 Anton Kamnev
Anton Kamnev
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 November 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.750537
This article is part of the Research Topic Immune Cell Migration in Health and Disease View all 14 articles
Motility is a crucial activity of immune cells allowing them to patrol tissues as they differentiate, sample or exchange information, and execute their effector functions. Although all immune cells are highly migratory, each subset is endowed with very distinct motility patterns in accordance with functional specification. Furthermore individual immune cell subsets adapt their motility behaviour to the surrounding tissue environment. This review focuses on how the generation and adaptation of diversified motility patterns in immune cells is sustained by actin cytoskeleton dynamics. In particular, we review the knowledge gained through the study of inborn errors of immunity (IEI) related to actin defects. Such pathologies are unique models that help us to uncover the contribution of individual actin regulators to the migration of immune cells in the context of their development and function.
Understanding how the diverse motility strategies of immune cells are controlled at the molecular level is of paramount importance when investigating immune cell responses in the context of health and disease and when designing cell-based immunotherapies. Motility is inherent to leukocyte development and differentiation for proper positioning in specific regions of lymphoid organs (1, 2). Moreover, motility is essential for mature immune cells to travel across organs and ensure their immuno-surveillance function (3, 4). Given the diversity of tissue environments and barriers crossed by any given leukocyte along its life cycle, motility needs to be regulated as a highly adaptable function (5). The intrinsic regulation of leukocyte motility relies on the integration of motility signals into adapted cell shape remodelling. This process is governed by the actin cytoskeleton that promotes the protrusive and contractile activities necessary for cell movement (6). Furthermore actin remodelling sustains other motility-related activities, such as organelle recycling, mitochondria positioning and nuclear envelope deformation (7). The molecular machinery responsible for actin remodelling comprises actin-binding proteins as well as upstream regulators accounting for a few hundreds of proteins (8, 9). In the context of leukocyte migration, the knowledge we have today about the specific roles of actin regulators stems in part from the study of rare inborn errors of immunity (IEI) caused by mutations in corresponding genes. The elucidation of molecular mechanisms underlying IEIs has revealed that more than 20 are either directly caused by, or associated with, defective actin cytoskeleton remodelling (10–14). These disease entities might therefore be considered as actinopathies specific to the immune system (from here onwards referred to as actinopathies). In this review, we present updated knowledge on actinopathies with a focus on leukocyte motility defects. In the first part of the review, we assemble knowledge on leukocyte circulation through the organism affected by actinopathies. In the second part, we turn to the cellular scale and present some of the motility challenges leukocytes face while executing their function. Finally, in the third part of the review, we zoom in on the subcellular scale and examine how actin remodelling shapes diverse cellular protrusions and ultrastructures to propel cell through dense environments following migration stimuli.
Table 1 presents an updated list of 23 actinopathies with a focus on the leukocyte motility defects characterized so far in each of these pathologies. In addition, we sorted identified defects in motility by the experimental model used in the study: primary material from patient versus cellular and animal models of the specific gene defect. The molecular mechanisms affected in actinopathies span multiple facets of the molecular machinery responsible for actin remodelling, as detailed in (14). Since multiple molecular layers operate upstream of actin remodelling, it is not obvious to define a threshold for the inclusion of gene defects falling under the umbrella of actinopathies. We here focus on actin itself (β-actin), actin-binding proteins or subunits of actin-binding protein complexes (ARPC1B, CORO1A, DIAPH1, HEM1, MKL1, MSN, MYH9, WASP, WDR1 and WIP), direct regulators of actin-binding proteins (CARMIL2, PSTPIP1 and STK4/MST1), RHO GTPases (CDC42, RAC2, RHOG and RHOH) and GTPase regulators (ARHGEF1, DOCK2, DOCK8, RASGRP1 and TTC7A). Table 1 highlights crucial contribution of actinopathy discovery to our understanding of the role of individual molecular regulators in the motility and specific functions of immune cells.

Table 1 Actin-related inborn errors of immunity and associated leukocyte motility defects.
The life journey of leukocytes is indissociable from their trafficking across the organism. From their differentiation in primary lymphoid organs to their homing and recirculation in secondary lymphoid organs and peripheral tissues, immune cells navigate through various tissues (Figure 1). They use both blood and lymphatic systems to commute between the organs they visit or colonise. To date, most actinopathies have been found to be associated with impaired migration of immune cells within and between organs (Figure 1). This section will review data collected on actinopathies and complementary animal models that have provided fundamental knowledge about leukocyte trafficking at the organism scale.
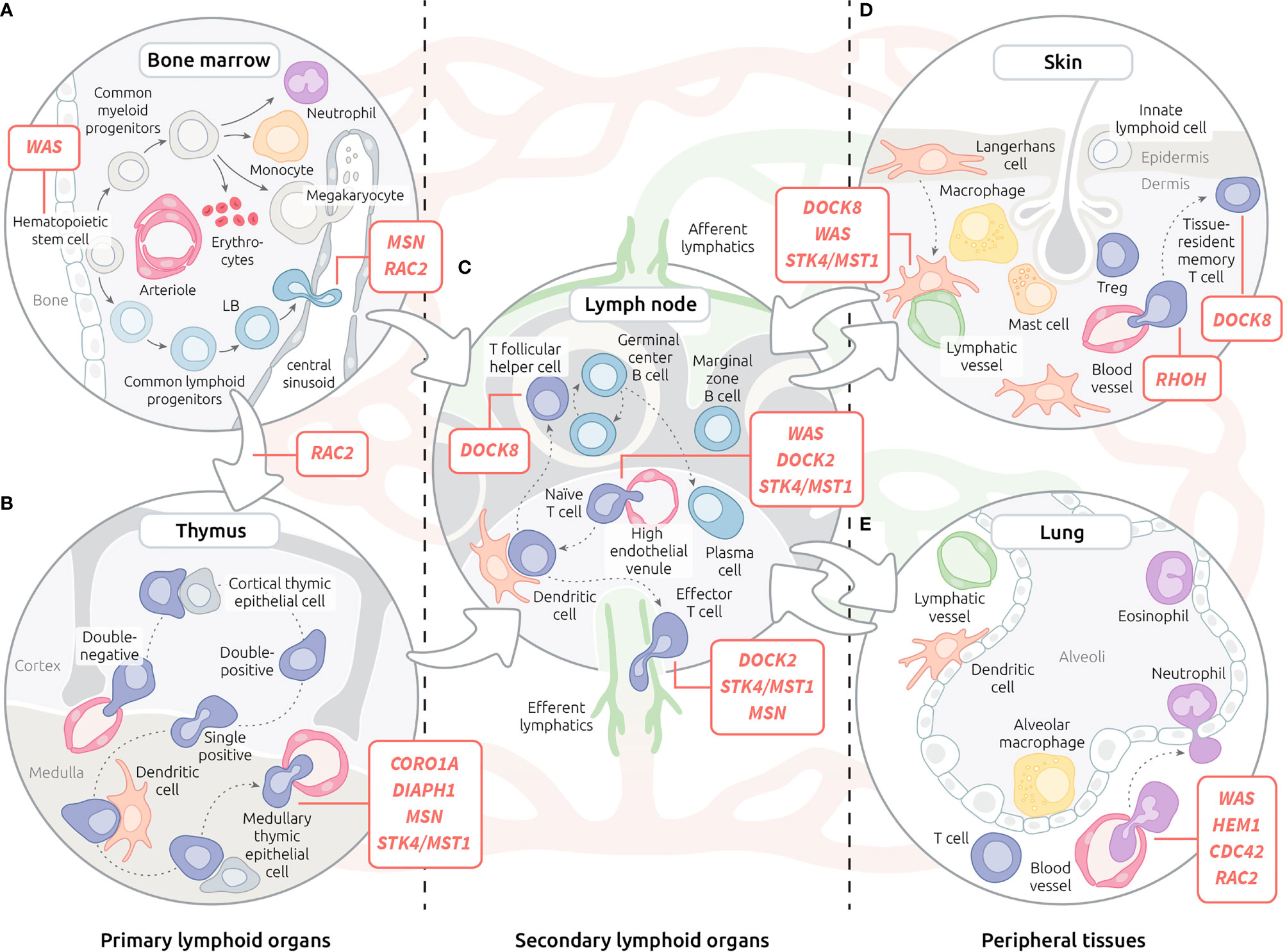
Figure 1 Motility defects in actinopathies at the organism level. (A) Leukocyte trafficking in the bone marrow. (B) T lymphocyte trafficking in the thymus. (C) Recirculation of leukocytes through blood, lymphatic system and lymph nodes. (D) Migration of leukocytes within the skin. (E) Migration of leukocytes within the lungs. Red lines indicate steps of leukocyte trafficking affected by actinopathies with affected genes displayed in corresponding bubble. Lymphatic vessels are depicted in green, while red vessels are depicted in red.
The foetal liver is the initial site of hematopoiesis. After the development of bones, hematopoietic stem cells migrate to the bone marrow (BM), which then becomes the major site of maturation of most immune cells (106). The precise positioning within BM niches of developing hematopoietic cell subsets (Figure 1A) is important for the tuning of differentiation (107). This is controlled, at least in part, by chemokine receptors, adhesion molecules and local concentrations of Ca2+ and oxygen (108).
The earliest motility defect in immune cell ontogeny reported in the context of actinopathies applies to the Wiskott-Aldrich syndrome (WAS). Indeed, hematopoietic progenitors from Was-KO mice displayed reduced migration from foetal liver to BM (93). Impaired colonisation of BM by WASP-deficient cells could explain biased X-inactivation observed in WAS female carriers. Although the complex orchestration of hematopoietic cell positioning within the BM is expected to require motility steps and acquisition of specific motility properties as cells differentiate, little is known about the function of actin regulators in these processes. In the context of actinopathies, reported bias in peripheral blood cell counts and immunophenotype in patients may reflect defects in BM migration and positioning. Indeed, in the context of WASP deficiency, the proportion of immature B cells in the BM is decreased, while that of transitional B cells in the periphery is increased (77). Such bias in B cell development has been proposed to result from the defective ability of B cells from WAS patients to respond to CXCL12, which plays a major role in immature B cell retention in the BM.
Differently, deficiency in MSN appears to be associated with a defective egress of B cells from the BM, at least in the murine KO model (39). Interestingly, in the context of B cell development, MSN expression peaks in immature B cells. Analysis of B cell subpopulations in BM and peripheral blood point to a defective egress of immature B cells from the BM parenchyma into the sinusoids.
Deficiency in WDR1, a key actin severing protein, causes an even more severe defect in B cell differentiation (99). Indeed, patient BM displayed very low frequency of CD20+ B cell precursors, which was accompanied by a marked peripheral B cell lymphopenia. However, in contrast to WASP deficiency, WDR1 deficiency did not appear to affect the ability of B cells to respond to CXCL12. Rather, defective activation and regulation of apoptosis upon BCR engagement might explain the early B cell development defect in this actinopathy.
As highlighted by the Wiskott-Aldrich syndrome, proper positioning of megakaryocytes in the BM is a key step in the control of platelet production. Megakaryocytes from WASP-deficient mice displayed impaired CXCL12-evoked migration upon interaction with fibrillar collagen I (95). This combined adhesion and motility defect was shown to be associated with an inability of these cells to assemble actin-rich podosomes (see chapter 4 for detailed description). As a result of these defects, WASP-deficient megakaryocytes appeared to shed platelets ectopically within the BM space, which might explain the severe thrombocytopenia characteristic of WAS.
Beyond the few reports cited above, we currently lack insight in the relevance of actinopathy-related proteins in the multiplicity of motility steps occurring in the context of hematopoietic development in the BM. Certainly, the application of intravital imaging (109) to relevant murine models is expected to help filling this knowledge gap.
Thymopoiesis is initiated upon the migration of progenitor T cells from the BM to the thymus. Then, the negative and positive selection steps of T cell differentiation occurring in the thymus are intimately associated with regulated trafficking from cortico-medullary junction to the cortex, followed by migration to the medulla (Figure 1B). This trafficking is governed by several chemokine gradients and parallel up-regulation of chemokine receptors throughout the differentiation process (2). Once T cells reach the single positive stage, they initiate expression of S1PR1, the receptor of sphingosine-1 phosphate, a molecule crucial for egress from the thymus (110).
To date, only one actinopathy, RAC2 deficiency, has been suggested to be associated with a defect of migration of T cell precursors to the thymus. Depending on the effect of the mutation, RAC2-deficient patients present with severe T cell lymphopenia, suggestive of a defective thymic function (111). Interestingly, the use of a Rac2-deficient zebrafish model has revealed defective migration of T cell progenitors from the caudal hematopoietic tissue to the thymus (63), pointing to an early migration defect as the reason for T cell lymphopenia.
The migration within the thymus, in particular from the thymic cortex to the medulla area (promoted by CCL21), has been studied in CD4+ T cells from Actb knock-out mice. An in vitro transwell migration assay revealed a lack of response of these cells to CCL21, suggesting that β-actin would be necessary for this aspect of T cell motility (16).
Defect of T cell egress from thymus has been documented for CORO1A deficiency (27). CORO1A deficiency was first described in a child with a T-B+NK+ severe combined immunodeficiency (SCID) phenotype. However, unlike many SCID patients with absent or undetectable thymus, the patient had a thymic image on CT-scan, suggesting defective thymic egress rather than developmental impairment. This finding agrees with previous research of CORO1A deficiency in murine models which documented defective thymic egress as well (29, 30). This defect has been shown to be related to impaired migration of T cells toward S1P, although low survival of the CORO1A-deficient T cells might contribute to the severity of the defective thymic output (27).
