 Yi-Yuan Ge1
Yi-Yuan Ge1 Li-Ye Yang
Li-Ye Yang- 1Guangzhou Hybribio Medicine Science and Technology Corporation, Guangzhou, Guangdong Province, China
- 2Precision Medical Lab Center, People’s Hospital of Yangjiang, Yangjiang, Guangdong Province, China
Objective: This study aimed to develop and assess a novel reverse dot blot assay for the simultaneous detection of 10 types of α-thalassemia alleles in the Chinese population, including six common variants of–SEA, -α3.7, -α4.2, αCS, αQS, and αWS, and four rare variants of αααanti−4.2, αααanti−3.7, --FIL deletion and--THAI deletion.
Methods: The novel thalassemia gene assay utilized a two-tier multiplex polymerase chain reaction amplification system and one round of hybridization. Genomic DNA samples were sourced from three hospitals in southern China. Each clinically validated DNA sample was re-evaluated using the new multiplex polymerase chain reaction/reverse dot blot assay Ⅲ (M-PCR/RDB Ⅲ).
Results: The study analyzed a total of 1,148 unrelated participants, consisting of 810 thalassemia patients and 338 healthy control subjects. Valid hybridization results were obtained for 1,147 samples, with one case (thalassemia carrier) being excluded from the study due to the poor quality of DNA. All 1,147 samples, including those with α heterozygous thalassemia, α homozygous thalassemia, α compound heterozygous thalassemia, and control subjects were accurately genotyped, showing 100% concordance with the reference assays.
Conclusion: The novel M-PCR/RDB Ⅲ assay proved to be simple, rapid, and precise, indicating its potential for genetic screening and clinical diagnosis of both common and rare α-thalassemia variants in Chinese populations.
1 Introduction
Thalassemia syndrome stands as the most prevalent single gene mutation disorder among humans (Weatherall.,1997; Taher et al., 2018). It encompasses two primary forms, namely α-thalassemia and β-thalassemia. β-thalassemia and α-thalassemia are inherited blood disorders characterized by reduced production of hemoglobin, the protein in red blood cells that carries oxygen throughout the body. β-thalassemia results from mutations in the HBB gene affecting β-globin, while α-thalassemia is caused by deletions or mutations in the HBA1 and HBA2 genes affecting α-globin, resulting in diminished or absent synthesis of the α-globin chain of hemoglobin (Hb) (Chen et al.,2002; Muncie and Campbell, 2009). These conditions manifest in varying degrees of anemia, with clinical severity ranging from mild to life-threatening. Understanding the genetic basis, types, clinical manifestations, diagnostic methods, and treatment options for both beta-thalassemia and alpha-thalassemia is crucial for effective management and care.
The prevalence of thalassemia is notably high in tropical and subtropical regions such as the Mediterranean basin, Africa, the Middle East, the Indian subcontinent, and Southeast Asia (Weatherall and Clegg, 2001; Viprakasit and Ekwattanakit,2018). Previous research has highlighted a marked incidence of thalassemia in southern China, particularly in the provinces of Guangxi, Guangdong, and Hainan (Xu et al.,2004; Xiong et al.,2010; Wang et al.,2023). In southern China, the--SEA deletion ranks as the most prevalent mutation in α-thalassemia, followed by -α3.7 and -α4.2 (Xu et al.,2004; Xiong et al.,2010; Lin, M et al.,2013; Liang et al.,2023).
Various techniques, including reverse dot blot and gap-polymerase chain reaction (gap-PCR), have been utilized in thalassemia screening (Xu et al.,2004; Xiong et al.,2010; Lin et al.,2013; Liang et al., 2023). Yet, these methods are known for their high costs and labor-intensive procedures (Lin et al., 2013; Liang et al.,2023). Our prior studies introduced two multiplex PCR/reverse dot blot assays (M-PCR/RDB I and M-PCR/RDB II) that have become widely adopted in clinical thalassemia diagnosis in southern China (Lin et al., 2012; Liang et al., 2022). Despite their effectiveness, these assays were unable to distinguish relatively rare α variants, such as αααanti−4.2, αααanti−3.7, --FIL deletion, and--THAI deletion.
This investigation presents a novel α-thalassemia genetic assay that employs a two-tier multiplex polymerase chain reaction (PCR) amplification system and a single round of hybridization. Additionally, the incorporation of αααanti−4.2, αααanti−3.7, --FIL deletion, and--THAI deletion enhances the assay’s capabilities. Developed to concurrently identify 10 types of α-thalassemia mutations, the updated M-PCR/RDB III assay showcases enhanced effectiveness and diagnostic accuracy. This research delineates the evolution and diagnostic efficacy of the M-PCR/RDB III assay in patients originating from southern China.
