cytotoxicity. Cancer Immunol Immunother (2011) 60(10):1405–18. doi: 10.1007/s00262-011-1040-4
65. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of Cd11b+Gr1+ cells. J Immunol (2011) 187(11):6120–9. doi: 10.4049/jimmunol.1101225
66. Umansky V, Shevchenko I, Bazhin AV, Utikal J. Extracellular adenosine metabolism in immune cells in melanoma. Cancer Immunol Immunother (2014) 63(10):1073–80. doi: 10.1007/s00262-014-1553-8
67. Zhang D, Wang Y, Shi Z, Liu J, Sun P, Hou X, et al. Metabolic reprogramming of cancer-associated fibroblasts by Idh3α downregulation. Cell Rep (2015) 10(8):1335–48. doi: 10.1016/j.celrep.2015.02.006
68. Rozen-Zvi B, Hayashida T, Hubchak SC, Hanna C, Platanias LC, Schnaper HW. Tgf-β/Smad3 activates mammalian target of rapamycin complex-1 to promote collagen production by increasing hif-1α expression. Am J Physiol Renal Physiol (2013) 305(4):F485–F94. doi: 10.1152/ajprenal.00215.2013
69. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. Cd39/Cd73 upregulation on myeloid-derived suppressor cells Via tgf-β-Mtor-Hif-1 signaling in patients with non-small cell lung cancer. Oncoimmunology (2017) 6(6):e1320011. doi: 10.1080/2162402X.2017.1320011
70. Graham DK, DeRyckere D, Davies KD, Earp HS. The Tam family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer (2014) 14(12):769–85. doi: 10.1038/nrc3847
71. Lemke G, Rothlin CV. Immunobiology of the Tam receptors. Nat Rev Immunol (2008) 8(5):327–36. doi: 10.1038/nri2303
72. Holtzhausen A, Harris W, Ubil E, Hunter DM, Zhao J, Zhang Y, et al. Tam Family receptor kinase inhibition reverses mdsc-mediated suppression and augments anti-Pd-1 therapy in melanoma. Cancer Immunol Res (2019) 7(10):1672–86. doi: 10.1158/2326-6066.CIR-19-0008
73. McMillan SJ, Crocker PR. Cd33-related sialic-Acid-Binding immunoglobulin-like lectins in health and disease. Carbohydr Res (2008) 343(12):2050–6. doi: 10.1016/j.carres.2008.01.009
74. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. Mdsc targeting with gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine (2019) 47:235–46. doi: 10.1016/j.ebiom.2019.08.025
75. Bejarano L, Jordāo MJC, Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Discovery (2021) 11(4):933–59. doi: 10.1158/2159-8290.CD-20-1808
76. Hack SP, Zhu AX, Wang Y. Augmenting anticancer immunity through combined targeting of angiogenic and pd-1/Pd-L1 pathways: Challenges and opportunities. Front Immunol (2020) 11:598877. doi: 10.3389/fimmu.2020.598877
77. Genova C, Dellepiane C, Carrega P, Sommariva S, Ferlazzo G, Pronzato P, et al. Therapeutic implications of tumor microenvironment in lung cancer: Focus on immune checkpoint blockade. Front Immunol (2021) 12:799455. doi: 10.3389/fimmu.2021.799455
78. Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and prevention of pancreatic cancer. Trends Cancer (2018) 4(6):418–28. doi: 10.1016/j.trecan.2018.04.001
79. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (Time) for effective therapy. Nat Med (2018) 24(5):541–50. doi: 10.1038/s41591-018-0014-x
80. Sharpe AH, Pauken KE. The diverse functions of the Pd1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
81. Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments pd-1 blockade in cholangiocarcinoma. J Clin Invest (2020) 130(10):5380–96. doi: 10.1172/JCI137110
82. Kim W, Chu TH, Nienhüser H, Jiang Z, Del Portillo A, Remotti HE, et al. Pd-1 signaling promotes tumor-infiltrating myeloid-derived suppressor cells and gastric tumorigenesis in mice. Gastroenterology (2021) 160(3):781–96. doi: 10.1053/j.gastro.2020.10.036
83. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. Pd-L1 is a novel direct target of hif-1α, and its blockade under hypoxia enhanced mdsc-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
84. Yu B, Luo F, Sun B, Liu W, Shi Q, Cheng S-Y, et al. Kat6a acetylation of Smad3 regulates myeloid-derived suppressor cell recruitment, metastasis, and immunotherapy in triple-negative breast cancer. Adv Sci (Weinh) (2021) 8(20):e2100014. doi: 10.1002/advs.202100014
85. Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature (2017) 543(7647):728–32. doi: 10.1038/nature21676
86. Klein S, Abraham M, Bulvik B, Dery E, Weiss ID, Barashi N, et al. Cxcr4 promotes neuroblastoma growth and therapeutic resistance through mir-15a/16-1-Mediated erk and Bcl2/Cyclin D1 pathways. Cancer Res (2018) 78(6):1471–83. doi: 10.1158/0008-5472.CAN-17-0454
87. Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent ipilimumab (Anti-Ctla-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother (2010) 33(8):828–33. doi: 10.1097/CJI.0b013e3181eec14c
88. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. Bl-8040, a Cxcr4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The combat trial. Nat Med (2020) 26(6):878–85. doi: 10.1038/s41591-020-0880-x
89. Warfel NA, Kraft AS. Pim kinase (and akt) biology and signaling in tumors. Pharmacol Ther (2015) 151:41–9. doi: 10.1016/j.pharmthera.2015.03.001
90. Broutian TR, Jiang B, Li J, Akagi K, Gui S, Zhou Z, et al. Human papillomavirus insertions identify the pim family of Serine/Threonine kinases as targetable driver genes in head and neck squamous cell carcinoma. Cancer Lett (2020) 476:23–33. doi: 10.1016/j.canlet.2020.01.012
91. Xin G, Chen Y, Topchyan P, Kasmani MY, Burns R, Volberding PJ, et al. Targeting Pim1-mediated metabolism in myeloid suppressor cells to treat cancer. Cancer Immunol Res (2021) 9(4):454–69. doi: 10.1158/2326-6066.CIR-20-0433
92. Wright GJ, Cherwinski H, Foster-Cuevas M, Brooke G, Puklavec MJ, Bigler M, et al. Characterization of the Cd200 receptor family in mice and humans and their interactions with Cd200. J Immunol (2003) 171(6):3034–46. doi: 10.4049/jimmunol.171.6.3034
93. Choueiry F, Torok M, Shakya R, Agrawal K, Deems A, Benner B, et al. Cd200 promotes immunosuppression in the pancreatic tumor microenvironment. J Immunother Cancer (2020) 8(1):e000189. doi: 10.1136/jitc-2019-000189
94. Porta C, Sica A, Riboldi E. Tumor-associated myeloid cells: New understandings on their metabolic regulation and their influence in cancer immunotherapy. FEBS J (2018) 285(4):717–33. doi: 10.1111/febs.14288
95. Adeshakin AO, Liu W, Adeshakin FO, Afolabi LO, Zhang M, Zhang G, et al. Regulation of ros in myeloid-derived suppressor cells through targeting fatty acid transport protein 2 enhanced anti-Pd-L1 tumor immunotherapy. Cell Immunol (2021) 362:104286. doi: 10.1016/j.cellimm.2021.104286
96. Mohamed E, Al-Khami AA, Rodriguez PC. The cellular metabolic landscape in the tumor milieu regulates the activity of myeloid infiltrates. Cell Mol Immunol (2018) 15(5):421–7. doi: 10.1038/s41423-018-0001-7
97. Olenchock BA, Rathmell JC, Vander Heiden MG. Biochemical underpinnings of immune cell metabolic phenotypes. Immunity (2017) 46(5):703–13. doi: 10.1016/j.immuni.2017.04.013
98. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity Via a specific cebpb isoform in triple-negative breast cancer. Cell Metab (2018) 28(1):87–103.e6. doi: 10.1016/j.cmet.2018.04.022
99. Hammami I, Chen J, Murschel F, Bronte V, De Crescenzo G, Jolicoeur M. Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC Cell Biol (2012) 13:18. doi: 10.1186/1471-2121-13-18
100. Jian S-L, Chen W-W, Su Y-C, Su Y-W, Chuang T-H, Hsu S-C, et al. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ros-mediated apoptosis. Cell Death Dis (2017) 8(5):e2779. doi: 10.1038/cddis.2017.192
101. Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, et al. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in tumor. Front Immunol (2019) 10:1399. doi: 10.3389/fimmu.2019.01399
102. Davies LC, Rice CM, McVicar DW, Weiss JM. Diversity and environmental adaptation of phagocytic cell metabolism. J Leukoc Biol (2019) 105(1):37–48. doi: 10.1002/JLB.4RI0518-195R
103. den Brok MH, Raaijmakers TK, Collado-Camps E, Adema GJ. Lipid droplets as immune modulators in myeloid cells. Trends Immunol (2018) 39(5):380–92. doi: 10.1016/j.it.2018.01.012
104. Yan D, Yang Q, Shi M, Zhong L, Wu C, Meng T, et al. Polyunsaturated fatty acids promote the expansion of myeloid-derived suppressor cells by activating the Jak/Stat3 pathway. Eur J Immunol (2013) 43(11):2943–55. doi: 10.1002/eji.201343472
Keywords: tumor, myeloid-derived suppressor cells, tumor microenvironment, immunotherapy, immune checkpoint blockade (ICB)
Citation: Sui H, Dongye S, Liu X, Xu X, Wang L, Jin CQ, Yao M, Gong Z, Jiang D, Zhang K, Liu Y, Liu H, Jiang G and Su Y (2022) Immunotherapy of targeting MDSCs in tumor microenvironment. Front. Immunol. 13:990463. doi: 10.3389/fimmu.2022.990463
Received: 10 July 2022; Accepted: 15 August 2022;
Published: 05 September 2022.
Edited by:
Peng Qu, National Institutes of Health (NIH), United StatesReviewed by:
Lukman Olalekan Afolabi, Shenzhen Institutes of Advanced Technology, (CAS), ChinaCopyright © 2022 Sui, Dongye, Liu, Xu, Wang, Jin, Yao, Gong, Jiang, Zhang, Liu, Liu, Jiang and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Liu, bGl1aHVpQHNkZm11LmVkdS5jbg==; Guomin Jiang, Z3VvbWluamlhbmdAaG90bWFpbC5jb20=; Yanping Su, c3UteWFucGluZ0AxNjMuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.