In addition to CORO1A deficiency, defects in T cell egress from the thymus have been documented in murine models defective in a number of actinopathy-related molecules: DOCK8 (112), DIAPH1 (33), STK4/MST1 (73, 74) and MSN (39, 52). T cells from DOCK2-deficient mice failed to migrate toward S1P, which resulted in defective thymic egress and peripheral lymphopenia (35). The T cell lymphopenia observed in DOCK8-deficient mice was associated with accumulation of mature single positive T cells in the thymus, as a result of increased chemotaxis in response to CXCL12 (112). Knock-out of Diaph1 in mice caused impaired chemotaxis toward CCL21 and CXCL12, associated with reduced T cell numbers in spleen and lymph node (LN), but normal cellularity and cell distribution in the thymus (33). A defective egress of Diaph1-/- thymocytes in response to CCL21 was identified using an organ culture of the thymus. Patients with STK4/MST1 deficiency have a profound CD4 lymphopenia with very low circulating naive CD4 and CD8 T cells. In addition, patient CD4 T cells displayed a defective migration toward CCL19 and CCL21 (72). STK4/MST1-deficient mice were shown to accumulate mature thymocytes in the thymus, which was associated with peripheral T cell lymphopenia. A transwell assay with thymic lobes was used to show that Mst1-/- T cells have impaired emigration from the thymus in response to CCL19 (73). In a complementary study, STK4/MST1-deficient thymocytes exhibited defective migration in response to CCL19, CCL21, CXCL12 and CCL25 but not S1P (74). Thus, STK4/MST1 could act as a signalling hub for several chemokines. Finally, Msn-/- mice exhibited an accumulation of mature single positive thymocytes with peripheral lymphopenia (39), in line with the finding that MSN expression is induced at the single positive stage at which it regulates the response to S1P via the downregulation of S1PR1 (52).
In conclusion, a number of actinopathies associated with T cell lymphopenia are associated with perturbations in the egress of mature T cells from the thymus. Whether some of the considered actinopathies might also be associated with more subtle alterations in the precise positioning of developing T cells in the different regions of the thymus remains to be investigated.
The architecture of secondary lymphoid organs is defined by specialized areas, the organization of which highly depends on the selective migration programs of immune cell subsets interacting in these areas (Figure 1C). Therefore, histological analysis of lymphoid organ biopsies in actinopathies, when available, can be informative to reveal leukocyte migration defects. This is the case for WAS patients in whom examination of LNs and spleen pointed out a reduction in T and B cell areas (78). This abnormal architecture was partly recapitulated in Was-KO mice that harbour reduced B cell areas and slower germinal centre reaction after immunization (76). This defect was found to be B cell intrinsic since homing capacity was impaired when WASP-deficient B cells were transferred into wild-type recipients.
Histological examination of the lamina propria of the colon of a child with DOCK2 deficiency suffering from colitis showed low density of B cells, plasma cells, and T cells (34). This was suggestive of a defective homing of lymphocytes to local lymphoid tissues in the context of inflammation. In agreement, data on isolated B and T cells from DOCK2-deficient patients have revealed defects in RAC1 activation, actin polymerization and migration towards chemokines. The major role of DOCK2 in driving leukocyte trafficking to the LN had previously been established in Dock2-/- mice (36). Indeed, the accumulation of Dock2-/- cells in LNs was reduced upon adoptive transfer. Multiphoton intravital microscopy has been successfully used to investigate the intra-nodal migratory behaviour of Dock2-/- T and B cells (35). Although these cells localized properly, they featured reduced motility with erratic oscillations in contrast to the random walk pattern observed in control cells. DOCK2-deficient T and B cells also exhibited a two-fold increase in dwelling time caused by a defect in LN egress with impaired response to S1P signalling.
DOCK8 deficiency is also associated with combined defects of lymphocyte subsets in the context of secondary lymphoid organs. Defective homing of Dock8-KO T cells to LNs was suggested to be attributable to the role of DOCK8 in activating WASP via CDC42 (40). A genetic screen revealed that DOCK8 is required for the formation of marginal zone B cells, the persistence of B cells in germinal centres and their affinity maturation. However, these B cell intrinsic defects were associated neither with homing nor with chemokine-induced motility, but rather with a defect in LFA-1 polarization at the immunological synapse (IS) (113). T cell defects in the Dock8-KO mice were also shown to contribute to the poor antibody responses to T cell dependent antigens. In particular the migration of T follicular helper cells to B cell follicles was severely reduced (41). The role of DOCK8 in T and B cell function is further highlighted by cases of somatic reversion showing clinical improvement as a consequence of partial functional restoration of the T and B cell memory compartments (114).
Patients with MSN deficiency typically have very low T cell count in their peripheral blood but do not experience as many severe infections as SCID patients (51). A possible explanation for this discrepancy might be an abnormal retention of MSN-deficient lymphocytes in LNs. In support of this possibility, lymphocyte entry into the spleen and LNs was documented to be only minimally impacted in the Msn-/- model, while lymphocytes egress from LNs was reduced (39). Such differential defect in LN entry as opposed to egress might be due to a preponderant role of MSN in regulating S1P-dependent egress, as suggested by the recent finding that ezrin-radixin-moesin (ERM) proteins are particularly important to spatially control a bleb-based motility mechanism, specifically triggered by S1P (53).
STK4/MST1 deficiency in humans is associated with reduced expression of the homing receptors CCR7 and CD62L on lymphocytes and impaired migration towards CCL19 and CCL21 (72). These defects are expected to severely impact lymphocyte homing to secondary lymphoid organs, in agreement with the reduced cellularity found in the secondary lymphoid organs of STK4/MST1-deficient mice at steady state and upon adoptive transfer of B and T cells (73). Furthermore, evidences suggest that STK4/MST1 contributes to LN and non-lymphoid tissue egress (74). In addition to its role as a transcriptional regulator, STK4/MST1 has been shown to exert a direct function on actin cytoskeleton remodelling in T cells through phosphorylation of L-plastin (115). It is therefore possible that the extended role of STK4/MST1 in regulating lymphocyte trafficking might result from its combined function as a regulator of both actin cytoskeleton and transcription.
Dendritic cells (DCs) are of critical importance for mounting an adaptive immune response. After antigen uptake in the peripheral tissue, these antigen-presenting cells migrate to the draining LNs through the lymph to activate specific T cells. It was shown in mice that WASP-deficient DCs exhibit a delayed migration from the skin to the draining LNs. In WASP-deficient animals, LNs do not increase in size and cellularity, reflecting the absence of lymphocyte traffic modification (91). Bone marrow-derived DCs were also studied in a mouse model of X-linked neutropenia (XLN), caused by gain-of-function mutations in the WAS gene (L272P). Unlike WAS-KO DC, WAS L272P DC show abnormal speed fluctuations and reduced global displacement. When tested in vivo, both seem to impair skin DC entry into the draining LN (98). In addition to WASP, a correct expression of DOCK8 in DCs seems to be a prerequisite for efficient T cell priming in vivo. Indeed, DOCK8 deficiency does not impact antigen uptake nor its presentation, but decreases DC trafficking to the draining LNs (38). A defect of skin DC migration to the draining LNs was also observed in STK4/MST1-deficient mice (73). Of note, STK4/MST1- and DOCK8-deficient patients share a common susceptibility to cutaneous warts, which could be explained by impairment of skin DC homing to the LNs.
In conclusion, a number of actinopathies are associated with alterations in the homing of lymphocytes and DCs to LNs (WASP, DOCK2, STK4/MST1) and/or in the egress of antigen-experienced lymphocytes from LNs (DOCK2, MSN, STK4/MST1). Interestingly, each examined deficiency appears to impact non-redundant mechanisms and steps accounting for LN homing and egress.
Because of their contact with the environment, the skin and the lungs represent the main sites of pathogen entry (Figures 1D, E). It is therefore not surprising that most IEIs are associated with increased susceptibility to skin and lung infections (116). In the context of actinopathies, defective recruitment to these sites of both innate immune cells and primed lymphocytes may account for reduced ability to fight local infections.
During the primary phase of an infection, innate cells (e.g., neutrophils and monocytes) migrate to the site of inflammation. The infection spectrum observed in WAS patients may be suggestive of a neutrophil defect. This was confirmed in WASP-deficient mice where impaired integrin-dependent function in neutrophils was linked to a delay in migration into an inflamed site (83). On the contrary, despite a severe neutropenia, XLN patients are not at high risk of infections (117). This could be explained by normal numbers of neutrophils in peripheral sites, as exemplified in XLN patients saliva (97). Interestingly, neutrophils from XLN mice exhibit an increased infiltrating capacity with a competitive advantage over WT neutrophils in mixed bone marrow chimeras. The study of HEM1-null macrophages and neutrophils suggested an impaired migration in vitro (46, 47). More precisely, the knock-down of nckap1l, the gene encoding for HEM1, in zebrafish was responsible for a defective neutrophil migration after tail injury along with a decrease in circulating neutrophils (45). In a model of lipopolysaccharide (LPS)-induced lung inflammation, a prominent role of CDC42 was discovered in neutrophil emigration. Murine marrow cells with inducible KO of Cdc42 were infused in irradiated mice. After LPS challenge, neutrophil counts in the lungs were significantly lower for Cdc42-/- reconstituted mice, revealing a defect in neutrophil infiltration (25). RAC2-deficient mice presented a decrease in cellular inflammatory exudate despite a persistent neutrophilic leukocytosis. This phenotype was reminiscent of leukocyte adhesion deficiency and thus suggested a defect in migration to the site of inflammation (66). To recapitulate the phenotype observed in humans, Deng and colleagues engineered a zebrafish model harbouring the inhibitory D57N mutation in Rac2 (64). This model allowed the observation of an impaired neutrophil migration to the site of infection with high neutrophil counts in peripheral blood. Interestingly, researchers discovered the role of RAC2 signalling in neutrophil retention in the bone marrow as Rac2 D57N mutation was able to partially rescue a zebrafish model of WHIM with constitutive CXCR4 signalling.
Once activated, effector T cells have to move to the site of infection (e.g., lung or skin) to exert their helper or cytolytic function. For this purpose, they express tissue homing chemokines receptors or specific integrin. Some of these T cells will acquire a tissue residency program to respond rapidly to a second antigen encounter (118). Interestingly, DOCK8-deficient patients suffer from numerous skin infections (119, 120). This peculiar infectious phenotype motivated studies on the role of DOCK8 in migration into the skin constrained environment, as we will detail in part 3. In DOCK8 deficiency, non-DC mononuclear phagocytes are prone to migration-induced cell death. This drives the skewing of the CD4+ T cell response to a Th2 profile in the context of respiratory tract infection with Cryptococcus neoformans (121). RHOH-deficient patients also display susceptibility for skin infections with persistent Epidermodysplasia verruciformis linked to human papillomavirus infections (EV-HPV), that could be explained by a defect in lymphocyte skin-homing (69). The study of RHOH-deficient patients evidenced several differences in tissue-homing markers compared to healthy donors. In particular, a defect in b7+ T cells was documented and confirmed in RhoH-/- mice.
In conclusion, the study of actinopathies has uncovered a number of key molecules driving effective trafficking and localization of immune cells within the organism. In these pathologies, defective recruitment of effector cells to tissues particularly exposed to infectious agents, such as the lungs and the skin, is at least in part accounting for the susceptibility of patients to infections.
To gain insight into the actual leukocyte motility defects underlying actinopathies, it is important to consider the precise steps accounting for the translocation of leukocytes to tissues and their navigation within those tissues [as reviewed in (122)]. As described in chapter 2 (organism scale), among the trafficking defects associated with actinopathies are navigation of naïve lymphocytes to secondary lymphoid organs and migration of activated lymphocytes to infection sites. Although each subset of immune cells tends to migrate along a specific route, four migration steps are shared as depicted in Figure 2: 1) adhesion to the blood vessel at the site of priming or infection, 2) trans-endothelial migration (TEM), 3) navigation through the interstitial space and 4) interaction with a target (e.g., pathogens, antigen presenting cell or other cells of the immune system).
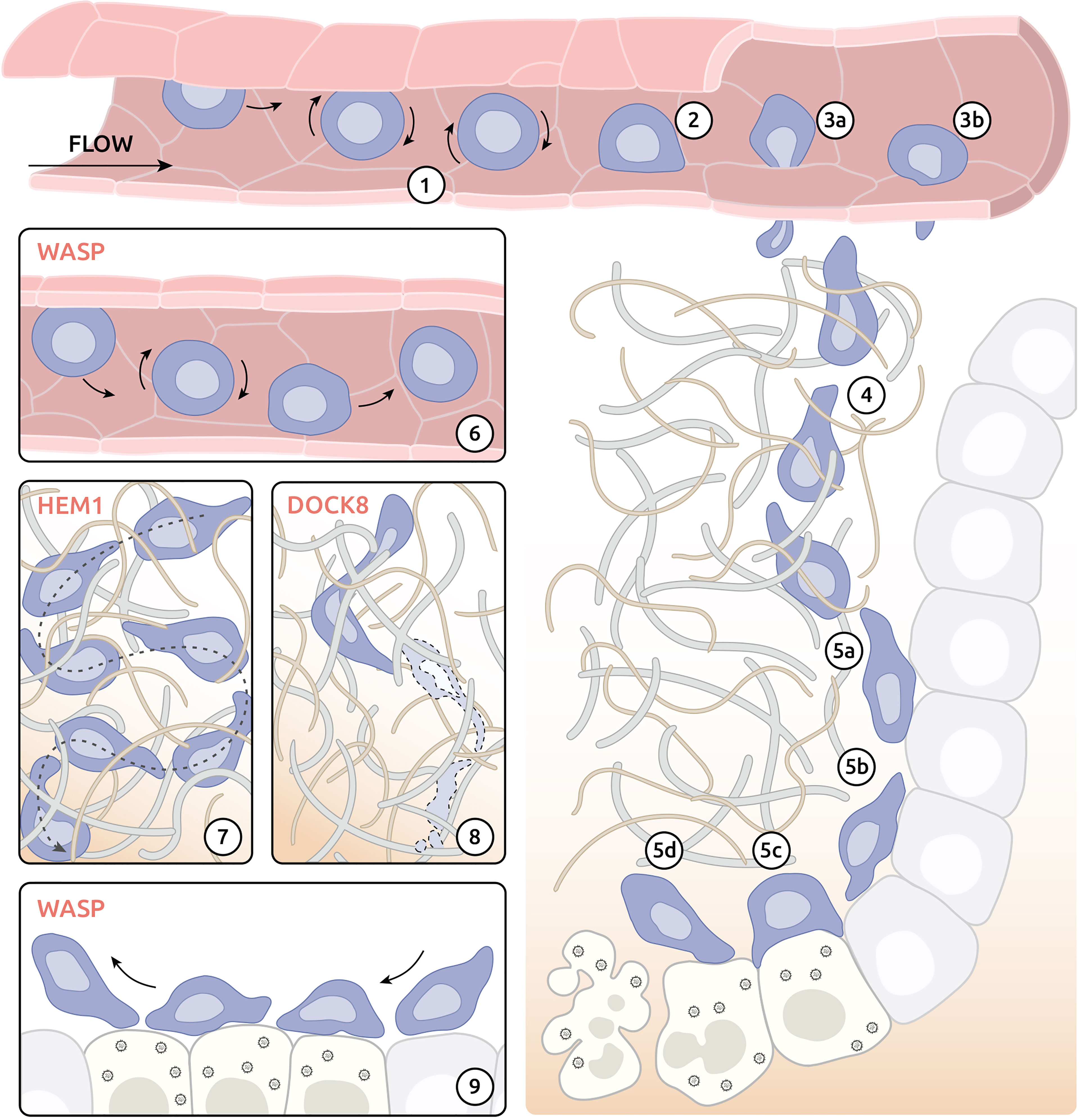
Figure 2 Migratory challenges and actinopathy-associated defects of effector CD8+ T cells on the tissue level. (1) Weak adhesion (rolling) of effector T cells after interaction with activated endothelium. (2) Adhesion at the site of exit. (3) Exit of the blood vessel by migration between (3a) or through (3b) endothelium cells. (4) Interstitial navigation following chemokine gradient. (5) Execution of cytolytic activity at the site of infection. (5a) Interaction with target cells. (5b) Kynapse-based scanning of target cells. (5c) Development of IS with infected cells and delivery of cytotoxic compounds. (5d) Destabilization of immune synapse (IS) and detachment from targeted cell. (6) Compromised attachment of WASP-deficient T cells to the endothelium at the exit site. (7) Reduced capacity of directional migration in tissue environment of HEM1-deficient T cells. (8) Cytothripsis of DOCK8-deficient T cells during migration through confined environment. (9) Impaired formation of IS by WASP-deficient T cells.