2 Patients and methods
2.1 Study population
This study collected samples from patients with thalassemia and normal controls who had previously been genotyped using the reference methods (described in the Reference Methods) between April 2021 and March 2024 from the People’s Hospital of Guangxi Zhuang Autonomous Region, Zhujiang Hospital of Southern Medical University, and Xiangya Reproductive and Genetic Hospital of Central South University. Approval for this study was obtained from the Ethics Committees of the three hospitals, with the approval numbers 2020-51, 2021-SJ-001-04, and SJ2023002, respectively. The patients with thalassemia were deliberately chosen, while the healthy controls were randomly selected from routine check-up volunteers. These samples were used to evaluate the specificity and accuracy of the newly developed assay in a double-blind manner. Written consent was obtained from the patients or their guardians, as well as from the healthy volunteers, for this investigation.
The study also received approval from the Ethics Committees of People’s Hospital of Yangjiang (No.2023003). The study adheres to the STROBE guidelines (von Elm et al., 2007). Patient information was de-identified to ensure that no patient’s identity could be disclosed in any manner.
2.2 DNA extraction
Genomic DNA extracted from peripheral blood leukocytes of the study participants was obtained using a DNA Prep Kit (Guangdong Hybribio Limited Corporation, Chaozhou, Guangdong Province, China). The DNA concentration was assessed using a NanoDrop™ One/One C Microvolume UV-Vis Spectrophotometer (Thermo Fisher Scientific, Rockford, IL, United States) at a wavelength of 260 nm, with DNA purity evaluated based on the 260/280 nm ratio. These DNA samples were utilized for subsequent PCR analysis. The clinical test (reference methods) and validation test (M-PCR/RDB III) were conducted in the three aforementioned hospitals.
2.3 Design of primers and probes
The thalassemia detection kit was designed and made by Guangzhou Hybribio Medicine Science and Technology Corporation. The detection kit included two PCR reaction systems as follows. Three sets of primers of the M-PCR assay were designed to amplify three α-thalassemia deletions (the Southeast Asian [--SEA], the rightward deletion (-α3.7) and the leftward deletion (-α4.2)] on chromosome 16; Three sets of primers of the M-PCR assay were designed to amplify the three common α-globin gene mutations: Hb Constant Spring [Hb CS (αCSα) HBA2: c.427T > C], Hb Quong Sze [Hb QS (αQSα), HBA2: c.377T > C], Hb Westmead [Hb WS (αWSα), HBA2: c.369C > G]; One 1800 base pair (bp) fragment of α-2 globin gene was amplified as a normal control (marked NP on the hybridization membrane). Four sets of primers of the M-PCR assay were designed to amplify four rare variants αααanti−4.2, αααanti−3.7, --FIL deletion and--THAI deletion, respectively. Oligo 6.31 (Molecular Biology Insights, Colorado Springs, CO, United States) software was used to design the primers and probes. Schematic representation of the ten types of α-thalassemia allele location in the α-gene cluster is shown in Figure 1. All probes were immobilized on a nylon membrane. Their localization in the membrane is shown in Figure 2I. The detailed information of the primers and probes are presented in Supplementary Tables S1, S2.
