 Riccardo Papa
Riccardo Papa Federica Penco
Federica Penco Marco Gattorno
Marco Gattorno- Center for Autoinflammatory Diseases and Immunodeficiencies, IRCCS Istituto Giannina Gaslini, Genoa, Italy
A growing number of monogenic immune-mediated diseases have been related to genes involved in pathways of actin cytoskeleton remodeling. Increasing evidences associate cytoskeleton defects to autoinflammatory diseases and primary immunodeficiencies. We reviewed the pathways of actin cytoskeleton remodeling in order to identify inflammatory and immunological manifestations associated to pathological variants. We list more than twenty monogenic diseases, ranging from pure autoinflammatory conditions as familial Mediterranean fever, mevalonate kinase deficiency and PAPA syndrome, to classic and novel primary immunodeficiencies as Wiskott-Aldrich syndrome and DOCK8 deficiency, characterized by the presence of concomitant inflammatory and autoimmune manifestations, such as vasculitis and cytopenia, to severe and recurrent infections. We classify these disorders according to the role of the mutant gene in actin cytoskeleton remodeling, and in particular as disorders of transcription, elongation, branching and activation of actin. This expanding field of rare immune disorders offers a new perspective to all immunologists to better understand the physiological and pathological role of actin cytoskeleton in cells of innate and adaptive immunity.
“Cosa bella e mortal passa e non dura.”
Francesco Petrarca
Introduction
Actin is a family of globular proteins that form microfilaments of cell cytoskeleton. In the past, the most important function of actin was related to the binding of myosin, collaborating to the muscle contraction with troponin. These properties can easily be tested adding pure myosin to water and actin, causing an increase in viscosity and birefringence of the liquid due to the formation of the actomyosin complex (1). Thus, the term of actinopathies was originally considered for a well-defined group of monogenic muscle diseases secondary to the actomyosin complex dysfunction (2). During the recent years, a growing number of disorders of the immune system have been linked to actin cytoskeleton abnormalities (numbers are related to the Table 1 and Figure 1) (3). Furthermore, evidences that actin cytoskeletal deregulation in immune cells causes inflammatory manifestations are increasing (4). In this review, we illustrate the inflammatory and immunological disorders associated with different pathways of actin-binding molecules.
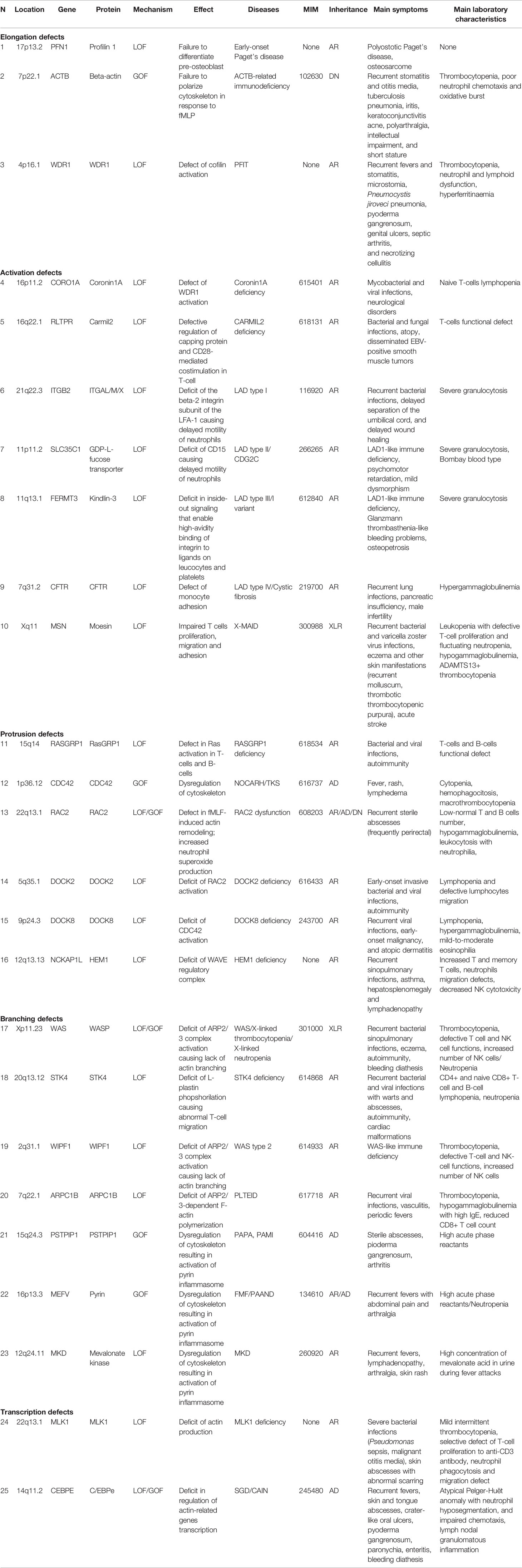
Table 1 Monogenic immune system diseases characterized by actin remodeling defects.
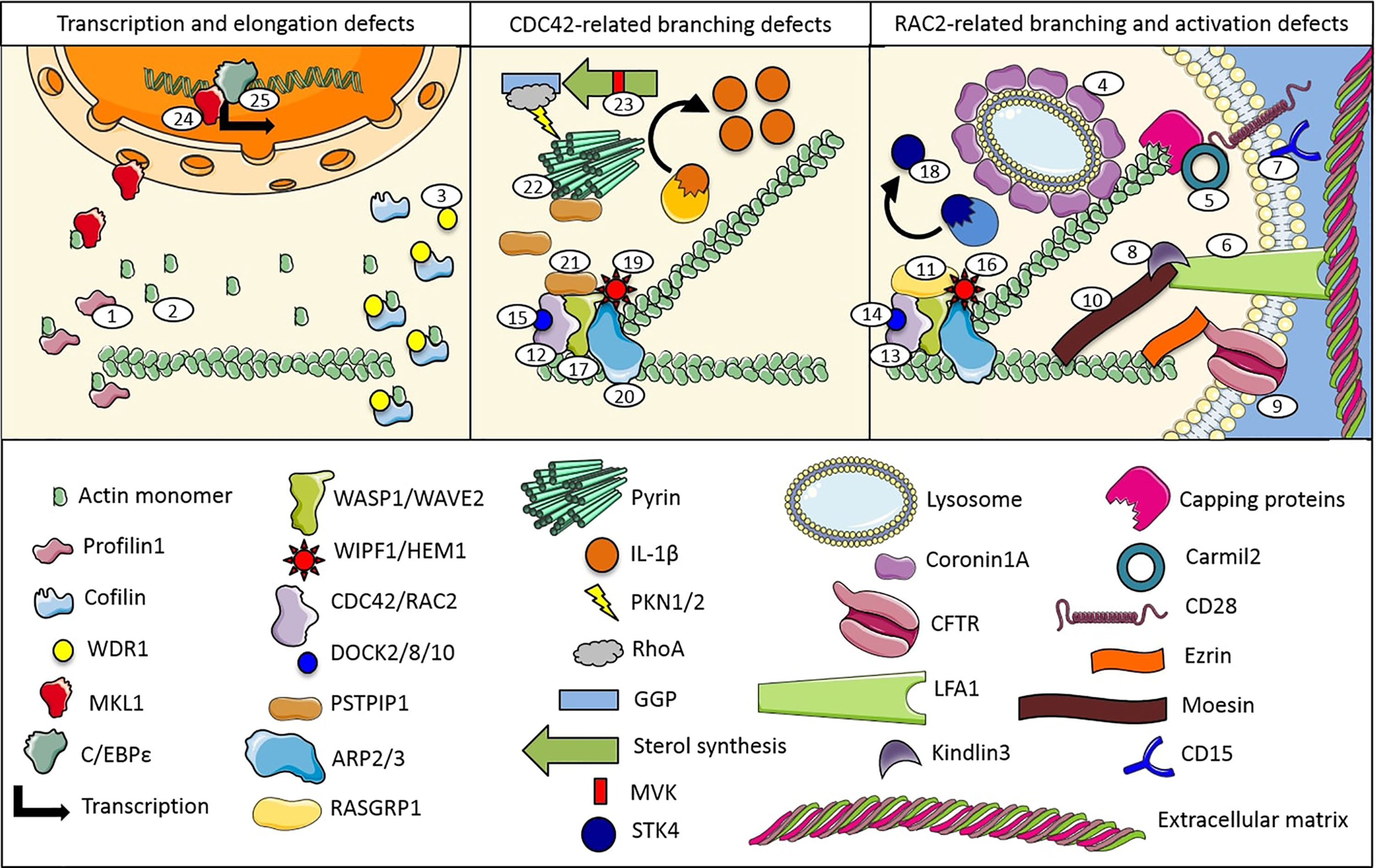
Figure 1 Proteins and pathways involved in monogenic immune system diseases characterized by actin remodeling defects (numbers are related to the manuscript and Table 1).
