 Wenzhi Wang1,2*
Wenzhi Wang1,2* Jungang Wang1
Jungang Wang1 Xiaoyan Feng1
Xiaoyan Feng1 Llinbo Shen1Cuilian Feng1Tingting Zhao1Hong Xiao3
Llinbo Shen1Cuilian Feng1Tingting Zhao1Hong Xiao3 Shifang Li3*Shuzhen Zhang1*
Shifang Li3*Shuzhen Zhang1*- 1Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences, Haikou, China
- 2State Key Laboratory of Crop Genetics and Germplasm Enhancement, Nanjing Agricultural University, Nanjing, China
- 3Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
Various RNA viral diseases on sugarcane result in yield loss and decreased sugar content. Breeding new varieties with virus resistance is the main goal of the sugarcane breeding program. Both single-stranded and double-stranded RNA viruses generated a double-stranded RNA replicative form (RF) during the replication cycle progress. While double-stranded RNA-specific ribonuclease (PAC1) encoded by the Pac1 gene (from Schizosaccharomyces pombe) can recognize and degrade double-stranded RNA specifically without any sequence, the expression of PAC1 in transgenic sugarcane may successfully develop virus-resistant sugarcane. In this research, we first expressed the PAC1 in prokaryotic cells. Then, double-stranded RNA RF of sugarcane's streak mosaic virus (SCSMV) was artificially synthesized. The degradation activity of the PAC1 was successfully tested by mixing the PAC1 protein and the double-stranded RNA RF. After that, the Pac1 gene was ligated to a plant expression vector and was then introduced into a virus-sensitive sugarcane cultivar by using Agrobacterium tumefaciens-mediated transformation method. Transgenic plants were challenged by inoculating with SCSMV. Results showed that although all the transgenic lines were infected by SCSMV, the mosaic symptoms that appeared on the leaves were significantly milder than that of the wild type. All transgenic shoots showed significantly lower viral loads and attained greater heights than wild-type shoots. This research provided a new pathway for breeding new varieties of sugarcane with virus resistance.
Introduction
Sugarcane is among the most important commercial crops and an important bioenergy crop. It is cultivated in large areas of tropical and subtropical countries; It contributes to 60–70% of annual sugar consumption worldwide (Manickavasagam et al., 2004; Suprasanna et al., 2011; ISMA, 2020) and about 90% in China. In addition, it is used as a raw material for ethanol production in Brazil, which is the largest producer of cane sugar in the world (Snyman, 2004; López-Ortega et al., 2021). Similar to other food crops, the yield of sugarcane is seriously affected by various viral diseases. These diseases cause increasingly serious harm to sugarcane and affect its yield (Singh et al., 2003; Viswanathan and Balamuralikrishnan, 2005; Rao et al., 2006). Currently, at least 10 species of viruses can infect commercial sugarcane cultivars. Sugarcane mosaic virus (SCMV), sorghum mosaic virus (SrMV), sugarcane streak mosaic virus (SCSMV), and sugarcane yellow leaf virus (SCYLV) are the most serious viruses that cause sugarcane diseases (Viswanathan et al., 2018). All these viruses are single-strained RNA viruses, and each species includes complicated virus strains. For example, SCMV contains six virus strains (Moradi et al., 2016), SrMV contains six virus strains (Luo et al., 2016), SCSMV contains four virus strains (Kasemsin et al., 2016), and SCYLV contains 10 virus strains (Lin et al., 2014). All these viruses mutate rapidly, and usually different species or strains mix and infect a single plant. Such combinations make the pathogens difficult to control, thereby, finally inducing huge economic losses to the sugarcane industry (Zhang et al., 2015; He et al., 2016). Efficient ways of controlling the viral diseases of sugarcane need to be developed by plant protection researchers. Considering the special pathogenic processes of viruses, some successful agronomic ways or drugs to control fungal, bacterial, and nematode diseases are useless in the control of viral diseases during agriculture production. Planting virus-free seeds seem to be a successful way to control viral disease (Dewanti et al., 2016), but it is not sustainable because virus-free plants can easily be infected again under open field conditions. So, creating cultivars that are immune or insensitive to viruses is an efficient way to stop the spread of viral diseases. The genetic engineering breeding approach is supposed to be an efficient way to create novel sugarcane cultivars (Wang et al., 2017b).
Both double-stranded RNA (ds-RNA) and single-stranded RNA viruses of plants contain a double-stranded RNA replicative form (RF) in the replication cycle. So, if the double-stranded RNA replicative form is degraded and the virus' replication process is stocked, then it may fail to finish the systemic infection in the hosts. Pac1 gene comes from the DNA of Schizosaccharomyces pombe, and its coding protein is a kind of ds-RNA-dependent ribonuclease. It has the potential to degrade the double-stranded RNA virus genome and the double-stranded replication form of single-stranded RNA virus and viroid genomes (the replication form; all are double-stranded RNA) (Lino et al., 1991; Rotondo and Frendwwey, 1996). So, integrating the Pac1 gene into host plants' genome and encoding ds-RNA-specific ribonuclease may give the RNA virus-sensitive plants resistance to most kinds of RNA viruses.
By now, many virus-resistant plants have been created by integrating the Pac1 gene through the genetic transformation strategy. In 1995, Watanabe from the University of Tokyo created transgenic tobacco plants with the Pac1 gene; these were resistant to tobacco mosaic virus (TMV), cucumber mosaic virus (CMV), and potato virus Y (PVY) (Watanabe et al., 1995). In 1997, Sano from the Hirosaki University created Pac1 gene transgenic potato plants, which were resistant to potato spindle tuber viroid (PSTVd). The Pac1 gene transgenic potato plants significantly reduced the spread of PSTVd from the mother potato to the child potato (Sano et al., 1997). In 2001, Zhang from the University of Nebraska-Lincoln created Pac1 gene-transgenic barley plants resistant to barley stripe mosaic virus (BSMV) (Zhang et al., 2001). In 2002, Ishida inserted the Pac1 gene into the chrysanthemum's genome, and the transgenic plants showed significant resistance to chrysanthemum stunt viroid (Ishida et al., 2002). In 2008, Li from the Institute of Plant Protection, Chinese Academy of Agricultural Sciences certified that ds-RNA specific ribonuclease encoded from the Pac1 gene can degrade the artificially synthesized double-stranded RNA of CMV, TMV, rice black-streaked dwarf (RBSDV), rice dwarf virus (RDV), apple scar skin viroid (ASSVd), coleus blumei viroid (CBVd), and hop stunt viroid (HSVd) in in vitro tests (Li et al., 2008).