The mechanism of exit from blood circulation used by immune cells is well-studied and has been reviewed in detail elsewhere (123). In case of infection, tissue-resident sentinel cells (e.g., macrophages, dendritic and mast cells) release cytokines (including TNF-α, IL-1β or histamine), which, in turn, activate endothelial cells in proximal blood vessels. Activated endothelium increases expression of key adhesion molecules, such as selectins and integrin ligands, at its surface. Selectins are responsible for initial low-affinity adhesion of immune cells leading to rolling of immune cells along the activated endothelium (Figure 2-1). Rolling helps immune cells to sample chemokines bound at the surface of endothelium and eventually leads to activation of integrin receptors (e.g., LFA-1) in immune cells. Activated integrin receptors bind to endothelium with much higher affinity and allow immune cells to resist the blood flow following initial adhesion at the exit site (Figure 2-2).
Studies in a murine model of WAS has revealed compromised ability of WASP-deficient T cells to adhere under shear flow (Figure 2-6) (96). Further study of XLN found that neutrophils in both XLN patients and XLN mouse models displayed dramatic increase in adhesion under shearing flow and increased migration into tissue (97). These studies suggest WASP to be important for proper adhesion of immune cells at the exit site of blood vessels. DOCK8 deficiency was also documented to result in defective attachment of T cells to ICAM-1 under flow, resulting in LN homing defects (40). In that study, DOCK8, WIP and WASP were reported to form a molecular complex positioning DOCK8 as a major guanine nucleotide-exchange factor to activate WASP.
MKL1 is a myocardin-related transcription factor, which regulates transcription of actin and multiple cytoskeleton-related genes (124, 125). Recent study of MKL1-deficient neutrophils demonstrated role of MKL1 in leukocyte adhesion under shear flow (49). Specifically, neutrophils lacking MKL1 displayed lower amount of F-actin following stimulation by an adhesive substrate.
Following arrest at the exit site, immune cells exit the blood vessel via the complex process of TEM. This can occur by two major mechanisms: para-cellular transmigration (Figure 2-3A) and trans-cellular transmigration (Figure 2-3B). During para-cellular TEM immune cells leave the blood vessel between endothelial cells using transient disruption of adherens junctions. In contrast, cells using trans-cellular TEM leave the blood vessel by pushing directly though endothelial cells using invasive podosomes (89). By probing the physical properties of the endothelium, leukocytes may opt for the path of least resistance, as shown by manipulating junctional integrity (126). Breaching of endothelial cell junctions or foraging through endothelial cells require protrusive activities and high deformability, both coordinated by actin remodelling (127).
Seminal study of trans-cellular TEM mechanism revealed a key role of WASP in T cell migration (89). Researchers demonstrated that invasive podosomes (reviewed in detail in chapter 4) are crucial for trans-cellular TEM of T cells. Furthermore, results showed that WASP-deficient lymphocytes were unable to form invasive podosomes and failed to migrate through endothelial cells by trans-cellular TEM. These findings suggest that immune cells lacking WASP are restricted to para-cellular TEM to exit blood vessel. It is, however, unclear if compromised adhesion of WASP-deficient immune cells under shear flow discussed in chapter 3.1 is linked to inability of these cells to form podosomes.
In addition to WASP deficiency, studies of immune cells from patients deficient in proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1), a scaffolding protein involved in the regulation of WASP activity (128, 129), showed defective podosome formation as well (58). Further experiments would be required to clarify whether PSTPIP1 deficiency compromises TEM in vivo.
Recently, 3 additional actin-related deficiencies have been added to the list of actinopathies affecting TEM of immune cells: MKL1 (49), HEM1 (47) and ERM (53) deficiencies. MKL1 deficiency led to poor adhesion and TEM of patient neutrophils (49). Studies of HEM1 deficiency in murine models showed drastic accumulation of HEM1-deficient myeloid cells both within and near the blood vessels (47). It is unclear, however, which step of immune cell migration (trans-endothelial or interstitial) might be affected in HEM1-deficient cells. Finally, recent study of ERM-deficient mice revealed crucial role of proper coupling between actin cytoskeleton and plasma membrane in TEM of murine T cells (53). Specifically authors showed impaired ability of T cells from MSN and moesin/ezrin-deficient mice to exit the blood stream.
After crossing the endothelial barrier, leukocytes migrate to their targets through interstitium and tissue environments (Figure 2-4). Immune cells may employ two major modes of migration: adhesion-dependent and adhesion-independent [reviewed in (122)]. Both modes of migration are extremely dependent on active and precise remodelling of actin cytoskeleton but differ in mechanism of action. Adhesion-dependent migration involves attachment of plasma membrane receptors at the leading edge (such as integrins) to the extracellular substrate followed by crosslinking of membrane receptors and actin cytoskeleton. Resulting link of extracellular matrix to actin cytoskeleton allows cells to use a molecular clutch mechanism for propulsion of the cell body forward (130, 131). In contrast, adhesion-independent migration uses combination of reward actin flow and contractility of actomyosin cortex to match the topology of surrounding 3D environment. Such mechanism allows for a “chimneying” type of directional motion that applies to lymphocytes, neutrophils and DCs (132). Although adhesion-dependent and adhesion-independent migration modalities have classically been opposed, leukocytes appear to be able to use a continuum of strategies, as shown by the use of channels with variable geometries (132).
To date, studies of virtually all actin-related IEIs revealed defects in immune cell migration in vitro (Table 1). In contrast, data on ability of immune cells to migrate in vivo is available for a handful of gene defects: DOCK8 (37), DOCK2 (35), NCKAP1L (47) and MYH9 (55). Deficiency of DOCK8, an atypical immune system specific guanine nucleotide-exchange factor, is probably one of the best described actin-related IEIs affecting interstitial migration of immune cells. Ex vivo studies of DOCK8-deficient T cells migrating in the skin revealed dramatic stretching of the cell leading to cell death by stretching-induced rupture termed cytothripsis (37) (Figure 2-8). The exact mechanism of DOCK8 deficiency induced cell rupture remains to be clarified. Interestingly, studies of other DOCK deficiencies (e.g., DOCK2) found normal interstitial migration of immune cells (35). Specifically, lymphocytes from DOCK2-null mice successfully navigated through complex 3D environment albeit with decreased velocity.
Recent studies of HEM1 (47)- and MYH9 (55)- deficient cells expanded the number of actin-related IEIs affecting interstitial migration of immune cells. Mouse models of both HEM1 and MYH9 deficiencies demonstrated compromised ability of leukocytes to migrate in a 3D chemokine gradient (Figure 2-7). Directionality of immune cell migration, however, was preserved in both deficiencies.
The immunological synapse (IS) describes a tight junction formed by a T cell with either an antigen-presenting cell or an infected cell. At the tissue level formation of IS consists of 4 major phases (Figures 2-5 A~D): a) initial contact, b) spreading, c) mature synapse and d) destabilisation and termination [reviewed in (14)]. All phases of IS formation are controlled by a complex network of activatory and inhibitory stimuli and require sophisticated orchestration of actin-cytoskeleton [reviewed in (133)]. In accordance, numerous actin-related IEIs have been associated with defects in morphology and functions of IS, such as T cell activation, cytokine secretion and cytolytic activity (reviewed in (14); summarized in Table 1). In addition to T cells, the other subsets of lymphocytes including B cells, NK cells and innate lymphoid cells (ILCs) also assemble IS with partner cells or target cells. Although the molecular composition of the IS might vary among those lymphocyte subsets, its dependence on actin remodelling is a shared property. Exemplified by WASP deficiency, it is therefore expected that IS defects across numerous lymphocyte subsets are associated with numerous actinopathies.
To date, little is known about the impact of actin-related IEIs on interaction dynamics between lymphocytes and their targets in the context of tissue environments. One may speculate that deficiencies in WASP and DOCK2 for example might alter lymphocyte scanning activity. Indeed the roles of WASP in the stabilization and termination of the IS (88, 134–136) are expected to translate into biased interaction of lymphocytes with antigen-presenting cells or target cells in peripheral organs (Figure 2-9). The over-stabilization of the IS observed between CD4+ T cells and DCs in the DOCK2-deficient mice (137) is also expected to alter the serial scanning behaviour of the CD4+ T cells. More in vivo studies however, would be needed to clarify the impact of the actinopathy-related proteins to the motility behaviour of various lymphocyte subsets in the context of antigen search and IS dynamics.
The highly adaptable motility property of leukocytes is supported by their ability to emit a variety of highly dynamic actin-rich protrusions (Figure 3). Protrusions such as leading-edge lamellipodia and filopodia may exert an exploratory function. Others like podosomes and microvilli may rather promote and regulate adhesion. The uropod at the trailing edge is the site of contractile activities that regulates cell detachment in the context of motility. In addition, the actin cytoskeleton is involved in controlling the squeezing of the nucleus in the context of migration through confined spaces. Section below summarizes cellular protrusions affected in actinopathies. These natural deficiencies thereby highlight the key function of actin regulators in controlling leukocyte motility at the ultrastructural level.
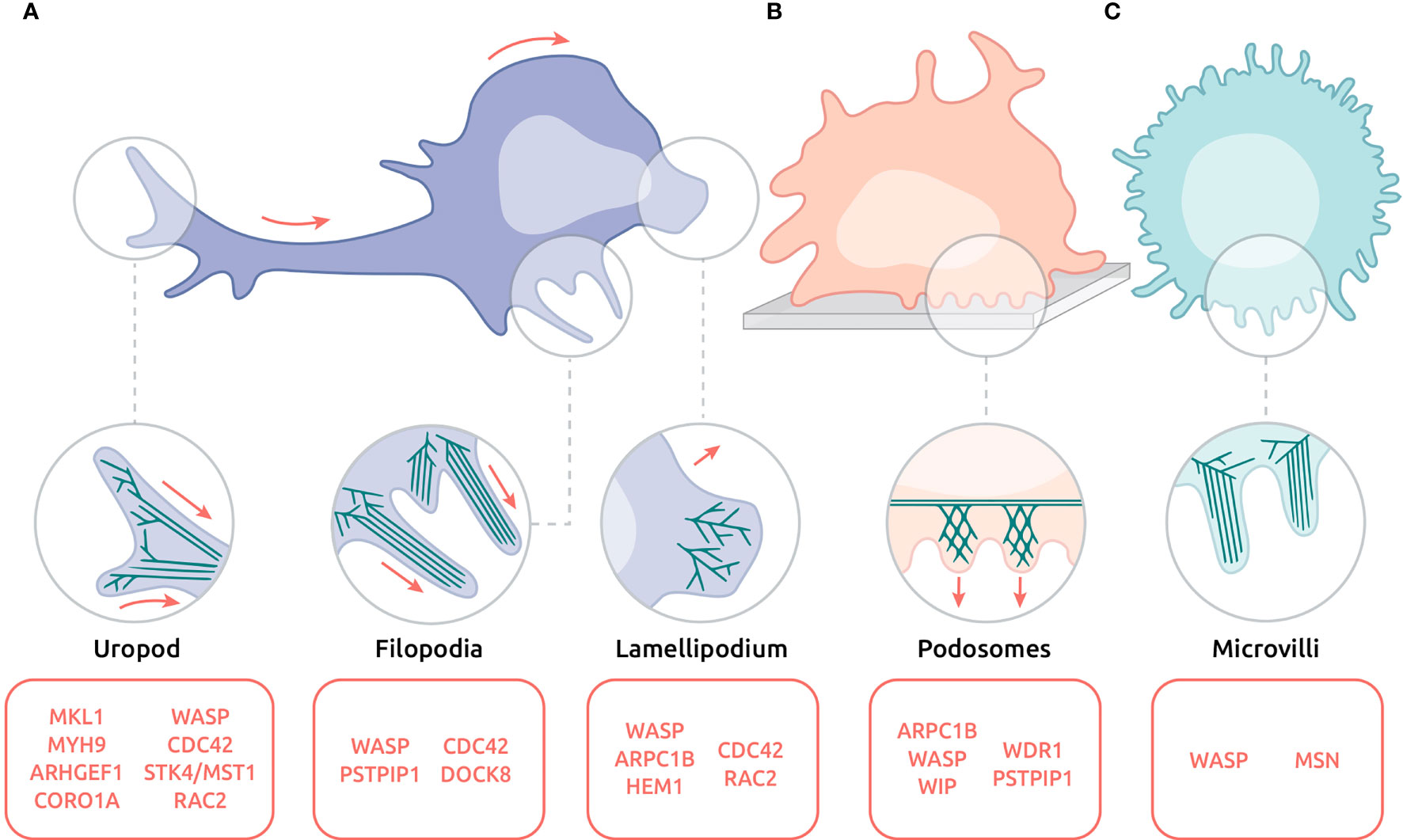
Figure 3 Actin-based protrusions affected in actinopathies on the ultrastructural level. (A) Migrating lymphocyte with uropod, filopodia and lamellipodium. (B) Adhering DC emitting an array of podosomes. (C) Circulating neutrophil decorated with microvilli. Red arrows indicate direction of movement. Protein names listed in red indicate their involvement in the corresponding protrusions.
