 Blessing Chidinma Nwachukwu
Blessing Chidinma Nwachukwu Olubukola Oluranti Babalola*
Olubukola Oluranti Babalola*- Food Security and Safety Focus Area, Faculty of Natural and Agricultural Sciences, North-West University, Mmabatho, South Africa
Microorganisms are immense in nature and exist in every imaginable ecological niche, performing a wide range of metabolic processes. Unfortunately, using traditional microbiological methods, most microorganisms remain unculturable. The emergence of metagenomics has resolved the challenge of capturing the entire microbial community in an environmental sample by enabling the analysis of whole genomes without requiring culturing. Metagenomics as a non-culture approach encompasses a greater amount of genetic information than traditional approaches. The plant root-associated microbial community is essential for plant growth and development, hence the interactions between microorganisms, soil, and plants is essential to understand and improve crop yields in rural and urban agriculture. Although some of these microorganisms are currently unculturable in the laboratory, metagenomic techniques may nevertheless be used to identify the microorganisms and their functional traits. A detailed understanding of these organisms and their interactions should facilitate an improvement of plant growth and sustainable crop production in soil and soilless agriculture. Therefore, the objective of this review is to provide insights into metagenomic techniques to study plant root-associated microbiota and microbial ecology. In addition, the different DNA-based techniques and their role in elaborating plant microbiomes are discussed. As an understanding of these microorganisms and their biotechnological potentials are unlocked through metagenomics, they can be used to develop new, useful and unique bio-fertilizers and bio-pesticides that are not harmful to the environment.
Introduction
The soil environment is a highly complex terrestrial environment and one of the main reservoirs for microbial diversity, harboring a wide range of different genomes. The soil is the hotspot of a myriad of agriculturally beneficial microorganisms. These microorganisms interact both with one another and with plants to improve soil quality and enhance crop productivity. The microorganisms therefore have positive influences on agricultural sustainability. Sustainable and innovative agriculture are among the solutions to the global challenges regarding the degeneration of the quality and quantity of natural resources for growing crops and identifying key agriculturally beneficial microorganisms and their function.
The relationships between agriculture, environmental sustainability and social systems suggest that the developments in agriculture are dependent upon many complex interactions including green technology and innovations, biodiversity conservation, plant and animal health, cultural acceptance, pollution management, economy, political stability (Goel et al., 2017) and urban agriculture. Moreover, some estimates suggest that the world population will reach 10 billion by 2050, and with such dramatic population growth there is a need to improve crop production for sustainable agriculture and food security (Levy and Lubell, 2018).
In 2015, the United Nations established 17 Sustainable Development Goals (SDGs) to improve the livelihood of human populations worldwide. The SDGs second goal termed Zero Hunger was proposed to solve the problem of hunger by advancing sustainable agriculture globally, hence, accomplishing global food security and increased global nutrition. The goal of this agenda is to seek sustainable solutions to end all kinds of hunger, thereby achieving food security in the world by 2030 (Grafton et al., 2015). The SDGs target is to ensure that everyone globally has good quality food and a healthy life.
To realize this goal, there should be demand for better access to quality food, widespread awareness of modern agriculture and the use of advanced technologies (Agler et al., 2016). To improve agricultural productivity governments and agricultural stakeholders can provide assistance to farmers including financial incentives for small scale farmers, promotion of equal access to land, trainings on the use of innovative technologies, creation of markets that are suitable food promotion systems, and good agricultural practices. Also, there is a need to increase investment and research through international bodies to boost agricultural production capacity in developing countries (Singh et al., 2019). Enabling environment for sustainable production of foods in bioponic systems, which is a type of urban agriculture should be encouraged. Urban agriculture is an emerging technology that aims to tackle the different aspects of “urban sustainability” which include but not limited to food security, waste management and equitable food supply (Lovell and Taylor, 2013; Ackerman et al., 2014; Russo et al., 2017). In developed nations, urban planning captured agriculture as a transient activity for empty geographical areas that can later be converted to more lucrative commercial, industrial and residential land uses (Van Veenhuizen and Danso, 2007). Nevertheless, urban agriculture has become a durable initiative by improving profits via mass production of perishable vegetables that are costly to transport from rural settlements, specific marketing approaches including plant growth using organic manure, crop selection diversification, and integrating non-agricultural services, e.g., tourism, establishment of decent and lively dwelling environment, cooking services, nature rehabilitation, and environmental enlightenment (Plakias, 2016; Pölling et al., 2016, 2017).
Almost two decades, the fight against world hunger has experienced little progress due to the gradual integration of advanced technologies into farming practices in the developing nations. Despite a global decrease in the number of undernourished persons in the past 15–20 years, over 790 million people still experience irregular access to an adequate food supply (Grafton et al., 2015). Thus, if this current trend continues the zero hunger mandates will not be achieved by 2030.
Therefore, the need to increase production strategies and overcome various challenges is critical. It is necessary to make the availability of nutrients a priority, and to give precedence to the cultivation of plants that are more tolerant of marginal lands, biotic factors, and environmental changes (Chadwick et al., 2015; Brader et al., 2017).
Studies have shown that improving soil health can increase crop productivity by 10–50%, and when it is combined with the use of microorganisms with plant growth-promoting traits there can be 50–60% productivity increase (Abram, 2015). This approach can reduce the use and dependence on chemical fertilizers. To reach sustainable agriculture goals many agricultural microorganisms are present as an alternative to chemicals. Unfortunately, it is estimated that 99% of microorganisms found in the natural environment are non-culturable using conventional techniques (Chadwick et al., 2015; Brader et al., 2017).
Microbial communities perform essential functions in the soil ecosystem despite the fact that the functions of the unculturable microorganisms are largely unknown. Microorganisms differ in the ways that they forage for food, harness energy, interact, compete, and associate with each other, however, the diversities that we are aware of are just a small part of the microbial population. To better understand the total soil microbial community, it is possible to employ metagenomics, the sequencing and analysis of DNA extracted from environmental samples. This approach may be referred to as ecogenomics, environmental genomics, or community genomics (Abram, 2015).
Metagenomics can contribute significantly to the study of agroecosystems. Metagenomics can facilitate an understanding of the huge microbial diversities available in the soil environment, for the production of novel molecules of therapeutic importance, biotechnological advancements, and for sustainable agriculture (Garrido-Oter et al., 2018). Until now, most previous research has focused on biological features of cultivable microorganisms and how these organisms can decrease environmental impacts. While this approach has met with some measure of success, it is intrinsically limited due to the fact that it ignores the role of the vast majority of soil microorganisms (Bevivino and Dalmastri, 2017).