In this research, we aimed to create antiviral transgenic sugarcane plants. First, double-stranded RNA-specific ribonuclease gene Pac1 was ligated into a prokaryotic expression vector, and Pac1 protein was expressed by prokaryotic expression. Double-stranded RNA replicative forms of SCSMV were artificially synthesized. Then, the double-stranded RNA-specific degradation ability of purified PAC1 protein was successfully tested. After that, a plant expression vector ligated with the Pac1 gene was constructed. The virus-sensitive commercial variety ROC22 was chosen as the breeding improvement receptor. The transformation process served as the transformation selection system that we established in our previous research (Wang et al., 2017a). After molecular analysis, positive transgenic plants were transplanted into the greenhouse, and virus resistance was tested. The results showed that we have created a novel sugarcane variety that is resistant to the virus.
Materials and methods
Pac1 gene isolation and sequencing
A plasmid vector harboring the Pac1 gene was presented by Prof. Li (Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China). According to the known sequence of Pac1 gene from NCBI, a pair of cloning primers, Pac1 1F and 1R, were designed and synthesized Primer sequence shows in Table 1. The forward primer was inserted with BamH I restriction enzyme cutting site and the reverse primer was inserted with Sac I restriction enzyme cutting site. The Pac1 gene was then isolated using the presented vector as the template. PCR production was purified with the Gel Extraction Kit (AP-GX-P-50G, Axygen, China), and cloning was performed by using the TA cloning vector (T-Vector pMD™19, Takara, China). Then, the positive cloning vector (pMD19T-Pac1) was certified by sequencing.
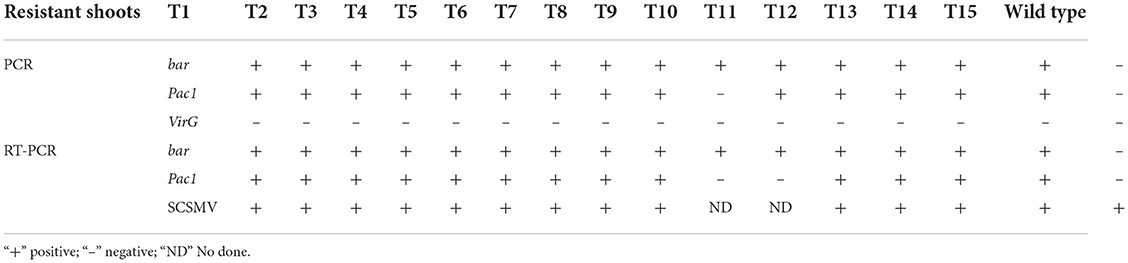
Table 1. Molecular analysis of resistant shoots.
PAC1 protein prokaryotic expression
Prokaryotic expression and purification of PAC1 protein were carried out by Detai Biologics Co., Ltd. (Nanjing, China). The main protocol was as follows. The Pac1 gene was synthesized artificially, and an N-His tag was added. Nde I and Hind III restriction enzyme cutting sites were added at the 5′ and 3′ sites, respectively. Then, by using Nde I and Hind III restriction enzymes, the Pac1 gene with the N-His tag was ligated to the prokaryotic expression vector (pET30a, Biodee, China). After verification, the positive prokaryotic expression vector pET30a-Pac1 was transformed into receptor bacteria BL21 (DE3) (EC1001S, WEIDI 2nd lab, China). The positive BL21 bacterial cell was collected after 16 h of induced cultivation (ITPG.6 mmol/L). BL21 bacterial cell harboring empty pET30a vector was cultured and collected in the same way and set as a negative control. The bacterial cell was re-suspended in the reaction buffer (20 mmol/L Tris-HCl (pH 7.6), 100 mmol/L KCl, 10 mmol/L MgCl2, and 0.1 mmol/L DTT) and destroyed by ultrasonic wave. After centrifugation, the supernatant was derived, and the crude protein was analyzed by the SDS-PAGE gel segregation method. Then, the PAC1 protein was purified by affinity chromatography (Ni-IDA resin) method. The concentration of the purified protein was tested by the Bradford Protein Assay Kit (P0006, Beyotime Biotechnology, China). Western blot was conducted to test the activity of artificially synthesized PAC1 protein by using the Anti-His antibody.
Double-stranded RNA synthesis
Double-stranded RNA was artificially synthesized in vitro by using an RNA synthesize kit (MEGAscript® RNAi Kit, Invitrogen, USA). PCR primers, SCSMV 1F and 1R, were synthesized to amplify part of the genomic fragment of SCSMV Primer sequence shows in Table 1. T7 promoter (23 bp) was added to the 5′ site of all PCR amplification primers. Each 25 μl of the PCR reactant contained 12.5 μl of PCR Master Mix buffer (2× Taq PCR Master Mix, ComWin Biotech, China), 1 μl of cDNA template synthesized from each kind of virus by reverse transcription reaction, 1 μl of forwarding primer (10 μmol/L), 1 μl reverse primer (10 μmol/L), and 9.5 μl of double-distilled (dd) H2O. PCR amplification was conducted as follows. Initial polymerase activation was performed at 105°C for 5 min; 5 cycles at 95°C for 30 s, 66°C for 30 s, 72°C for 1 min; 25 cycles at 95°C for the 30 s, 70°C for 30 s, and 72°C for 1 min; and final extension at 72°C for 10 min. PCR production was purified by the Gel Extraction Kit (AP-GX-P-50G, Axygen, China). The purified PCR product and proteinase K (200 μg/mL) were mixed with the enzyme reaction buffer (10 mmol/L Tris-HCl at pH 8.0, 20 mmol/L EDTA at pH 8.0, 5% SDS, and 0.4 mmol/L NaCl), held at 50°C, and reacted for 30 min. Then, the phenol-chloroform (25:24) was added and maintained for 10 min. After centrifugation (13,000 r/min, 10 min), the supernatant was removed into a new tube (DNAse/RNAse free). Two-fold cold absolute ethyl alcohol was added and held at −20°C in the refrigerator for 1 h. After centrifugation (13,000 r/min, 10 min), the DNA sediment was washed with 70% DNAse/RNAse-free alcohol twice. After air drying, DEPC water was used to dilute the DNA sediment. By then, the DNA sediment was a double-stranded DNA that was connected with the T7 promoter.
