 Lily Gierke
Lily Gierke- 1Department of Biological Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI, United States
- 2Department of Computational and Systems Biology, Pittsburgh Center for Evolutionary Biology and Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 3Pacific Northwest National Laboratories, Seattle, WA, United States
- 4Retired, Olympia, WA, United States
Introduction: In temperate regions, one of the most critical determinants of present range-wide genetic diversity was the Pleistocene climate oscillations, the most recent one created by the last glacial maximum (LGM). This study aimed to describe N. luetkeana genetic structure across its entire range (Alaska to California) and test different models of population connectivity within the Salish Sea. This region was colonized after the LGM and has been under increased disturbance in recent decades.
Methods: We utilized microsatellite markers to study N. luetkeana genetic diversity at 53 sites across its range. Using higher sampling density in the Salish Sea, we employed a seascape genetics approach and tested isolation by hydrodynamic transport and environment models.
Results: At the species distribution scale, we found four main groups of genetic co-ancestry, Alaska; Washington with Vancouver Island’s outer coast and Juan de Fuca Strait; Washington’s inner Salish Sea; and Oregon with California. The highest allelic richness (AR) levels were found in California, near the trailing range edge, although AR was also high in Alaska. The inner Salish Sea region had the poorest diversity across the species distribution. Nevertheless, a pattern of isolation by hydrodynamic transport and environment was supported in this region.
Discussion: The levels of allelic, private allele richness and genetic differentiation suggest that during the LGM, bull kelp had both northern and southern glacial refugia in the Prince of Wales Island-Haida Gwaii region and Central California, respectively. Genetic diversity in Northern California sites seems resilient to recent disturbances, whereas the low levels of genetic diversity in the inner Salish Sea are concerning.
Introduction
The range-wide distribution of species’ genetic diversity results from evolutionary, ecological, and biogeographic processes. In temperate regions, the most important determinant of current range-wide genetic diversity was the Pleistocene glacial-interglacial climate oscillations, the most recent one created by the last glacial maximum (LGM) 20,000 years ago (Clark et al., 2009), which resulted in range contraction and expansions. Under the expansion-contraction model (ECM), when the glaciers retreated, wide-spread recolonization into higher latitudes (the leading-edge) occurred across many taxa of species (Blanchette et al., 2008; Kelly and Palumbi, 2010; Lindstrom et al., 2011). For many marine taxa, where fossil records are rare or absent, the present patterns of genetic structure and diversity are the single indicator of how the LGM shaped the species range (Marko, 2004). The ECM posits that glacial refugia were located in one or several regions at lower latitudes. Depending on the taxa dispersal capacity, these LGM refugia (the trailing-edge) hold currently higher genetic diversity than found in regions colonized after the glaciers retreated. While genetic diversity patterns according to ECM expectations are found across terrestrial and aquatic taxa in many regions of the world (Huntley and Birks, 1983; Hewitt, 1996; Williams et al., 1998; Hewitt, 2003; Provan and Bennett, 2008), there are many exceptions. Several higher latitude coastal regions in the Northeast Pacific coast remained unglaciated, serving as glacial refugia for many species (Hickerson and Ross, 2001; Hickerson and Cunningham, 2005; Marko et al., 2010). These higher-latitude glacial refugia are detectable today by the additional hotspots of genetic diversity observed across the latitudinal range of species distributions.
It is generally accepted that high genetic diversity, often found at the trailing-edge, can confer resilience to rapidly changing environments by providing the necessary variation for an evolutionary response (Reusch et al., 2005; Sgrò et al., 2011; Bernhardt and Leslie, 2013). At the leading-edge, the strength of the founder effect (Excoffier et al., 2009) will dictate how much genetic diversity is lost due to drift and inbreeding (Saccheri et al., 1998) or kept allowing for adaptation to new environments (Hughes et al., 2008). Studying the current partition of range-wide genetic diversity and differentiation informs our predictions on how species will respond to increased Anthropocene pressure (Thuiller et al., 2008). Marine ecosystems face an increased risk of degradation and species loss due to global climate change (Doney et al., 2012; Pörtner et al., 2022). Habitat destruction has already led to declines in the size of populations and increased fragmentation, resulting in reductions and shifts in species range (Poloczanska et al., 2013; Smale and Wernberg, 2013; Pinsky et al., 2020). These impacts can also result in rapid losses of intraspecific genetic diversity (Bálint et al., 2011; Pauls et al., 2013) that are better understood when the biogeographic patterns built over a much longer time-scaler are known. Although both leading and trailing-edge are predicted to shift polewards, the latter contracts the species range outside the current lower latitude distribution limit. Where genetic refugia occurred only at lower latitudes, contractions of the current trailing-edge might disproportionately impact a higher fraction of a species’ genetic diversity. With climate change predictions, populations at the trailing-edges that cannot acclimatize or adapt will depend on dispersal to higher latitudes or face extirpation. How much will species be able to naturally shift their trailing-edge and maintain the levels of genetic diversity they hold is a timely but complex question (Pinsky et al., 2020). A non-exhaustive list of determinants will be habitat size, suitability at higher latitudes, and the interplay between dispersal capacity and climate velocity, i.e., how fast changes will occur (Burrows et al., 2011; Pinsky et al., 2013). Understanding the partition of genetic diversity and differentiation will assist in the decision-making related to assisted evolution (van Oppen et al., 2015; Coleman and Goold, 2019) and migration (Schlaepfer et al., 2009; Aitken and Whitlock, 2013), as well as determine ex-situ conservation priorities (Schoen and Brown, 2001; Hoban et al., 2020).
The dispersal potential of a species can be inferred from estimates of population genetic differentiation - a measure of how gene flow across long temporal scales shapes genetic structure (Pritchard et al., 2000; Waples and Gaggiotti, 2006). This genetic structure can result from historical barriers to gene flow, range shifts, or expansions from glacial refugia (Maggs et al., 2008; Assis et al., 2014). The most apparent feature controlling gene flow is the geographic distance between populations; dispersal is typically limited by distance, and thus, geographically closer individuals are often more genetically similar than individuals further away (Wright, 1943). Geographic distance and genetic differentiation share a positive linear relationship under the isolation by distance (IBD) or the stepping stone model of IBD (Wright, 1943; Kimura and Weiss, 1964). In complex systems, however, more than geography alone is needed to predict patterns of genetic structure, and IBD models may have a poor fit for empirical data. In marine species, ocean currents are an essential means of dispersal for many marine species with pelagic dispersal and often brake the linear association of gene flow and geography (Selkoe et al., 2008; Selkoe et al., 2016). The model of isolation by oceanographic distance (IBOD) describes a pattern in which ocean currents influence gene flow more than spatial distance alone (Selkoe et al., 2008; White et al., 2010; Alberto et al., 2011; Riginos and Liggins, 2013). Therefore, modeling the oceanographic transport by ocean currents can result in a better understanding of population structure and connectivity in marine species and is particularly useful in species with limited dispersal life histories.
Although ocean currents may extend the dispersal of gametes, heterogeneity between source and destination environments can affect the establishment of migrants and decrease gene flow between different environments (Sexton et al., 2014). This pattern of isolation by environment (IBE) is characterized by an association between genetic differentiation and environmental distance independent of geographic distance (Wang and Bradburd, 2014). IBE alone does not explain the forces that drive this pattern, but IBE can be used to describe the pattern. Under certain conditions, IBE may lead to, or be caused by, isolation by adaptation, where fitness differences prevent some genotypes from persisting in some non-native environments (Nosil et al., 2008).
In the temperate reefs of the world, kelp forests provide a complex three-dimensional structure characterized by a dynamic canopy that further influences the physical conditions and ecosystem processes. This engineering role is probably the most important factor explaining the high diversity supported by kelp forests (Miller et al., 2018). Globally, recent marine heatwaves and climate forecasts have sparked concerns about the persistence of kelp forests under climate change (Wernberg et al., 2016; Assis et al., 2018; Smale et al., 2020). In the Northeast Pacific, the bull kelp, Nereocystis luetkeana, is one of two large kelp species with a canopy that reaches the surface, providing structure for complex habitat niches (Siddon et al., 2008). Due to its fast growth rate, bull kelp forests have high productivity and play a key role in nutrient cycling in coastal marine ecosystems (Foster and Schiel, 1985; Graham et al., 2008). Recent declines in bed density have caused concern for some populations, specifically in northern California (Rogers-Bennett and Catton, 2019; Arafeh-Dalmau et al., 2023), and the Puget Sound (Berry et al., 2021).
Bull kelp’s role as an ecosystem engineer, the geological history of the range it inhabits, and the regional extinction threats it faces all call for an overdue study of its genetic diversity. Our study had two main goals: 1) describe N. luetkeana genetic structure across its entire range (Alaska to California) using microsatellite markers, and 2) test different models of population connectivity within the Salish Sea, a region that was colonized after the LGM and faces increased disturbance in recent decades.
Materials and methods
Study species; the bull kelp
Nereocystis luetkeana, or bull kelp, is an annual brown alga in the order Laminariales with distribution limited to the Northeast Pacific Ocean. Like other brown algae, it has a haplodiplontic life cycle in which adult diploid sporophytes release from sori haploid zoospores that produce unisexual, haploid gametophytes. The microscopic gametophyte stage develops from the germinating zoospores after these settle on the benthos. Gametophytes reproduce sexually producing the sporophyte stage. N. luetkeana differs from other kelps in that spore-filled tissues, called sori, abscise completely from the blade and fall to the benthos releasing haploid zoospores (Walker, 1980). The blades of bull kelp are held near the surface of the water by a gas-filled, spherical pneumatocyst at the end of a long, slim stipe (~1/3 inch in diameter), attached to the substratum with a hapterous holdfast. Sori are produced near the proximal end of the blade and their abscission happens between June and November, with the species reproducing annually. Spore dispersal from sori begins shortly before abscission and continues up to an hour post-abscission (Amsler and Neushul, 1989). Although zoospores are flagellated, mobility is minimal and thus negligible in dispersal (Norton, 1992). Because sori are located near the water surface, after their abscission, the proportion of zoospores released in the water column before reaching the seafloor, which is presently unknown, may limit their dispersal potential. However, other kelp species demonstrate that long-distance dispersal can be mediated by ocean currents transporting drifting sporophytes released from the substrate during storms. This ‘rafting’ is due to the pneumatocysts that afford positive buoyancy and facilitate floating. For example, in giant kelp, Macrocystis pyrifera, adult sporophytes were observed with viable sori after traveling over long distances (Reed, 1987; Hernández-Carmona et al., 2006), and sporophylls were found to maintain buoyancy for up to 21 days (Macaya et al., 2005). Rafts of N. luetkeana have been found far from both the southern and northern edges of the species distribution (Miller and Estes, 1989; Selivanova and Zhigadlova, 1997; Gibson et al., 2010).
Sample collections and DNA extraction
We sampled fifty-three sites across the geographic range of N. luetkeana, ranging from Herring Island, Alaska (59.65°N, 151.59°W) to Cambria Bay, California (35.53°N, 121.09°W), from May 2016 to August 2017. We collected a higher sampling site density inside the Salish Sea, the inner water body composed of the Strait of Georgia in British Columbia, Canada, and Puget Sound in Washington, USA (Figure 1, Table 1). Within each site, the average number of specimens collected was 40, ranging from 7 to 51, resulting in 2,188 individuals. We sampled specimens haphazardly, separated by at least 2 meters, by cutting 2-4 cm pieces of blade tissue in a non-destructive manner. We wiped the sampled blade tissue to remove epiphytes before storing the tissue in silica gel desiccant for preservation until DNA extraction. Next, we used a Tissue Lyser II (Qiagen, Valencia, CA) to homogenize the silica-dried tissue to a fine powder before extracting DNA using the DNeasy Nucleospin 96 Plant Kit II (Machery-Nagel, Duren, Germany) following the kit protocol.
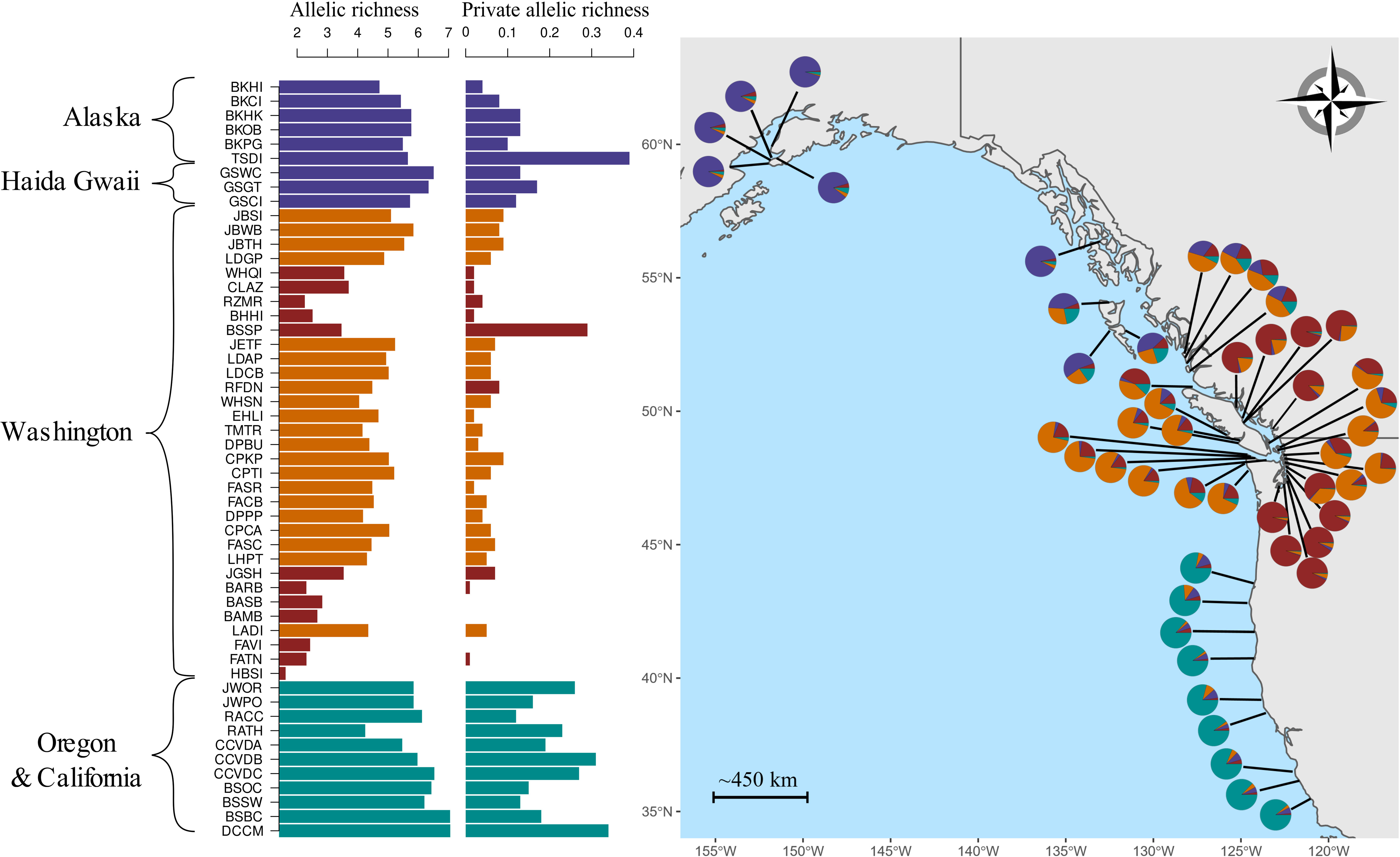
Figure 1 Genetic diversity of Nereocystis luetkeana across its distribution range in the northeast Pacific. On the right side, the sample locations are indicated on the map. For each site, a pie chart shows the average proportions of individual assignment to the four groups of genetic co-ancestry estimated with STRUCTURE. The barplots on the left side show within-population genetic diversity as measured by microsatellite allelic richness and private allele richness, standardized by a sample size of seven. Sites are ordered by decreasing latitude; the codes for each site are given with additional information provided in Table 1. The color for different bars represents the group of genetic co-ancestry (STRUCTURE) with the maximum proportion of average individual assignment for that site.
