 Song Yang
Song Yang Jun Zhou
Jun Zhou Dengwen Li
Dengwen Li- 1State Key Laboratory of Medicinal Chemical Biology, College of Life Sciences, Nankai University, Tianjin, China
- 2Institute of Biomedical Sciences, Shandong Provincial Key Laboratory of Animal Resistance Biology, Collaborative Innovation Center of Cell Biology in Universities of Shandong, College of Life Sciences, Shandong Normal University, Jinan, China
The retinal pigment epithelium is a fundamental component of the retina that plays essential roles in visual functions. Damage to the structure and function of the retinal pigment epithelium leads to a variety of retinopathies, and there is currently no curative therapy for these disorders. Therefore, studying the relationship between the development, function, and pathobiology of the retinal pigment epithelium is important for the prevention and treatment of retinopathies. Here we review the function of the retinal pigment epithelium and its relevance to the pathobiology, and discuss potential strategies for the treatment of retinopathies. In doing so, we provide new viewpoints outlining new ideas for the future study and treatment of retinopathies.
Introduction
Retinal pigment epithelium (RPE) is formed from a single layer of regular polygonal cells arranged at the outermost layer of the retina. The outer side of the RPE is connected to Bruch’s membrane and the choroid, while the inner side is connected to the outer segment of photoreceptor cells. The outer side exhibits basal infolding, which increases cell surface area and facilitates substance exchange. The basement membrane is closely connected to the basal folds by half desmosomes located in the innermost layer of Bruch’s membrane. The inside of RPE cells harbors microvillous structures extending between photoreceptor outer segments (POS), which participate in the phagocytic function of the RPE (Song and Zhou, 2020; Zhou and Zhou, 2020; Yang et al., 2021). The tight junction formed between the single-layer RPE and the gap junction control the movement of substances and at the same time forms the choroid-blood-retinal barrier with Bruch’s membrane and choroid at the lateral retina (Xie et al., 2020). The RPE appears dark brown due to its melanin content, which reduces damage to the retina and internal nerves from ultraviolet light (Tian et al., 2021). The RPE also harbors a complex metabolic system that reduces excessive accumulation of reactive oxygen species (ROS) and consequent oxidative damage.
Therefore, RPE structure and function are essential to normal vision, and alterations in the RPE can impair function and lead to retinopathy. For example, retinitis pigmentosa (RP), age-related macular degeneration (AMD), and Stargardt disease (SD) are degenerative retinal diseases in which RPE dysfunction has been implicated in their pathogenesis [for an excellent review, see Zarbin (Zarbin, 2016)]. RP afflicts 100,000 people in the United States and usually causes visual loss in childhood or young adulthood. AMD afflicts 1.75 million people in the United States alone, is the leading cause of blindness in individuals over 55 years of age in the United States and Europe, and was estimated to affect ∼196 million people worldwide in 2020. SD is the most common form of inherited juvenile macular degeneration, with a prevalence of 1 in 10,000 births. There are currently no cures for these degenerative diseases, so understanding the role of RPE in their pathogenesis is important for the development of new approaches to manage these common and debilitating disorders. Here we review the functions and diseases of the RPE to provide a theoretical basis for the treatment and prevention of associated diseases.
Retinal Development and Structure
The human eye begins to develop at embryonic day (E)18. The visual groove is formed at E22 before continuing to sag to form the visual vesicle, which expands to form the inner and outer layers of the optic cup. The RPE layer begins to differentiate around E30, with pigment particles found in RPE cells at E35. A set of genes (including PAX6, LHX2, RAX, and SIX3) expressed in the neural plate before E8 are involved in eye determination and eventually form the optic cup (Hoon et al., 2014). In general, vertebrate RPE cells develop and differentiate from optic vesicles. During embryonic development, early optic vesicle cells have bidirectional potential and can develop into the retinal neurocortical layer or the RPE layer. The cell fate decision and differentiation of RPE precursor cells is not spontaneous but rather influenced by a variety of microenvironmental factors. Under the influence of extracellular signals, differentiation is guided in strict temporal and spatial order through the regulation of transcription factors and intracellular signaling pathways. In particular, the transcription factor MITF (microphthalmia-associated transcription factor) has been confirmed to be involved in the normal RPE development, and Mitf knockout results in abnormal retinal development in mice (Bharti et al., 2008; Ma et al., 2019).
Light entering the eye is focused on the retina, which converts light signals into electrical signals that travel through the optic nerve to the visual center of the brain (Grossniklaus et al., 2015). The retina is located in the fundus of the eye and, as an important tissue forming vision, has a complex structure. The retina has multiple layers containing various cell types: the RPE lies at the boundary, while the retinal nerve layer contains five main neuronal cells including photoreceptor cells (rods and cones), horizontal cells, bipolar cells, amacrine cells, and ganglion cells. RPE cells are located between photoreceptor cells and the choroid membrane, with the basal side connected to Bruch’s membrane and tip microvilli connected to the outer segment of photoreceptor cells (Figure 1). The RPE is located in a specific position and has important functions, and the cells have no regenerative potential (Masland, 2012; Silverman and Wong, 2018). Therefore, studying the relationships between its structure, function, and associated diseases is important for the prevention and treatment of RPE lesions.