Double-stranded DNA promoted by the T7 promoter was used as the template for the double-stranded RNA synthesis in vitro. According to the protocol that the RNA synthesis kit (MEGAscript® RNAi Kit, Invitrogen, USA) provided, the double-stranded RNA synthesis reactant was found to contain 1–2 μg double-stranded DNA template, 2 μl 10 × T7 Reaction Buffer, 2 μl ATP Solution, 2 μl CTP Solution, 2 μ GTP Solution, 2 μ UTP Solution, and 2 μl T7 Enzyme mix. DNAse/RNAse-free water was added to achieve a total volume of 20 μl. The mixture was mixed gently and kept for 4 h at 37°C. Then, reactants were placed in a PCR device, and the temperature conditions were set as follows: 95°C for 3 min, 75°C for 3 min, 50°C for 3 min, and finally, room temperature for 5 min. Then, 5 μl of 10× Digestion Buffer, 2 μl of DNase I (2 U/μl), 2 μl of RNase (2 U/μl), and 21 μl of Nuclease-free water were added. The mixture was maintained at 37°C for 1 h to degrade all double-stranded DNA templates and any residue that the single-stranded RNA may have formulated during the synthesis process of the double-stranded RNA. After 1 h of degradation, 50 μl of 10× Binding Buffer, 250 μl of 100% Ethanol, and 150 μl of nuclease-free water were added. The 500-μl mixture was moved into the filter element provided by the RNA synthesis kit. After centrifugation (13,000 r/min, 10 min), the filtrate was removed, and 500 μl of 2× wash solution was added to the filter element. Centrifugation (13,000 r/min, 10 min) was performed, and the wash solution was removed. Pre-warmed (90°C) DEPC water (100 μl) was added to the filter element to elute the double-stranded RNA. Centrifugation was performed (13,000 r/min, 10 min), and the elution was transferred into a new nuclease-free tube. The elution (0.5 μl) was verified by 1% agarose gel electrophoresis. The concentration of the double-stranded RNA was tested by spectrophotometry.
PAC1 protein functional analysis
Purified PAC1 protein and double-stranded RNA were mixed with enzyme reaction buffer (10 mmol/L of Tris-HCl at pH 8.0, 20 mmol/L of EDTA at pH 8.0, 5% SDS, and 0.4 mmol/L of NaCl) to achieve a total volume of 1,500 μl. The mixture was left to stand at 37°C for the degradation reaction. After 10 and 30 min, half of the reactant (750 μl) was removed, and 750 μl of phenol-chloroform was added; a ratio of 25:24 was obtained. Centrifugation was performed (12,000 r/min, 10 min), and the supernatant was transferred to a new nuclease-free tube. A second extraction was performed to eliminate PAC1 protein and to block the degradation reaction. NaAc (3 mol/L, pH 5.2) at 35 μl and 2-folds of cold 100% ethanol were added to the supernatant, and the mixture was left to stand at −20°C in the refrigerator for 1 h. Centrifugation was performed (13,000 r/min, 10 min), and the supernatant was removed. The sediment was washed twice by using 70% ethanol. The sediment was dissolved in 20-μl nuclease-free water. The degradation level of double-stranded RNA was checked by 1% agarose gel electrophoresis. The double-stranded RNA that was not mixed with purified PAC1 protein but subjected to the same process was set as the negative control.
Plant expression vector construction
By using the BamH I and Sac I sites, the Pac1 gene was isolated from the TA cloning vector (pMD19T-Pac1), ligated to the plant binary vector (pCAMBIA-3300-Ubi-GFP), and replaced the GFP gene on the vector. The binary vector was constructed in our previous research (Wang et al., 2017a,b). It comprised a selectable marker gene of the bar gene. The binary vector pCAMBIA3300 contained the npt II gene for bacterial selection. After recombination, the Pac1 gene was promoted by Ubi 1 promoter. After PCR and enzyme validation, the recombination binary vector (pCAMBIA-3300-Ubi-Pac1) was mobilized into Agrobacterium EHA105 via freeze-thawing with liquid nitrogen.
Genetic transformation
Transgenic plants were obtained by the bar/Basta transformation selection system, which was established in our previous research (Wang et al., 2017a,b). ROC22, which is the main variety cultivated in China, was chosen as the receptor for molecular genetic improvement. After two times of sub-culturing, light yellow and compact calli were selected and fragmented before the transformation. After streaking on the solid YEP medium in the Petri dish, a single colony of Agrobacterium EHA105 that harbored the target vector was cultured again in fresh liquid YEP medium at 28°C overnight. Bacterial cells were collected via centrifugation (5,000 r/min, 10 min) and then resuspended in a starter culture of liquid initiating medium and diluted to an optical density of ~0.6 at 600 nm. The starter culture was vortexed at 100 r/min for 2 h at 28°C before infection. The finely prepared calli were first air-dried and then transferred to an Erlenmeyer flask. An appropriate amount of initiated Agrobacterium suspension was added into the Erlenmeyer flask and mixed gently with the calli. After 30 min of infection, the Agrobacterium suspension was then pipetted out. The calli were transferred to a Petri dish, blotted dry with filter paper to remove the excess Agrobacterium suspension, and air-dried for approximately 30 min on the clean bench. The infected calli were transferred to a new empty Petri dish and co-cultivated in the dark at 22°C for 3 days. After co-cultivation, all calli were transferred for resting cultivation for ~7 days. Subsequently, all calli were transferred for selection cultivation for 30 days in the dark at 28°C. After about 30 days of selection by glufosinate, resistant calli were transferred for regeneration cultivation (14 days), and, finally, for elongation cultivation (28 days). Resistant shoots were then sampled for molecular analysis, and positive shoots were transferred to the soil. More details on the transformation procedure and the medium recipe have been presented in our published article (Wang et al., 2017a,b).