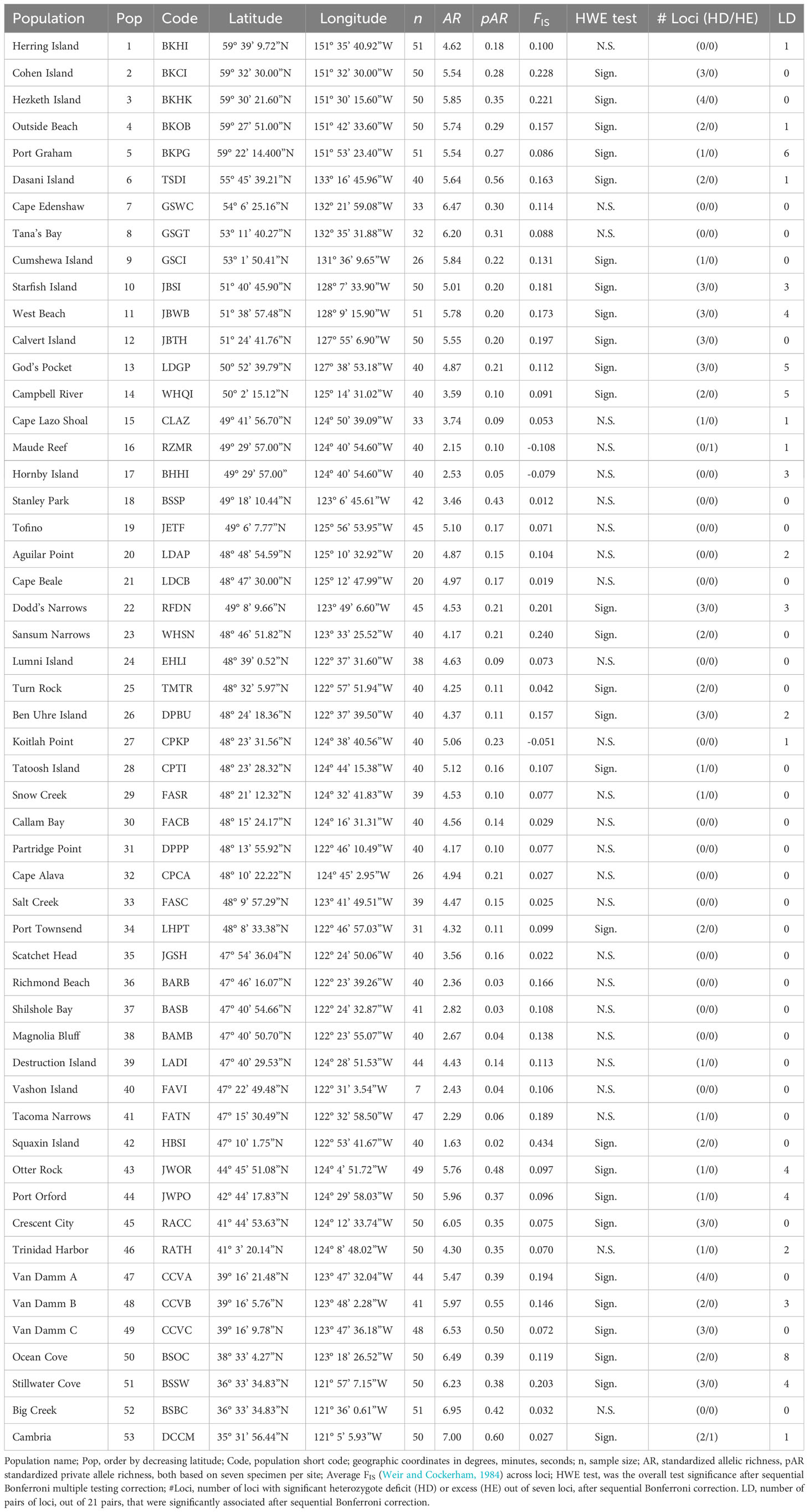
Table 1 Population genetics summary statistics for sampled sites of Nereocytis luetkeana.
Microsatellite loci genotyping
We characterized microsatellite regions for N. luetkeana (see supporting material) and used seven of the resulting microsatellite loci (Ner-2, Ner-4, Ner-6, Ner-9, Ner-11, Ner-13, and Ner-14, Table S2). We prepared PCRs in a total reaction volume of 15 µL comprised of 10 µM primer, 10 mM dNTP’s per base (Promega, Madison, WI), 25 mM MgCl2, 3.0 µl 5X PCR buffer, and 0.5 U GoTaq Polymerase. Thermocycler conditions consisted of a 5-minute denaturation step at 95°C, followed by 33 cycles of 20 seconds each at 95°C, 20 seconds at an annealing temperature of 57°C - 61°C, 30 seconds at 72°C followed by a final elongation step of 20 minutes at 72°C using an Eppendorf thermocycler (Eppendorf, USA). We sized microsatellite PCR fragments using fragment analysis on a 96-capillary DNA sequencer ABI 3730xl at the Madison Biotechnologies Center. We scored the resulting microsatellite fragments with STRand (https://www.vgl.ucdavis.edu/informatics/strand.php) and binned them into discrete allele sizes with the R (R Core Team, 2021) package “MsatAllele” (Alberto, 2009). The presence of null alleles was evaluated with MICRO-CHECKER v.2.2.3 (Van Oosterhout et al., 2004).
Population genetics summary statistics
Genepop version 4.2 (Rousset, 2008) was used to estimate and test linkage disequilibrium between loci (LD), test for Hardy Weinberg Equilibrium (HWE) within all populations, and estimate genetic differentiation between populations (pairwise FST). HWE and LD tests were interpreted after a sequential Bonferroni correction for multiple testing was applied to α values (Holm, 1979). An additional genetic differentiation metric was estimated, Jost’s DEST, using the R package “diversity” (Keenan et al., 2013). Jost’s DEST is included because it is not affected by differences in within-population heterozygosity like FST (Jost, 2008). Finally, we estimated allelic richness and private allele richness standardized for a sample size of seven individuals (the minimum among sampled sites) using HP-RARE (Kalinowski, 2005). Using the same approach, regional allele and private allele diversity were estimated for the genetically differentiated groups estimated below.
Range-wide genetic differentiation
We used the program STRUCTURE to characterize large-scale patterns of genetic differentiation across the entire distribution of N. luetkeana (Pritchard et al., 2000). We used the admixture model for all runs with allele frequencies correlated among populations with an initial burn-in of 250 000 steps and 750 000 (Monte Carlo Markov Chain) repetitions. We ran ten runs per k (number of genetic co-ancestry groups) and let k range from 1 to 8. We then used the Puechmaille method in StructureSelector to determine k (Puechmaille, 2016; Li and Liu, 2018). This method accounts for varying sample sizes between populations. Finally, STRUCTURE outputs were submitted to CLUMPAK to align runs for each k value (Kopelman et al., 2015). To visualize the relatedness between different groups of genetic co-ancestry, we produced a network based on Cavalli-Sforza and Edwards chord distances between sites, using R packages “adegenet” (Jombart, 2008) and “igraph” (Csardi and Nepusz, 2006). The network was pruned by sequentially removing edges with decreasing genetic distance up to one step before two separate networks of connected sites formed.
Additionally, to test for isolation-by-environment (Wang and Bradburd, 2014) that could explain the genetic clusters estimated by STRUCTURE analysis, we ran a canonical correspondence analysis (CCA) in R package “CCA” (González et al., 2008). The response variables were the assignment probabilities (Q-values) for each site to belong to each of four genetic co-ancestry groups detected by STRUCTURE (see results). The environmental predictor variables were site-specific values obtained from NASA’s MODIS AQUIS satellite for photosynthetically active radiation (PAR), particulate organic carbon (POC), summer sea surface temperature (SST), spring SST, fall SST, winter SST, light attenuation (measured as kd490) and chlorophyll-a (Chl a). We obtained monthly environmental data from 2002 to 2016, collected by NASA’s MODIS satellite and accessed through ERDDAP (https://coastwatch.pfeg.noaa.gov/erddap). We used long-term average and seasonal variation (for SST) conditions, hypothesized to affect the physiology of bull kelp. In addition, as predictor variables, we included the latitude and longitude of each site to control for spatial autocorrelation. We used a step-wise model-building approach in the R package “vegan” (Oksanen et al., 2022) to select the best model for the data and used variance partitioning to quantify the variation explained by the tested variables (Borcard et al., 1992; Økland and Eilertsen, 1994).
Seascape genetics in the Salish Sea
The seascape genetics analysis detailed below was only performed for the Salish Sea and adjacent North East Pacific coast sites where we had a higher sampling density and an available hydrodynamic transport model (Yang and Khangaonkar, 2010).
Defining an environmental distance between sites
Environmental data were obtained from NASA’s MODIS satellite and accessed through ERDDAP. We characterized pairwise environmental distances using the absolute difference between sites for each of eight variables: photosynthetically active radiation (PAR), particulate organic carbon (POC), particulate inorganic carbon (PIC), summer sea surface temperature (SST), spring SST, fall SST, winter SST, light attenuation (kd490) and chlorophyll-a (Chl a).
Characterizing an oceanographic distance
We used the Salish Sea Model (Yang and Khangaonkar, 2010) to simulate particle transport times in the General NOAA Operational Modeling Environment, GNOME (Zelenke et al., 2012), with the intent of estimating oceanographic distance. The average distance between sampled sites often exceeds the species dispersal in a single generation. This constitutes a challenge when using hydrodynamic models to estimate single-generation dispersal because most pairwise site connectivity values would be zero, thus not informative on pairwise site genetic differentiation. One way to tackle this limitation is to consider multiple-generation stepping-stone gene flow to estimate connectivity appropriately, i.e., two sites may be connected through intermediate site(s) across an undetermined numbers of generations (Yang et al., 2016). This approach requires simulating transport from additional kelps beds than just those sampled, hereafter referred to as stepping stone connectivity sites (SSC-sites). We selected SSC-sites using historical kelp bed maps provided by the British Columbia Coastal Resource Information Management System (https://forms.gov.bc.ca/databc-data-request/). Using QGIS Geographic Information System (http://www.qgis.org), we overlaid a 25 km grid over the map of historical kelp bed cover and sampling sites. We selected SSC-sites from the center of grid cells that contained kelp but where no sample had been collected for this study. Therefore, 42 SSC-sites and 33 sample sites were considered (Supplementary Figure S1). Using the GNOME modeling platform, one thousand particles were released from each site and tracked for 14 days, assuming the same spore longevity as Macrocystis pyrifera (Macaya et al., 2005). Spore releases were done separately for 13 periods (every 14 days long) from June to November to cover the season when sori are observed in N. luetkeana (Maxell and Miller, 1996). We estimated the probability of transport between pairs of sites as the proportion of particles released from the source site i that made it to the destination site j. From these transport probabilities, we estimated stepping stone connectivity between sampling sites using a directed network created using R package “igraph” (Csardi and Nepusz, 2006). Network nodes were composed of sample and SSC-sites while edges (the links between nodes) had weights representing the log probability of transport between sites. The shortest path along the network between all pairs of sample sites was found using Dijkstra’s algorithm (Dijsktra, 1959) in igraph. Finally, the directional stepping-stone connectivity between site i and j was measured by summing the log-transformed transport probabilities along the shortest path (Hock and Mumby, 2015). Because this is an asymmetric distance (both i to j and j to i), we used the mean value to obtain a single pairwise distance value.
Modeling genetic differentiation
We fitted a multiple linear regression model to estimate how pairwise genetic differentiation, as measured by FST, could be explained by over-the-water Euclidean distance (isolation by distance, IBD), environmental distance (isolation by environment, IBE) and oceanographic distance (isolation by oceanographic distance, IBOD). Multiple linear regression models were run in R and Akaike information criterion (AIC) to select the best model by eliminating all predictors that had a poor fit with genetic differentiation. A hierarchical partition of variance was used to estimate the proportion variance in genetic differentiation explained independently by the significant predictors remaining in the optimized model. Multicollinearity among predictor variables was checked using the variance inflation factor (VIF).
Results
Allele diversity and within-population genetic analysis
On average, the seven microsatellite markers amplified 25 alleles across the 53 populations, with a maximum of 40 for Ner-11 and a minimum of 14 for Ner-19. Mean allelic richness (AR), standardized to 7 individuals per site, ranged between 1.6 and 7.0 alleles·locus-1, with the highest values observed in central California at the southern limit of the species distribution (Figure 1). Lower AR values were observed in the inner regions of the Salish Sea, with the lowest value of 1.6 found in Squaxin Island (HBSI), the Puget Sound site farthest from the ocean. Generally, a decreasing AR pattern was observed with increasing isolation from the ocean into the Salish Sea (Figure 2). We found intermediate AR for the oceanic coast of British Columbia and Washington. At lower latitudes in Oregon and California, AR increased, reaching maximum levels at the southernmost sites in California (Figure 1). AR values also increased north from the Washington state region in Haida Gwaii, where it was near maximum, and in Alaska at the northern extent of the distribution. Genetic differentiation for all populations, as measured by global FST, was 0.1567.
