 Tian Lan
Tian Lan- 1Department of Genetics, Metabolism and Endocrinology, Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2GrandOmics Biosciences Co, Ltd., Beijing, China
Background: 21-Hydroxylase deficiency (21-OHD) is caused by mutations in the CYP21A2 gene. Due to the complex structure and the high genetic heterogeneity of the CYP21A2 gene, genetic testing for 21-OHD is currently facing challenges. Moreover, there are no comparative studies on detecting CYP21A2 mutations by both second-generation sequencing and long-read sequencing (LRS, also known as third-generation sequencing).
Objective: To detect CYP21A2 variations in 21-OHD patients using targeted capture with LRS method based on the PacBio (Pacific Biosciences) Sequel II platform.
Methods: A total of 67 patients with 21-OHD were admitted in Wuhan Children’s Hospital. The full sequence of CYP21A2 gene was analyzed by targeted capture combined with LRS based on the PacBio Sequel II platform. The results were compared with those of long-polymerase chain reaction (Long-PCR) combined with multiplex ligation probe amplification (MLPA) detection. Based on the in vitro study of 21-hydroxylase activity of common mutations, the patient genotypes were divided into groups of Null, A, B, and C, from severe to mild. The correlation between different genotype groups and clinical typing was observed.
Results: The study analyzed a total of 67 patients. Among them, 44 (65.67%) were males and 23 (34.33%) were females, with a male-to-female ratio of approximately 1.9:1. A total of 27 pathogenic variants were identified in the 67 patients, of which micro-conversion accounted for 61.9%, new variants of CYP21A2 accounted for 8.2%; deletion accounted for 22.4% (CYP21A2 single deletion and chimeric TNXA/TNXB accounted for 12.7%, chimeric CYP21A1P/CYP21A2 accounted for 9.7%); and duplication accounted for 3.0% (CYP21A2 Gene Duplication). I2G was the most common variant (26.9%). Targeted capture LRS and MLPA combined with Long-PCR detection of CYP21A2 mutations showed 30 detection results with differences. The overall genotype-phenotype correlation was 82.1%. The positive predictive rate of the Null group for salt wasting (SW) type was 84.6%, the A group for SW type was 88.9%, the group B for simple virilization (SV) type was 82.4%, and the group C for SV type was 62.5%. The correlation coefficient rs between the severity of the phenotype and the genotype group was 0.682 (P < 0.05).
Conclusion: Targeted capture combined with LRS is an integrated approach for detecting CYP21A2 mutations, allowing precise determination of connected sites for multiple deletions/insertions and cis/trans configurations without analyzing parental genomic samples. The overall genotype-phenotype correlation for 21-OHD is generally strong, with higher associations observed between genotype and phenotype for group Null, A, and B mutations, and larger genotype-phenotype variation in group C mutations. Targeted capture with LRS sequencing offers a new method for genetic diagnosis in 21-OHD patients.
1 Introduction
21-hydroxylase deficiency (21-OHD, OMIM #201910) is an autosomal recessive genetic disease, accounting for more than 95% of congenital adrenal hyperplasia (CAH) (Pignatelli et al., 2019). The root cause of the condition is the deficiency of 21-hydroxylase in the adrenal corticosteroid synthesis pathway, resulting in impaired production of cortisol and aldosterone. Based on differences in genotype and residual 21-hydroxylase activity, 21-hydroxylase deficiency (21-OHD) can be classified into classic and non-classic types (Hannah-Shmouni et al., 2017). The classic type is further divided two subtypes: the salt wasting (SW) type with almost complete loss of enzyme activity, and the simple virilization (SV) type that retains 1%–2% of enzyme activity. Non-classic 21-OHD typically presents with milder clinical symptoms.
The 21-hydroxylase (21-OH), is encoded by the CYP21A2 gene, which is located in the main histocompatibility complex III region of chromosome 6p21.3, with a full length of 3.35 kb and composed of 10 exons (Higashi et al., 1986). The CYP21A2 is located within the tandem repeat sequence RCCX module. The RCCX module is one of the most complex copy number variation (CNV) loci in humans, and gene misalignment may occur during meiosis, leading to gene conversion, unequal crossing, deletion, and the formation of non-functional chimeric genes (White et al., 1986). About 30 kb from the CYP21A2 gene, there is a highly conserved pseudogene, CYP21A1P, which has up to 98% and 96% sequence identity with the real gene respectively (Higashi et al., 1986). The genes in this region vary significantly in size and copy number (Parajes et al., 2008). Currently, the Human Gene Mutation Database (HGMD) Professional Edition (Stenson et al., 2017) has reported nearly 500 mutations in the CYP21A2 gene, of which 388 disease-causing mutations (DM) are associated with the 21-OHD phenotype (http://www.hgmd.cf.ac.uk/). About 75% of these are due to micro-conversion of the non-functional CYP21A1P gene, 20%–25% are deletions or duplications, and 1%–2% are novel CYP21A2 variants (Hannah-Shmouni et al., 2017; Tusie-Luna and White, 1995).
At present, 21-OHD is screened by testing 17-hydroxyprogesterone (17-OHP) levels, and the false positive rate fluctuates between 0.4% and 9.3% (Auer et al., 2023; Hayashi et al., 2017). The genetic testing is the gold standard for determining the cause of CAH. Previous generation sequencing used Sanger combined with quantitative polymerase chain reaction (QPCR) to detect point mutations and deletion duplications (CNVs), with read lengths of only 600–1,000 bp. Currently, the commonly used method is next-generation sequencing (NGS), which employs Whole Exome Sequencing (WES) combined with multiplex ligation probe amplification (MLPA) detection. NGS-WES detects variations in genes that cause CAH besides CYP21A2. CYP21A2 mutations are detected using long-polymerase chain reaction (Long-PCR) to obtain target gene detection point mutations. MLPA detects deletions and duplications. MLPA has higher sensitivity and better reproducibility than QPCR. However, the read length of NGS is about 200 base pairs, which can only fuzzily match the reference sequence and can not detect pseudo-genes that are highly similar to both the target region and homologous sequences. At the same time, in the process of PCR amplification in NGS sequencing, GC bias will affect the detection accuracy. NGS can’t determine specific types of chimeras caused by complex rearrangements or large deletions. LRS, also known as third-generation sequencing, can achieve read lengths ranging from 15 kb to 2 Mb for DNA sequencing, which is nearly ten-thousand-fold improvement over NGS. At the same time, the capture process can reduce GC bias, which can significantly improve the detection accuracy. A single read of LRS can span the entire CYP21A2 gene (3.35 kb), accurately mapping to the reference genome and distinguishing it from its pseudogene CYP21A1P. The unique advantage of LRS in long read length enables comprehensive detection of single nucleotide variations (SNVs), deletions/insertions, tandem duplications, structural variations, differentiation between true gene and pseudogenes, methylation, and cis/trans configurations. This approach provides a new solution for detecting pathogenic variants in the CYP21A2 gene. This study uses targeted capture and LRS techniques to sequence the genes of 67 patients with 21-OHD, to achieve a comprehensive genetic analysis of all CAH-related gene variants.
2 Materials and methods
2.1 Research object
This study included 67 patients diagnosed with 21-OHD who visited the Department of Genetic Metabolism and Endocrinology, Wuhan Children’s Hospital from January 2013 to October 2023. The diagnostic criteria refer to “Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline” (Speiser et al., 2018). The patients were divided into three groups: SW, SV, and NC. This study was approved by the Ethics Committee of Wuhan Children’s Hospital (Ethical Review Number 2023R054-E02). All legal guardians of the patients signed informed consent forms.
