 Tinh-Hai Collet1,2
Tinh-Hai Collet1,2 Valerie Schwitzgebel2,3,4*
Valerie Schwitzgebel2,3,4*- 1Service of Endocrinology, Diabetes, Nutrition, and Therapeutic Education, Geneva University Hospitals, Geneva, Switzerland
- 2Faculty of Medicine, Diabetes Center, University of Geneva, Geneva, Switzerland
- 3Pediatric Endocrine and Diabetes Unit, Department of Pediatrics, Obstetrics, and Gynecology, Geneva University Hospitals, Geneva, Switzerland
- 4Institute of Genetics and Genomics in Geneva (iGE3), University of Geneva, Geneva, Switzerland
The prevalence of obesity is increasing worldwide, affecting both children and adults. This obesity epidemic is mostly driven by an increase in energy intake (abundance of highly palatable energy-dense food and drinks) and to a lesser degree a decrease in energy expenditure (sedentary lifestyle). A small proportion of individuals with obesity are affected by genetic forms of obesity, which often relate to mutations in the leptin-melanocortin pathway or are part of syndromes such as the Bardet-Biedl syndrome. These rare forms of obesity have provided valuable insights into the genetic architecture of obesity. Recent advances in understanding the molecular mechanisms that control appetite, hunger, and satiety have led to the development of drugs that can override genetic defects, enabling precision treatment. Leptin deficiency is uniquely treated with recombinant human metreleptin, while those with LEPR, PCSK1, or POMC deficiency can now be treated with the MC4R agonist setmelanotide. This review highlights the most frequent monogenic and syndromic forms of obesity, and the future outlook of precision treatment for these conditions.
Introduction
As global obesity prevalence continues to climb, rare forms of obesity remain underdiagnosed and insufficiently recognized, despite their classification as orphan diseases. This review delves into the epidemiology of common and rare obesity, highlighting the underlying mechanisms. It also explores recent advances in targeted therapies, such as the melanocortin 4 receptor (MC4R) agonist setmelanotide, underscoring the critical need for personalized approaches to address these unique and often overlooked conditions effectively.
Epidemiology
One in eight people is affected by obesity worldwide, translating into over 1 billion people globally, including approximately 890 million adults, 160 million children and adolescents (aged 5–19 years) (1). When adding those who are overweight, the numbers are far greater with an estimated 43% of adults and 18% children with overweight in 2022.
Hundreds of genes have been associated with obesity-related traits, while fewer genes are recognized as causally implicated in obesity (2–5). In terms of causal genes, around 20–30 genes have been identified as having a clear role in the development of monogenic forms of obesity. A small proportion of individuals are affected by genetic forms of obesity (3.9–9.3%) (6), as first highlighted by twin studies among adults (7, 8) and in childhood (9) showing a high heritability of body mass index (BMI). Monogenic forms of obesity are rare disorders, some of which are registered as orphan diseases. While being rare taken individually, collectively they can affect up to 5–7% of children with severe obesity (10, 11). In this review, we explore forms of monogenic obesity for which precision treatments are now available.
Monogenic and syndromic forms of obesity
Monogenic obesity
The most common monogenic form of obesity is associated with mutations in the MC4R gene (12), followed by mutations in the LEPR, POMC, PCSK1, and LEP genes. The prevalence of loss of function MC4R variants in the UK population is estimated at 1 in 340 (13). This prevalence rises to 0.5–1.7% among obese adults (BMI > 30 kg/m2) and around 5% in those with severe obesity (12–15). In cases of severe childhood-onset obesity, the prevalence can be even higher, varying by ethnic group (16, 17). The specific variant is also significant; highly pathogenic variants typically result in early childhood obesity, while variants with milder effects may contribute to common polygenic obesity. In addition to monogenic obesity, certain syndromes are linked to obesity. Bardet-Biedl Syndrome (BBS) has an estimated prevalence of 1 in 160,000 in northern Europe, 1 in 100,000 in the U.S., and 1 in 13,500 in some Middle Eastern populations (18). Although epidemiological data is limited in Europe, Denmark has an estimated prevalence of 1 in 59,000, while Reunion Island, France, reports rates of 1 in 45,000 to 66,000, likely due to a founder effect. Alström Syndrome (ALMS), caused by homozygous or compound heterozygous mutations in the ALMS1 gene, has a prevalence of approximately 1 in 1,000,000.1 However, higher frequencies have been reported in populations with high consanguinity or geographic isolation, with over 950 cases identified worldwide.
Genes involved in the leptin-melanocortin pathway
Leptin, produced by adipocytes, correlates with body fat and serves as a key signal for the hypothalamic arcuate nucleus. Here, it stimulates pro-opio-melanocortin (POMC) expression, which is cleaved into α- and β-melanocyte-stimulating hormones (MSH) (19). These hormones act on neurons in the paraventricular nuclei to reduce appetite and increase fat oxidation via the sympathetic nervous system. The leptin-melanocortin pathway is central to energy metabolism and body weight regulation. Mutations in MC4R or upstream genes, discussed below, can disrupt α- and β-MSH functions, leading to increased energy intake and early-onset obesity (12) (Figure 1).