Located at the leading edge, the lamellipodium is a large expansion of the plasma membrane, generated by actin polymerization and branching (Figure 3A-Lamellipodium) [see (138) for review]. In lymphocytes it supports not only the exploratory behaviour during migration but also IS assembly during APC encounter (139). In accordance with the major role of actin cytoskeleton remodelling in lamellipodium formation, defects in its development and organization have been reported in deficiencies affecting the ARP2/3 complex-dependent process of actin branching. ARPC1B is an essential subunit of the ARP2/3 complex that is critical for its assembly and maintenance (140). Upon interaction with a surface coated with ICAM-1 and α-CD3 antibodies to mimic APC encounter, ARPC1B-deficient T cells fail to assemble a circular lamellipodium and instead emit aberrant thin filopodia (20, 21, 90). ARPC1B deficiency in human T cells is also associated with defective migration in response to chemokines (20), presumably because of a defective lamellipodium. Indeed, ARP3 subunit knock-down, which also leads to a destabilization of the ARP2/3 complex, results in a reduced exploratory behaviour of murine T cells in both 3D collagen matrices and zebrafish embryos (141). Interestingly, reduced motility of ARP3-deficient T cells is associated with reduced cortical tension and a switch from the leading-edge lamellipodia to blebs.
Although WASP operates as an ARP2/3 activator, its involvement in the regulation of lamellipodial protrusion is not as prominent as it is for ARP2/3 (142, 143). Indeed, WASP is not essential for lamellipodium emergence, but rather controls its stability and dynamics, as shown in T cells in the context of IS assembly (134, 135). Instead of emitting a radially distributed lamellipodium, WASP-deficient CD8+ T cells display a disrupted lamellipodium that irradiates in different directions (87, 90). Alternation of arrest phases at the contact with APC and motility phases to search for new APC is key to the tuning of lymphocyte activation and function. In this context WASP appears to play a particularly central role since its ubiquitination-dependent degradation activated upon TCR ligation sets the turnover of the IS (136). In line with the role of WASP in stabilizing the lamellipodia of T cells and DCs isolated from WAS patients emitted unstable lamellipodia (79) and failed to maintain polarization at the leading edge (80). On the other hand, a study of neutrophils carrying a gain-of-function mutation in WASP (L270P) showed an increase of the spreading area of the lamellipodia compared with normal neutrophils, which was correlated with an increase in the adhesion footprint (97).
In agreement with its function as a chaperone of WASP (144), WIP plays a prominent role in lamellipodial assembly as well. Upon interaction with surfaces coated with ICAM-1 and α-CD3 antibodies or fibronectin and chemokines, T cells derived from a WIP-deficient patient failed to assemble lamellipodia (103). Furthermore, upon exposure to a CCL19 gradient, WIP-depleted B cells emitted multipolar filopodia and pseudopodia, which were associated with reduced speed and directional migration (103). Comparably, WIP-deficient DCs displayed defective polarization due to unstable lamellae and ruffles in place of the leading edge (104).
The pentameric WAVE complex is a key ARP2/3 activator in addition to WASP, albeit sustaining distinct cellular activities (61, 145, 146). HEM1 is a hematopoietic-specific subunit of the WAVE complex. Recent studies found deficiency in HEM1 to be a novel form of immune-related actinopathy (43–45). In the absence of HEM1 the other subunits of the WAVE complex are unstable and degraded. Deficiency of the WAVE complex severely affects actin branching and consequently lamellipodial dynamics. HEM1-deficient T cells struggle to spread and lack lamellipodial protrusions. In addition, HEM1 deficiency leads to a decrease in F-actin density at the leading edge (43, 44). Upon stimulation with ICAM-1, HEM1-deficient T cells isolated from patients, present lack of a leading edge or an abnormal distribution of F-actin at the leading edge instead of the lamellipodium (45). HEM1-deficient B cells also displayed an aberrant morphology associated with defective directional migration when exposed to a CCL19 gradient (44). As reported above for ARP3 knock-down, HEM1-deficient B cells adopted a bleb-driven type of motility. Before the discovery of HEM1 deficiency in humans, its role had been studied in the context of murine DC migration (48). Interestingly, the lack of lamellipodia in Nckap1l-/- immature DCs was associated with increased speed and unusually straight migration paths in 3D collagen gels. Preserved motility of HEM1-deficient cells suggests that the lamellipodium of leukocytes acts as an exploratory device allowing change in direction rather than as a force generating structure (48). This notion is sustained by TIRF analysis of the actin cytoskeleton in migratory DCs, neutrophils, T and B cells which showed that the lamellipodium undulates at the front of the cells and makes only transient and limited contacts with the substrate (48, 147). A recent study widened the role of HEM1 in supporting lamellipodia assembly in macrophages and platelets. In particular, HEM1-deficient macrophages adopted a spiky shape with filopodia emitted instead of lamellipodia, which was associated with reduced motility in collagen gels (47). Taken together, studies on HEM1 deficiency indicate that the WAVE complex is a crucial driver of ARP2/3-dependent lamellipodia assembly in hematopoietic cells.
The RHO GTPases RAC1 and RAC2 act as upstream regulators of lamellipodia assembly by activating the WAVE complex. Studies of patients suffering from severe disease associated predominantly with neutrophil dysfunction identified dominant negative mutations in RAC2, altering the activity of both RAC2 and RAC1 (148, 149). Upon stimulation with fMLP, patient-derived neutrophils failed to assemble a lamellipodium and to polarize. Similarly, neutrophils with mutated RAC2 failed to migrate in response to zymosan-activated serum. Study of a loss-of-function mutation in RAC2 in patients suffering from common variable immunodeficiency revealed impaired chemotaxis of RAC2-deficient neutrophils (150). Preserved activity of RAC1 in these patients could result in residual neutrophil function and, thus, explain the distinct clinical phenotype of RAC2 deficiency. Furthermore, recent study of 3 patients with a gain-of-function mutation in RAC2 showed defective migration of neutrophils to fMLP as well (62).
Filopodia are usually described as thin protrusions, composed of bundles of parallel actin filaments, that emerge from the tip of the lamellipodia of migrating cells (Figure 3A-Filopodia) [see (151) for review]. Lamellipodia-associated filopodia are considered as sensors. In leukocytes, filopodia are more often dissociated from the lamellipodia and may emerge as micrometre-long protrusions from different parts of the cell body. In addition to their role as environment sensors, filopodia play a role in phagocytosis in macrophages (152).
CDC42 is considered the main RHO GTPase involved in filopodia assembly (153). Recent study of the distinct immunological syndrome with immune dysregulation and inflammation identified mutation in CDC42 (R186C) leading to impaired ability of the protein to interact with its binding partners: IQGAP1 and, to a lesser extent, WASP (24). In the affected patients, both fibroblasts and immune cells displayed lack of cell polarization associated with aberrant density and distribution of filopodia. In addition, hematopoietic stem cells, peripheral blood mononuclear cells (PBMCs) and NK cells from affected patients displayed reduced migratory capacity towards CXCL12.
An early study on the CDC42 activator DOCK8 (42), showed that disruption of actin polymerisation induced by Cytochalasin D treatment of mice microglia led to accumulation of DOCK8 at the sites of filopodia formation. However, similar treatment of cells from DOCK8-deficient mice led to a 30% reduction in number of cells with filopodia compared to cells from WT mice.
As a further support of the role of the CDC42 axis in regulating filopodia assembly and dynamics, WASP deficiency was reported to result in a decrease in the number of filopodia in macrophages. The remaining filopodia occupied a large distribution around the cell instead of being concentrated in one specific edge (84). Moreover, a reduction of the length of filopodia-type protrusions in B cells has been observed in the context of WASP deficiency (76).
The uropod is a highly contractile structure at the rear of migrating cells, and plays a decisive role in cellular migration [see (154) for review] (Figure 3A-Uropod). The cell propulsion relies on myosin-based contractions at the uropod that allow detachment from the substrate and rearward squeezing (155).
A number of studies in cell lines and murine models have assessed the role of MYH9 in leukocyte motility. A study on human lymphocytes observed that the uropod of migrating T cells is enriched in MYH9 (57). Chemical inhibition of MYH9 activity with blebbistatin and genetic inhibition with siRNA resulted in aberrant elongation of the uropod, suggesting that the de-adhesion process was impaired upon the loss of MYH9 activity. Other studies showed that T lymphocytes can rapidly switch between an adhesion-dependent sliding motility and an amoeboid walking motility that depends on MyoIIA (156). Myh9 conditional knockout in T cells impaired the turnover of adhesion sites, resulting in increased adhesion and impaired interstitial migration in vivo (56). Interestingly, a downregulation of MYH9 in mice neutrophils showed no defect in term of cellular shape (with a rear comparable to a normal uropod), but caused decrease in migration (55). MYH9 mutations in humans have been found to be the cause of heterogeneous group of diseases, mostly characterized by macrothrombocytopenia, hearing loss and renal disease (157). Whether defects in the motility of leukocytes and in particular uropod dynamics might contribute to yet uncharacterized immune cell defects in individuals with MYH9 mutations remains to be investigated.
Although CDC42 is mainly localized at the cell leading edge, it is also involved in the modulation of the myosin light-chain pathway at the uropod (26). Via its control over WASP activation, CDC42 has also been reported to regulate CD11b reorganization at the uropod (25). In line with a role of the CDC42-WASP axis in uropod dynamics, WASP-deficient leukocytes displayed extreme elongation of uropods (80). Abnormally elongated uropods in T cells have also been described in the context of deficiency in ARHGEF1, a regulator of RHOA GTPase. This defect was associated with a decrease in the mean square displacement of migrating T cells (17). A failure of uropod retraction was also reported in MKL1-deficient neutrophil-like HL-60 cells, presumably because of a reduced expression of myosin light chain 9 (MYL9), a component of the myosin II complex (50). Deficiency in CORO1A in mice has also been reported to impair uropod assembly in T cells. Instead of this structure, CORO1A-deficient T cells formed several patch-like talin-rich clusters, abnormally distributed around the cell cortex (30). The uropod has also been reported to be affected in RAC2 deficiency (148) and STK4/MST1 deficiency (73), highlighting the complexity of the molecular control of actin dynamics in the context of this cellular structure.
Podosomes are actin-driven micron-scale cellular adhesive protrusions and are crucial for adhesion, migration and degradation of the extracellular matrix. The actin cytoskeleton within podosomes is mostly composed of branched actin filaments. Moreover, podosomes are linked together by a network of unbranched actin filaments [see (158) for review] (Figure 3B-Podosomes).
In this context, the WASP-ARP2/3 pathway appears crucial in the assembly of podosomes, while the WAVE complex is dispensable. Indeed both WASP and ARPC1B deficiencies are associated with the defective assembly of podosomes by monocyte-derived macrophages (19, 84). In contrast, macrophages from Nckap1l-KO mice have a preserved ability to assemble podosomes although they display multiple defects in lamellipodia, focal adhesions and endocytosis (47). The specificity of WASP in activating ARP2/3 towards podosome assembly might be related to its specific location in podosome priming areas and its ability to recruit ARP2/3 at such sites (84). Reconstitution experiments in WAS patient-derived macrophages using micro-injections of full-length human WASP sequence suggested that the chemotactic response to the cytokine colony stimulating factor-1 is dependent on WASP-driven assembly of podosomes (159). To date, studies of WASP revealed its key role in podosome assembly in multiple cellular systems beyond macrophages: immature DCs (80), THP-1 (19) and T cells (89). Moreover, experiments using RNA interference to induce partial down-regulation of the intracellular WASP level in DCs led to compromised podosome formation (160), indicating that a precise regulation of WASP expression might be crucial for assembly of podosomes.
As an essential partner of WASP, WIP has been reported to be essential for podosome assembly as well. In WIP-deficient mice, DCs formed few podosomes associated with loss of podosomal structure, including the actin core and the vinculin rings (104). Moreover, the authors found that lack of podosome formation in WIP-deficient DCs is compensated by assembly of large vinculin-rich focal adhesion contacts. In human macrophages, WIP localizes to the core of podosomes, where it co-localizes with F-actin (105). To date, however, defects in podosome formation have not been examined in WIP-deficient patient cells.
Studies of patients with mutations in PSTPIP1 found decreased numbers of macrophages emitting podosomes as well (58). Similar to WASP-deficient cells, deficiency in PSTPIP1 led to increase in the number and the size of vinculin-containing focal complexes compared to normal cells. A follow-up study proposed that defect of migration of PSTPIP1-deficient macrophages resulted in a switch from formation of podosomes to filopodia (59).
MKL1 deficiency is associated with a severe defect in podosome formation resulting in impaired adhesion (50). Indeed, a complete absence of podosomes has been reported in MKL1-deficient DCs on fibronectin. Conjointly, the spreading of MKL1-deficient cells was reduced, and the F-actin staining was severely decreased compare to normal DCs.
As opposed to deficiencies in ARPC1B, WASP, WIP, PSTPIP1, and MKL1, deficiency in WDR1 is associated with a reinforcement of podosomes in myeloid cells (100). WDR1, together with cofilin, promotes actin severing and is important for actin filament turnover. Its impact on leukocyte podosomes has been initially described on monocyte-derived DCs (100). In these cells, the number of podosomes was close to that of normal cells, but their volume and F-actin content were increased. A complementary study on WDR1-deficient monocytes on fibronectin found an abnormally high number of actin-rich podosome-like structures compared to normal monocytes (99). These findings support the notion that WDR1-mediated actin filament turnover is particularly important for podosome formation.
Microvilli are very thin finger-like protrusions that tend to decorate most of the cell surface (Figure 3C-Microvilli). In leukocytes, they have initially been described by scanning electron microscopy in B and T cells (161), macrophages (162) and DCs (163). Their function in leukocytes is to help migration and assist antigen sensing [see (164) for review]. In particular, in T cells most of the TCR molecules are localized at the surface of the microvilli, increasing the probability to detect antigen (165).