Molecular analysis
Total genomic DNA extracted from resistant shoots was first used for traditional PCR analysis. Primers Bar 1F and 1R, Pac1 2F and 2R, and VirG 1F and 1R were designed and synthesized for PCR detection of Bar gene, Pac1 gene, and VirG, respectively Primer sequence shows in Table 1. Genomic DNA from wild-type plants was used as the negative control, and binary vector (pCAMBIA-3300-Ubi-Pac1) was used as the positive control.
Total RNA of resistant shoots was extracted by an RNA extraction kit (E.Z.N.ATM Plant RNA Kit, Omega, USA). The first strand's cDNA synthesis was conducted using a first strand cDNA synthesis kit (RevertAid First Strand cDNA Synthesis Kit, Thermo Fisher, USA). In the nuclease-free tube, 6 μl of the total RNA mix with 1 μl of Oligo (dT)18 Primer and 5 μl nuclease-free water were mixed. The mixture was subjected to a temperature of 65°C for 5 min and then rapidly transferred to the ice for 2–3 min. The 4 μl of 5× Reaction Buffer, 1 μl of RiboLock RNase Inhibitor (20 U/μl), 2 μl of 10 mmol/L dNTP Mix, and 1 μl of RevertAid M-MuLV RT (200 U/μl) were added to the tube and mixed gently. The mixture was maintained at 42°C for 1 h and then at 70°C to end the reverse transcript reaction. Then, 2 μl of the reactant was used as the cDNA template for RT-PCR detection. Negative and positive control settings were the same as those used in traditional PCR detection.
Virus resistance analysis
Each traditional PCR and RT-PCR positive shoot were transplanted into a plastic pot for virus inoculation. SCSMV is a stronger pathogen compared with the other pathogens that induce sugarcane mosaic disease. So, single SCSMV strain infected sugarcane leaves were collected as the original pathogen for the transgenic shoot virus resistance testing. The inoculation protocol was described by Li et al. (2008). The SCSMV infected leaves were cut into small pieces and ground fully into muddy status with the help of silica sand. Phosphate buffer at 3:1 (v/g) (pH 7.2) was added to the ground leaves, and then, the mixture was filtrated through a cotton gauze. The filtered juice was used for artificial inoculation. Inoculation was conducted thrice in a gap of 1 week to increase the efficiency of successful inoculation. Wild-type plants were inoculated by the same pathogen and served as the negative control. Eight months later, leaf samples were collected from each transgenic shoot for total RNA extraction. RT-PCR detection of SCSMV was conducted to analyze the positive inoculation. Then, three axillary buds were cut from the shoots showing positive infection and propagated in plastic pots. Three months later, the symptoms and viral loads of the propagated plants were observed and interpreted. Symptoms on the leaves were recorded by a digital camera. RT-qPCR was conducted to compare the viral loads among different transgenic shoots. Ten months later (late growing period), the heights of all axillary buds of propagated shoots were determined.
Results
Pac1 gene isolation and sequencing
Pac1 gene PCR production was purified by the Gel Extraction Kit and then successfully integrated into the TA cloning vector. The sequencing result shows that the target fragment ligated in the TA cloning vector was 1,092 bp in size, and the sequence showed 100% identity with the sequence that Prof. Li submitted in the GenBank (Sequence ID: AY695822.1). Thus, we successfully isolated the Pac1 gene from the presented plasmid vector from that of Prof. Li's. Otherwise, the sequencing result showed that BamH I and Sac I restriction enzyme-cutting sites had been added at the 5′ and 3′ sites, respectively, which could be utilized for plant expression vector construction.
PAC1 protein prokaryotic expression
By utilizing Nde I and Hind III restriction enzymes, the Pac1 gene with an N-His tag was ligated to the prokaryotic expression vector (pET30a, Biodee, China). Positive plasmid pET30a-Pac1 was successfully identified by restriction enzyme digestion (Figure 1A) and then transformed into receptor bacteria BL21 (DE3). After 16 h of inducing cultivation, crude protein was extracted from the positive BL21 bacterial cell and the negative control bacterial cell. After segregation by SDS-PAGE and in comparison with negative control, the BL21 bacterial cell harboring the pET30a-Pac1 vector expressed a specific protein that is similar to the target size 43 kDa (Figure 1B). Thus, we successfully expressed PAC1 protein by prokaryotic expression. Then, the PAC1 protein was purified by affinity chromatography (Ni-IDA resin) method (Figure 1C). The concentration of the purified protein was 0.151 mg/ml, which was tested by Bradford Protein Assay Kit (P0006, Beyotime Biotechnology, China). Western blot shows prokaryotic expressed PAC1 protein interacted with Anti-His Antibody strongly demonstrated the fine activity of purified PAC1 protein.