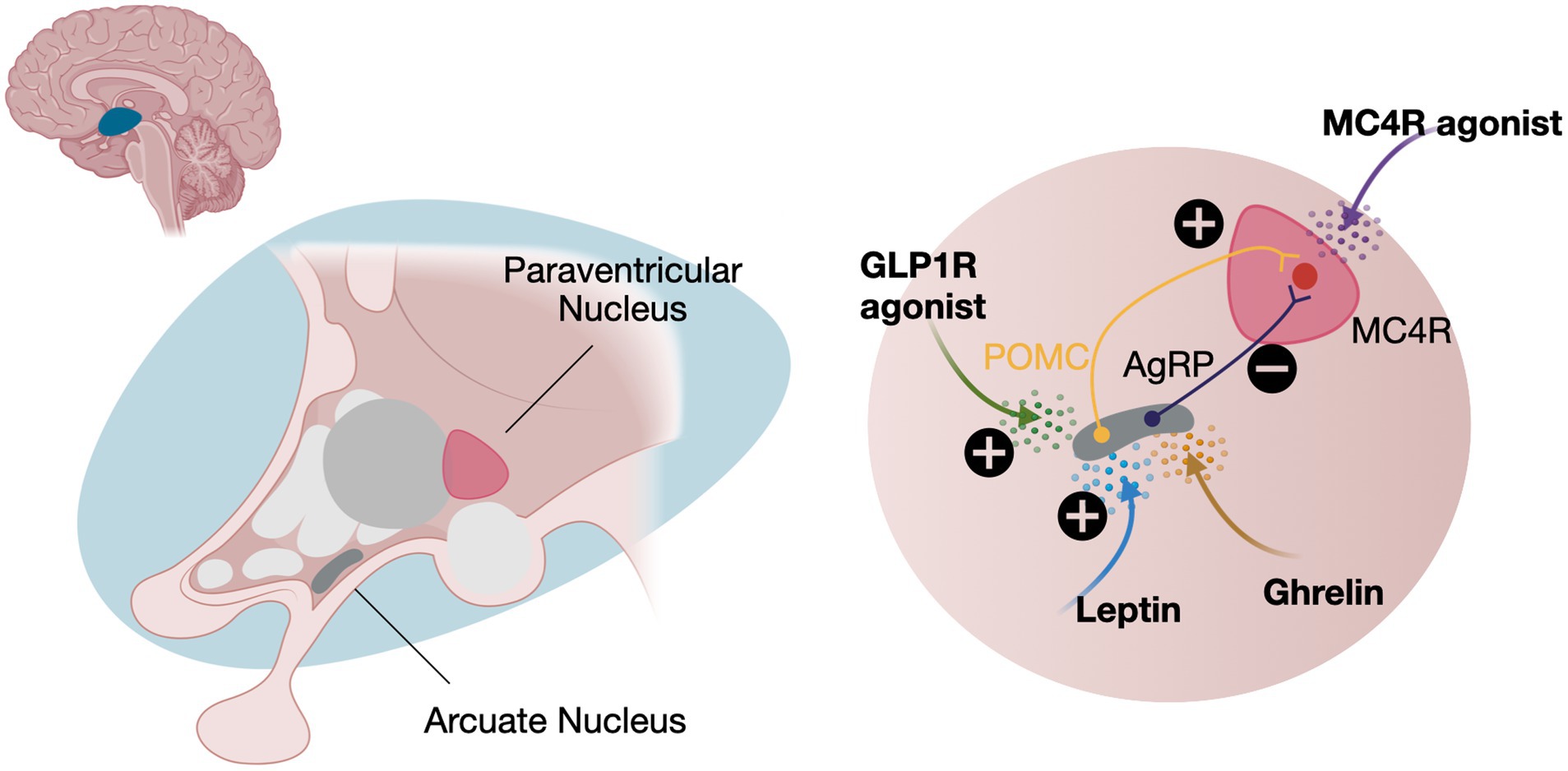
Figure 1. The central role of the leptin-melanocortin pathway. Leptin and ghrelin have opposing effects on appetite regulation. Leptin inhibits appetite by activating POMC neurons, which stimulate MC4R. In contrast, ghrelin, secreted by the stomach during fasting, activates AgRP neurons, inhibiting MC4R signaling and increasing appetite. Treatment with either a GLP1R or a MC4R agonists can help decrease appetite. POMC, Pro-opio-melanocortin expressing neurons; AgRP, Agouti related-protein expressing neurons; MC4R, Melanocortin receptor expressing neurons; GLP1R, Glucagon-like peptide 1 receptors. Created in BioRender. Schwitzgebel, V. (2024) https://BioRender.com/y38z865
Leptin and leptin receptor
The human leptin gene (LEP) is located on chromosome 7q32.1 and encodes the 16 kDa hormone leptin, which has a four-helix bundle structure typical of cytokines. Leptin binding to its receptor triggers dimerization and activates the JAK–STAT signaling pathway, influencing metabolism, appetite, and energy expenditure (20). Mutations in LEP can lead to congenital leptin deficiency, resulting in severe obesity (21). The leptin receptor is encoded by the LEPR gene on chromosome 1p31.3 and exists in multiple isoforms, with the long form (Ob-Rb) being crucial for leptin signaling in the hypothalamus (22). Mutations in LEPR can cause receptor deficiency, leading to severe obesity and hyperphagia due to improper leptin signaling (23, 24).
Pro-opiomelanocortin
The POMC gene, located on chromosome 2p23.3, encodes a precursor, pro-opiomelanocortin, that is processed into several peptides involved in energy homeostasis, adrenal function, and pigmentation. POMC is mainly expressed in the anterior pituitary, hypothalamus, and skin, and is cleaved into active peptides such as adrenocorticotropic hormone (ACTH), α−/β-MSH, and β-endorphin by specific prohormone convertases (such as PCSK1 and PCSK2). α-MSH is crucial for appetite suppression, while β-endorphin modulates pain and reward pathways. Mutations in POMC can lead to early-onset obesity, adrenal insufficiency, and pigmentation disorders (25).
Proprotein convertase subtilisin/kexin-type 1
The PCSK1 gene on chromosome 5q15-q21 encodes an enzyme vital for converting prohormones into active forms. Primarily expressed in neuroendocrine cells of the pancreas, intestines, and brain, PCSK1 processes key hormones like insulin, glucagon, and POMC, which are crucial for glucose metabolism, energy balance, and appetite regulation. Mutations can result in enzyme deficiency, leading to obesity, hyperphagia, and endocrine dysfunction (26, 27).
Melanocortin 4 receptor
The MC4R gene, located on chromosome 18q21.32, encodes a G protein-coupled receptor essential for energy homeostasis and appetite regulation, primarily in the hypothalamus (28). MC4R mediates the effects of neuropeptides from POMC, and when α-MSH binds, it activates G proteins that reduce food intake and increase energy expenditure (29, 30). MC4R variants also affect endocytosis, trafficking and dimerization highlighting various cellular mechanisms in weight regulation. Conversely, the agouti-related peptide acts as an antagonist, blocking α-MSH binding and promoting increased appetite and reduced energy expenditure. Mutations in MC4R or upstream signaling, can prevent α- and β-MSH from exerting their effects, leading to increased energy intake and weight gain from early childhood and into adulthood (12).