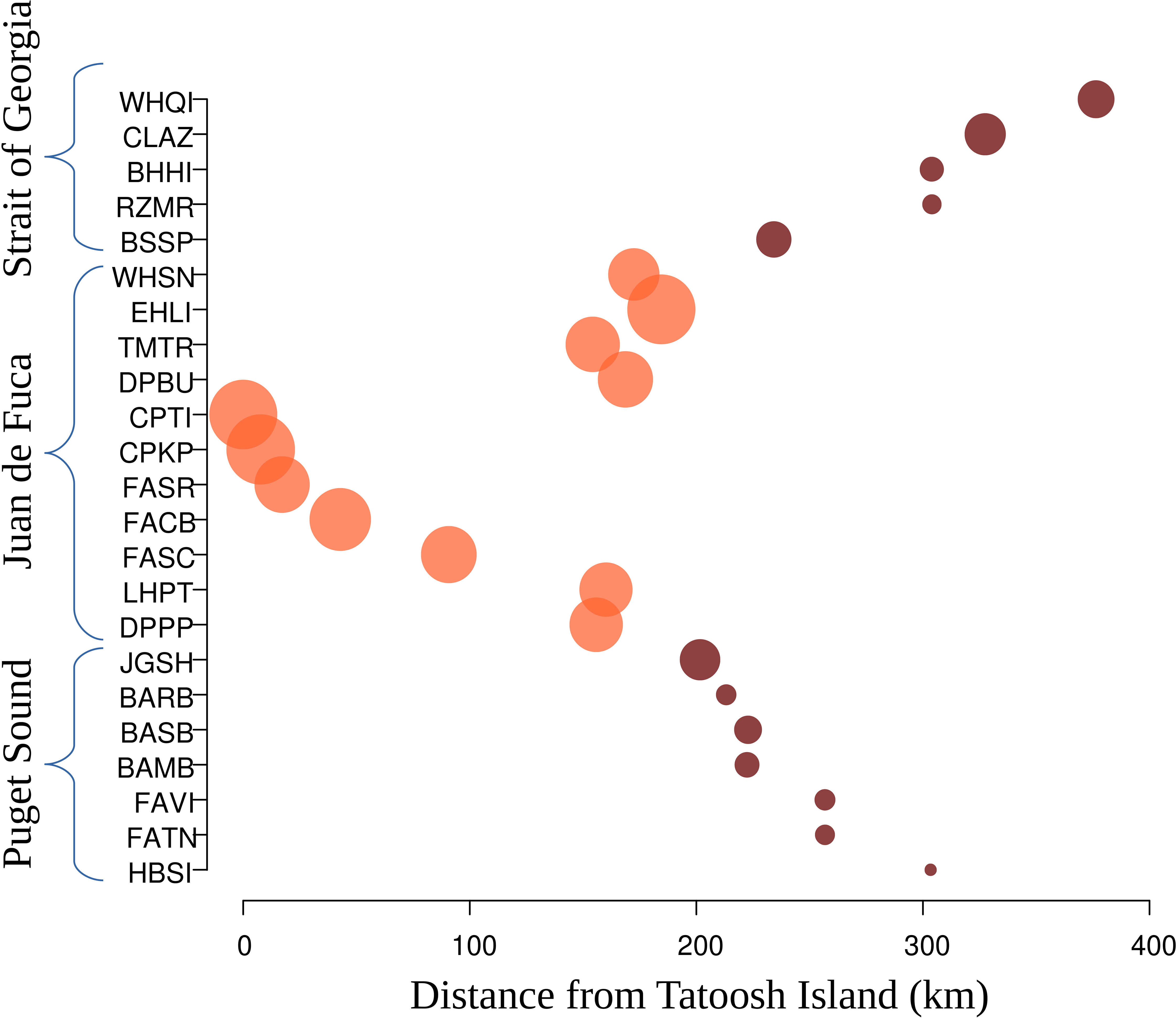
Figure 2 Loss of microsatellite allelic richness (circle area), across Nereocystis luetkeana Salish Sea sites, with increasing site distance from the ocean. Population names are ordered by latitude on the y-axis. Distances on the x-axis are measured over the water from Tatoosh Island, located at the entry of the Strait of Juan de Fuca. Circle colors represent the dominant STRUCTURE group of genetic co-ancestry for each population (see Figure 3). In Puget Sound, allelic richness decreases with increasing distance into the system to reach a minimum (across all sampled populations) in Squaxin Island. In the Strait of Georgia, allelic richness is lowest in the middle region and recovers in the northernmost sites, likely due to the open connectivity with the ocean provided by Discovery Passage.
Given the patterns of genetic differentiation reported below, standardized regional private allele richness, using three populations, was estimated in HP-RARE for Alaska; Haida Gwaii; Pacific coast of Washington including the Strait of Juan de Fuca; Salish Sea; and Oregon combined with California. California and Oregon had the highest average number of private alleles (1.67), followed by Alaska (1.23), Haida Gwaii (1.00), coastal Washington and Juan de Fuca (0.54), and Salish Sea (0.36). Private allele richness (pAR) was also estimated at the site level (Figure 1). The only site sampled in southern Alaska, Dasani Island, part of the larger Prince of Wales Island (TSDI), showed high pAR that exceeded the values observed in all Haida Gwaii sites (Figure 1). In the Salish Sea, the kelp bed in Stanley Park (Vancouver) also had higher pAR than all other Salish Sea sites. Cambria, the site closest to the southern distribution edge in California, had the highest pAR across all sites.
About half of the sites (50.9%) had significant heterozygote deficiency, as indicated after Bonferroni correction was applied to the exact test results. Within these sites, the number of markers with significant FIS ranged from one to four out of seven (Table 1). The correlation coefficient (r) between the number of makers showing positive FIS and flagged for potentially having null alleles (MICRO-CHECKER) was 0.74, while r between the former and the number of loci pairs with significant linkage disequilibrium was only 0.27. After sequential Bonferroni correction, the maximum number of pairs of loci with significant LD within a population was 8 (out of 21), observed in BSOC. Only eight sites had more than three pairs of loci in LD, whereas 31 sites had zero. The pair of loci Ner-11 and Ner 14 had the most significant LD cases in 11 sites (out of 53), whereas Ner-11 and Ner-13 were significant in 13 sites. Only four pairs of loci (out of 21 pairs) had significant LD in more than four sites.
Range-wide genetic differentiation
STRUCTURE analysis estimated four (k) groups of genetic co-ancestry distributed geographically coherently along the entire sampling distribution (Figures 1, 3). An exception to the continuous distribution of these groups was a genetic break observed between the group made by the innermost regions of the Strait of Georgia and Puget Sound (Figure 1). This inner Salish Sea genetic group, characterized by low genetic diversity, was interrupted by sites located in the adjacent eastern areas to the Strait of Juan de Fuca (SJF), which were assigned to the SJF and outer coast dominant group (Figure 4). In addition, we observed two other groups of genetic co-ancestry, one with all sampled Alaska sites and another with sites from Oregon and California. This southernmost group span over 1,000 km across areas of discontinuous habitat. An admixture of genetic co-ancestry was apparent from Haida Gwaii Island to northern Vancouver Island (Figure 1).
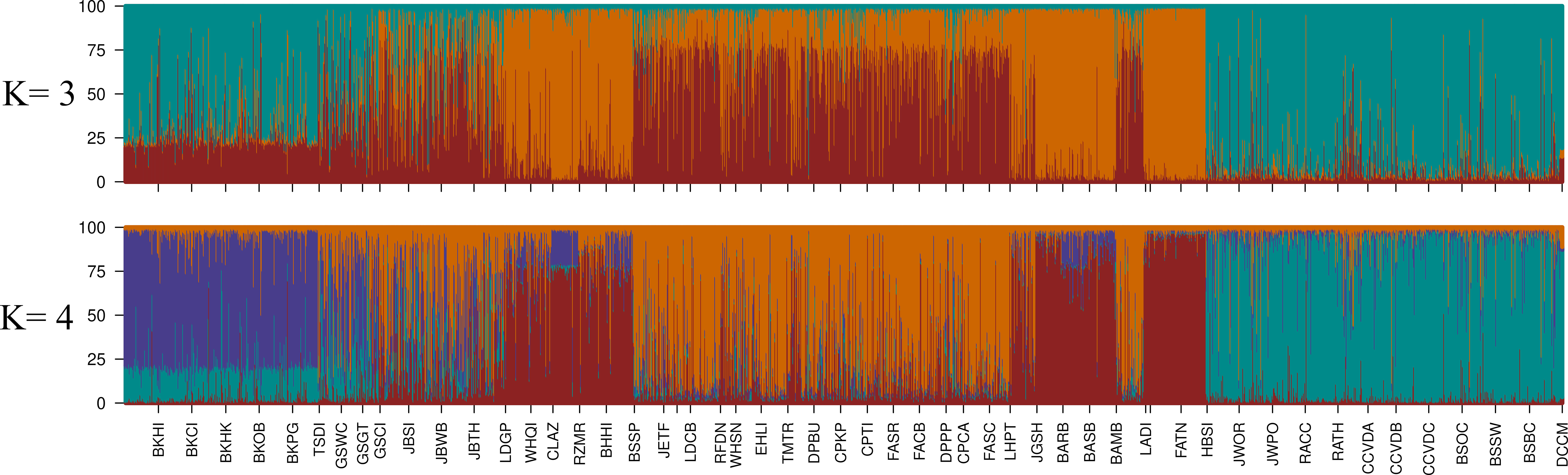
Figure 3 STRUCTURE plots for k=3 and k=4 where k is the number of co-ancestry groups, each represented by a different color. Each specimen genotyped has a stacked vertical bar indicating the proportion of its genome assigned to the different co-ancestry groups. Sites are ordered left to right according to their latitude. The Puechmaille method (see methods) provided support for k=4 co-ancestry groups. Each bar represents genetic co-ancestry membership for an individual, and the solid bar at the bottom indicates the co-ancestry group with the maximum mean individual assignment for each site.
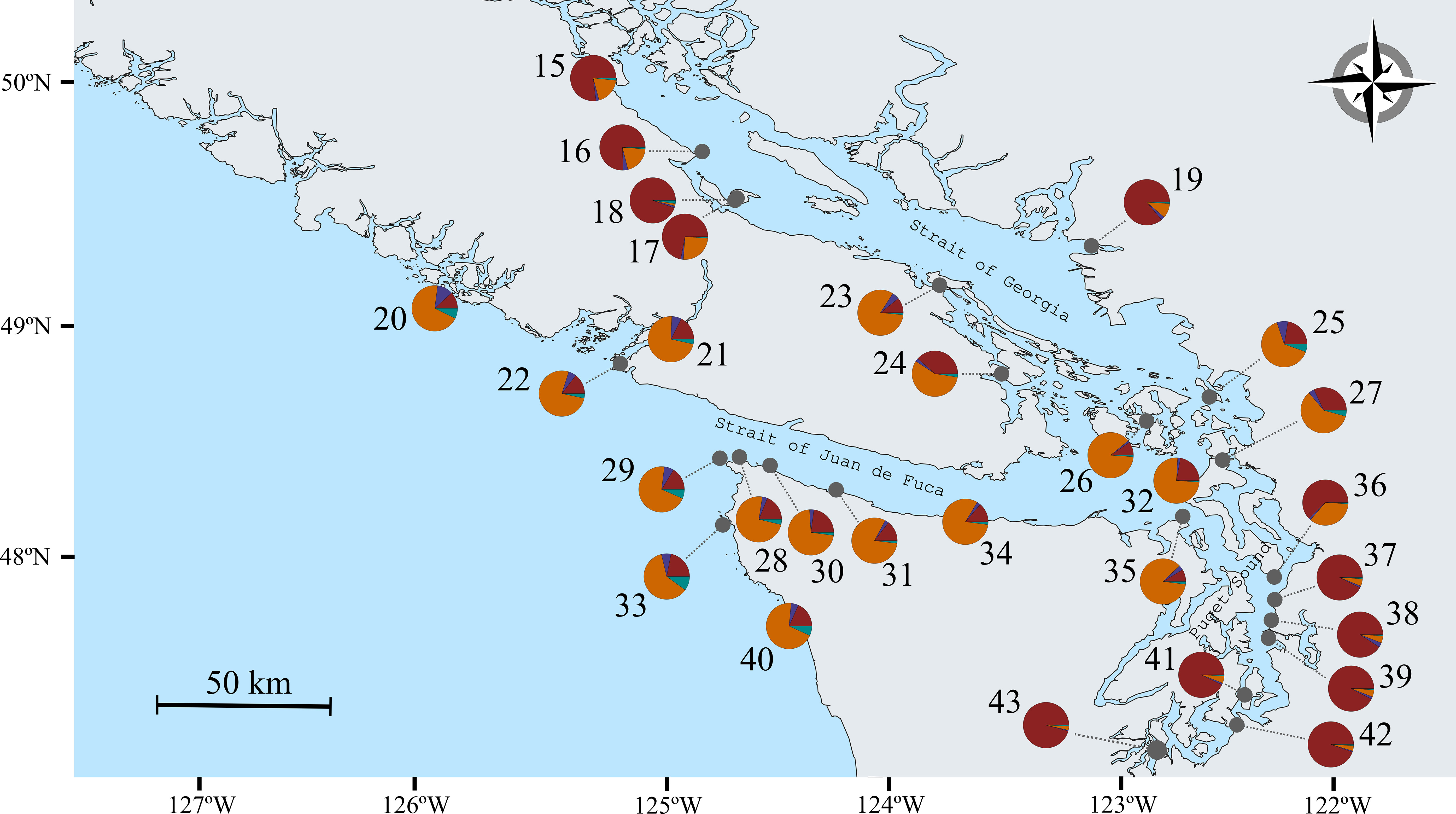
Figure 4 Detail of sampling locations of Nereocystis luetkeana in the Salish Sea and adjacent areas. For each sample site, a pie chart shows the average proportions of individual assignment to the four groups of genetic co-ancestry estimated with STRUCTURE. A pattern of genetic differentiation between the inner regions of the Salish Sea and the areas adjacent, or belonging to, the Strait of Juan de Fuca and the outer coast was apparent.