Elongation Defects
Actin is the most abundant protein in the majority of eukaryotic cells, contributing to acquire and maintain cell structure and functions. Vertebrates express three actin isoforms, including the α-isoform of skeletal, cardiac, and smooth muscles cells, and the β- and γ-isoforms (5). The conformation of actin monomer, called globular (G)-actin, is the same among different isoforms. G-actin assembles into polarized filaments, called filamentous (F)-actin, that form cortical actin network (CAcN) and cell protusions (6). Monomer binding proteins, such as the Profilin-1, control polymerization. Individual filaments lifetime can be as short as ten seconds or lasting for days, depending on the extracellular stimulus duration and intracellular conditions (7). Inhibiting the actin polymerization through activity of the capping proteins, or stimulating actin disassembly through the Cofilin/actin depolarizing factor (ADF) influences the intracellular concentration of G-actin, usually relatively equal throughout the cell cytoplasm.
Profilin-1 is ubiquitously expressed in human cells (8). Its main function is to chaperone G-actin to the positive-charged barbed end of F-actin in response to an increased concentration of the phosphatidylinositol (4,5)-bi-phosphate (PIP2). Mutation of the PFN1 gene coding for the Profilin-1 causes the familial form of amyotrophic lateral sclerosis (9) and deletions have been recently related to an early-onset form of Paget’s disease (no. 1 in Table 1 and Figure 1) (10). This condition is characterized by anomalies of the appendicular bone, favoring malign tumors. Pre-osteoblasts lacking Profilin-1 lose their differentiation and adhesion capability and fail to mineralize efficiently the appendicular bone, acquiring invasive properties. Depletion of the Profilin-1 in breast tumor cells causes defects in formation of filopodia, limiting cell motility and favoring proliferation through upregulation of the transcriptional factor SMAD3 (11). On the other hand, deficiency of Profilin-1 acts against invasion of cytotoxic T lymphocytes in tumors and haploinsufficiency of Profilin-1 seems protective against subcutaneous inflammation induced by high fat diet (12). Furthermore, activation of the Profilin-1 pathway has been related to the inflammatory vascular damage in patients with diabetic retinopathy (13, 14).
Heterozygous gain-of-function (GoF) variant of the ACTB gene, coding for the β-isoform of actin, has been reported in a female with recurrent infections and defect of neutrophil chemotaxis and oxidative burst (no. 2 in Table 1 and Figure 1) (15). The patient also presented a short stature and intellectual disabilities. No other patients have been reported to date. The authors showed that the mutant β-isoform binds Profilin-1 less efficiently, despite a normal actin polymerization. Loss-of-function (LoF) variants of the ACTB gene, as well as of the ACTG1 gene, coding for the γ-isoform of actin, have been related to the highly variable spectrum of the Baraitser-Winter syndrome, a rare condition without relevant immunological manifestations (16).
Cofilin/ADF activation is dependent by phospholipase Cγ (PLCγ) in tumors and Rac2 signaling in neutrophils (17). Reduction of Cofilin/ADF expression in leukocytes is associated with abnormal chemotaxis (18). In neurons, Cofilin/ADF controls axon elongation and regeneration (19) and serum levels are significantly higher in patient with Alzheimer’s disease (20). Cofilin/ADF is also upregulated in patients with Friedreich’s ataxia, whose mutations correlate with an altered immune-related genes transcription (21, 22).
Proteins containing a short structural motif of approximately 40 amino acids, often terminating in a tryptophan-aspartic acid (WD) dipeptide, called WD40 repeat, can accelerate the Cofilin/ADF activity. The best-known example is the WD40 repeat protein 1 (WDR1), also known as Actin interacting protein 1 (AIP1). Homozygous LoF mutations of the WDR1 gene cause a monogenic autoinflammatory disease characterized by periodic fever, immunodeficiency, and thrombocytopenia (PFIT; no. 3 in Table 1 and Figure 1) (23, 24). Patients display recurrent fever attacks lasting 3–7 days, every 6–12 weeks, with high acute phase reactants and hyperferritinaemia. Recurrent mucosal inflammation, causing a peculiar acquired microstomia, may resemble the Behcet’s disease’s attacks during childhood (25). Lymphocytes of patients with PFIT show adhesion and motility defects (26). Coronin-1A is another WD40 repeat-containing protein whose LoF mutants have been related to a severe combined immunodeficiency characterized by increased susceptibility to viral and mycobacterial infections (no. 4 in Table 1 and Figure 1) (27–30). Patients usually present with mucocutaneous manifestations, sinopulmonary diseases and neurocognitive disorders without inflammatory manifestations.
On the other hand, the capping proteins are heterodimers composed by two unrelated subunits with highly conserved amino acid sequences. The RGD, leucine-rich repeat, tropomodulin and proline-rich containing protein (RLTPR), also called CARMIL2, is a cytosolic protein that acts as scaffold between the nuclear factor kappa-light-chain-enhancer of activated B cells (NFkB) and CD28 (31, 32). Autosomal recessive (AR) LoF mutations of the RLTPR gene cause a primary immunodeficiency (PID) characterized by allergy, increased incidence of bacterial and fungal infections, and virus-related tumors (no. 5 in Table 1 and Figure 1) (33). The abnormal cytoskeleton of T-cell in patients with CARMIL2 deficiency causes defects of activation and is related to an abnormal activity of the capping proteins (34).
Activation Defects
Over 40 years ago, studies on the ligand-induced movement of immunoglobulin on the surface of lymphocytes called attention to a special relationship between CAcN and antigen-presenting cells (35). A specialized cell–cell junction, the immune synapse (36), is required for the activation of lymphocytes and begin with the formation of thousands of transient, low affinity interactions between antigens and integrins, such as the lymphocyte function-associated antigen 1 (LFA-1) (37). These interactions require a minimum distance of 40 nm, while the major histocompatibility complexes require 15 nm. The consequent antigen-induced CAcN rearrangements leads to morphological changes that are crucial for adhesion, migration, endocytosis, division, gene expression, and calcium flux, as well as for the releasing of cytokines and cytotoxic granules in lymphocytes, neutrophils and monocytes (38).