Syndromic forms of obesity
Bardet-Biedl syndrome (BBS)
BBS is a heterogeneous disorder caused by mutations in over 25 different genes (31). While the mechanism underlying hyperphagia in BBS remains unclear, reduced ciliary length impairs leptin signaling (32). Cilia are essential sensory organelles on the surface of POMC neurons, and studies show that ciliary defects in specific hypothalamic neurons can induce obesity and hyperphagia in mice (33). BBS is characterized by six primary features: retinal degeneration, truncal obesity, postaxial polydactyly, hypogonadism, intellectual disability, and renal abnormalities (34). Obesity is a prominent feature, affecting 72–92% of patients, with significant weight gain typically observed early in life. By age 2, 33% of children are overweight and 23% are obese; by age 5, 90% are either overweight or obese (31). Additionally, the prevalence of type 2 diabetes among adolescents with BBS is 6% (35), along with hypertension and hypertriglyceridemia, which increase the risk of cardiovascular disease (36, 37).
Alström syndrome (ALMS)
ALMS is caused by mutations in the ALMS1 gene, located on chromosome 2p13.1. This gene is essential for cilia function—hair-like structures on cell surfaces that play critical roles in signaling and sensory functions. Mutations in ALMS1 lead to a rare genetic disorder characterized by progressive vision and hearing loss, obesity, type 2 diabetes, heart disease, and kidney dysfunction, among other symptoms (38).
Hyperphagia, hunger and satiety
Hyperphagia is a common feature of all monogenic and syndromic forms of obesity (39). It is characterized by an abnormally intense and persistent sensation of hunger or urge to eat, often leading to overeating. Unlike typical hunger, this condition does not diminish after eating, frequently resulting in the rapid consumption of excessive amounts of food. Hyperphagia is not a disorder in itself but a symptom of an underlying medical issue, such as a genetic disruption in the leptin-melanocortin signaling pathway (40). This pathway plays a key role in the homeostatic regulation of eating, as opposed to the hedonic pathway, which is associated with the more common polygenic form of obesity.
Often confused with hyperphagia but distinct from it, hunger is a sensation that drives the consumption of food. Hunger typically arises a few hours after eating and is generally considered unpleasant.
Satiety, which usually occurs 15–20 min after eating, is a state of fullness that extends beyond mere satisfaction, representing the opposite of hunger. After satiation (the point at which a meal ends), satiety persists as a feeling of fullness until the next meal (41). When food is still present in the gastrointestinal tract after a meal, satiety signals suppress hunger signals, but as time passes, satiety gradually fades while hunger increases.
How to measure hunger: different hunger scales
Like other patient-reported outcomes such as pain and fatigue, hunger can only be assessed through self-report rather than clinical or laboratory evaluations. A widely used tool for this purpose is the visual analog scale (VAS), which features a horizontal line, usually 100 mm long, with endpoints labeled “Not at all hungry” and “Extremely hungry” (42). Patients mark or slide along this line to indicate their hunger level, providing a quantifiable score that is easy to administer, reproducible, and sensitive to short-term changes, much like VAS tools for other symptoms. Another popular tool is the Likert scale, where participants rate statements on hunger or satiety using a graded scale (e.g., 1–5 or 1–7) (41, 43).
Burden on families and patients
Numerous studies highlight the substantial burden of genetically related hyperphagia on patients and caregivers, particularly in cases involving POMC, PCSK1, and LEPR deficiencies, as well as ALMS and BBS (44). Patients often experience intense emotional distress, including sadness, frustration, anxiety, and guilt, driven by a relentless preoccupation with food and an inability to control their hunger (45). Caregivers, especially parents, share this emotional strain, facing feelings of guilt, helplessness, and frustration as they navigate their child’s behaviors, such as food sneaking and hoarding (46). The persistent focus on food intrudes on daily life, affecting patients’ school and work performance while limiting social engagement. This ongoing challenge severely diminishes their quality of life, as highlighted in a multi-country survey (47). These findings underscore the multifaceted burden that hyperphagia places on patients, siblings and caregivers, highlighting the urgent need for precision therapies to address this debilitating condition.
Need of early detection and management: screening program, genetic confirmation, to decrease complications
In patients with early-onset obesity, especially when it begins before the age of 5, physicians should strongly consider the possibility of a genetic cause (48). Pediatricians play a key role in identifying cases of monogenic obesity, using characteristic BMI trajectories as a diagnostic aid, since these patterns differ from those seen in polygenic obesity (49). For confirmation and expert evaluation, referral to tertiary obesity clinics is recommended, where specialists have the expertise to conduct and interpret the necessary genetic tests, such as dedicated obesity gene panels or whole exome/genome sequencing. The French INSERM NutriOmics group has developed an online diagnostic support tool called ObsGen to help practitioners diagnose monogenic obesity more effectively (50).2 Early treatment of patients with genetic obesity is crucial, as it helps to limit the condition’s progression during adolescence, prevents related complications (51), and reduces the stigmatization and suffering these individuals often experience (52).
Lack of response to conventional therapies—failure of diet and lifestyle interventions, no sustained response to bariatric surgery
Patients with mutations in the leptin-melanocortin pathway and BBS often receive dietary and exercise counseling similar to those with polygenic obesity, yet they show limited response to these lifestyle changes, as seen in MC4R deficiency (53). While bariatric surgery is effective for common obesity (54), its success relies on a functional leptin-melanocortin pathway (55, 56). Thus, alternative treatments are necessary for this genetic population, as bariatric surgery is typically ineffective unless carefully considered for select adults (57, 58).
Glucagon-like peptide-1 (GLP-1) receptor agonists (RAs) show promise in monogenic and syndromic obesity, with real-world evidence of effectiveness in ALMS and BBS patients (59, 60). A study of liraglutide (3.0 mg for 16 weeks) reported weight loss in patients with MC4R deficiency (6.8 kg ± 1.8 kg) compared to controls (6.1 kg ± 1.2 kg) (61), suggesting that GLP-1 RAs may work independently of a fully functional leptin-melanocortin pathway. Ongoing trials of newer GLP-1 RAs and dual/triple agonists are awaited for further insights.