2.2 Genetic testing methods
2.2.1 Long-read sequencing
Collect 2 mL of peripheral blood from the patient and send it to Beijing Grandomics Biosciences Co., Ltd. for targeted capture sequencing of CAH-related genes including CYP21A2, CYP11A1, CYP11B1, CYP17A1, HSD3B2, StAR, POR, SRD5A2, CYP11B2, and TNXB using LRS. Firstly, the high-quality DNA is extracted using a blood extraction kit (Meijibio, Guangzhou). The extracted DNA is fragmented using a g-tube (Covaris, United States) and repaired with an end repair enzyme. The repaired DNA is then ligated with Barcode adapters, followed by the purification of the ligation products using Agencourt AMPure XP beads (Beckman Coulter, United States). After the library amplification, the PCR products are purified with Agencourt AMPure XP beads (Beckman Coulter, United States) and quantified with Qubit. Nanodrop is used to assess DNA purity and agarose gel electrophoresis is used to check the degradation and size of DNA fragments. The above products are captured by hybridization using CAH probes, followed by washing and purifying the hybridized products using Agencourt AMPure XP beads (Beckman Coulter, United States). Concentrations are measured using the Qubit dsDNA HS Assay Kit.
Qualified DNA samples are used to construct libraries following the instructions provided with the PacBio Single Molecule Real-Time (SMRT) bellTM Express Template Prep Kit 2.0 (PacBio, United States) kit. The resulting products are purified with Agencourt AMPure XP beads (Beckman Coulter, United States), and the concentrations are determined using the Qubit dsDNA HS Assay Kit. The library fragment sizes are detected using the Agilent 2100 Bioanalyzer (Agilent Technologies, United States). The constructed DNA libraries are then sequenced using the PacBio Sequel II platform (PacBio, United States). After the raw sequencing data is qualified by SMRT Link (version 12.0) to obtain HiFi reads, proceed with subsequent bioinformatics analysis. Circular Consensus Sequence (CCS) reads were automatically generated by the PacBio SMRT analysis module and were mapped to the reference genome GRCh38/hg38 by using Minimap2 (version 2.24). SNVs were called by DeepVariant (version 1.3.0) and annotated by Vep (version 107). SVs were detected by Sniffles (version 2.0.7) and Cutesv (version 1.0.12). SVs were annotated by AnnotSV (version 3.1.1).
2.2.2 MLPA combined with long-PCR sequencing
2.2.2.1 MLPA
Each sample takes 2 mL of blood, and DNA is extracted u sing a blood DNA extraction kit and dissolved in 1 * TE buffer. Using the MLPA kit of MRC-Holland to carry out DNA denaturation, probe hybridization, ligation reaction, amplification and data analysis. 1) Take 50 ng of DNA, add 1*TE buffer to make up to 5 ul, and denature at 98°C for 5 min. 2) After DNA denaturation, add the corresponding probe mixture (1.5 μL MLPA buffer, 1.5 μL probemix), mix well and incubate overnight for 18 h. 3) Add 32 μL ligase mixture (25 μL dH2O, 3 μL Ligase Buffer A, 3 μL Ligase Buffer B, 1 μL Ligase-65 enzyme), mix well and perform ligation reaction. 4) Add PCR mixture (7.5 μL Ultrapure water, 2 μL SALSA PCR primer mix, 0.5 μL SALSA Polymerase), gently mix and perform PCR reaction. 5) The PCR products are detected by ABI 3730XL. 6) The test results are analyzed by COFFALYSER.NET software. COFFALYSER.NET software.
2.2.2.2 Long-PCR
The target gene is amplified by specific PCR amplification primers. The amplification reaction procedure is: 94°C, 5 min; 98°C for 1 min, 68°C for 10 min, 25 cycles; 68°C for 20 min. The PCR product is purified by Ampure magnetic beads and quantified by Qubit. The amplified DNA is broken up by ultrasound, and Ampure magnetic bead fragments are selected to obtain DNA products of 300–400 bp. The second-generation DNA library is constructed using the Rapid Plus DNA Lib Prep Kit for Illumina kit and the Dual DNA Adapter 96 Kit for Illumina kit. The constructed DNA library is subjected to Illumina NovaSeq high-throughput sequencing. After the sequencing data is evaluated to be qualified by Illumina Sequence Control Software (SCS), data reading and bioinformatics analysis are performed. After the sequencing data is evaluated to be qualified by Illumina Sequence Control Software (SCS), data reading and bioinformatics analysis are performed.
2.3 CYP21A2 pathogenic variant types
Based on the different pathogenic variants of the CYP21A2 gene, it is divided into microconversion events, novel CYP21A2 variants, deletions (CYP21A2 Gene Deletion and Chimeric TNXA/TNXB), Chimeric (CYP21A1P/CYP21A2), and CYP21A2 Gene Duplications (chimeric CYP21A2/CYP21A1P chimeric and chimeric TNXB/TNXA (Concolino and Costella, 2018).
2.3.1 Microconversion events
Microconversion refers to the variation formed when the non-functional pseudogene CYP21A1P is transferred to the functional gene CYP21A2 through microconversion events. Microconversion events mainly include: splicing mutations that occur at the end of intron2 (c.293-13A/C > G, also known as I2G); deletion of 8 bp in exon 3 (p.G111Vfs), insertion of 1 nucleotide in exon 7 (p.L308Ffs), a nonsense mutation in exon 8 (p.Q319X), 3 missense mutations (p.P31L, p.I173N and p.R357W), and 1 cluster conversion in exon 6 (p.I237N, p.V238E and p.M240K) (Choi et al., 2016; Concolino and Costella, 2018).
2.3.2 Novel CYP21A2 variants
Novel CYP21A2 variants occur without gene conversion, and the mutation sites are not present in CYPA21A1P, primarily consisting of private and missense mutations (Pignatelli et al., 2019).
2.3.3 CYP21A2 gene deletions
The CYP21A2 gene deletions include complete and partial deletions of the CYP21A2 gene. Complete absence of the gene can be manifested as either a solo deletion of the CYP21A2 gene or a chimeric TNXA/TNXB. The chimeric TNXA/TNXB results in the absence of the complete CYP21A2 and part of the TNXB, with three most common types being CAH-X CH1 to CH3 (Carrozza et al., 2021). CAH-X CH1 is caused by the complete CYP21A2 and a 120 bp deletion in the exon 35 of TNXB. CAH-X CH2 has a complete exon 35 of TNXB but with a complete absence of CYP21A2 and a p.C4058W mutation in exon 40. CAH-X CH3 has a cluster of three pseudo-genes (p.R4073H in exon 41, p.D4172N and p.S4175N in exon 43) derived mutations in TNXB and a complete absence of the CYP21A2 (Marino et al., 2021). Chimeric CYP21A1P/CYP21A2 gene leads to a partial replacement of the true gene CYP21A2 with the pseudogene CYP21A1P. Nine different chimeric CYP21A1P/CYP21A2 genes have been identified based on their chimeric junctions (Chen et al., 2012). Depending on whether the junction site is upstream or downstream of the I2G mutation, they can be divided into seven chimeric molecules carrying the I2G mutation (CH1, CH2, CH3, CH5, CH6, CH7, and CH8 (Chen et al., 2012; Concolino and Costella, 2018). Two chimeric subjects with CYP21A1P promoter and p.P31L mutation (CH4 and CH9) (Chen et al., 2012).
2.3.4 CYP21A2 gene duplications
The CYP21A2 gene is generated through genetic recombination during meiosis, producing the RCCX trimeric fragment, which carries one CYP21A1P pseudogene copy and two CYP21A2 genes. It includes the chimeric CYP21A2/CYP21A1P and the chimeric TNXB/TNXA (Concolino and Costella, 2018).
2.4 Genotype group
The clinical phenotype of 21-OHD is primarily influenced by mutations in alleles that have less impact on enzyme activity (Xu et al., 2013). Based on in vitro data of 21-hydroxylase activity in Table 1, CYP21A2 mutations are categorized from severe to mild as group Null, A, B, and C (Merke and Auchus, 2020; Wedell et al., 1994).
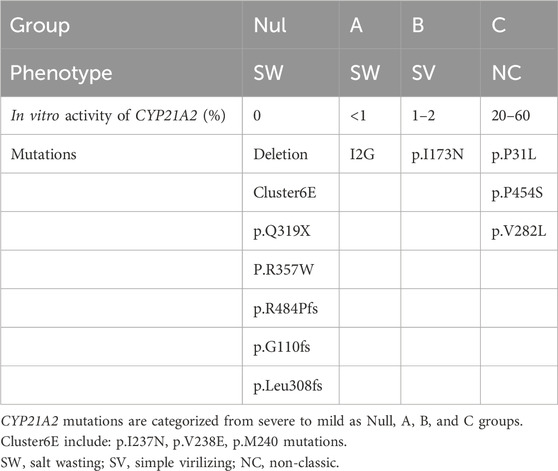
Table 1. The classification of common CYP21A2 mutations based on in vitro data.