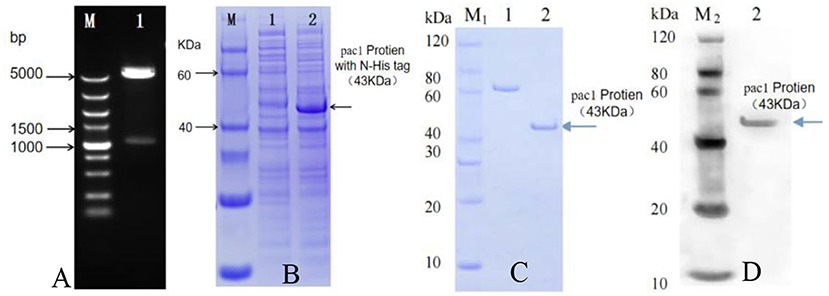
Figure 1. Pac1 protein prokaryotic expression and purification. (A) pET30a-Pac1 enzyme digestion by Nde I and Hind III. Lane M: DNA maker ladder; Lane 1: pET30a-Pac1 plasmid. (B) Prokaryotic expression of Pac1 protein. Lane M: Protein maker ladder; 1: negative control bacterial cell protein; 2: pET30a-Pac1 bacterial cell protein. (C) Quality assurance of purified Pac1 protein by SDS-PAGE. M1: SDS-PAGE Marker; 1: BSA (1.2 μg); 2: Pac1 protein (1.2 μg) purified affinity chromatography (Ni-IDA resin) method. (D) Activity assurance of purified Pac1 protein by Western blot. M2: Western blot marker; 2: purified Pac1 protein (1.2 μg) interacted with Anti-His Antibody.
PAC1 protein functional analysis
After the artificial synthesis, 985 bp of double-stranded RNA was obtained by using the genome of SCSMV as the original synthesis template. Mixing of 7.5 μg purified PAC1 protein with 3 μg of double-stranded RNA was conducted in the nuclease-free tube. After 10 and 30 min of degradation, 10 μl of the degraded double-stranded RNA was checked by 1% agarose gel electrophoresis, respectively. Results in Figure 2 showed that double-stranded RNA was degraded clearly by PAC1 protein in 10 min. However, the control double-stranded RNA maintained stability throughout the same treatment process. So, it means that the purified PAC1 protein could degrade double-stranded RNA efficiently.
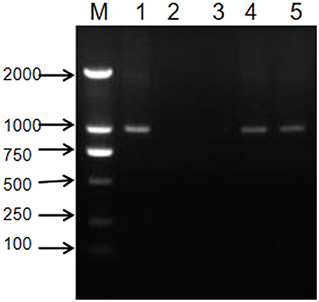
Figure 2. Functional analysis of PAC1 protein. M: DNA maker ladder; 1: artificially synthesized double-stranded RNA; 2: 10 min after degradation; 3: 30 min after degradation; 4: 10 min without degradation but with the same treatment process; 5: 30 min without degradation but with the same treatment process.
Plant expression vector construction and verification
After successful construction, the recombination Pac1 gene was promoted by the Ubi promoter and ended by the tNOS terminator (Figure 3). The selectable marker bar gene was promoted by CaMV 35S promoter and ended with 35S poly A. The positive recombination binary vector (pCAMBIA-3300-Ubi-Pac1) was identified by restriction enzyme analysis (Figure 4A). Then, the recombination binary vector was mobilized into Agrobacterium EHA105. The single colonies of the transformed bacterium were successfully analyzed by PCR assay individually (Figure 4B). Thus, the Pac1 gene plant expression vector was successfully constructed and transformed to the Agrobacterium EHA105 strain.
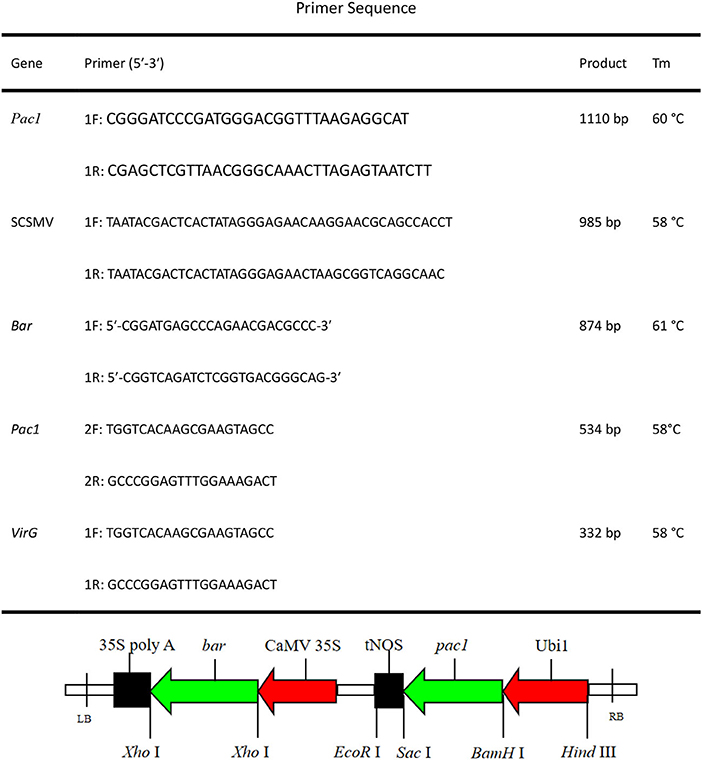
Figure 3. Pac1 gene plant expression vector (pCAMBIA-3300-Ubi-Pac1). Ubi 1, maize Ubi 1 promoter; tNOS, nopaline synthase terminator; CaMV 35S, cauliflower mosaic virus 35S promoter; 35S poly A, cauliflower mosaic virus 35S poly A terminator.
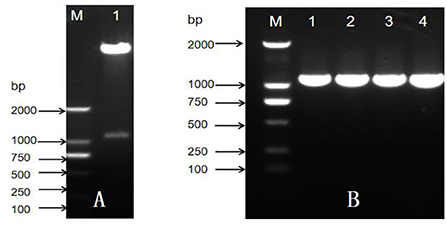
Figure 4. Construction and verification of plant expression vector. (A) Restriction enzyme analysis of plant expression vector, M: DNA maker ladder; 1: plasmid vector. (B) PCR assay of Agrobacterium EHA105 single colony, M: DNA maker ladder; 1–4: EHA105 single colony.