Precision treatment approach
Metreleptin
Leptin deficiency due to LEP mutations is uniquely treated with recombinant human leptin (metreleptin). This synthetic analog is administered subcutaneously at 0.03 mg/kg of lean body mass daily, leading to significant weight loss and reduced hyperphagia (62, 63). In one case, a 9-year-old patient lost 16.4 kg in the first year and achieved BMI reduction over 4 years, despite weight remaining above the 98th percentile by age 14 (62, 63). The development of metreleptin-neutralizing antibodies can lead to hyperphagia recurrence and weight regain. This therapy is ineffective for patients with downstream leptin-melanocortin pathway mutations.
Setmelanotide
Setmelanotide, a synthetic cyclic peptide, binds with high affinity to human MC4R (64). Given the central role of the leptin-melanocortin pathway, this MC4R agonist can effectively “rescue” downstream signaling, even without upstream LEPR, PCSK1, or POMC activity. However, treatment is appropriate only for cases with validated loss-of-function mutations — either homozygous or specific heterozygous autosomal dominant mutations of LEPR, PCSK1 or POMC. Missense mutations with neutral or partial deleterious effects are unlikely to benefit (29, 30, 65, 66). This approach heralds a future of precision medicine, where treatment is tailored to variant-specific responses based on genotype.
What are the therapeutic goals of setmelanotide?
The therapeutic goals of setmelanotide for treating obesity linked to POMC, PCSK1, or LEPR deficiencies and BBS include weight stabilization, hyperphagia control, quality of life improvement, safety and tolerability, enhanced metabolic and cardiovascular health, and sustained long-term efficacy.
Efficacy of setmelanotide
In infants and children, weight stabilization—not weight loss—is prioritized to prevent adverse effects on growth (24, 67–70). Weight control is achieved primarily by reducing hyperphagia, as children with LEPR and POMC mutations consume three times more calories per kg of lean mass than controls (24). Additionally, children with homozygous MC4R mutations consume more than those with partial loss-of-function variants (64). Setmelanotide has shown promising results in clinical trials and in countries where it is marketed. So far, a relatively small number of patients have been tested with this MC4R agonist, but setmelanotide efficiently leads to weight control in patients with LEPR and POMC deficiencies and BBS (71, 72) (Table 1). Hunger reduction has been measured on an 11-point Likert scale, though some studies used retrospective self-reports, introducing potential recall bias (45, 46). Nonetheless, most studies indicate a reduction in hyperphagia with setmelanotide (46, 73).
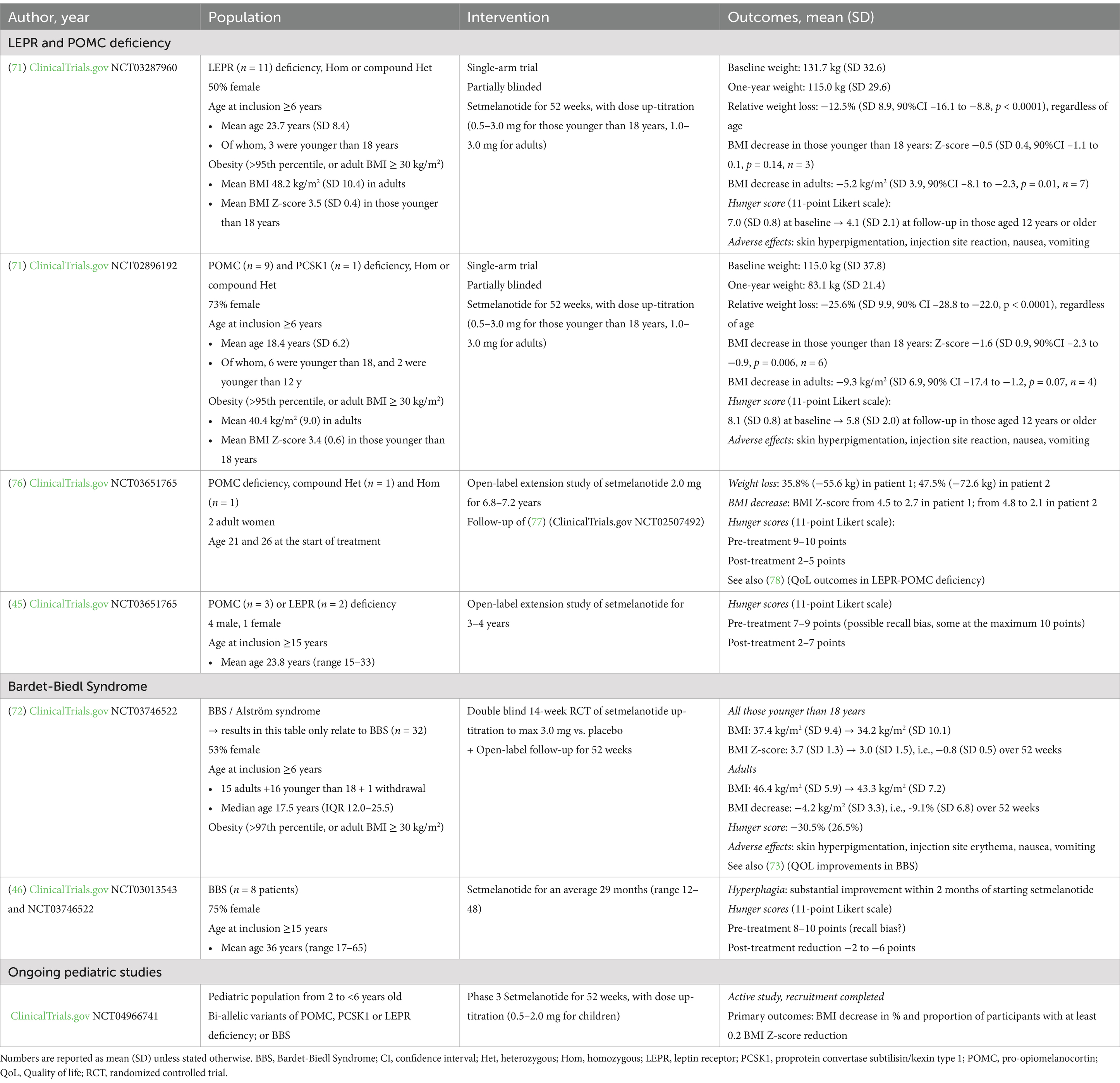
Table 1. Summary of studies on setmelanotide efficacy.