To test the isolation by environment model, we used CCA to explore associations between environmental predictors and membership to genetic co-ancestry groups as determined by STRUCTURE analysis for k=4 co-ancestry groups (Figure 5). Model selection resulted in latitude, longitude, PAR, light attenuation, and Chl a as significant predictors associated with mean population proportion assignments to groups of genetic co-ancestry. Light attenuation was not associated with latitude and longitude, while PAR was. However, when spatial auto-correlation (latitude and longitude) was controlled for, light attenuation, PAR, and POC were important variables separating the two STRUCTURE clusters from the Salish Sea and outer coastal area. Variance partitioning showed that environmental predictors and geography explained 70% of the variance. Of this explained variance, 3% was explained by geography, 19% by environmental variables, and 75% was jointly explained by both. When geography was controlled, POC, PAR, light attenuation, and Chl a remained significant predictors of population assignment to the different clusters. Under the same model controlling for spatial auto-correlation, Alaska and Oregon/California co-ancestry groups clustered closer to each other, despite being the most geographically separated groups, whereas the inner Salish Sea and outer coastal Washington/British Columbia remained separated (Figure 5B). The relative genetic relatedness between Alaska and the Oregon/California groups, the two groups with the highest AR, was also apparent in a genetic distance network (Figure 6).
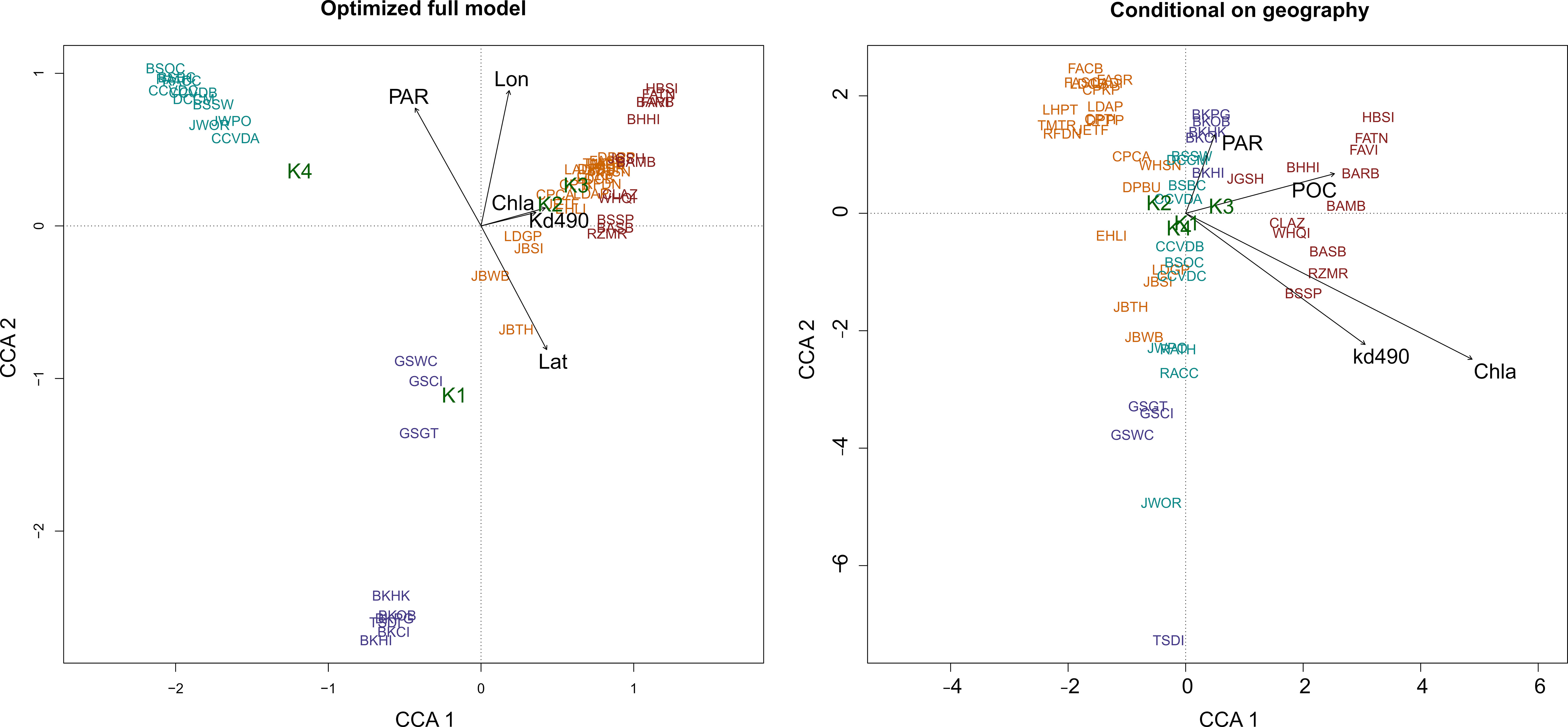
Figure 5 Canonical correspondence analysis showing the association of STRUCTURE assignments to the four groups of bull kelp genetic co-ancestry (response variable) and the following predictor variables: photosynthetically active radiation (PAR), particulate organic carbon (POC), summer sea surface temperature (SST), spring SST, fall SST, winter SST, light attenuation (kd490) and chlorophyll-a (chl a). Site names are colored by the dominant group of STRUCTURE assignment: Alaska (purple), outer coastal Washington, British Columbia, Strait of Juan de Fuca, and adjacent areas of Salish Sea (orange), inner Salish Sea (red); and Oregon and California (teal). Site name abbreviations are found in Table 1, and information on predictor variables is found in the Methods.
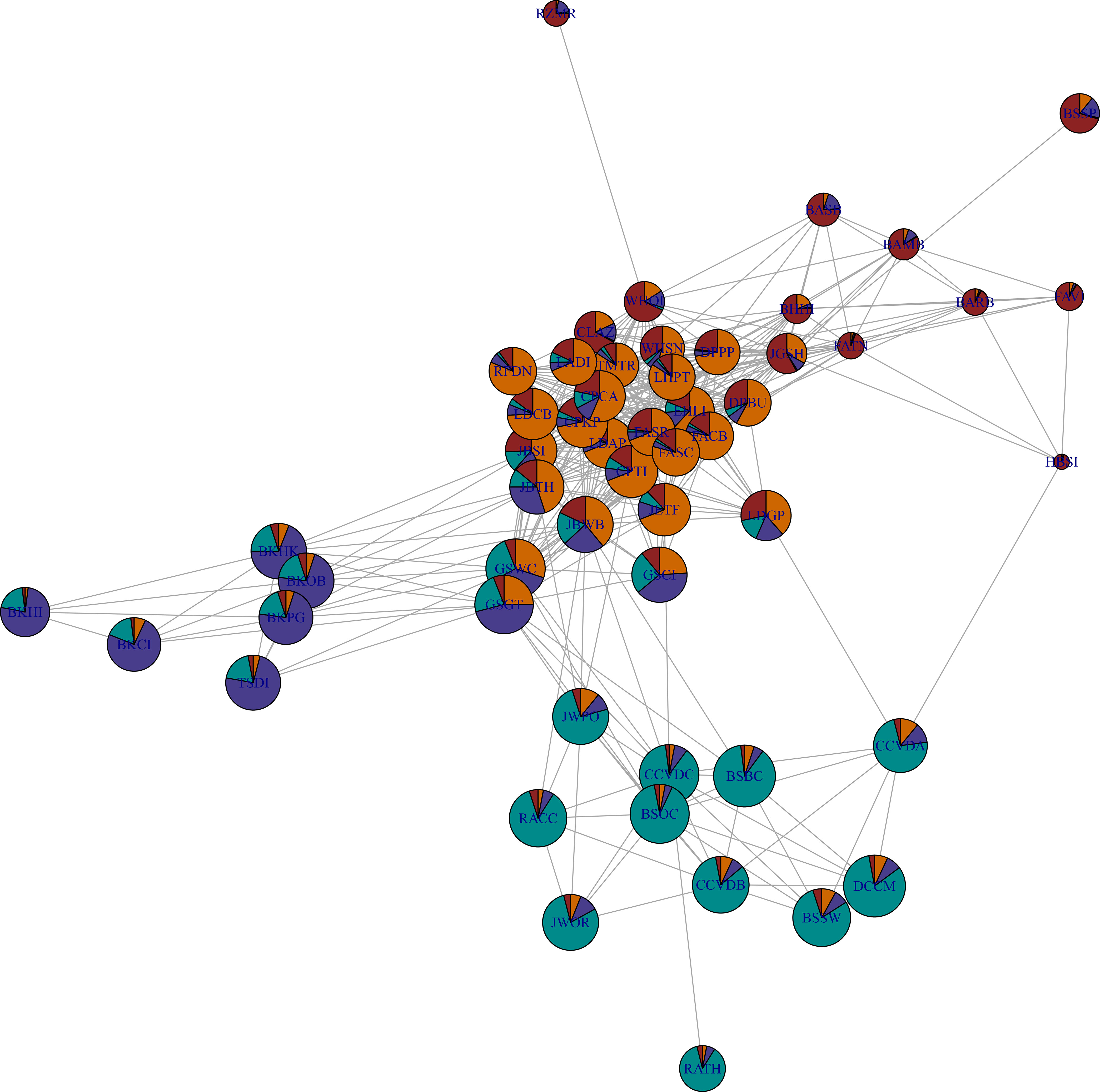
Figure 6 Genetic differentiation network for Nereocystis luetkeana sites sampled across the species distribution range. Network topology representing pair-wise Cavalli-Sforza and Edwards chord distance between sites. The network was pruned (removal of edges with higher genetic distances) until one step before two clusters of connected sites would be formed. Pie charts for each site show the mean individual assignment to four groups of genetic co-ancestry (same colors as used in other figures). The pie-chart area represents site allelic richness (Figure 1). Genetic distances were calculated with R package adegenet and network produced with R package igraph.
Seascape genetics in the Salish Sea and adjacent areas
Multiple regression analysis identified oceanographic connectivity, spatial distances, and environmental variables as predictors of genetic differentiation within the Salish Sea (Table 2). Temporal variability in oceanographic connectivity revealed slight differences between the different 14-day periods evaluated (Supplementary Table S1). The final optimized multiple regression model included spatial and oceanographic (from July 15th to the 29th period) pairwise distances between sites as predictors of FST. Likewise, light attenuation (kd490); Chl-a; summer, fall and winter SST; PIC; and POC were the environmental distances retained in the final model (Figure 7). Hierarchical partitioning of the model explained variance showed that the largest contributions were made by oceanographic distance (21.4%), PIC (16.7%), summer SST (13.6%), spatial distance (12.2%), POC (10.5%) and light penetration (9.3%). These predictors were positively associated with increased genetic differentiation (FST), whereas PAR, fall SST and winter SST were negatively associated. The predictors with negative partial regression slopes explained small % of the total variance (< 6%). There was no multicollinearity among the predictors retained in the final model (VIF ranged from 1.2 to 2.5). The final multiple regression model explained 40% of the genetic differentiation in pairwise FST between sites in the Salish Sea and adjacent areas.
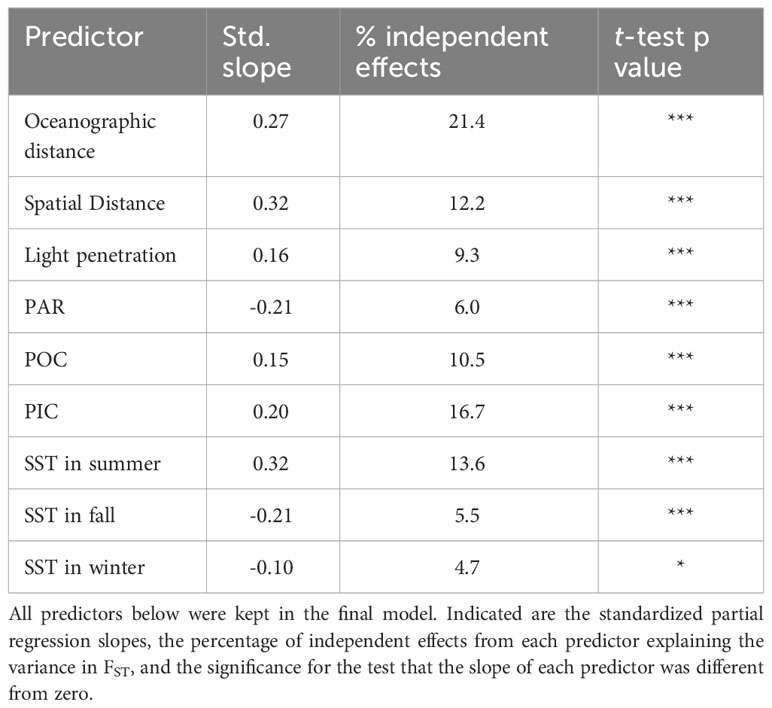
Table 2 Optimized multiple regression model for Nereocystis luetkeana pair-wise genetic differentiation (FST).
