 Kexun Zhou
Kexun Zhou Shuo Li3,4†
Shuo Li3,4† Ke Cheng
Ke Cheng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 February 2023
Sec. Cancer Immunity and Immunotherapy
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1127071
This article is part of the Research TopicBiomarkers of Immune Checkpoint Inhibitors in Lung Cancer: Dusk or Dawn?View all 8 articles
Immune checkpoint inhibitors (ICIs) in the form of anti-CTLA-4 and anti-PD-1/PD-L1 have become the frontier of cancer treatment and successfully prolonged the survival of patients with advanced non-small cell lung cancer (NSCLC). But the efficacy varies among different patient population, and many patients succumb to disease progression after an initial response to ICIs. Current research highlights the heterogeneity of resistance mechanisms and the critical role of tumor microenvironment (TME) in ICIs resistance. In this review, we discussed the mechanisms of ICIs resistance in NSCLC, and proposed strategies to overcome resistance.
Lung cancer is currently the second most frequently diagnosed cancer and the leading cause of cancer mortality globally (1). Immune checkpoint inhibitors (ICIs) have changed the cancer treatment paradigm and become the standard care for many cancers, including lung cancer. Whether as monotherapy or in combination, ICIs have shown inspiring efficacy in advanced non-oncogene-driven non-small cell lung cancer (NSCLC), extensive-stage small cell cancer, as well as unresectable stage III NSCLC (2, 3). In addition, the role of ICIs in neoadjuvant chemotherapy for resectable lung cancer is gradually being explored (4–7). To date, ICIs targeting three different immune checkpoints have been approved by the US Food and Drug Administration (FDA) for cancer therapy, namely cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibody, programmed cell death protein 1 (PD-1) antibody, and programmed death ligand 1 (PD-L1) antibody. Over years, the application of anti-PD-1/PD-L1 antibodies has greatly surpassed anti-CTLA-4, due to superior efficacy and safety profiles (8).
Although ICIs have significantly improved the survival in patients, the efficacy varied in different patient population, as a large proportion of patients had poor response to ICIs and succumb to disease progression, especially with ICIs monotherapy. The disparities represented a critical knowledge gap and attracted widespread attention, driving efforts to address this issue. Therefore, it is important to review the current mechanism of resistance and reveal how to ‘activate’ non-responders. In this review, we summarized the mechanisms of ICIs resistance in lung cancer and the factors which were associated with the clinical effects, aiming to promote better use of ICIs in lung cancer treatment.
Initiation of the anti-tumor immune response occurs when cells of the innate immune system become alerted to the presence of tumor, the signal of which may be conveyed by pro-inflammatory molecules, chemokines of the tumor site, and cells of the innate immune system (9). Tumor cells are recognized by the intrinsic immune cells, and the recognition mechanism is inconclusive (10). Once recognized, the intrinsic immune cells (e.g., natural killer (NK) T cells, γδ T cells, and macrophages) will trigger initial intrinsic immunity and interferon (IFN) secretion, which promotes chemokines production, and the recruitment of immune cells. IFN could activate a series of IFN-dependent processes (e.g., anti-proliferative, pro-apoptotic, anti-angiogenic) to kill tumor cells (9). The dead tumor cells during this process makes tumor antigens available, driving specific immunity.
Immature dendritic cells (DCs) are recruited to the tumor and triggered by cytokine exposure or intercellular interaction. Mature DCs take up tumor antigens, which finally presented as major histocompatibility complex class I (MHC-I) antigenic tumor peptides complex on DCs surface, migrate to the draining lymph nodes, where initial tumor-specific T helper type 1 (Th1) CD4+ T cells and CD8+ cytotoxic T lymphocytes (CTLs) are induced by antigen cross-presentation (9). Other antigen-presenting cells (APCs) are also involved in the process. CD4+ and CD8+ T cells migrate to the tumor site. CD4+ T cells produce IL-2, which, together with IL-15 produced by the host cells, helps to maintain CTLs production and viability. T cells can also release large amounts of IFN, which, together with intrinsic immune cells, kill tumors and induce tumor regression through numerous mechanisms (9, 11) (Figure 1A).
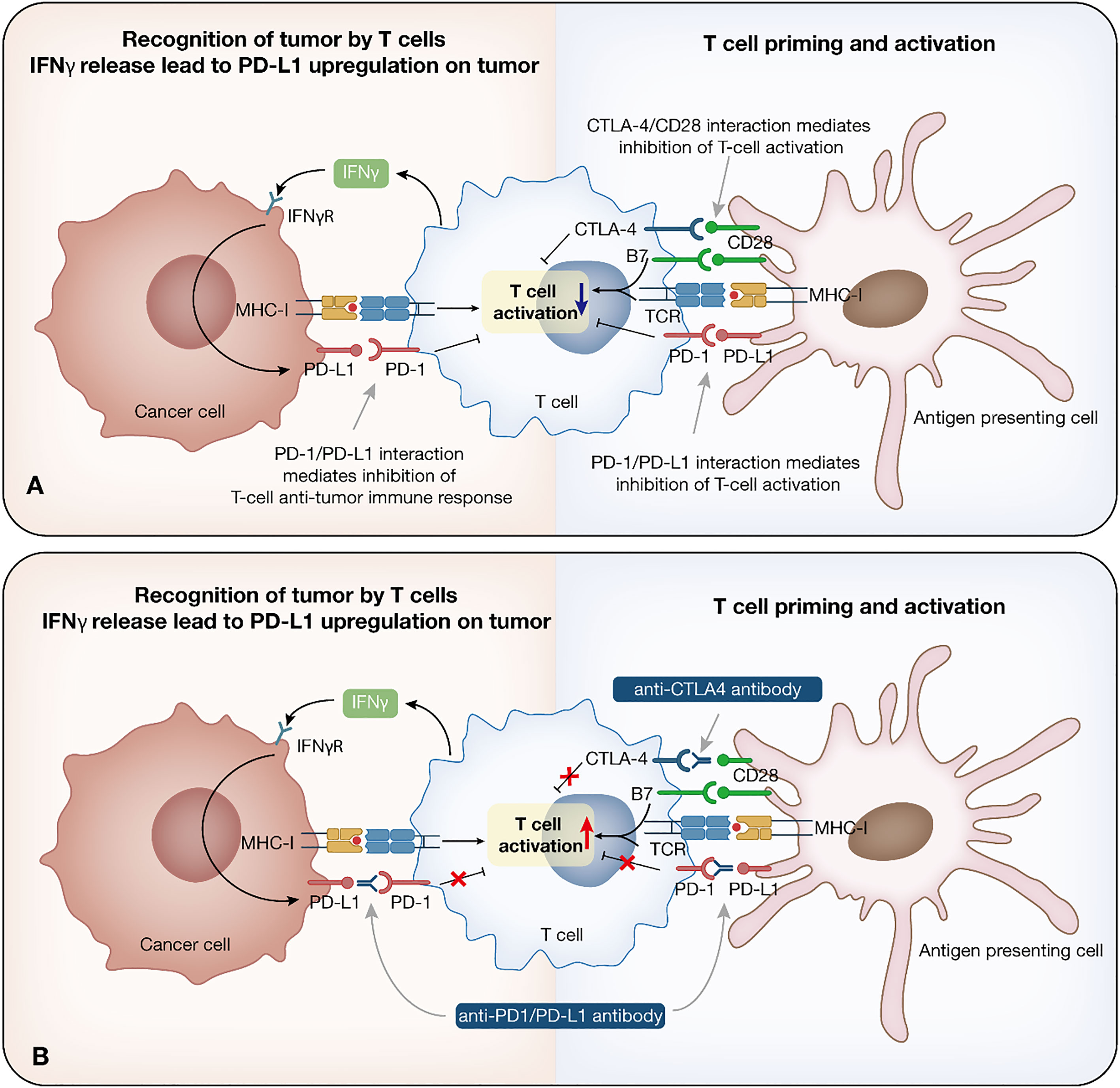
Figure 1 Immune checkpoint inhibitor therapy. Immune response to cancer and relationship among cancer cells, T cells and antigen presenting cells. (A) Tumor cells are recognized by the intrinsic immune cells. Once recognized, immune cells trigger initial intrinsic immunity and IFN secretion, promoting chemokines production, and the recruitment of immune cells. Mature DCs take up tumor antigens, which finally presented as MHC-I antigenic tumor peptides complex, migrate to the draining lymph nodes, where initial tumor-specific Th1 CD4+ T cells and CTLs are induced by antigen cross-presentation. IL-2 and IL-15 help to maintain CTLs production and viability. (B) Mechanism of anti-PD-1/PD-L1/CTLA-4 antibody. PD-1 is expressed on T cells, while PD-L1 is expressed on tumor cells. These two form the PD-1/PD-L1 axis that mediates T cell function inhibition. The function of anti-PD-1/PD-L1 antibodies lies in restoring the T cell effect by blocking the PD-1/PD-L1 interaction. CTLA-4 on T cells binds to B7 ligand on the APC, leading to immunosuppression. Anti-CTLA-4 antibodies inhibit the binding between CTLA-4 and B7, which prolongs T cell activation, restore T cell proliferation, and establish an immune response to tumor-associated antigens. IFN, interferon; DC, dendritic cell; MHC-I, major histocompatibility complex class I; CTL, cytotoxic T lymphocyte; APC, antigen-presenting cell.
Tumor regression announces the victory of the immune system in the fight against tumors, but this is not the end. The ‘3Es’ hypothesis describes the continuous dynamic changes of immune cells and tumor cells in the three sequential processes of elimination, equilibrium, and escape (9, 12). Some tumor variants can escape from killing and enter dynamic equilibrium, a phase in which killing factors within the tumor exert a solid and continuous selective pressure on tumor cells. Due to genomic instability, although some of the escaped variants are destroyed, new variants carrying different mutations re-emerge and enhance resistance to immune attack. The result of the equilibrium is the generation of a new population of tumor clones with reduced immunogenicity and nearly ineffective anti-cancer defense from heterogeneous parents. It is well established that anti-cancer defenses are ineffective in solid tumors (13).
To survive and proliferate, tumors employ various strategies to escape anti-tumor immunity. Bypassing, overriding, or abolishing is the key mechanisms to restart tumor elimination, and restore T cell-mediated anti-tumor immunity in current cancer immunotherapies (14). T cells exerting immune effects requires multiple sequential steps, including successful antigen presentation and recognition, activation and proliferation, transport, and execution of killing effects. Each step is regulated by the balance between stimulatory and inhibitory signals.
Immune checkpoints are a range of inhibitory molecules that physiologically balance co-stimulatory pathways to fine-tune immune effects and prevent further immune responses. They involve inhibitory receptors and their ligands. Under normal physiological conditions, immune checkpoints are essential for maintaining self-tolerance, regulating the duration and intensity of normal physiological immune responses, and minimizing collateral damage to healthy tissues (8). Tumors can also express immune checkpoints to escape from the elimination phase and promote progression. Meanwhile, it also provides a chance to restore the anti-tumor immunity of T cells through antibodies that bind to immune checkpoints, and block inhibitory receptor-ligand interactions. These mechanisms established a cornerstone for the development of ICIs.
Currently, ICIs that have been approved by the FDA target three different immune checkpoints, PD-1, PD-L1, and CTLA-4. The bind between CD28 on T cells and B7 ligand on MHC cells is essential for T cell function, further induction, and amplification of the immune response (15). In response to T cell activation, CTLA-4 is induced to upregulate on T cells, competing with higher affinity for binding to B7 ligand on the APC, which leads to downregulation of T cells and immunosuppression (16). Anti-CTLA-4 antibodies inhibit the binding between CTLA-4 and B7, which could prolong T cell activation, restore T cell proliferation, and establish an immune response to tumor-associated antigens (TAAs) (17). Ipilimumab is the currently approved ICI targeting CTLA-4.
PD-1/PD-L1 inhibitors mainly block the feedback mechanism between T cells and tumor cells in TME. PD-1 is expressed on T cells, while PD-L1 is expressed on tumor cells. These two form the PD-1/PD-L1 axis that mediates T cell function inhibition. The function of anti-PD-1/PD-L1 antibodies lies in restoring the T cell effect by blocking the PD-1/PD-L1 interaction (18). Currently, Nivolumab and Pembrolizumab target PD-1, while Atezolizumab, Durvalumab, and Avelumab target PD-L1 (2) (Figure 1B).
To date, clinically approved ICIs targeting PD-1/PD-L1 and CTLA-4 have shown promising efficacy and became the standard treatments for various cancers, such as advanced melanoma, NSCLC, and solid tumors with microsatellite instability (2, 12, 19). However, substantial evidence indicated that responses to ICIs varied widely among different patient population. For patients with NSCLC, the efficacy of ICIs was largely associated with the expression of PD-L1, as well as tumor percentage score (TPS). For example, the results of CheckMate-227 study suggested, treated with nivolumab in combination with ipilimumab, a response rate (ORR) of 35.9% was achieved in NSCLC patients with PD-L1 TPS ≥1% (20). These findings were born out in KEYNOTE-598 study, as pembrolizumab combined with ipilimumab provided an ORR of up to 45.4% in patients with PD-L1 TPS ≥50%. Some candidates did not benefit from the ICIs, and even suffered progression during the administration of ICIs (21).
Extensive studies have been conducted to explore the mechanisms of ICIs resistance. From the perspective of the anti-tumor immunity process, ICIs resistance can arise at all stages, from antigen uptake and presentation to T cell killing. For example, antigen loss, impaired antigen presentation, reduced T cell infiltration, lack of PD-L1 expression, and other immune co-suppressive molecules were associated with response to ICIs. In addition, intrinsic immunity components, especially IFN, which plays a crucial role in anti-tumor immunity, are related to ICIs resistance. Other components in tumor microenvironment (TME), such as immunosuppressive molecules, immunosuppressive cells, and neutralizing antibodies for PD-1 antibodies, can also influence ICIs response. In addition, the intestinal microbiota is involved in such progress. However, due to the lack of sufficient data the mechanisms remain unclear at present (22, 23) (Figure 2).