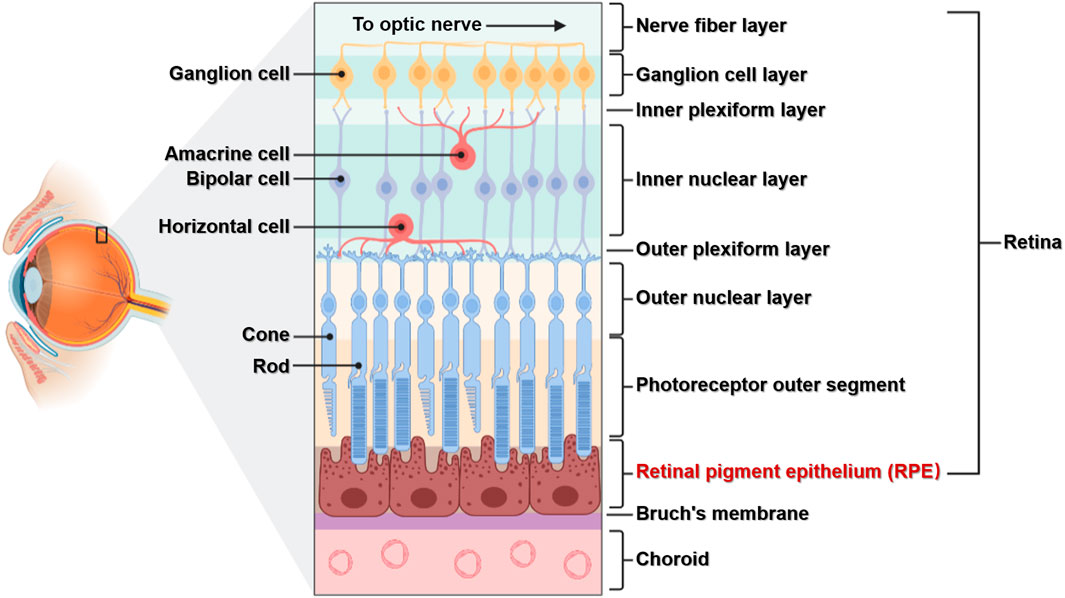
FIGURE 1. The structure of the retina. The retina is composed of multiple layers and different cell types. The RPE is composed of a single layer of RPE cells, which are connected to the choroid membrane through Bruch’s membrane.
Function of the Retinal Pigment Epithelium
Maintaining the Visual Cycle and Phagocytosis
The RPE plays an important role in maintaining visual function and the visual cycle. RPE cells are phagocytic, with the ability to engulf and eliminate exfoliated POS and maintain the normal renewal of visual cells (Ran et al., 2020; Ran and Zhou, 2020). In mammals, each RPE cell is responsible for about 30 photoreceptors, and of all cell types RPE cells consume the most material in a mammal’s lifetime (Young, 1967; Young, 1971; Penberthy et al., 2018). RPE cell phagocytosis is divided into three stages: binding, endocytosis, and elimination. During binding, the inner microvilli cell membranes of RPE cells bind to the shed outer segment of the visual cell before being endocytosed into the cell and finally being transported by the cytoskeleton and vesicles to lysosomes for elimination. The TAM receptor tyrosine kinase MerTK is expressed by RPE cells and is crucial for RPE function, mediating the recognition and endocytosis of the POS by RPE cells. RPE cells without MerTK cannot engulf the POS, causing complete degeneration of the photoreceptor and blindness after birth (Prasad et al., 2006; Burstyn-Cohen et al., 2012). Other studies have shown that mice lacking αvβ5 integrin have gradually reduced retinal phagocytic capacity with age (Nandrot et al., 2004; C. Yu et al., 2019a) via a mechanism by which αvβ5 integrin acts as a receiver for the POS (Figure 2A). In addition, the RPE65 gene encodes all-trans retinol ester isomerase, which is essential for the retinoid cycle. Mutation of the RPE65 allele has been found to destroy optic cells and cause clinical manifestations of Leber congenital amaurosis type 2 (LCA2) and early-onset retinal dystrophy, eventually leading to complete blindness (Gu et al., 1997; Marlhens et al., 1997; Aguirre et al., 1998).