The group Null of mutations includes homozygous or compound heterozygous mutations of deletion, p.G110fs, Cluster6E (p.I237N, p.V238E, p.M240K), p.Q319X, p.R357W, or p.R484P. Group A includes homozygous mutations of I2G or compound heterozygous mutations of I2G and group Null. Group B includes homozygous mutations of p.I173N or compound heterozygous mutations of p.I173N and group Null/A. Group C includes homozygous mutations of p.P31L, p.V281L, p.P453S or compound heterozygous mutations with group Null/A/B. Group D is mutations with unclear effects on 21-hydroxylase activity, including homozygous mutations of Group D or compound heterozygous mutations formed between Group D and other groups.
2.5 Statistical analysis
The statistical analysis was performed using SPSS 22.1 software. The normality of the data was tested, and the results of non-normally distributed quantitative data were expressed as Mean ± SD, M (P25, P75). Kruskal-Wallis H and Bonferroni tests were used to compare differences among multiple groups. Spearman’s rank correlation analysis was used to determine the relationship between the severity of the disease (SW, SV, NC) and the genotype groups. P < 0.05 indicated a statistically significant difference, and rs > 0 indicated a positive correlation.
3 Results
3.1 The clinical features of 21-OHD patients
A total of 67 patients were analyzed in this study. Among them, 44 were male (65.67%), and 23 were female (34.33%). The male-to-female ratio was approximately 1.9:1, and the age of detection ranged from 0.01 to 11.5 years old, with an average age of 1.7 years old. The patients were divided into three clinical types: SW in 38 cases, SV in 24 cases, and NC in 5 cases. The hormone levels and clinical manifestations of patients in different typing groups are shown in Table 2. The age at initial diagnosis of the SW group was significantly younger than that of the SV and NC groups, and the androstenedione level in the NC group was significantly lower than that in the SW and SV groups, and the testosterone and progesterone levels in the SV group were significantly lower than those in the SW group.
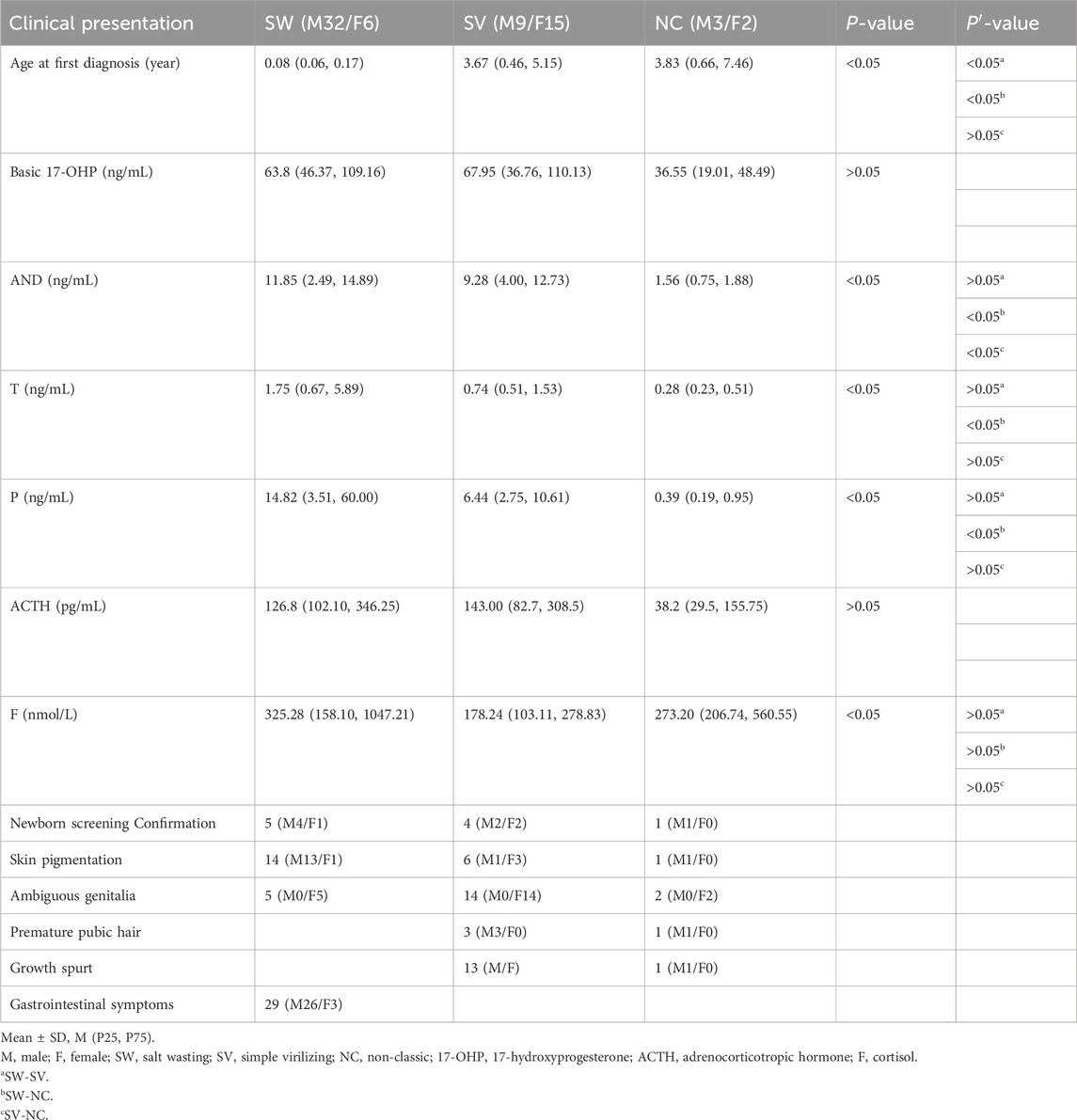
Table 2. Hormone levels and clinical manifestations at initial diagnosis in 67 patients with 21-OHD.
3.2 Results of CYP21A2 gene testing in 67 patients
The total number of CYP21A2 allele pathogenic variants detected in 67 patients is 134, while the copy number is 136 (Table 3). Among them, micro-conversions accounted for 61.9%; Novel Variants of CYP21A2 accounted for 8.2%; deletions accounted for 22.4%, including 12.7% for chimeric TNXA/TNXB, and 9.7% for chimeric CYP21A1P/CYP21A2; and CYP21A2 gene duplications accounted for 3.0%. The results of LRS, MLPA combined with Long-PCR detection, genotype classification, and clinical typing in 67 patients are shown in Table 3. The distribution of mutation sites in the CYP21A2 alleles of all patients is shown in Figure 1. A total of 29 different mutations were identified in this study. The frequency of CYP21A2 allele gene mutations are shown in Table 4. The comparison of different detection results between targeted capture LRS and MLPA combined with Long-PCR is shown in Table 5.