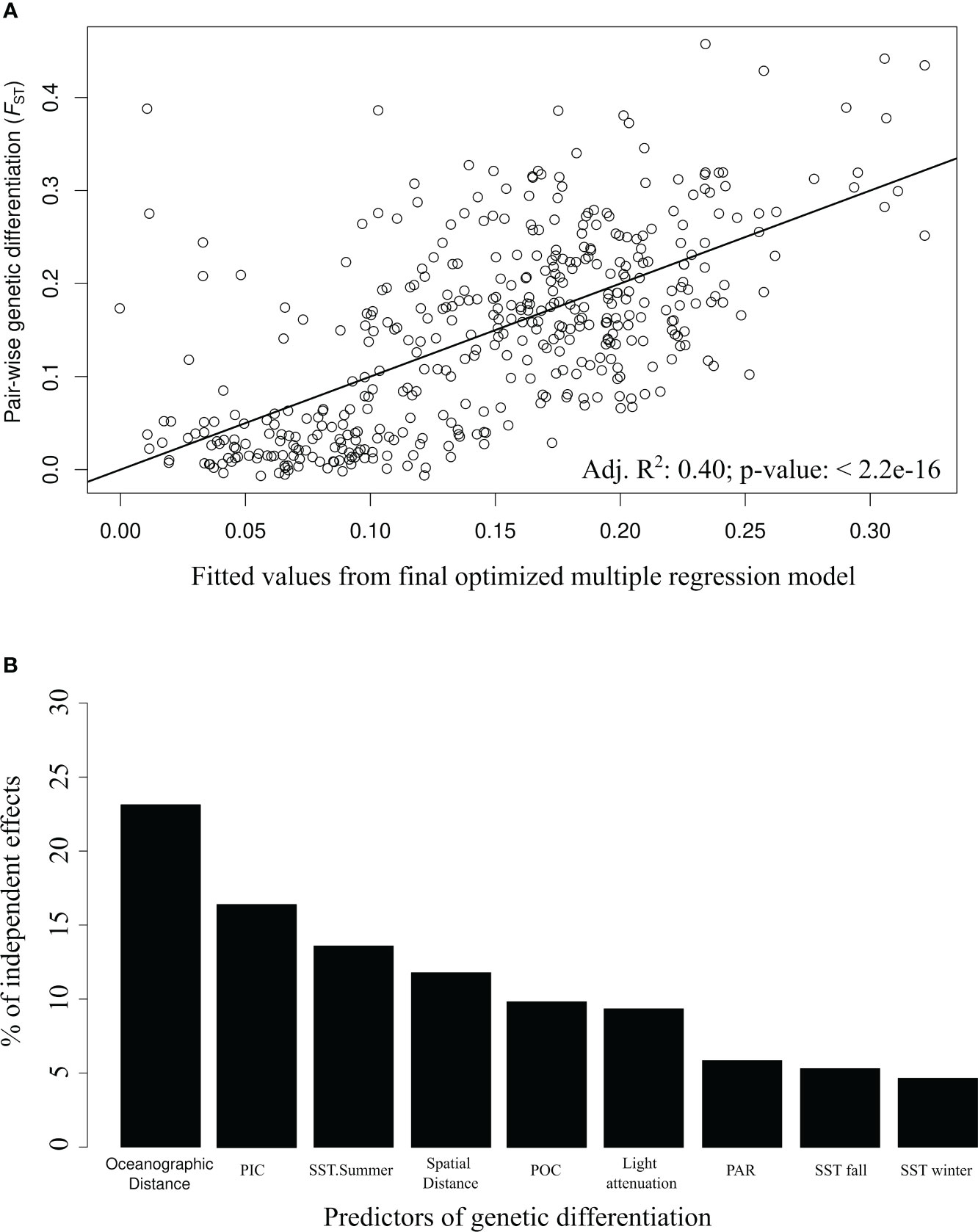
Figure 7 Optimized regression model for Nereocystis luetkeana pair-wise genetic differentiation (FST) in the Salish Sea and adjacent areas (A). Only predictors with significant associations remained in the final model. Their percentage of independent effects on the response variable was estimated using R package hier.part (B).
Discussion
Range-wide genetic structure and diversity
The species distribution level analysis of Nereocystis luetkeana revealed four genetic co-ancestry groups with coherent geographic distribution across the species range. Maximum allelic and private allele richness were observed within the southernmost of these groups, at the southern range of the distribution in central California. However, high allelic and private allele richness were also found in the northern part of the species range in Prince of Wales Island (southern Alaska) and Haida Gwaii. These patterns of genetic structure and diversity support the hypothesis that during the last glacial maximum (LGM) N. luetkeana had both southern and northern glacial refugia. During this period, much of the coastal Pacific Northwest was glaciated (Mann and Hamilton, 1995; Jacobs et al., 2004) aside from small areas of glacial refugia where species could persist (Blanchette et al., 2008; Shafer et al., 2010; Ager, 2019). At the southern edge of the Alaska group, Haida Gwaii had close to maximum values of allele richness, while the relatively close Prince of Wales Island site of Dasani Island had the highest private allele diversity in the northern group and second highest across all sites. Similar patterns have been recorded in other species and used to support the hypothesis of a northern refugium (Warner et al., 1982; Byun et al., 1997; Soltis et al., 1997; Jacobs et al., 2004; Carrara et al., 2007; Lindstrom, 2009; Shafer et al., 2010). This hypothesis is supported by the likelihood of ice-free conditions (Warner et al., 1982; Mann and Hamilton, 1995) and animal and plant morphological characteristics distinct from those of mainland counterparts (Clarke et al., 2001). The northern part of Vancouver Island also showed high allelic richness. The continental shelf between Haida Gwaii and Vancouver Island remained ice-free and terrestrial (Guilbault et al., 2003) up until the late Pleistocene when glaciers retreated, and sea level rise filled the area between Haida Gwaii and Vancouver Island. The documented ice-free conditions (Mann and Hamilton, 1995) and similar genetic admixture found in our study between these two islands suggested historic connectivity and a potential recolonization pathway originating in Haida Gwaii and extending into the Salish Sea. Higher allelic richness found further north in the Alaskan Kenai Peninsula, could have resulted from an earlier deglaciation (16,000 ybp) of this area when compared to Washington (14,000-13,000 ybp), allowing for an earlier range expansion north from the Prince of Wales Island and Haida Gwaii refugia (Mann and Hamilton, 1995). Based on molecular evidence, northern refugia for other kelp species have been recently proposed further north in the Gulf of Alaska Bay (Grant and Bringloe, 2020; Grant et al., 2020; Grant and Chenoweth, 2021).
The highest allelic richness values observed in central California just north of Point Conception, at the southern limit of the species distribution, are likely due to a southern glacial refugium resulting from the extension and contraction of species distribution during the glacial-interglacial cycles. This pattern of maximum genetic diversity at the southern ranges of species distribution is well documented in the Northern Hemisphere across many taxa (Bennett et al., 1991; Hewitt, 1996), including seaweed species, such as Fucus serratus (Hoarau et al., 2007), Macrocystis pyrifera (Johansson et al., 2015), Saccorhiza polyschides (Assis et al., 2016), and across taxa that maintained southern refugia during the LGM (Maggs et al., 2008). A single genetic co-ancestry group was estimated for California and Oregon sites. Interestingly, the separation between this group and Washington sites matches the southern limit of continental glaciation (Dyke, 2004). On land, a secondary biogeographic transition zone, the “Soltis line”, has been found across many taxa (Soltis et al., 1997; Wilke and Duncan, 2004; Miller et al., 2006; Jaramillo-Correa et al., 2009). The possible causes for this discontinuity have been proposed to be northwards dispersal during the interglacial and the existence of genetically distinct glacial populations, including the Haida Gwaii islands (Shafer et al., 2010).
When the number of STRUCTURE co-ancestry groups was constrained to three (one below the most supported number of four), Alaska, Oregon and California sites all grouped together. This higher genetic relatedness between the southern and northernmost groups, than between them separately with the Washington groups, was also evident from the CCA, and network analyses. Allelic richness was higher in the California-Oregon and the Alaska-northern British Columbia groups than in Washington, where the inner Salish Sea had the poorest allelic richness observed across the distribution range. Private allele richness was also the lowest in the Salish Sea with an exception found for Stanley Park site (Vancouver). This pattern supports that outer and inner Salish Sea bull kelp descend from a post-glaciation colonization event and thus only represent a subset of the historical diversity (a founder’s effect). The high private allele richness at Stanley Park could be evidence that the overall genetic diversity representative of the post-glacial colonization might have been higher and has since been lost due to the small population size and isolation in the Salish Sea.
The coastal extent covered by bull kelp populations sharing the same dominant group of genetic co-ancestry was around ~1,000km for both Alaska and Oregon-California groups. We note that these represent groups sharing a more recent common ancestor, not present-day panmictic populations. The giant kelp, Macrocystis pyrifera, a species with similar life history characteristics (e.g., haplodiplontic with spore dispersal) and northern hemisphere distribution, is also characterized by long swats of coast where a single dominant ancestral genetic group dominates (Johansson et al., 2015). These spatially extensive genetic co-ancestry groups spanning discontinuous habitat suggest that multiple-generation, stepping-stone dispersal distances are sufficient to balance the effects of genetic drift that otherwise further isolate populations (Reed et al., 1988; Alberto et al., 2010). This extended dispersal capability may be due to the rafting of adult sporophytes – a mechanism proposed for several kelps that could also occur in N. luetkeana (Macaya et al., 2005; Hernández-Carmona et al., 2006; Waters et al., 2018; Fraser et al., 2022). Rafts of N. luetkeana have been found far from both the southern and northern edges of the species distribution (Miller and Estes, 1989; Selivanova and Zhigadlova, 1997). In Macrocystis pyrifera, adult sporophytes have viable sori after traveling over long distances (Reed, 1987; Hernández-Carmona et al., 2006), and sporophylls were found to maintain buoyancy for up to 21 days (Macaya et al., 2005). Raft dispersal has been suggested across other kelps (Waters et al., 2018; Fraser et al., 2022).
In general, patterns of genetic differentiation for intertidal species in the Pacific Northeast are associated with environmental variation and oceanographic transport. Modeling by Fenberg et al. (2015) identified biogeographic structure for rocky intertidal species, including a main biogeographic break where the North Pacific Current diverges into the California Current and Alaska Current just south of Haida Gwaii Island, which is consistent with data for the California sea cucumber (Xuereb et al., 2018). This diverging current does not appear to be a barrier to gene flow in N. luetkeana, as Haida Gwaii and Vancouver Island share similar genetic co-ancestry and are admixture centers, suggesting successful gene flow from multiple populations. Particle transport models by Robinson et al. (2005) found that sea surface currents would allow drifting individuals to be carried between the two islands.
Bull kelp dispersal and mating system
Bull kelp in Alaska and Oregon-California sustain high genetic diversity and low within-region genetic differentiation, suggesting an association between dispersal capacity, large effective population sizes, and outcrossing. At the same time, selfing was supported by significant heterozygote deficiencies at many sites (positive FIS). Generally, there was no regional association with FIS, except in the Salish Sea, where many non-significant values were observed. Given the extremely low diversity in the inner sites within the Salish Sea, higher biparental selfing is expected in the region. However, the fact that many loci are fixed for dominant alleles at these sites precludes the detection of heterozygote deficiency. Selfing in this mixed mating system species was expected from the unique abscission of the entire ripe sori from blades when spore release starts and the fast rate at which spores are released compared to related species (Amsler and Neushul, 1989). However, this mechanism also makes it possible for spores to be released along a sporophyte’s entire vertical distribution, which will maximize the dispersal distance of a fraction of spores. Amsler and Neushul (1989) suggested that bull kelp could disperse some propagules while maintaining others near the environment where the parents have successfully matured. This prediction is supported by the within and between-site genetic diversity pattern found here. The bull kelp sporophyte is largely annual, and its mixed mating system is likely a critical reproductive assurance trait important for species coping with high environmental and demographic variation (Bell et al., 2023; Cavanaugh et al., 2023).
The association between dispersal and mating system is often described between two opposite evolutionary syndromes. On the one hand, a selfing and not dispersing strategy is proposed for species with low or absent inbreeding depression and a predictable environment where locally adapted gene associations are maintained. On the other hand, an outcrossing and dispersing strategy is proposed for species facing inbreeding depression and environmental heterogeneity (Auld and Rubio de Casas, 2013, Cheptou, 2012). Additionally, a “bet-hedging” strategy has been proposed for species with intermediate levels of self-fertilization and predominant local dispersal but maintaining some long-distance dispersal (Cohen, 1966, Simons, 2011). Inbreeding depression is central to the theory behind the maintenance of mixed mating systems due to selection against reduced fitness (Charlesworth and Charlesworth, 1987). Thus, understanding the evolutionary advantages of the unique bull kelp sori abscission, maintenance of a mixed mating system, and long-distance dispersal will require studying inbreeding depression (Raimondi et al., 2004; Barner et al., 2011), as well as the pattern of local adaptation and reproductive assurance at the metapopulation scale (Dornier et al., 2008). In other related kelp species, contrasting results have been found regarding inbreeding depression. The sea palm Postelsia palmaeformis has strong genetic differentiation between populations, a mating system strongly reliant on selfing for reproductive assurance and no inbreeding depression (Kusumo et al., 2006; Barner et al., 2011). In giant kelp, Macrocystis pyrifera, the mating system is more reliant on outcrossing and dispersal capacity, as suggested by lower FIS in most sites (Johansson et al., 2015) and evidence for inbreeding depression (Raimondi et al., 2004). Given our results and bull kelp life history traits we posit that its mating system is intermediate to P. palmaeformis and M. pyrifera. Future studies on the species inbreeding depression will inform our fundamental understanding of kelp life history variation and directly apply to the growing restoration and breeding efforts targeting this kelp.
Patterns of genetic differentiation within the Salish Sea
In the Salish Sea, we observed a disjunct population structure where the inner Puget Sound and Strait of Georgia sites were genetically differentiated (and had lower genetic diversity) from those in the outer coast, Strait of Juan de Fuca, and adjacent Salish Sea region. This pattern does not follow a simple isolation by distance (IBD) model of genetic differentiation because some sites directly adjacent (e.g., Admiralty Inlet and Suan Juan Islands) transition sharply between co-ancestry groups. This disjunctive pattern within the Salish Sea is inconsistent with similar studies on fish species that have mobile larval stages, such as the Pacific hake, Merluccius productus (Iwamoto et al., 2015) and copper rockfish, Sebastes caurinus (Buonaccorsi et al., 2002). These studies support the hypothesis that currents in the Strait of Juan de Fuca are a barrier to gene flow between these inner waterways. The Strait of Juan de Fuca is a highly dynamic environment with large fluxes of water movement each day (Khangaonkar et al., 2018) that could limit gene flow between the Strait of Georgia and the inner regions of Puget Sound.
The inconsistency between fish models and N. luetkeana is that within the Puget Sound and Strait of Georgia bull kelp colonization post-glaciation resulted in a founder’s effect and reduction of genetic diversity. This hypothesis is supported by allelic richness values that show a stepwise reduction from Alaska southward and into the Salish Sea. There is a further reduction of diversity with distance from the outer coast moving inward within the Salish Sea, with the lowest values found in the southernmost site, Squaxin Island (Figure 2). Founder effects have been suggested for other species with distributions that span the outer coast of Washington and the Salish Sea, such as the Pacific hake M. productus (Iwamoto et al., 2015). A founder effect for bull kelp in the Salish Sea is likely accentuated by increased isolation and habitat fragmentation further into Puget Sound and parts of the Strait of Georgia. In the inner regions of the Puget Sound, bull kelp has declined dramatically over the past few decades, more so recently. In south Puget Sound, bull kelp canopy decreased 63% basin-wide from the 1870s to 2017, with up to 96% loss in smaller sub-basins (Berry et al., 2021). Several factors, including increased temperature, may be causing the recent declines and increased fragmentation (Pfister et al., 2017; Beas-Luna et al., 2020; Supratya et al., 2020; Starko et al., 2022). Recent bottlenecks could be why a single Salish Sea site (Stanley Park) had high private allele richness.