Initial studies have reported WASP as a key regulator of microvilli. Lymphocytes from WAS patients were shown to harbour reduced density of what appeared as blunted microvilli (166, 167). However, in a more recent study microvilli density was found to be unaltered in fresh WASP-deficient human lymphocytes (168). Furthermore, Was-KO B cells were observed to have aberrant microvilli formation upon anti-CD40 plus IL-4 stimulation, but not upon LPS stimulation (76). These studies suggest that the WASP-dependent assembly of microvilli in lymphocytes is dependent on the activation status of these cells. In addition to WASP, MSN appear to also regulate microvilli assembly. Indeed, experiments using scanning electron microscopy on murine MSN-deficient lymphocytes revealed a decrease of microvilli density, as well as defective development of these structures (39).
Deformability is an essential property of leukocytes that allows probing narrow interstices and migrating through very constrained environments. Whereas the plasma membrane is extremely deformable, the nucleus is more rigid, thereby imposing a physical checkpoint for leukocyte motility. The squeezing of the nucleus is well described in multiple cell types [see (169–171) for reviews]. In leukocytes, this process is associated with an active mechanism of pore-size discrimination facilitated by frontward positioning of the nucleus (172). Indeed, to facilitate crossing of the endothelial barrier, T cells use MyoIIA-driven contractility to squeeze the nucleus through the endothelial junctions (173). In addition, a formin-dependent actin polymerization mechanism was shown to push the nucleus at the back of effector T cells migrating in constrained inflamed tissues (174). In particular, FMNL1 was shown to promote actin polymerization at the back of the cell to enable translation of the rigid nucleus through restrictive barriers of extracellular matrix. DIAPH1, a formin highly expressed in leukocytes, has recently been shown to be associated to immune cell defects in humans when mutated (32). Whether DIAPH1 also contributes to nucleus squeezing has not yet been investigated. A further striking evidence for the role of actin remodelling in the integrity of the nucleus in the context of leukocyte migration stems from the DOCK8 deficiency. As described in chapter 3, DOCK8-deficient T cells migrating in dense environments display abnormally elongated shapes leading to cytothripsis. Interestingly, pieces of deformed nuclei were observed in the cellular fragments resulting from this atypical cell death process. This suggests that DOCK8 plays a key role in nucleus integrity by regulating the mechanical forces imposed on migrating T cells (37).
Multiple aspects of intracellular vesicle trafficking are sustaining cell polarity and motility (175). These mechanisms, such as endo- and exocytic pathways, are increasingly recognized as being dependent on vesicle-associated actin remodelling activities (176).
In the context of actin-related IEI, we currently lack direct evidence that the involved molecules might control immune cell motility via the regulation of vesicle trafficking.
However, given the fact that proteins such as WASP regulate endosome trafficking (177), it would be interesting to further investigate the possible contribution of vesicle trafficking to the actinopathy-associated cell motility defects.
The generalization of exome sequencing as an approach to identify the genetic aetiology of rare IEIs has led to the recent characterization of multiple actinopathies. This review highlights 23 gene defects related to actin cytoskeleton remodelling in immune cells and highlighted the motility impairments associated with these defects. Although alteration of leukocyte motility is not the sole explanation for the complex immune dysfunction associated with actinopathies, it certainly plays a major role. Leukocyte motility, when viewed through the prism of actinopathies highlights gaps in our knowledge of molecules and pathways crucial for the integrity of the immune system. Many actinopathies share defects in leukocyte motility (e.g., T cell egress from the thymus, homing of lymphocytes to secondary lymphoid organs, migration towards chemokines, assembly of the uropod of migrating lymphocytes) while others display unique defects specific to certain molecules (e.g., cytothripsis in DOCK8-deficient T cells during interstitial migration). Partial overlap of defects in immune cell motility among different actinopathies suggests that multiple leukocyte motility steps require the combination of multiple actin remodelling activities.
Research on actinopathies provided unique insight into the regulation of leukocyte motility. However, our current picture of the role of individual disease-related molecules remains very fragmented. Lack of detailed understanding is related to both novelty of many of the deficiencies and the amount of work it takes to apprehend the multiplicity of motility steps that immune cells undergo. Moreover, studies of immune cell motility are further complicated by the diversification of motility strategies adopted by the different immune cell subsets and restricted amount of patient material available. In an effort to address some of those limitations we have recently introduced morphological profiling of leukocytes via high-content cell imaging (90). Such approach can be applied to restricted sample sizes, can be used in standardizing the comparison of morphological defects in primary leukocytes from patients and has proven to be efficient at discriminating defects in closely related deficiencies such as ARPC1B and WASP deficiencies. Beyond high-content cell imaging, recent advances in super-resolution microscopy and design of probes and micro-patterned surfaces for live imaging are expected to probe cell migration more precisely and identify novel actinopathy-related leukocyte defects. Furthermore, a more systematic application of live tissue imaging in murine or zebrafish models is expected to complement in vitro studies with patient cells and fill the gap in our understanding of leukocyte motility across the different cellular and tissue scales.
AK, MF, CL, and LD conceptualized the review and the figures. All authors contributed substantially to the text of the manuscript. All authors approved the submitted version.
This work received support from WWTF (PrecisePID project LS16-060 to LD) and CNRS (International Research Project SysTact to LD). MF is supported by the Fondation pour la recherche médicale (FDM202006011216).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We wish to thank Tatjana Hirschmugl (Scillustration, Graz and Vienna, Austria) for design and production of the artwork.
1. Mazo IB, Massberg S, von Andrian UH. Hematopoietic Stem and Progenitor Cell Trafficking. Trends Immunol (2011) 32:493–503. doi: 10.1016/j.it.2011.06.011
2. Takahama Y. Journey Through the Thymus: Stromal Guides for T-Cell Development and Selection. Nat Rev Immunol (2006) 6:127–35. doi: 10.1038/nri1781
3. Nourshargh S, Hordijk PL, Sixt M. Breaching Multiple Barriers: Leukocyte Motility Through Venular Walls and the Interstitium. Nat Rev Mol Cell Biol (2010) 11:366–78. doi: 10.1038/nrm2889
4. Krummel MF, Bartumeus F, Gérard A. T Cell Migration, Search Strategies and Mechanisms. Nat Rev Immunol (2016) 16:193–201. doi: 10.1038/nri.2015.16
5. Friedl P, Wolf K. Plasticity of Cell Migration: A Multiscale Tuning Model. J Cell Biol (2010) 188:11–9. doi: 10.1083/jcb.200909003
6. Vicente-Manzanares M, Sánchez-Madrid F. Role of the Cytoskeleton During Leukocyte Responses. Nat Rev Immunol (2004) 4:110–22. doi: 10.1038/nri1268
7. Mastrogiovanni M, Juzans M, Alcover A, Di Bartolo V. Coordinating Cytoskeleton and Molecular Traffic in T Cell Migration, Activation, and Effector Functions. Front Cell Dev Biol (2020) 8:591348. doi: 10.3389/fcell.2020.591348
8. Rottner K, Faix J, Bogdan S, Linder S, Kerkhoff E. Actin Assembly Mechanisms at a Glance. J Cell Sci (2017) 3427–35. doi: 10.1242/jcs.206433
9. Svitkina T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb Perspect Biol (2018) 10:1–21. doi: 10.1101/cshperspect.a018267
10. Burns SO, Zarafov A, Thrasher AJ. Primary Immunodeficiencies Due to Abnormalities of the Actin Cytoskeleton. Curr Opin Hematol (2017) 24:16–22. doi: 10.1097/MOH.0000000000000296
11. Janssen E, Geha RS. Primary Immunodeficiencies Caused by Mutations in Actin Regulatory Proteins. Immunol Rev (2019) 287:121–34. doi: 10.1111/imr.12716
12. Tangye SG, Bucciol G, Casas-Martin J, Pillay B, Ma CS, Moens L, et al. Human Inborn Errors of the Actin Cytoskeleton Affecting Immunity: Way Beyond WAS and WIP. Immunol Cell Biol (2019) 97:389–402. doi: 10.1111/imcb.12243
13. Papa R, Penco F, Volpi S, Gattorno M. Actin Remodeling Defects Leading to Autoinflammation and Immune Dysregulation. Front Immunol (2021) 11:604206. doi: 10.3389/fimmu.2020.604206
14. Dupré L, Boztug K, Pfajfer L. Actin Dynamics at the T Cell Synapse as Revealed by Immune-Related Actinopathies. Front Cell Dev Biol (2021) 9:665519. doi: 10.3389/fcell.2021.665519
15. Nunoi H, Yamazaki T, Tsuchiya H, Kato S, Malech HL, Matsuda I, et al. A Heterozygous Mutation of -Actin Associated With Neutrophil Dysfunction and Recurrent Infection. Proc Natl Acad Sci (1999) 96:8693–8. doi: 10.1073/pnas.96.15.8693
16. Bunnell TM, Burbach BJ, Shimizu Y, Ervasti JM. β-Actin Specifically Controls Cell Growth, Migration, and the G-Actin Pool. Mol Biol Cell (2011) 22:4047–58. doi: 10.1091/mbc.E11-06-0582
17. Bouafia A, Lofek S, Bruneau J, Chentout L, Lamrini H, Trinquand A, et al. Loss of ARHGEF1 Causes a Human Primary Antibody Deficiency. J Clin Invest (2019) 129:1047–60. doi: 10.1172/JCI120572
18. Muppidi JR, Schmitz R, Green JA, Xiao W, Larsen AB, Braun SE, et al. Loss of Signalling via G α 13 in Germinal Centre B-Cell-Derived Lymphoma. Nature (2014) 516:254–8. doi: 10.1038/nature13765
19. Rivers E, Rai R, Lötscher J, Hollinshead M, Markelj G, Thaventhiran J, et al. Wiskott Aldrich Syndrome Protein Regulates Non-Selective Autophagy and Mitochondrial Homeostasis in Human Myeloid Cells. Elife (2020) 9:1–23. doi: 10.7554/eLife.55547
20. Brigida I, Zoccolillo M, Cicalese MP, Pfajfer L, Barzaghi F, Scala S, et al. T-Cell Defects in Patients With ARPC1B Germline Mutations Account for Combined Immunodeficiency. Blood (2018) 132:2362–74. doi: 10.1182/blood-2018-07-863431
21. Randzavola LO, Kuijpers TW, Gillian M, Randzavola LO, Strege K, Juzans M, et al. Loss of ARPC1B Impairs Cytotoxic T Lymphocyte Maintenance and Cytolytic Activity. J Clin Invest (2019) 129:1–15. doi: 10.1172/JCI129388
22. Kahr WHA, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, et al. Loss of the Arp2/3 Complex Component ARPC1B Causes Platelet Abnormalities and Predisposes to Inflammatory Disease. Nat Commun (2017) 8:1–14. doi: 10.1038/ncomms14816
23. Schober T, Magg T, Laschinger M, Rohlfs M, Linhares ND, Puchalka J, et al. A Human Immunodeficiency Syndrome Caused by Mutations in CARMIL2. Nat Commun (2017) 8:1–13. doi: 10.1038/ncomms14209
24. Lam MT, Coppola S, Krumbach OHF, Prencipe G, Insalaco A, Cifaldi C, et al. A Novel Disorder Involving Dyshematopoiesis, Inflammation, and HLH Due to Aberrant CDC42 Function. J Exp Med0 (2019). p. 2778–99. doi: 10.1084/jem.20190147
25. Kumar S, Xu J, Perkins C, Guo F, Snapper S, Finkelman FD, et al. Cdc42 Regulates Neutrophil Migration via Crosstalk Between WASp, CD11b, and Microtubules. Blood (2012) 120:3563–74. doi: 10.1182/blood-2012-04-426981
26. Szczur K, Zheng Y, Filippi MD. The Small Rho GTPase Cdc42 Regulates Neutrophil Polarity via CD11b Integrin Signaling. Blood (2009) 114:4527–37. doi: 10.1182/blood-2008-12-195164
27. Shiow L, Roadcap D, Paris K, Watson S, Cyster J. The Actin Regulator Coronin-1A Is Mutated in a Thymic Egress Deficient Mouse Strain and in a T–B+ NK+ SCID Patient. Nature (2008) 9:1307–15. doi: 10.1038/ni.1662.The
28. Yee CS, Massaad MJ, Bainter W, Ohsumi TK, Föger N, Chan AC, et al. Recurrent Viral Infections Associated With a Homozygous CORO1A Mutation That Disrupts Oligomerization and Cytoskeletal Association. J Allergy Clin Immunol (2016) 137:879–88.e2. doi: 10.1016/j.jaci.2015.08.020
29. Yagi H, Matsumoto M, Nakamura M, Makino S, Suzuki R, Harada M, et al. Defect of Thymocyte Emigration in a T Cell Deficiency Strain (CTS) of the Mouse. J Immunol (1996) 157:3412–9.