In particular, on resting leucocytes, LFA-1 is maintained in a low activity state by an inhibitory interaction with the CAcN (39, 40). Therefore, activation of leucocytes requires the release of CAcN-integrin interactions, so that LFA-1 can diffuse in the cell membrane and start binding activities (37). The essential role of CAcN in phagocyte function can be highlighted during chronic infections (41). In fact, microbes are able to lose their integrin ligands in order to escape the immune response (42). The abnormal rolling of leukocytes seems the main affected mechanism in patients with PID caused by LFA-1 defects (nos. 6–9 in Table 1 and Figure 1) (43). The deficiency of the β2 integrin subunit of the LFA-1 causes the leukocyte adhesion deficiency (LAD) type I, and the defective activation of LFA-1 subunits has been related to the LAD type III, both nowadays effectively treated with the hematopoietic stem cells transplantation (44, 45). On the other side, LAD type II is caused by mutations of a fucose transporter gene leading to cell membrane glycans lacking fucosylation. The administration of oral fucose did not seem effective to control the LAD type II clinical manifestations (46, 47).
Finally, a monocyte-selective adhesion defect has been recently noted in patients with cystic fibrosis (CF) and called LAD type IV (48–50). CFTR heterozygous LoF variants cause hyper activation of the small G-proteins Rho family that controls integrins activation (51). Interestingly, these small G-proteins are also well-known inhibitor of the pyrin inflammasome (52). Furthermore, CFTR interacts with Ezrin protein via its C-terminal domain. Ezrin is the most prominent members of the Ezrin-Radixin-Moesin (ERM) domain-containing protein family that links CAcN to the cell membrane, regulating tension during motility and endocytosis (53, 54). In hematopoietic cells, Ezrin and Moesin are highly expressed, whereas Radixin is mostly absent. Hemizygous LoF mutations of the MSN gene coding for Moesin is associated to a PID called X-linked MSN-associated immunodeficiency (X-MAID; no. 10 in Table 1 and Figure 1) (55). Patient T cells displayed impaired proliferative responses after activation by certain mitogens, and a variable defects in cell migration and adhesion, whereas the formation of immunologic synapses is normal. Thus, CAcN dysfunctions impair epithelial tight junction formation as well as lymphocytes adhesion capability in X-MAID patients.
Protrusions Defects
The collapse of CAcN to the side of cells occupied by microtubule organizing centers creates an opening for new actin polymerization to form membrane protrusions at the leading edge. This process is controlled by the small G-proteins Rho family, including the Cell division control protein 42 homolog (Cdc42) and Rac2 (56).
Small G-proteins are a superfamily of ubiquitously expressed cytosolic hydrolase enzymes that can independently bind and hydrolyze guanosine triphosphate (GTP) to guanosine diphosphate (GDP), becoming inactive (57). The best-known subfamily members are the Ras GTPases that are divided into five main families: Ras, Rho, Ran, Rab, and Arf. The Ras family is generally responsible for cell proliferation, Rho for cell morphology, Ran for nuclear transport and Rab and Arf for vesicle transport. The Ras guanyl nucleotide-releasing protein 1 (RASGRP1) is a diacylglycerol-regulated nucleotide exchange factor specifically activating Ras and regulating T and B cells development, homeostasis and differentiation. Rasgrp1 deregulation in mice results in a systemic lupus erythematosus-like disorder (58) and RASGRP1 deficiency in humans causes a PID characterized by impaired cytoskeletal dynamics (no. 11 in Table 1 and Figure 1) (59). Patients with RASGRP1 deficiency suffer from recurrent bacterial and viral infections especially affecting the lung with a severe failure to thrive and can develop EBV-related lymphomas.
The localization of small G-proteins on the cell membrane is due to their prenylation, a post-translational modification characterized by the addition of twenty-carbon lipophilic isoprene units to the cysteine residues at the C-terminus (60). Furthermore, most of the Rho family members contain a cluster of positively charged residues (i.e., polybasic domain), directly preceding their geranylgeranyl moiety that serves to fine-tune their localization among different cell membrane sites. Overall, the prenylation of small G-proteins is involved in the regulation of cytokines production (61) and can be regulated by statins in monocytes and macrophages (62).
On 2D surfaces, activated Cdc42 and Rac2 generate filopodia and lamellipodia, respectively. The formation of these membrane protrusions consents leucocytes to reach the damaged tissue passing through an intact vessel wall, a process called diapedesis. The local concentration of the complement system C3 fraction also contributes to this process (63). However, in 3D environment, the blebbing motility seems the more common migratory strategy of blood cells (64, 65). Stop-codon variants of the CDC42 gene has been recently associated with a novel autoinflammatory disease characterized by neonatal-onset of cytopenia, rash, and hemophagocytosis (NOCARH), successfully treated with interleukin-1β inhibition (no. 12 in Table 1 and Figure 1) (66). Furthermore, heterozygous CDC42 missense variants have been related to the Takenouchi-Kosaki syndrome (TKS) (67–69). TKS patients do not usually display autoinflammatory manifestations but hematologic and/or lymphatic defects, including macrothrombocytopenia, lymphedema, intestinal lymphangiectasia and recurrent infections. Characteristics of platelets and B cells have been recently described (70–72). A recent extensive genotype-phenotype correlation study allows to classify three groups of the CDC42 variants regarding involved protein domain (73). Based on these evidences, the NOCARH-associated variants occur at the C-terminus that usually allows PIP2 interaction, whereas variants associated with TKS resembling Noonan syndrome occurs at the N-terminus. Thus, different roles of the Cdc42 protein may be subverted in these conditions with different clinical manifestations.
The Rho guanosine triphosphatases Rac2 is expressed only in hematopoietic cells. Patients with Rac2 dysfunction secondary to dominant negative or homozygous LoF mutations present early-onset recurrent abscesses, neutrophilia, and defective wound healing, whereas monoallelic germline GoF mutations of the same RAC2 gene cause a severe combined immunodeficiency (no. 13 in Table 1 and Figure 1) (74–77). Interestingly, Rac2 activation in neutrophils is primarily mediated by the dedicator of cytokinesis (DOCK) 2, an atypical guanine nucleotide exchange factor (GEF) that rapidly translocate to the plasma membrane in a phosphatidylinositol 3,4,5-trisphosphate (PIP3)-dependent manner upon stimulation, resulting in increased local CAcN polymerization (78, 79). DOCK2 is mainly expressed in peripheral blood leukocytes and DOCK2 deficiency causes an early-onset PID characterized by a T-cell defective chemotactic responses with bacterial and viral infections (no. 14 in Table 1 and Figure 1) (80).
On the other side, DOCK8 is a Cdc42-specific GEF that regulates interstitial migration of dendritic cells and DOCK8 deficiency causes the AR Hyper-IgE syndrome (HIES), a combined immunodeficiency characterized by recurrent viral infections, early-onset malignancy and atopic dermatitis (no. 15 in Table 1 and Figure 1). Patients with DOCK8 deficiency display severe viral skin infections, such as chronic anogenital ulcers, multiple acral warts, and disfiguring molluscum contagiosum (81–84). Selective loss of group 3 innate lymphoid cell has been described in these patients (85).
Branching Defects
Cdc42 and Rac2 transmit many signals through the GTP-dependent binding of effector proteins containing the Cdc42/Rac interactive binding (CRIB) motif, such as the Wiskott-Aldrich syndrome (WAS) protein (WASP) (86). WASP is restricted to hematopoietic cells, while neuronal WASP (N-WASP), closely related in amino acids sequence, is more widely expressed (87). Other members of this proteins family include the Scar/WAVE proteins. N-WASP has been implicated in filopodia formation downstream of Cdc42, and the Scar/WAVE proteins family has been shown to contribute to the formation of lamellipodia downstream of Rac2. Recently, an immune dysregulation disorders characterized by deficit of the hematopoietic-specific WAVE complex regulator HEM1, coded by the NCKAP1L gene, has been characterized (no. 16 in Table 1 and Figure 1) (88). Patients with HEM1 deficiency suffer from recurrent infections, asthma and lymphoproliferation.