Side effects/contraindication of setmelanotide
As with any injectable medication, hypersensitivity to setmelanotide or its excipients can be observed. Additionally, due to cross-stimulation of MC1R in melanocytes, some patients experience skin hyperpigmentation and darkening of naevi (51, 72). For safety, regular skin monitoring and restricted prescription through specialized centers are recommended. The clinical and research community now awaits long-term data on this second-generation MC4R agonist and the potential development of new therapies.
Broader indications for setmelanotide?
Setmelanotide has been approved by the FDA for patients over 6 years of age with POMC, PCSK1, LEPR deficiencies and BBS, while the EMA has approved it for biallelic POMC, PCSK1, LEPR deficiencies and BBS in Europe (51, 74). Recently, its approval expanded to include patients as young as 2 years old (Clinicaltrials.gov no. NCT04966741). Trials are also underway to assess its efficacy in acquired hypothalamic obesity (75) and in other leptin-melanocortin pathway genes, including SH2B1, CPE, and 16p11.2 chromosomal rearrangements (51).
Upcoming developments and outlook
With the high prevalence of obesity and advancing insights into its mechanisms, we anticipate drug developments targeting additional genes, alongside investigations into how different mutation types (e.g., null, frameshift, missense) affect treatment outcomes. Distinctions in receptor function—complete versus partial loss—may also yield varied drug responses (66). Given the diversity of outcomes in prior studies (Table 1), a standardized hunger scale, trial design, and extended follow-up are essential. In the development of MC4R agonists, we expect next-generation drugs to avoid MC1R cross-activity, thereby minimizing skin and naevi darkening, which currently necessitates regular monitoring. With the strong development GLP-1/GIP/glucagon receptor agonists, we expect some efficacy for individuals with monogenic and syndromic obesity in terms of weight loss and metabolic improvement (51).
Conclusion
Obesity is a heterogenous disorder, involving single genes to hundreds of genes (2, 4, 5). Improved and precision treatment is now required. Recent advances in understanding the mechanisms leading to obesity and controlling appetite, hunger, and satiety have led to the development of drugs that can override genetic defects, enabling precision treatment.
Author contributions
TC: Conceptualization, Writing – original draft, Writing – review & editing. VS: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Open access funding was provided by the University of Geneva.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
1. ^https://www.orpha.net/en/disease/detail/64?name=Alstrom%20syndrome%20&mode=name
2. ^https://redc.integromics.fr/surveys/index.php?s=XEFPD474YT
References
1. NCD Risk Factor Collaboration. Worldwide trends in underweight and obesity from 1990 to 2022: a pooled analysis of 3663 population-representative studies with 222 million children, adolescents, and adults. Lancet. (2024) 403:1027–50. doi: 10.1016/S0140-6736(23)02750-2
2. Akbari, P, Gilani, A, Sosina, O, Kosmicki, JA, Khrimian, L, and Fang, Y-Y. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science. (2021) 373:eabf8683. doi: 10.1126/science.abf8683
3. Locke, AE, Kahali, B, Berndt, SI, Justice, AE, Pers, TH, Day, FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. (2015) 518:197–206. doi: 10.1038/nature14177
4. Turcot, V, Yingchang, L, Highland, HM, Schurmann, C, Justice, AE, Fine, RS, et al. Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat Genet. (2018) 50:26–41. doi: 10.1038/s41588-017-0011-x
5. Zhao, Y, Chukanova, M, Kentistou, KA, Fairhurst-Hunter, Z, Siegert, AM, Jia, RY, et al. Protein-truncating variants in BSN are associated with severe adult-onset obesity, type 2 diabetes and fatty liver disease. Nat Genet. (2024) 56:579–84. doi: 10.1038/s41588-024-01694-x
6. Kleinendorst, L, Massink, MPG, Cooiman, MI, Savas, M, van der Baan-Slootweg, OH, Roelants, RJ, et al. Genetic obesity: next-generation sequencing results of 1230 patients with obesity. J Med Genet. (2018) 55:578–86. doi: 10.1136/jmedgenet-2018-105315
7. Silventoinen, K, Magnusson, PKE, Tynelius, P, Kaprio, J, and Rasmussen, F. Heritability of body size and muscle strength in young adulthood: a study of one million Swedish men. Genet Epidemiol. (2008) 32:341–9. doi: 10.1002/gepi.20308
8. Stunkard, AJ, Harris, JR, Pedersen, NL, and McClearn, GE. The body-mass index of twins who have been reared apart. N Engl J Med. (1990) 322:1483–7. doi: 10.1056/NEJM199005243222102
9. Wardle, J, Carnell, S, Haworth, CM, and Plomin, R. Evidence for a Strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. (2008) 87:398–404. doi: 10.1093/ajcn/87.2.398
10. Ramachandrappa, S, and Sadaf Farooqi, I. Genetic approaches to understanding human obesity. J Clin Invest. (2011) 121:2080–6. doi: 10.1172/JCI46044
11. Styne, DM, Arslanian, SA, Connor, EL, Farooqi, IS, Hassan Murad, M, Silverstein, JH, et al. Pediatric obesity-assessment, treatment, and prevention: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. (2017) 102:709–57. doi: 10.1210/jc.2016-2573
12. Farooqi, IS, Keogh, JM, Yeo, GSH, Lank, EJ, Cheetham, T, and O’Rahilly, S. Clinical Spectrum of obesity and mutations in the Melanocortin 4 receptor gene. N Engl J Med. (2003) 348:1085–95. doi: 10.1056/NEJMoa022050
13. Wade, KH, Lam, BYH, Melvin, A, Pan, W, Corbin, LJ, Hughes, DA, et al. Loss-of-function mutations in the Melanocortin 4 receptor in a UK birth cohort. Nat Med. (2021) 27:1088–96. doi: 10.1038/s41591-021-01349-y
14. Alharbi, KK, Spanakis, E, Tan, K, Smith, MJ, Aldahmesh, MA, O’Dell, SD, et al. Prevalence and functionality of Paucimorphic and private MC4R mutations in a large, unselected European British population, scanned by meltMADGE. Hum Mutat. (2007) 28:294–302. doi: 10.1002/humu.20404
15. Stutzmann, F, Tan, K, Vatin, V, Dina, C, Jouret, B, Tichet, J, et al. Prevalence of Melanocortin-4 receptor deficiency in Europeans and their age-dependent penetrance in multigenerational pedigrees. Diabetes. (2008) 57:2511–8. doi: 10.2337/db08-0153
16. Kleinendorst, L, Abawi, O, van der Voorn, B, Jongejan, MHTM, Brandsma, AE, Visser, JA, et al. Identifying underlying medical causes of pediatric obesity: results of a systematic diagnostic approach in a pediatric obesity center. PLoS One. (2020) 15:e0244508. doi: 10.1371/journal.pone.0244508
17. Trier, C, Hollensted, M, Schnurr, TM, Lund, MAV, Nielsen, TRH, Gao, R, et al. Obesity treatment effect in Danish children and adolescents carrying Melanocortin-4 receptor mutations. In J Obesity. (2021) 45:66–76. doi: 10.1038/s41366-020-00673-6
18. Forsythe, E, and Beales, PL. Bardet-Biedl syndrome. Eur J Hum Genet. (2012) 21:8–13. doi: 10.1038/ejhg.2012.115
19. Ayers, KL, Glicksberg, BS, Garfield, AS, et al. Melanocortin 4 Receptor Pathway Dysfunction in Obesity: Patient Stratification Aimed at MC4R Agonist Treatment. J Clin Endocrinol Metab. (2018) 103:2601–12.