30. Foger N, Rangell L, Danilenko D, Chan A. Requirement for Coronin 1 in T Lymphocyte Trafficking and Cellular Homeostasis. Sci (80- ) (2006) 313:839–42. doi: 10.1016/b978-1-4832-3292-8.50018-7
31. Pick R, Begandt D, Stocker TJ, Salvermoser M, Thome S, Böttcher RT, et al. Coronin 1A, a Novel Player in Integrin Biology, Controls Neutrophil Trafficking in Innate Immunity. Blood (2017) 130:847–58. doi: 10.1182/blood-2016-11-749622
32. Kaustio M, Nayebzadeh N, Hinttala R, Tapiainen T, Åström P, Mamia K, et al. Loss of DIAPH1 Causes SCBMS, Combined Immunodeficiency, and Mitochondrial Dysfunction. J Allergy Clin Immunol (2021) 148:599–611. doi: 10.1016/j.jaci.2020.12.656
33. Sakata D, Taniguchi H, Yasuda S, Adachi-Morishima A, Hamazaki Y, Nakayama R, et al. Impaired T Lymphocyte Trafficking in Mice Deficient in an Actin-Nucleating Protein, Mdia1. J Exp Med (2007) 204:2031–8. doi: 10.1084/jem.20062647
34. Dobbs K, Domínguez Conde C, Zhang S-Y, Parolini S, Audry M, Chou J, et al. Inherited DOCK2 Deficiency in Patients With Early-Onset Invasive Infections. N Engl J Med (2015) 372:2409–22. doi: 10.1056/nejmoa1413462
35. Nombela-Arrieta C, Mempel TR, Soriano SF, Mazo I, Wymann MP, Hirsch E, et al. A Central Role for DOCK2 During Interstitial Lymphocyte Motility and Sphingosine-1-Phosphate-Mediated Egress. J Exp Med (2007) 204:497–510. doi: 10.1084/jem.20061780
36. Nombela-Arrieta C, Lacalle RA, Montoya MC, Kunisaki Y, Megías D, Marqués M, et al. Differential Requirements for DOCK2 and Phosphoinositide-3-Kinase γ During T and B Lymphocyte Homing. Immunity (2004) 21:429–41. doi: 10.1016/j.immuni.2004.07.012
37. Zhang Q, Dove CG, Hor JL, Murdock HM, Strauss-Albee DM, Garcia JA, et al. DOCK8 Regulates Lymphocyte Shape Integrity for Skin Antiviral Immunity. J Exp Med (2014) 211:2549–66. doi: 10.1084/jem.20141307
38. Harada Y, Tanaka Y, Terasawa M, Pieczyk M, Habiro K, Katakai T, et al. DOCK8 Is a Cdc42 Activator Critical for Interstitial Dendritic Cell Migration During Immune Responses. Blood (2012) 119:4451–61. doi: 10.1182/blood-2012-01-407098
39. Hirata T, Nomachi A, Tohya K, Miyasaka M, Tsukita S, Watanabe T, et al. Moesin-Deficient Mice Reveal a Non-Redundant Role for Moesin in Lymphocyte Homeostasis. Int Immunol (2012) 24:705–17. doi: 10.1093/intimm/dxs077
40. Janssen E, Tohme M, Hedayat M, Leick M, Kumari S, Ramesh N, et al. A DOCK8-WIP-WASp Complex Links T Cell Receptors to the Actin Cytoskeleton. J Clin Invest (2016) 126:3837–51. doi: 10.1172/JCI85774
41. Janssen E, Tohme M, Butts J, Giguere S, Sage PT, Velázquez FE, et al. DOCK8 Is Essential for LFA-1–Dependent Positioning of T Follicular Helper Cells in Germinal Centers. JCI Insight (2020) 5:1–13. doi: 10.1172/jci.insight.134508
42. Ruusala A, Aspenström P. Isolation and Characterisation of DOCK8, a Member of the DOCK180-Related Regulators of Cell Morphology. FEBS Lett (2004) 572:159–66. doi: 10.1016/j.febslet.2004.06.095
43. Cook SA, Comrie WA, Poli MC, Similuk M, Oler AJ, Faruqi AJ, et al. HEM1 Deficiency Disrupts Mtorc2 and F-Actin Control in Inherited Immunodysregulatory Disease. Sci (80- ) (2020) 369:202–7. doi: 10.1126/science.aay5663
44. Salzer E, Zoghi S, Kiss MG, Kage F, Rashkova C, Stahnke S, et al. The Cytoskeletal Regulator HEM1 Governs B Cell Development and Prevents Autoimmunity. Sci Immunol (2020) 5:1–16. doi: 10.1126/sciimmunol.abc3979
45. Castro CN, Rosenzwajg M, Carapito R, Shahrooei M, Konantz M, Khan A, et al. NCKAP1L Defects Lead to a Novel Syndrome Combining Immunodeficiency, Lymphoproliferation, and Hyperinflammation. J Exp Med (2020) 217:1–18. doi: 10.1084/JEM.20192275
46. Park H, Staehling-Hampton K, Appleby MW, Brunkow ME, Habib T, Zhang Y, et al. A Point Mutation in the Murine Heml Gene Reveals an Essential Role for Hematopoietic Protein 1 in Lymphopoiesis and Innate Immunity. J Exp Med (2008) 205:2899–913. doi: 10.1084/jem.20080340
47. Stahnke S, Doering H, Kusch C, Nieswandt B, Rottner K, Stradal TEB. Loss of Hem1 Disrupts Macrophage Function and Impacts Migration, Phagocytosis, and Integrin- Mediated Adhesion. Curr Biol (2021) 31:1–14. doi: 10.1016/j.cub.2021.02.043
48. Leithner A, Eichner A, Müller J, Reversat A, Brown M, Schwarz J, et al. Diversified Actin Protrusions Promote Environmental Exploration But Are Dispensable for Locomotion of Leukocytes. Nat Cell Biol (2016) 18:1253–9. doi: 10.1038/ncb3426
49. Sprenkeler EGG, Henriet SSV, Tool ATJ, Kreft IC, van der Bijl I, Aarts CEM, et al. MKL1 Deficiency Results in a Severe Neutrophil Motility Defect Due to Impaired Actin Polymerization. Blood (2020) 135:2171–81. doi: 10.1182/blood.2019002633
50. Record J, Malinova D, Zenner HL, Plagnol V, Nowak K, Syed F, et al. Immunodeficiency and Severe Susceptibility to Bacterial Infection Associated With a Loss-of-Function Homozygous Mutation of MKL1. Blood (2015) 126:1527–35. doi: 10.1182/blood-2014-12-611012
51. Lagresle-Peyrou C, Luce S, Ouchani F, Soheili TS, Sadek H, Chouteau M, et al. X-Linked Primary Immunodeficiency Associated With Hemizygous Mutations in the Moesin (MSN) Gene. J Allergy Clin Immunol (2016) 138:1681–1689.e8. doi: 10.1016/j.jaci.2016.04.032
52. Nomachi A, Yoshinaga M, Liu J, Kanchanawong P, Tohyama K, Thumkeo D, et al. Moesin Controls Clathrin-Mediated S1PR1 Internalization in T Cells. PloS One (2013) 8:1–13. doi: 10.1371/journal.pone.0082590
53. Robertson TF, Chengappa P, Gomez Atria D, Wu CF, Avery L, Roy NH, et al. Lymphocyte Egress Signal Sphingosine-1-Phosphate Promotes ERM-Guided, Bleb-Based Migration. J Cell Biol (2021) 220:1–17. doi: 10.1083/jcb.202007182
54. Matsumoto M, Hirata T. Moesin Regulates Neutrophil Rolling Velocity In Vivo. Cell Immunol (2016) 304–305:59–62. doi: 10.1016/j.cellimm.2016.04.007
55. Zehrer A, Pick R, Salvermoser M, Boda A, Miller M, Stark K, et al. Begandt D. A Fundamental Role of Myh9 for Neutrophil Migration in Innate Immunity. J Immunol (2018) 201:1748–64. doi: 10.4049/jimmunol.1701400
56. Jacobelli J, Friedman RS, Conti MA, Lennon-Dumenil AM, Piel M, Sorensen CM, et al. Confinement-Optimized Three-Dimensional T Cell Amoeboid Motility Is Modulated via Myosin IIA-Regulated Adhesions. Nat Immunol (2010) 11:953–61. doi: 10.1038/ni.1936
57. Morin NA, Oakes PW, Hyun YM, Lee D, Chin EY, King MR, et al. Nonmuscle Myosin Heavy Chain IIA Mediates Integrin LFA-1 De-Adhesion During T Lymphocyte Migration. J Exp Med (2008) 205:195–205. doi: 10.1084/jem.20071543
58. Cortesio CL, Wernimont SA, Kastner DL, Cooper KM, Huttenlocher A. Impaired Podosome Formation and Invasive Migration of Macrophages From Patients With a PSTPIP1 Mutation and PAPA Syndrome. Arthritis Rheum (2010) 62:2556–8. doi: 10.1002/art.27521.Impaired
59. Starnes TW, Bennin DA, Bing X, Eickhoff JC, Grahf DC, Bellak JM, et al. The F-BAR Protein PSTPIP1 Controls Extracellular Matrix Degradation and Filopodia Formation in Macrophages. Blood (2014) 123:2703–14. doi: 10.1182/blood-2013-07-516948
60. Janssen WJM, Grobarova V, Leleux J, Jongeneel L, van Gijn M, van Montfrans JM, et al. Proline-Serine-Threonine Phosphatase Interacting Protein 1 (PSTPIP1) Controls Immune Synapse Stability in Human T Cells. J Allergy Clin Immunol (2018) 142:1947–55. doi: 10.1016/j.jaci.2018.01.030
61. Takenawa T, Suetsugu S. The WASP-WAVE Protein Network: Connecting the Membrane to the Cytoskeleton. Nat Rev Mol Cell Biol (2007) 8:37–48. doi: 10.1038/nrm2069
62. Hsu AP, Donkó A, Arrington ME, Swamydas M, Fink D, Das A, et al. Dominant Activating RAC2 Mutation With Lymphopenia, Immunodeficiency, and Cytoskeletal Defects. Blood (2019) 133:1977–88. doi: 10.1182/blood-2018-11-886028
63. Lu X, Zhang Y, Liu F, Wang L. Rac2 Regulates the Migration of T Lymphoid Progenitors to the Thymus During Zebrafish Embryogenesis. J Immunol (2020) 204:2447–54. doi: 10.4049/jimmunol.1901494
64. Deng Q, Yoo SK, Cavnar PJ, Green JM, Huttenlocher A. Dual Roles for Rac2 in Neutrophil Motility and Active Retention in Zebrafish Hematopoietic Tissue. Dev Cell (2011) 21:735–45. doi: 10.1016/j.devcel.2011.07.013
65. Croker BA, Handman E, Hayball JD, Baldwin TM, Voigt V, Cluse LA, et al. Rac2-Deficient Mice Display Perturbed T-Cell Distribution and Chemotaxis, But Only Minor Abnormalities in TH1 Responses. Immunol Cell Biol (2002) 80:231–40. doi: 10.1046/j.1440-1711.2002.01077.x
66. Roberts AW, Chaekyun K, Zhen L, Lowe JB, Kapur R, Petryniak B, et al. Deficiency of the Hematopoietic Cell-Specific Rho Family GTPase Rac2 Is Characterized by Abnormalities in Neutrophil Function and Host Defense. Immunity (1999) 10:183–96. doi: 10.1016/S1074-7613(00)80019-9
67. Salzer E, Cagdas D, Hons M, Mace EM, Garncarz W, Petronczki ÖY, et al. RASGRP1 Deficiency Causes Immunodeficiency With Impaired Cytoskeletal Dynamics. Nat Immunol (2016) 17:1352–60. doi: 10.1038/ni.3575
68. Kalinichenko A, Perinetti Casoni G, Dupré L, Trotta L, Huemer J, Galgano D, et al. RhoG Deficiency Abrogates Cytotoxicity of Human Lymphocytes and Causes Hemophagocytic Lymphohistiocytosis. Blood (2021) 137:2033–45. doi: 10.1182/blood.2020008738
69. Crequer A, Troeger A, Patin E, Ma CS, Picard C, Pedergnana V, et al. Human RHOH Deficiency Causes T Cell Defects and Susceptibility to EV-HPV Infections. J Clin Invest (2012) 122:3239–47. doi: 10.1172/JCI62949
70. Chae HD, Lee KE, Williams DA, Gu Y. Cross-Talk Between RhoH and Rac1 in Regulation of Actin Cytoskeleton and Chemotaxis of Hematopoietic Progenitor Cells. Blood (2008) 111:2597–605. doi: 10.1182/blood-2007-06-093237
71. Dang TS, Willet JDP, Griffin HR, Morgan NV, O’Boyle G, Arkwright PD, et al. Defective Leukocyte Adhesion and Chemotaxis Contributes to Combined Immunodeficiency in Humans With Autosomal Recessive MST1 Deficiency. J Clin Immunol (2016) 36:117–22. doi: 10.1007/s10875-016-0232-2
72. Nehme NT, Schmid JP, Debeurme F, André-Schmutz I, Lim A, Nitschke P, et al. MST1 Mutations in Autosomal Recessive Primary Immunodeficiency Characterized by Defective Naive T-Cell Survival. Blood (2012) 119:3458–68. doi: 10.1182/blood-2011-09-378364
73. Katagiri K, Katakai T, Ebisuno Y, Ueda Y, Okada T, Kinashi T. Mst1 Controls Lymphocyte Trafficking and Interstitial Motility Within Lymph Nodes. EMBO J (2009) 28:1319–31. doi: 10.1038/emboj.2009.82
74. Dong Y, Du X, Ye J, Han M, Xu T, Zhuang Y. Tao W. A Cell-Intrinsic Role for Mst1 in Regulating Thymocyte Egress. J Immunol (2009) 183:3865–72. doi: 10.4049/jimmunol.0900678
75. Lemoine R, Pachlopnik-Schmid J, Farin HF, Bigorgne A, Debré M, Sepulveda F, et al. Immune Deficiency-Related Enteropathy-Lymphocytopenia-Alopecia Syndrome Results From Tetratricopeptide Repeat Domain 7A Deficiency. J Allergy Clin Immunol (2014) 134:1354–64.e6. doi: 10.1016/j.jaci.2014.07.019
76. Westerberg L, Larsson M, Hardy SJ, Fernández C, Thrasher AJ, Severinson E. Wiskott-Aldrich Syndrome Protein Deficiency Leads to Reduced B-Cell Adhesion, Migration, and Homing, and a Delayed Humoral Immune Response. Blood (2005) 105:1144–52. doi: 10.1182/blood-2004-03-1003
77. Castiello MC, Bosticardo M, Pala F, Catucci M, Chamberlain N, Van Zelm MC, et al. Wiskott-Aldrich Syndrome Protein Deficiency Perturbs the Homeostasis of B-Cell Compartment in Humans. J Autoimmun (2014) 50:42–50. doi: 10.1016/j.jaut.2013.10.006
78. Snover DC, Frizzera G, Spector BD, Perry GS, Kersey JH. Wiskott-Aldrich Syndrome: Histopathologic Findings in the Lymph Nodes and Spleens of 15 Patients. Hum Pathol (1981) 12:821–31. doi: 10.1016/S0046-8177(81)80085-8
79. Binks M, Jones GE, Brickell PM, Kinnon C, Katz DR, Thrasher AJ. Intrinsic Dendritic Cell Abnormalities in Wiskott-Aldrich Syndrome. Eur J Immunol (1998) 28:3259–67. doi: 10.1002/(SICI)1521-4141(199810)28:10<3259::AID-IMMU3259>3.0.CO;2-B
80. Burns S, Thrasher AJ, Blundell MP, Machesky L, Jones GE. Configuration of Human Dendritic Cell Cytoskeleton by Rho GTPases, the WAS Protein, and Differentiation. Blood (2001) 98:1142–9. doi: 10.1182/blood.V98.4.1142
81. Badolato R, Sozzani S, Malacarne F, Bresciani S, Fiorini M, Borsatti A, et al. Monocytes From Wiskott-Aldrich Patients Display Reduced Chemotaxis and Lack of Cell Polarization in Response to Monocyte Chemoattractant Protein-1 and Formyl-Methionyl-Leucyl-Phenylalanine. J Immunol (1998) 161:1026–33.