N-WASP deficiency increases the production of inflammatory cytokine (89, 90). Human LoF mutations of WAS gene cause severe defects in hematopoietic cell functions, leading to the well-known triad of microthrombocytopenia, immunodeficiency, and eczema (no. 17 in Table 1 and Figure 1) (91). The cytoskeletal defects of megakaryocytes are responsible for the low number of platelets in patients with WAS (92). WASP deficiency promotes T-cell cytoskeletal tension decay and phosphorylation of a serine/threonine protein kinase 4 (STK4) that usually increase T-cell migration, therefore promoting immune synapse breaking and secondary B cells dysfunction (93, 94). WASP-deficient lymphocytes fails to differentiate into memory cells (95) and are more prone to develop DNA damages due to the loss of the Golgi-dispersal response, a recently described mechanism of cell survival after ionized radiation exsposure (96). The STK4 deficiency causes a PID characterized by B and T cell lymphopenia, neutropenia, and cardiac malformations (no. 18 in Table 1 and Figure 1) (97). STK4 phosphorylates the Forkhead box O1 transcription factor, increasing NFkB-mediated production of interleukin 12 in dendritic cells and limiting the oxidative stress susceptibility (98). No platelets anomalies have been described in patients with STK4 deficiency. Equally, deficiency of the WASP interacting protein family member 1 (WIPF1) causes a WAS-like syndrome with normal platelet volume (no. 19 in Table 1 and Figure 1). WIPF1 is able to stabilize WASP, preventing its degradation in lymphocytes (99).
ASP controls the rate of actin branching by activating the actin related protein (ARP) 2/3 complex constituted by seven subunits. Two of them, the ARP2 and 3, closely resemble the structure of the G-actin, allowing the formation of a thermodynamically stable dimer that serves as a nucleation site for the new actin filaments at 70° angle from the main filament. Homozygous LoF variants of the ARPC1B gene, coding for the p41 regulatory subunits of the ARP2/3 complex, cause the platelet abnormalities with eosinophilia and immune-mediated inflammatory disease (PLTEID; no. 20 in Table 1 and Figure 1) (100–104). Patients with PLTEID usually present systemic inflammation with lymphoproliferation and immunodeficiency resembling WAS, with early onset vasculitis, severe infections, and eczema. A functional test has been recently described to detect asymptomatic carriers (105).
Additional WASP activators include the proline–serine–threonine phosphatase-interacting protein 1 (PSTPIP1), PIP2, and the c-Src protein-tyrosine kinases family. Heterozygous GoF mutation of the PSTPIP1 gene causes the pyogenic sterile arthritis, pyoderma gangrenosum, and acne (PAPA) syndrome and the PSTPIP1-associated myeloid-related proteinemia inflammatory (PAMI) syndrome (no. 21 in Table 1 and Figure 1) (106, 107). PAMI syndrome is caused by variants that substantially alter electrostatic properties of the PSTPIP1 critical region for auto-inhibiting dimerization, resulting in a GoF mutant protein that constitutively activates the underlying Pyrin inflammasome (108). Pyrin is the pivotal protein of the related inflammasome, a member of cytosolic multiprotein oligomers family responsible for the activation of inflammatory responses in human cells. The Pyrin-associated autoinflammation with neutrophilic dermatosis (PAAND) and familial Mediterranean fever (FMF) are well-known monogenic autoinflammatory diseases both related to GoF variants at different locus sites of the MEFV gene and associated with an excessive activation of the Pyrin inflammasome (no. 22 in Table 1 and Figure 1). Recently, the mevalonate kinase deficiency (MKD) caused by homozygous or compound heterozygous LoF mutations in the MVK gene has been related to the constitutive activation of Pyrin (no. 23 in Table 1 and Figure 1) (109).
Production Defects
Megakaryoblastic leukemia 1 (MKL1) is a member of the myocardin-related transcription factors and usually held in an inactive state in the cytoplasm in a reversible complex with G-actin (110). Stimulation of the small Rho GTPases promotes incorporation of G-actin into F-actin, allowing MLK1 to enter into the nucleus, stimulating transcription of actin and other cytoskeletal proteins genes. Homozygous LoF mutation in the MKL1 gene result in a PID characterized by susceptibility to severe bacterial infection and recurrent skin abscesses (no. 24 in Table 1 and Figure 1) (111). MKL1 deficiency causes reduced phagocytosis and almost complete abrogation of neutrophils spreading properties (112). MLK1 participates in differentiation of megakaryocytes and mild thrombocytopenia has been noted in patients with MKL1 deficiency (113).
Finally, LoF variants of the gene coding for the transcription factor CCAAT enhancer binding protein epsilon (C/EBPϵ) cause a PID called AR neutrophil-specific granule deficiency-1 (SGD) (114), whereas heterozygous GoF variants have been recently related to an autoinflammatory disease called the C/EBPϵ-associated autoinflammation and immune impairment of neutrophils (CAIN; no. 25 in Table 1 and Figure 1). Patients with CAIN display recurrent fevers characterized by abdominal pain, lasting 4–5 days, and skin inflammatory manifestations, such as sterile abscesses, pyoderma gangrenosum and oral ulcerations. The mutant C/EBPϵ causes deregulated transcription of interleukins and interferon response genes in neutrophils (115).
Discussion
The field of autoinflammation is moving from a gene-centric view of innate immune-mediated diseases towards a systems-based concept, which describes how various convergent molecular pathways, including actin cytoskeleton, contribute to the autoinflammatory process (116) and to a number of conditions characterized by the coexistence of inflammation, autoimmunity and defective immune response. Indeed, the complex regulation of the actin remodeling represents an example of autoinflammatory diseases merging with immunodeficiencies. Despite the wide range of symptoms associated with these disorders, some features may suggest the diagnosis, such as recurrent fevers or infections, atypical skin manifestations (from severe viral infections to eczema and sterile abscesses), cytopenias and defects of chemotaxis and lymphocytes proliferation. Cytopenias may be secondary to the abnormal release of immune cells from the bone marrow and/or impairments in the immune synapsis, while the abnormal diapedesis associated with an altered vessels wall and the increased cell apoptosis in the skin matrix, called cytothripsis, may favor cutaneous manifestations (86). Cytoskeleton-targeted therapies, such as colchicine, may play new roles in these disorders. The study of the molecular and modular diversity of these immune responses to the changing conditions has only recently become possible through the development of the new “omics”-based screening technologies (117). The adoption of “omics” and systems-based concepts will have implications for the discovery of novel diseases and for the possible development of targeted diagnostic tests and treatment options.
Author Contributions
RP drafted the manuscript. FP, SV, and MG reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The study was supported with public funds granted by the Italian Ministry of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are members of the European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Diseases—Project ID No 739543.