In the soil environment, the interactions between microbial community members help in maintaining the soil ecosystem integrity, nutrient recycling, conservation of natural habitats, balance of the food chains, and important physiological activities in the plants, and animals. A highly diverse microbial taxa are also linked with plants in a singular entity that is known as holobiont (O'brien et al., 2019; Suárez and Stencel, 2020); these plants associated microbial taxa interactions could be neutral, pathogenic or beneficial (Igiehon and Babalola, 2018). Thus, all agricultural production depends on closely related biological organisms that co-inhabit and interact with microorganisms and their internal and external environments (Martínez-Porchas and Vargas-Albores, 2017).
The study of microbial communities associated with agricultural practices is not yet well-studied (Bevivino and Dalmastri, 2017). However, the emergence of metagenomics has offered exceptional opportunities to understand the microbial community structure in a specific microniche of interest. This approach is important, since microbial community structure provide insights into the characteristics of microbes (e.g., bacteria, archaea, and eukaryotes) as determined by functional gene composition including the microbial composition (that is, the identity and relative abundances of all taxa) in the community (Philippot et al., 2013). Therefore, understanding the structural profile of bacterial, archaeal, and fungal communities and their compositions is now possible (Martínez-Porchas and Vargas-Albores, 2017) in soil and soilless agriculture.
The Problematics of Urban Agriculture: A Motivation for the Use of Omics Tools to Ensure Sustainable Agriculture
Urban agriculture involves food production and consumption within cities and it's practiced by different individuals motivated and driven by different socio-economic conditions (Simon Rojo et al., 2014). It can range from “urban gardening” to “large scale urban farming” (DuŽí et al., 2017).
It is sacrosanct to mention some of the adverse effects of urban agriculture irrespective of its positive benefits (DuŽí et al., 2017), as such knowledge will help to decide on the way to go to ensure environmental and agricultural sustainability. Some urban areas are presently not conducive to live in since they are characterized with a particular amount of air pollution cum soil contamination from activities such as urban agriculture. Also, urban soils especially soils situated close to industrial clime or heavy traffic roadways may habour organic wastes that may pose some health and environmental hazards (Nehls et al., 2015; Schwarz et al., 2016). Urban agriculture can also result in the degradation of urban milieu by contributing to increase in carbon footprint if it is not practiced in an eco-friendly manner (DuŽí et al., 2014; Mok et al., 2014). Therefore, despite the stern motivation to encourage urban agriculture, the environmental and health implications of this practice should not be undermined (DuŽí et al., 2017). Also agricultural practices in the rural areas have also come with their shortcomings. Some of these challenges can be addressed by harnessing the microbiome of such environments as it is believed that some microbes are endowed with the capacity to remove and/or detoxify pollutants emanating from agricultural and industrial sites (Igiehon, 2015; Igiehon and Babalola, 2019). Such microbes with novel ability to create a pollutant-free environment can be detected by exploring omics tools (Guerra et al., 2018; Ma et al., 2020; Ruiz et al., 2021).
Omics Approaches Used for Soil and Soilless Microbiome Analyses in the Context of Sustainable Agriculture
In the mid-1980s, scientists began to sequence the 16S rRNA genes from isolated bacterial samples (Head et al., 1998; Handelsman, 2004; Brader et al., 2017; Fadiji and Babalola, 2020). In 1998, the term metagenome came into general use when Handelsman et al. (1998) described soil microorganisms as essential sources of novel natural compounds. In general, studies conducted using a metagenomic approach are conducted by collecting environmental samples, DNA extraction of the total DNA which can be done using DNA extraction kits (Table 1). The DNA samples are enzymatically fragmented with the use of commercially available library preparation kits such as Nextera Tagmentation (Illumina) kits, and Fragmentase (New England Biolabs) which uses either endonucleases or transposases (Sabale et al., 2019). Other DNA fragmentation methods that generates DNA fragments of many kb in length include the use of acoustic shearing, centrifugal shearing, sonication, needle shearing and point-sink shearing. Furthermore, the concentration and purity level of the DNA libraries are determined by using Qubit and Nanodrop spectrophotometer. Also, the integrity of the sampled DNA are evaluated by loading DNA sample on agarose gel and differentiating size distribution to a suitable marker, such as lambda DNA (Farouk et al., 2020). On the other hand, DNA samples can be run on an Agilent TapeStation® using a Genomic DNA ScreenTape.

Table 1. Widely used DNA extraction kit for identifying rhizospheric microorganisms.
Further, the extracted DNA fragments can then be cloned into a suitable bacterial plasmid that acts as a vector. The ideal plasmid vectors are characterized by their small size, origin of replication (Ori site), unique restriction site, selective marker (antibiotics resistance), and multiple cloning sites (Granjou and Phillips, 2019). Finally, the fragments are analyzed using fragment analyzer system (a parallel capillary electrophoresis machine that automates the analysis of the quantity and quality of library preparations), and normalized. Sequencing is performed using different sequencing platforms such as Illumina, Pyrosequencing, ABI SOLiD sequencing system, and Nanopore sequencing (Martin, 2011; Zhang et al., 2021), then the short reads are de-multiplexed, and data are analyzed (Mahmoud et al., 2019).
The advancement of next-generation sequencing techniques (e.g., amplicon sequencing and shotgun metagenomics) has great potential in alleviating the challenges of capturing the whole profile of the microbial communities (Jansson and Hofmockel, 2018). Shotgun metagenomics is used extensively to study the microbial diversity and functional genes in various environmental samples (Table 2). This sequencing method is a recent replacement of the phylogenetic marker (taxonomic) or targeted genes sequencing (Zhang et al., 2021). For instance, recent studies have shown that Illumina Hiseq and MiSeq sequencing have been used to study microorganisms from farmland soil. Babalola et al. (2020) studied the diversity of microbial species from sunflower rhizosphere soil from Bloemhof, South Africa. These researchers found that the microbial community consist of mainly six genera Conexibacter, Streptomyces, Methylobacterium, Burkholderia, Geodermatophilu, and Nocardioides. Wang et al. (2019) identified Chloroflexi, Actinobacteria, Acidobacteria, Gemmatimonadetes, and Proteobacteria using Illumina HiSeq from farmland soils in Jilin province, China.

Table 2. Some advantages and disadvantages of amplicon and shotgun metagenomic sequencing.
Also, Wu et al. (2018b) identified different bacterial phyla from Rehmannia glutinosa rhizosphere and bulk soil samples using 454 pyrosequencing. The major bacterial phyla reported were Proteobacteria, Bacteiodetes, Actinobacteria, and Firmicutes. Bacillus subtilis, Alternaria alternata, and other microbial species have also been identified in plant rhizosphere. The limitation in this method is that it requires up-to-date databases for accurate microbial identification (Wu et al., 2018a).