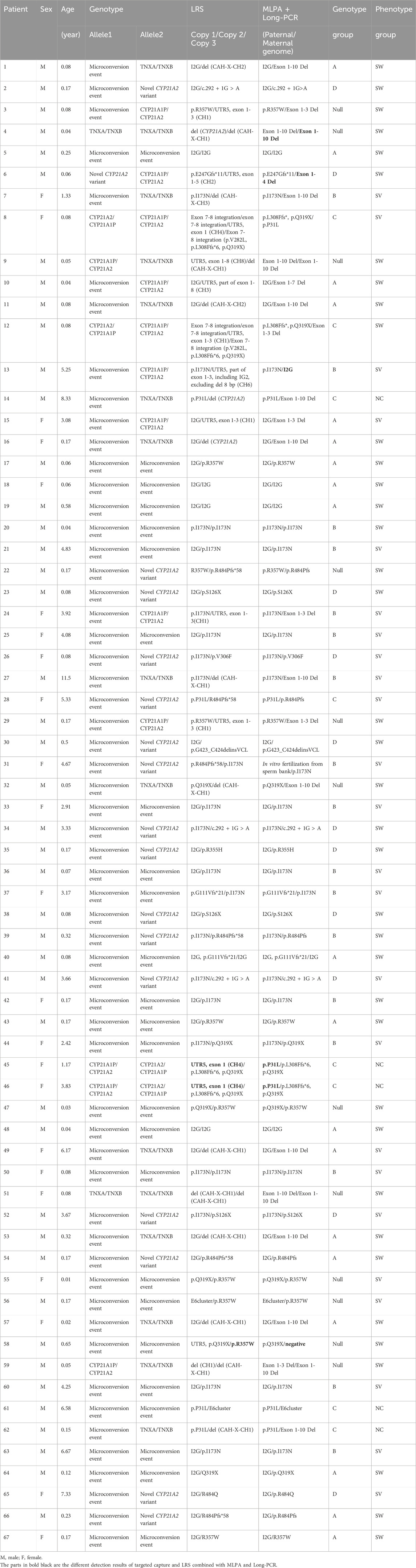
Table 3. Results of LRS and MLPA combined with long-PCR sequencing for CYP21A2 in 21-OHD.
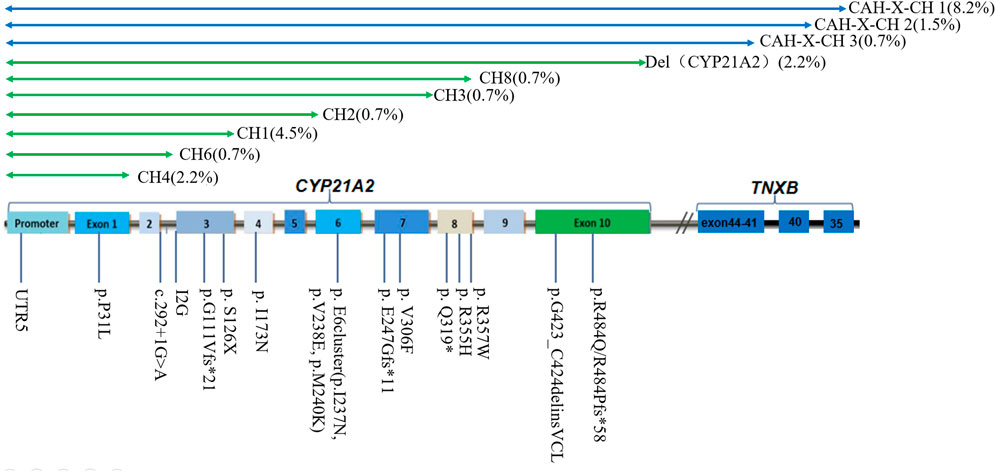
Figure 1. Distribution of CYP21A2 allelic mutation sites in 67 patients of this study. CH1-9 belongs to chimeric CYP21A1P/CYP21A2; CAH-X CH1, CAH-X CH2 and CAH-X CH3 belong to chimeric TNXA/TNXB.
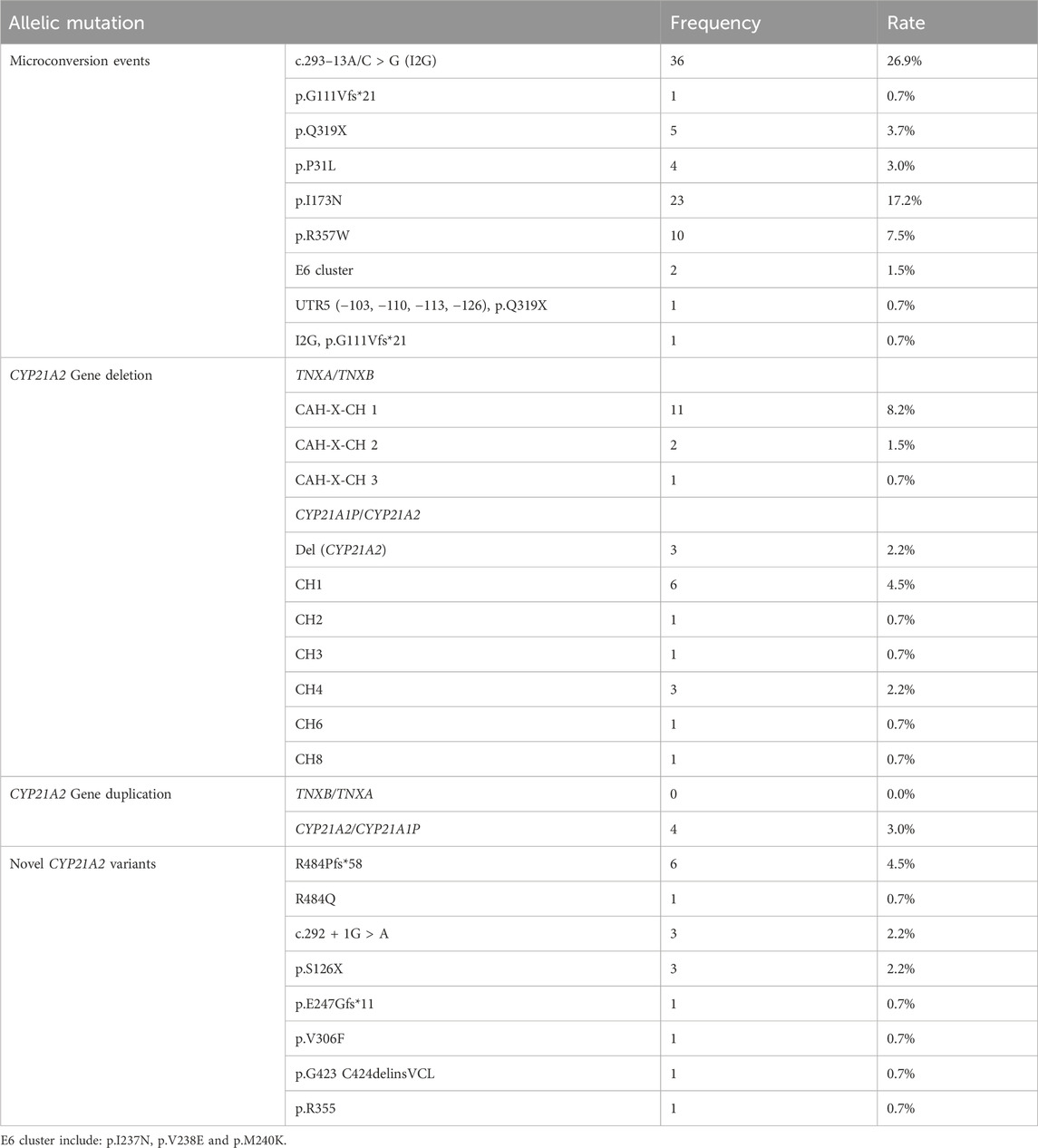
Table 4. Mutation frequency of CYP21A2 allele in this study.