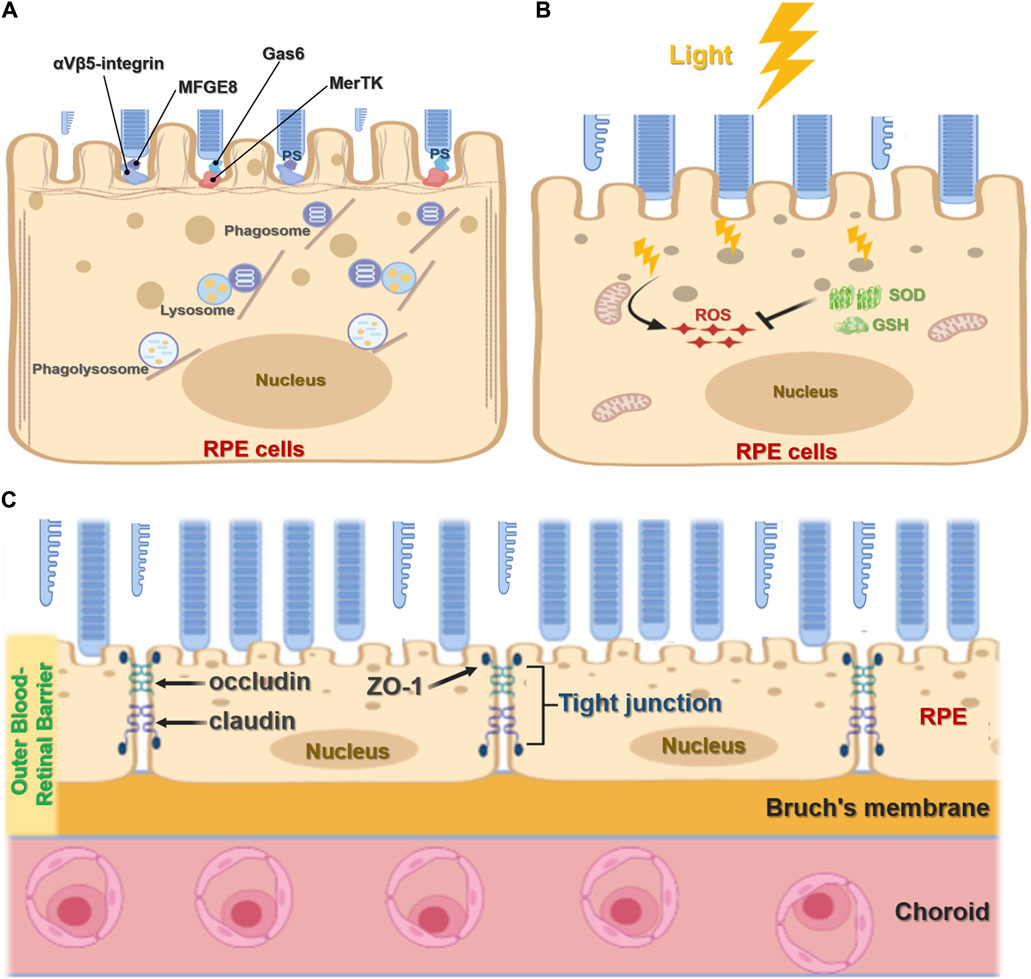
FIGURE 2. Function of retinal pigment epithelial cells. (A), The phagocytic function of RPE cells. RPE cells recognize and bind phosphatidylserine (PS) exposed by POS through MerTK/Gas6 and αVβ5-integrin/MFGE8 pathways to initiate phagocytosis. It further forms phagosomes and binds with lysosomes to form the phagolysosome, which digests POS. (B), Antioxidant function of RPE cells. Light stress produces ROS. RPE cells absorb light through melanin or eliminate ROS accumulation through antioxidants such as superoxide dismutase (SOD) and glutathione (GSH). (C), RPE cell barrier function. The RPE forms an outer blood-retinal barrier between the interior of the retina and the choroid. The RPE cells form tight junctions, including ZO-1, occludin, and claudin, which act as barriers.
Protection and Anti-Oxidative Functions
Located in the outermost layer of the retina, the RPE is rich in pigment particles including melanin and lipofuscin, which prevent light damage. These pigment particles are formed in utero and are no longer synthesized after birth. RPE melanin absorbs and filters natural light and protects the neural parts of the retina. Since the eyes are exposed to various light stimuli, they exist in a physiological state of photooxidation, accumulating high levels of oxygen free radicals that threaten oxidative damage. As a result, RPE cells contain many antioxidants such as superoxide dismutase and glutathione (Figure 2B). Melanosomes also participate in the antioxidant process, scavenging oxygen free radicals. Several mechanisms have been shown to underpin the antioxidant capacity and regulation of RPE cells including the ERK signaling pathway (Chong and Zheng, 2016; Chen et al., 2021); MMP-14 and TIMP-2 (Alcazar et al., 2007); micro(mi)RNA-23 (Lin et al., 2011); and toll-like receptor 3 (TLR3) (Patel and Hackam, 2013).
Barrier and Substance Transport Functions
RPE cells are terminally differentiated in a mitotic quiescent phase. As a typical barrier cells, they guard both the inside and outside of the retina and strictly control substance entry and exit. RPE cells form tight junctions through ZO-1, occludin, and claudin, acting as the outer blood barrier between choroidal pore capillaries and the retinal photosensitive layer (Figure 2C). RPE cells use membrane pumps, endocytosis, passive diffusion, and other mechanisms to complete transport and play a key role in nutrient, water, and electrolyte transport between the choroid and retinal cells (Danesh-Meyer et al., 2016; Sun and Zhou, 2020). To achieve epithelium-specific functions, tight junction permeability and selectivity must match the epithelium-specific extracellular transport mechanism. At the same time, the rich membrane pump system in the retina, including the Na-K-ATPase system, enables ion concentrations on both sides to reach a dynamic balance and maintain normal retinal function. In addition, the rich RPE cell transporter system facilitates the transport of substances inside and outside the retina (Sugasawa et al., 1994).
Retinal Pigment Epithelial Diseases and Pathogenesis
Oxidative Stress and Inflammation
The cornea has a transparent structure and the RPE is exposed to light for long periods of time, has a rich oxygen supply, and consequently large amounts of reactive oxygen are easily generated. In addition, increased systemic glucose levels, such as in diabetics, can facilitate excessive ROS accumulation (F. Yu et al., 2019b; Yu et al., 2020). In degenerative retinopathy, antioxidant levels decrease in cells; that is, the capacity of RPE cells to remove ROS variably reduces, resulting in a large accumulation of POS (Mitter et al., 2014; Campello et al., 2020). Many studies have been conducted on retinal epithelial damage caused by oxidative stress and inflammation, and recent studies have shown that the redox-sensitive microRNA (miR)-144 plays an important role in the regulation of antioxidant signaling pathways in human and mouse RPE. Oxidative stress enhanced the expression of miR-144-3p and mir-144-5p, decreased expression of Nrf2 and downstream antioxidant target genes Nqo1 and Gclc, decreased glutathione levels, and increased RPE cell death (Yam et al., 2019; Jadeja and Martin, 2020). In summary, oxidative stress and inflammation cause RPE cell damage, which in turn causes retinal dysfunction and even blindness.