82. Stabile H, Carlino C, Mazza C, Giliani S, Morrone S, Notarangelo LD, et al. Impaired NK-Cell Migration in WAS/XLT Patients: Role of Cdc42/WASp Pathway in the Control of Chemokine-Induced β2 Integrin High-Affinity State. Blood (2010) 115:2818–26. doi: 10.1182/blood-2009-07-235804
83. Zhang H, Schaff UY, Green CE, Chen H, Sarantos MR, Hu Y, et al. Impaired Integrin-Dependent Function in Wiskott-Aldrich Syndrome Protein-Deficient Murine and Human Neutrophils. Immunity (2006) 25:285–95. doi: 10.1016/j.immuni.2006.06.014
84. Linder S, Nelson D, Weiss M, Aepfelbacher M. Wiskott-Aldrich Syndrome Protein Regulates Podosomes in Primary Human Macrophages. Proc Natl Acad Sci USA (1999) 96:9648–53. doi: 10.1073/pnas.96.17.9648
85. Haddad E, Zugaza JL, Louache F, Debili N, Crouin C, Schwarz K, et al. The Interaction Between Cdc42 and WASP Is Required for SDF-1-Induced T-Lymphocyte Chemotaxis. Blood (2001) 97:33–8. doi: 10.1182/blood.V97.1.33
86. Trifari S, Scaramuzza S, Catucci M, Ponzoni M, Mollica L, Chiesa R, et al. Revertant T Lymphocytes in a Patient With Wiskott-Aldrich Syndrome: Analysis of Function and Distribution in Lymphoid Organs. J Allergy Clin Immunol (2010) 125:439–48.e8. doi: 10.1016/j.jaci.2009.11.034
87. Houmadi R, Guipouy D, Rey-Barroso J, Vasconcelos Z, Cornet J, Manghi M, et al. The Wiskott-Aldrich Syndrome Protein Contributes to the Assembly of the LFA-1 Nanocluster Belt at the Lytic Synapse. Cell Rep (2018) 22:979–91. doi: 10.1016/j.celrep.2017.12.088
88. Lafouresse F, Cotta-de-Almeida V, Malet-Engra G, Galy A, Valitutti S, Dupré L. Wiskott-Aldrich Syndrome Protein Controls Antigen-Presenting Cell-Driven CD4 + T-Cell Motility by Regulating Adhesion to Intercellular Adhesion Molecule-1. Immunology (2012) 137:183–96. doi: 10.1111/j.1365-2567.2012.03620.x
89. Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HDD, et al. Transcellular Diapedesis Is Initiated by Invasive Podosomes. Immunity (2007) 26:784–97. doi: 10.1016/j.immuni.2007.04.015
90. German Y, Vulliard L, Kamnev A, Pfajfer L, Huemer J, Mautner A-K, et al. Morphological Profiling of Human T and NK Lymphocytes by High-Content Cell Imaging. Cell Rep (2021) 36:1–14. doi: 10.1016/j.celrep.2021.109318
91. De Noronha S, Hardy S, Sinclair J, Blundell MP, Strid J, Schulz O, et al. Impaired Dendritic-Cell Homing In Vivo in the Absence of Wiskott-Aldrich Syndrome Protein. Blood (2005) 105:1590–7. doi: 10.1182/blood-2004-06-2332
92. Bouma G, Burns S, Thrasher AJ. Impaired T-Cell Priming In Vivo Resulting From Dysfunction of WASp-Deficient Dendritic Cells. Blood (2007) 110:4278–84. doi: 10.1182/blood-2007-06-096875
93. Lacout C, Haddad E, Sabri S, Svinarchouk F, Garçon L, Capron C, et al. A Defect in Hematopoietic Stem Cell Migration Explains the Nonrandom X-Chromosome Inactivation in Carriers of Wiskott-Aldrich Syndrome. Blood (2003) 102:1282–9. doi: 10.1182/blood-2002-07-2099
94. Dovas A, Gevrey JC, Grossi A, Park H, Abou-Kheir W, Cox D. Regulation of Podosome Dynamics by WASp Phosphorylation: Implication in Matrix Degradation and Chemotaxis in Macrophages. J Cell Sci (2009) 122:3873–82. doi: 10.1242/jcs.051755
95. Sabri S, Foudi A, Boukour S, Franc B, Charrier S, Jandrot-Perrus M, et al. Deficiency in the Wiskott-Aldrich Protein Induces Premature Proplatelet Formation and Platelet Production in the Bone Marrow Compartment. Blood (2006) 108:134–40. doi: 10.1182/blood-2005-03-1219
96. Snapper SB, Meelu P, Nguyen D, Stockton BM, Bozza P, Alt FW, et al. WASP Deficiency Leads to Global Defects of Directed Leukocyte Migration In Vitro and In Vivo. J Leukoc Biol (2005) 77:993–8. doi: 10.1189/jlb.0804444
97. Keszei M, Record J, Kritikou JS, Wurzer H, Geyer C, Thiemann M, et al. Constitutive Activation of WASp in X-Linked Neutropenia Renders Neutrophils Hyperactive. J Clin Invest (2018) 128:4115–31. doi: 10.1172/JCI64772
98. Oliveira MMS, Kung S, Moreau HD, Maurin M, Record J, Sanséau D, et al. The WASp L272P Gain-of-Function Mutation Alters Dendritic Cell Coordination of Actin Dynamics for Migration and Adhesion. J Leukoc Biol (2021), 1–11. doi: 10.1002/jlb.1ab0821-013rr
99. Pfajfer L, Mair NK, Gulez N, Rey-barroso J, Ijspeert H, Tangye SG, et al. Mutations Affecting the Actin Regulator WD Repeat – Containing Protein 1 Lead to Aberrant Lymphoid Immunity. J Allergy Clin Immunol (2018) 142:1589–604.e11. doi: 10.1016/j.jaci.2018.04.023
100. Standing ASI, Malinova D, Hong Y, Record J, Moulding D, Blundell MP, et al. Autoinflammatory Periodic Fever, Immunodeficiency, and Thrombocytopenia (PFIT) Caused by Mutation in Actinregulatory Gene WDR1. J Exp Med (2017) 214:59–71. doi: 10.1084/jem.20161228
101. Kuhns DB, Fink DL, Choi U, Sweeney C, Lau K, Priel DL, et al. Cytoskeletal Abnormalities and Neutrophil Dysfunction in WDR1 Deficiency. Blood (2016) 128:2135–43. doi: 10.1182/blood-2016-03-706028
102. Kile BT, Panopoulos AD, Stirzaker RA, Hacking DF, Tahtamouni LH, Willson TA, et al. Mutations in the Cofilin Partner Aip1/Wdr1 Cause Autoinflammatory Disease and Macrothrombocytopenia. Blood (2007) 110:2371–80. doi: 10.1182/blood-2006-10-055087
103. Pfajfer L, Seidel MG, Houmadi R, Boztug K, Dupré L. WIP Deficiency Severely Affects Human Lymphocyte Architecture During Migration and Synapse Assembly. Blood (2018) 130:1949–54. doi: 10.1182/blood-2017-04-777383
104. Chou HC, Antón IM, Holt MR, Curcio C, Lanzardo S, Worth A, et al. WIP Regulates the Stability and Localization of WASP to Podosomes in Migrating Dendritic Cells. Curr Biol (2006) 16:2337–44. doi: 10.1016/j.cub.2006.10.037
105. Tsuboi S. Requirement for a Complex of Wiskott-Aldrich Syndrome Protein (WASP) With WASP Interacting Protein in Podosome Formation in Macrophages. J Immunol (2007) 178:2987–95. doi: 10.4049/jimmunol.178.5.2987
106. Ciriza J, Thompson H, Petrosian R, Manilay JO, García-Ojeda ME. The Migration of Hematopoietic Progenitors From the Fetal Liver to the Fetal Bone Marrow: Lessons Learned and Possible Clinical Applications. Exp Hematol (2013) 41:411–23. doi: 10.1016/j.exphem.2013.01.009
107. Bonaud A, Lemos JP, Espéli M, Balabanian K. Hematopoietic Multipotent Progenitors and Plasma Cells: Neighbors or Roommates in the Mouse Bone Marrow Ecosystem? Front Immunol (2021) 12:658535. doi: 10.3389/fimmu.2021.658535
108. Lévesque JP, Helwani FM, Winkler IG. The Endosteal Osteoblastic Niche and Its Role in Hematopoietic Stem Cell Homing and Mobilization. Leukemia (2010) 24:1979–92. doi: 10.1038/leu.2010.214
109. Upadhaya S, Krichevsky O, Akhmetzyanova I, Sawai CM, Fooksman DR, Reizis B. Intravital Imaging Reveals Motility of Adult Hematopoietic Stem Cells in the Bone Marrow Niche. J Clean Prod (2020) 27:336–345.e4. doi: 10.1016/j.stem.2020.06.003
110. James KD, Jenkinson WE. Anderson G. T-Cell Egress From the Thymus: Should I Stay or Should I Go? J Leukoc Biol (2018) 104:275–84. doi: 10.1002/JLB.1MR1217-496R
111. Lougaris V, Baronio M, Gazzurelli L, Benvenuto A, Plebani A. RAC2 and Primary Human Immune Deficiencies. J Leukoc Biol (2020) 108:687–96. doi: 10.1002/JLB.5MR0520-194RR
112. Lambe T, Crawford G, Johnson AL, Crockford TL, Bouriez-Jones T, Smyth AM, et al. DOCK8 Is Essential for T-Cell Survival and the Maintenance of CD8 + T-Cell Memory. Eur J Immunol (2011) 41:3423–35. doi: 10.1002/eji.201141759
113. Randall KL, Lambe T, Johnson A, Treanor B, Kucharska E, Domaschenz H, et al. Dock8 Mutations Cripple B Cell Immunological Synapses, Germinal Centers and Long-Lived Antibody Production. Nat Immunol (2009) 10:1283–91. doi: 10.1038/ni.1820
114. Pillay BA, Fusaro M, Gray PE, Statham AL, Burnett L, Bezrodnik L, et al. Somatic Reversion of Pathogenic DOCK8 Variants Alters Lymphocyte Differentiation and Function to Effectively Cure DOCK8 Deficiency. J Clin Invest (2021) 131:1–17. doi: 10.1172/JCI142434
115. Xu X, Wang X, Todd EM, Jaeger ER, Vella JL, Mooren OL, et al. Mst1 Kinase Regulates the Actin-Bundling Protein L-Plastin To Promote T Cell Migration. J Immunol (2016) 197:1683–91. doi: 10.4049/jimmunol.1600874
116. Notarangelo LD. Primary Immunodeficiencies. J Allergy Clin Immunol (2010) 125:S182–94. doi: 10.1016/j.jaci.2009.07.053
117. Beel K, Cotter MM, Blatny J, Bond J, Lucas G, Green F, et al. A Large Kindred With X-Linked Neutropenia With an I294T Mutation of the Wiskott-Aldrich Syndrome Gene. Br J Haematol (2009) 144:120–6. doi: 10.1111/j.1365-2141.2008.07416.x
118. Fowell DJ, Kim M. The Spatio-Temporal Control of Effector T Cell Migration. Nat Rev Immunol (2021) 21:582–96. doi: 10.1038/s41577-021-00507-0
119. Aydin SE, Kilic SS, Aytekin C, Kumar A, Porras O, Kainulainen L, et al. DOCK8 Deficiency: Clinical and Immunological Phenotype and Treatment Options - a Review of 136 Patients. J Clin Immunol (2015) 35:189–98. doi: 10.1007/s10875-014-0126-0
120. Su HC, Jing H, Angelus P, Freeman AF. Insights Into Immunity From Clinical and Basic Science Studies of DOCK8 Immunodeficiency Syndrome. Immunol Rev (2019) 287:9–19. doi: 10.1111/imr.12723
121. Schneider C, Shen C, Gopal AA, Douglas T, Forestell B, Kauffman KD, et al. Migration-Induced Cell Shattering Due to DOCK8 Deficiency Causes a Type 2–Biased Helper T Cell Response. Nat Immunol (2020) 21:1528–39. doi: 10.1038/s41590-020-0795-1
122. Weninger W, Biro M, Jain R. Leukocyte Migration in the Interstitial Space of Non-Lymphoid Organs. Nat Rev Immunol (2014) 14:232–46. doi: 10.1038/nri3641
123. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat Rev Immunol (2007) 7:678–89. doi: 10.1038/nri2156
124. Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin Dynamics Control SRF Activity by Regulation of Its Coactivator MAL. Cell (2003) 113:329–42. doi: 10.1016/S0092-8674(03)00278-2
125. Vartiainen MK, Guettler S, Larijani B, Treisman R. Nuclear Actin Regulates Dynamic Subcellular Localization and Activity of the SRF Cofactor MAL. Sci (80- ) (2007) 316:1749–52. doi: 10.1126/science.1141084
126. Martinelli R, Zeiger AS, Whitfield M, Sciuto TE, Dvorak A, van Vliet KJ, et al. Probing the Biomechanical Contribution of the Endothelium to Lymphocyte Migration: Diapedesis by the Path of Least Resistance. J Cell Sci (2014) 127:3720–34. doi: 10.1242/jcs.148619
127. Alon R, van Buul JD. Leukocyte Breaching of Endothelial Barriers: The Actin Link. Trends Immunol (2017) 38:606–15. doi: 10.1016/j.it.2017.05.002
128. Côté JF, Chung PL, Théberge JF, Hallé M, Spencer S, Lasky LA, et al. PSTPIP Is a Substrate of PTP-PEST and Serves as a Scaffold Guiding PTP-PEST Toward a Specific Dephosphorylation of WASP. J Biol Chem (2002) 277:2973–86. doi: 10.1074/jbc.M106428200
129. Badour K, Zhang J, Shi F, Leng Y, Collins M, Siminovitch KA. Fyn and PTP-PEST-Mediated Regulation of Wiskott-Aldrich Syndrome Protein (WASp) Tyrosine Phosphorylation Is Required for Coupling T Cell Antigen Receptor Engagement to WASp Effector Function and T Cell Activation. J Exp Med (2004) 199:99–111. doi: 10.1084/jem.20030976