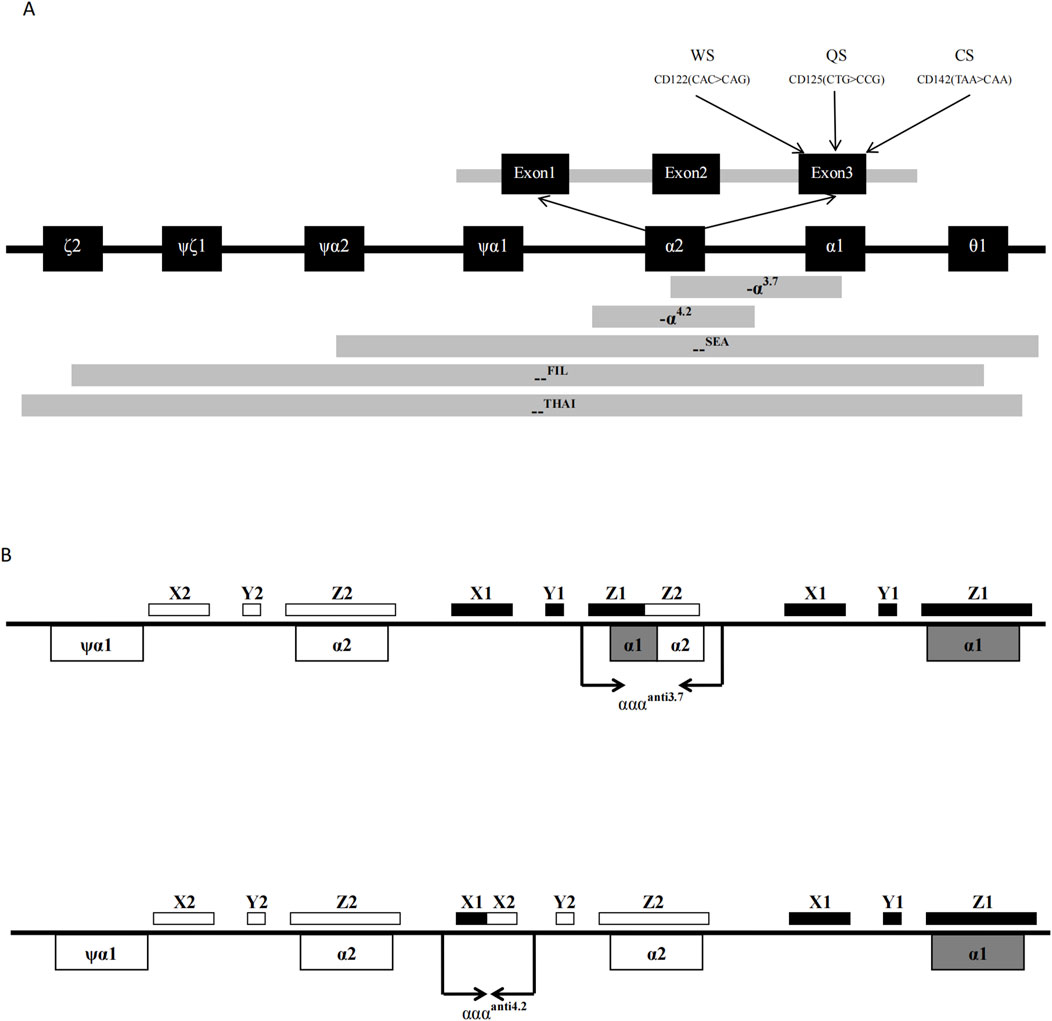
Figure 1. Schematic representation of the 6 kinds of common α-thalassemia allele (A) and 4 kinds of rare α-thalassemia allele; (B) location in the α-gene cluster.
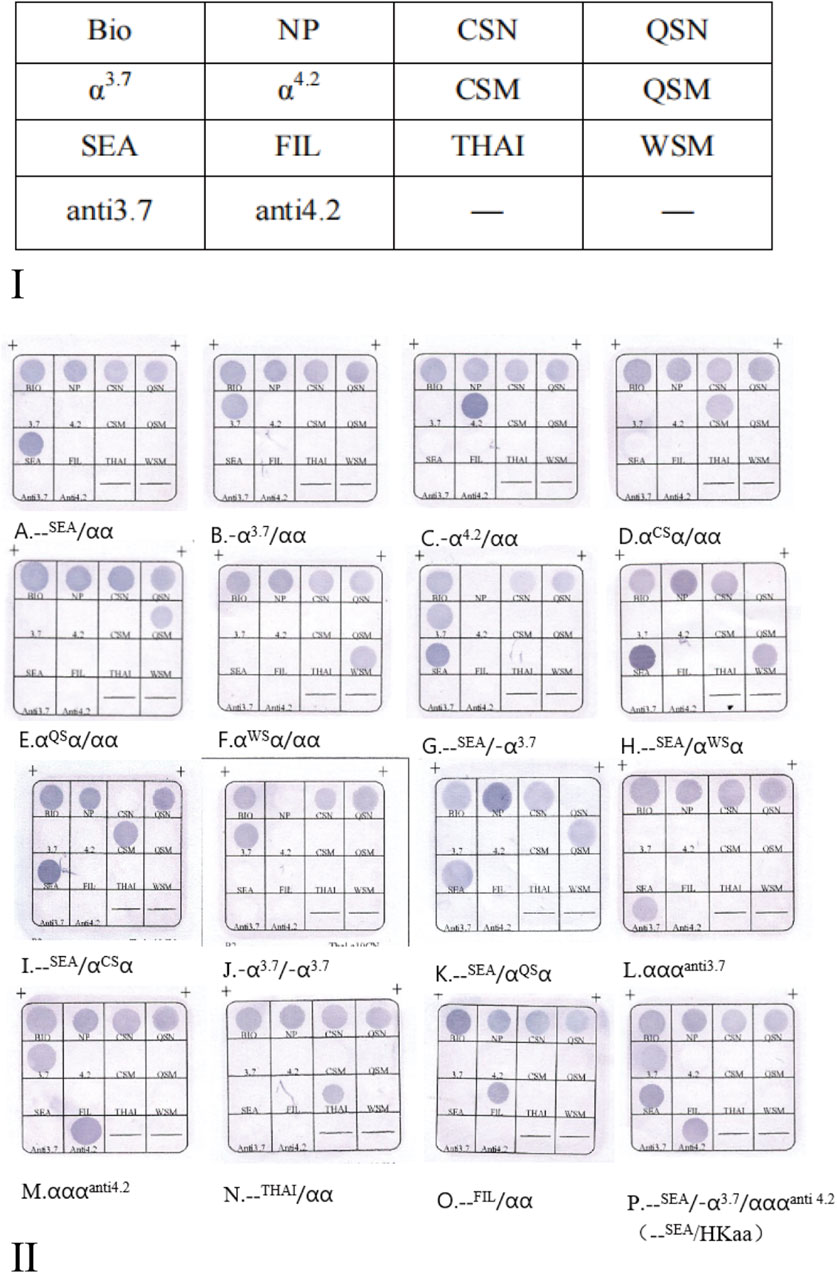
Figure 2. (Ⅰ) The probes location in the gene chip. Note: N, Normal control; M, Mutation. NP indicates a fragment for α2 gene, as the control for --SEA, -α3.7, and -α4.2, --FIL and--THAI; QSN is the control for QSM and WSM, CSN is the control for CSM, respectively. αααanti3.7 (referred to as anti3.7) and αααanti4.2 (referred to as anti4.2) have no normal controls; Bio refers to biotin points used to monitor the hybridization process. (Ⅱ) Hybridization results of M-PCR/RDB Ⅲ assay. (A. --SEA/αα; B. -α3.7/αα; C. -α4.2/αα; D. αCSα/αα; E. αQSα/αα; F. αWSα/αα; G. --SEA/-α3.7; H. --SEA/αWSα; I. --SEA/αCSα; J. -α3.7/-α3.7; K. --SEA/αQSα; L.αα/αααanti3.7; M. αα/αααanti4.2; N. --Thai/αα; O. --Fil/αα; P. –SEA/-α3.7αααanti4.2 (HKαα/--SEA).
2.4 Multiplex PCR amplification
The assay was conducted following the manufacturer’s protocol (Guangzhou Hybribio Medicine Science and Technology Corporation, Guangzhou, China). In brief, α-thalassemia PCRs were amplified in two tubes. The A group included the detected genotypes: --SEA, -α3.7, -α4.2, αCS, αQS, αWS; while the B group included the detected