Apoptosis and Autophagy
AMD is a serious neurodegenerative disease and a major cause of blindness in developed countries. Transcriptional profiling has shown that diseased RPE exhibits increased apoptosis, autophagy, and endoplasmic reticulum stress levels than normal cells. Other studies have shown that retinopathy is associated with disrupted cellular homeostasis and increased apoptosis, endoplasmic reticulum stress, and autophagy. Moreover, RPE cell death via apoptosis and endoplasmic reticulum stress has been observed in AMD and other retinal degenerative diseases (Dunaief et al., 2002; Feher et al., 2006; Bazan, 2007). In a study of RPE cells cultured ex vivo from AMD and normal donors, RPE cells from AMD donors accumulated lipid droplets and glycogen particles, harbored disintegrated mitochondria, and had increased numbers of autophagosomes. In addition, compared to RPE cells cultured from normal donors, RPE cells from AMD donors showed increased sensitivity to oxidative stress and decreased mitochondrial activity. The impaired autophagy function of AMD donor RPE was also demonstrated through measurement of the ratio of autophagy markers LC3-II/LC3-I (Golestaneh et al., 2017). These findings indicate a potential pathological mechanism for AMD through abnormal apoptosis and autophagy, thereby providing new targets for novel therapeutic strategies.
Cell Polarity and Interactions
The polarity and cell junctions of the RPE play critical roles in the blood-retinal barrier, maintaining the stability of the internal photoreceptor microenvironment and supporting the choroidal system. Disrupted cell polarity and cell junctions significantly increase the risk of retinal degenerative disease (Caceres and Rodriguez-Boulan, 2020). RPE polarity and cell junction stability are related to the unique basal and apical structures of the retina, which affect phagocytosis and material exchange. For example, cholesterol efflux is mediated by the ABCA1 transport protein at the top and basolateral aspects of the cell (Storti et al., 2017). RPE phagocytic defects are related to photoreceptor degeneration, so further study of the RPE endocytic pathway may help establish new mechanisms of retinal diseases (Anderson et al., 2017; Kaur et al., 2018; Ran et al., 2021). In addition, patients with RP have been shown to have RPE polarity and functional defects, and the cilial mRNA splicing factor PRPF was mutated in these RPE cells (Buskin et al., 2018). In summary, abnormalities in RPE polarity, barrier destruction, and retinal stability may contribute to the pathogenesis of blinding retinal diseases.
Treatment of Retinal Pigment Epithelial Diseases
Cell Therapy
Stem cell-derived RPE and photoreceptors have restored vision in pre-clinical models of human retinal degenerative diseases. Stem cell transplantation may therefore be an effective future approach to treat RPE diseases. The sources of cells used for retinal cell therapy include stem cells such as embryonic stem cells (ESCs), adult stem cells, and induced pluripotent stem cells (iPSCs). Currently, ESCs, and iPSCs are mainly used for differentiation into RPE, but these cell types still have some limitations including allogeneic rejection and carrying donor pathogenic genes. Stem cell transplantation is feasible for the treatment of retinal epithelial lesions (Zamiri et al., 2006). Indeed, allogeneic fetal retina-RPE transplants under the retina were not rejected in people with RP and advanced AMD (Enzmann et al., 1999). However, the immune privilege of the RPE is not absolute (Zamiri et al., 2004). In another study, patients with AMD undergoing CNV resection received subretinal allogeneic RPE transplantation, and there was immune rejection after immunosuppressive therapy was stopped (Tezel et al., 2007). The clinical application of cell therapy for retinal degenerative diseases faces some important challenges including cell manufacturing, delivery, survival, and physiological behavior; immune responses; and a risk of cancer development.
Gene and Drug Therapy
Thanks to many years of research into retinal diseases, many genes and signal transduction pathways have been identified as potential targets for gene therapy or other therapeutics. For example, ITH12674 is a melatonin and sulforaphane hybrid drug that induces expression of the transcription factor Nrf2, which can alleviate retinal degeneration leading to blindness (Campello et al., 2020). The lipid molecule ELV blocks the CB1 receptor and PLD2 in the eye to delay the development of degenerative and inflammatory retinal pathology (Bermudez et al., 2019). Emixustat is a non-retinal small molecule hydrochloride that acts as a highly efficient and selective visual cycle modulator targeting visual cycle isomerase. In the AMD animal model, emixustat can reduce A2E levels, protect the retina from light-mediated damage, and reduce neovascularization in premature retinopathy models (Kubota et al., 2020). Humanin (HNG), a 2.7 kDa 24 amino acid polypeptide, was discovered in a cDNA library derived from the brains of patients with familial Alzheimer’s disease. HNG protects primary RPE cells from oxidative damage (Nashine et al., 2017). Retinal cell mitochondria are severely damaged in AMD patients, and HNG is an important cell survival factor that can protect ARPE-19 RPE cell line mitochondria, making it an exciting target in AMD (Gong et al., 2018). In addition, as the first approved target for ophthalmological treatment, recombinant adeno-associated viruses (AAVs) have been used to deliver the RPE65 gene into RPE cells with mutations or absence of RPE65 to prevent and treat inherited retinal diseases, such as LCA2 and inherited retinal dystrophy (Acland et al., 2001; Russell et al., 2017).