The genetic similarity between the inner regions of the Strait of Georgia and Puget Sound found with this set of microsatellite markers might mask some differentiation across the entire genome. When only looking at a limited number of microsatellite markers, the two regions could have been assigned to the same genetic co-ancestry group simply due to the chance of fixing the same dominant alleles in a common ancestor population during the population bottleneck. Any further divergence between the Strait of Georgia and Puget Sound might go undetectable if populations are highly homozygous or may require scanning a larger region of the genome (e.g., SNP analysis).
Oceanographic distance was a better predictor of genetic differentiation between sites than over-the-water spatial distance. The Salish Sea is influenced by coastal oceanographic currents predominantly through the Strait of Juan de Fuca and less so by the northern limit of the Strait of Georgia. Khangaonkar et al. (2018) showed that less than 12% of the water that flows from the Pacific into the Strait of Juan de Fuca makes it into the Puget Sound; in comparison, up to 42% reaches the Strait of Georgia, and the remaining 46% is returned into the Pacific Ocean. This suggests that ocean currents affect the Salish Sea more directly in the Juan de Fuca and Georgia Straits than in Puget Sound. The importance of oceanographic currents as a driver of gene flow has been found across a variety of marine dispersed taxa, including other kelps (White et al., 2010; Alberto et al., 2011; Truelove et al., 2017; Wesselmann et al., 2018; Xuereb et al., 2018; Gouvêa et al., 2023).
The multiple regression model and the CCA showed an association between environmental and genetic variation within the Salish Sea. Light attenuation, chl a, summer SST and POC were all significant predictors of genetic differentiation in the region. Despite a vigorous exchange flow with the Ocean (Khangaonkar et al., 2018), the Salish Sea is a region with substantial environmental variation from the outer Ocean, with the largest gradients for the above variables (in addition to PIC and light penetration) across the species range. In addition, other interacting sources of environmental variation are associated with anthropogenic factors (Sobocinski et al., 2018). For example, the major cities of Seattle and Vancouver provide a high influx of pollutants, such as sewage and other runoff, often in the form of suspended sediment in the water column. Sedimentation reduces light penetration in the water column and negatively affects the recruitment of N. luetkeana zoospores and juvenile sporophytes (Carney, 2003).
Temperatures above 19°C inhibit the formation of germ tubes in N. luetkeana zoospores, effectively halting the life cycle before gametophyte development (Schiltroth et al., 2018). Other experiments on the effects of temperature on bull kelp production of gametophytes and embryonic sporophytes suggest that adverse effects might start at temperatures as low as 15°C (Korabik et al., 2022). The observed association of genetic differentiation with Summer SST is therefore expected under a model of localized adaptation to a warmer environment. However, such a model will require validation from direct fitness measurements from reciprocal transplant studies. We are currently using a genome-wide molecular approach combined with reciprocal transplants to investigate if the isolation by environment (IBE) found within the Salish Sea has an adaptive basis.
Bull kelp conservation
We observed that near maximum N. luetkeana allelic richness persisted in northern California despite the recent dramatic population declines (Rogers-Bennett and Catton, 2019). Samples in this study from this region were collected in 2016, two years after the documented decline, while the sporophyte density was still extremely low. A recent study using high-resolution CubSats imagery showed that despite the regional kelp forest collapse, there were small remnant pockets where bull kelp sporophytes could survive (Cavanaugh et al., 2023). Therefore, despite near-complete canopy loss, genetic diversity was still comparable to maximal values recorded further south in California, where these declines were not observed (Bell et al., 2023). A future study using population genetics simulations, guided by satellite-derived population size census (Cavanaugh et al., 2023; Saccomanno et al., 2023) and dispersal estimates obtained from oceanographic transport models (Gaylord et al., 2006) can inform if the high genetic diversity found in the region could have been maintained by these remnant populations. A non-mutual exclusive explanation for genetic diversity resilience is the presence of a bank of gametophytes with delayed development, acting as a reservoir of the effective population size while the sporophyte generation is down (Carney, 2013, Edwards, 2022). More species-specific studies on delayed development of gametophytes are needed (Veenhof et al., 2022); new eDNA methods could assist in this regard (Schoenrock et al., 2021).
The reduction in genetic diversity within the inner regions of the Salish Sea, combined with environmental stressors due to proximity to metropolitan areas, raises concerns for the future of bull kelp in this region. Low genetic diversity is associated with decreased resilience to increasing temperatures in seagrasses (Reusch et al., 2005; Hughes et al., 2008; Duarte et al., 2018). Currently, we hold ex-situ collections of gametophytes from California and Puget Sound that are sufficiently large (> 1,800 haploid genotypes) to contain a large fraction of the extant genetic diversity and can be used to restore threatened areas (Wade et al., 2020).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://datadryad.org/stash, DOI: 10.5061/dryad.g4f4qrfvt and https:/github.com/falberto73/NereoFMSpaper.
Author contributions
FA: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Visualization, Writing – review & editing. LG: Formal Analysis, Investigation, Methodology, Software, Writing – original draft. NC: Methodology, Project administration, Resources, Writing – review & editing. TK: Funding acquisition, Investigation, Methodology, Resources, Software, Writing – review & editing. TM: Funding acquisition, Methodology, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding was provided by the California Sea Grant, University of California, San Diego, Grant/Award Number: C0874002. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
The authors would like to thank Makah tribe, and Squaxin Island Tribe for collections in their land. We thank B. Konar and T. Stephens for collections in Alaska. We thank C. Brooks and G. Saunders sampling Haida Gwaii, using their Discovery funding from the Natural Sciences and Engineering Research Council of Canada. We thank J. Edwards, L. Druehl, B. Heath, B Weeks, A. Weeks, S. Bisgrove, B. Schiltroth for sampling sites in British Columbia. We thank, C. Pfister, J. Wootton, L. Antrim, L. Heart, E. Hines, D. Parros, B. Allen, H. Berry, B. Peabody, J. Gaeckle, and M. Calloway for sampling sites in Washington, and J. Watson for sampling Oregon sites. We thank R. Alvarez, C. Catton, B. San Miguel, D. Canestro, and P. Raimondi for sampling sites in California. This work was part of L. Gierke MsC thesis (Gierke, 2019).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1275905/full#supplementary-material
References
Ager T. A. (2019). Late Quaternary vegetation development following deglaciation of northwestern Alexander Archipelago, Alaska. Front. Earth Sci. 31 (7). doi: 10.3389/feart.2019.00104
Aitken S. N., Whitlock M. C. (2013). Assisted gene flow to facilitate local adaptation to climate change. Annu. Rev. Ecol. Evol. Syste. 44, 367–388. doi: 10.1146/annurev-ecolsys-110512-135747
Alberto F. (2009). MsatAllele-1.0: An R package to visualize the binning of microsatellite alleles. J. Heredity 100, 394–397. doi: 10.1093/jhered/esn110
Alberto F., Raimondi P. T., Reed D. C., Coelho N. C., Leblois R., Whitmer A., et al. (2010). Habitat continuity and geographic distance predict population genetic differentiation in giant kelp. Ecology 91 (1), 49–56. doi: 10.1890/09-0050.1
Alberto F., Raimondi P. T., Reed D. C., Watson J. R., Siegel D. A., Mitarai S., et al. (2011). Isolation by oceanographic distance explains genetic structure for Macrocystis pyrifera in the Santa Barbara Channel. Mol. Ecol. 20, 2543–2554. doi: 10.1111/j.1365-294X.2011.05117.x
Amsler C. D., Neushul M. (1989). Diel periodicity of spore release from the kelp Nereocystis luetkeana (Mertens) Postels et Ruprecht. J. Exp. Mar. Biol. Ecol. 134, 117–127. doi: 10.1016/0022-0981(90)90104-K
Arafeh-Dalmau N., Olguín-Jacobson C., Bell T. W., Micheli F., Cavanaugh K. C. (2023). Shortfalls in the protection of persistent bull kelp forests in the USA. Biol. Conserv. 283, p.110133. doi: 10.1016/j.biocon.2023.110133
Assis J., Araújo M. B., Serrão. E. A. (2018). Projected climate changes threaten ancient refugia of kelp forests in the North Atlantic. Global Change Biol. 24, 55–66. doi: 10.1111/gcb.13818
Assis J., Coelho N. C., Lamy T., Valero M., Alberto F., Serrão E. A. (2016). Deep reefs are climatic refugia for genetic diversity of marine forests. J. Biogeogr. 43 (4), 833–844. doi: 10.1111/jbi.12677
Assis J., Serrão E. A., Claro B., Perrin C., Pearson G. A. (2014). Climate-driven range shifts explain the distribution of extant gene pools and predict future loss of unique lineages in a marine brown alga. Mol. Ecol. 23, 2797–2810. doi: 10.1111/mec.12772
Auld J. R., Rubio de Casas R. (2013). The correlated evolution of dispersal and mating-system traits. Evolution. Biol. 40, 185–193. doi: 10.1007/s11692-012-9202-7
Bálint M., Domisch S., Engelhardt C. H. M., Haase P., Lehrian S., Sauer J., et al. (2011). Cryptic biodiversity loss linked to global climate change. Nat. Climate Change 1 (6), 313–318. doi: 10.1038/nclimate1191
Barner A. K., Pfister C. A., Wootton J. T. (2011). The mixed mating system of the sea palm kelp Postelsia palmaeformis: few costs to selfing. Proc. R. Soc. B: Biol. Sci. 278 (1710), 1347–1355. doi: 10.1098/rspb.2010.1928
Beas-Luna R., Micheli F., Woodson C. B., Carr M., Malone D., Torre J., et al. (2020). Geographic variation in responses of kelp forest communities of the California Current to recent climatic changes. Global Change Biol. 26, 6457–6473. doi: 10.1111/gcb.15273
Bell T. W., Cavanaugh K. C., Saccomanno V. R., Cavanaugh K. C., Houskeeper H. F., Eddy N., et al. (2023). Kelpwatch: A new visualization and analysis tool to explore kelp canopy dynamics reveals variable response to and recovery from marine heatwaves. PloS One 18 (3), e0271477. doi: 10.1371/journal.pone.0271477
Bennett K. D., Tzedakis P. C., Willis K. J. (1991). Quaternary refugia of north European trees. J. biogeogr. 18, 103–115. doi: 10.2307/2845248
Bernhardt J. R., Leslie H. M. (2013). Resilience to climate change in coastal marine ecosystems. Annu. Rev. Mar. Sci. 5, 371–392. doi: 10.1146/annurev-marine-121211-172411
Berry H. D., Mumford T. F., Christiaen B., Dowty P., Calloway M., VanArendonk N. R. (2021). Long-term changes in kelp forests in an inner basin of the Salish Sea. PloS One 16 (2), e0229703. doi: 10.1371/journal.pone.0229703
Blanchette C. A., Miner C. M., Raimondi P. T., Lohse D., Heady K. E. K., Broitman. B. R. (2008). Biogeographical patterns of rocky intertidal communities along the Pacific coast of North America. J. Biogeogr. 35, 1593–1607. doi: 10.1111/j.1365-2699.2008.01913.x
Borcard D., Legendre P., Drapeau. P. (1992). Partialling out the spatial component of ecological variation. Ecology 73, 1045–1055. doi: 10.2307/1940179
Buonaccorsi V. P., Kimbrell C. A., Lynn E. A., Vetter. R. D. (2002). Population structure of copper rockfish (Sebastes caurinus) reflects postglacial colonization and contemporary patterns of larval dispersal. Can. J. Fish. Aquat. Sci. 59, 1374–1384. doi: 10.1139/f02-101
Burrows M. T., Schoeman D. S., Buckley L. B., Moore J., Poloczanska E. S., Brander K. M., et al. (2011). The pace of shifting climate in marine and terrestrial ecosystems. Science 334, 652–655. doi: 10.1126/science.1210288
Byun S. A., Koop B. F., Reimchen. T. E. (1997). North American black bear mtDNA phylogeography: implications for morphology and the haida gwaii glacial refugium controversy. Evolution 51 (5), 1647–1653. doi: 10.2307/2411216
Carney L. T. (2003). 17 Restoration techniques for Nereocystis luetkeana (Mertens) Postels Et Ruprecht (Bull Kelp). J. Phycol. 39, 7–7. doi: 10.1111/j.0022-3646.2003.03906001_17.x
Carney L. T., Bohonak A. J., Edwards M. S., Alberto F. (2013). Genetic and experimental evidence for a mixed age and mixed origin bank of microscopic stages in a southern California kelp forest. Ecology 94 (9), 1955–1965. doi: 10.1890/13-0250.1
Carrara P. E., Ager T. A., Baichtal. J. F. (2007). Possible refugia in the Alexander Archipelago of southeastern Alaska during the late Wisconsin glaciation. Can. J. Earth Sci. 44, 229–244. doi: 10.1139/e06-081
Cavanaugh K. C., Cavanaugh K. C., Pawlak C. C., Bell T. W., Saccomanno V. R. (2023). CubeSats show persistence of bull kelp refugia amidst a regional collapse in California. Remote Sens. Environ. 290, 113521. doi: 10.1016/j.rse.2023.113521
Charlesworth D., Charlesworth B. (1987). Inbreeding depression and its evolutionary consequences. Annu. Rev. Ecol. Evol. Syste. 18, 237–268. doi: 10.1146/annurev.es.18.110187.001321
Clark P. U., Dyke A. S., Shakun J. D., Carlson A. E., Clark J., Wohlfarth B., et al. (2009). The last glacial maximum. Science 325 (5941), 710–714. doi: 10.1126/science.1172873
Clarke T. E., Levin D. B., Kavanaugh D. H., Reimchen. T. E. (2001). Rapid evolution in the Nebria gregaria group (Coleoptera: Carabidae) and the paleogeography of the queen charlotte islands. Evolution 55, 1408–1418. doi: 10.1111/j.0014-3820.2001.tb00662.x
Cohen D. (1966). Optimizing reproduction in a randomly varying environment. J. Theor. Biol. 12, 119–129. doi: 10.1016/0022-5193(66)90188-3
Coleman M. A., Goold H. D. (2019). Harnessing synthetic biology for kelp forest conservation. J. Phycol. 55 (4), 745–751. doi: 10.1111/jpy.12888
Csardi G., Nepusz T. (2006). The igraph software package for complex network research. InterJournal Complex Syst. 1695.