genotypes: --FIL, --THAI, αααanti3.7, and αααanti4.2. PCRs were carried out using a PCR appliance (GeneTouch; BIOER, Hangzhou, China), with a reaction volume of 50 μL comprising 6 μL of DNA template, 0.5 μL DNA polymerase, and 43.5 μL PCR MIX (100 μmol/L Primer MIX: 1.25 μL, 25 mmol/L MgCl2: 3 μL, 10 × PCR buffer: 5 μL, 5 × PCR enhancer: 7 μL, 25 mmol/L dNTPs: 0.8 μL, H2O: 26.45 μL). The cycling program included an initial denaturation at 95°C for 15 min, followed by 35 cycles consisting of denaturation at 98°C for 40 s, annealing at 64°C for 70 s, elongation at 72°C for 150 s, and a final elongation step at 72°C for 5 min. Subsequently, the amplicons from the reaction system (two tubes) were denatured and subjected to hybridization.
2.5 Flow-through hybridization
Hybridization reactions were performed using α thalassemia gene diagnostic kit and flow-through hybridization kit (Guangzhou Hybribio Medicine Science and Technology Corporation, Guangzhou, China), the reaction and hybridization condition was the same as described previously (Lin et al., 2012; Liang et al., 2022). The assay utilized flow-through hybridization technology (HB2012A; Guangdong Hybribio Limited Corporation). After hybridization, a blue-purple precipitate at the probe dot could be discerned. The results were interpreted by direct visualization.