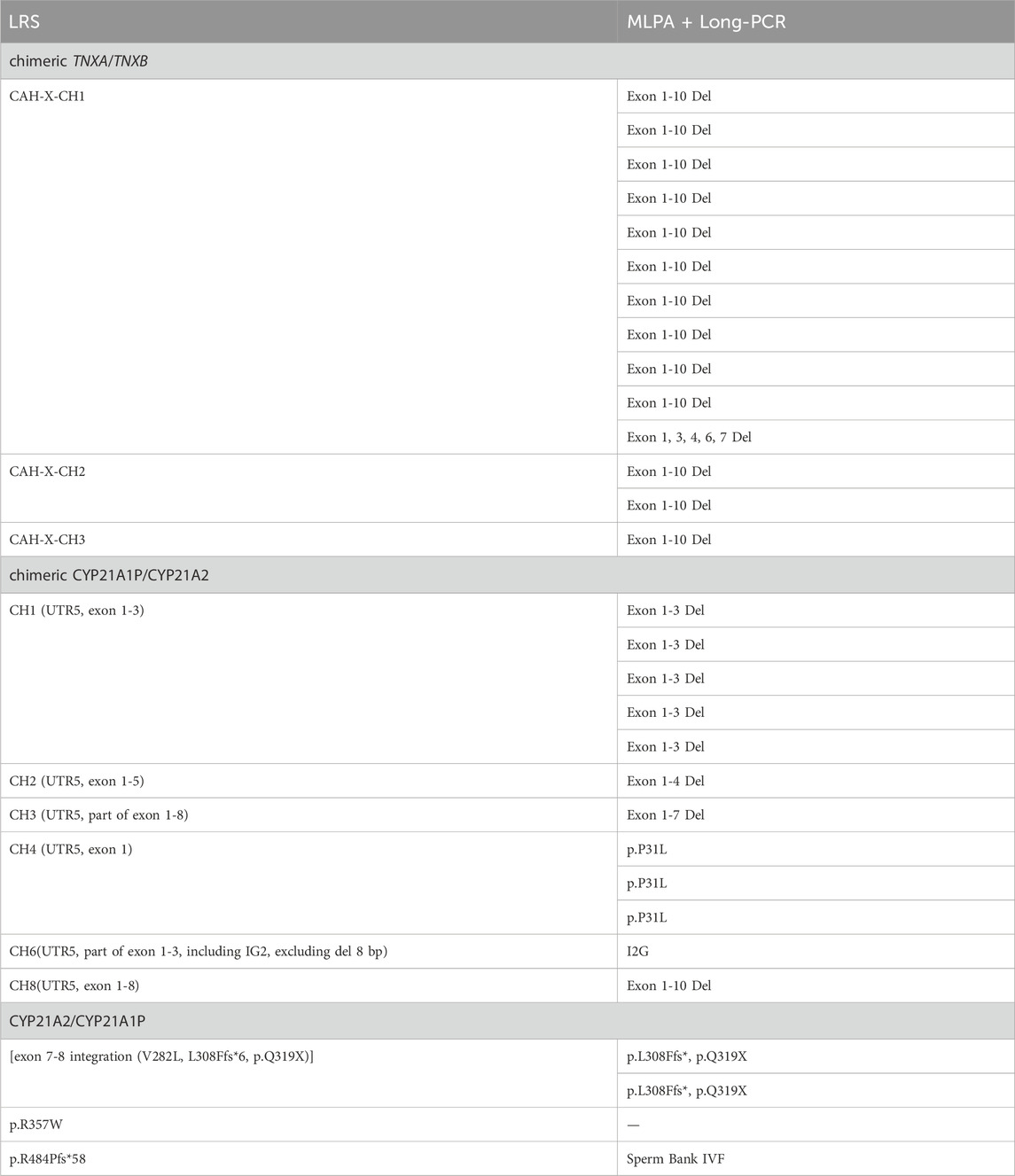
Table 5. The comparison of different detection results between LRS and MLPA combined with Long-PCR.
The overall genotype-phenotype correlation was 82.1%, with a positive predictive rate of 83.3% of the Null group in SW, 89.47% of Group A in SW, 82.4% of Group B in SV, and 62.5% of Group C in NC. The correlation coefficient rs between the severity of the phenotype and the genotype grouping was 0.682 (P < 0.05) (Table 6).
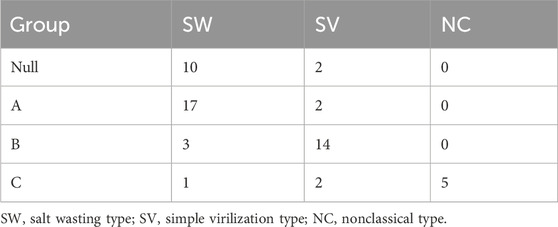
Table 6. Relationship between genotype and clinical phenotype.
3.2.1 Microconversion events
In this cohort, 83 variants were derived from the transformation of the CYP21A1P pseudogene, with mutation frequencies as follows: I2G (36/134, 26.9%), p.I173N (23/134, 17.2%), p.R357W (10/134, 7.5%), p.Q319X (5/134, 3.7%), p.P31L (4/134, 3.0%), p.G111Vfs*21 (1/134, 0.7%), E6 cluster (p.I237N, p.V238E, and p.M240K) (2/134, 1.5%), UTR5 and p.Q319X (1/134, 0.7%), I2G and p.G111Vfs*21 (1/134, 0.7%).
3.2.2 Novel CYP21A2 variants
Novel CYP21A2 variants were detected in 17 alleles. Seven rare variants of CYP21A2 include R484Pfs*58, c.292 + 1G > A, S126X, E247Gfs*11, V306F, p.G423_C424delinsVCL (c.1268_1275delinsTGTGCCTGGGC), and R355H. Notably, p.G423_C424delinsVCL (c.1268_1275delinsTGTGCCTGGGC) is a newly discovered mutation in this study and has not been reported in the HGMD pro database.
3.2.3 CYP21A2 gene deletions
A total of 30 allelic mutations were identified in the patients in this cohort. Among them, 17 were complete losses of CYP21A2, and 13 were partial losses. These included chimeric TNXA/TNXB CAH-X-CH1 accounted for 8.2% (11/134), CAH-X-CH2 accounted for 1.5% (2/134), CAH-X-CH3 accounted for 0.7% (1/134). In chimeric CYP21A1P/CYP21A2, deletion (Del) (CYP21A2) accounted for 2.2% (3/134), followed by CH1 accounted for 4.5% (6/134), CH4 accounted for 2.2% (3/134), and CH2 0.7% (1/134), CH3 0.7% (1/134), CH6 0.7% (1/134), and CH8 0.7% (1/134), respectively.
LRS detected the chimeric TNXA/TNXB CAH-X-CH1 mutation, and the results of second-generation sequencing combined with MLPA showed Exon 1, 3, 4, 6, 7 Del or Exon 1-10 Del; for the CAH-X-CH2 mutation, the results showed Exon 3 Del; for the CAH-X-CH3 mutation, the results showed Exon 1-10 Del. For the chimeric CYP21A1P/CYP21A2 CH1 mutation, the results showed Exon 1-3 Del or p.P31L; the CH2 showed Exon 1-5 Del; the CH3 showed Exon 1-6 Del; the CH4 showed p.P31L, but UTR5 was not detected; the CH6 showed I2G, and UTR5 and Exon 1-3 partial Del were not detected. The results above were detected using a combination of second-generation sequencing and MLPA.
3.2.4 CYP21A2 gene duplications
Gene recombination during meiosis can lead to duplication of the CYP21A2 gene, including CYP21A2/CYP21A1P chimeras and TNXB/TNXA chimeras. In this cohort of patients, a total of four CYP21A2 gene duplications were detected, all of which were chimeric CYP21A2/CYP21A1P, with no chimeric TNXB/TNXA identified. The combined results of next-generation sequencing and MLPA for patients 08 and 12 showed p.L308Ffs*, p.Q319X mutations, but no p.V282L mutation was detected.
4 Discussion
21-OHD has high clinical and genetic heterogeneity and seriously impacts the quality of life for patients. Neonatal screening for 21-OHD has been implemented in some regions, but traditional screenings based on 17-hydroxyprogesterone (17-OHP) concentrations have a certain false-positive rate, limiting their diagnostic value (Guran et al., 2020; Lind-Holst et al., 2022; Tippabathani et al., 2023; White, 2009). Currently, genetic testing is recommended as a secondary screening tool for 21-OHD (Merke and Auchus, 2020). Genetic testing technology for 21-OHD has progressed from Sanger sequencing combined with QPCR, and NGS combined with MLPA, to LRS. LRS can obtain target genes through either specific long-range PCR amplification or probe capture methods. The LRS based on specific long PCR amplification has been reported to detect five genes: CYP21A2, CYP11B1, CYP17A1, HSD3B2, and StAR, and can amplify highly homologous sequences that are difficult to amplify by conventional PCR. The length of the single amplification can reach 5–20 kb. However, long-range PCR amplification is mainly used for prenatal screening and can only detect common variants that have been reported (Li et al., 2023; Liu et al., 2022; Zhang et al., 2023). In this study, LRS used probe capture to achieve the capture and to enrich target genes. The method can detect ten genes: CYP21A2, CYP11B1, CYP17A1, HSD3B2, StAR, CYP11A1, POR, CYP11B2, SRD5A2, and TNXB, with a wider range of detection that is not limited to previously reported variations. Compared with Sanger and NGS sequencing methods, LRS can accurately distinguish between true genes and pseudogenes, tandem repeats, and directly detect structural variants, such as microrearrangements of CYP21A2, CAH-X mutations from CH1 to CH3, CYP21A1P/CYP21A2 chimeras (CH1-CH9), CYP21A2/CYP21A1P chimeras, and TNXB/TNX chimeras, etc. In addition, the method can accurately determine the cis/trans configurations of the variants without parental gene validation.