Dijsktra E. W. (1959). A note on two problems in connection with graphs. Numberische Mathematik 1, 269–271. doi: 10.1145/3544585.3544600
Doney S. C., Ruckelshaus M., Emmett Duffy J., Barry J. P., Chan F., English C. A., et al. (2012). Climate change impacts on marine ecosystems. Annu. Rev. Mar. Sci. 4, 11–37. doi: 10.1146/annurev-marine-041911-111611
Dornier A., Munoz F., Cheptou P.-O. (2008). Allee effect and self-fertilizationn in hermaphrodites: reproductive assurance in a structured metapopulation. Evolution 62, 2558–2569. doi: 10.1111/j.1558-5646.2008.00464.x
Duarte B., Martins I., Rosa R., Matos A. R., Roleda M. Y., Reusch T. B., et al. (2018). Climate change impacts on seagrass meadows and macroalgal forests: an integrative perspective on acclimation and adaptation potential. Front. Mar. Sci. 5. doi: 10.3389/fmars.2018.00190
Dyke A. S. (2004). An outline of North American deglaciation with emphasis on central and northern Canada. Developments in Quaternary Sciences 2, 373–424.
Edwards M. S. (2022). It’s the little things: the role of microscopic life stages in maintaining kelp populations. Front. Mar. Sci. 9. doi: 10.3389/fmars.2022.871204
Excoffier L., Foll M., Petit R. J. (2009). Genetic consequences of range expansions. Annu. Rev. Ecol. Evol. Syste. 40 (1), 481–501. doi: 10.1146/annurev.ecolsys.39.110707.173414
Fenberg P. B., Menge B. A., Raimondi P. T., Rivadeneira M. M. (2015). Biogeographic structure of the northeastern Pacific rocky intertidal: The role of upwelling and dispersal to drive patterns. Ecography 38, 83–95. doi: 10.1111/ecog.00880
Foster M. S., Schiel D. R. (1985). The Ecology of Giant Kelp Forests in California: A Community Profile (US Fish and Wildlife Service).
Fraser C. I., Dutoit L., Morrison A. K., Pardo L. M., Smith S. D., Pearman W. S., et al. (2022). Southern Hemisphere coasts are biologically connected by frequent, long-distance rafting events. Curr. Biol. 32 (14), 3154–3160. doi: 10.1016/j.cub.2022.05.035
Gaylord B., Reed D. C., Raimondi P. T., Washburn L. (2006). Macroalgal spore dispersal in coastal environments: mechanistic insights revealed by theory and experiment. Ecol. Monogr. 76 (4), 481–502. doi: 10.1890/0012-9615(2006)076[0481:MSDICE]2.0.CO;2
Gibson R. N., Atkinson R. J. A., Gordon J. D. M. (2010). Toward ecosystem-based management of marine macroalgae - the bull kelp, Nereocystis luetkeana. Oceanogr. Mar. Biol.: Annu. Rev. 48, 1–42.
Gierke L. (2019). A seascape genetics approach to studying genetic differentiation in the bull kelp, Nereocystis luetkeana. (Milwaukee (WI: UW-Milwaukee).
González I., Déjean S., Martin P. G. P., Baccini A. (2008). CCA: An R package to extend canonical correlation analysis. J. Stat. Softw. 23, 1–14. doi: 10.18637/jss.v023.i12
Gouvêa L. P., Fragkopoulou E., Cavanaugh K., Serrão E. A., Araújo M. B., Costello M. J., et al. (2023). Oceanographic connectivity explains the intra-specific diversity of mangrove forests at global scales. Proc. Natl. Acad. Sci. 120 (14), 2209637120. doi: 10.1073/pnas.2209637120
Graham M. H., Halpern B., Carr M. H. (2008). “Diversity and dynamics of Californian subtidal kelp forests,” in Food webs and the dynamics of marine reefs. Eds. McClanahan T., Branch G. (Oxford: Oxford University Press). doi: 10.1093/acprof:oso/9780195319958.003.0005
Grant W. S., Bringloe T. T. (2020). Pleistocene ice ages created new evolutionary lineages, but limited speciation in Northeast Pacific winged kelp. J. Heredity 111, 593–605. doi: 10.1093/jhered/esaa053
Grant W. S., Chenoweth E. (2021). Phylogeography of sugar kelp: Northern ice-age refugia in the Gulf of Alaska. Ecol. Evol. 11 (9), 4670–4687. doi: 10.1002/ece3.7368
Grant W. S., Lydon A., Bringloe T. T. (2020). Phylogeography of split kelp Hedophyllum nigripes: Northern ice-age refugia and trans-Arctic dispersal. Polar Biol. 43, 1829–1841. doi: 10.1007/s00300-020-02748-6
Guilbault J. P., Barrie J. V., Conway K., Lapointe M., Radi T. (2003). Paleoenvironments of the Strait of Georgia, British Columbia during the last deglaciation: microfaunal and microfloral evidence. Quaternary Sci. Rev. 22 (8-9), 839–857. doi: 10.1016/S0277-3791(02)00252-4
Hernández-Carmona G., Hughes B., Graham M. H. (2006). Reproductive longevity of drifting kelp Macrocystis pyrifera (Phaeophyceae) in Monterey Bay, USA. J. Phycol. 42, 1199–1207. doi: 10.1111/j.1529-8817.2006.00290.x
Hewitt G. M. (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 58, 247–276. doi: 10.1006/bijl.1996.0035
Hewitt G. M. (2003). “Ice ages: their impact on species distributions and evolution,” in Evolution on Planet Earth. Eds. Rothschild L. J., Lister A. M. (London: Academic Press), 339–361. doi: 10.1006/bijl.1996.0035
Hickerson M. J., Cunningham C. W. (2005). Contrasting Quaternary histories in an ecologically divergent sister pair of low-dispersing intertidal fish (Xiphister) revealed by multilocus DNA analysis. Evolution 59, 344–360. doi: 10.1111/j.0014-3820.2005.tb00994.x
Hickerson M. J., Ross J. R. P. (2001). Post-glacial population history and genetic structure of the northern clingfish (Gobbiesox maeandricus), revealed from mtDNA analysis. Mar. Biol. 138, 407–419. doi: 10.1007/s002270000465
Hoarau G., Coyer J. A., Vedsink J. H., Stam W. T., Olsen J. L. (2007). Glacial refugia and recolonization pathways in the brown seaweed Fucus serratus. Mol. Ecol. 16, 3606–3616. doi: 10.1111/j.1365-294X.2007.03408.x
Hoban S., Bruford M., Jackson J. D. U., Lopes-Fernandes M., Heuertz M., Hohenlohe P. A., et al. (2020). Genetic diversity targets and indicators in the CBD post-2020 Global Biodiversity Framework must be improved. Biol. Conserv. 248, 108654. doi: 10.1016/j.biocon.2020.108654
Hock K., Mumby P. J. (2015). Quantifying the reliability of dispersal paths in connectivity networks. J. R. Soc. Interface 12, 20150013–20150013. doi: 10.1098/rsif.2015.0013
Holm S. (1979). A simple sequentially rejective multiple test procedure. Scand. J. Statist. 6, 65–70.
Hughes R. A., Inouye B. D., Johnson M. T. J., Underwood N., Vellend M. (2008). Ecological consequences of genetic diversity. Ecol. Lett. 11 (6), 609–623. doi: 10.1111/j.1461-0248.2008.01179.x
Huntley B., Birks H. J. B. (1983). An Atlas of Past and Present Pollen Maps for Europe: 0-13,000 Years Ago (Cambridge: Cambridge University Press).
Iwamoto E. M., Elz A. E., García-De León F. J., Silva-Segundo C. A., Ford M. J., Palsson W. A., et al. (2015). Microsatellite DNA analysis of Pacific hake Merluccius productus population structure in the Salish Sea. ICES J. Mar. Sci. 72, 2720–2731. doi: 10.1093/icesjms/fsv146
Jacobs D. K., Haney T. A., Louie K. D. (2004). Genes, diversity, and geologic process on the pacific coast. Annu. Rev. Earth Planet. Sci. 32, 601–652. doi: 10.1146/annurev.earth.32.092203.122436
Jaramillo-Correa J. P., Beaulieu J., Khasa D. P., Bousquet. J. (2009). Inferring the past from the present phylogeographic structure of North American forest trees: seeing the forest for the genes. Can. J. For. Res. 39 (2), 286–307. doi: 10.1139/X08-181
Johansson M. L., Alberto F., Reed D. C., Raimondi P. T., Coelho N. C., Young M. A., et al. (2015). Seascape drivers of Macrocystis pyrifera population genetic structure in the northeast Pacific. Mol. Ecol. 24, 4866–4885. doi: 10.1111/ec.13371
Jombart T. (2008). Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24 (11), 1403–1405. doi: 10.1093/bioinformatics/btn129
Jost L. (2008). GST and its relatives do not measure differentiation. Mol. Ecol. 17, 4015–4026. doi: 10.1111/j.1365-294X.2008.03887.x
Kalinowski S. T. (2005). HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes. 5, 187–189. doi: 10.1111/j.1471-8286.2004.00845.x
Keenan K., McGinnity P., Cross T. F., Crozier W. W., Prodöhl P. A. (2013). diveRsity: An R package for the estimation of population genetics parameters and their associated errors. Methods Ecol. Evol. 4 (8), 782–788. doi: 10.1111/2041-210X.12067
Kelly R. P., Palumbi S. R. (2010). Genetic structure among 50 species of the northeastern pacific rocky intertidal community. PloS One 5 (1), e8594. doi: 10.1371/journal.pone.0008594
Khangaonkar T., Nugraha. A., Xu W., Long W., Bianucci L., Ahmed A., et al. (2018). Analysis of hypoxia and sensitivity to nutrient pollution in salish sea. J. Geophys. Res. – Oceans 123 (7), 4735–4761. doi: 10.1029/2017JC013650
Kimura M., Weiss G. H. (1964). The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 49, 561–576. doi: 10.1093/genetics/49.4.561
Kopelman N. M., Mayzel J., Jakobsson M., Rosenberg N. A., Mayrose I. (2015). Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191. doi: 10.1111/1755-0998.12387
Korabik A. R., Winquist T., Grosholz E. D., Hollarsmith J. A. (2022). Examining the reproductive success of bull kelp (Nereocystis luetkeana) in climate change conditions. biorxiv, 2022–2011. doi: 10.1101/2022.11.01.514766
Kusumo H. T., Pfister C. A., Wootton J. T. (2006). Small-scale genetic structure in the sea palm Postelsia palmaeformis Ruprecht (Phaeophyceae). Mar. Biol. 149, 731–742. doi: 10.1007/s00227-006-0254-z
Li Y. L., Liu J. X. (2018). StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 18, 176–177. doi: 10.1111/1755-0998.12719
Lindstrom S. C. (2009). The biogeography of seaweeds in Southeast Alaska. J. Biogeogr. 36, 401–409. doi: 10.1111/j.1365-2699.2007.01855.x
Lindstrom S. C., Olsen J. L., Stam W. T. (2011). Postglacial recolonization and the biogeography of Palmaria mollis (Rhodophyta) along the Northeast Pacific coast. Can. J. Bot. 75, 1887–1896. doi: 10.1139/b97-900
Macaya E. C., Boltaña S., Hinojosa I. A., Macchiavello J. E., Valdivia N. A., Vasquez N. R., et al. (2005). Presence of sporophylls in floating kelp rafts of Macrocystis spp. (Phaeophyceae) along the Chilean Pacific Coast. J. Phycol. 41, 913–922. doi: 10.1111/j.1529-8817.2005.00118.x
Maggs C. A., Castilho R., Foltz D., Henzler C., Jolly M. T., Kelly J., et al. (2008). Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology 89, 108–122. doi: 10.1890/08-0257.1
Mann D. H., Hamilton T. D. (1995). Late Pleistocene and Holocene paleoenvironments of the North Pacific coast. Quaternary Sci. Rev. 14, 449–471. doi: 10.1016/0277-3791(95)00016-I
Marko P. B. (2004). What’s larvae got to do with it? Disparate patterns of post-glacial population structure in two benthic marine gastropods with identical dispersal potential. Mol. Ecol. 13, 597–611. doi: 10.1046/j.1365-294X.2004.02096.x
Marko P. B., Hoffman J. M., Emme S. A., McGovern T. M., Keever C. C., Nicole Cox L. (2010). The ‘Expansion–Contraction’model of Pleistocene biogeography: rocky shores suffer a sea change? Mol. Ecol. 19 (1), 146–169. doi: 10.1111/j.1365-294X.2009.04417.x
Maxell B. A., Miller K. A. (1996). Demographic studies of the annual kelps nereocystis luetkeana and Costaria costata (Laminariales, Phaeophyta) in Puget Sound, Washington. Botanica Marina 39, 479–489. doi: 10.1515/botm.1996.39.1-6.479
Miller K. A., Estes J. A. (1989). Western range extension for Nereocystis luetkeana in the North Pacific Ocean. Botanica Marina 32, 535–538. doi: 10.1515/botm.1989.32.6.535
Miller M. P., Haig S. M., Wagner R. S. (2006). Phylogeography and spatial genetic structure of the Southern torrent salamander: implications for conservation and management. J. Heredity 97, 561–570. doi: 10.1093/jhered/esl038
Miller R. J., Lafferty K. D., Lamy T., Kui L., Rassweiler A., Reed D. C. (2018). Giant kelp, Macrocystis pyrifera, increases faunal diversity through physical engineering. Proc. R. Soc. B: Biol. Sci. 285 (1874), 20172571. doi: 10.1098/rspb.2017.2571
Norton T. A. (1992). Dispersal by macroalgae. Br. Phycol. J. 27, 293–301. doi: 10.1080/00071619200650271
Nosil P., Egan S. P., Funk D. J. (2008). Heterogeneous genomic differentiation between walking-stick ecotypes: “Isolation by adaptation” and multiple roles for divergent selection. Evolution 62, 316–336. doi: 10.1111/j.1558-5646.2007.00299.x
Økland R. H., Eilertsen O. (1994). Canonical Correspondence Analysis with variation partitioning: some comments and an application. J. Vegetation Sci. 5, 117–126. doi: 10.2307/3235645
Oksanen J., Simpson G., Blanchet F., Kindt R., Legendre P., Minchin P., et al. (2022). vegan: Community Ecology Package. R package version 2.6-4.