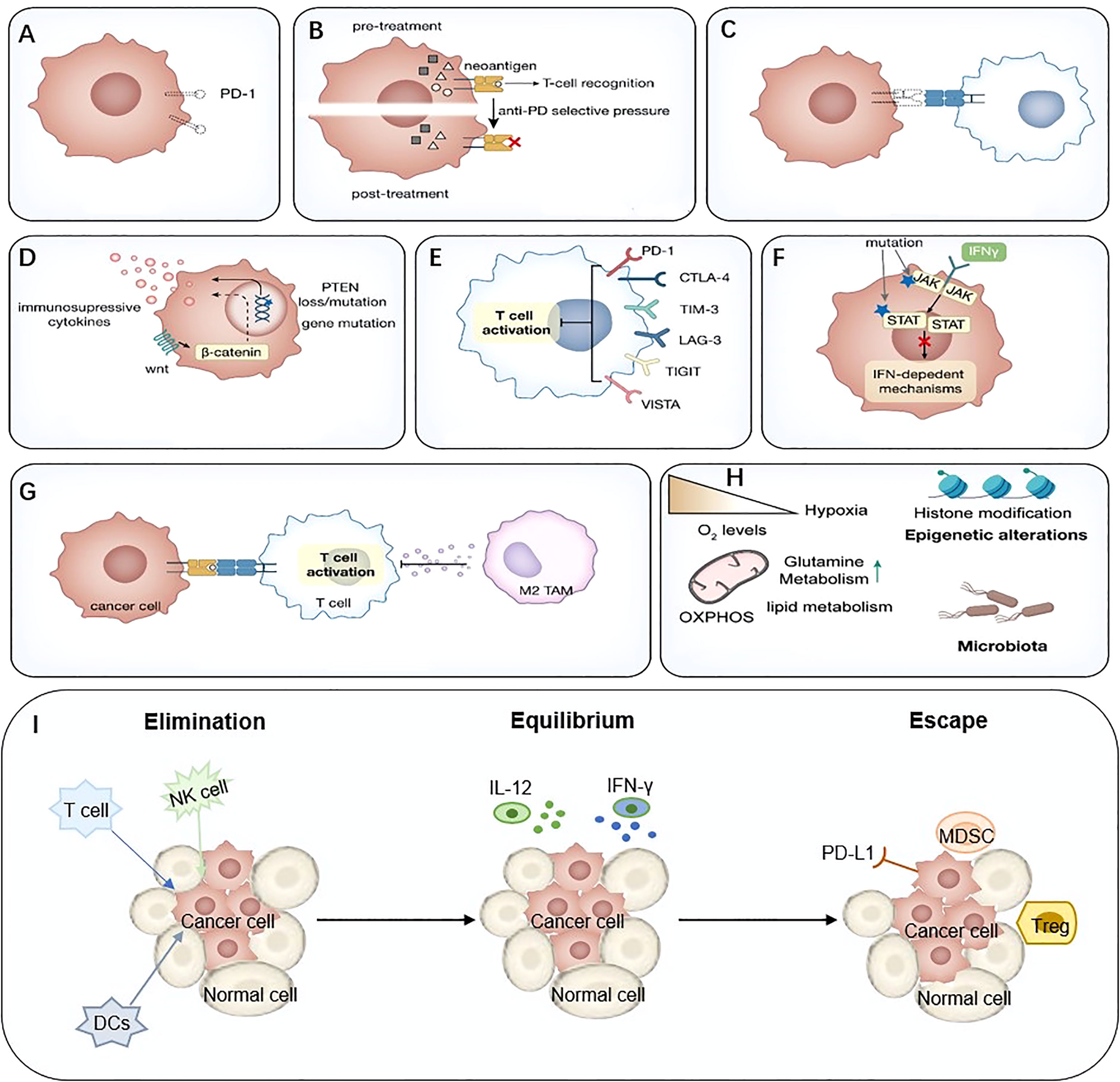
Figure 2 Mechanisms of immune-mediated resistance to ICIs. (A) Lack of PD-L1, (B) Neoantigen-depletion, (C) Antigen presentation defect, (D) Tumor-mediated immune-suppression or –exclusion, (E) Immune inhibitory receptors, (F) Abnormal IFN signaling pathway, (G) Immunosuppressive cells, (H) Metabolic reprogramming, (I) The ‘3Es’ hypothesis. IFN, interferon; ICIs, immune checkpoint inhibitor.
The main effect of PD-1/PD-L1 inhibitors is to block the PD-1/PD-L1 axis. Based on the expression pattern of tumor-infiltrating T cells (TIL) and B7-H1 (PD-L1), the tumor immune microenvironment was classified as four types: T1 (B7-H1−, TIL−), T2 (B7-H1+, TIL+), T3 (B7-H1−, TIL+), and T4 (B7-H1+, TIL−) (24, 25), among of which, the Type II was most likely to respond to ICIs. This hypothesis is supported by clinical practice, as patients with PD-L1 positive were associated with better response to anti-PD-1 therapy. Despite of this, but the efficacy of ICIs limited in a subset of these patients, suggesting the existence of other immunosuppressive pathways. For the other three types, it can be inferred that the T1 type should be the least effective, as it is lack of both TIL and B7-H1. And for T3 type, it is identified as infiltrated with TIL but lack of PD-L1 expression, which possibly due to the lack of IFN-γ production by effector T cells ((Teffs), a sign of cellular dysfunction (24). Unfortunately, the proportion of patients with T2 is not high. Data from previous studies suggest that only 17% of NSCLC patients have type II tumor immune microenvironment. Furthermore, more than half of tumors lack PD-L1 expression or TIL infiltration (25–27). Interestingly, some studies suggest that neither knockout nor overexpression of PD-L1 in tumor cells affected the efficacy of PD-L1 inhibitors, but it is the PD-L1 located on immune cells (DCs, macrophages) that correlates with the efficacy of anti-PD-1 alone or in combination with anti-CTLA-4. Although this finding remains to be further validated, it suggests a role of other cellular components within TME in the efficacy of ICIs (28).
Assessment of tissue PD-L1 expression has predictive utility for treatment outcome (29). Notably, current clinical trials largely examined the predictive value of PD-L1 expression on tumor cells. This remains some limitations. For example, due to the dynamic progression of tumors, this method can only specify tumors’ PD-L1 profile at a particular time. In addition, due to the limitation of biopsy sample size and tumor spatial, as well as temporal heterogeneity, biopsies may miss tumor tissues with high PD-L1 expression. This may explain the response of a proportion of PD-L1-negative patients to ICIs (30, 31). Existing evidence suggested, besides of tumor cells, the PD-L1 expression on tumor-infiltrating immune cells (e.g., macrophages, DCs, neutrophils, and T cells) or elsewhere could also be assessed. By using immunohistochemistry (IHC), Taube JM et al. found the expression of PD-L1 was also observed in the TILs and associated macrophages/histiocytes, which was associated with the outcome of patients with metastatic melanoma (32). Meanwhile, it has been established that PD-L1-expressing tumor-associated macrophages (TAMs) could impair the function of NK cells, and contribute to the inefficient sensitivity to PD-1 inhibitors (33). Furthermore, study performed by Hinterleitner C and their co-workers have found the interaction between the blood platelets and lung cancer cells. Specially, PD-L1 expression on platelets could not only reflect that on tumor, but also inhibit CD4+ and CD8+ T cells, which provided a new perspective to overcome the limitations of traditional quantification of PD-L1 expression on tumor cells (34). Further understanding the host immune system and TME will better identify patients who will benefit from these agents. For patients with high PD-L1 expression but insensitive to ICIs, other resistance mechanisms may play a leading role. For example, EGFR and HER-2 mutations, and the fusion of ALK, ROS1, RET, MET could define NSCLC subsets with minimal benefit from ICIs, despite of high PD-L1 expression (35).
On the other hand, in addition to membrane-bound form, PD-L1 could be secreted as a truncated form, which called soluble PD-L1. Preclinical evidence suggested that this form also contributed to the resistance to immunotherapy (36, 37). In a retrospective study, blood samples from patients treated with ICIs were analyzed. The results revealed that soluble PD-L1 levels could be regarded as an independent prognostic factor, as inferior outcomes were observed in patients with high soluble PD-L1 (38). This finding has been conquered in other studies (39–41). But the potential mechanism needs to be further investigated.
Tumor cells may have defects in the processing and presentation of antigen and antigen, resulting in the inability of the immune system to detect new antigens and initiate an effective immune response. Each human leukocyte antigen class I (HLA-I) molecule binds specific peptides derived from intracellular proteins and is expressed on the cell surface for presentation to CD8+ T cells. β2 microglobulin (B2M) is necessary to maintain the stability of HLA-I molecules, and mutations in B2M can lead to MHC instability and failure of antigen presentation. This mechanism has received more attention. In a study of NSCLC, acquired homozygous loss of B2M led to a lack of HLA-I molecule expression in tumors, and mouse models also exhibited resistance after the knockdown of B2M, suggesting that disruption of HLA-I molecule-associated antigen presentation can lead to ICIs resistance (42). The lack of HLA-I expression in small-cell lung cancer shows minimal ICIs activity, and HLA-I upregulation through IL-27/STAT3 activation can enhance ICIs efficacy (43). In addition, some studies suggest that 40% of NSCLC patients have a heterozygous loss of HLA, which is associated with tumor immune escape, but it is not clear how it relates to ICIs drug resistance (44).
The genotype of HLA may also influence the tumor response to ICIs. The anti-tumor activity of ICIs depends on CD8+ T cells and HLA-I-dependent immune activity (45). The HLA-I molecule is highly polymorphic, and each variant binds a particular peptide ligand. Thus, if HLA-I locus is heterozygous, it is most likely to bind the largest combinations of peptide ligands. In contrast, more presence of homozygous genes at the HLA-I locus theoretically is expected to provide relatively fewer combinations of MHC complex to CTLs (46). Studies have shown that maximal heterozygosity of HLA-I A/B/C improves overall survival (OS) after ICIs therapy, compared to patients with at least one homozygous HLA locus. Among NSCLC patients treated with nivolumab, those not expressing HLA-A*02 and alleles had inferior outcomes with progression-free survival (PFS) of 7.5 months, while HLA-A*-01 positive patients had the best outcome with a PFS of 22.6 months. Regarding the impact of heterozygosity on prognosis, heterozygotes in HLA-A were associated with worse OS, while heterozygotes in DRB1 prolonged OS, which highlights the influence of host germline genetics (47). Moreover, tumor cells may also express non-classical MHC-I antigens (like HLA-G, HLA-E), which can bind to inhibitory receptors on T cells and other immune cells to suppress cell function, causing ICIs resistance (23).
The expression of co-suppressor molecules in TME is the underlying mechanism of tumor immune escape, and targeting the PD-1/PD-L1 co-inhibitory axis demonstrates that blocking co-inhibitory signaling can reactivate T cells to mediate tumor elimination or control. However, the expression of co-inhibitory molecules other than PD-L1 on cancer cells could potentially lead to the failure of ICIs therapy.
Following the first series of inhibitory receptors (PD-1, PD-L1, CTLA-4), three new inhibitory receptors stood out: lymphocyte activation gene 3 (LAG-3), T cell immunoglobulin and mucin domain 3 (TIM-3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT). LAG-3 is expressed on activated NK cells, Teffs, regulatory T cells (Tregs), B cells, and plasma cell-like DCs, preventing immune overactivation (48–50), TIM-3 has been identified on Tregs, DCs, NK cells, and monocytes, and has similar effects to LAG-3. TIGIT is also a co-inhibitory receptor on various immune cells (49). These molecules are promising therapeutic targets, and there is some evidence that they are associated with anti-PD-1 resistance. In 90 patients with advanced NSCLC treated with anti-PD-1 therapy, elevated LAG-3 expression was associated with PD-1 blockade insensitivity and resulted in shorter PFS (51). And in histology, LAG-3 enrichment suggests an immune contexture represented by CD8+ T cell dysfunction, which is associated with a worse prognosis (52). Another study also suggested anti-PD-1 resistant NSCLC harbored higher expression levels of TIM-3, LAG-3, and CTLA-4 on CD4+ and CD8+ T cells in, but only TIM-3 was significantly increased, suggesting the TIM-3 upregulation surrogate mechanism (53). It is noteworthy that studies on renal cell carcinoma (RCC) suggest that the expression of LAG-3, TIM-3, and TIGIT is exclusive at the single-cell level, with most suppressive costimulatory receptor-positive cells expressing mainly one of them, and no circumstance of three molecules are all low expressed. Among the three members, RCC with high LAG-3 expression had the worst survival rates and the most severe immunosuppressive microenvironment. It was resistant to anti-angiogenic therapy and ICIs, suggesting that tumor classification and outcome prediction can be based on dominant immunosuppressive molecules (54). TIGIT can also be elevated in NSCLC after resistance to anti-PD-1 therapies, but this remained controversial (42). Some clinical trials combining anti-PD-1 with anti-LAG-3/TIM-3/TIGIT have shown improved anti-PD-1 efficacy, providing strategies to overcome drug resistance (48, 55–57).
V-domain immunoglobulin suppressor of T cell activation (VISTA), or PD-1 homolog (PD-1H), is a novel inhibitory immune checkpoint, which is expressed on myeloid cells, lymphoid cells, and tumoral cells (58, 59). A study showed that VISTA was detected in 99% of NSCLC cases and predominantly expressed on stromal cells. But its expression was correlated spatially with immune infiltration and PD-1/PD-L1 expression, and regulated by local cytokines and IFN, suggesting that VISTA may not induce PD-1 resistance (60). However, upregulation of VISTA on infiltrating immune cells has been reported in prostate cancer treated with ipilimumab (anti-CTLA-4), suggesting its role in anti-CTLA-4 resistance (61). In addition, HMBD-002, an anti-VISTA antibody, can reduce myeloid-derived suppression of T cell activity and prevent neutrophil migration, stimulating a pro-inflammatory phenotype characterized by a Th1/Th17 response, making it a promising therapeutic target for ICIs combination (62).