Genetic transformation and molecular analysis
Figure 5A shows post-subculturing of light yellow, and compact calli were obtained and selected for transformation. Then, transformed calli were screened by selection medium supplemented with 2 mg/L of Basta. Figure 5B shows that after 30 days of selection, resistant calli sprouted and showed significant resistance on the screening medium. Figure 5C shows that after 14 days of regeneration, the resistant calli emerged quickly on the regeneration medium supplemented with 2 mg/L of Basta. Figure 5D shows that after more than 28 days of rooting cultivation, the resistant shoots were dark green and strong enough for transplanting. In the end, fifteen resistant shoots were obtained and successfully transplanted in the greenhouse (Figure 5E). All resistant shoots were sampled and evaluated for bar gene and Pac1 gene integration by PCR and RT-PCR assay.

Figure 5. Genetic transformation and resistant shoot selection. (A) Embryogenic calli prepared for transformation. (B) After 30 days of calli selection, resistant calli sprouted and grew healthily on the selection medium. (C) After 14 days of regeneration, healthy resistant calli emerged rapidly. (D) After 28 days of elongation, dark-green shoots grew quickly. (E) Resistant shoots were transplanted in the greenhouse.
Molecular assay results in Table 1 show that the 15 resistant shoots were positive for the bar gene. PCR detection showed that 14 resistant shoots were positive for the Pac1 gene. All the resistant shoots were PCR negative for the VirG gene, thereby suggesting that the resistant sugarcane plants were free from Agrobacterium. RT-PCR assay results showed that 13 resistant shoots were both PCR and RT-PCR positive for bar and Pac1 genes. Then, 13 resistant shoots that were positive for bar and Pac1 genes according to the PCR and RT-PCR results were chosen for further research.
Virus resistance analysis
After inoculation by strong pathogen SCSMV and cultivation in the greenhouse for 8 months, samples were collected from each transgenic shoot for the RT-PCR assay. The result in Table 1 shows that all the transgenic and wild-type shoots were successfully infected by the same pathogen SCSMV. Also, classic leaf mosaic symptoms were visible on all axillary buds of propagation shoots. However, upon closer observation, the mosaic symptoms on all transgenic shoots' leaves were mild compared with the serious mosaic symptoms on the wild-type shoots (Figure 6). All the transgenic shoots significantly attained greater heights compared with the wild-type shoots (Figures 6, 7). Transgenic shoots T5 and T7 were significantly shorter than normal uninfected shoots (without viral infection). However, other transgenic shoots T1, T2, T3, T4, T5, T6, T9, T12, T13, T14, and T15 were not significantly different from normal shoots. By comparison, wild-type shoots infected by the pathogen SCSMV were significantly shorter than normal shoots. RT-qPCR test results showed that all transgenic shoots had lower viral loads than wild-type shoots (Figure 8).

Figure 6. Phenotypic observation of axillary bud propagation shoots (Three months after transplant in green house). (A) Serious mosaic symptoms on the wild-type shoots' leaves. (B) Mild mosaic symptoms on the transgenic shoots' leaves. (C) Normal shoots' leaves (without SCSMV infection). (D) Wild-type shoots infected by SCSMV were shorter and more slender than transgenic shoots (T5). (E) Transgenic shoots (T5) infected by SCSMV significantly attained greater heights than wild-type shoots but were still significantly shorter than normal shoots. (F) Normal shoots (without SCSMV infection).
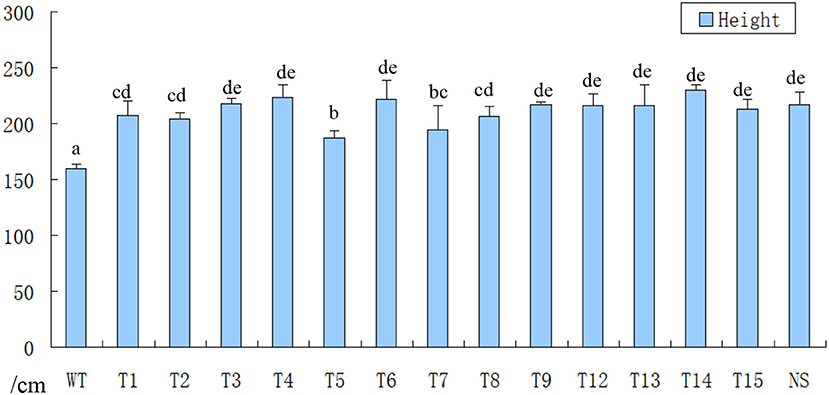
Figure 7. Height measurement of axillary bud propagation shoots (Ten months after transplant in green house). WT, infected wild-type shoots were significantly shorter than all transgenic shoots; T1–T15, infected transgenic shoots attained greater heights than wild-type shoots, and some transgenic shoots were shorter than normal shoots; NS, normal shoots (without SCSMV infection).
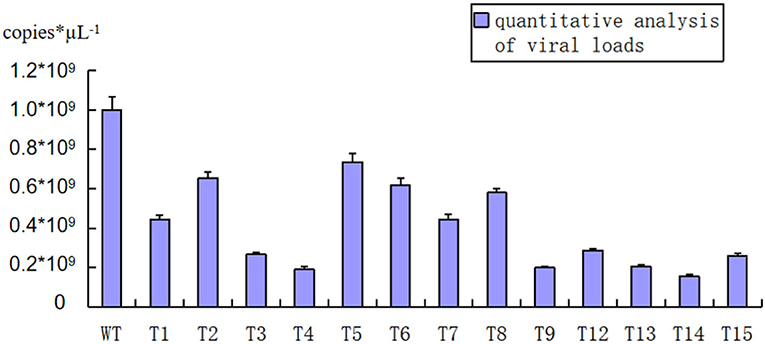
Figure 8. RT-qPCR testing of SCSMV viral loads for each transgenic and wild-type shoot (Three months after transplant in green house). WT, wild-type shoots showed higher SCSMV viral loads than transgenic shoots; T1-T15, transgenic shoots showed lower SCSMV viral loads than wild-type shoots significantly.