This study used targeted capture LRS, MLPA combined with Long-PCR to detect CYP21A2 mutations. There were 30 differences in the results of the detection. Patient 31 was conceived through in vitro fertilization (IVF) using donor sperm from a sperm bank. The MLPA combined with Long-PCR could not determine the origin of his mutation. LRS could identify the cis-trans configuration of patients without family verification, helping patients with genetic diagnosis. MLPA combined with Long-PCR did not detect the p.R357W mutation in patient 58, which was inherited from the father, but LRS did, aiding in identifying the pathogenic gene. LRS detected the integration of exon 7-8 including p.V282L (exon 7), p.L308Ffs * (exon 7), p.Q319X (exon 8) mutations in patients 8 and 12, while MLPA combined with Long-PCR failed to detect p.V282L. This may be related to Long-PCR’s tendency to off-target.
Additionally, CYP21A2 is highly homologous to CYP21A1P, with only 65 nucleotide differences between the active gene CYP21A2 and the pseudogene CYP21A1P across the exons and introns regions (White et al., 1986). If NGS sequencing is used, the resulting gene fragments are only about 200 bp in size. The high similarity between the target and the homologous regions can lead to ambiguous matching of the reference sequence, making it difficult to fully distinguish between the true gene and pseudogenes. At the same time, the specific probes used by MLPA are also difficult to detect fixed complex rearrangements. Furthermore, LRS can detect the specific variations in each copy. Patients 08 and 12 have three copy number variations with exon 7-8 fusion. The fusion of exon 7-8 is one type of chimeric CYP21A2/CYP21A1P, with a carriage rate of approximately 7%. This is not a complete duplication of CYP21A2, but rather a partial duplication of certain exons within the CYP21A2 gene (Parajes et al., 2008). p.Q319X is usually associated with gene duplication of CYP21A2 and a functional loss of CYP21A2 (Kleinle et al., 2009). This study identified a total of 14 chimeric TNXA/TNXB, but the results of MLPA combined with Long-PCR detection were all lack of exon 1-10 of CYP21A2. This indicates that CAH-X-CH1, CAH-X-CH2 and CAH-X-CH3 cannot be distinguished from each other. This is related to the probe sets in CAH-MLPA which are composed of four areas. Area 1 contains four probes of CYP21A1P, area 2 contains eight probes of CYP21A2 exon 1-7, area 3 contains six probes of TNXB, and area 4 is a reference probe. The six probes of TNXB include two sites on exon 35 and one site on exon 19, 20, 29 and 31. Therefore, it cannot detect deletions in specific exons from TNXB exon 40-44. In other words, it cannot distinguish different structural variations from CAH-X-CH1 to CAH-X-CH3 in chimeric TNXA/TNXB. However, the BAM figure of LRS can clearly show the fusion situation of active gene CYP21A2 and the pseudogene CYP21A1P that occurred in patients. This study identified 12 instances of chimeric CYP21A1P/CYP21A2. The LRS detected recombinations in CH1 (UTR5, exon 1-3), CH2 (UTR5, exon 1-5), CH3 (UTR5, part of exon 1-8), CH4 (UTR5, exon 1), CH6 (UTR5, part of exon 1-3, including IG2, excluding del 8 bp), and CH8 (UTR5, exon 1-8). The results of combined MLPA and Long-PCR were Exon 1-3 Del, Exon 1-4 Del, Exon 1-7 Del, p.P31L, I2G, and Exon 1-10 Del. The discrepancies in the detection results were due to the limited probe coverage of CYP21A2 in region 2 by MLPA, which only contained probes for exons 1, 3, 4, 6, 7, and I2G. In CH1, the mutation in exon 1-3 detected by combined MLPA combined with Long-PCR was inferred to be a deletion of exon 1-3 based on the detection of missing CYP21A2 probes for exon 1 and exon 3. In CH2, MLPA combined with Long-PCR failed to detect a mutation in exon 5 because no probe was set for it, thus the deletion of exon 5 could not be detected. Similarly, in CH3, MLPA combined with Long-PCR failed to detect a partial deletion in exon 8 due to the absence of a probe for CYP21A2 exon 8. The deletion in exon 1 of CH4 was consistent with the detection of p.P31L by MLPA combined with Long-PCR. In CH6, the failure of MLPA combined with Long-PCR to completely distinguish between true and false genes led to the undetected mutation in exon 1. In CH8, MLPA combined with Long-PCR, which did not include probes for CYP21A2 exons 8, 9, and 10, failed to detect deletions in these exons. The reported Exon 1-10 Del result was merely an inference based on the absence of exons 1, 3, 4, 6, 7.
This study found a concordance rate of 82.1% (46/56 cases) between genotype and phenotype, which is generally similar to other reports (Finkielstain et al., 2011; New et al., 2013; Riedl et al., 2019; Stikkelbroeck et al., 2003). The mutations in Group Null and Group A affect the key functions of 21-hydroxylase, causing alterations in membrane anchoring, heme binding, or enzyme stability, leading to the complete loss of 21-hydroxylase function. The mutations in Group B affect the transmembrane regions or conserved hydrophobic patches, managing to retain only 1%–2% of the normal enzyme activity (Higashi et al., 1988; Tusie-Luna et al., 1990). The mutation in Group C disrupts the interaction of redox enzymes, salt bridges, and hydrogen bond networks, retaining 20%–60% activity of 21-hydroxylase (Helmberg et al., 1992; Tusie-Luna et al., 1991; Tusie-Luna and White, 1995). Previous studies have shown a stronger correlation between genotype and phenotype for group Null and A mutations, while patients with group B and C mutations exhibit greater genotype-phenotype variability (Riedl et al., 2019). The genotype-phenotype variability rate in group B patients is 17.6%, which may be associated with alterations in transcriptional regulation of the protein or downstream protein translation (New et al., 2013). The positive prediction rate of group C for NC is only 62.5%. Overall, the p.V283L and p.P454S mutation genotypes in group C have a relatively good correlation with the phenotype, while the genotype-phenotype variation for the p.P31L genotype is greater (Krone et al., 2000; New et al., 2013). The sequencing results of patients 08 and 12 in group C of this study showed the p.P31L mutation, but their corresponding LRS results were actually CH4 and CH1, respectively. In patient 08, the chimeric CYP21A1P/CYP21A2 CH4 had a deletion at a site upstream of the I2G mutation. CH4 carried two mutations: the CYP21A1P promoter and p.P31L, which had a relatively weak impact on the activity of 21-hydroxylase, presenting with the clinical phenotype of SV. In patient 12, the deletion site of CH1 was downstream of the I2G mutation, containing I2G and multiple pseudogene mutations. This mutation had a greater impact on the 21-hydroxylase, with the clinical phenotype being SW. The LRS test results can better assist in the clinical typing of 21-OHD. Of course, the genotype-phenotype variation in 21-OHD patients is caused by various factors. In addition to CYP21A2 mutations, the length of the CAG repeats in the androgen receptor (Kaupert et al., 2013; Moura-Massari et al., 2016), the high polymorphism of the protein P450 oxidoreductase, and the splicing mutations in RNA (Buchner et al., 2003; L’Allemand et al., 2000), can all affect the patient’s phenotype. Genotype-phenotype inconsistencies require further study.