Illumina MiSeq has also been used to sequence microbiome of plant roots and organic matter obtained from bioponic system. In the study, it was observed that the plant roots and organic matter contain different microbial taxa (Wongkiew et al., 2022). Management practices, plant type and substrate qualities of another type of urban agriculture initiative, e.g., rooftop farming have been reported to significantly impact the abundance and composition of microbial community and other living organisms of the rooftop farming environment, stressing the need of experimental and/or practical studies unique to rooftop agriculture (Ksiazek et al., 2014; Williams et al., 2014; Macivor and Ksiazek, 2015; Mcguire et al., 2015; Bretzel et al., 2017; Aloisio et al., 2019). Metagenomics has been used to detect microbiome genomic characteristics that are involved in plant-microbial interactions (Bulgarelli et al., 2015; Xu et al., 2018). These microbiome genomic characteristics include abiotic and biotic stress response, production and use of different metabolites, chemotaxis, ability to produce toxin, and cellular mobility. Beneficial characteristics that are also useful to plant growth such as phosphate solubilization, siderophore production, nitrogen fixation and mobilization and indole-acetic acid production capacities are embedded in some microorganisms associated with plants in soil (Sessitsch et al., 2012; Xu et al., 2018; Carrión et al., 2019; Li et al., 2019), bioponic and hydroponic environments. These microbial functional traits needed for plant growth and sustainable agriculture have been studied using NGS technique (Igiehon et al., 2019). Although, metagenomic analyses provide in-depth understanding of the functions of plant microbial community in the soil, evaluation of the associations between plant and microorganisms will only be comprehended when these functional characteristics or traits are studied in-situ. This can, however, be accomplished when metagenomics techniques are combined with other techniques such as transcriptomics and proteomics (Trivedi et al., 2021).
Even though, the most commonly used next-generation sequencing technique is the Illumina system, recently, the attention of researchers is shifting toward the application of MinION (Nanopore) and PacBio (Oulas et al., 2015). The new sequencing approaches are the latest third-generation sequencing technology. The main advantage of the third generation sequencing approach is that the reads are completed in between 4 and 6 h rather than in days. Further, this technique can obtain ultra-long reads up to hundreds of thousand bases (Goodwin et al., 2016). This approach is less expensive, can be connected to a laptop or computer, and does not require any special computer training or equipment for data analysis (Kerkhof et al., 2017). The third-generation technology generates a bacterial and archaeal population profile derived from the full length of the 16S rRNA gene sequence to provide high taxonomic resolution (e.g., at the genus level) (Johnson et al., 2019; Winand et al., 2019).
After sequencing and obtaining the metagenome data, it is essential to preprocess the raw data in order to have high quality reads for further analysis, and this can be achieved by using UCHIME, MG-RAST, and RDP tools (Bolger et al., 2014), as well as Trim Galore and Trimmomatic software (Sun, 2020). The platforms frequently used for deionizing metagenome data are MOTHUR and QIIME 2, while UCHIME is employed for cross-checking and expulsion of chimeras from the raw sequenced metagenome datasets (Santamaria et al., 2018). At the end of pretreatment and processing, the reads are grouped depending on their discrete barcodes, and then the primers are removed (Sabale et al., 2019; Kumar Awasthi et al., 2020). A number of computer software for categorizing reads into taxonomic and functional groups are available including daSILVAta (Dröge et al., 2015) and MEGAHIT (Liu et al., 2015).
Also, De Novo assemblers including Meta-IDBA, metaSPAdes, Genovo, Ray Meta, and Contig Extender have emerged recently and they are employed in sequencing novel microbial genomes where there is no reference metagenome sequence available for alignment (Table 3) (Kumar et al., 2018). Metagenome sequence reads are assembled as contigs, and the coverage quality of the sequence data depends on the size and continuity of the number of gaps in the data. Generally, the problem of de novo assembly is confirmed to be NP-Hard and as a result this problem cannot be resolved effectively (Medvedev et al., 2007). Some challenges of employing the de novo assemblers are memory usage, higher assembling time, and CPU on paired-end or joined data sets is higher than single-end data sets, for example ABySS (Khan et al., 2018). Contigs can be separated using SqueezeMeta. SqueezeMeta employs different procedures to separate the contigs. Essentially, binning algorithms classify contigs emanating from the same genomes because their nucleotide structure is simila (Tamames and Puente-Sánchez, 2019). Also, SqeezeMeta uses DAS Tool to combine the numerous binning results to form a single set (Sieber et al., 2018).

Table 3. Some popular phylogenomic analytical tools for phylogenetic and taxonomic classification.
Phylogenomic Analysis of Environmental Microorganisms That Are of Importance in Sustainable Agriculture
Phylogenomics involves evolutionary reconstructions which gives insights into the relationships that exist between organisms by comparing the entire genomes and their functions. The result from a phylogeny (species tree) is primarily required for many analyses with the purpose of gaining more insights into biological process by reorganizing the evolutionary history (Figure 1) (Emms and Kelly, 2019). Although, this strategy is among the best and commonly accepted computational techniques in analyzing sequenced metagenomic data, but there are still many limitations affecting its usage, for instance the species tree. The main areas that fall under phylogenomics are establishment and clarification of evolutionary relationships, prediction of gene functions, and prediction and retracing of lateral gene transfer (Raymann, 2014).
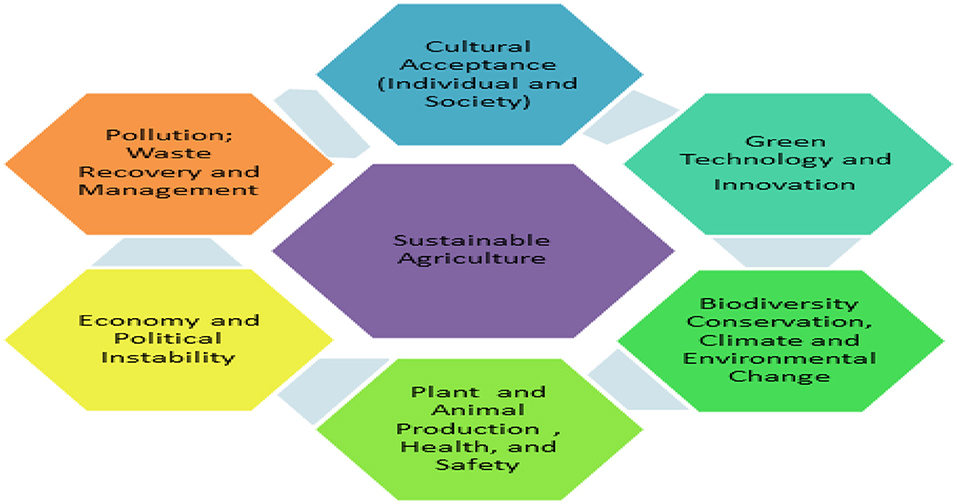
Figure 1. Integration of sustainable agriculture into the society.