2.6 Reference methods
For the reference methods, all thalassemia alleles were characterized using a combination of techniques, including M-PCR/RDB Ⅱ,M-PCR/RDB I, Sanger’s sequencing, and a two-round nested PCR strategy. The analysis involved three types of α-thalassemia deletions (--SEA, -α3.7, and -α4.2) and three types of α-thalassemia mutations (Hb CS, Hb WS, and Hb QS) using M-PCR/RDB I or M-PCR/RDB II (Guangdong Hybribio Limited Corporation) (Lin et al., 2012; Liang et al., 2022). Additionally, a two-round nested PCR strategy was implemented to detect the potential presence of HK αα, a rare α-thalassemia mutation reported in southern Chinese subjects (Wu et al., 2015). The--FIL deletion and--THAI deletion were identified using gap-PCR, as previously described (Eng et al., 2000). Furthermore, the testing for αααanti−3.7 and αααanti−4.2 triplications followed established protocols (Wang et al., 2003). Finally, --FIL deletion, --THAI deletion, αααanti−3.7, and αααanti−4.2 were subsequently validated through Sanger sequencing.
2.7 Statistical analyses
All statistical analyses were conducted using the SPSS® statistical package, version 16.0 (SPSS Inc., Chicago, IL, United States), on Windows®. The agreement between M-PCR/RDB III and the reference methods was assessed using kappa statistics and McNemar’s χ -test. Statistical significance was defined as a P-value <0.05.
3 Results
This study collected samples from 1,148 unrelated participants, including 810 patients with thalassemia and 338 healthy control subjects as follows: People’s Hospital of Guangxi Zhuang Autonomous Region (380 cases), Zhujiang Hospital of Southern Medical University (385 cases), and Xiangya Reproductive and Genetic Hospital of Central South University (383 cases). The detection panel included common--SEA, -α3.7, -α4.2, αCS, αQS, αWS, and rare variants αααanti−4.2, αααanti−3.7, --FIL deletion, and--THAI deletion in Chinese.
In a verification test, a total of 1,148 genomic DNA pre-characterized samples (810 patients with thalassemia and 338 healthy control subjects) were analyzed with the M-PCR/RDB Ⅲ assay. Valid hybridization results were obtained for 1,147 samples, with one case (thalassemia carrier) being excluded from the study due to the poor quality of DNA. The new kit successfully diagnosed 809 patients with thalassemia and 338 healthy control subjects. The hybridization results of the M-PCR/RDB Ⅲ assay are presented in Supplementary Table S3, which included α heterozygous thalassemia, α homozygous thalassemia and α compound heterozygous thalassemia (Figure 2Ⅱ).
The agreement between the M-PCR/RDB Ⅲ assay and the reference methods was found to be in absolute concordance (kappa = 1, P < 0.001) for detecting the three α-globin deletions (--SEA, -α3.7, -α4.2) (Figure 2ⅡA–C) and three α-globin mutations (αCS, αQS, αWS) (Figure 2ⅡD–F). Additionally, rare variants including αααanti 3.7 (n = 25) (Figure 2ⅡL), αααanti 4.2 (n = 24) (Figure 2ⅡM), --THAI (n = 12) (Figure 2ⅡN), and--FIL (n = 4) (Figure 2ⅡO) were successfully identified using the M-PCR/RDB Ⅲ assay with 100% concordance with the reference methods (Supplementary Table S3; Figure 2Ⅱ).
The current study could also identify compound combinations such as--SEA/-α3.7(Figure 2ⅡG), --SEA/αWSα (Figure 2ⅡH), --SEA/αCSα (Figure 2ⅡI), -α3.7/-α3.7(Figure 2ⅡJ), --SEA/αQSα (Figure 2ⅡK), -α3.7/αααanti 4.2 (n = 7), --SEA/-α3.7αααanti4.2 (HKαα/--SEA, n = 2) (Figure 2ⅡP), -α3.7/αααanti3.7(n = 1), -α3.7/αCSα/αααanti 4.2(n = 1), and--SEA/αααanti 3.7(n = 1).
For cases with--SEA/-α3.7αααanti4.2 (Figure 2ⅡP) genotypes, where one chromosome has been deleted (--SEA deletion), the -α3.7 and αααanti4.2 alleles are expected to be on the same chromosome. We can confirm that the genotype is HKαα/--SEA.
4 Discussion
Thalassemia is a prevalent genetic disease in southern China, significantly impacting public health in these areas with high prevalence. While a screening test involving a complete blood count, hemoglobin quantification via capillary electrophoresis and/or high performance liquid chromatography, may not identify all thalassemia subtypes, laboratory diagnosis necessitates molecular analysis. With advancements in molecular diagnostics, genetic diagnosis of thalassemia can be easily achieved through gap-PCR and PCR-RDB of the affected globin genes. These methods have demonstrated precision and sensitivity in identifying thalassemia genotypes, leading to their widespread adoption in clinical settings for the molecular diagnosis of thalassemia (Lin et al., 2012; Liang et al., 2022; Wang et al., 2003). However, despite their efficacy, these assays were unable to distinguish relatively rare α variants, such as anti−4.2, anti−3.7, --FIL deletion, and--THAI deletion (Lin et al., 2012; Liang et al., 2022).