References
1. von der Ecken J, Heissler SM, Pathan-Chhatbar S, Manstein DJ, Raunser S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature (2016) 534(7609):724–8. doi: 10.1038/nature18295
2. Goebel HH. Congenital Myopathies in the New Millennium. J Child Neurol (2005) 20(2):94–101. doi: 10.1177/08830738050200020201
3. Etzioni A, Ochs HD. Lazy Leukocyte Syndrome—an Enigma Finally Solved? J Clin Immunol (2020) 40(1):9–12. doi: 10.1007/s10875-019-00718-0
4. Papa R, Picco P, Gattorno M. The expanding pathways of autoinflammation: a lesson from the first 100 genes related to autoinflammatory manifestations. Adv Protein Chem Struct Biol (2020) 120:1–44. doi: 10.1016/bs.apcsb.2019.11.001
5. Dominguez R, Holmes KC. Actin Structure and Function. Annu Rev Biophys (2011) 40(1):169–86. doi: 10.1146/annurev-biophys-042910-155359
6. Skruber K, Read T-A, Vitriol EA. Reconsidering an active role for G-actin in cytoskeletal regulation. J Cell Sci (2018) 131(1):jcs203760. doi: 10.1242/jcs.203760
7. Merino F, Pospich S, Raunser S. Towards a structural understanding of the remodeling of the actin cytoskeleton. Semin Cell Dev Biol (2020) 102:51–64. doi: 10.1016/j.semcdb.2019.11.018
8. Alkam D, Feldman EZ, Singh A, Kiaei M. Profilin1 Biology and its Mutation, Actin(g) in Disease. Cell Mol Life Sci (2017) 74(6):967–81. doi: 10.1007/s00018-016-2372-1
9. Wu C-H, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, et al. Mutations in the Profilin 1 Gene Cause Familial Amyotrophic Lateral Sclerosis. Nature (2012) 488(7412):499–503. doi: 10.1038/nature11280
10. Scotto di Carlo F, Pazzaglia L, Esposito T, Gianfrancesco F. The Loss of Profilin 1 Causes Early Onset Paget’s Disease of Bone. J Bone Miner Res (2020) 35(8):1387–98. doi: 10.1002/jbmr.3964
11. Chakraborty S, Jiang C, Gau D, Oddo M, Ding Z, Vollmer L, et al. Profilin-1 deficiency leads to SMAD3 upregulation and impaired 3D outgrowth of breast cancer cells. Br J Cancer (2018) 119(9):1106–17. doi: 10.1038/s41416-018-0284-6
12. Schoppmeyer R, Zhao R, Cheng H, Hamed M, Liu C, Zhou X, et al. Human profilin 1 is a negative regulator of CTL mediated cell-killing and migration. Eur J Immunol (2017) 47(9):1562–72. doi: 10.1002/eji.201747124
13. Romeo GR, Pae M, Eberle D, Lee J, Shoelson SE. Profilin-1 Haploinsufficiency Protects Against Obesity-Associated Glucose Intolerance and Preserves Adipose Tissue Immune Homeostasis. Diabetes (2013) 62(11):3718–26. doi: 10.2337/db13-0050
14. Qianyi L, Peirong L, Wei C, Li L, Zhi Z. ANGPTL-4 induces diabetic retinal inflammation by activating Profilin-1. Exp Eye Res (2017) 166:140–50. doi: 10.1016/j.exer.2017.10.009
15. Nunoi H, Yamazaki T, Tsuchiya H, Kato S, Malech HL, Matsuda I, et al. A heterozygous mutation of beta-actin associated with neutrophil dysfunction and recurrent infection. Proc Natl Acad Sci USA (1999) 96(15):8693–8. doi: 10.1073/pnas.96.15.8693
16. Cuvertino S, Stuart HM, Chandler KE, Roberts NA, Armstrong R, Bernardini L, et al. ACTB Loss-of-Function Mutations Result in a Pleiotropic Developmental Disorder. Am J Hum Genet (2017) 101(6):1021–33. doi: 10.1016/j.ajhg.2017.11.006
17. van Rheenen J, Condeelis J, Glogauer M. A common cofilin activity cycle in invasive tumor cells and inflammatory cells. J Cell Sci (2009) 122(3):305–11. doi: 10.1242/jcs.031146
18. Bamburg JR, Wiggan OP. ADF/cofilin and actin dynamics in disease. Trends Cell Biol (2002) 12(12):598–605. doi: 10.1016/S0962-8924(02)02404-2
19. Tedeschi A, Dupraz S, Curcio M, Laskowski CJ, Schaffran B, Flynn KC, et al. ADF/Cofilin-Mediated Actin Turnover Promotes Axon Regeneration in the Adult CNS. Neuron (2019) 103(6):1073–85.e6. doi: 10.1016/j.neuron.2019.07.007
20. Sun Y, Liang L, Dong M, Li C, Liu Z, Gao H. Cofilin 2 in Serum as a Novel Biomarker for Alzheimer’s Disease in Han Chinese. Front Aging Neurosci (2019) 11:214. doi: 10.3389/fnagi.2019.00214
21. Muñoz-Lasso DC, Mollá B, Calap-Quintana P, García-Giménez JL, Pallardo FV, Palau F, et al. Cofilin dysregulation alters actin turnover in frataxin-deficient neurons. Sci Rep (2020) 10(1):5207. doi: 10.1038/s41598-020-62050-7
22. Haugen AC, Di Prospero NA, Parker JS, Fannin RD, Chou J, Meyer JN, et al. Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: cellular model of pathology. PloS Genet (2010) 6(1):e1000812. doi: 10.1371/journal.pgen.1000812
23. Kuhns DB, Fink DL, Choi U, Sweeney C, Lau K, Priel DL, et al. Cytoskeletal abnormalities and neutrophil dysfunction in WDR1 deficiency. Blood (2016) 128(17):2135–43. doi: 10.1182/blood-2016-03-706028
24. Standing ASI, Malinova D, Hong Y, Record J, Moulding D, Blundell MP, et al. Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin-regulatory gene WDR1. J Exp Med (2017) 214(1):59–71. doi: 10.1084/jem.20161228
25. Papadopoulou C, Omoyinmi E, Standing A, Pain CE, Booth C, D’Arco F, et al. Monogenic mimics of Behçet’s disease in the young. Rheumatol (Oxford) (2019) 58(7):1227–38. doi: 10.1093/rheumatology/key445
26. Pfajfer L, Mair NK, Jiménez-Heredia R, Genel F, Gulez N, Ardeniz Ö, et al. Mutations affecting the actin regulator WD repeat-containing protein 1 lead to aberrant lymphoid immunity. J Allergy Clin Immunol (2018) 142(5):1589–604.e11. doi: 10.1016/j.jaci.2018.04.023
27. Shiow LR, Roadcap DW, Paris K, Watson SR, Grigorova IL, Lebet T, et al. The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nat Immunol (2008) 9(11):1307–15. doi: 10.1038/ni.1662
28. Shiow LR, Paris K, Akana MC, Cyster JG, Sorensen RU, Puck JM. Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a Coronin-1A mutation and a chromosome 16p11.2 deletion. Clin Immunol (2009) 131(1):24–30. doi: 10.1016/j.clim.2008.11.002
29. Moshous D, Martin E, Carpentier W, Lim A, Callebaut I, Canioni D, et al. Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J Allergy Clin Immunol (2013) 131(6):1594–603. doi: 10.1016/j.jaci.2013.01.042
30. Stray-Pedersen A, Jouanguy E, Crequer A, Bertuch AA, Brown BS, Jhangiani SN, et al. Compound heterozygous CORO1A mutations in siblings with a mucocutaneous-immunodeficiency syndrome of epidermodysplasia verruciformis-HPV, molluscum contagiosum and granulomatous tuberculoid leprosy. J Clin Immunol (2014) 34(7):871–90. doi: 10.1007/s10875-014-0074-8
31. Matsuzaka Y, Okamoto K, Mabuchi T, Iizuka M, Ozawa A, Oka A, et al. Identification, expression analysis and polymorphism of a novel RLTPR gene encoding a RGD motif, tropomodulin domain and proline/leucine-rich regions. Gene (2004) 343(2):291–304. doi: 10.1016/j.gene.2004.09.004