Phylogenomic analyses usually involve different steps, including comparing the pairwise sequence among the entire genomes, that is, resolution of homologous relationships among gene-coding genes, like the gene families inference or orthologous groups (Rabiee et al., 2019). This places restrictions on the number of data set through which de novo inference of similar relationship can use (Emms and Kelly, 2019). Actually, the step prefers culture-independent and model organisms, as well as clinical isolates which bring about bias in sampling, resulting in unbalanced data sets, and also without reflecting the correct microbial diversity in the sampled community thus could give misleading information on the phylogenetic inferences. Lastly, the widely accepted clonal approach for microbial replication stops gene mutations in numerous genetic backgrounds, hence make it extremely difficult to identify and differentiate the impact of mutation and homogenous genetic background (Hollister et al., 2015).
Among the challenges of phylogenomic analyses of genes of interest in large microbial community include failure to recognize the actual original genetic determinants, partial assembly, assembly errors, redundancy, and inaccurate positive results (Yu et al., 2017; Young and Gillung, 2020). To overcome the challenge of relatedness, it is better to focus more on the observable changes that occur during evolutionary histories rather than only the genomes genetic features (Earle et al., 2016). For most experiments involving microbial community analyses, it is essential to focus on evolutionary history of the microbial community of study against the host phylogency background (Kumar et al., 2018). Unfortunately, proving the hypothesis is not feasible when studying microbial taxonomic diversity and genomes between different host species because the significant differences are not reasonable between niche adaptation and true co-evolution. In spite that the correlation between host species phylogenic tree and microbial community member phylogenies can be employed as evidence of co-diversification, however, it does not provide convincing knowledge of the basis for genetic interrelatedness (Markowitz et al., 2012; Earle et al., 2016).
What Can Metagenomics Do?
Metagenomic Applications for Sustainable Agriculture
Meta-omic approaches as next-generation sequencing methods are used to explore metagenomes from the environmental samples and are used to identify essential genes of special functions and/or bioactive compounds. Meta-omics supports different scientific research areas in agricultural sciences and provides an understanding of the functional potential of genomes in different species that are of agricultural benefits (Handelsman, 2004).
Currently, many molecular information arising from proteome (proteins) and transcriptome (all sets of RNA) sequencing is important for better comprehension of gene content, specific functions, and metabolic networks that are then translated to livestock or crop breeding, disease resistance, health, and yields. Consequently, they have led to several advancements in agriculture and biological sciences (Prosser, 2015). Also, different pieces of evidence reported have shown the contributions of genomic studies in agriculture that have span beyond identifying or manipulating genes from samples associated with a particular trait and genomic breeding (Goel et al., 2017; Pinu et al., 2017).
Agricultural genomics is focused on finding novel solutions through studying livestock or crop genomes. For this reason, researchers have obtained information that would deal with issues of protecting the crops or livestock, and increase of food products in the food industry, so that food security is achieved (Makonde et al., 2015). Microbial communities from the soil and plants are key players in agriculture, determining plant health, soil features, and biogeochemical characteristics, in like manner influence yields and ultimately quality traits. The knowledge of the soil environment is sparsely available. For example, researchers have recorded that on earth, soil as the largest reservoir of carbon, while the prokaryotic microorganisms are richly abundant in the soil environment. The traditional techniques over the years have only identified a few of these microorganisms (Pinu et al., 2017).
The application of metagenomics in agriculture has not only profiled plant growth promoting rhizobacteria (PGPR) but also has described their functional characteristics. The approach has proved to be an appropriate technique in understanding the different interactions occurring in the soil environment, rhizosphere, and plant tissues (Gupta et al., 2018). The metagenomic approach has successfully identified diazotrophs from the rhizosphere soil of red kidney beans by targeting nitrogen fixing gene (nifH) (Sabale et al., 2019).
Correspondingly, metagenomics is revealed as a useful tool in tracing the shift in taxonomic component and functional redundancy of rhizobiomes. Soil microorganisms are unlimited and critical members of the ecosystem playing integral roles in triggering plant growth, development, defense, and stress responses in plants (Gupta et al., 2018). Therefore, for progressive sustainable agriculture production, the knowledge of the relationship between plants and soil microbial community using metagenomics will be helpful in designing crop systems. Applying metagenomics techniques in the study of soils inoculated with organic manures would be useful in formulating strategies for fertilization and reduce farmers' dependence on chemical-based fertilizers (Manjula and Narsimha, 2015).
At this present time, the toxic impacts of chemical-based fertilizers and pesticides on the agricultural ecosystem, plant health, and development are having a noticeable damaging influence on the environment. Among the abundance of microorganisms are beneficial agricultural microorganisms that can act as a critical option to attaining agricultural sustainability production. On the contrary, the full potentials of these microbial communities are still untapped (Goel et al., 2017).
Another key point is that considering the intriguing resources that could be unraveled from the microorganisms, hence the need for developing novel genes with plant growth-promoting traits that serve as bioinoculants which can contribute to agricultural sustainability (Gupta et al., 2018). Ordinarily, identifying and characterizing the rich microbial community becomes unavoidable. According to Goel et al. (2017), metagenomics can be used for sustainable agriculture advancement programs that can regularly assess the plant growth-promoting microorganisms. For this reason, it requires a comprehensive, correct analysis and characterization of essential beneficial plant growth-promoting microorganisms and their functionalities rather than just cataloging.
Similarly, functional metagenomics can be employed for reshaping and readdressing of microbial activities in the rhizosphere, otherwise termed rhizosphere engineering (Goel et al., 2017; Louca et al., 2018). In recent whole-genome studies, metagenomic applications have unlocked the diverse metabolic, genomic, and phylogenetic resources stored in the metagenome of soil microbial communities (Garrido-Oter et al., 2018). Agricultural soil is reported as the most challenging and diversified terrestrial environment that is richly embedded with unlimited resources.