Outlook
The RPE promotes normal retinal function and is an integral part of the retinal system, arising at the earliest stage of retinal development. As specialized phagocytes, RPE cells undertake the most intensive phagocytic task in the body through POS phagocytosis and maintaining the normal renewal and function of rod and cone cells. Its function in the blood-retinal barrier plays an important role in nutrient transport, maintenance of ion homeostasis, and the steady state of the microenvironment inside and outside the retina. The excessive accumulation of ROS and consequent retinopathy due to RPE dysfunction mean that these cells remain an important research focus. The visual circulation maintenance, barrier, substance transport, protection, and antioxidant functions of the RPE are all impaired to varying degrees in retinal degenerative diseases, highlighting the importance and necessity to protect the RPE and maintain or restore its function. Multiple mutations associated with AMD have been identified using sequencing techniques of different strategies. The genes in the complement system, such as complement C3 (C3), complement C9 (C9), complement factor I (CFI), and complement factor H (CFH), are associated with the development of AMD. When the membrane cofactor protein CD46 was knocked out, the mice developed lesions consistent with human dry AMD. In addition, mutations in the promoter of the HTRA1 (high-temperature requirement protein A1) gene lead to increased HTRA1 expression, resulting in the occurrence of AMD. FGD6 (FYVE, Rho GEF and PH domain-containing 6) gene mutation leads to increased expression of HTRA1 protein, which increases the risk of wet AMD (Lyzogubov et al., 2016; de Breuk et al., 2020).
At present, there is no effective and feasible method for the treatment of retinal degenerative diseases, although our increased understanding of the cellular biology and molecular genetics of retinal diseases might provide new avenues for prevention and treatment. Given the importance of the RPE in normal retinal function, this cell type provides an excellent focus for the development of treatments for retinopathy. In that regard, stem cell transplantation and gene therapy are currently hot research topics. While directing stem cell differentiation into RPE cells to restore function is an exciting and promising research direction, application to humans might be hampered by poor efficacy or immune reactions; further research is necessary. Encouragingly, as the study of the genes involved in the development of retinopathy and their signaling pathways increases, various inhibitors designed to target pathogenic genes and mutations are emerging, particularly for the treatment of RP. However, it still remains to be determined whether these targets are applicable to humans and whether modulation of these signaling pathways has off-target effects that might result in adverse reactions.
Author Contributions
SY wrote the manuscript and drew the figures. JZ revised the manuscript. DL conceived the study and edited the manuscript.
Funding
This work was supported by grants from National Natural Science Foundation of China (31771542).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Acland, G. M., Aguirre, G. D., Ray, J., Zhang, Q., Aleman, T. S., Cideciyan, A. V., et al. (2001). Gene Therapy Restores Vision in a Canine Model of Childhood Blindness. Nat. Genet. 28 (1), 92–95. doi:10.1038/ng0501-92
Aguirre, G. D., Baldwin, V., Pearce-Kelling, S., Narfström, K., Ray, K., and Acland, G. M. (1998). Congenital Stationary Night Blindness in the Dog: Common Mutation in the RPE65 Gene Indicates Founder Effect. Mol. Vis. 4, 23. Available at: https://www.ncbi.nlm.nih.gov/pubmed/9808841. (Accessed June 04, 2021).
Alcazar, O., Cousins, S. W., and Marin-Castano, M. E. (2007). MMP-14 and TIMP-2 Overexpression Protects Against Hydroquinone-Induced Oxidant Injury in RPE: Implications for Extracellular Matrix Turnover. Invest. Ophthalmol. Vis. Sci. 48 (12), 5662–5670. doi:10.1167/iovs.07-0392
Anderson, D. M. G., Ablonczy, Z., Koutalos, Y., Hanneken, A. M., Spraggins, J. M., Calcutt, M. W., et al. (2017). Bis(Monoacylglycero)Phosphate Lipids in the Retinal Pigment Epithelium Implicate Lysosomal/Endosomal Dysfunction in a Model of Stargardt Disease and Human Retinas. Sci. Rep. 7 (1), 17352. doi:10.1038/s41598-017-17402-1