Approximately 95% of disease-causing variants arise from microconversions during meiosis, unequal crossing-over, deletions, and the formation of non-functional chimeric genes in the CYP21A2 gene (Merke and Auchus, 2020; Werkmeister et al., 1986). In this study, targeted capture and LRS on the PacBio Sequel II platform were employed to detect 28 types of CYP21A2 pathogenic mutations in 67 21-OHD patients. Microconversions accounted for 61.9%, novel CYP21A2 variants for 8.2%, deletions for 22.4%, and CYP21A2 gene duplication for 3.0%. Microconversions, chimeric TNXA/TNX, chimeric CYP21A1P/CYP21A2, and chimeric CYP21A2/CYP21A1P mutations altogether accounted for 91.8% of the mutations. This proportion is roughly similar to other reported studies. The I2G mutation had the highest proportion (26.9%), followed by p.I173N (17.2%) and CAH-X-CH1 (8.2%). I2G is currently the most common mutation affecting splicing, located in the most polymorphic region of the CYP21A2 gene (Concolino and Costella, 2018). I2G is also the most common in Western Alaska Eskimos and Iranians (Wilson et al., 2007). No suspicious point mutations were detected in exons 2, 5, and 9 during this test, which may be related to the fact that these regions contain non-differential sequences of pseudo-genes and are less likely to undergo recombination or conversion.
Although LRS has longer sequencing read length and higher accuracy, it also has limitations. LRS has higher requirements for sample quality, and the sample cellular DNA samples are prone to degradation. In addition, the price of the LRS sequencer is high, and the single-molecule sequencing chip is not reusable, and the reagent price is relatively expensive. At the same time, the HiFi sequencing obtains Circular Consensus Sequencing (CCS) sequences, which can greatly improve the accuracy of the test, but at the same time, the amount of data generated after the test is large, and bioinformatics analysis requires a lot of manpower and computing resources, resulting in high costs.
The targeted capture based on the PacBio Sequel II platform and LRS detection technology will become an important tool for molecular diagnosis in the near future. LRS detection can achieve precise genotyping of candidate genes for CAH in one step, including microtransformation, new variations, deletions, and duplications. It can determine the trans/cis position of the variation without detecting the CYP21A2 gene of the patient’s parents. However, the sample size of this study is relatively limited, and the advantages of other types of CAH have not been fully demonstrated. Large-scale, multicenter prospective studies are still needed to maximize the advantages of LRS.
Data availability statement
The data presented in the study are deposited in the NCBI repository, accession number PRJNA1152331.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Wuhan Children’s Hospital (Ethical Review Number: 2023R054-E02). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
TL: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing–original draft, Writing–review and editing. JW: Visualization, Writing–original draft, Writing–review and editing. KC: Resources, Validation, Visualization, Writing–review and editing. JZ: Data curation, Methodology, Writing–review and editing. XC: Investigation, Methodology, Software, Writing–review and editing. HY: Formal Analysis, Funding acquisition, Project administration, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by Wuhan Knowledge Innovation Program (Grant number: 22022020801020570) and China Children’s Growth and Development Academic Exchange Special Fund (Z-2019-41-2303).
Acknowledgments
We are grateful for the patient and her family. We also thank the staff in GrandOmics Biosciences Co., Ltd. (Beijing, China) for their technical assist.
Conflict of interest
Author JZ was employed by GrandOmics Biosciences Co, Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1472516/full#supplementary-material
References
Auer, M. K., Nordenstrom, A., Lajic, S., and Reisch, N. (2023). Congenital adrenal hyperplasia. Lancet 401, 227–244. doi:10.1016/S0140-6736(22)01330-7
Buchner, D. A., Trudeau, M., and Meisler, M. H. (2003). Scnm1, a putative rna splicing factor that modifies disease severity in mice. Science 301, 967–969. doi:10.1126/science.1086187
Carrozza, C., Foca, L., De Paolis, E., and Concolino, P. (2021). Genes and pseudogenes: complexity of the rccx locus and disease. Front. Endocrinol. (Lausanne) 12, 709758. doi:10.3389/fendo.2021.709758
Chen, W., Xu, Z., Sullivan, A., Finkielstain, G. P., Van Ryzin, C., Merke, D. P., et al. (2012). Junction site analysis of chimeric cyp21a1p/cyp21a2 genes in 21-hydroxylase deficiency. Clin. Chem. 58, 421–430. doi:10.1373/clinchem.2011.174037
Choi, J. H., Kim, G. H., and Yoo, H. W. (2016). Recent advances in biochemical and molecular analysis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Ann. Pediatr. Endocrinol. Metab. 21, 1–6. doi:10.6065/apem.2016.21.1.1
Concolino, P., and Costella, A. (2018). Congenital adrenal hyperplasia (cah) due to 21-hydroxylase deficiency: a comprehensive focus on 233 pathogenic variants of cyp21a2 gene. Mol. Diagn Ther. 22, 261–280. doi:10.1007/s40291-018-0319-y
Finkielstain, G. P., Chen, W., Mehta, S. P., Fujimura, F. K., Hanna, R. M., Van Ryzin, C., et al. (2011). Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 96, E161–E172. doi:10.1210/jc.2010-0319
Guran, T., Tezel, B., Cakir, M., Akinci, A., Orbak, Z., Keskin, M., et al. (2020). Neonatal screening for congenital adrenal hyperplasia in Turkey: outcomes of extended pilot study in 241,083 infants. J. Clin. Res. Pediatr. Endocrinol. 12, 287–294. doi:10.4274/jcrpe.galenos.2020.2019.0182
Hannah-Shmouni, F., Chen, W., and Merke, D. P. (2017). Genetics of congenital adrenal hyperplasia. Endocrinol. Metab. Clin. North Am. 46, 435–458. doi:10.1016/j.ecl.2017.01.008
Hayashi, G. Y., Carvalho, D. F., de Miranda, M. C., Faure, C., Vallejos, C., Brito, V. N., et al. (2017). Neonatal 17-hydroxyprogesterone levels adjusted according to age at sample collection and birthweight improve the efficacy of congenital adrenal hyperplasia newborn screening. Clin. Endocrinol. (Oxf) 86, 480–487. doi:10.1111/cen.13292
Helmberg, A., Tusie-Luna, M. T., Tabarelli, M., Kofler, R., and White, P. C. (1992). R339h and p453s: cyp21 mutations associated with nonclassic steroid 21-hydroxylase deficiency that are not apparent gene conversions. Mol. Endocrinol. 6, 1318–1322. doi:10.1210/mend.6.8.1406709
Higashi, Y., Tanae, A., Inoue, H., Hiromasa, T., and Fujii-Kuriyama, Y. (1988). Aberrant splicing and missense mutations cause steroid 21-hydroxylase [p-450(c21)] deficiency in humans: possible gene conversion products. Proc. Natl. Acad. Sci. U. S. A. 85, 7486–7490. doi:10.1073/pnas.85.20.7486
Higashi, Y., Yoshioka, H., Yamane, M., Gotoh, O., and Fujii-Kuriyama, Y. (1986). Complete nucleotide sequence of two steroid 21-hydroxylase genes tandemly arranged in human chromosome: a pseudogene and a genuine gene. Proc. Natl. Acad. Sci. U. S. A. 83, 2841–2845. doi:10.1073/pnas.83.9.2841
Kaupert, L. C., Lemos-Marini, S. H., De Mello, M. P., Moreira, R. P., Brito, V. N., Jorge, A. A., et al. (2013). The effect of fetal androgen metabolism-related gene variants on external genitalia virilization in congenital adrenal hyperplasia. Clin. Genet. 84, 482–488. doi:10.1111/cge.12016