The enhanced M-PCR/RDB Ⅲ, as described, offers a more comprehensive coverage compared to the previously developed M-PCR/RDB I/M-PCR/RDB Ⅱ and other existing clinical methods. It successfully detects rare variants such as anti−4.2, anti−3.7, --FIL deletion, and--THAI deletion. Typically, α-globin gene deletions are diagnosed using gap-PCR, which can be time-consuming due to post-PCR work (electrophoresis). The improved M-PCR/RDB Ⅲ simplifies and expands the analysis panel.
Our new kit has the capability to identify intricate and rare α variant combinations, including -α3.7/αααanti−4.2, --SEA/-α3.7αααanti−4.2, -α3.7/αααanti−4.2, --SEA/αααanti−3.7, and -α3.7/αCSαααanti−4.2. However, it is noted that the new M-PCR/RDB Ⅲ assay does have a specific limitation. A rare α-thalassemia variant, the HKαα genotype (-α3.7/αααanti−4.2), previously reported in China (Wu et al., 2015), can only be detected when it is concurrent with--SEA (as seen in an individual with the--SEA/-α3.7αααanti−4.2 genotype). In cases where an individual is identified with -α3.7 and αααanti−4.2 using the M-PCR/RDB Ⅲ, certainty regarding their location on the same allele (HKαα) cannot be established.
Hong Kong type α-thalassemia (HKαα) is a recombinant gene formed by non-homologous recombination of the α-globin gene, resulting in a gene that contains both the -α3.7 and αααanti4.2 segments, with two functional α-globin genes. Individuals with HKαα/--SEA, due to the presence of a functional α2 gene, have a hematological phenotype similar to that of--SEA heterozygotes, indicating a mild form of α-thalassemia (Wu et al., 2015). This suggests that if one parent is HKαα/αα and the other is a--SEA heterozygote, prenatal diagnosis is not necessary. However, if a mistaken diagnosis labels HKαα/αα as a -α3.7 heterozygote and the other parent is a--SEA heterozygote, there is a 25% chance of the offspring having Hb H (intermediate α-thalassemia), prompting the need for informed consent and allowing the pregnant woman to choose whether to undergo prenatal diagnosis. This increases the difficulty of clinical genetic counseling and unnecessarily adds anxiety to the pregnant woman and her family. Therefore, accurate screening and differentiation between -α3.7 and HKαα have important clinical significance. A previous study conducted HKαα genotype diagnosis on 507 samples, identified as -α3.7/αα through Gap-PCR gene testing. Subsequent nested PCR analysis revealed that 7.27% of the samples had the HKαα genotype (Zhang et al., 2019). In another study on Chinese carriers of silent deletional α-thalassemia, the frequencies of the HKαα and anti-HKαα alleles were 2.27% and 0.35% in -α3.7 and -α4.2 carriers, respectively (Zhong et al., 2018).
The α-globin triplication is the result of unequal exchange of homologous sequences in the α-globin gene cluster of chromosome 16 during meiotic pairing. The homologous region of the α-globin gene cluster includes X, Y, and Z boxes, with two Z homologous boxes spaced 3.7 kb apart and two X homologous boxes spaced 4.2 kb apart. Recombination at the Z homologous boxes can lead to a 3.7 kb deletion on one chromosome (-α3.7), forming the α-globin triplication (αααanti3.7) on the other chromosome. Recombination at the X homologous boxes can result in a 4.2 kb deletion (-α4.2) and another type of α-globin triplication (αααanti4.2) (Wang et al., 2003).
There are relevant literature reports on the prevalence of α-globin triplication in populations both domestically and internationally. In 2018, Iranian scholar Seyedeh (Abedini et al., 2018) conducted α-globin triplication detection on 4,010 individuals from different provinces, revealing a carrier rate of 1.7% in the population. In China, a study on 1,169 newborn umbilical cord blood samples suggested a carrier rate of 1.2% for two types of α-globin triplication (Xie et al., 2015). Research data from 20,222 individuals across five southern provinces of China conducted in 2017 revealed a high carrier rate of α-globin triplication at 1.67% (Shang et al., 2017).