CD91, also known as low-density lipoprotein receptor-related protein-1 (LRP-1), was a receptor expressed on antigen-presenting cells (APCs). Recent studies explored its role in immunosurveillance and found it was critical to trigger immune responses. Sedlacek AL et demonstrated that CD91 could not only activate NK cells through APCs in vivo, but also cytokines in NK cells. This procedure was essential for T cell and APC function (63). In addition, by stimulating the secretion of cytokines from macrophages, CD91 induced the activation of DCs to produce co-stimulation. When the expression of CD91 on DCs was depleted, mice were more likely to suffer the development of tumors induced by chemical compound. And in clinical practice, patients with advanced melanoma harboring high CD91 expression had better prognosis. Based on this, authors highlighted the potential function of CD91 in immunotherapy (64). However, it still needs further investigation. For example, heat shock protein (HSP) gp96 could combine with CD91. But the dose of gp96 had opposite roles, as low dose contributed to Th1 immune responses, and high dose induced immunosuppression via Tregs (65–67). Despite all this, the importance of CD19-mediated immune responses possess a variety of research potential.
Lymphocyte infiltration and PD-L1 expression are vital factors for effective ICIs, and most tumors with anti-tumor immunodeficiency was lack of CD8+ T cell infiltration and tend to be resistant (68). Several signaling pathway abnormalities are associated with lymphocyte infiltration in TME and PD-L1 expression on the surface of cancer cells.
Existing evidence indicated that Wnt/β-catenin pathway activation can lead to non-inflammatory TME and cause anti-PD-1/PD-L1 resistance. In detail, β-catenin activation can lead to failure of DCs and effector T cell recruitment, resulting in impaired immune response (69–71). In addition, β-catenin can regulate Treg cell infiltration (72). PTEN is a negative regulator of the PI3K/AKT pathway, and PTEN loss leading to anti-PD1 resistance has been reported in various cancers, such as lung cancer and melanoma (70, 73). PTEN loss in tumor cells increases the expression of immunosuppressive cytokines, leading to reduced T cell infiltration in tumors and inhibition of autophagy. This could further reduce T-cell-mediated cytotoxicity and cause ICIs resistance (73–75). Moreover, patients carrying PTEN mutations could not benefit from ICIs, even if they possess high tumor mutation burden (TMB) and positive PD-L1 expression. But combined with temsirolimus (mTOR inhibitor), synergic therapeutic effects can be reached, which provided a combination option (76).
The RAS/MAPK pathway is a significant factor driving PD-L1 expression (77, 78), but its activation in triple-negative breast cancer is associated with reduced TIL infiltration (79, 80), which may cause immune escape and ICIs resistance. If MAPK activation causes low TIL infiltration and high PD-L1 expression simultaneously, it is ambiguous whether ICIs resistance could occur. In contrast, studies in melanoma suggest that MAPK pathway inhibition with targeting agents can lead to cross-resistance between targeted therapies and ICIs, following reduced infiltration of CD103 DCs (81), suggesting multiple functions of MAPK activation. In conclusion, the impact of MAPK on anti-PD-1 effects in NSCLC needs to be further investigated.
The PI3K/AKT/mTOR pathway was also associated with the expression of PD-L1. For example, by activating β-catenin and the downstream mTOR signaling, AKT pathway increased PD-L1 expression (82). Meanwhile, Liu M et al. found that PD-1 on myeloid-driven suppressor cells (MDSCs) could bind to PD-L1, and trigger PI3K/AKT pathway in B cells, which further impaired the function of T cells and led to tumor immune escape (83). It has been confirmed in a study that the combination of mTOR inhibitor (rapamycin) and anti-PD-1 blocked the progression of NSCLC (82). Currently, a phase I trial is active, which aims to evaluate the efficacy and safety of mTOR inhibitor combined with durvalumab for stage I-IIIA NSCLC (NCT04348292). We are looking forward to the results.
IFNs are essential for anti-tumor immune response and critical cytokines in cancer elimination. IFNs are classified into three types: type I (IFN-α, IFN-β, IFN-ϵ, IFN-κ, and IFN-ω), type II (IFN-γ), and type III (IFN-λ), which have multiple roles in anti-tumor cell proliferation, promotion of apoptosis, anti-angiogenesis, increasing antigen processing and presentation of APCs. Through multiple pathways, IFN can ultimately exert anti-tumor immune effects by increasing intrinsic and adaptive immune cell functions, and IFN mainly induces intracellular changes through the JAK/STAT pathway (84). Despite these positive effects of IFN, chronic IFN exposure exerts solid selective pressure on tumors and promotes tumor immune escape. In this process, tumors can not only upregulate the expression of immunosuppressive receptors and metabolites (PD-L1, CYLA-4, indoleamine 2, 3-dioxygenase (IDO)). but also downregulate JAK inhibitors, leading to sustained activation of the JAK/STAT3 pathway and promoting tumor growth (85). Sustained IFN exposure also attracts Tregs and suppressive MDSCs infiltration, leading to immunosuppression. Therefore, it is reasonable to discuss the effect of IFN on ICIs resistance.
The contribution of IFN to anti-PD-1 resistance is currently evaluated. On the one hand, the inactivation of the IFN effector pathway could impair IFN effects. For example, loss-of-function mutations in JAK1/2 can lead to a lack of PD-L1 expression, resulting in no response to ICIs, regardless of high TMB (86, 87). In addition, studies suggest that ICI-resistant melanomas contain B2M loss, which mediates resistance to CD8+ TIL, and JAK mutations create a severe pan-T cell immune escape phenotype (88). The second aspect is that IFN can initially induce anti-tumor effects but develop a range of mechanisms leading to resistance during ICIs treatment. Sustained IFN-γ signaling can lead to STAT1-related epigenomic changes to enhance the expression of IFN-stimulated genes (ISG), which leads to PD-L1 non-dependent resistance and multiple T cell inhibitory receptor ligands (89). Notably, another study suggests that STAT pathway silencing can lead to MHC II inhibition and immune escape, which may also be associated with ICIs resistance (88). Further exploration is necessary. In a mouse lung cancer model, IFN-γ was observed to cause phase separation of Yes-associated protein (YAP) to form condensation and translocate to the nucleus to form a potent transcription center, promoting the expression of multiple genes, including CD155 (ligand for TIGIT), leading to CTLs function inhibition and anti-PD-1 resistance (90). In addition, chronic IFN-I has been shown to trigger lipid peroxidation of terminal CD8+ T cells and accelerate their depletion, suggesting an effect of IFN on immune cell metabolism, which may also contribute to ICIs resistance (91).
TME is a highly heterogeneous environment composed of cancer cells, stromal tissue, extracellular matrix, and immune cells, characterized by high vascularization, glucose depletion, and hypoxia, where collective molecular signaling of immune cells affects tumor outcome by influencing the balance of inhibitory signaling and cytotoxic responses in the vicinity of tumor cells (23, 92). The acquisition and maintenance of cancer features, such as proliferative signaling, resistance to cell death, induction of angiogenesis, activation of invasion and metastasis, triggering tumor-promoting inflammation, and avoidance of immune destruction, rely to varying degrees on TME (93, 94). The immune system is an essential determinant of TME, and complex interactions between tumor cells and host immune responses are related to multiple components, including tumor parenchymal cells, fibroblasts, mesenchymal cells, blood, lymphatic vessels, TIL, chemokines, and cytokines.
Various components in TME can shape the immunosuppressive microenvironment and lead to ICIs resistance (23). Tregs negatively regulates the function of T cell (95). In cancer, Tregs mediate the dysfunction of the CD8+ T cells, which is characterized by upregulation of surface inhibitory receptors (e.g., CTLA-4). Furthermore, Treg-mediated IL-2 deprivation can exacerbate this dysregulation, leading to immune escape (96). Whether Tregs can cause ICI resistance is inconclusive, but some studies suggest its possible role in ICIs resistance. Co-blockade of PD-1 and PD-L1 in triple-negative breast cancer can upregulate the expression of immune checkpoints (e.g., CTLA-4, TIM-3, LAG-3) on Tregs, which could potentially lead to more severe T cell dysfunction through other co-inhibitory molecules expression (97). Moreover, selective inhibition of TGF-β1 released from Treg cells can overcome anti-PD-1 resistance (98), suggesting that TGF-β may be one of the reasons for Treg-mediated resistance (99). In addition, oxidative stress in TME can lead to Treg cell apoptosis. Apoptotic Tregs can release large amounts of ATP that further convert to adenosine via CD39 and CD73, exacerbating immunosuppression and eliminating PD-L1 blockade-mediated T cell activation (100).
TAMs are abundantly distributed and highly plastic in TME, and they are often defined as M1 and M2 subpopulations (101). M1 TAMs are the primary to trigger inflammation in the early stage of cancer. After that, TAMs undergo M2 polarization in response to signals in the TME (102). In contrast to tumor cells, PD-L1 expression on TAMs is mainly independent on local IFN-γ level. TAMs residing in TME have been shown to inhibit the anti-tumor effects of ICIs through multiple mechanisms (103), and TAMs enrichment-mediated anti-PD-1 resistance is independent of PD-L1 status (104). First, M2 TAMs can lead to T cell exclusion. In lung adenocarcinoma (LUAD), CD8+ T cell can interact with TAMs, leading to its poor migration and low CTLs infiltration in the TME. TAMs depletion reverses the circumstance and restore ICIs resistance (105). Additionally, induction of TAMs reprogramming to the M1 type could restore ICIs resistance, indirectly proving the role of M2 TAMs in ICIs resistance. Meanwhile, studies suggest that the re-polarization of M2 to M1 may cause PD-L1 upregulation, so ICIs combined with TAMs reprogramming may be a novel therapeutic strategy (106). Secondly, IDO is an essential immunomodulatory enzyme that inhibits T cell proliferation, promotes Treg cell differentiation, and induces TAMs and DCs to differentiate to suppressive phenotypes, producing potent immunosuppression (107). TAMs could in turn induce M2 transformation, exacerbating the immunosuppressive phenotype (89). Although various studies have demonstrated the contribution of IDO to T cell dysfunction, how M2 TAM-derived IDO-induced T cell tolerance contributes to PD-1/PD-L1 blockade resistance still needs to be thoroughly investigated.
MDSCs are heterogeneous bone marrow cells recruited to immunosuppressive TME. Some studies suggest that MDSCs may serve as a prognostic marker for ICIs, but the relationship between MDSCs and ICIs resistance has not been fully revealed. In NSCLC patients, the peripheral blood of non-responders had a higher frequency of MDSCs and fewer NK cells, compared with responders at the end of one round of PD-1 treatment (108). And high MDSCs in the peripheral blood was negatively associated with anti-PD-1 efficacy and prognosis (109, 110). Some studies also suggest that inhibition of MDSCs migration to TME may enhance the efficacy of anti-PD-1 and overcome PD-1 resistance (111–115). This evidence suggests that MDSCs may be involved in anti-PD-1 resistance. Theoretically, MDSCs, as a class of suppressive immune cell population, can interact with other immune cell components, secrete negative immune regulatory molecules, and are associated with resistance to chemoresistance and targeted therapy. Their relevant mechanisms in immunotherapy resistance deserve to be investigated (116–118).
It has been shown that genomic alterations in cancer cells can lead to ICIs resistance. Homozygous deletion of the 9p21.3 gene is one of the common genomic defects, accounting for approximately 13% of all cancers. 9p21 deletion is associated with a ‘cold’ tumor immune phenotype. This phenotype is characterized as reduced abundance of TIL (especially T/B/NK cells) immune cell trafficking and activation, PD-L1 positivity, and activation of immunosuppressive signaling. Clinical studies have demonstrated that patients with 9p21 loss have significantly lower response rates to ICIs and worse outcomes (119). In addition, KRAS mutations are common oncogenic mutations in NSCLC, and KRAS G12C mutations are associated with higher levels of inflammation in TME and better ICIs response (35, 120). However, conversely, KRAS-G12D mutations can suppress PD-L1 expression through the PI3K/AKT pathway and reduce chemokine CXCL10/CXCL11 levels through downregulation of high-mobility group AT-hook 2 (HMGA2), leading to reduced CTLs recruitment and ICIs resistance (121).
Serine/threonine kinase 11 (STK11) is a critical oncogene, and STK11 mutation is a common mutation secondary to KRAS mutation. Compared with wild-type STK11, NSCLC patients carrying STK11 mutations have a significantly lower OS rate (122). Genetic ablation of STK11 in animal models directly promotes resistance to anti-PD-1 therapies, and in a cohort of NSCLC patients, STK11mut was associated with PD-L1 deletion, suggesting STK11 is related to PD-L1 expression and ICIs resistance. Moreover, STK11-deficient tumors were associated with a low density of CTLs. However, the negative impacts of STK11/LKB1 genomic alterations on resistance to ICIs are also seen in PD-L1 positive NSCLC, suggesting that PD-L1 expression is partially dependent on STK11 (123). In the periphery, STK11mut tumors have lower NK cells and CD4+/CD8+ effector memory T cells. In TME, there is a significant upregulation of neutrophil-related markers (CXCL2, IL-6, CSF3) and inhibitory immune checkpoint killer Ig-like receptor (KIR), indicating the poor outcome of STK11 mutation may be attributed to enrichment of immunosuppressive mechanisms (124).
Moreover, kelch-like ECH-associated protein 1 (KEAP1) mutation can be seen in 20% of NSCLC and is the third most frequently mutated gene in LUAD, with a common substrate of neuropilin 2 (NRP2) (125). And KEAP1 is like to co-mutate with STK11 and KRAS (126). A study suggests that KEAP1 mutation in NSCLC is correlated with inferior ICIs response (127), and patients carrying both STK11 and KEAP1 mutations had worse outcomes compared with those harboring one mutation, suggesting an additive effect of these mutations (128, 129). Despite KEAP1 show its role in chemotherapy resistance (130), the contribution of KEAP1 mutation to ICIs resistance is unclear. The study shows that patients who harbored KEAP1 mutation had higher PD-L1 expression, compared with wild type, which may assist ICIs efficacy (131). But other studies also point out that the contribution of KEAP1 mutation to ‘cold’ microenvironment was characterized by failure in IFN expression, innate immune cell recruitment and TIL infiltration (132–136).