Kleinle, S., Lang, R., Fischer, G. F., Vierhapper, H., Waldhauser, F., Fodinger, M., et al. (2009). Duplications of the functional cyp21a2 gene are primarily restricted to q318x alleles: evidence for a founder effect. J. Clin. Endocrinol. Metab. 94, 3954–3958. doi:10.1210/jc.2009-0487
Krone, N., Braun, A., Roscher, A. A., Knorr, D., and Schwarz, H. P. (2000). Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J. Clin. Endocrinol. Metab. 85, 1059–1065. doi:10.1210/jcem.85.3.6441
L’Allemand, D., Tardy, V., Gruters, A., Schnabel, D., Krude, H., and Morel, Y. (2000). How a patient homozygous for a 30-kb deletion of the c4-cyp 21 genomic region can have a nonclassic form of 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 85, 4562–4567. doi:10.1210/jcem.85.12.7018
Li, H., Zhu, X., Yang, Y., Wang, W., Mao, A., Li, J., et al. (2023). Long-read sequencing: an effective method for genetic analysis of cyp21a2 variation in congenital adrenal hyperplasia. Clin. Chim. Acta 547, 117419. doi:10.1016/j.cca.2023.117419
Lind-Holst, M., Baekvad-Hansen, M., Berglund, A., Cohen, A. S., Melgaard, L., Skogstrand, K., et al. (2022). Neonatal screening for congenital adrenal hyperplasia in Denmark: 10 years of experience. Horm. Res. Paediatr. 95, 35–42. doi:10.1159/000522230
Liu, Y., Chen, M., Liu, J., Mao, A., Teng, Y., Yan, H., et al. (2022). Comprehensive analysis of congenital adrenal hyperplasia using long-read sequencing. Clin. Chem. 68, 927–939. doi:10.1093/clinchem/hvac046
Marino, R., Garrido, N. P., Ramirez, P., Notaristefano, G., Moresco, A., Touzon, M. S., et al. (2021). Ehlers-danlos syndrome: molecular and clinical characterization of tnxa/tnxb chimeras in congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 106, e2789–e2802. doi:10.1210/clinem/dgab033
Merke, D. P., and Auchus, R. J. (2020). Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N. Engl. J. Med. 383 (13), 1248–1261. doi:10.1056/NEJMra1909786
Moura-Massari, V. O., Cunha, F. S., Gomes, L. G., Bugano, D. G. D., Marcondes, J. A., Madureira, G., et al. (2016). The presence of clitoromegaly in the nonclassical form of 21-hydroxylase deficiency could be partially modulated by the cag polymorphic tract of the androgen receptor gene. Plos One 11, e0148548. doi:10.1371/journal.pone.0148548
New, M. I., Abraham, M., Gonzalez, B., Dumic, M., Razzaghy-Azar, M., Chitayat, D., et al. (2013). Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc. Natl. Acad. Sci. U. S. A. 110, 2611–2616. doi:10.1073/pnas.1300057110
Parajes, S., Quinteiro, C., Dominguez, F., and Loidi, L. (2008). High frequency of copy number variations and sequence variants at cyp21a2 locus: implication for the genetic diagnosis of 21-hydroxylase deficiency. Plos One 3, e2138. doi:10.1371/journal.pone.0002138
Pignatelli, D., Carvalho, B. L., Palmeiro, A., Barros, A., Guerreiro, S. G., and Macut, D. (2019). The complexities in genotyping of congenital adrenal hyperplasia: 21-hydroxylase deficiency. Front. Endocrinol. (Lausanne) 10, 432. doi:10.3389/fendo.2019.00432
Riedl, S., Rohl, F. W., Bonfig, W., Bramswig, J., Richter-Unruh, A., Fricke-Otto, S., et al. (2019). Genotype/phenotype correlations in 538 congenital adrenal hyperplasia patients from Germany and Austria: discordances in milder genotypes and in screened versus prescreening patients. Endocr. Connect. 8, 86–94. doi:10.1530/EC-18-0281
Speiser, P. W., Arlt, W., Auchus, R. J., Baskin, L. S., Conway, G. S., Merke, D. P., et al. (2018). Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 103, 4043–4088. doi:10.1210/jc.2018-01865
Stikkelbroeck, N. M., Hoefsloot, L. H., de Wijs, I. J., Otten, B. J., Hermus, A. R., and Sistermans, E. A. (2003). Cyp21 gene mutation analysis in 198 patients with 21-hydroxylase deficiency in The Netherlands: six novel mutations and a specific cluster of four mutations. J. Clin. Endocrinol. Metab. 88, 3852–3859. doi:10.1210/jc.2002-021681
Stenson, P. D., Mort, M, Ball, E. V., Evans, K., Hayden, M., and Heywood, S. (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136 (6), 665–677. doi:10.1007/s00439-017-1779-6
Tippabathani, J., Seenappa, V., Murugan, A., Phani, N. M., Hampe, M. H., Appaswamy, G., et al. (2023). Neonatal screening for congenital adrenal hyperplasia in indian newborns with reflex genetic analysis of 21-hydroxylase deficiency. Int. J. Neonatal Screen 9, 9. doi:10.3390/ijns9010009
Tusie-Luna, M. T., Speiser, P. W., Dumic, M., New, M. I., and White, P. C. (1991). A mutation (pro-30 to leu) in cyp21 represents a potential nonclassic steroid 21-hydroxylase deficiency allele. Mol. Endocrinol. 5, 685–692. doi:10.1210/mend-5-5-685
Tusie-Luna, M. T., Traktman, P., and White, P. C. (1990). Determination of functional effects of mutations in the steroid 21-hydroxylase gene (cyp21) using recombinant vaccinia virus. J. Biol. Chem. 265, 20916–20922. doi:10.1016/s0021-9258(17)45304-x
Tusie-Luna, M. T., and White, P. C. (1995). Gene conversions and unequal crossovers between cyp21 (steroid 21-hydroxylase gene) and cyp21p involve different mechanisms. Proc. Natl. Acad. Sci. U. S. A. 92, 10796–10800. doi:10.1073/pnas.92.23.10796
Wedell, A., Thilén, R. J., Ritzén, E. M., Stengler, B., and Luthman, H. (1994). Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: implications for genetic diagnosis and association with disease manifestation. J. Clin. Endocrinol. Metab. 78 (5), 1145–1152. doi:10.1210/jcem.78.5.8175971
Werkmeister, J. W., New, M. I., Dupont, B., and White, P. C. (1986). Frequent deletion and duplication of the steroid 21-hydroxylase genes. Am. J. Hum. Genet. 39, 461–469.
White, P. C. (2009). Neonatal screening for congenital adrenal hyperplasia. Nat. Rev. Endocrinol. 5, 490–498. doi:10.1038/nrendo.2009.148
White, P. C., New, M. I., and Dupont, B. (1986). Structure of human steroid 21-hydroxylase genes. Proc. Natl. Acad. Sci. U. S. A. 83, 5111–5115. doi:10.1073/pnas.83.14.5111
Wilson, R. C., Nimkarn, S., Dumic, M., Obeid, J., Azar, M. R., Najmabadi, H., et al. (2007). Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol. Genet. Metab. 90, 414–421. doi:10.1016/j.ymgme.2006.12.005
Xu, Z., Chen, W., Merke, D. P., and McDonnell, N. B. (2013). Comprehensive mutation analysis of the cyp21a2 gene: an efficient multistep approach to the molecular diagnosis of congenital adrenal hyperplasia. J. Mol. Diagn 15, 745–753. doi:10.1016/j.jmoldx.2013.06.001
Keywords: 21-hydroxylase deficiency (21-OHD), CYP21A2, long-read sequencing (LRS), targeted capture, multiplex ligation probe amplification (MLPA), genotypes
Citation: Lan T, Wang J, Chen K, Zhang J, Chen X and Yao H (2024) Comparison of long-read sequencing and MLPA combined with long-PCR sequencing of CYP21A2 mutations in patients with 21-OHD. Front. Genet. 15:1472516. doi: 10.3389/fgene.2024.1472516
Received: 29 July 2024; Accepted: 14 October 2024;
Published: 01 November 2024.
Edited by:
Jordi Pérez-Tur, Spanish National Research Council (CSIC), SpainReviewed by:
Baosheng Zhu, The First People’s Hospital of Yunnan Province, ChinaSemra Gürsoy, Dokuz Eylül University, Türkiye
Copyright © 2024 Lan, Wang, Chen, Zhang, Chen and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Yao, huiyaomail@126.com
†These authors have contributed equally to this work and share first authorship