In terms of clinical phenotype and hematological changes, most current views still suggest that the phenotype becomes more severe after the compound of β-thalassemia mutations with α-globin triplication. This is mainly due to the increased copy number of α-globin genes leading to an imbalance in the ratio of α-globin peptide chains to β-globin peptide chains, resulting in moderate to severe anemia, skin pallor, and hepatosplenomegaly, as well as hematological characteristic changes in most thalassemia patients (Moosavi et al., 2011), some of whom require intermittent blood transfusions for treatment.
In this scenario, our M-PCR/RDB III assay was especially beneficial for a couple where one partner carries a β-thalassemia mutation, necessitating α-globin triplication analysis for the other individual. If α-globin triplication is confirmed in the latter, they should be notified, and if desired, prenatal diagnosis could be pursued. In conclusion, this enhanced multiplex-PCR-based RDB assay has the potential to greatly enhance the screening panel for both common and rare genotypes of α-thalassemia in southern China.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the People’s Hospital of Yangjiang (No. 2023003), the Ethics Committee of Peoples’ Hospital of Guangxi Zhuang Autonomous Region (No. 2020-51), the Ethics Committee of Zhujiang Hospital of Southern Medical University (No. 2021-SJ-001-04), and the Ethics Committee of Xiangya Reproductive and Genetic Hospital of Central South University (No. SJ2023002). The studies were conducted in accordance with the local legislation and institutional requirements.
Author contributions
Y-YG: Investigation, Methodology, Resources, Supervision, Validation, Writing–review and editing. JX: Investigation, Methodology, Resources, Software, Validation, Writing–review and editing. Y-WL: Methodology, Writing–review and editing. L-XX: Supervision, Writing–review and editing.L-YY: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the High-level Key Medical and Health Research Project of Yangjiang (No.2023001). The funders had no role in the study design and data analysis, decision to publish or submission of the manuscript.
Conflict of interest
Authors Y-YG, JX, and L-XX were employed by Guangzhou Hybribio Medicine Science and Technology Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1457248/full#supplementary-material
References
Abedini, S. S., Forouzesh Pour, F., Karimi, K., Ghaderi, Z., Farashi, S., Tavakoli Koudehi, A., et al. (2018). Frequency of α-globin gene triplications and coinheritance with β-globin gene mutations in the Iranian population. Hemoglobin 42 (4), 252–256. doi:10.1080/03630269.2018.1526192
Chen, T. P., Liu, T. C., Chang, C. S., Chang, J. G., Tsai, H. J., and Lin, S. F. (2002). PCR-based analysis of alpha-thalassemia in Southern Taiwan. Int. J. Hematol. 75 (3), 277–280. doi:10.1007/BF02982041
Eng, B., Patterson, M., Borys, S., Chui, D. H., and Waye, J. S. (2000). PCR-based diagnosis of the Filipino (--(FIL)) and Thai (--(Thai)) alpha-thalassemia-1 deletions. Am. J. Hematol. 63 (1), 54–56. doi:10.1002/(sici)1096-8652(200001)63:1<54::aid-ajh12>3.0.co;2-b
Liang, H. F., Li, L. J., Yang, H., Zheng, X. B., Lu, M., Ge, Y. Y., et al. (2022). Clinical validation of a single-tube PCR and reverse dot blot assay for detection of common α-thalassaemia and β-thalassaemia in Chinese. J. Int. Med. Res. 50 (2), 3000605221078785. doi:10.1177/03000605221078785
Liang, H. F., Liang, W. M., Xie, W. G., Lin, F., Liu, L. L., Li, L. J., et al. (2023). The gene spectrum of thalassemia in Yangjiang of western Guangdong Province. Front. Genet. 14, 1126099. doi:10.3389/fgene.2023.1126099
Lin, M., Wen, Y. F., Wu, J. R., Wang, Q., Zheng, L., Liu, G. R., et al. (2013). Hemoglobinopathy: molecular epidemiological characteristics and health effects on Hakka people in the Meizhou region, southern China. PloS one 8 (2), e55024. doi:10.1371/journal.pone.0055024
Lin, M., Zhu, J. J., Wang, Q., Xie, L. X., Lu, M., Wang, J. L., et al. (2012). Development and evaluation of a reverse dot blot assay for the simultaneous detection of common alpha and beta thalassemia in Chinese. Blood Cells Mol. Dis. 48 (2), 86–90. doi:10.1016/j.bcmd.2011.12.001
Moosavi, S. F., Amirian, A., Zarbakhsh, B., Kordafshari, A., Mirzahoseini, H., Zeinali, S., et al. (2011). The carrier frequency of α-globin gene triplication in an Iranian population with normal or borderline hematological parameters. Hemoglobin 35 (4), 323–330. doi:10.3109/03630269.2011.571527
Muncie, H. L., and Campbell, J. (2009). Alpha and beta thalassemia. Am. Fam. Physician 80 (4), 339–344.