While, enabling many soil microbiologists to overcome the complex challenges of culturing microorganisms in a pure state, and capturing the uncultured, and unculturable percentage. The metagenomic approaches for screening libraries from soil genomes are sequence-driven and function-driven techniques that are suitable, flexible, and feasible. Employing one or both of the methods helps researchers to evaluate a larger number of new products important for agricultural purposes (Garrido-Oter et al., 2018) (Figure 2).
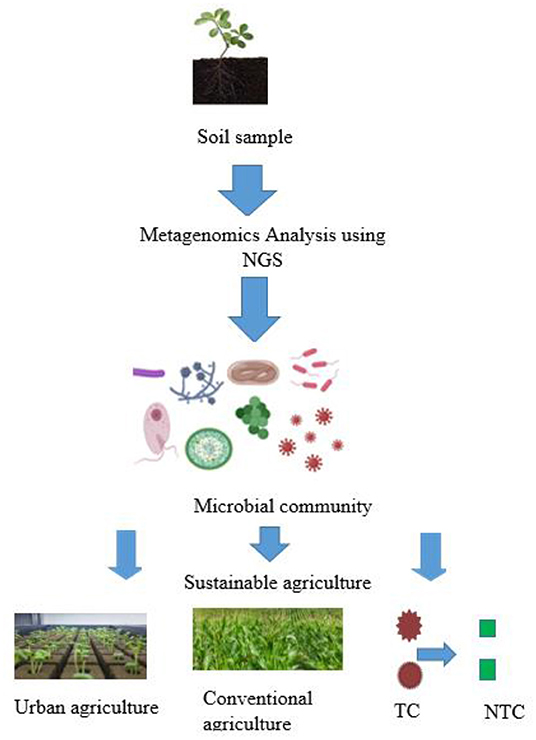
Figure 2. Soil microbial genetic diversity and their applications in sustainable agriculture. Microbes obtained and identified from rhizosphere soil can be harnessed to enhance plant growth and productivity in both urban and conventional agriculture. Some of such microbes may also play a role in converting toxic contaminants to their respective non-toxic forms. NGS, next generation sequencing; TC, toxic contaminants; NTC, non-toxic contaminants.
Altogether, metagenomics provides information on culture-independent soil microbial communities that are influenced by environmental conditions, agricultural management, and land-use practices, like the use of pesticides, fertilizers, and tillage systems. Different biomolecules are synthesized due to the response of diverse microorganisms to edaphic changes, including other essential factors. To sum it up, the concentration of metabolites that are released differs owing to different factors, namely, nutrient concentration, pH, and temperature affecting the uptake and secretion of metabolites in the soil environment (Gupta et al., 2018), and even in bioponic systems.
Microorganisms present in bioponic systems aid in the breakdown of organic matter to the forms that can be utilized by plants (Sharma et al., 2017; Cáceres et al., 2018). Knowing that bioponics is driven and facilitated by microbial activities, understanding microbial interactions and functions are key for the assessment and enhancement of the efficiency of bioponic environments. Metagenomic studies involving microbial compositions and functions, co-occurrence microbiome network as well as “discriminant analysis” have provided insights into the microbial community of bioponic systems (Thomas et al., 2011; Langille et al., 2013; Hu et al., 2017). For instance, symbiotic interconnectivity and keystone taxa can be detected using co-occurrence microbiome networks, while discriminant analysis can unveil microbial biomarkers that are significantly present under certain conditions (Thomas et al., 2011; Tan et al., 2020). Metagenomics investigations, when coupled with efficiency assessment may, therefore, help to enhance bioponic systems (Bao et al., 2021) and sustainably improve urban agriculture.
Plant-Microorganism Interactions: Soil Health
Soil agro-ecosystem is a well-known hotspot of network systems with different biodiversity because it is a special environment. Soil hosts plant and diverse microbial communities performing many diverse interactions. The soil at the plant root region (rhizosphere) is colonized by numerous microbial populations (rhizobiomes) coexisting and performing a variety of beneficial functions and therefore, make the rhizosphere soil a habitat where there are exchanges of energy among microbial communities. The rhizobiomes are the notable driving force that influences the soil structure and physical-chemical compositions that guide the ecosystem and for that reason contribute to the organization.
Rhizosphere soil has a special capability to conform to environmental changes due to the microbial communities inhabiting the soil that is sensitive to climate changes, land use, and management. The microbial ecologist has documented that the natural ecosystem including soil environment has the capacity of self-healing that is, returning to its initial state after disturbance. Over the years, soil has been used for the cultivation of crops and food production. The soil is the most biologically active terrestrial niche with a large number of cultivable, un-cultivable, unknown, and known microbial communities, it becomes challenging studying it (Jansson and Hofmockel, 2018). Soni and Goel (2011) employed the metagenomic approach to evaluate the microbial population from different rhizosphere soil samples. They reported metagenome from rhizosphere soil with nif H genes having nitrogen-fixing ability. Metagenomic studies on soil biochemistry have revealed many functions that are not known to be of importance to life sustenance (Goel et al., 2017).
Along with the use of various intense agricultural management practices that affected the soil microbiome without positive influence, many practices, including the use of agrochemicals, excessive use of chemical fertilizers, erosions, saline run-off on soil environments, economical use of land for housing, and multiple cropping are the leading cause of soil degradation over a long period that has led to the challenges of sustainable agriculture (Maropola et al., 2015). Together with the use, and overuse of machinery that deteriorate the fertility, structure, and vigor. Under those consequences, microbial life cannot be sustained. In that case, to sustain soil health and improve soil characteristics is of necessity to the agricultural ecosystem and survival of humans.
Metagenomics has improved the knowledge of the microbial community that is essential to the plant environment and the interactions that occur in the soil environments that are negatively impacted by land preparation practices. In a report by Chávez-Romero et al. (2016) different microbial genera, including Bacillus, Sinorhizobium, Agromyces, Lysobacter, and Streptomyces from soil impacted by tillage-residue management were documented. Similarly, metagenomics was employed to identify Firmicutes, and Actinobacteria from wheat plant materials that were used in soil amendment, as a result, improved the soil by increasing the abundance of microbial communities (Chávez-Romero et al., 2016).
Presently, different modern biotechnological strategies have been employed to identify the essential microbial community that can improve soil health, nutrient availability, and increased crop yields. Microbial diversity identified from soil using the shotgun metagenomic technique was incorporated into the soil through agricultural practices (soil amendment). The soil amendment increased soil fertility, improved plant growth, and crop productivity. The frequently identified microorganisms from the soil with the potential of impacting soil health, include Spirochetes, Norcadia, Chloroflexi, and many more microbial communities have been reported (Manjula and Narsimha, 2015).