There are some ‘driver mutations’ in cancer that are responsible for both the initiation and maintenance of the malignancy. Receptor protein tyrosine kinases like EGFR and ALK are typical examples, whose mutations are related to ICIs resistance (137). The EGFR signaling pathway is one of the most important oncogenic pathways in NSCLC (138). Recent studies demonstrated their impact on the immune system was apart from tumor biology, and relationships between activation of EGFR and PD-L1 upregulation have been widely validated clinically in NSCLC patients (139, 140). But under high PD-L1 circumstances, EGFR-mutant cancer is insensitive to ICIs. Studies show that EGFR-mutant and ALK-positive NSCLCs lack concurrent PD-L1 expression and high levels of CD8+ TILs (141). EGFR mutation is correlated with uninflamed TME with immunological tolerance and weak immunogenicity, resulting in an inferior response to PD-1 blockade (142).
Tumors carrying oncogenic mutations usually follow the principle of oncogenic addiction, which means that the evolution of these tumors depends on the specific activity of an activated oncogene that confers a survival advantage to tumor cells (143, 144). Consequently, these tumors were usually characterized as low TMB and ‘cold’ TME, which is related to ICIs resistance (144, 145). For driver mutation-positive NSCLC, target therapy is currently the optimal choice. Considering the effect of driver mutations on the immune system, combining targeted therapy and immunotherapy may be feasible. Unfortunately, the therapeutic role of ICIs in oncogene-driven NSCLC remains unclear. The level of evidence supporting the use of immunotherapy for patients with NSCLC harboring driver mutations is quite low, which is because patients with EGFR or ALK alterations are often excluded in most ICIs trails (144). Some clinical trials that expect to find rationale to combine tyrosine kinase inhibitor (TKI) and ICIs show limited clinical benefits but with more adverse events (146, 147) (Table 1). Therefore, more factors need to be considered to determine the feasibility of combining ICIs and targeted therapy.
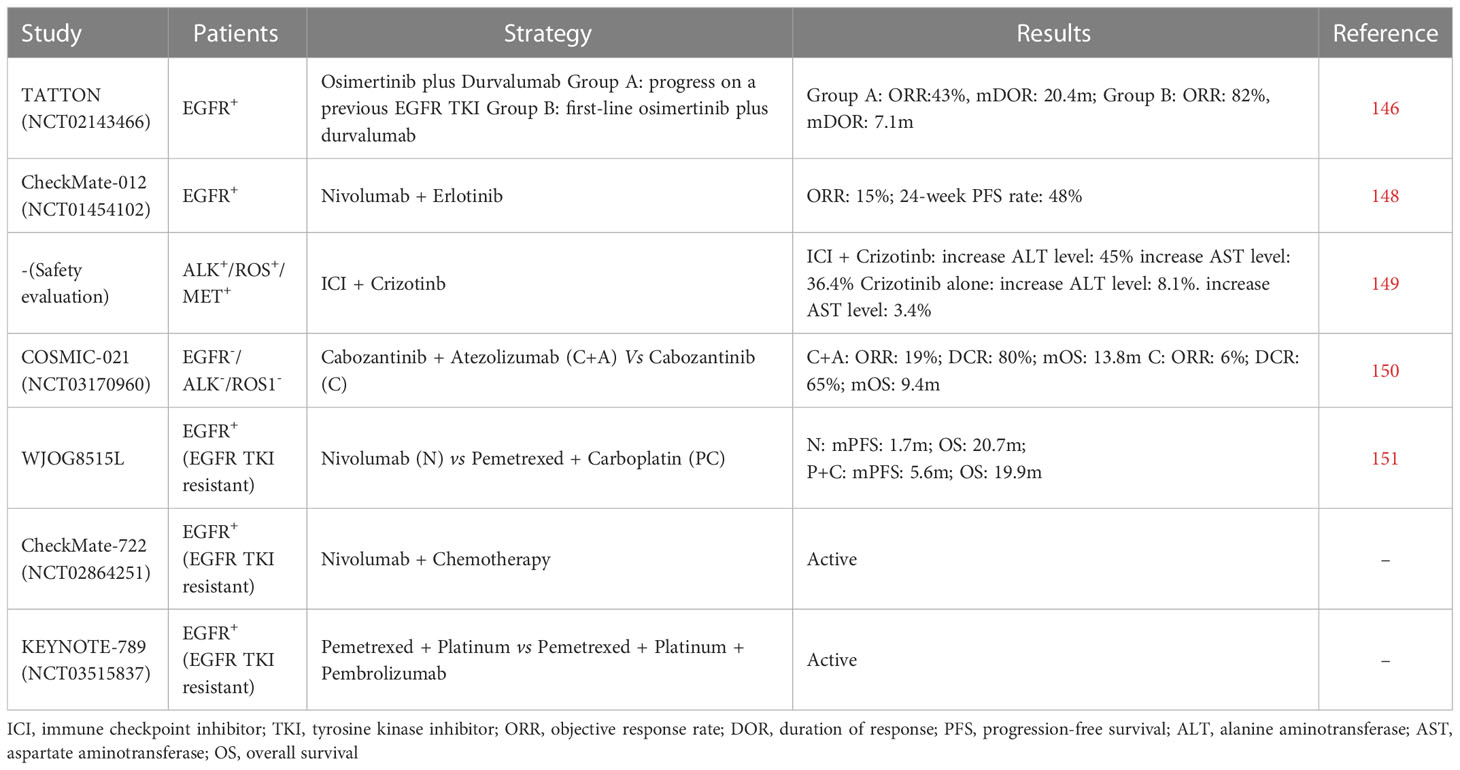
Table 1 Clinical trials investigating the effects of ICIs in driver mutation-positive NSCLC.
Metabolic reprogramming is considered a new hallmark in cancer (152). Metabolic reprogramming was essential for the proliferation, differentiation, and various functional assays of immune cells (153, 154). In addition, nutrient depletion, hypoxia, reactive nitrogen, and oxygen intermediates production in TME force immune cells to undergo metabolism reprogramming, which further impair immune cell function and cultivate an immunosuppressive TME (155–157). Metabolites in the TME can, in turn, affect immune cell differentiation and their function (158). It has been shown that PD-1 can lead to an immunosuppressive landscape through immune cell metabolism reprogramming, and ICIs application can reinvigorate the immune response (159, 160). Based on this, it is rationale to discuss the relation between ICIs resistance and cancer metabolism.
Existing evidence suggested that oxidative metabolism and hypoxia may contributed to ICIs resistance. In head and neck squamous carcinoma, tumors resistant to ICIs showed a more active oxidative metabolism, leading to TME hypoxia and a decrease of CD8+ T cells (161). Studies in melanoma also shown that enhanced tumor oxidative metabolic activity contributed to increased oxygen consumption, which correlated with T cell depletion and reduced immune activity (162, 163). In conclusion, tumors resistant to ICIs was characterized as a ‘hypermetabolic’ phenotype, with an upregulation of oxidative phosphorylation activity. On the one hand, such phenotype exacerbated hypoxia, which contributed to T cell apoptosis by inhibition of CCR7 and NK cells (164). Meanwhile, tumor cell hypermetabolism may lead to T cell metabolic starvation, resulting in loss of glycolytic potential (163). Although how PD-1 treatment impacts tumor oxidative metabolism is unknown, these studies suggest that TME hypoxic state after ICIs treatment could be served as a potential prognostic biomarker.
T cell metabolism may relate to ICIs resistance. It was established that PD-1 signaling activation can attenuate T cell glycolysis, glutaminolysis, and branched-chain amino acid metabolism. It can also affect mitochondrial sub-microstructure, leading to reduced mitochondrial depolarization and mitochondrial dysfunction, which could impair cellular oxidative phosphorylation. In contrast, fatty acid oxidation was enhanced during the management of ICIs (165, 166). Overall, PD-1 signaling activation altered T cell metabolic activity, which prevents terminal differentiation and glycolysis-induced death of T cells (166). Consequently, blockading PD-1 signaling with ICIs may accelerate apoptosis and deletion of Teffs, resulting in no response to ICIs.
Glutamine metabolism is essential for tumors, while glutamate is necessary for T cell activation and expansion (167). When tumor cells competitively intake glutamine could lead to T cell dysfunction and ICIs resistance (168). A subset of lung squamous carcinoma resistant to ICIs was defined as the hypermetabolic, which was dependent on glucose and glutamine. When glutamine intake was inhibited, the effect of ICIs was restored. These findings established a cornerstone that the inhibition of glutamine metabolism may enhance the efficacy of ICIs (169). In addition, after inhibiting the glutamate/cystine reverse transporter, melanoma cells secrete large amounts of PD-L1 in the form of exosomes to induce M2 polarization of TAM, leading to ICIs resistance (170). Combined with the evidence above, high glutamate in tumor cells is suggested to be detrimental to ICIs response.
Moreover, glutamate has novel atypical functions. Cancer cells with abnormal glutamate decarboxylase (GAD) expression could synthesize γ-aminobutyric acid (GABA). Subsequently, GABA binds and activates GABAB receptors to inhibit GSK-3β and enhance β-catenin signaling, stimulating tumor cell proliferation, and inhibiting CD8+ T cell infiltration, leading to anti-PD-1 resistance (171).
Disturbances of lipid metabolism also contribute to the development of cancer, as well as ICIs resistance. First, it has been mentioned above that chronic IFN can lead to lipid peroxidation in terminal CD8+ T cells and accelerate their depletion (91). Second, oxidized low-density lipoprotein (LDL) in serum not only suppresses T cell function but also upregulates heme oxygenase 1 (HO-1) expression, which induces a cytoprotective stress response to avoid cancer cell apoptosis and leads to ICIs resistance (172). Furthermore, cholesterol is vital in T cell function and ICIs sensitivity. Elevated cholesterol in T cells upregulates the expression of various immunosuppressive receptors (e,g, PD-1, TIM-3, LAG-3) and impairs ferroptosis of tumor (173). It also modulates the endoplasmic reticulum stress pathway to inactivate T cells (174), leading to immune depletion. Moreover, inhibiting cholesterol esterification in T cells by acetyl-CoA acetyltransferase (ACAT1, a cholesterol esterase) enhances the effector function and proliferation of CD8+ T cells. It enhances ICIs effect due to elevated cholesterol levels in the CD8+ T cell plasma membrane, which enhances T cell receptor aggregation and synapse formation (175).
Although many studies suggest a role for metabolism in immunosuppression, and targeting metabolism can improve ICIs effect, they do not demonstrate that metabolic profiles can distinguish ICIs responders from non-responders (176). In addition, current studies are preclinical, it is unclear whether targeting metabolic pathways will promote ICIs response in clinical practice. More definitive mechanisms of resistance remain to be explored.
Microbiota, has a vital role in various aspects of human physiology. The gut microbiota has a significant role in regulating host metabolism and directing the development and function of immune system. Studies suggest that the primary response to ICIs in patients with epithelial tumors and melanoma may benefit from the gut microbial ecosystem (177–180). Several clinical and preclinical evidence has validated the gut microbiota’s impact on ICIs efficacy (178–183).
Microbiota composition is related to ICIs sensitivity. Combining anti-PD-1 with fecal microbiota transplantation (FMT) from healthy donors could enhance sensitivity to anti-PD-1 treatment (184). The antibiotic application which could change microbiota composition may impair the efficacy of ipilimumab. Moreover, anti-CTLA-4-induced TIL infiltration was significantly reduced in germ-free mice (180). Clinical trials of FMT in combination with anti-PD-1/PD-L1 also showed promising results, as 6 of 15 patients benefitted from it. Increased CD8+ T cell activation and reduced myeloid cell were identified (185). These suggest that microbiota is necessary for ICIs treatment. Moreover, CTLA-4 blockade reverses the gut microbiota repertoire, leading to an increase of bacteria, which diminishes the effects of ipilimumab. And this provided new insights into acquired resistance to anti-CTLA-4 (186).
The efficacy of ICIs in epithelial tumors was correlated with the abundance of Akkermansia muciniphila (AKK) (179), and the study in advanced NSCLC conquered the conclusion (177). AKK-free feces can confer resistance to anti-PD-1 in mice, and the baseline expression of AKK is positively correlated with increased ORR and OS. on the other hand, studies suggest that inosine, a metabolite of Bifidobacterium pseudolongum, can interact with T cell A2A receptors to enhance anti-CTLA-4 effects (187). In advanced NSCLC, patients with abundant Bifidobacterium breve have superior PFS than patients with undetectable bifidobacteria (188).
In colon cancer, intestinal innate lymphoid cell 3 (ILC-3) was severely dysregulated. ILC-3 can interact with T cells via MHC-II to induce microbiota colonization, eliciting type I immune responses. Transferring microbiota from patients with dysregulated intestinal ILC-3 to mice induces ICIs resistance, suggesting that immune cells can directly influence intestinal microbiota colonization, which in turn affects the anti-tumor immune response (189).
In all, current evidence shows that bacterial species and microbiota composition are associated with ICIs effect. Moreover, an increasing number of studies suggest that microorganisms may be present in tumor tissues, and involved in tumor progression and ICIs resistance (190, 191). The mechanisms remain unclear. However, some mechanisms have been proposed, such as cross-reaction between cancer antigens and microbial peptides, direct presentation of intracellular bacterial peptides by cancer cells (25, 187, 189, 192). Based on this, relative therapeutic approaches have been proposed, such as fecal microbiota transplantation, bacterial isolates and consortia, probiotic therapy (190). More research is still needed to determine microbiota’s role in immunotherapy and ICIs resistance.
Epigenetics has a vital role in regulating gene expression (193). Anti-Silencing Function 1A Histone Chaperone (ASF1A) encodes a member of the H3/H4 family of histone chaperone proteins and is a critical component of the histone donor complex during nucleosome assembly. It also binds to bivalent promoters and transcription factors to regulate gene expression (194, 195). ASF1A deficiency sensitizes LUAD to anti-PD-1 by promoting M1 TAM polarization and T cell activation, suggesting that ASF1A may be a regulator of anti-PD-1 therapeutic sensitivity (194).