130. Gardel ML, Schneider IC, Aratyn-schaus Y, Waterman CM, Engineering B, Biology C, et al. Mechanical Integration of Actin and Adhesion Dynamics in Cell Migration. Annu Rev Cell Dev Biol (2010) 26:315–33. doi: 10.1146/annurev.cellbio.011209.122036.Mechanical
131. Plotnikov SV, Waterman CM. Guiding Cell Migration by Tugging. Curr Opin Cell Biol (2013) 25:619–26. doi: 10.1016/j.ceb.2013.06.003
132. Reversat A, Gaertner F, Merrin J, Stopp J, Tasciyan S, Aguilera J, et al. Cellular Locomotion Using Environmental Topography. Nature (2020) 582:582–5. doi: 10.1038/s41586-020-2283-z
133. Blumenthal D, Burkhardt JK. Multiple Actin Networks Coordinate Mechanotransduction at the Immunological Synapse. J Cell Biol (2020) 219:1–12. doi: 10.1083/jcb.201911058
134. Sims TN, Soos TJ, Xenias HS, Dubin-Thaler B, Hofman JM, Waite JC, et al. Opposing Effects of Pkcθ and WASp on Symmetry Breaking and Relocation of the Immunological Synapse. Cell (2007) 129:773–85. doi: 10.1016/j.cell.2007.03.037
135. Calvez R, Lafouresse F, de Meester J, Galy A, Valitutti S, Dupré L. The Wiskott-Aldrich Syndrome Protein Permits Assembly of a Focused Immunological Synapse Enabling Sustained T-Cell Receptor Signaling. Haematologica (2011) 96:1415–23. doi: 10.3324/haematol.2011.040204
136. Kumari S, Mak M, Poh Y, Tohme M, Watson N, Melo M, et al. Cytoskeletal Tension Actively Sustains the Migratory T-Cell Synaptic Contact. EMBO J (2020) 39:1–18. doi: 10.15252/embj.2019102783
137. Ackerknecht M, Gollmer K, Germann P, Ficht X, Abe J, Fukui Y, et al. Antigen Availability and DOCK2-Driven Motility Govern CD4 + T Cell Interactions With Dendritic Cells In Vivo. J Immunol (2017) 199:520–30. doi: 10.4049/jimmunol.1601148
138. Krause M, Gautreau A. Steering Cell Migration: Lamellipodium Dynamics and the Regulation of Directional Persistence. Nat Rev Mol Cell Biol (2014) 15:577–90. doi: 10.1038/nrm3861
139. Dustin ML, Long EO. Cytotoxic Immunological Synapses. Immunol Rev (2010) 235:24–34. doi: 10.1111/j.0105-2896.2010.00904.x
140. Welch MD, DePace AH, Verma S, Iwamatsu A, Mitchison TJ. The Human Arp2/3 Complex Is Composed of Evolutionarily Conserved Subunits and Is Localized to Cellular Regions of Dynamic Actin Filament Assembly. J Cell Biol (1997) 138:375–84. doi: 10.1083/jcb.138.2.375
141. Obeidy P, Ju LA, Oehlers SH, Zulkhernain NS, Lee Q, Ni JLG, et al. Partial Loss of Actin Nucleator Actin Related Protein 2/3 Activity Triggers Blebbing in Primary T Lymphocytes. Immunol Cell Biol (2020) 98:93–113. doi: 10.1111/imcb.12304
142. Machesky LM, Insall RH. Scar1 and the Related Wiskott-Aldrich Syndrome Protein, WASP, Regulate the Actin Cytoskeleton Through the Arp2/3 Complex. Curr Biol (1998) 8:1347–56. doi: 10.1016/S0960-9822(98)00015-3
143. Padrick SB, Doolittle LK, Brautigam CA, King DS, Rosen MK. Arp2/3 Complex Is Bound and Activated by Two WASP Proteins. Proc Natl Acad Sci USA (2011) 108:E472-9. doi: 10.1073/pnas.1100236108
144. De La Fuente MA, Sasahara Y, Calamito M, Antón IM, Elkhal A, Gallego MD, et al. WIP Is a Chaperone for Wiskott-Aldrich Syndrome Protein (WASP). Proc Natl Acad Sci USA (2007) 104:926–31. doi: 10.1073/pnas.0610275104
145. Higgs HN, Pollard TD. Regulation of Actin Filament Network Formation Through Arp2/3 Complex: Activation by a Diverse Array of Proteins. Annu Rev Biochem (2001) 70:649–76. doi: 10.1146/annurev.biochem.70.1.649
146. Pollitt AY, Insall RH. WASP and SCAR/WAVE Proteins: The Drivers of Actin Assembly. J Cell Sci (2009) 122:2575–8. doi: 10.1242/jcs.023879
147. Rey-Barroso J, Calovi DS, Combe M, German Y, Moreau M, Canivet A, et al. Switching Between Individual and Collective Motility in B Lymphocytes Is Controlled by Cell-Matrix Adhesion and Inter-Cellular Interactions. Sci Rep (2018) 8:1–16. doi: 10.1038/s41598-018-24222-4
148. Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, et al. Human Neutrophil Immunodeficiency Syndrome Is Associated With an Inhibitory Rac2 Mutation. Proc Natl Acad Sci USA (2000) 97:4654–9. doi: 10.1073/pnas.080074897
149. Accetta D, Syverson G, Bonacci B, Reddy S, Bengtson C, Surfus J, et al. Human Phagocyte Defect Caused by a Rac2 Mutation Detected by Means of Neonatal Screening for T-Cell Lymphopenia. J Allergy Clin Immunol (2011) 127:535–38.e2. doi: 10.1016/j.jaci.2010.10.013
150. Alkhairy OK, Rezaei N, Graham RR, Abolhassani H, Borte S, Hultenby K, et al. RAC2 Loss-of-Function Mutation in 2 Siblings With Characteristics of Common Variable Immunodeficiency. J Allergy Clin Immunol (2015) 135:1380–4.e5. doi: 10.1016/j.jaci.2014.10.039
151. Mattila PK, Lappalainen P. Filopodia: Molecular Architecture and Cellular Functions. Nat Rev Mol Cell Biol (2008) 9:446–54. doi: 10.1038/nrm2406
152. Kress H, Stelzer EHK, Holzer D, Buss F, Griffiths G, Rohrbach A. Filopodia Act as Phagocytic Tentacles and Pull With Discrete Steps and a Load-Dependent Velocity. Proc Natl Acad Sci USA (2007) 104:11633–8. doi: 10.1073/pnas.0702449104
153. Krugmann S, Jordens I, Gevaert K, Driessens M, Vandekerckhove J, Hall A. Cdc42 Induces Filopodia by Promoting the Formation of an IRSp53:Mena Complex. Curr Biol (2001) 11:1645–55. doi: 10.1016/S0960-9822(01)00506-1
154. Sánchez-Madrid F, Serrador JM. Bringing Up the Rear: Defining the Roles of the Uropod. Nat Rev Mol Cell Biol (2009) 10:353–9. doi: 10.1038/nrm2680
155. Smith LA, Aranda-Espinoza H, Haun JB, Dembo M, Hammer DA. Neutrophil Traction Stresses Are Concentrated in the Uropod During Migration. Biophys J (2007) 92:L58–60. doi: 10.1529/biophysj.106.102822
156. Jacobelli J, Bennett FC, Pandurangi P, Tooley AJ, Krummel MF. Myosin-IIA and ICAM-1 Regulate the Interchange Between Two Distinct Modes of T Cell Migration. J Immunol (2009) 182:2041–50. doi: 10.4049/jimmunol.0803267
157. Syndrome Consortium T. Mutations in MYH9result in the May-Hegglin Anomaly, and Fechtner and Sebastian Syndromes. Nat Genet (2000) 26:103–5. doi: 10.1038/79063
158. Murphy DA, Courtneidge SA. The “Ins” and “Outs” of Podosomes and Invadopodia: Characteristics, Formation and Function. Nat Rev Mol Cell Biol (2011) 12:413–26. doi: 10.1038/nrm3141
159. Jones GE, Zicha D, Dunn GA, Blundell M, Thrasher A. Restoration of Podosomes and Chemotaxis in Wiskott-Aldrich Syndrome Macrophages Following Induced Expression of WASp. Int J Biochem Cell Biol (2002) 34:806–15. doi: 10.1016/S1357-2725(01)00162-5
160. Olivier A, Jeanson-Leh L, Bouma G, Compagno D, Blondeau J, Seye K, et al. A Partial Down-Regulation of WASP Is Sufficient to Inhibit Podosome Formation in Dendritic Cells. Mol Ther (2006) 13:729–37. doi: 10.1016/j.ymthe.2005.11.003
161. Alexander E, Sanders S, Braylan R. Purported Difference Between Human T- and B-Cell Surface Morphology Is an Artefact. Nature (1976) 261:239–41. doi: 10.1038/261239a0
162. Parakkal P, Pinto J, Hanifin JM. Surface Morphology of Human Mononuclear Phagocytes During Maturation and Phagocytosis. J Ultrasructure Res (1974) 48:216–26. doi: 10.1016/S0022-5320(74)80078-X
163. Fisher PJ, Bulur PA, Vuk-Pavlovic S, Prendergast FG, Dietz AB. Dendritic Cell Microvilli: A Novel Membrane Structure Associated With the Multifocal Synapse and T-Cell Clustering. Blood (2008) 112:5037–45. doi: 10.1182/blood-2008-04-149526
164. Orbach R, Su X. Surfing on Membrane Waves: Microvilli, Curved Membranes, and Immune Signaling. Front Immunol (2020) 11:2187. doi: 10.3389/fimmu.2020.02187
165. Ghosh S, Di Bartolo V, Tubul L, Shimoni E, Kartvelishvily E, Dadosh T, et al. ERM-Dependent Assembly of T Cell Receptor Signaling and Co-Stimulatory Molecules on Microvilli Prior to Activation. Cell Rep (2020) 30:3434–3447.e6. doi: 10.1016/j.celrep.2020.02.069
166. Kenney DM, Cairns L, Remold-O’donnell E, Peterson J, Rosen FS, Parkman R. Morphological Abnormalities in the Lympcytes of Patients the Wiskott-Aldrich Syndrome. Blood (1986) 68:1329–32. doi: 10.1182/blood.V68.6.1329.1329
167. Molina IJ, Kenney DM, Rosen FS, Remold-O’donnell E. T Cell Lines Characterize Events in the Pathogenesis of the Wiskott-Aldrich Syndrome. J Exp Med (1992) 176:867–74. doi: 10.1084/jem.176.3.867
168. Majstoravich S, Zhang J, Nicholson-Dykstra S, Linder S, Friedrich W, Siminovitch KA, et al. Lymphocyte Microvilli Are Dynamic, Actin-Dependent Structures That do Not Require Wiskott-Aldrich Syndrome Protein (WASp) for Their Morphology. Blood (2004) 104:1396–403. doi: 10.1182/blood-2004-02-0437
169. McGregor AL, Hsia CR, Lammerding J. Squish and Squeeze - the Nucleus as a Physical Barrier During Migration in Confined Environments. Curr Opin Cell Biol (2016) 40:32–40. doi: 10.1016/j.ceb.2016.01.011
170. Sneider A, Hah J, Wirtz D, Kim DH. Recapitulation of Molecular Regulators of Nuclear Motion During Cell Migration. Cell Adhes Migr (2019) 13:50–62. doi: 10.1080/19336918.2018.1506654
171. Yamada KM, Sixt M. Mechanisms of 3D Cell Migration. Nat Rev Mol Cell Biol (2019) 20:738–52. doi: 10.1038/s41580-019-0172-9
172. Renkawitz J, Kopf A, Stopp J, de Vries I, Driscoll MK, Merrin J, et al. Nuclear Positioning Facilitates Amoeboid Migration Along the Path of Least Resistance. Nature (2019) 568:546–50. doi: 10.1038/s41586-019-1087-5
173. Jacobelli J, Estin Matthews M, Chen S, Krummel MF. Activated T Cell Trans-Endothelial Migration Relies on Myosin-IIA Contractility for Squeezing the Cell Nucleus Through Endothelial Cell Barriers. PloS One (2013) 8:1–13. doi: 10.1371/journal.pone.0075151
174. Thompson SB, Sandor AM, Lui V, Chung JW, Waldman MM, Long RA, et al. Formin-Like 1 Mediates Effector T Cell Trafficking to Inflammatory Sites to Enable T Cell-Mediated Autoimmunity. Elife (2020) 9:1–27. doi: 10.7554/eLife.58046
175. Maritzen T, Schachtner H, Legler DF. On the Move: Endocytic Trafficking in Cell Migration. Cell Mol Life Sci (2015) 72:2119–34. doi: 10.1007/s00018-015-1855-9
176. Zech T, Calaminus SDJ, Machesky LM. Actin on Trafficking: Could Actin Guide Directed Receptor Transport? Cell Adhes Migr (2012) 6:476–81. doi: 10.4161/cam.21373
Keywords: leukocytes, cell migration, chemotaxis, actin, cytoskeleton, actin regulators, inborn errors of immunity, IEI
Citation: Kamnev A, Lacouture C, Fusaro M and Dupré L (2021) Molecular Tuning of Actin Dynamics in Leukocyte Migration as Revealed by Immune-Related Actinopathies. Front. Immunol. 12:750537. doi: 10.3389/fimmu.2021.750537
Received: 30 July 2021; Accepted: 12 October 2021;
Published: 15 November 2021.
Edited by:
Hélène D. Moreau, INSERM U932 Immunité et Cancer, FranceReviewed by:
Hassan Abolhassani, Karolinska University Hospital, SwedenCopyright © 2021 Kamnev, Lacouture, Fusaro and Dupré. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Loïc Dupré, loic.dupre@rud.lbg.ac.at
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.