Furthermore, the interactions that occur between plants and microorganisms in the rhizosphere is ever dynamic and complex (Philippot et al., 2013; Turner et al., 2013). Some of these interactions have been shown to sustainably improve food production in agriculture, while others are not. Even though, plant-microbial interactions can enhance aboveground crop yield, compounds generated in the aboveground region may have either positive or negative impacts on the rhizosphere microbial community structure and vice versa (Igiehon and Babalola, 2018). For instance, there was a decline in the diversity of belowground endophytes owing to the action of salicylic acid which triggered the defense mechanism of Arabidopsis thaliana in the phyllosphere region, whereas, plants devoid of jasmonate-elicited defense mechanism showed greater epiphytic diversity. These further showed that specific plant immune system differentially affect belowground microbial communities (Philippot et al., 2013). Also, symbiotic interactions have been reported to occur between plants and microorganisms. A typical example of such is the interaction between plant and mycorrhizal fungi, in which the fungi aid in the supply of nutrient to the plant hosts, while the plants supply carbon substrates to the fungi for survival (Zarik et al., 2016). For example, a consortium of arbuscular mycorrhizal fungi (AMF) co-inoculated with Rhizobium species also observed to improve the yield of soybean plants in North West Province of South Africa (Igiehon et al., 2021).
Symbiotic interactions do not only arise between plant and microorganisms, but there are also microorganism-microorganism interactions. In particular, the interaction between some plant growth promoting bacteria and arbuscular mycorrhizal fungi (AMF) is symbiotic as the bacterial species help the AMF in establishing symbiotic interaction with their host plant while AMF enhance the invasion capacity and diversity of the bacteria. Nevertheless, the bacteria and AMF benefit in other ways, from the symbiosis. Symbiosis has also been observed between Burkholderia species (a bacterium) and the fungus Rhizopus, which depends on metabolites produced by the bacterium to produce spores and survive (Braga et al., 2016).
During plant growth, there may be modification of the surrounding soil, nutrient content and soil microbial community (Bennett et al., 2017; Fujii et al., 2018). Such modifications might feed-back on plant survival and growth (that is, plant-soil feedback), thereby changing plant population (Bennett et al., 2017). The impacts of mutualists on plant-soil feedback are mainly positive since they pacify the negative plant-soil feedback caused by nutrient depletion or presence of natural enemies. Positive plant-soil feedback is said to occur when, for instance, there is an increase in the amount of nutrient and population of mutualists in the environment, while negative plant-soil feedback might occur as a result of nutrient depletion, release of autotoxic chemicals and increase in the population of natural enemies (Bennett et al., 2017; Kulmatiski et al., 2017; Smith-Ramesh and Reynolds, 2017). AMF can facilitate positive plant-soil feedback and this might be influenced by the relative quantities of various natural enemies and nutrients since AMF helps more in the absorption of phosphorus than nitrogen and protect host plants against pathogens, but not root herbivores (Delavaux et al., 2017; Teste et al., 2017). Thus, AMF can drive positive plant-soil feedback when there is a limited amount of phosphorous or when plant pathogens are more than root herbivores. Rhizobacteria can cause positive plant-soil feedback in symbiotic relationships with leguminous plants by enhancing the growth of the legumes and feedback as free living bacteria (Revillini et al., 2016).
Rhizosphere Soil Microbial Genetic Diversity Assessment in Relation to Sustainable Agriculture
Soil is considered as one of the most diverse environments containing billions of microorganisms. The soil environment is mainly composed of bacterial and fungal species that are made of thousands of taxa. Again, large organisms, like nematodes, ants, and moles cohabits in the soil (Sergaki et al., 2018). Soil microbial ecosystem is composed of microflora and microfauna that co-exists while playing different essential roles in improving plant growth and all-round developments. This is mostly dependent on soil diversity and integrated nutrient management systems (Priya et al., 2018).
Soil from the rhizosphere could contain numerous taxa up-to 4x106 (Bulgarelli et al., 2015). The common traditional cultivation of rhizospheric microorganisms has hampered microbiological studies. In effect, a large number of bacteria and other groups of microorganisms are yet to be cultivated in the laboratory. Microorganisms identified using the common traditional culture methods are less abundant in the soil micro-niches. Besides, the well-known microbial species isolated from soil habitat are mostly from Proteobacteria, Bacteroidetes, Firmicutes, and Actinobacteria, these are the few major phyla that can be cultivated easily in the laboratory under growth conditions (Babalola, 2010).
Previous studies have shown that metagenomic approaches have given a more comprehensive picture of the rhizospheric microbiome, and many new rhizosphere microorganisms will be discovered in the shortest space of time (Figure 3). Most of these microorganisms have been implicated to be useful for the enhancement of crops in urban agriculture, while others have the potential to remove contaminates from the environment. Moreover, other researchers identified Proteobacteria, Acidobacteria, Bacteroidetes, BRCI, Chloroflexi, Actinobacteria, Firmicutes, Cyanobacteria, Planctomycetes, Chlorobi, and Nitrospira from rhizosphere soil samples with the aid of metagenomic applications. Plants have relied on the beneficial symbiotic cohabitation of microorganisms widely used in sustainable agriculture. Metagenomic applications have explored and unlocked the access to rhizosphere microbial communities of many plants by targeting beneficial functional genes present in the microorganisms. Some of such genes are associated with nitrogen fixation (nif H) and Cold Shock Protein (CSP) (Goel et al., 2017).
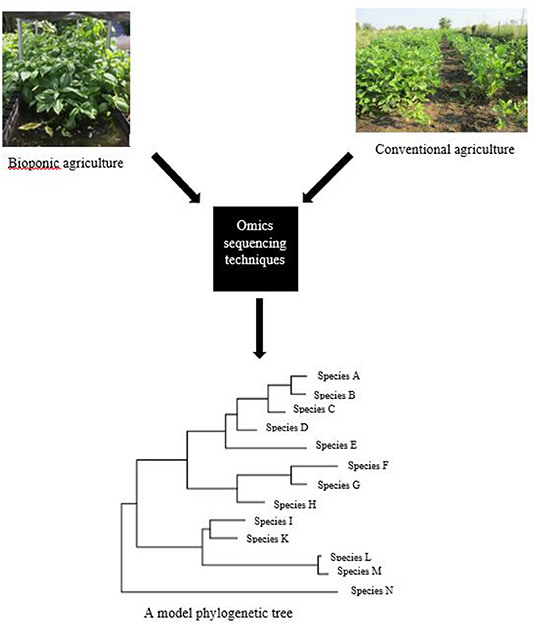
Figure 3. A phylogenetic tree of microbial species obtained from omics analyses of urban and conventional agriculture. Root associated samples of bioponic plants and rhizosphere soil samples can be obtained and sequenced using omics techniques, followed by construction of a phylogenetic tree of the microbial species that are of importance in achieving sustainable agriculture.