In addition, it has been shown that a secreted PD-L1 splicing variant, which lacks a transmembrane structural domain, competitively binds anti-PD-L1 monoclonal antibodies, neutralizing their effects and leading to drug resistance in NSCLC (196).
A classification has been proposed based on the clinical response to ICIs treatment. Tumors are considered ‘hot’ if they respond to ICIs and ‘cold’ was characterized as the lack of HLA-I expression, low PD-L1 expression, and infiltration of immunomodulatory cells (197). The third category of ‘warm’ tumors has also been proposed, but this group lacks sufficient clinical information details (12, 198). However, this classification remains controversial. For example, tumor can have a low initial antigen presentation effect by reducing neoantigen expression, resulting in reduced T cell activation, or tumor overexpressing multiple suppressive immune checkpoint molecules. Both of them were identified as ‘cold’ tumors. but the response to ICIs cannot be generalized. Therefore, how to fire up ‘cold’ tumors is the key to overcoming ICIs resistance.
Combination therapy is the most common treatment, but it should be based on an in-depth understanding of the mechanisms of ICIs resistance, rather than simply combining existing therapies (25). Radiotherapy/chemotherapy combined with anti-PD-1/PD-L1 therapy has become the standard treatment for advanced NSCLC. Its rationale has been elaborated, such as enhancing tumor antigenicity, modulating suppressive immune cells, promoting pro-inflammatory factor release and antigen presentation (11, 199, 200). A large number of clinical trials have demonstrated its effectiveness (201, 202). In addition, some studies suggest that ICIs therapy is ineffective after first-line chemotherapy, and transcriptional analysis shows significant downregulation of gene expression related to antigen processing and presentation, as well as IFN and chemokine-related pathways, suggesting that immunotherapy after chemotherapy is not beneficial (203). In addition, anti-PD-1/PD-L1 combined with anti-CTLA-4 has complementary anti-tumor mechanisms. Ipilimumab combined with nivolumab has been approved for the treatment of advanced melanoma, renal cell carcinoma, and metastatic colon cancer (204). Clinical trials in NSCLC have also shown promising results. Checkmate-227 showed superior outcomes of nivolumab in combination with ipilimumab, compared to chemotherapy, especially for patients with TPS ≥ 50% (205). Another phase II clinical trial investigated the impact of durvalumab in combination with tremelimumab in NSCLC patients resistant to anti-PD-1, with 10% overall response rate of and 30% disease control rate (206).
In addition, there are many novel drugs for combination therapy, such as targeted therapies (207–210), lysing viruses (211), cancer vaccines (212), Chinese herbal medicine (213, 214), and nanoparticles (215, 216). Meanwhile, metabolism and microbiome with immunotherapy is ongoing. For example, inhibition of mitochondrial metabolism using atovaquone reduces MDSCs and Tregs infiltration, while CD4+ TIL infiltration increases (217). Targeting the fatty acid metabolizing enzyme Stearoyl-CoA desaturase-1 (SCD1) combined with anti-PD-1 also have a synergistic anti-tumor effect, by reducing Wnt/β-catenin signaling and promoting DCs recruitment (218). Although some studies showed promising results, there are still many challenges, such as the double-edged impact of glutaminase antagonism on T cell (167, 219, 220). The combination of IDO inhibitors with ICIs did not provide clinical benefit for patients (221). Moreover, several combination therapies are partly achieved by intratumor injection, limiting its clinical application. Therefore, more exploration is necessary.
Although ICIs have led to a paradigm shift in cancer treatment, resistance to ICIs has limited its application and the mechanism represents a critical knowledge gap. Here, we discussed ICIs resistance mechanisms. Briefly, factors such as neoantigen presentation, epigenetic modifications, tumor heterogeneity, quantity, and quality of immune cells in TME simultaneously determine the response to ICIs therapy (2). However, the interaction of these factors increases the complexity of the study, and there are still many mechanisms need to be explored.
Although significant efforts have been made to overcome the resistance to ICIs, but up to the present they remain investigational and need to be confirmed in the future.
KZ, SL and KC contributed to conception and design of the study. KZ wrote the first draft of the manuscript. SL and YZ wrote sections of the manuscript. KC reviewed the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This study was supported by the Sichuan Science and Technology Department Key Research and Development Project (2022YFS0336).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin (2021) 71:7–33. doi: 10.3322/caac.21654
2. Zhou F, Qiao M, Zhou C. The cutting-edge progress of immune-checkpoint blockade in lung cancer. Cell Mol Immunol (2021) 18:279–93. doi: 10.1038/s41423-020-00577-5
3. Walsh RJ, Soo RA. Resistance to immune checkpoint inhibitors in non-small cell lung cancer: Biomarkers and therapeutic strategies. Ther Adv Med Oncol (2020) 12:1758835920937902. doi: 10.1177/1758835920937902
4. Wang S, Yuan P, Mao B, Li N, Ying J, Tao X, et al. Genomic features and tumor immune microenvironment alteration in NSCLC treated with neoadjuvant PD-1 blockade. NPJ Precis Oncol (2022) 6:2. doi: 10.1038/s41698-021-00244-6
5. Rocha P, Zhang J, Laza-Briviesca R, Cruz-Bermúdez A, Bota-Rabassedas N, Sanchez-Espiridon B, et al. Distinct immune gene programs associated with host tumor immunity, neoadjuvant chemotherapy, and chemoimmunotherapy in resectable NSCLC. Clin Cancer Res Off J Am Assoc Cancer Res (2022) 28:2461–73. doi: 10.1158/1078-0432.CCR-21-3207
6. Reuss JE, Anagnostou V, Cottrell TR, Smith KN, Verde F, Zahurak M, et al. Neoadjuvant nivolumab plus ipilimumab in resectable non-small cell lung cancer. J Immunother Cancer (2020) 8. doi: 10.1136/jitc-2020-001282
7. Cascone T, William WNJ, Weissferdt A, Leung CH, Lin HY, Pataer A, et al. Neoadjuvant nivolumab or nivolumab plus ipilimumab in operable non-small cell lung cancer: the phase 2 randomized NEOSTAR trial. Nat Med (2021) 27:504–14. doi: 10.1038/s41591-020-01224-2
8. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: Clinical impact and mechanisms of response and resistance. Annu Rev Pathol (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
9. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
10. Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature (2019) 574:45–56. doi: 10.1038/s41586-019-1593-5
11. Domagala-Kulawik J. The role of the immune system in non-small cell lung carcinoma and potential for therapeutic intervention. Transl Lung Cancer Res (2015) 4:177–90. doi: 10.3978/j.issn.2218-6751.2015.01.11
12. Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol (2019) 20:1100–9. doi: 10.1038/s41590-019-0433-y
13. Tartour E, Zitvogel L. Lung cancer: potential targets for immunotherapy. Lancet Respir Med (2013) 1:551–63. doi: 10.1016/S2213-2600(13)70159-0
14. Carosella ED, Ploussard G, LeMaoult J, Desgrandchamps F. A systematic review of immunotherapy in urologic cancer: Evolving roles for targeting of CTLA-4, PD-1/PD-L1, and HLA-G. Eur Urol (2015) 68:267–79. doi: 10.1016/j.eururo.2015.02.032
15. Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol (2006) 18:206–13. doi: 10.1016/j.coi.2006.01.011
16. Pulluri B, Kumar A, Shaheen M, Jeter J, Sundararajan S. Tumor microenvironment changes leading to resistance of immune checkpoint inhibitors in metastatic melanoma and strategies to overcome resistance. Pharmacol Res (2017) 123:95–102. doi: 10.1016/j.phrs.2017.07.006
17. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
18. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
19. Akhbariyoon H, Azizpour Y, Esfahani MF, Firoozabad MSM, Rad MR, Esfahani KS, et al. Immune checkpoint inhibition for the treatment of cancers: An update and critical review of ongoing clinical trials. Clin Immunol Orlando Fla (2021) 232:108873. doi: 10.1016/j.clim.2021.108873
20. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim S-W, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-Small-Cell lung cancer. N Engl J Med (2019) 381:2020–31. doi: 10.1056/NEJMoa1910231
21. Boyer M, Şendur MAN, Rodríguez-Abreu D, Park K, Lee DH, Çiçin I, et al. Pembrolizumab plus ipilimumab or placebo for metastatic non-Small-Cell lung cancer with PD-L1 tumor proportion score ≥ 50%: Randomized, double-blind phase III KEYNOTE-598 study. J Clin Oncol Off J Am Soc Clin Oncol (2021) 39:2327–38. doi: 10.1200/JCO.20.03579
22. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
23. Peters S, Paz-Ares L, Herbst RS, Reck M. Addressing CPI resistance in NSCLC: targeting TAM receptors to modulate the tumor microenvironment and future prospects. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2022-004863
24. Zhang Y, Chen L. Classification of advanced human cancers based on tumor immunity in the MicroEnvironment (TIME) for cancer immunotherapy. JAMA Oncol (2016) 2:1403–4. doi: 10.1001/jamaoncol.2016.2450
25. Vesely MD, Zhang T, Chen L. Resistance mechanisms to anti-PD cancer immunotherapy. Annu Rev Immunol (2022) 40:45–74. doi: 10.1146/annurev-immunol-070621-030155
26. Schalper KA, Carvajal-Hausdorf D, McLaughlin J, Altan M, Velcheti V, Gaule P, et al. Differential expression and significance of PD-L1, IDO-1, and B7-H4 in human lung cancer. Clin Cancer Res (2017) 23:370–8. doi: 10.1158/1078-0432.CCR-16-0150
27. Kim TK, Herbst RS, Chen L. Defining and understanding adaptive resistance in cancer immunotherapy. Trends Immunol (2018) 39:624–31. doi: 10.1016/j.it.2018.05.001
28. Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade–mediated tumor regression. J Clin Invest (2018) 128:805–15. doi: 10.1172/JCI96113
29. Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther (2015) 14:847–56. doi: 10.1158/1535-7163.MCT-14-0983
30. Gaule P, Smithy JW, Toki M, Rehman J, Patell-Socha F, Cougot D, et al. A quantitative comparison of antibodies to programmed cell death 1 ligand 1. JAMA Oncol (2017) 3:256–9. doi: 10.1001/jamaoncol.2016.3015
31. Hong L, Negrao MV, Dibaj SS, Chen R, Reuben A, Bohac JM, et al. Programmed death-ligand 1 heterogeneity and its impact on benefit from immune checkpoint inhibitors in NSCLC. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2020) 15:1449–59. doi: 10.1016/j.jtho.2020.04.026
32. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med (2012) 4:127ra37. doi: 10.1126/scitranslmed.3003689
33. Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood (2018) 131:1809–19. doi: 10.1182/blood-2017-07-796342
34. Hinterleitner C, Strähle J, Malenke E, Hinterleitner M, Henning M, Seehawer M, et al. Platelet PD-L1 reflects collective intratumoral PD-L1 expression and predicts immunotherapy response in non-small cell lung cancer. Nat Commun (2021) 12:7005. doi: 10.1038/s41467-021-27303-7
35. Negrao MV, Skoulidis F, Montesion M, Schulze K, Bara I, Shen V, et al. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J Immunother Cancer (2021) 9:e002891. doi: 10.1136/jitc-2021-002891
36. Gong B, Kiyotani K, Sakata S, Nagano S, Kumehara S, Baba S, et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non-small cell lung cancer. J Exp Med (2019) 216:982–1000. doi: 10.1084/jem.20180870
37. Mahoney KM, Shukla SA, Patsoukis N, Chaudhri A, Browne EP, Arazi A, et al. A secreted PD-L1 splice variant that covalently dimerizes and mediates immunosuppression. Cancer Immunol Immunother (2019) 68:421–32. doi: 10.1007/s00262-018-2282-1