20. Saxton, RA, Caveney, NA, Moya-Garzon, MD, Householder, KD, Rodriguez, GE, Burdsall, KA, et al. Structural insights into the mechanism of leptin receptor activation. Nat Commun. (2023) 14:1797. doi: 10.1038/s41467-023-37169-6
21. Montague, CT, Farooqi, IS, Whitehead, JP, Soos, MA, Rau, H, Wareham, NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. (1997) 387:903–8. doi: 10.1038/43185
22. Mao, H, Kim, GH, Pan, L, and Qi, L. Regulation of leptin signaling and diet-induced obesity by SEL1L-HRD1 ER-associated degradation in POMC expressing neurons. Nat Commun. (2024) 15:8435. doi: 10.1038/s41467-024-52743-2
23. Clément, K, Vaisse, C, Lahlou, N, Cabrol, S, Pelloux, V, Cassuto, D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. (1998) 392:398–401. doi: 10.1038/32911
24. Farooqi, IS, Wangensteen, T, Collins, S, Kimber, W, Matarese, G, Keogh, JM, et al. Clinical and molecular genetic Spectrum of congenital deficiency of the leptin receptor. N Engl J Med. (2007) 356:237–47. doi: 10.1056/NEJMoa063988
25. Krude, H, Biebermann, H, Luck, W, Horn, R, Brabant, G, and Gruters, A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. (1998) 19:155–7. doi: 10.1038/509
26. Frank, GR, Fox, J, Candela, N, Jovanovic, Z, Bochukova, E, Levine, J, et al. Severe obesity and diabetes insipidus in a patient with PCSK1 deficiency. Mol Genet Metab. (2013) 110:191–4. doi: 10.1016/j.ymgme.2013.04.005
27. Jackson, RS, Creemers, JW, Ohagi, S, Raffin-Sanson, ML, Sanders, L, Montague, CT, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. (1997) 16:303–6. doi: 10.1038/ng0797-303
28. Morton, GJ, Cummings, DE, Baskin, DG, Barsh, GS, and Schwartz, MW. Central nervous system control of food intake and body weight. Nature. (2006) 443:289–95.
29. Brouwers, B, Mendes, E, de Oliveira, M, Marti-Solano, FBF, Monteiro, S-AL, Keogh, JM, et al. Human MC4R variants affect endocytosis, trafficking and dimerization revealing multiple cellular mechanisms involved in weight regulation. Cell Rep. (2021) 34:108862. doi: 10.1016/j.celrep.2021.108862
30. Rondón, R, Alejandra, V, Welling, MS, van den Akker, ELT, van Rossum, EFC, Boon, EMJ, et al. MC4R variants modulate α-MSH and Setmelanotide induced cellular signaling at multiple levels. J Clin Endocrinol Metab. (2024) 3:dgae210. doi: 10.1210/clinem/dgae210
31. Tomlinson, JW. Bardet-Biedl syndrome: a focus on genetics, mechanisms and metabolic dysfunction. Diabetes Obes Metab. (2024) 26:13–24. doi: 10.1111/dom.15480
32. Mi, HY, Kang, GM, Byun, K, Ko, HW, Kim, J, Shin, M-S, et al. Leptin-promoted cilia assembly is critical for Normal energy balance. J Clin Invest. (2014) 124:2193–7. doi: 10.1172/JCI69395
33. Lee, CH, Song, DK, Park, CB, Choi, J, Kang, GM, Shin, SH, et al. Primary cilia mediate early life programming of adiposity through lysosomal regulation in the developing mouse hypothalamus. Nat Commun. (2020) 11:5772. doi: 10.1038/s41467-020-19638-4
34. Khan, SA, Muhammad, N, Khan, MA, Kamal, A, Rehman, ZU, and Khan, S. Genetics of human Bardet–Biedl syndrome, an updates. Clin Genet. (2016) 90:3–15. doi: 10.1111/cge.12737
35. Baig, S, Wanninayake, S, Foggensteiner, L, Elhassan, YS, Manolopoulos, K, Ali, S, et al. Adipose tissue function and insulin sensitivity in syndromic obesity of Bardet-Biedl syndrome. Int J Obesity. (2023) 47:382–90. doi: 10.1038/s41366-023-01280-x
36. Feuillan, PP, Ng, D, Han, JC, Sapp, JC, Wetsch, K, Spaulding, E, et al. Patients with Bardet-Biedl syndrome have Hyperleptinemia suggestive of leptin resistance. J Clin Endocrinol Metab. (2011) 96:E528–35. doi: 10.1210/jc.2010-2290
37. Mujahid, S, Hunt, KF, Cheah, YS, Forsythe, E, Hazlehurst, JM, Sparks, K, et al. The endocrine and metabolic characteristics of a large Bardet-Biedl syndrome clinic population. J Clin Endocrinol Metab. (2018) 103:1834–41. doi: 10.1210/jc.2017-01459
38. Tahani, N, Pietro, M, Hélène, D, Richard, P, Diana, V, Gabriella,, et al. Consensus Clinical Management Guidelines for Alström Syndrome. Orphanet Journal of Rare Diseases. (2020) 15:253. doi: 10.1186/s13023-020-01468-8
39. Heymsfield, SB, Avena, NM, and Baier, L. Hyperphagia: current concepts and future directions proceedings of the 2nd international conference on hyperphagia. Obesity (Silver Spring). (2014) 1:S1–17.