Exploring Beneficial Microorganisms of Biogeochemical Cycles
Agricultural sustainability relies on the biogeochemical cycle and diverse microbial communities play important functional roles in biogeochemical cycles in soils. Biogeochemical cycles often rely on the type of biodiversity accessible in a specific soil environment and the variety of crops planted. Application of metagenomics approaches has explored and identified novel genomes involve in biogeochemical cycles. The genomes of Kuenenia stuttgartiens a bacterium involved in anaerobic ammonium fixing have been documented (Van Niftrik et al., 2008).
Similarly, novel protein cytochrome an important component for iron oxidation involved in the formation of acid mine drainage was identified using the combined shotgun and MS-based proteomics with genomic analysis of soil microbial community (Bevivino and Dalmastri, 2017). In brief, nif H genes involve in nitrogen cycling were identified using the metagenomic approach. In another event, a new complete genome sequence of Ferroplasma type II and Leptospirillum group II. Correspondingly, the partial genome sequence of Ferroplasma type I, Leptospirillum group III, and G- plasma linked to biogeochemical cycles were identified with the aid of metagenomics (Goel et al., 2017).
Identifying Novel Biocatalysts From the Soil Environment
Living or biological systems are classified as essential proteins that speed up metabolic reactions are biocatalysts or enzymes. In that case, they play significant roles in all biological organisms. Metagenomic approaches are adopted in identifying entirely novel genes encoding new classes, types of enzymes, and their substrate specificity. Indeed, metagenomics enables researchers to understand the cells participating in metabolic processes and identification of new bioactive compounds (Murphy et al., 2015). Specifically, new enzymes of agricultural importance are being identified and developed with metagenomic applications. Its application in agricultural soils reduces the dependence on chemical-based fertilizers and promotes environmental sustainability.
Functional metagenomics with compelling evidence is an effective tool to discover new enzymes from the soil, and to understand the enzyme activities and functions. The demand for production of enzymes and biocatalysts in commercial scale for agricultural purposes are rapidly growing, and presently, metagenomics is the technology to provide the needed enzyme molecules on a massive scale (Ghosh et al., 2018). Few of the main enzymes used to improve soil fertility, crop growth, and yield are sulfatases, dehydrogenase, and phosphatases are known to reduce the use of pesticides for agricultural productions too (Peng et al., 2018). Extremophilic esterases with a contrasting active site compared to the one formed by culturable microorganisms capable of increasing the crop performance have been identified from soil using metagenomics (Murphy et al., 2015).
Metagenomic applications have unlocked novel halotolerant enzymes from the soil which have great potentials in tolerating high salinity in the soil. It is equally important in industrial applications for bioethanol production and lignocellulosic deconstruction (Salem et al., 2020). Soil microbial sources with attention to bacteria, fungi, and yeast are the reservoir of many enzymes. Eventually, it is attracting special attention for their tremendous agricultural benefits (Ghosh et al., 2018). Enzymes employed in the food and agrochemicals productions were reported by Ahmed et al. (2018). Above all, agricultural applications of enzymes are advantageous in enriching and fertilizing the soil, as well as ensuring continuous crop production (Ahmed et al., 2018).
Furthermore, some biotechnologically important enzymes, including amylases, xylanases, lipases, cellulases, proteases, and other several enzymes have been unlocked from genetically untapped soil resources through metagenomic approaches. Lipases were discovered from novel soil microorganisms (Salem et al., 2020). Currently, there is a record of lipases identified from species of plants and rhizosphere metagenomes. Equivalently, a new alkaline stable lipase was identified from a metagenomic library constructed from agricultural soil (Peng et al., 2018). Cellulase a naturally occurring and the most abundant biopolymer on earth is recorded as the third largest used industrial enzyme globally for catalyzing the hydrolysis of cellulose (Salem et al., 2020).
Metagenomic approaches have captured new cellulases from compost soils, soils from regions, and other natural environments by creating the libraries and screening all active clones (Ahmed et al., 2018). New cellulase was reported from the soil of sugar cane farm by screening functional genes of the metagenomic libraries (Peng et al., 2018). Cellulase has the potential for the biological conversion of crops, like sugar came, sugar beet, corn, and wheat biomass into renewable liquid biofuels. Lots of successes have been met by several bioenergy research centers (López-López et al., 2014). Again, in animal farming, cellulases are added to animal feeds which improve digestibility and promotes agricultural sustainability through animal production (Ghosh et al., 2018).
Specifically, the metagenomic approach has identified novel xylanases from metagenome of compost soil that showed thermostability and alkali stability properties with the potential (Alves et al., 2018). Furthermore, two novel phytases enzymes of industrial benefits were identified from soil microbial communities with the aid of function-based metagenomic approaches. The two phytases belonging to the phytase family were identified for their phytases activities.
Interestingly, the first phytase enzyme has an uncommon histidine acid phosphatase as a conserved motif of the active site of PhyX possessing extra amino acid residue, while the second, member of a new type phytase is encoded by multiple open reading frames (ORFs) differentiating it from all known phytases that are encoded by a single ORF (Tan et al., 2015). Generally, the enzymes identified from soil environment as mentioned above have different potentials for increasing soil fertility, influencing crop productivity, and promoting sustainable agriculture.
Paleogenomic Survey
Among the many emerging applications of metagenomics is paleogenomics, alternatively called genome-wide ancient DNA analysis. This is a new field in science that is based on the analyzing, and reconstruction of genomes in the extinct plant, and animal species (Tan et al., 2015). As it is known, that soil is a habitat with unlimited species of organisms, including plants with a variety of genetic materials and functions. Metagenomic approach has provided insights to what means traits evolve, and the relationship between extinct organisms, and living species. It has been established that DNA from samples is not limited to one species of a gene (Kim et al., 2014).
The ancient DNA of plants is fragmented, destroyed, and mixed with genomes of the abundant microbial variations (Novak, 2018). Gupta et al. (2018) stated that palegenomic applications with high- throughput sequencing have provided access and understanding of the nuclear genomes of the extinct organisms. These approaches could revolutionize the field of paleobiology, and in deciphering genome researches focusing on extinct species.