38. Oh SY, Kim S, Keam B, Kim TM, Kim DW, Heo DS. Soluble PD-L1 is a predictive and prognostic biomarker in advanced cancer patients who receive immune checkpoint blockade treatment. Sci Rep (2021) 11:19712. doi: 10.1038/s41598-021-99311-y
39. Mahoney KM, Ross-Macdonald P, Yuan L, Song L, Veras E, Wind-Rotolo M, et al. Soluble PD-L1 as an early marker of progressive disease on nivolumab. J Immunother Cancer (2022) 10:e003527. doi: 10.1136/jitc-2021-003527
40. Cheng Y, Wang C, Wang Y, Dai L. Soluble PD-L1 as a predictive biomarker in lung cancer: a systematic review and meta-analysis. Future Oncol (2022) 18:261–73. doi: 10.2217/fon-2021-0641
41. Sagawa R, Sakata S, Gong B, Seto Y, Takemoto A, Takagi S, et al. Soluble PD-L1 works as a decoy in lung cancer immunotherapy via alternative polyadenylation. JCI Insight (2022) 7:e153323. doi: 10.1172/jci.insight.153323
42. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discovery (2017) 7:1420–35. doi: 10.1158/2159-8290.CD-17-0593
43. Carbotti G, Nikpoor AR, Vacca P, Gangemi R, Giordano C, Campelli F, et al. IL-27 mediates HLA class I up-regulation, which can be inhibited by the IL-6 pathway, in HLA-deficient small cell lung cancer cells. J Exp Clin Cancer Res (2017) 36:140. doi: 10.1186/s13046-017-0608-z
44. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell (2017) 171:1259–1271.e11. doi: 10.1016/j.cell.2017.10.001
45. Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science (2018) 359:582–7. doi: 10.1126/science.aao4572
46. Doherty PC, Zinkernagel RM. A biological role for the major histocompatibility antigens. Lancet Lond Engl (1975) 1:1406–9. doi: 10.1016/s0140-6736(75)92610-0
47. Correale P, Saladino RE, Giannarelli D, Giannicola R, Agostino R, Staropoli N, et al. Distinctive germline expression of class I human leukocyte antigen (HLA) alleles and DRB1 heterozygosis predict the outcome of patients with non-small cell lung cancer receiving PD-1/PD-L1 immune checkpoint blockade. J Immunother Cancer (2020) 8:e000733. doi: 10.1136/jitc-2020-000733
48. Schöffski P, Tan DSW, Martín M, Ochoa-de-Olza M, Sarantopoulos J, Carvajal RD, et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. J Immunother Cancer (2022) 10:e003776. doi: 10.1136/jitc-2021-003776
49. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
50. Maruhashi T, Sugiura D, Okazaki I, Shimizu K, Maeda TK, Ikubo J, et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity (2022) 55:912–924.e8. doi: 10.1016/j.immuni.2022.03.013
51. Datar I, Sanmamed MF, Wang J, Henick BS, Choi J, Badri T, et al. Expression analysis and significance of PD-1, LAG-3, and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin Cancer Res Off J Am Assoc Cancer Res (2019) 25:4663–73. doi: 10.1158/1078-0432.CCR-18-4142
52. Zeng H, Zhou Q, Wang Z, Zhang H, Liu Z, Huang Q, et al. Stromal LAG-3 + cells infiltration defines poor prognosis subtype muscle-invasive bladder cancer with immunoevasive contexture. J Immunother Cancer (2020) 8:e000651. doi: 10.1136/jitc-2020-000651
53. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
54. Takamatsu K, Tanaka N, Hakozaki K, Takahashi R, Teranishi Y, Murakami T, et al. Profiling the inhibitory receptors LAG-3, TIM-3, and TIGIT in renal cell carcinoma reveals malignancy. Nat Commun (2021) 12:5547. doi: 10.1038/s41467-021-25865-0
55. Niu J, Maurice-Dror C, Lee DH, Kim D-W, Nagrial A, Voskoboynik M, et al. First-in-human phase 1 study of the anti-TIGIT antibody vibostolimab as monotherapy or with pembrolizumab for advanced solid tumors, including non-small-cell lung cancer. Ann Oncol (2022) 33:169–80. doi: 10.1016/j.annonc.2021.11.002
56. Hollebecque A, Chung HC, de Miguel MJ, Italiano A, Machiels J-P, Lin C-C, et al. Safety and antitumor activity of α-PD-L1 antibody as monotherapy or in combination with α-TIM-3 antibody in patients with microsatellite instability-High/Mismatch repair-deficient tumors. Clin Cancer Res Off J Am Assoc Cancer Res (2021) 27:6393–404. doi: 10.1158/1078-0432.CCR-21-0261
57. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/Ib clinical trial of sabatolimab, an anti-TIM-3 antibody, alone and in combination with spartalizumab, an anti-PD-1 antibody, in advanced solid tumors. Clin Cancer Res Off J Am Assoc Cancer Res (2021) 27:3620–9. doi: 10.1158/1078-0432.CCR-20-4746
58. Hosseinkhani N, Derakhshani A, Shadbad MA, Argentiero A, Racanelli V, Kazemi T, et al. The role of V-domain ig suppressor of T cell activation (VISTA) in cancer therapy: Lessons learned and the road ahead. Front Immunol (2021) 12:676181. doi: 10.3389/fimmu.2021.676181
59. Huang X, Zhang X, Li E, Zhang G, Wang X, Tang T, et al. VISTA: an immune regulatory protein checking tumor and immune cells in cancer immunotherapy. J Hematol OncolJ Hematol Oncol (2020) 13:83. doi: 10.1186/s13045-020-00917-y
60. Villarroel-Espindola F, Yu X, Datar I, Mani N, Sanmamed M, Velcheti V, et al. Spatially resolved and quantitative analysis of VISTA/PD-1H as a novel immunotherapy target in human non–small cell lung cancer. Clin Cancer Res (2018) 24:1562–73. doi: 10.1158/1078-0432.CCR-17-2542
61. Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med (2017) 23:551–5. doi: 10.1038/nm.4308
62. Thakkar D, Paliwal S, Dharmadhikari B, Guan S, Liu L, Kar S, et al. Rationally targeted anti-VISTA antibody that blockades the c-c’ loop region can reverse VISTA immune suppression and remodel the immune microenvironment to potently inhibit tumor growth in an fc independent manner. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2021-003382
63. Sedlacek AL, Kinner-Bibeau LB, Binder RJ. Phenotypically distinct helper NK cells are required for gp96-mediated anti-tumor immunity. Sci Rep (2016) 6:29889. doi: 10.1038/srep29889
64. Sedlacek AL, Younker TP, Zhou YJ, Borghesi L, Shcheglova T, Mandoiu II, et al. CD91 on dendritic cells governs immunosurveillance of nascent, emerging tumors. JCI Insight (2019) 4:e127239. doi: 10.1172/jci.insight.127239
65. Kinner-Bibeau LB, Sedlacek AL, Messmer MN, Watkins SC, Binder RJ. HSPs drive dichotomous T-cell immune responses via DNA methylome remodelling in antigen presenting cells. Nat Commun (2017) 8:15648. doi: 10.1038/ncomms15648
66. Liu Z, Li X, Qiu L, Zhang X, Chen L, Cao S, et al. Treg suppress CTL responses upon immunization with HSP gp96. Eur J Immunol (2009) 39:3110–20. doi: 10.1002/eji.200939593
67. Li X, Liu Z, Yan X, Zhang X, Li Y, Zhao B, et al. Induction of regulatory T cells by high-dose gp96 suppresses murine liver immune hyperactivation. PloS One (2013) 8:e68997. doi: 10.1371/journal.pone.0068997
68. Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer (2017) 5:16. doi: 10.1186/s40425-017-0218-5
69. de Galarreta MR, Bresnahan E, Molina-Sánchez P, Lindblad KE, Maier B, Sia D, et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discovery (2019) 9:1124–41. doi: 10.1158/2159-8290.CD-19–0074
70. Trujillo JA, Luke JJ, Zha Y, Segal JP, Ritterhouse LL, Spranger S, et al. Secondary resistance to immunotherapy associated with β-catenin pathway activation or PTEN loss in metastatic melanoma. J Immunother Cancer (2019) 7:295. doi: 10.1186/s40425-019-0780-0
71. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature (2015) 523:231–5. doi: 10.1038/nature14404
72. Dai W, Liu F, Li C, Lu Y, Lu X, Du S, et al. Blockade of wnt/β-catenin pathway aggravated silica-induced lung inflammation through tregs regulation on Th immune responses. Mediators Inflammation (2016) 2016:6235614. doi: 10.1155/2016/6235614
73. Cretella D, Digiacomo G, Giovannetti E, Cavazzoni A. PTEN alterations as a potential mechanism for tumor cell escape from PD-1/PD-L1 inhibition. Cancers (2019) 11:1318. doi: 10.3390/cancers11091318
74. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discovery (2016) 6:202–16. doi: 10.1158/2159-8290.CD-15-0283
75. Gkountakos A, Sartori G, Falcone I, Piro G, Ciuffreda L, Carbone C, et al. PTEN in lung cancer: Dealing with the problem, building on new knowledge and turning the game around. Cancers (2019) 11:1141. doi: 10.3390/cancers11081141
76. Parikh AR, Ali SM, Schrock AB, Albacker LA, Miller VA, Stephens PJ, et al. Response to rapamycin analogs but not PD-1 inhibitors in PTEN-mutated metastatic non-small-cell lung cancer with high tumor mutational burden. Lung Cancer Targets Ther (2018) 9:45–7. doi: 10.2147/LCTT.S161738
77. Stutvoet TS, Kol A, de Vries EG, de Bruyn M, Fehrmann RS, Terwisscha van Scheltinga AG, et al. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J Pathol (2019) 249:52–64. doi: 10.1002/path.5280
78. Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol Off J Eur Soc Med Oncol (2016) 27:409–16. doi: 10.1093/annonc/mdv615
79. Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res Off J Am Assoc Cancer Res (2016) 22:1499–509. doi: 10.1158/1078-0432.CCR-15-1125
80. Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. Correction: RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin Cancer Res Off J Am Assoc Cancer Res (2019) 25:1437. doi: 10.1158/1078-0432.CCR-18-4264
81. Haas L, Elewaut A, Gerard CL, Umkehrer C, Leiendecker L, Pedersen M, et al. Acquired resistance to anti-MAPK targeted therapy confers an immune-evasive tumor microenvironment and cross-resistance to immunotherapy in melanoma. Nat Cancer (2021) 2:693–708. doi: 10.1038/s43018-021-00221-9
82. Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res (2016) 76:227–38. doi: 10.1158/0008-5472.CAN-14-3362
83. Liu M, Wei F, Wang J, Yu W, Shen M, Liu T, et al. Myeloid-derived suppressor cells regulate the immunosuppressive functions of PD-1-PD-L1+ bregs through PD-L1/PI3K/AKT/NF-κB axis in breast cancer. Cell Death Dis (2021) 12:465. doi: 10.1038/s41419-021-03745-1
84. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol (2006) 6:836–48. doi: 10.1038/nri1961
85. von Locquenghien M, Rozalén C, Celià-Terrassa T. Interferons in cancer immunoediting: sculpting metastasis and immunotherapy response. J Clin Invest (2021) 131:e143296. doi: 10.1172/JCI143296
86. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discovery (2017) 7:188–201. doi: 10.1158/2159-8290.CD-16-1223
87. Nguyen T-T, Ramsay L, Ahanfeshar-Adams M, Lajoie M, Schadendorf D, Alain T, et al. Mutations in the IFNγ-JAK-STAT pathway causing resistance to immune checkpoint inhibitors in melanoma increase sensitivity to oncolytic virus treatment. Clin Cancer Res (2021) 27:3432–42. doi: 10.1158/1078-0432.CCR-20-3365
88. Nielsen M, Presti M, Sztupinszki Z, Jensen AWP, Draghi A, Chamberlain CA, et al. Coexisting alterations of MHC class I antigen presentation and IFNγ signaling mediate acquired resistance of melanoma to post–PD-1 immunotherapy. Cancer Immunol Res (2022) 10:1254–62. doi: 10.1158/2326-6066.CIR-22-0326
89. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell (2016) 167:1540–1554.e12. doi: 10.1016/j.cell.2016.11.022
90. Yu M, Peng Z, Qin M, Liu Y, Wang J, Zhang C, et al. Interferon-γ induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol Cell (2021) 81:1216–1230.e9. doi: 10.1016/j.molcel.2021.01.010
91. Chen W, Teo JMN, Yau SW, Wong MY-M, Lok C-N, Che C-M, et al. Chronic type I interferon signaling promotes lipid-peroxidation-driven terminal CD8+ T cell exhaustion and curtails anti-PD-1 efficacy. Cell Rep (2022) 41:111647. doi: 10.1016/j.celrep.2022.111647
92. Sellars MC, Wu CJ, Fritsch EF. Cancer vaccines: Building a bridge over troubled waters. Cell (2022) 185:2770–88. doi: 10.1016/j.cell.2022.06.035
93. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther (2021) 221:107753. doi: 10.1016/j.pharmthera.2020.107753
94. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