Bazan, N. G. (2007). Homeostatic Regulation of Photoreceptor Cell Integrity: Significance of the Potent Mediator Neuroprotectin D1 Biosynthesized from Docosahexaenoic Acid the Proctor Lecture. Invest. Ophthalmol. Vis. Sci. 48 (11), 4866–4881. biography 4864-4865. doi:10.1167/iovs.07-0918
Bermúdez, V., Tenconi, P. E., Giusto, N. M., and Mateos, M. V. (2019). Lipid Signaling in Retinal Pigment Epithelium Cells Exposed to Inflammatory and Oxidative Stress Conditions. Molecular Mechanisms Underlying Degenerative Retinal DiseasesMolecular Mechanisms Underlying Degenerative Retinal Diseases. Adv. Exp. Med. Biol. 1185, 289–293. doi:10.1007/978-3-030-27378-1_47
Bharti, K., Liu, W., Csermely, T., Bertuzzi, S., and Arnheiter, H. (2008). Alternative Promoter Use in Eye Development: The Complex Role and Regulation of the Transcription Factor MITF. Development 135 (6), 1169–1178. doi:10.1242/dev.014142
Burstyn-Cohen, T., Lew, E. D., Través, P. G., Burrola, P. G., Hash, J. C., and Lemke, G. (2012). Genetic Dissection of TAM Receptor-Ligand Interaction in Retinal Pigment Epithelial Cell Phagocytosis. Neuron 76 (6), 1123–1132. doi:10.1016/j.neuron.2012.10.015
Buskin, A., Zhu, L., Chichagova, V., Basu, B., Mozaffari-Jovin, S., Dolan, D., et al. (2018). Disrupted Alternative Splicing for Genes Implicated in Splicing and Ciliogenesis Causes PRPF31 Retinitis Pigmentosa. Nat. Commun. 9 (1), 4234. doi:10.1038/s41467-018-06448-y
Caceres, P. S., and Rodriguez-Boulan, E. (2020). Retinal Pigment Epithelium Polarity in Health and Blinding Diseases. Curr. Opin. Cel Biol. 62, 37–45. doi:10.1016/j.ceb.2019.08.001
Campello, L., Kutsyr, O., Noailles, A., Michalska, P., Fernández-Sánchez, L., Martínez-Gil, N., et al. (2020). New Nrf2-Inducer Compound ITH12674 Slows the Progression of Retinitis Pigmentosa in the Mouse Model Rd10. Cell Physiol Biochem 54 (1), 142–159. doi:10.33594/000000210
Chen, M., Wang, J., Yang, Y., Zhong, T., Zhou, P., Ma, H., et al. (2021). Redox-dependent Regulation of End-Binding Protein 1 Activity by Glutathionylation. Sci. China Life Sci. 64 (4), 575–583. doi:10.1007/s11427-020-1765-6
Chong, C.-M., and Zheng, W. (2016). Artemisinin Protects Human Retinal Pigment Epithelial Cells from Hydrogen Peroxide-Induced Oxidative Damage through Activation of ERK/CREB Signaling. Redox Biol. 9, 50–56. doi:10.1016/j.redox.2016.06.002
de Breuk, A., Acar, I. E., Kersten, E., Schijvenaars, M. M. V. A. P., Colijn, J. M., Haer-Wigman, L., et al. (2020). Development of a Genotype Assay for Age-Related Macular Degeneration. Ophthalmology S0161-6420 (20), 30725–30729. doi:10.1016/j.ophtha.2020.07.037
Dunaief, J. L., Dentchev, T., Ying, G. S., and Milam, A. H. (2002). The Role of Apoptosis in Age-Related Macular Degeneration. Arch. Ophthalmol. 120 (11), 1435–1442. doi:10.1001/archopht.120.11.1435
Enzmann, V., Stadler, M., Wiedemann, P., and Kohen, L. (1999). Down-Regulation of MHC Class II Expression on Bovine Retinal Pigment Epithelial Cells by Cytokines. Ophthalmic Res. 31 (4), 256–266. doi:10.1159/000055545
Feher, J., Kovacs, I., Artico, M., Cavallotti, C., Papale, A., and Balacco Gabrieli, C. (2006). Mitochondrial Alterations of Retinal Pigment Epithelium in Age-Related Macular Degeneration. Neurobiol. Aging 27 (7), 983–993. doi:10.1016/j.neurobiolaging.2005.05.012
Golestaneh, N., Chu, Y., Xiao, Y.-Y., Stoleru, G. L., and Theos, A. C. (2017). Dysfunctional Autophagy in RPE, a Contributing Factor in Age-Related Macular Degeneration. Cell Death Dis 8 (1), e2537. doi:10.1038/cddis.2016.453
Gong, Z., Tasset, I., Diaz, A., Anguiano, J., Tas, E., Cui, L., et al. (2018). Humanin Is an Endogenous Activator of Chaperone-Mediated Autophagy. J. Cel Biol 217 (2), 635–647. doi:10.1083/jcb.201606095
Grossniklaus, H. E., Geisert, E. E., and Nickerson, J. M. (2015). Introduction to the Retina. Prog. Mol. Biol. Transl Sci. 134, 383–396. doi:10.1016/bs.pmbts.2015.06.001
Gu, S.-m., Thompson, D. A., Srikumari, C. R. S., Lorenz, B., Finckh, U., Nicoletti, A., et al. (1997). Mutations in RPE65 Cause Autosomal Recessive Childhood-Onset Severe Retinal Dystrophy. Nat. Genet. 17 (2), 194–197. doi:10.1038/ng1097-194