Shang, X., Peng, Z., Ye, Y., Zhang, X., Chen, Y., Zhu, B., et al. (2017). Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine 23, 150–159. doi:10.1016/j.ebiom.2017.08.015
Taher, A. T., Weatherall, D. J., and Cappellini, M. D. (2018). Thalassaemia. Lancet 391 (10116), 155–167. doi:10.1016/S0140-6736(17)31822-6
Viprakasit, V., and Ekwattanakit, S. (2018). Clinical classification, screening and diagnosis for thalassemia. Hematol. Oncol. Clin. North Am. 32 (2), 193–211. doi:10.1016/j.hoc.2017.11.006
von Elm, E., Altman, D. G., Egger, M., Pocock, S. J., Gøtzsche, P. C., and Vandenbroucke, J. P.STROBE Initiative (2007). The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet 370 (9596), 1453–1457. doi:10.1016/S0140-6736(07)61602-X
Wang, W., Ma, E. S., Chan, A. Y., Prior, J., Erber, W. N., Chan, L. C., et al. (2003). Single-tube multiplex-PCR screen for anti-3.7 and anti-4.2 alpha-globin gene triplications. Clin. Chem. 49 (10), 1679–1682. doi:10.1373/49.10.1679
Wang, W. D., Hu, F., Zhou, D. H., Gale, R. P., Lai, Y. R., Yao, H. X., et al. (2023). Thalassaemia in China. Blood Rev. 60, 101074. doi:10.1016/j.blre.2023.101074
Weatherall, D. J. (1997). The thalassaemias. BMJ Clin. Res. ed. 314 (7095), 1675–1678. doi:10.1136/bmj.314.7095.1675
Weatherall, D. J., and Clegg, J. B. (2001). Inherited haemoglobin disorders: an increasing global health problem. Bull. World Health Organ. 79 (8), 704–712.
Wu, M. Y., Li, J., Li, S. C., Li, Y., and Li, D. Z. (2015). Frequencies of HKαα and anti-hkαα alleles in Chinese carriers of silent deletional α-thalassemia. Hemoglobin 39 (6), 407–411. doi:10.3109/03630269.2015.1071268
Xie, X. M., Wu, M. Y., and Li, D. Z. (2015). Evidence of selection for the α-globin gene deletions and triplications in a southern Chinese population. Hemoglobin 39 (6), 442–444. doi:10.3109/03630269.2015.1072551
Xiong, F., Sun, M., Zhang, X., Cai, R., Zhou, Y., Lou, J., et al. (2010). Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang Autonomous Region of southern China. Clin. Genet. 78 (2), 139–148. doi:10.1111/j.1399-0004.2010.01430.x
Xu, X. M., Zhou, Y. Q., Luo, G. X., Liao, C., Zhou, M., Chen, P. Y., et al. (2004). The prevalence and spectrum of alpha and beta thalassaemia in Guangdong Province: implications for the future health burden and population screening. J. Clin. Pathol. 57 (5), 517–522. doi:10.1136/jcp.2003.014456
Zhang, M., Huang, H., Chen, M., Chen, L., Wang, Y., Lin, N., et al. (2019). Variant analysis for patients from Fujian area with Hong Kong αα type thalassemia, 36(4), 297–300. doi:10.3760/cma.j.issn.1003-9406.2019.04.002
Keywords: reverse dot blot (RDB), α-thalassemia, polymerase chain reaction (PCR), China, αααanti−4.2, αααanti−3.7
Citation: Ge Y-Y, Xie J, Liao Y-W, Xie L-X and Yang L-Y (2024) Development and clinical validation of a novel detection kit for α-thalassemia in southern Chinese. Front. Genet. 15:1457248. doi: 10.3389/fgene.2024.1457248
Received: 30 June 2024; Accepted: 22 August 2024;
Published: 05 September 2024.
Edited by:
Roberta Russo, University of Naples Federico II, ItalyReviewed by:
Ahlem Achour, Charles Nicolle Hospital, TunisiaMichela Grosso, University of Naples Federico II, Italy
Fernando Marqués-García, Hospital Germans Trias i Pujol, Spain
Copyright © 2024 Ge, Xie, Liao, Xie and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Long-Xu Xie, bHh4aWVAaHlicmliaW8uY24=; Li-Ye Yang, eWFuZ2xlZXllZUBzaW5hLmNvbQ==