95. Masugi Y, Nishihara R, Yang J, Mima K, da Silva A, Shi Y, et al. Tumour CD274 (PD-L1) expression and T cells in colorectal cancer. Gut (2017) 66:1463–73. doi: 10.1136/gutjnl-2016-311421
96. Noyes D, Bag A, Oseni S, Semidey-Hurtado J, Cen L, Sarnaik AA, et al. Tumor-associated tregs obstruct antitumor immunity by promoting T cell dysfunction and restricting clonal diversity in tumor-infiltrating CD8+ T cells. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2022-004605
97. Saleh R, Toor SM, Khalaf S, Elkord E. Breast cancer cells and PD-1/PD-L1 blockade upregulate the expression of PD-1, CTLA-4, TIM-3 and LAG-3 immune checkpoints in CD4+ T cells. Vaccines (2019) 7:149. doi: 10.3390/vaccines7040149
98. de Streel G, Bertrand C, Chalon N, Liénart S, Bricard O, Lecomte S, et al. Selective inhibition of TGF-β1 produced by GARP-expressing tregs overcomes resistance to PD-1/PD-L1 blockade in cancer. Nat Commun (2020) 11:4545. doi: 10.1038/s41467-020-17811-3
99. Koh J, Hur JY, Lee KY, Kim MS, Heo JY, Ku BM, et al. Regulatory (FoxP3+) T cells and TGF-β predict the response to anti-PD-1 immunotherapy in patients with non-small cell lung cancer. Sci Rep (2020) 10:18994. doi: 10.1038/s41598-020-76130-1
100. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol (2017) 18:1332–41. doi: 10.1038/ni.3868
101. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
102. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci (2021) 22:6995. doi: 10.3390/ijms22136995
103. Li W, Wu F, Zhao S, Shi P, Wang S, Cui D. Correlation between PD-1/PD-L1 expression and polarization in tumor-associated macrophages: A key player in tumor immunotherapy. Cytokine Growth Factor Rev (2022) 67:49–57. doi: 10.1016/j.cytogfr.2022.07.004
104. Larroquette M, Guegan J-P, Besse B, Cousin S, Brunet M, Le Moulec S, et al. Spatial transcriptomics of macrophage infiltration in non-small cell lung cancer reveals determinants of sensitivity and resistance to anti-PD1/PD-L1 antibodies. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2021-003890
105. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc Natl Acad Sci (2018) 115. doi: 10.1073/pnas.1720948115
106. Cai H, Zhang Y, Wang J, Gu J. Defects in macrophage reprogramming in cancer therapy: The negative impact of PD-L1/PD-1. Front Immunol (2021) 12:690869. doi: 10.3389/fimmu.2021.690869
107. Munn DH, Mellor AL. IDO in the tumor microenvironment: Inflammation, counter-regulation, and tolerance. Trends Immunol (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
108. Youn J-I, Park S-M, Park S, Kim G, Lee H-J, Son J, et al. Peripheral natural killer cells and myeloid-derived suppressor cells correlate with anti-PD-1 responses in non-small cell lung cancer. Sci Rep (2020) 10:9050. doi: 10.1038/s41598-020-65666-x
109. Limagne E, Richard C, Thibaudin M, Fumet J-D, Truntzer C, Lagrange A, et al. Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. OncoImmunology (2019) 8:e1564505. doi: 10.1080/2162402X.2018.1564505
110. Koh J, Kim Y, Lee KY, Hur JY, Kim MS, Kim B, et al. MDSC subtypes and CD39 expression on CD8+ T cells predict the efficacy of anti-PD-1 immunotherapy in patients with advanced NSCLC. Eur J Immunol (2020) 50:1810–9. doi: 10.1002/eji.202048534
111. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med (2014) 6. doi: 10.1126/scitranslmed.3007974
112. Li R, Salehi-Rad R, Ong S, Momcilovic M, Liu B, Lim R, et al. Abstract 2710: Depletion of CXCR2-dependent myeloid-derived suppressor cells (MDSCs) overcomes anti-PD-1 resistance in a murine model of LKB1-deficient non-small cell lung cancer (NSCLC) with high mutational load. Cancer Res (2019) 79:2710–0. doi: 10.1158/1538-7445.AM2019-2710
113. Ghonim MA, Ibba SV, Tarhuni AF, Errami Y, Luu HH, Dean MJ, et al. Targeting PARP-1 with metronomic therapy modulates MDSC suppressive function and enhances anti-PD-1 immunotherapy in colon cancer. J Immunother Cancer (2021) 9:e001643. doi: 10.1136/jitc-2020-001643
114. DeVito N, Nguyen Y-V, Yarla N, Plebanek M, Theivanthiran B, Aksu M, et al. 513 overcoming hedgehog mediated anti-PD-1 resistance in melanoma through prostaglandin inhibition. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2022-SITC2022.0513
115. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell (2019) 35:559–572.e7. doi: 10.1016/j.ccell.2019.02.008
116. Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol (2018) 9:1310. doi: 10.3389/fimmu.2018.01310
117. Tesi RJ. MDSC; the most important cell you have never heard of. Trends Pharmacol Sci (2019) 40:4–7. doi: 10.1016/j.tips.2018.10.008
118. Feng P-H, Yu C-T, Chen K-Y, Luo C-S, Wu SM, Liu C-Y, et al. S100A9+ MDSC and TAM-mediated EGFR-TKI resistance in lung adenocarcinoma: the role of RELB. Oncotarget (2018) 9:7631–43. doi: 10.18632/oncotarget.24146
119. Han G, Yang G, Hao D, Lu Y, Thein K, Simpson BS, et al. 9p21 loss confers a cold tumor immune microenvironment and primary resistance to immune checkpoint therapy. Nat Commun (2021) 12:5606. doi: 10.1038/s41467-021-25894-9
120. Liu C, Zheng S, Jin R, Wang X, Wang F, Zang R, et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett (2020) 470:95–105. doi: 10.1016/j.canlet.2019.10.027
121. Liu C, Zheng S, Wang Z, Wang S, Wang X, Yang L, et al. KRAS-G12D mutation drives immune suppression and the primary resistance of anti-PD-1/PD-L1 immunotherapy in non-small cell lung cancer. Cancer Commun Lond Engl (2022) 42:828–47. doi: 10.1002/cac2.12327
122. Rosellini P, Amintas S, Caumont C, Veillon R, Galland-Girodet S, Cuguillière A, et al. Clinical impact of STK11 mutation in advanced-stage non-small cell lung cancer. Eur J Cancer Oxf Engl 1990 (2022) 172:85–95. doi: 10.1016/j.ejca.2022.05.026
123. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discovery (2018) 8:822–35. doi: 10.1158/2159-8290.CD-18-0099
124. Pore N, Wu S, Standifer N, Jure-Kunkel M, de Los Reyes M, Shrestha Y, et al. Resistance to durvalumab and durvalumab plus tremelimumab is associated with functional STK11 mutations in patients with non-small cell lung cancer and is reversed by STAT3 knockdown. Cancer Discovery (2021) 11:2828–45. doi: 10.1158/2159-8290.CD-20-1543
125. The cancer genome atlas research network. comprehensive molecular profiling of lung adenocarcinoma. Nature (2014) 511:543–50. doi: 10.1038/nature13385
126. Papillon-Cavanagh S, Doshi P, Dobrin R, Szustakowski J, Walsh AM. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open (2020) 5:e000706. doi: 10.1136/esmoopen-2020-000706
127. Frank R, Scheffler M, Merkelbach-Bruse S, Ihle MA, Kron A, Rauer M, et al. Clinical and pathological characteristics of KEAP1 - and NFE2L2 -mutated non–small cell lung carcinoma (NSCLC). Clin Cancer Res (2018) 24:3087–96. doi: 10.1158/1078-0432.CCR-17-3416
128. Di Federico A, De Giglio A, Parisi C, Gelsomino F. STK11/LKB1 and KEAP1 mutations in non-small cell lung cancer: Prognostic rather than predictive? Eur J Cancer (2021) 157:108–13. doi: 10.1016/j.ejca.2021.08.011
129. Laktionov KK, Kazakov AM, Sarantseva KA, Scherbo DS, Koval AP. Mutation in the kras gene as a predictor of the effectiveness of immunotherapy for non-small cell lung cancer. Sib J Oncol (2022) 21:115–21. doi: 10.21294/1814-4861-2022-21-1-115-121
130. Barrera-Rodríguez R. Importance of the Keap1-Nrf2 pathway in NSCLC: Is it a possible biomarker? (Review). BioMed Rep (2018) 9:375–82. doi: 10.3892/br.2018.1143
131. Shang X, Li Z, Sun J, Zhao C, Lin J, Wang H. Survival analysis for non-squamous NSCLC patients harbored STK11 or KEAP1 mutation receiving atezolizumab. Lung Cancer (2021) 154:105–12. doi: 10.1016/j.lungcan.2021.02.010
132. Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun (2016) 7:11624. doi: 10.1038/ncomms11624
133. Kitamura H, Onodera Y, Murakami S, Suzuki T, Motohashi H. IL-11 contribution to tumorigenesis in an NRF2 addiction cancer model. Oncogene (2017) 36:6315–24. doi: 10.1038/onc.2017.236
134. Scalera S, Mazzotta M, Cortile C, Krasniqi E, De Maria R, Cappuzzo F, et al. KEAP1-mutant NSCLC: The catastrophic failure of a cell-protecting hub. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2022) 17:751–7. doi: 10.1016/j.jtho.2022.03.011
135. Jiang T, Shi J, Dong Z, Hou L, Zhao C, Li X, et al. Genomic landscape and its correlations with tumor mutational burden, PD-L1 expression, and immune cells infiltration in Chinese lung squamous cell carcinoma. J Hematol OncolJ Hematol Oncol (2019) 12. doi: 10.1186/s13045-019-0762-1
136. Marzio A, Kurz E, Sahni JM, Di Feo G, Puccini J, Jiang S, et al. EMSY inhibits homologous recombination repair and the interferon response, promoting lung cancer immune evasion. Cell (2022) 185:169–183.e19. doi: 10.1016/j.cell.2021.12.005
137. Bronte G, Rizzo S, La Paglia L, Adamo V, Siragusa S, Ficorella C, et al. Driver mutations and differential sensitivity to targeted therapies: a new approach to the treatment of lung adenocarcinoma. Cancer Treat Rev (2010) 36:S21–9. doi: 10.1016/S0305-7372(10)70016-5
138. Li X, Lian Z, Wang S, Xing L, Yu J. Interactions between EGFR and PD-1/PD-L1 pathway: Implications for treatment of NSCLC. Cancer Lett (2018) 418:1–9. doi: 10.1016/j.canlet.2018.01.005
139. Tang Y, Fang W, Zhang Y, Hong S, Kang S, Yan Y, et al. The association between PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced non-small cell lung cancer patients treated with EGFR-TKIs. Oncotarget (2015) 6:14209–19. doi: 10.18632/oncotarget.3694
140. Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol (2014) 25:1935–40. doi: 10.1093/annonc/mdu242
141. Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non–small cell lung cancer: A retrospective analysis. Clin Cancer Res (2016) 22:4585–93. doi: 10.1158/1078-0432.CCR-15-3101
142. Dong Z-Y, Zhang J-T, Liu S-Y, Su J, Zhang C, Xie Z, et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. OncoImmunology (2017) 6:e1356145. doi: 10.1080/2162402X.2017.1356145
143. Weinstein IB. Addiction to oncogenes–the Achilles heal of cancer. Science (2002) 297:63–4. doi: 10.1126/science.1073096
144. Addeo A, Passaro A, Malapelle U, Banna GL, Subbiah V, Friedlaender A. Immunotherapy in non-small cell lung cancer harbouring driver mutations. Cancer Treat Rev (2021) 96:102179. doi: 10.1016/j.ctrv.2021.102179
145. Nagahashi M, Sato S, Yuza K, Shimada Y, Ichikawa H, Watanabe S, et al. Common driver mutations and smoking history affect tumor mutation burden in lung adenocarcinoma. J Surg Res (2018) 230:181–5. doi: 10.1016/j.jss.2018.07.007
146. Ahn MJ, Cho BC, Ou X, Walding A, Dymond AW, Ren S, et al. Osimertinib plus durvalumab in patients with EGFR-mutated, advanced NSCLC: A phase 1b, open-label, multicenter trial. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2022) 17:718–23. doi: 10.1016/j.jtho.2022.01.012
147. Lisberg A, Cummings A, Goldman JW, Bornazyan K, Reese N, Wang T, et al. A phase II study of pembrolizumab in EGFR-mutant, PD-L1+, tyrosine kinase inhibitor naïve patients with advanced NSCLC. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2018) 13:1138–45. doi: 10.1016/j.jtho.2018.03.035
148. Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol (2017) 18:31–41. doi: 10.1016/S1470-2045(16)30624-6
149. Lin JJ, Chin E, Yeap BY, Ferris LA, Kamesan V, Lennes IT, et al. Increased hepatotoxicity associated with sequential immune checkpoint inhibitor and crizotinib therapy in patients with non-small cell lung cancer. J Thorac Oncol (2019) 14:135–40. doi: 10.1016/j.jtho.2018.09.001
150. 9610–poster-Cabozantinib in combination with atezolizumab in non-small cell lung cancer (NSCLC) patients previously treated with an immune checkpoint inhibitor: Results from cohort 7 of the COSMIC-021 study. Presented at ASCO congress (2020).