Hoon, M., Okawa, H., Della Santina, L., and Wong, R. O. L. (2014). Functional Architecture of the Retina: Development and Disease. Prog. Retin. Eye Res. 42, 44–84. doi:10.1016/j.preteyeres.2014.06.003
Jadeja, R. N., and Martin, P. M. (2020). Data on the Role of miR-144 in Regulating Fetal Hemoglobin Production in Retinal Pigmented Epithelial Cells. Data in Brief 28, 104874. doi:10.1016/j.dib.2019.104874
Kaur, G., Tan, L. X., Rathnasamy, G., La Cunza, N., Germer, C. J., Toops, K. A., et al. (2018). Aberrant Early Endosome Biogenesis Mediates Complement Activation in the Retinal Pigment Epithelium in Models of Macular Degeneration. Proc. Natl. Acad. Sci. USA 115 (36), 9014–9019. doi:10.1073/pnas.1805039115
Kubota, R., Gregory, J., Henry, S., and Mata, N. L. (2020). Pharmacotherapy for Metabolic and Cellular Stress in Degenerative Retinal Diseases. Drug Discov. Today 25 (2), 292–304. doi:10.1016/j.drudis.2019.11.013
Lin, H., Qian, J., Castillo, A. C., Long, B., Keyes, K. T., Chen, G., et al. (2011). Effect of miR-23 on Oxidant-Induced Injury in Human Retinal Pigment Epithelial Cells. Invest. Ophthalmol. Vis. Sci. 52 (9), 6308–6314. doi:10.1167/iovs.10-6632
Lyzogubov, V. V., Bora, P. S., Wu, X., Horn, L. E., de Roque, R., Rudolf, X. V., et al. (2016). The Complement Regulatory Protein CD46 Deficient Mouse Spontaneously Develops Dry-type Age-Related Macular Degeneration-Like Phenotype. Am. J. Pathol. 186 (8), 2088–2104. doi:10.1016/j.ajpath.2016.03.021
Ma, X., Li, H., Chen, Y., Yang, J., Chen, H., Arnheiter, H., et al. (2019). The Transcription Factor MITF in RPE Function and Dysfunction. Prog. Retin. Eye Res. 73, 100766. doi:10.1016/j.preteyeres.2019.06.002
Marlhens, F., Bareil, C., Griffoin, J.-M., Zrenner, E., Amalric, P., Eliaou, C., et al. (1997). Mutations in RPE65 Cause Leber's Congenital Amaurosis. Nat. Genet. 17 (2), 139–141. doi:10.1038/ng1097-139
Masland, R. H. (2012). The Neuronal Organization of the Retina. Neuron 76 (2), 266–280. doi:10.1016/j.neuron.2012.10.002
Mitter, S. K., Song, C., Qi, X., Mao, H., Rao, H., Akin, D., et al. (2014). Dysregulated Autophagy in the RPE Is Associated with Increased Susceptibility to Oxidative Stress and AMD. Autophagy 10 (11), 1989–2005. doi:10.4161/auto.36184
Nandrot, E. F., Kim, Y., Brodie, S. E., Huang, X., Sheppard, D., and Finnemann, S. C. (2004). Loss of Synchronized Retinal Phagocytosis and Age-Related Blindness in Mice Lacking αvβ5 Integrin. J. Exp. Med. 200 (12), 1539–1545. doi:10.1084/jem.20041447
Nashine, S., Cohen, P., Chwa, M., Lu, S., Nesburn, A. B., Kuppermann, B. D., et al. (2017). Humanin G (HNG) Protects Age-Related Macular Degeneration (AMD) Transmitochondrial ARPE-19 Cybrids from Mitochondrial and Cellular Damage. Cel Death Dis 8 (7), e2951. doi:10.1038/cddis.2017.348
Patel, A. K., and Hackam, A. S. (2013). Toll-like Receptor 3 (TLR3) Protects Retinal Pigmented Epithelium (RPE) Cells from Oxidative Stress through a STAT3-Dependent Mechanism. Mol. Immunol. 54 (2), 122–131. doi:10.1016/j.molimm.2012.11.005
Penberthy, K. K., Lysiak, J. J., and Ravichandran, K. S. (2018). Rethinking Phagocytes: Clues from the Retina and Testes. Trends Cel Biol. 28 (4), 317–327. doi:10.1016/j.tcb.2018.01.004
Prasad, D., Rothlin, C. V., Burrola, P., Burstyn-Cohen, T., Lu, Q., Garcia de Frutos, P., et al. (2006). TAM Receptor Function in the Retinal Pigment Epithelium. Mol. Cell Neurosci. 33 (1), 96–108. doi:10.1016/j.mcn.2006.06.011
Ran, J., Li, H., Zhang, Y., Yu, F., Yang, Y., Nie, C., et al. (2021). A Non-Mitotic Role for Eg5 in Regulating Cilium Formation and Sonic Hedgehog Signaling. Sci. Bull. doi:10.1016/j.scib.2021.02.001
Ran, J., Liu, M., Feng, J., Li, H., Ma, H., Song, T., et al. (2020). ASK1-Mediated Phosphorylation Blocks HDAC6 Ubiquitination and Degradation to Drive the Disassembly of Photoreceptor Connecting Cilia. Develop. Cel 53 (3), 287–299. doi:10.1016/j.devcel.2020.03.010
Ran, J., and Zhou, J. (2020). Targeting the Photoreceptor Cilium for the Treatment of Retinal Diseases. Acta Pharmacol. Sin 41 (11), 1410–1415. doi:10.1038/s41401-020-0486-3
Russell, S., Bennett, J., Wellman, J. A., Chung, D. C., Yu, Z.-F., Tillman, A., et al. (2017). Efficacy and Safety of Voretigene Neparvovec (AAV2-hRPE65v2) in Patients with RPE65 -Mediated Inherited Retinal Dystrophy: A Randomised, Controlled, Open-Label, Phase 3 Trial. The Lancet 390 (10097), 849–860. doi:10.1016/S0140-6736(17)31868-8