32. Roncagalli R, Cucchetti M, Jarmuzynski N, Grégoire C, Bergot E, Audebert S, et al. The scaffolding function of the RLTPR protein explains its essential role for CD28 co-stimulation in mouse and human T cells. J Exp Med (2016) 213(11):2437–57. doi: 10.1084/jem.20160579
33. Schober T, Magg T, Laschinger M, Rohlfs M, Linhares ND, Puchalka J, et al. A human immunodeficiency syndrome caused by mutations in CARMIL2. Nat Commun (2017) 8:14209. doi: 10.1038/ncomms14209
34. Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, et al. Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med (2016) 213(11):2413–35. doi: 10.1084/jem.20160576
35. Unanue ER, Perkins WD, Karnovsky MJ. Ligand-induced movement of lymphocyte membrane macromolecules. I. Analysis by immunofluorescence and ultrastructural radioautography. J Exp Med (1972) 136(4):885–906. doi: 10.1084/jem.136.4.885
36. Li J, Yin W, Jing Y, Kang D, Yang L, Cheng J, et al. The Coordination Between B Cell Receptor Signaling and the Actin Cytoskeleton During B Cell Activation. Front Immunol (2018) 9:3096. doi: 10.3389/fimmu.2018.03096
37. Dustin ML, Cooper JA. The immunological synapse and the actin cytoskeleton: molecular hardware for T cell signaling. Nat Immunol (2000) 1(1):23–9. doi: 10.1038/76877
38. Seppänen MRJ. Novel cytoskeletal mutations with immunodeficiency: Why is the raven like a writing desk? J Allergy Clin Immunol (2018) 142(5):1444–6. doi: 10.1016/j.jaci.2018.08.020
39. Das A, Bhat A, Sknepnek R, Köster D, Mayor S, Rao M. Stratification relieves constraints from steric hindrance in the generation of compact actomyosin asters at the membrane cortex. Sci Adv (2020) 6(11):eaay6093. doi: 10.1126/sciadv.aay6093
40. Schroer CFE, Baldauf L, van Buren L, Wassenaar TA, Melo MN, Koenderink GH, et al. Charge-dependent interactions of monomeric and filamentous actin with lipid bilayers. Proc Natl Acad Sci USA (2020) 117(11):5861–72. doi: 10.1073/pnas.1914884117
41. Demirdjian S, Hopkins D, Cumbal N, Lefort CT, Berwin B. Distinct Contributions of CD18 Integrins for Binding and Phagocytic Internalization of Pseudomonas aeruginosa. Infect Immun (2020) 88(5):e00011–20. doi: 10.1128/IAI.00011-20
42. Dos Santos MA, Alves Martins F, Borges BC, de Gouveia Santos J, Nascimento Alves R, Dias MH, et al. Human B cells infected by Trypanosoma cruzi undergo F-actin disruption and cell death via caspase-7 activation and cleavage of phospholipase Cγ1. Immunobiology (2020) 225(3):151904. doi: 10.1016/j.imbio.2020.151904
43. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
44. Essa MF, Elbashir E, Alroqi F, Mohammed R, Alsultan A. Successful hematopoietic stem cell transplant in leukocyte adhesion deficiency type III presenting primarily as malignant infantile osteopetrosis. Clin Immunol (2020) 213:108365. doi: 10.1016/j.clim.2020.108365
45. Qian X, Wang P, Wang H, Jiang W, Sun J, Wang X, et al. Successful umbilical cord blood transplantation in children with leukocyte adhesion deficiency type I. Transl Pediatr (2020) 9(1):34–42. doi: 10.21037/tp.2020.01.06
46. Etzioni A, Frydman M, Pollack S, Avidor I, Phillips ML, Paulson JC, et al. Recurrent Severe Infections Caused by a Novel Leukocyte Adhesion Deficiency. N Eng J Med (1992) 327(25):1789–992. doi: 10.1056/NEJM199212173272505
47. Wolach B, Gavrieli R, Wolach O, Stauber T, Abuzaitoun O, Kuperman A, et al. Leucocyte adhesion deficiency-A multicentre national experience. Eur J Clin Invest (2019) 49(2):e13047. doi: 10.1111/eci.13047
48. Fan Z, Ley K. Leukocyte Adhesion Deficiency IV. Monocyte Integrin Activation Deficiency in Cystic Fibrosis. Am J Respir Crit Care Med (2016) 193(10):1075–7. doi: 10.1164/rccm.201512-2454ED
49. Sorio C, Montresor A, Bolomini-Vittori M, Caldrer S, Rossi B, Dusi S, et al. Mutations of cystic fibrosis transmembrane conductance regulator gene cause a monocyte-selective adhesion deficiency. Am J Respir Crit Care Med (2016) 193(10):1123–33. doi: 10.1164/rccm.201510-1922OC
50. Das J, Sharma A, Jindal A, Aggarwal V, Rawat A. Leukocyte adhesion defect: Where do we stand circa 2019? Genes Dis (2020) 7(1):107–14. doi: 10.1016/j.gendis.2019.07.012
51. Castellani S, Guerra L, Favia M, Di Gioia S, Casavola V, Conese M. NHERF1 and CFTR restore tight junction organisation and function in cystic fibrosis airway epithelial cells: role of ezrin and the RhoA/ROCK pathway. Lab Invest (2012) 92(11):1527–40. doi: 10.1038/labinvest.2012.123
52. Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol (2016) 17(8):914–21. doi: 10.1038/ni.3457
53. Di Pietro C, Zhang P, O’Rourke TK, Murray TS, Wang L, Britto CJ, et al. Ezrin links CFTR to TLR4 signaling to orchestrate anti-bacterial immune response in macrophages. Sci Rep (2017) 7(1):1–11. doi: 10.1038/s41598-017-11012-7
54. Slifer ZM, Blikslager AT. The Integral Role of Tight Junction Proteins in the Repair of Injured Intestinal Epithelium. Int J Mol Sci (2020) 21(3):972–83. doi: 10.3390/ijms21030972
55. Lagresle-Peyrou C, Luce S, Ouchani F, Soheili TS, Sadek H, Chouteau M, et al. X-linked primary immunodeficiency associated with hemizygous mutations in the moesin (MSN) gene. J Allergy Clin Immunol (2016) 138(6):1681–9. doi: 10.1016/j.jaci.2016.04.032
56. Ghose D, Lew D. Mechanistic insights into actin-driven polarity site movement in yeast. Mol Biol Cell (2020) 31(10):1085–102. doi: 10.1091/mbc.E20-01-0040
57. Murali A, Rajalingam K. Small Rho GTPases in the control of cell shape and mobility. Cell Mol Life Sci (2014) 71(9):1703–21. doi: 10.1007/s00018-013-1519-6
58. Yasuda S, Stevens RL, Terada T, Takeda M, Hashimoto T, Fukae J, et al. Defective Expression of Ras Guanyl Nucleotide-Releasing Protein 1 in a Subset of Patients with Systemic Lupus Erythematosus. J Immunol (2007) 179(7):4890–900. doi: 10.4049/jimmunol.179.7.4890
59. Salzer E, Cagdas D, Hons M, Mace EM, Garncarz W, Petronczki ÖY, et al. RASGRP1 deficiency causes immunodeficiency with impaired cytoskeletal dynamics. Nat Immunol (2016) 17(12):1352–60. doi: 10.1038/ni.3575
60. Johnson JL, Erickson JW. Cerione RA. C-terminal Di-arginine Motif of Cdc42 Protein Is Essential for Binding to Phosphatidylinositol 4,5-Bisphosphate-containing Membranes and Inducing Cellular Transformation. J Biol Chem (2012) 287(8):5764–74. doi: 10.1074/jbc.M111.336487
61. Akula MK, Ibrahim MX, Ivarsson EG, Khan OM, Kumar IT, Erlandsson M, et al. Protein prenylation restrains innate immunity by inhibiting Rac1 effector interactions. Nat Commun (2019) 10(1):1–13. doi: 10.1038/s41467-019-11606-x