Pauls S. U., Nowak C., Bálint M., Pfenninger M. (2013). The impact of global climate change on genetic diversity within populations and species. Mol. Ecol. 22, 925–946. doi: 10.1111/mec.12152
Pfister C. A., Berry H. D., Mumford T. (2017). The dynamics of kelp forests in the northeast pacific ocean and the relationship with environmental drivers. J. Ecol. 106 (4), 1520–1533. doi: 10.1111/1365-2745.12908
Pinsky M. L., Selden R. L., Kitchel Z. J. (2020). Climate-driven shifts in marine species ranges: Scaling from organisms to communities. Annu. Rev. Mar. Sci. 12, 153–179. doi: 10.1146/annurev-marine-010419-010916
Pinsky M. L., Worm B., Fogarty M. J., Sarmiento J. L., Levin S. A. (2013). Marine taxa track local climate velocities. Science 341, 1239–1242. doi: 10.1126/science.1239352
Poloczanska E. S., Brown C. J., Sydeman W. J., Kiessling W., Schoeman D. S., Moore P. J., et al. (2013). Global imprint of climate change on marine life. Nat. Climate Change 3 (10), 919–925. doi: 10.1038/nclimate1958
Pörtner H. O., Roberts D. C., Adams H., Adler C., Aldunce P., Ali E., et al. (2022). Climate change 2022: Impacts, adaptation and vulnerability (Geneva, Switzerland: IPCC), 3056.
Pritchard J. K., Stephens M., Donnelly P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
Provan J., Bennett K. D. (2008). Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 23, 564–571. doi: 10.1016/j.tree.2008.06.010
Puechmaille S. J. (2016). The program structure does not reliably recover the correct population structure when sampling is uneven: Subsampling and new estimators alleviate the problem. Mol. Ecol. Resour. 16, 608–627. doi: 10.1111/1755-0998.12512
R Core Team. (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available at: https://www.R-project.org/.
Raimondi P. T., Reed D. C., Gaylord B., Washburn L. (2004). Effects of self-fertilization in the giant kelp, Macrocystis pyrifera. Ecology 85 (12), 3267–3276. doi: 10.1890/03-0559
Reed D. C. (1987). Factors affecting the production of sporophylls in the giant kelp. J. Exp. Mar. Biol. Ecol. 113, 61–69. doi: 10.1016/0022-0981(87)90082-7
Reed D. C., Laur D. R., Ebeling. A. W. (1988). Variation in algal dispersal and recruitment: the importance of episodic events. Ecol. Monogr. 58, 321–335. doi: 10.2307/1942543
Reusch T. B. H., Ehlers A., Hämmerli A., Worm B. (2005). Ecosystem recovery after climatic extremes enhanced by genotypic diversity. Proc. Natl. Acad. Sci. United States America 102, 2826–2831. doi: 10.1073/pnas.0500008102
Riginos C., Liggins L. (2013). Seascape genetics: populations, individuals, and genes marooned and adrift. Geogr. Compass 7, 197–216. doi: 10.1111/gec3.12032
Robinson C. L. K., Morrison J., Foreman M. G. G. (2005). Oceanographic connectivity among marine protected areas on the north coast of British Columbia, Canada. Canadian. J. Fish. Aquat. Sci. 62, 1350–1362. doi: 10.1139/f05-088
Rogers-Bennett L., Catton C. A. (2019). Marine heat wave and multiple stressors tip bull kelp forest to sea urchin barrens. Sci. Rep. 9 (1), 15050. doi: 10.1038/s41598-019-51114-y
Rousset F. (2008). GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106. doi: 10.1111/j.1471-8286.2007.01931.x
Saccheri I., Kuussaari M., Kankare M., Vikman P., Hanski I. (1998). Inbreeding and extinction in a butterfly metapopulation. Nature 45, 1996–1999. doi: 10.1038/33136
Saccomanno V. R., Bell T., Pawlak C., Stanley C. K., Cavanaugh K. C., Hohman R., et al. (2023). Using unoccupied aerial vehicles to map and monitor changes in emergent kelp canopy after an ecological regime shift. Remote Sens. Ecol. Conserv. 9 (1), 62–75. doi: 10.1002/rse2.295
Schiltroth B., Bisgrove S., Heath B. (2018). “Effects of warm ocean temperatures on bull kelp forests in the Salish Sea,” in Salish Sea Ecossystem Conference, Seatle, WA.
Schlaepfer M. A., Helenbrook W. D., Searing K. B., Shoemaker K. T. (2009). Assisted colonization: evaluating contrasting management actions (and values) in the face of uncertainty. Trends Ecol. Evol. 24, 471–472. doi: 10.1016/j.tree.2009.05.008
Schoen D. J., Brown A. H. (2001). The conservation of wild plant species in seed banks: attention to both taxonomic coverage and population biology will improve the role of seed banks as conservation tools. BioScience 51 (11), 960–966. doi: 10.1641/0006-3568(2001)051[0960:TCOWPS]2.0.CO;2
Schoenrock K. M., McHugh T. A., Krueger-Hadfield S. A. (2021). Revisiting the ‘bank of microscopic forms’ in macroalgal-dominated ecosystems. J. Phycol. 57 (1), 14–29. doi: 10.1111/jpy.13092
Selivanova O. N., Zhigadlova G. G. (1997). Marine algae of the Commander Islands. Preliminary remarks on the revision of the flora II. (Phaeophyta). Botanica Marina 40, 9–13. doi: 10.1515/botm.1997.40.1-6.9
Selkoe K. A., D’Aloia C. C., Crandall E. D., Iacchei M., Liggins L., Puritz J. B., et al. (2016). A decade of seascape genetics: Contributions to basic and applied marine connectivity. Mar. Ecol. Prog. Ser. 554, 1–19. doi: 10.3354/meps11792
Selkoe A. K., Henzler C. M., Gaines S. D. (2008). Seascape genetics and the spatial ecology of marine populations. Fish Fish. 9, 363–377. doi: 10.1111/j.1467-2979.2008.00300.x
Sexton J. P., Hangartner S. B., Hoffmann A. A. (2014). Genetic isolation by environment or distance: Which pattern of gene flow is most common? Evolution 68, 1–15. doi: 10.1111/evo.12258
Sgrò C. M., Lowe A. J., Hoffmann A. A. (2011). Building evolutionary resilience for conserving biodiversity under climate change. Evolution. Appl. 4, 326–337. doi: 10.1111/j.1752-4571.2010.00157.x
Shafer A. B. A., Cullingham C. I., Côté S. D., Coltman D. W. (2010). Of glaciers and refugia: A decade of study sheds new light on the phylogeography of northwestern North America. Mol. Ecol. 19, 4589–4621. doi: 10.1111/j.1365-294X.2010.04828.x
Siddon E. C., Siddon C. E., Stekoll M. S. (2008). Community level effects of Nereocystis luetkeana in southeastern Alaska. J. Exp. Mar. Biol. Ecol. 361, 8–15. doi: 10.1016/j.jembe.2008.03.015
Simons A. M. (2011). Modes of response to environmental change and the elusive empirical evidence for bet-hedging. Proc. R. Soc. B: Biol. Sci. 278 (1712), 1601–1609. doi: 10.1098/rspb.2011.0176
Smale D. A., Wernberg T. (2013). Extreme climatic event drives range contraction of a habitat-forming species. Proc. R. Soc. B: Biol. Sci. 280 (1754), 20122829. doi: 10.1098/rspb.2012.2829
Smale D. A., Epstein G., Hughes E., Mogg A. O., Moore P. J. (2020). Patterns and drivers of understory macroalgal assemblage structure within subtidal kelp forests. Biodivers. Conserv. 29, 4173–4192. doi: 10.1007/s10531-020-02070-x
Sobocinski K. L., Greene C. M., Schmidt M. W. (2018). Using a qualitative model to explore the impacts of ecosystem and anthropogenic drivers upon declining marine survival in Pacific salmon. Environ. Conserv. 45 (3), 278–290. doi: 10.1017/S0376892917000509
Soltis D. E., Gitzendanner M. A., Strenge D. D., Soltis P. S. (1997). Chloroplast DNA intraspecific phylogeography of plants from the Pacific Northwest of North America. Plant Syste. Evol. 206, 353–373. doi: 10.1007/BF00987957
Starko S., Neufeld C. J., Gendall L., Timmer B., Campbell L., Yakimishyn J., et al. (2022). Microclimate predicts kelp forest extinction in the face of direct and indirect marine heatwave effects. Ecol. Appl. 32 (7), e2673. doi: 10.1002/eap.2673
Supratya V. P., Coleman L. J. M., Martone P. T. (2020). Elevated temperature affects phenotypic plasticity in the bull kelp (Nereocystis luetkeana, Phaeophyceae). J. Phycol. 56, 1534–1541. doi: 10.1111/jpy.13049
Thuiller W., Albert C., Araújo M. B., Berry P. M., Cabeza M., Guisan A., et al. (2008). Predicting global change impacts on plant species’ distributions: future challenges. Perspect. Plant Ecol. Evol. Syste. 9 (3-4), 137–152. doi: 10.1016/j.ppees.2007.09.004
Truelove N. K., Box S. J., Aiken K. A., Blythe-Mallett A., Boman E. M., Booker C. J., et al. (2017). Isolation by oceanic distance and spatial genetic structure in an overharvested international fishery. Diversity Distrib. 23 (11), 1292–1300. doi: 10.1111/ddi.12626
Van Oosterhout C., Hutchinson W. F., Wills D. P. M., Shipley P. (2004). Micro-Checker: Software for identifying and correcting genotyping rrrors in microsatellite data. Mol. Ecol. Notes 4, 535–538. doi: 10.1111/j.1471-8286.2004.00684.x
van Oppen M. J. H., Oliver J. K., Putnam H. M., Gates R. D. (2015). Building coral reef resilience through assisted evolution. Proc. Natural Acad. Sci. U.S.A. 112, 2307–2313. doi: 10.1073/pnas.1422301112
Veenhof R. J., Champion C., Dworjanyn S. A., Wernberg T., Minne A. J., Layton C., et al. (2022). Kelp gametophytes in changing oceans. Oceanogr. Mar. Biol.: Annu. Rev. 60, 335–371. doi: 10.1201/9781003288602-7
Wade R., Augyte S., Harden M., Nuzhdin S., Yarish C., Alberto F. (2020). Macroalgal germplasm banking for conservation, food security, and industry. PloS Biol. 18 (2), e3000641. doi: 10.1371/journal.pbio.3000641
Walker D. C. (1980). Sorus abscission from laminae of Nereocystis luetkeana (Mert.) Post. and Rupr. Ph.D. Dissertation (Vancouver: University of British Columbia), 464.
Wang I. J., Bradburd G. S. (2014). Isolation by environment. Mol. Ecol. 23, 5649–5662. doi: 10.1111/mec.12938
Waples R. S., Gaggiotti O. (2006). What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol. Ecol. 15 (6), 1419–1439. doi: 10.1111/j.1365-294X.2006.02890.x
Warner B. G., Mathewes R. W., Clague J. J. (1982). Ice-free conditions on the Queen Charlotte Islands, British Columbia, at the height of Late Wisconsin glaciation. Science 218, 675–677. doi: 10.1126/science.218.4573.675
Waters J. M., King T. M., Fraser C. I., Craw D. (2018). An integrated ecological, genetic and geological assessment of long-distance dispersal by invertebrates on kelp rafts. Front. Biogeogr. 10 (3-4). doi: 10.21425/F5FBG40888
Weir B. S., Cockerham C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370. doi: 10.2307/2408641
Wernberg T., Bennett S., Babcock R. C., De Bettignies T., Cure K., Depczynski M., et al. (2016). Climate-driven regime shift of a temperate marine ecosystem. Science 353 (6295), 169–172. doi: 10.1126/science.aad8745
Wesselmann M., González-Wangüemert M., Serrão E. A., Engelen A. H., Renault L., García-March J. R., et al. (2018). Genetic and oceanographic tools reveal high population connectivity and diversity in the endangered pen shell Pinna nobilis. Sci. Rep. 8 (1), 4770. doi: 10.1038/s41598-018-23004-2
White C., Selkoe K. A., Watson J., Siegel D. A., Zacherl D. C., Toonen R. J. (2010). Ocean currents help explain population genetic structure. Proc. R. Soc. B: Biol. Sci. 277, 1685–1694. doi: 10.1098/rspb.2009.2214
Wilke T., Duncan N. (2004). Phylogeographical patterns in the American Pacific Northwest: lessons from the arionid slug Prophysaon coeruleum. Mol. Ecol. 13, 2303–2315. doi: 10.1111/j.1365-294X.2004.02234.x
Williams D., Dunkerley D., DeDekker P., Kershaw P., Chappell M. (1998). Quaternary Environments (Arnold: New York).
Xuereb A., Benestan L., Normandeau É., Daigle R. M., Curtis J. M. R., Bernatchez L., Fortin M. J. (2018). Asymmetric oceanographic processes mediate connectivity and population genetic structure, as revealed by RADseq, in a highly dispersive marine invertebrate (Parastichopus californicus). Mol. Ecol. 27, 2347–2364. doi: 10.1111/mec.14589
Yang D., Song Y., Ma J., Li P., Zhang H., Price M. R. S., et al. (2016). Stepping-stones and dispersal flow: establishment of a metapopulation of Milu (Elaphurus davidianus) through natural re-wilding. Sci. Rep. 6 (1), 27297. doi: 10.1038/srep27297
Yang Z., Khangaonkar T. (2010). Multi-scale modeling of Puget Sound using an unstructured-grid coastal ocean model: from tide flats to estuaries and coastal waters. Ocean Dynam. 60 (6), 1621–1637. doi: 10.1007/s10236-010-0348-5
Zelenke B., O'Connor C., Barker C. H., Beegle-Krause C. J., Eclipse L. (2012). General NOAA operational modeling environment (GNOME) technical documentation. (NOAA). Available at: https://repository.library.noaa.gov/view/noaa/2621.
Keywords: kelp forest, genetic diversity, glacial refugia, mating system, dispersal, microsatellite markers, bull kelp
Citation: Gierke L, Coelho NC, Khangaonkar T, Mumford T and Alberto F (2023) Range wide genetic differentiation in the bull kelp Nereocystis luetkeana with a seascape genetic focus on the Salish Sea. Front. Mar. Sci. 10:1275905. doi: 10.3389/fmars.2023.1275905
Received: 10 August 2023; Accepted: 26 October 2023;
Published: 14 November 2023.
Edited by:
Christophe William Vieira, Jeju National University, Republic of KoreaReviewed by:
Marie-Laure Guillemin, Austral University of Chile, ChileZi-Min Hu, Yantai University, China
Copyright © 2023 Gierke, Coelho, Khangaonkar, Mumford and Alberto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Filipe Alberto, albertof@uwm.edu