40. Šket, R, Kotnik, P, and Bizjan, BJ. Heterozygous Genetic Variants in Autosomal Recessive Genes of the Leptin-Melanocortin Signalling Pathway Are Associated With the Development of Childhood Obesity. Front Endocrinol (Lausanne). (2022) 13:832911
41. Gibbons, C, Hopkins, M, Beaulieu, K, Oustric, P, and Blundell, JE. Issues in Measuring and Interpreting Human Appetite (Satiety/Satiation) and Its Contribution to Obesity. Curr Obes Rep. (2019) 8:77–87. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6517339/
42. Flint, A, Raben, A, Blundell, JE, and Astrup, A. Reproducibility, power and validity of visual analogue scales in assessment of appetite sensations in single test meal studies. Int J Obes Relat Metab Disord. (2000) 24:38–48.
43. Stubbs, RJ, Hughes, DA, and Johnstone, AM. The use of visual analogue scales to assess motivation to eat in human subjects: a review of their reliability and validity with an evaluation of new hand-held computerized systems for temporal tracking of appetite ratings. Br J Nutr. (2000) 84:405–15.
44. Dollfus, H, Lilien, MR, Maffei, P, Verloes, A, Muller, J, Bacci, GM, et al. Bardet-Biedl syndrome improved diagnosis criteria and management: inter European reference networks consensus statement and recommendations. Eur. J. Hum. Genet. (2024) 32:1347–60. doi: 10.1038/s41431-024-01634-7
45. Wabitsch, M, Fehnel, S, Mallya, UG, Sluga-O’Callaghan, M, Richardson, D, Price, M, et al. Understanding the patient experience of hunger and improved quality of life with Setmelanotide treatment in POMC and LEPR deficiencies. Adv Ther. (2022) 39:1772–83. doi: 10.1007/s12325-022-02059-8
46. Ervin, C, Norcross, L, Mallya, UG, Fehnel, S, Mittleman, RS, Webster, M, et al. Interview-based patient- and caregiver-reported experiences of hunger and improved quality of life with Setmelanotide treatment in Bardet-Biedl syndrome. Adv Ther. (2023) 40:2394–411. doi: 10.1007/s12325-023-02443-y
47. Forsythe, E, Mallya, UG, and Yang, M. Burden of hyperphagia and obesity in Bardet-Biedl syndrome: a multicountry survey. Orphanet J Rare Dis. (2023) 18:182.
48. Ruiz, I, Bouthors, T, Borloz, S, Hauschild, M, Maggio, A, and Schwitzgebel, VM. Obesity in infancy: new precision treatment. Rev Med Suisse. (2023) 19:374–9.
49. Wabitsch, M, Farooqi, S, and Flück, CE. Natural History of Obesity Due to POMC, PCSK1, and LEPR Deficiency and the Impact of Setmelanotide. J Endocr Soc. (2022) 6:bvac057.
50. Dubern, B, Mosbah, H, Pigeyre, M, Clément, K, and Poitou, C. Rare genetic causes of obesity: Diagnosis and management in clinical care. Annales d’Endocrinologie [Internet]. (2022) 83:63–72. Available at: https://www.sciencedirect.com/science/article/pii/S0003426621011094
51. Hinney, A, Körner, A, and Fischer-Posovszky, P. The promise of new anti-obesity therapies arising from knowledge of genetic obesity traits. Nat. Rev. Endocrinol. (2022) 18:623–37. doi: 10.1038/s41574-022-00716-0
52. Dubern, B, Lourdelle, A, and Clément, K. Beneficial Effects of Setmelanotide in a 5-Year-Old Boy With POMC Deficiency and on His Caregivers. JCEM Case Reports [Internet].(2023). 1:luad041. doi: 10.1210/jcemcr/luad041
53. Reinehr, T, Hebebrand, J, Friedel, S, et al. Lifestyle intervention in obese children with variations in the melanocortin 4 receptor gene. Obesity (Silver Spring). (2009) 17:382–9.
54. Carlsson, LMS, Sjöholm, K, and Jacobson, P. Life Expectancy after Bariatric Surgery in the Swedish Obese Subjects Study. N Engl J Med [Internet]. (2020) 383:1535–43. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7580786/
55. Hatoum, IJ, Stylopoulos, N, and Vanhoose, AM. Melanocortin-4 receptor signaling is required for weight loss after gastric bypass surgery. J Clin Endocrinol Metab. (2012) 97:E1023–31.
56. Cooiman, MI, Alsters, SIM, and Duquesnoy, M. Long-Term Weight Outcome After Bariatric Surgery in Patients with Melanocortin-4 Receptor Gene Variants: a Case-Control Study of 105 Patients. Obes Surg. (2022) 32:837–44.
57. Vos, N, Oussaada, SM, and Cooiman, MI. Bariatric Surgery for Monogenic Non-syndromic and Syndromic Obesity Disorders. Curr Diab Rep [Internet]. (2020) 20:44. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7391392/
58. Poitou, C, Puder, L, and Dubern, B. Long-term outcomes of bariatric surgery in patients with bi-allelic mutations in the POMC, LEPR, and MC4R genes. Surg Obes Relat Dis. (2021) 17:1449–56.