62. Fu H, Alabdullah M, Großmann J, Spieler F, Abdosh R, Lutz V, et al. The differential statin effect on cytokine production of monocytes or macrophages is mediated by differential geranylgeranylation-dependent Rac1 activation. Cell Death Dis (2019) 10(12):880. doi: 10.1038/s41419-019-2109-9
63. Kolev M, West EE, Kunz N, Chauss D, Moseman EA, Rahman J, et al. Diapedesis-Induced Integrin Signaling via LFA-1 Facilitates Tissue Immunity by Inducing Intrinsic Complement C3 Expression in Immune Cells. Immunity (2020) 52(3):513–527.e8. doi: 10.1016/j.immuni.2020.02.006
64. Yamada KM, Collins JW, Cruz Walma DA, Doyle AD, Morales SG, Lu J, et al. Extracellular matrix dynamics in cell migration, invasion and tissue morphogenesis. Int J Exp Pathol (2019) 100(3):144–52. doi: 10.1111/iep.12329
65. Guzman A, Avard RC, Devanny AJ, Kweon OS, Kaufman LJ. Membrane blebs play a critical role in a hybrid mode of cancer cell invasion in three-dimensional environments. J Cell Sci (2020) 133(8):jcs236778. doi: 10.1242/jcs.236778
66. Gernez Y, de Jesus AA, Alsaleem H, Macaubas C, Roy A, Lovell D, et al. Severe autoinflammation in 4 patients with C-terminal variants in cell division control protein 42 homolog (CDC42) successfully treated with IL-1β inhibition. J Allergy Clin Immunol (2019) 144(4):1122–5.e6. doi: 10.1016/j.jaci.2019.06.017
67. Takenouchi T, Kosaki R, Niizuma T, Hata K, Kosaki K. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: Yet another locus for thrombocytopenia and developmental delay. Am J Med Genet A (2015) 167A(11):2822–5. doi: 10.1002/ajmg.a.37275
68. Takenouchi T, Okamoto N, Ida S, Uehara T, Kosaki K. Further evidence of a mutation in CDC42 as a cause of a recognizable syndromic form of thrombocytopenia. Am J Med Genet A (2016) 170A(4):852–5. doi: 10.1002/ajmg.a.37526
69. Motokawa M, Watanabe S, Nakatomi A, Kondoh T, Matsumoto T, Morifuji K, et al. A hot-spot mutation in CDC42 (p.Tyr64Cys) and novel phenotypes in the third patient with Takenouchi-Kosaki syndrome. J Hum Genet (2018) 63(3):387–90. doi: 10.1038/s10038-017-0396-5
70. Pleines I, Eckly A, Elvers M, Hagedorn I, Eliautou S, Bender M, et al. Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood (2010) 115(16):3364–73. doi: 10.1182/blood-2009-09-242271
71. Burbage M, Keppler SJ, Gasparrini F, Martínez-Martín N, Gaya M, Feest C, et al. Cdc42 is a key regulator of B cell differentiation and is required for antiviral humoral immunity. J Exp Med (2015) 212(1):53–72. doi: 10.1084/jem.20141143
72. Uehara T, Suzuki H, Okamoto N, Kondoh T, Ahmad A, O’Connor BC, et al. Pathogenetic basis of Takenouchi-Kosaki syndrome: Electron microscopy study using platelets in patients and functional studies in a Caenorhabditis elegans model. Sci Rep (2019) 9(1):1–9. doi: 10.1038/s41598-019-40988-7
73. Martinelli S, Krumbach OHF, Pantaleoni F, Coppola S, Amin E, Pannone L, et al. Functional Dysregulation of CDC42 Causes Diverse Developmental Phenotypes. Am J Med Genet A (2018) 102(2):309–20. doi: 10.1016/j.ajhg.2017.12.015
74. Williams DA, Tao W, Yang F, Kim C, Gu Y, Mansfield P, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood (2000) 96(5):9. doi: 10.1182/blood.V96.5.1646
75. Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci USA (2000) 97(9):4654–9. doi: 10.1073/pnas.080074897
76. Alkhairy OK, Rezaei N, Graham RR, Abolhassani H, Borte S, Hultenby K, et al. RAC2 loss-of-function mutation in 2 siblings with characteristics of common variable immunodeficiency. J Allergy Clin Immunol (2015) 135(5):1380–4.e5. doi: 10.1016/j.jaci.2014.10.039
77. Lougaris V, Chou J, Beano A, Wallace JG, Baronio M, Gazzurelli L, et al. A monoallelic activating mutation in RAC2 resulting in a combined immunodeficiency. J Allergy Clin Immunol (2019) 143(4):1649–53.e3. doi: 10.1016/j.jaci.2019.01.001
78. Nishikimi A, Fukuhara H, Su W, Hongu T, Takasuga S, Mihara H, et al. Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Science (2009) 324(5925):384–7. doi: 10.1126/science.1170179
79. Kunimura K, Uruno T, Fukui Y. DOCK family proteins: key players in immune surveillance mechanisms. Int Immunol (2020) 32(1):5–15. doi: 10.1093/intimm/dxz067
80. Dobbs K, Domínguez Conde C, Zhang S-Y, Parolini S, Audry M, Chou J, et al. Inherited DOCK2 Deficiency in Patients with Early-Onset Invasive Infections. N Engl J Med (2015) 372(25):2409–22. doi: 10.1056/NEJMoa1413462
81. Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined Immunodeficiency Associated with DOCK8 Mutations. N Engl J Med (2009) 361(21):2046–55. doi: 10.1056/NEJMoa0905506
82. Engelhardt KR, Gertz ME, Keles S, Schäffer AA, Sigmund EC, Glocker C, et al. The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol (2015) 136(2):402–12. doi: 10.1016/j.jaci.2014.12.1945
83. Buchbinder D, Kirov I, Danielson J, Shah NN, Freeman AF, Chavan RS, et al. Compound Heterozygous DOCK8 Mutations in a Patient with B Lymphoblastic Leukemia and EBV-Associated Diffuse Large B Cell Lymphoma. J Clin Immunol (2019) 39(6):592–5. doi: 10.1007/s10875-019-00663-y
84. Zhang Q, Dove CG, Hor JL, Murdock HM, Strauss-Albee DM, Garcia JA, et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. J Exp Med (2014) 211(13):2549–66. doi: 10.1084/jem.20141307
85. Eken A, Cansever M, Okus FZ, Erdem S, Nain E, Azizoglu ZB, et al. ILC3 deficiency and generalized ILC abnormalities in DOCK8-deficient patients. Allergy (2020) 75(4):921–32. doi: 10.1111/all.14081
86. Amato C, Thomason PA, Davidson AJ, Swaminathan K, Ismail S, Machesky LM, et al. WASP Restricts Active Rac to Maintain Cells’ Front-Rear Polarization. Curr Biol (2019) 29(24):4169–82.e4. doi: 10.1016/j.cub.2019.10.036
87. Abdul-Manan N, Aghazadeh B, Liu GA, Majumdar A, Ouerfelli O, Siminovitch KA, et al. Structure of Cdc42 in complex with the GTPase-binding domain of the ‘Wiskott–Aldrich syndrome’ protein. Nature (1999) 399(6734):379–83. doi: 10.1038/20726
88. Cook SA, Comrie WA, Poli MC, Similuk M, Oler AJ, Faruqi AJ, et al. HEM1 deficiency disrupts mTORC2 and F-actin control in inherited immunodysregulatory disease. Science (2020) 369(6500):202–7. doi: 10.1126/science.aay5663
89. Volpi S, Santori E, Abernethy K, Mizui M, Dahlberg CIM, Recher M, et al. N-WASP is required for B-cell–mediated autoimmunity in Wiskott-Aldrich syndrome. Blood (2016) 127(2):216–20. doi: 10.1182/blood-2015-05-643817
90. Jain N, Thanabalu T. Molecular difference between WASP and N-WASP critical for chemotaxis of T-cells towards SDF-1α. Sci Rep (2015) 5:15031. doi: 10.1038/srep15031