151. Hayashi H, Sugawara S, Fukuda Y, Fujimoto D, Miura S, Ota K, et al. A randomized phase II study comparing nivolumab with carboplatin-pemetrexed for EGFR-mutated NSCLC with resistance to EGFR tyrosine kinase inhibitors (WJOG8515L). Clin Cancer Res (2022) 28:893–902. doi: 10.1158/1078-0432.CCR-21-3194
152. Ocaña MC, Martínez-Poveda B, Quesada AR, Medina MÁ.Metabolism within the tumor microenvironment and its implication on cancer progression: An ongoing therapeutic target. Med Res Rev (2019) 39:70–113. doi: 10.1002/med.21511
153. Sk B. Metabolic reprogramming of immune cells in cancer progression. Immunity (2015) 43. doi: 10.1016/j.immuni.2015.09.001
154. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
155. Terry S, Engelsen AST, Buart S, Elsayed WS, Venkatesh GH, Chouaib S. Hypoxia-driven intratumor heterogeneity and immune evasion. Cancer Lett (2020) 492:1–10. doi: 10.1016/j.canlet.2020.07.004
156. Huang B, Song B-L, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab (2020) 2:132–41. doi: 10.1038/s42255-020-0174-0
157. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer (2021) 20:28. doi: 10.1186/s12943-021-01316-8
158. Karayama M, Masuda J, Mori K, Yasui H, Hozumi H, Suzuki Y, et al. Comprehensive assessment of multiple tryptophan metabolites as potential biomarkers for immune checkpoint inhibitors in patients with non-small cell lung cancer. Clin Transl Oncol (2021) 23:418. doi: 10.1007/s12094-020-02421-8
159. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity (2016) 45:358–73. doi: 10.1016/j.immuni.2016.07.008
160. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi: 10.1038/ncomms7692
161. Zandberg DP, Menk AV, Velez M, Normolle D, DePeaux K, Liu A, et al. Tumor hypoxia is associated with resistance to PD-1 blockade in squamous cell carcinoma of the head and neck. J Immunother Cancer (2021) 9:e002088. doi: 10.1136/jitc-2020-002088
162. Najjar YG, Menk AV, Sander C, Rao U, Karunamurthy A, Bhatia R, et al. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight (2019) 4:e124989. doi: 10.1172/jci.insight.124989
163. Jaiswal AR, Liu AJ, Pudakalakatti S, Dutta P, Jayaprakash P, Bartkowiak T, et al. Melanoma evolves complete immunotherapy resistance through the acquisition of a hypermetabolic phenotype. Cancer Immunol Res (2020) 8:1365–80. doi: 10.1158/2326-6066.CIR-19-0005
164. Jiang Z, Hsu JL, Li Y, Hortobagyi GN, Hung MC. Cancer cell metabolism bolsters immunotherapy resistance by promoting an immunosuppressive tumor microenvironment. Front Oncol (2020) 10:1197. doi: 10.3389/fonc.2020.01197
165. Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8+ T cells and facilitates anti-PD-1 therapy. Cancer Immunol Res (2018) 6:1375–87. doi: 10.1158/2326-6066.CIR-18-0095
166. Kumar A, Chamoto K. Immune metabolism in PD-1 blockade-based cancer immunotherapy. Int Immunol (2021) 33:17–26. doi: 10.1093/intimm/dxaa046
167. Best SA, Gubser PM, Sethumadhavan S, Kersbergen A, Negrón Abril YL, Goldford J, et al. Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metab (2022) 34:874–887.e6. doi: 10.1016/j.cmet.2022.04.003
168. Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest (2021) 131:140100. doi: 10.1172/JCI140100
169. Brady M, Momcilovic M, Montemurro C, Yokoyama Y, Christofk H, Politi K, et al. Abstract 1572: Broad glutamine pathway inhibition by DRP-104 results in anti-tumor activity in hypermetabolic lung tumors resistant to PD-1 or osimertinib therapy. Cancer Res (2021) 81:1572–2. doi: 10.1158/1538-7445.AM2021-1572
170. Liu N, Zhang J, Yin M, Liu H, Zhang X, Li J, et al. Inhibition of xCT suppresses the efficacy of anti-PD-1/L1 melanoma treatment through exosomal PD-L1-induced macrophage M2 polarization. Mol Ther (2021) 29:2321–34. doi: 10.1016/j.ymthe.2021.03.013
171. Huang D, Wang Y, Thompson JW, Yin T, Alexander PB, Qin D, et al. Cancer-cell-derived GABA promotes β-catenin-mediated tumour growth and immunosuppression. Nat Cell Biol (2022) 24:230–41. doi: 10.1038/s41556-021-00820-9
172. Khojandi N, Kuehm LM, Piening A, Donlin MJ, Hsueh EC, Schwartz TL, et al. Oxidized lipoproteins promote resistance to cancer immunotherapy independent of patient obesity. Cancer Immunol Res (2021) 9:214–26. doi: 10.1158/2326-6066.CIR-20-0358
173. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab (2019) 30:143–156.e5. doi: 10.1016/j.cmet.2019.04.002
174. Picarda E, Ren X, Zang X. Tumor cholesterol up, T cells down. Cell Metab (2019) 30. doi: 10.1016/j.cmet.2019.06.007
175. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531. doi: 10.1038/nature17412
176. Jiang Z, Lim S-O, Yan M, Hsu JL, Yao J, Wei Y, et al. TYRO3 induces anti–PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J Clin Invest (2021) 131. doi: 10.1172/JCI139434
177. Derosa L, Routy B, Thomas AM, Iebba V, Zalcman G, Friard S, et al. Intestinal akkermansia muciniphila predicts clinical response to PD-1 blockade in patients with advanced non-small-cell lung cancer. Nat Med (2022) 28:315–24. doi: 10.1038/s41591-021-01655-5
178. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science (2018) 359:97–103. doi: 10.1126/science.aan4236
179. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (2018) 359:91–7. doi: 10.1126/science.aan3706
180. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (2015) 350:1079–84. doi: 10.1126/science.aad1329
181. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science (2015) 350:1084–9. doi: 10.1126/science.aac4255
182. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol Off J Eur Soc Med Oncol (2017) 28:1368–79. doi: 10.1093/annonc/mdx108
183. Borgerding JN, Shang J, Britton GJ, Salmon H, Bigenwald C, Maier B, et al. Human microbial transplant restores T cell cytotoxicity and anti-tumor response to PD-L1 blockade in gnotobiotic mice. Immunology (2020). doi: 10.1101/2020.08.07.242040
184. Routy B, Lenehan J, Daisley B, Messaoudene M, Al K, Richard C, et al. 614 microbiome modification with fecal microbiota transplant from healthy donors before anti-PD1 therapy reduces primary resistance to immunotherapy in advanced and metastatic melanoma patients. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2022-SITC2022.0614
185. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science (2021) 371. doi: 10.1126/science.abf3363
186. Shui L, Yang X, Li J, Yi C, Sun Q, Zhu H. Gut microbiome as a potential factor for modulating resistance to cancer immunotherapy. Front Immunol (2020) 10:2989. doi: 10.3389/fimmu.2019.02989
187. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science (2020) 369:1481–9. doi: 10.1126/science.abc3421
188. Zhao H, Li D, Liu J, Zhou X, Han J, Wang L, et al. Bifidobacterium breve predicts the efficacy of anti-PD-1 immunotherapy combined with chemotherapy in Chinese NSCLC patients. Cancer Med (2022). doi: 10.1002/cam4.5312
189. Goc J, Lv M, Bessman NJ, Flamar A-L, Sahota S, Suzuki H, et al. Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell (2021) 184:5015–5030.e16. doi: 10.1016/j.cell.2021.07.029
190. Matson V, Chervin CS, Gajewski TF. Cancer and the microbiome–influence of the commensal microbiota on cancer, immune responses, and immunotherapy. Gastroenterology (2021) 160:600–13. doi: 10.1053/j.gastro.2020.11.041
191. Li Q, Goggin KE, Seo S, Warawa JM, Egilmez NK. Anti-PD-1 antibody-activated Th17 cells subvert re-invigoration of antitumor cytotoxic T-lymphocytes via myeloid cell-derived COX-2/PGE2. Cancer Immunol Immunother (2022). doi: 10.1007/s00262-022-03285-3
192. Kalaora S, Nagler A, Nejman D, Alon M, Barbolin C, Barnea E, et al. Identification of bacteria-derived HLA-bound peptides in melanoma. Nature (2021) 592:138–43. doi: 10.1038/s41586-021-03368-8
193. Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, et al. Early transcriptional and epigenetic regulation of CD8+ T cell differentiation revealed by single-cell RNA sequencing. Nat Immunol (2017) 18:422–32. doi: 10.1038/ni.3688
194. Li F, Huang Q, Luster TA, Hu H, Zhang H, Ng W-L, et al. In vivo epigenetic CRISPR screen identifies Asf1a as an immunotherapeutic target in kras -mutant lung adenocarcinoma. Cancer Discovery (2020) 10:270–87. doi: 10.1158/2159-8290.CD-19-0780
195. Gao Y, Gan H, Lou Z, Zhang Z. Asf1a resolves bivalent chromatin domains for the induction of lineage-specific genes during mouse embryonic stem cell differentiation. Proc Natl Acad Sci U.S.A. (2018) 115:E6162–71. doi: 10.1073/pnas.1801909115
196. Gong B, Kiyotani K, Sakata S, Nagano S, Kumehara S, Baba S, et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non–small cell lung cancer. J Exp Med (2019) 216:982–1000. doi: 10.1084/jem.20180870
197. Sabbatino F, Marra A, Liguori L, Scognamiglio G, Fusciello C, Botti G, et al. Resistance to anti-PD-1-based immunotherapy in basal cell carcinoma: a case report and review of the literature. J Immunother Cancer (2018) 6:126. doi: 10.1186/s40425-018-0439-2
198. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol (2018) 15:422–42. doi: 10.1038/s41571-018-0003-5
199. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al. Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell (2021) 39:193–208.e10. doi: 10.1016/j.ccell.2020.11.005
200. Gao Q, Wang S, Chen X, Cheng S, Zhang Z, Li F, et al. Cancer-cell-secreted CXCL11 promoted CD8(+) T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J Immunother Cancer (2019) 7:42. doi: 10.1186/s40425-019-0511-6
201. Theelen WSME, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts JGJV, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: Results of the PEMBRO-RT phase 2 randomized clinical trial. JAMA Oncol (2019) 5:1276–82. doi: 10.1001/jamaoncol.2019.1478
202. Shirasawa M, Yoshida T, Matsumoto Y, Shinno Y, Okuma Y, Goto Y, et al. Impact of chemoradiotherapy on the immune-related tumour microenvironment and efficacy of anti-PD-(L)1 therapy for recurrences after chemoradiotherapy in patients with unresectable locally advanced non-small cell lung cancer. Eur J Cancer Oxf Engl 1990 (2020) 140:28–36. doi: 10.1016/j.ejca.2020.08.028
203. He Y, Chen L, Zhao L, Dang S, Liu G, Sasada S, et al. Genomic and transcriptional alterations in first-line chemotherapy exert a potentially unfavorable influence on subsequent immunotherapy in NSCLC. Theranostics (2021) 11:7092–109. doi: 10.7150/thno.58039
204. Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res CR (2019) 38:255. doi: 10.1186/s13046-019-1259-z
205. Paz-Ares LG, Ramalingam SS, Ciuleanu T-E, Lee J-S, Urban L, Caro RB, et al. First-line nivolumab plus ipilimumab in advanced NSCLC: 4-year outcomes from the randomized, open-label, phase 3 CheckMate 227 part 1 trial. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer (2022) 17:289–308. doi: 10.1016/j.jtho.2021.09.010
206. Schoenfeld JD, Giobbie-Hurder A, Ranasinghe S, Kao KZ, Lako A, Tsuji J, et al. Durvalumab plus tremelimumab alone or in combination with low-dose or hypofractionated radiotherapy in metastatic non-small-cell lung cancer refractory to previous PD(L)-1 therapy: an open-label, multicentre, randomised, phase 2 trial. Lancet Oncol (2022) 23:279–91. doi: 10.1016/S1470-2045(21)00658-6
207. Lai AY, Sorrentino JA, Dragnev KH, Weiss JM, Owonikoko TK, Rytlewski JA, et al. CDK4/6 inhibition enhances antitumor efficacy of chemotherapy and immune checkpoint inhibitor combinations in preclinical models and enhances T-cell activation in patients with SCLC receiving chemotherapy. J Immunother Cancer (2020) 8. doi: 10.1136/jitc-2020-000847
208. Limagne E, Nuttin L, Thibaudin M, Jacquin E, Aucagne R, Bon M, et al. MEK inhibition overcomes chemoimmunotherapy resistance by inducing CXCL10 in cancer cells. Cancer Cell (2022) 40:136–152.e12. doi: 10.1016/j.ccell.2021.12.009
209. Zhao Y, Guo S, Deng J, Shen J, Du F, Wu X, et al. VEGF/VEGFR-targeted therapy and immunotherapy in non-small cell lung cancer: Targeting the tumor microenvironment. Int J Biol Sci (2022) 18:3845–58. doi: 10.7150/ijbs.70958
210. Wang J, Zhang R, Lin Z, Zhang S, Chen Y, Tang J, et al. CDK7 inhibitor THZ1 enhances antiPD-1 therapy efficacy via the p38α/MYC/PD-L1 signaling in non-small cell lung cancer. J Hematol OncolJ Hematol Oncol (2020) 13:99. doi: 10.1186/s13045-020-00926-x
211. Sun F, Guo ZS, Gregory AD, Shapiro SD, Xiao G, Qu Z. Dual but not single PD-1 or TIM-3 blockade enhances oncolytic virotherapy in refractory lung cancer. J Immunother Cancer (2020) 8. doi: 10.1136/jitc-2019-000294
212. Pan J, Zhang Q, Palen K, Wang L, Qiao L, Johnson B, et al. Potentiation of kras peptide cancer vaccine by avasimibe, a cholesterol modulator. EBioMedicine (2019) 49:72–81. doi: 10.1016/j.ebiom.2019.10.044
213. Jiang Z-B, Huang J-M, Xie Y-J, Zhang Y-Z, Chang C, Lai H-L, et al. Evodiamine suppresses non-small cell lung cancer by elevating CD8(+) T cells and downregulating the MUC1-C/PD-L1 axis. J Exp Clin Cancer Res CR (2020) 39:249. doi: 10.1186/s13046-020-01741-5
214. Zhu X, Yu Z, Feng L, Deng L, Fang Z, Liu Z, et al. Chitosan-based nanoparticle co-delivery of docetaxel and curcumin ameliorates anti-tumor chemoimmunotherapy in lung cancer. Carbohydr Polym (2021) 268:118237. doi: 10.1016/j.carbpol.2021.118237
215. Su W-P, Chang L-C, Song W-H, Yang L-X, Wang L-C, Chia Z-C, et al. Polyaniline-based glyco-condensation on au nanoparticles enhances immunotherapy in lung cancer. ACS Appl Mater Interfaces (2022) 14:24144–59. doi: 10.1021/acsami.2c03839
216. Kuerban K, Gao X, Zhang H, Liu J, Dong M, Wu L, et al. Doxorubicin-loaded bacterial outer-membrane vesicles exert enhanced anti-tumor efficacy in non-small-cell lung cancer. Acta Pharm Sin B (2020) 10:1534–48. doi: 10.1016/j.apsb.2020.02.002
217. Huang M, Xiong D, Pan J, Zhang Q, Wang Y, Myers CR, et al. Prevention of tumor growth and dissemination by In situ vaccination with mitochondria-targeted atovaquone. Adv Sci Weinh Baden-Wurtt Ger (2022) 9:e2101267. doi: 10.1002/advs.202101267
218. Katoh Y, Yaguchi T, Kubo A, Iwata T, Morii K, Kato D, et al. Inhibition of stearoyl-CoA desaturase 1 (SCD1) enhances the antitumor T cell response through regulating β-catenin signaling in cancer cells and ER stress in T cells and synergizes with anti-PD-1 antibody. J Immunother Cancer (2022) 10. doi: 10.1136/jitc-2022-004616
219. Varghese S, Pramanik S, Williams LJ, Hodges HR, Hudgens CW, Fischer GM, et al. The glutaminase inhibitor CB-839 (Telaglenastat) enhances the antimelanoma activity of T-Cell-Mediated immunotherapies. Mol Cancer Ther (2021) 20:500–11. doi: 10.1158/1535-7163.MCT-20-0430
220. Huang M, Xiong D, Pan J, Zhang Q, Sei S, Shoemaker RH, et al. Targeting glutamine metabolism to enhance immunoprevention of EGFR-driven lung cancer. Adv Sci Weinh Baden-Wurtt Ger (2022) 9:e2105885. doi: 10.1002/advs.202105885
221. Jung KH, LoRusso P, Burris H, Gordon M, Bang Y-J, Hellmann MD, et al. Phase I study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) administered with PD-L1 inhibitor (Atezolizumab) in advanced solid tumors. Clin Cancer Res Off J Am Assoc Cancer Res (2019) 25:3220–8. doi: 10.1158/1078-0432.CCR-18-2740
Keywords: immunotherapy, resistance, mechanism, tumor microenvironment, lung cancer
Citation: Zhou K, Li S, Zhao Y and Cheng K (2023) Mechanisms of drug resistance to immune checkpoint inhibitors in non-small cell lung cancer. Front. Immunol. 14:1127071. doi: 10.3389/fimmu.2023.1127071
Received: 19 December 2022; Accepted: 30 January 2023;
Published: 08 February 2023.
Edited by:
Zichao Luo, National University of Singapore, SingaporeReviewed by:
Shahnaz Majid Qadri, Texas A&M University, United StatesCopyright © 2023 Zhou, Li, Zhao and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke Cheng, MTgzODE4MTI4QHFxLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.