Silverman, S. M., and Wong, W. T. (2018). Microglia in the Retina: Roles in Development, Maturity, and Disease. Annu. Rev. Vis. Sci. 4, 45–77. doi:10.1146/annurev-vision-091517-034425
Song, T., and Zhou, J. (2020). Primary Cilia in Corneal Development and Disease. Zool Res. 41 (5), 495–502. doi:10.24272/j.issn.2095-8137.2020.109
Storti, F., Raphael, G., Griesser, V., Klee, K., Drawnel, F., Willburger, C., et al. (2017). Regulated Efflux of Photoreceptor Outer Segment-Derived Cholesterol by Human RPE Cells. Exp. Eye Res. 165, 65–77. doi:10.1016/j.exer.2017.09.008
Sugasawa, K., Deguchi, J., Okami, T., Yamamoto, A., Omori, K., Uyama, M., et al. (1994). Immunocytochemical Analyses of Distributions of Na, K-ATPase and GLUT1, Insulin and Transferrin Receptors in the Developing Retinal Pigment Epithelial Cells. Cell Struct. Funct. 19 (1), 21–28. doi:10.1247/csf.19.21
Tezel, T. H., Del Priore, L. V., Berger, A. S., and Kaplan, H. J. (2007). Adult Retinal Pigment Epithelial Transplantation in Exudative Age-Related Macular Degeneration. Am. J. Ophthalmol. 143 (4), 584–595. doi:10.1016/j.ajo.2006.12.007
Tian, X., Cui, Z., Liu, S., Zhou, J., and Cui, R. (2021). Melanosome Transport and Regulation in Development and Disease. Pharmacol. Ther. 219, 107707. doi:10.1016/j.pharmthera.2020.107707
Xie, W., Li, D., Dong, D., Li, Y., Zhang, Y., Duan, L., et al. (2020). HIV-1 Exposure Triggers Autophagic Degradation of Stathmin and Hyperstabilization of Microtubules to Disrupt Epithelial Cell Junctions. Sig Transduct Target. Ther. 5 (1), 79. doi:10.1038/s41392-020-0175-1
Yam, M., Engel, A. L., Wang, Y., Zhu, S., Hauer, A., Zhang, R., et al. (2019). Proline Mediates Metabolic Communication Between Retinal Pigment Epithelial Cells and the Retina. J. Biol. Chem. 294 (26), 10278–10289. doi:10.1074/jbc.RA119.007983
Yang, Y., Chen, M., Li, J., Hong, R., Yang, J., Yu, F., et al. (2021). A Cilium-Independent Role for Intraflagellar Transport 88 in Regulating Angiogenesis. Sci. Bull. 66 (7), 727–739. doi:10.1016/j.scib.2020.10.014
Young, R. W. (1967). The Renewal of Photoreceptor Cell Outer Segments. J. Cel Biol 33 (1), 61–72. doi:10.1083/jcb.33.1.61
Young, R. W. (1971). The Renewal of Rod and Cone Outer Segments in the Rhesus Monkey. J. Cel Biol 49 (2), 303–318. doi:10.1083/jcb.49.2.303
Yu, C., Muñoz, L. E., Mallavarapu, M., Herrmann, M., and Finnemann, S. C. (2019a). Annexin A5 Regulates Surface αvβ5 Integrin for Retinal Clearance Phagocytosis. J. Cel Sci 132 (20), jcs232439. doi:10.1242/jcs.232439
Yu, F., Guo, S., Li, T., Ran, J., Zhao, W., Li, D., et al. (2019b). Ciliary Defects Caused by Dysregulation of O-GlcNAc Modification are Associated with Diabetic Complications. Cell Res 29 (2), 171–173. doi:10.1038/s41422-018-0114-7
Yu, F., Li, T., Sui, Y., Chen, Q., Yang, S., Yang, J., et al. (2020). O-GlcNAc Transferase Regulates Centriole Behavior and Intraflagellar Transport to Promote Ciliogenesis. Protein Cell 11 (11), 852–857. doi:10.1007/s13238-020-00746-2
Zamiri, P., Masli, S., Streilein, J. W., and Taylor, A. W. (2006). Pigment Epithelial Growth Factor Suppresses Inflammation by Modulating Macrophage Activation. Invest. Ophthalmol. Vis. Sci. 47 (9), 3912–3918. doi:10.1167/iovs.05-1267
Zamiri, P., Zhang, Q., and Streilein, J. W. (2004). Vulnerability of Allogeneic Retinal Pigment Epithelium to Immune T-Cell-Mediated Damage In Vivo and In Vitro. Invest. Ophthalmol. Vis. Sci. 45 (1), 177–184. doi:10.1167/iovs.03-0211
Zarbin, M. (2016). Cell-Based Therapy for Degenerative Retinal Disease. Trends Mol. Med. 22 (2), 115–134. doi:10.1016/j.molmed.2015.12.007
Keywords: retina, retinal pigment epithelium, development, function, disease, retinopathy, therapy
Citation: Yang S, Zhou J and Li D (2021) Functions and Diseases of the Retinal Pigment Epithelium. Front. Pharmacol. 12:727870. doi: 10.3389/fphar.2021.727870
Received: 20 June 2021; Accepted: 19 July 2021;
Published: 28 July 2021.
Edited by:
Wenru Su, Sun Yat-sen University, ChinaReviewed by:
Xianjun Zhu, University of Electronic Science and Technology of China, ChinaKai Yao, Wuhan University of Science and Technology, China
Copyright © 2021 Yang, Zhou and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dengwen Li, ZHdsaUBuYW5rYWkuZWR1LmNu