59. Ali, S, Baig, S, and Wanninayake, S. Glucagon-like peptide-1 analogues in monogenic syndromic obesity: Real-world data from a large cohort of Alström syndrome patients. Diabetes Obes Metab. (2024) 26:989–96.
60. Ganawa, S, Santhosh, SH, Parry, L, and Syed, AA. Weight loss with glucagon-like peptide-1 receptor agonists in Bardet-Biedl syndrome. Clin Obes. (2022) 12:e12546
61. Iepsen, EW, Zhang, J, and Thomsen, HS. Patients with Obesity Caused by Melanocortin-4 Receptor Mutations Can Be Treated with a Glucagon-like Peptide-1 Receptor Agonist. CMET. (2018) 28:23–32.e3.
62. Farooqi, IS, Jebb, SA, and Langmack, G. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. (1999) 341:879–84.
63. Farooqi, IS, Matarese, G, and Lord, GM. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. (2002) 110:1093–103.
64. Collet, T-H, Dubern, B, Mokrosinski, J, Connors, H, Keogh, JM, Mendes, E, et al. Evaluation of a Melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol. Metab. (2017) 6:1321–9. doi: 10.1016/j.molmet.2017.06.015
65. Folon, L, Baron, M, Toussaint, B, Vaillant, E, Boissel, M, Scherrer, V, et al. Contribution of heterozygous PCSK1 variants to obesity and implications for precision medicine: a case-control study. Lancet Diab. Endocrinol. (2023) 11:182–90. doi: 10.1016/S2213-8587(22)00392-8
66. Collen, L, Lauriane, BD, Poitou, C, Vaxillaire, M, Toussaint, B, Dechaume, A, et al. Heterozygous pathogenic variants in POMC are not responsible for monogenic obesity: implication for MC4R agonist use. Genet Med Off J Am College Med Genet. (2023) 25:100857. doi: 10.1016/j.gim.2023.100857
67. Amador, M, Ramos, LT, Moroño, M, and Hermelo, MP. Growth rate reduction during energy restriction in obese adolescents. Exp Clin Endocrinol. (1990) 96:73–82. doi: 10.1055/s-0029-1210991
68. Rogol, AD, Clark, PA, and Roemmich, JN. Growth and pubertal development in children and adolescents: effects of diet and physical Activity1234. Am J Clin Nutr. (2000) 72:521S–8S. doi: 10.1093/ajcn/72.2.521S
69. Sothern, Null, RMS, J. NUdall, Vargas, A, and Blecker, U. Weight loss and growth velocity in obese children after very low calorie diet, exercise, and behavior modification. Acta Paediatr. (2000) 89:1036–43. doi: 10.1111/j.1651-2227.2000.tb03347.x
70. Dietz, WH, and Hartung, R. Changes in height velocity of obese preadolescents during weight reduction. Am. J. Dis. Children. (1985) 139:705–7. doi: 10.1001/archpedi.1985.02140090067031
71. Clément, K, van den Akker, E, Argente, J, Bahm, A, Chung, WK, Connors, H, et al. Efficacy and safety of Setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: single-arm, open-label, multicentre, phase 3 trials. Lancet Diab. Endocrinol. (2020) 8:960–70. doi: 10.1016/S2213-8587(20)30364-8
72. Haqq, AM, Chung, WK, Dollfus, H, Haws, RM, Martos-Moreno, GÁ, Poitou, C, et al. Efficacy and safety of Setmelanotide, a Melanocortin-4 receptor agonist, in patients with Bardet-Biedl syndrome and Alström syndrome: a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial with an open-label period. Lancet Diab. Endocrinol. (2022) 10:859–68. doi: 10.1016/S2213-8587(22)00277-7
73. Forsythe, E, Haws, RM, Argente, J, Beales, P, Martos-Moreno, GÁ, Dollfus, H, et al. Quality of life improvements following one year of Setmelanotide in children and adult patients with Bardet-Biedl syndrome: phase 3 trial results. Orphanet J Rare Dis. (2023) 18:12. doi: 10.1186/s13023-022-02602-4
74. Barbosa, F, Bárbara, FC, de Moraes, A, Barbosa, CB, Santos, PTKP, Pereira, I, et al. Efficacy and safety of Setmelanotide, a Melanocortin-4 receptor agonist, for obese patients: a systematic review and meta-analysis. J. Pers. Med. (2023) 13:1460. doi: 10.3390/jpm13101460
75. Roth, CL, Scimia, C, Shoemaker, AH, Gottschalk, M, Miller, J, Yuan, G, et al. Setmelanotide for the treatment of acquired hypothalamic obesity: a phase 2, open-label, multicentre trial. Lancet Diab Endocrinol. (2024) 12:380–9. doi: 10.1016/S2213-8587(24)00087-1
76. Kühnen, P, and Clément, K. Long-term MC4R agonist treatment in POMC-deficient patients. N Engl J Med. (2022) 387:852–4. doi: 10.1056/NEJMc2207442
77. Kühnen, P, Clément, K, Wiegand, S, Blankenstein, O, Gottesdiener, K, Martini, LL, et al. Proopiomelanocortin deficiency treated with a Melanocortin-4 receptor agonist. N Engl J Med. (2016) 375:240–6. doi: 10.1056/NEJMoa1512693
Keywords: monogenic obesity, melanocortin-4 receptor (MC4R), proprotein convertase subtilisin/kexin-type 1 (PCSK1), pro-opio-melanocortin (POMC), leptin receptor (LEPR), leptin-melanocortin pathway, Bardet-Biedel syndrome, precision medicine
Citation: Collet T-H and Schwitzgebel V (2024) Exploring the therapeutic potential of precision medicine in rare genetic obesity disorders: a scientific perspective. Front. Nutr. 11:1509994. doi: 10.3389/fnut.2024.1509994
Edited by:
Xiaohua Wang, Soochow University, ChinaReviewed by:
Céline Cruciani-Guglielmacci, Université Paris Cité, FranceCopyright © 2024 Collet and Schwitzgebel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valérie Schwitzgebel, dmFsZXJpZS5zY2h3aXR6Z2ViZWxAdW5pZ2UuY2g=