91. Malik MA, Masab M. Wiskott-Aldrich Syndrome. Treasure Island (FL): StatPearls Publishing (2020).
92. Ghalloussi D, Dhenge A, Bergmeier W. New insights into cytoskeletal remodeling during platelet production. J Thromb Haemost (2019) 17(9):1430–9. doi: 10.1111/jth.14544
93. Huang L, Sun X, Yang D, Dai X, Jiang P, Bai X, et al. WASP and Mst1 coregulate B-cell development and B-cell receptor signaling. Blood Adv (2020) 4(3):573–85. doi: 10.1182/bloodadvances.2018027870
94. Kumari S, Mak M, Poh Y-C, Tohme M, Watson N, Melo M, et al. Cytoskeletal tension actively sustains the migratory T-cell synaptic contact. EMBO J (2020) 39(5):e102783. doi: 10.15252/embj.2019102783
95. Liu Q, Zhang L, Shu Z, Yu T, Zhou L, Song W, et al. WASp Is Essential for Effector-to-Memory conversion and for Maintenance of CD8+T Cell Memory. Front Immunol (2019) 10:2262. doi: 10.3389/fimmu.2019.02262
96. Wen K-K, Han S-S, Vyas YM. Wiskott-Aldrich syndrome protein senses irradiation-induced DNA damage to coordinate the cell-protective Golgi dispersal response in human T and B lymphocytes. J Allergy Clin Immunol (2020) 145(1):324–34. doi: 10.1016/j.jaci.2019.09.026
97. Xu X, Wang X, Todd EM, Jaeger ER, Vella JL, Mooren OL, et al. Mst1 Kinase Regulates the Actin-Bundling Protein L-Plastin To Promote T Cell Migration. J Immunol (2016) 197(5):1683–91. doi: 10.4049/jimmunol.1600874
98. Cho K-M, Kim MS, Jung H-J, Choi E-J, Kim TS. Mst1-Deficiency Induces Hyperactivation of Monocyte-Derived Dendritic Cells via Akt1/c-myc Pathway. Front Immunol (2019) 10:2142. doi: 10.3389/fimmu.2019.02142
99. Lanzi G, Moratto D, Vairo D, Masneri S, Delmonte O, Paganini T, et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med (2012) 209(1):29–34. doi: 10.1084/jem.20110896
100. Kuijpers TW, Tool ATJ, van der Bijl I, de Boer M, van Houdt M, de Cuyper IM, et al. Combined immunodeficiency with severe inflammation and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol (2017) 140(1):273–7.e10. doi: 10.1016/j.jaci.2016.09.061
101. Kahr WHA, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun (2017) 8:14816. doi: 10.1038/ncomms14816
102. Volpi S, Cicalese MP, Tuijnenburg P, Tool ATJ, Cuadrado E, Abu-Halaweh M, et al. A combined immunodeficiency with severe infections, inflammation, and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol (2019) 143(6):2296–9. doi: 10.1016/j.jaci.2019.02.003
103. Randzavola LO, Strege K, Juzans M, Asano Y, Stinchcombe JC, Gawden-Bone CM, et al. Loss of ARPC1B impairs cytotoxic T lymphocyte maintenance and cytolytic activity. J Clin Invest (2019) 129(12):5600–14. doi: 10.1172/JCI129388
104. Brigida I, Zoccolillo M, Cicalese MP, Pfajfer L, Barzaghi F, Scala S, et al. T-cell defects in patients with ARPC1B germline mutations account for combined immunodeficiency. Blood (2018) 132(22):2362–74. doi: 10.1182/blood-2018-07-863431
105. Kopitar AN, Markelj G, Oražem M, Blazina Š, Avčin T, Ihan A, et al. Flow Cytometric Determination of Actin Polymerization in Peripheral Blood Leukocytes Effectively Discriminate Patients With Homozygous Mutation in ARPC1B From Asymptomatic Carriers and Normal Controls. Front Immunol (2019) 10:1632. doi: 10.3389/fimmu.2019.01632
106. Holzinger D, Fassl SK, de Jager W, Lohse P, Röhrig UF, Gattorno M, et al. Single amino acid charge switch defines clinically distinct proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1)-associated inflammatory diseases. J Allergy Clin Immunol (2015) 136(5):1337–45. doi: 10.1016/j.jaci.2015.04.016
107. Klötgen H-W, Beltraminelli H, Yawalkar N, van Gijn ME, Holzinger D, Borradori L. The expanding spectrum of clinical phenotypes associated with PSTPIP1 mutations: from PAPA to PAMI syndrome and beyond. Br J Dermatol (2018) 178(4):982–3. doi: 10.1111/bjd.16136
108. Hashmi SK, Bergstrom K, Bertuch AA, Despotovic JM, Muscal E, Xia F, et al. PSTPIP1-associated myeloid-related proteinemia inflammatory syndrome: A rare cause of childhood neutropenia associated with systemic inflammation and hyperzincemia. Pediatr Blood Cancer (2019) 66(1):e27439. doi: 10.1002/pbc.27439
109. Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol (2016) 17(8):914–21. doi: 10.1038/ni.3457
110. Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature (2013) 497(7450):507–11. doi: 10.1038/nature12105
111. Sprenkeler EGG, Henriet S, Tool A, Kreft IC, van der Bijl I, Aarts CCEM, et al. MKL1 deficiency results in a severe neutrophil motility defect due to impaired actin polymerization. Blood (2020) 135(24):2171–81. doi: 10.1182/blood.2019002633
112. Record J, Malinova D, Zenner HL, Plagnol V, Nowak K, Syed F, et al. Immunodeficiency and severe susceptibility to bacterial infection associated with a loss-of-function homozygous mutation of MKL1. Blood (2015) 126(13):1527–35. doi: 10.1182/blood-2014-12-611012
113. Smith EC, Teixeira AM, Chen RC, Wang L, Gao Y, Hahn KL, et al. Induction of megakaryocyte differentiation drives nuclear accumulation and transcriptional function of MKL1 via actin polymerization and RhoA activation. Blood (2013) 121(7):1094–101. doi: 10.1182/blood-2012-05-429993
114. Gombart AF, Shiohara M, Kwok SH, Agematsu K, Komiyama A, Phillip Koeffler H. Neutrophil-specific granule deficiency: Homozygous recessive inheritance of a frameshift mutation in the gene encoding transcription factor CCAAT/enhancer binding protein-ϵ. Blood (2001) 97(9):2561–7. doi: 10.1182/blood.V97.9.2561
115. Göös H, Fogarty CL, Sahu B, Plagnol V, Rajamäki K, Nurmi K, et al. Gain-of-function CEBPE mutation causes noncanonical autoinflammatory inflammasomopathy. J Allergy Clin Immunol (2019) 144(5):1364–76. doi: 10.1016/j.jaci.2019.06.003
116. Savic S, Caseley EA, McDermott MF. Moving towards a systems-based classification of innate immune-mediated diseases. Nat Rev Rheumatol (2020) 16(4):222–37. doi: 10.1038/s41584-020-0377-5
Keywords: pyrin, Wiskott-Aldrich syndrome, autoinflammatory diseases, cytoskeleton, actin
Citation: Papa R, Penco F, Volpi S and Gattorno M (2021) Actin Remodeling Defects Leading to Autoinflammation and Immune Dysregulation. Front. Immunol. 11:604206. doi: 10.3389/fimmu.2020.604206
Received: 08 September 2020; Accepted: 19 November 2020;
Published: 07 January 2021.
Edited by:
Fabio Candotti, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandReviewed by:
Lisa S. Westerberg, Karolinska Institutet (KI), SwedenAlexandra Freeman, National Institutes of Health (NIH), United States
Copyright © 2021 Papa, Penco, Volpi and Gattorno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Gattorno, bWFyY29nYXR0b3Jub0BnYXNsaW5pLm9yZw==