Discussion
Many RNA virus diseases on sugarcane decrease its yield and sugar content (Wu et al., 2012). Mixed infection of multiple RNA viruses results in increasingly serious damage. Breeding new varieties with broad-spectrum virus resistance is one of the main goals of sugarcane disease resistance breeding. For both single-stranded and double-stranded RNA viruses, a double-stranded RNA replicative form (RF) exists in the replication cycle. Double-stranded RNA-specific ribonuclease encoded by the Pac1 gene can specifically recognize and degrade double-stranded RNA (Lino et al., 1991; Rotondo and Frendwwey, 1996). The expression of double-stranded RNA-specific ribonuclease in sugarcane varieties leads to the degradation of double-stranded RNA RF produced by the RNA virus in its replication cycle, thereby, blocking the virus replication process so that it cannot finish the systemic infection in sugarcane and resulting in the development of broad-spectrum virus-resistant sugarcane novel germplasm.
In this research, we expressed the PAC1 protein by prokaryotic expression, and a purified protein was obtained. SCSMV double-stranded RNA was synthesized by using a cDNA template from SCSMV. In vitro, functional assay showed that the purified PAC1 protein degraded the double-stranded RNA in 10 min.
After the successful functional assay, we integrated the Pac1 gene into the genome of a virus-sensitive variety ROC22. Fifteen resistant shoots were obtained after selection by glufosinate. After the molecular assay, 13 transgenic shoots remained and were used for further research. SCSMV, which induces sugarcane mosaic disease, was chosen to test the virus resistance of transgenic shoots. After three times of inoculation, all 13 transgenic shoots were infected by the pathogen, and classic symptoms appeared on the leaves. This finding meant that the transgenic shoots integrated with the Pac1 gene were still sensitive to the strong pathogen SCSMV. However, a good result was obtained, because the classic mosaic symptom and viral loads were better than those of non-transgenic shoots. Considering that they had lower viral loads and mild mosaic symptoms, all transgenic shoots grew better than non-transgenic shoots, although their growth was still poor compared with that of the healthy non-transgenic shoots without infection. So, if the transgenic shoots and the non-transgenic were planted in the field and were both infected by the same RNA virus pathogen, the transgenic shoots would surely produce a higher yield. For further research, the promoter for the Pac1 gene may be changed to increase the concentration of PAC1 protein in the transgenic shoots or ligated with a tag that can guide the PAC1 protein to the sub-cellular organelles, where the virus' double-strand RNA replicative form (RF) can be found. Through this strategy, the resistance level of the transgenic shoots to the RNA viral disease can be promoted. This research provided a new pathway for breeding new varieties of sugarcane with virus resistance.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/nucleotide/, AY695822.
Author contributions
WW take charge of the operation of the entire research and paper writing. SL provide the original plasmid harboring the pac1 gene. JW and HX take charge of the construction of the plant expression vector. XF take charge of virus resistant testing of the transgenic shoots. LS take charge of the planting and management of the transgenic shoots. CF helped to the tissue culture of the transformation of sugarcane. TZ and HW helped to finish the molecular assay of the transgenic shoots. SZ provide the financial support and is the director of the original research and paper writing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Hainan Yazhou Bay Seed Lab (B21HJ0302), Hainan Provincial Natural Science Foundation of China (2019RC300) and Central Public-interest Scientific Institution Basal Research Fund for Chinese Academy of Tropical Agricultural Sciences (1630052019019).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Butterfield, M. K., Irvine, J. E., Valdez Garza, M., and Mirkov, T. E. (2002). Inheritance and segregation of virus and herbicide resistance transgenes in sugarcane. Theor. Appl. Genet. 104, 797–803. doi: 10.1007/s00122-001-0830-z
Dewanti, P., Widuri, L. I., Ainiyati, C., Okviandari, P., and Maisaro Sugiharto, B. (2016). Elimination of SCMV (sugarcane mozaik virus) and rapid propagation of virus-free sugarcane (Saccharum officinarum L.) using somatic embryogenesis. Procedia Chem. 18, 96–102 doi: 10.1016/j.proche.2016.01.016
He, Z., Yasaka, R., Li, W. F., Li, S. F., and Ohshima, K. (2016). Genetic structure of populations of sugarcane streak mosaic virus in China: comparison with the populations in India. Virus Res. 211, 103–116. doi: 10.1016/j.virusres.2015.09.020
Ishida, I., Takahara, M., Yoshioka, M., Ogawa, T., Kakitani, M., and Toguri, T. (2002). Production of anti-virus, viroid plants bygenetic manipulations. Pest Manage. Sci. 58, 1132–1136. doi: 10.1002/ps.536
Kasemsin, P., Chiemsombat, P., and Hongprayoon, R. (2016). Characterization and genetic variation of sugarcane streak mosaic virus, a poacevirus infecting sugarcane in Thailand [J]. Modern Appl. Sci. 10:137. doi: 10.5539/mas.v10n4p137
Li, L., Li, S. F., Guo, L. H., and Wang, H. Q. (2008). In vitro activity assay of dsRNA nuclease Pac1 to digest dsRNAs of 4 plant viruses and 3 viroids, and Pac1 transgenic tobacco mediated by agrobacterium tumefaciens. Acta Phytopathol. Sinica 38, 489–495. doi: 10.13926/j.cnki.apps.2008.05.006
Lin, Y. H., Gao, S. J., Damaj, M. B., Fu, H. Y., Chen, R. K., and Mirkov, T. E. (2014). Genome characterization of sugarcane yellow leaf virus from China reveals a novel recombinant genotype. Arch. Virol. 159, 1421–1429. doi: 10.1007/s00705-013-1957-3
Lino, Y., Sugimoto, A., and Yamamoto, M. (1991). S.pombe Pac1, whose overexpression inhibits sexual development, encodes a ribonuclease III-like RNase. EMBO 10, 221–226. doi: 10.1002/j.1460-2075.1991.tb07939.x
López-Ortega, M. G., Guadalajara, Y., Junqueira, T. L., Sampaio, I. L. M., Bonomi, A., and Sánchez, A. (2021). Sustainability analysis of bioethanol production in Mexico by a retrofitted sugarcane industry based on the Brazilian expertise. Energy 232:121056. doi: 10.1016/j.energy.2021.121056
Luo, Q., Ahmad, K., Fu, H. Y., Wang, J. D., Chen, R. K., and Gao, S. J. (2016). Genetic diversity and population structure of Sorghum mosaic virus infecting Saccharum spp. hybrids. Annals Appl. Biol. 169, 398–407. doi: 10.1111/aab.12310
Manickavasagam, M., Ganapathi, A., Anbazhagan, V. R., Sudhakar, B., Selvaraj, N., Vasudevan, A., et al. (2004). Agrobacterium-mediated genetic transformation and development of herbicide-resistant sugarcane (Saccharum species hybrids) using axillary buds. Plant Cell Rep. 23, 134–143. doi: 10.1007/s00299-004-0794-y
Moradi, Z., Mehrvar, M., Nazifi, E., and Zakiaghl, M. (2016). The complete genome sequences of twonaturally occurring recombinant isolates of Sugarcane mosaic virus, from Iran. Virus Genes 52, 1–11. doi: 10.1007/s11262-016-1302-5
Rao, G. P., Chatenet, M., Girard, J. G., and Rott, P. (2006). Distribution of sugarcane mosaic and sugarcane streak mosaic virus in India. Sugar Tech. 8, 79–81. doi: 10.1007/BF02943747
Rotondo, G., and Frendwwey, D. (1996). Purification and characterization of the pac 1 ribonuclease of Schizosaccharomyces pombe. Nuclei Acids Res. 24, 23377–22386. doi: 10.1093/nar/24.12.2377
Sano, T., Nagayama, A., Ogawa, T., Insida, I., and Okada, Y. (1997). Transgenic potato expressing dsRNA specific ribonuclease is resistant to potato spindle tuber viroid. Nat. Biotechnol. 15, 1290–1294. doi: 10.1038/nbt1197-1290
Singh, V., Sinha, O. K., and Kumar, R. (2003). Progressive decline in yield and quality of sugarcane due to Sugarcane mosaic virus. Indian Phytopathol. 56, 500–502. doi: 10.1046/j.1365-2052.1999.00445-6.x
Snyman, S. J. (2004). Sugarcane transformation. Transgenic Crops of the World 2004, 103–114. doi: 10.1007/978-1-4020-2333-0_8
Suprasanna, P., Patade, V. Y., Desai, N. S., Devarumath, R. M., Kawar, P. G., Pagariya, M. C., et al. (2011). Biotechnological developments in sugarcane improvement: an overview. Sugar Tech 13, 322–335. doi: 10.1007/s12355-011-0103-3
Viswanathan, R., and Balamuralikrishnan, M. (2005). Impact of mosaic infection on growth and yield of sugarcane. Sugar Tech 7, 61–65. doi: 10.1007/BF02942419
Viswanathan, R., Parameswari., B, and Nithya, K. (2018). Molecular characterization of sugarcane viruses and their diagnostics. Crop Improvement Through Microbial Biotechnol. 2018, 175–193. doi: 10.1016/B978-0-444-63987-5.00008-6
Wang, W. Z., Yang, B. P., Feng, C. L., Wang, J. G., Xiong, G. R., Zhao, T. T., et al. (2017a). Efficient sugarcane transformation via bar gene selection. Tropical Plant Biol. 10, 77–85. doi: 10.1007/s12042-017-9186-7
Wang, W. Z., Yang, B. P., Feng, X. Y., Cao, Z. Y., Feng, C. L., Wang, J. G., et al. (2017b). Development and characterization of transgenic sugarcane with insect resistance and herbicide tolerance. Front. Plant Sci. 8, 1535–1545. doi: 10.3389/fpls.2017.01535
Watanabe, Y., Ogawa, T., Takahashi, H., Insida, I., Takeuchi, Y., Yamamoto, M., et al. (1995). Resitance against multiple plant viruses in plants mediated by double strant-RNA specific ribonuclease. FEBS Lett. 372, 165–168. doi: 10.1016/0014-5793(95)00901-K
Wu, L., Zu, X., Wang, S., and Chen, Y. (2012). Sugarcane mosaic virus–long history but still a threat to industry. Crop. Prot. 42, 74–78 doi: 10.1016/j.cropro.2012.07.005
Zhang, L., French, R., Langenberg, W. G., and Mitra, A. (2001). Accumulation of barley stripe mosaic virus is significantly reduced in transgenic wheat plants expressing a bacterial ribonuclease. Transgenic Res. 10, 13–19. doi: 10.1023/A:1008931706679
Keywords: sugarcane, double-stranded RNA-specific ribonuclease, PAC1 gene, broad-spectrum virus-resistant, genetic engineering
Citation: Wang W, Wang J, Feng X, Shen L, Feng C, Zhao T, Xiao H, Li S and Zhang S (2022) Breeding of virus-resistant transgenic sugarcane by the integration of the Pac1 gene. Front. Sustain. Food Syst. 6:925839. doi: 10.3389/fsufs.2022.925839
Received: 22 April 2022; Accepted: 14 July 2022;
Published: 15 August 2022.
Edited by:
Zhengnan Li, Inner Mongolia Agricultural University, ChinaReviewed by:
De-jie Cheng, Guangxi University, ChinaXingan Hao, Northwest Agriculture & Forest University, China
Copyright © 2022 Wang, Wang, Feng, Shen, Feng, Zhao, Xiao, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenzhi Wang, d2FuZ3dlbnpoaUBpdGJiLm9yZy5jbg==; Shifang Li, c2ZsaUBpcHBjYWFzLmNu; Shuzhen Zhang, emhhbmdzaHV6aGVuQGl0YmIub3JnLmNu