Conclusion and Future Prospects
The relevance of microorganisms in plant health and the development of the plant environment are well-established, but the majority of the rhizosphere microbial communities is yet to be characterized and is not explored. The combination of the conventional method with metagenomic approaches to assess the function and structure of the microbial community has provided insights into soil microbiome. Meanwhile, to identify plant signals, a wealth of resources and fundamental factors in the rhizosphere will provide microbial and chemical markers to describe how plants attract and stimulate advantageous microorganisms in soil and soilless agriculture.
Metagenomics has contributed to sustainable agricultural development by bringing to light many untapped soil microorganisms, their functionalities, genes for various applications, improving crop productivity, phytopathogen resistance, and nutrient cycling. In addition, shotgun metagenomics and bioinformatics tools have facilitated data interpretation of metagenomic data and resolved the challenges of pooling metagenomic data to establish the entire metagenomes of individual microbial community members, then correlating the diversity of different environments, and analyzing the challenges in diversity.
Author Contributions
BN searched the literatures and prepared the tables and figures, drafted the first manuscript, and read and approved the final draft version of the manuscript. OB evaluated the manuscript, edited the content, and read and approved the final draft version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by National Research Foundation, South Africa Grants (UID123634 and UID132595) provided to OB.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
BN appreciates the National Research Foundation South Africa/ The World Academy of Science (NRF-TWAS) (UID121772) for a stipend, which is of great help for her Ph.D. studies. OB acknowledges the National Research Foundation, South Africa for a grant (UID123634) that supported research in her laboratory.
References
Abram, F. (2015). Systems-based approaches to unravel multi-species microbial community functioning. Comput. Struct. Biotechnol. J. 13, 24–32. doi: 10.1016/j.csbj.2014.11.009
Ackerman, K., Conard, M., Culligan, P., Plunz, R., Sutto, M. P., and Whittinghill, L. (2014). Sustainable food systems for future cities: The potential of urban agriculture. Econ. Soc. Rev. 45, 189–206. Available online at: http://www.esr.ie/issue/archive
Agler, M. T., Ruhe, J., Kroll, S., Morhenn, C., Kim, S. T., Weigel, D., et al. (2016). Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 14, 1–20. doi: 10.1371/journal.pbio.1002352
Ahmed, V., Verma, M. K., Gupta, S., Mandhan, V., and Chauhan, N. S. (2018). Metagenomic profiling of soil microbes to mine salt stress tolerance genes. Front. Microbiol. 9, 159. doi: 10.3389/fmicb.2018.00159
Aloisio, J. M., Palmer, M. I., Tuininga, A. R., and Lewis, J. D. (2019). Plant colonization of green roofs is affected by composition of established native plant communities. Front. Ecol. Evol. 6, 238. doi: 10.3389/fevo.2018.00238
Alves, L. D. F., Westmann, C. A., Lovate, G. L., De Siqueira, G. M. V., Borelli, T. C., and Guazzaroni, M. E. (2018). Metagenomic approaches for understanding new concepts in microbial science. Int. J. Genomics. 2018, 1–15. doi: 10.1155/2018/2312987
Babalola, O. O. (2010). Beneficial bacteria of agricultural importance. Biotechnol. Lett. 32, 1559–1570. doi: 10.1007/s10529-010-0347-0
Babalola, O. O., Alawiye, T. T., Lopez, C. R., and Ayangbenro, A. S. (2020). Shotgun metagenomic sequencing data of sunflower rhizosphere microbial community in South Africa. Data Br. 31, 105831. doi: 10.1016/j.dib.2020.105831
Bao, Y., Feng, Y., Qiu, C., Zhang, J., Wang, Y., and Lin, X. (2021). Organic matter-and temperature-driven deterministic assembly processes govern bacterial community composition and functionality during manure composting. J. Waste Manag. 131, 31–40. doi: 10.1016/j.wasman.2021.05.033
Benítez-Páez, A., and Sanz, Y. (2017). Multi-locus and long amplicon sequencing approach to study microbial diversity at species level using the MinION™ portable nanopore sequencer. Gigascience. 6, 7–12. doi: 10.1093/gigascience/gix043
Bennett, J. A., Maherali, H., Reinhart, K. O., Lekberg, Y., Hart, M. M., and Klironomos, J. (2017). Plant-soil feedbacks and mycorrhizal type influence temperate forest population dynamics. Science 355, 181–184. doi: 10.1126/science.aai8212
Bevivino, A., and Dalmastri, C. (2017). “Impact of agricultural land management on soil bacterial community: A case study in the mediterranean area,” in Soil Biological Communities and Ecosystem Resilience, eds P. G. Lukax and M. Gamboni (New York, NY: Springer), 77–95.
Bolger, A. M., Lohse, M., and Usade, L. B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brader, G., Compant, S., Vescio, K., Mitter, B., Trognitz, F., Ma, L. J., et al. (2017). Ecology and genomic insights into plant-pathogenic and plant-nonpathogenic endophytes. Annu. Rev. Phytopathol. 55, 61–83. doi: 10.1146/annurev-phyto-080516-035641
Braga, R. M., Dourado, M. N., and Araújo, W. L. (2016). Microbial interactions: ecology in a molecular perspective. Braz. J. Microbiol. 47, 86–98. doi: 10.1016/j.bjm.2016.10.005
Bretzel, F., Vannucchi, F., Benvenuti, S., and Rumble, H. (2017). “Biodiversity of flora and fauna,” in Rooftop Urban Agriculture, eds F. Orsini, M. Dubbeling, H. de Zeeuw, and G. Gianquinto (New York, NY: Springer), 183–252.
Bulgarelli, D., Garrido-Oter, R., Münch, P. C., Weiman, A., Dröge, J., Pan, Y., et al. (2015). Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 17, 392–403. doi: 10.1016/j.chom.2015.01.011
Cáceres, R., Malińska, K., and Marfà, O. (2018). Nitrification within composting: a review. J. Waste Manag. 72, 119–137. doi: 10.1016/j.wasman.2017.10.049
Carrión, V. J., Perez-Jaramillo, J., Cordovez, V., Tracanna, V., De Hollander, M., Ruiz-Buck, D., et al. (2019). Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 366, 606–612. doi: 10.1126/science.aaw9285
Chadwick, D., Wei, J., Yan'an, T., Guanghui, Y., Qirong, S., and Qing, C. (2015). Improving manure nutrient management towards sustainable agricultural intensification in China. Agric. Ecosyst. Environ. 209, 34–46. doi: 10.1016/j.agee.2015.03.025
Chávez-Romero, Y., Navarro-Noya, Y. E., Reynoso-Martínez, S. C., Sarria-Guzmán, Y., Govaerts, B., Verhulst, N., et al. (2016). 16S metagenomics reveals changes in the soil bacterial community driven by soil organic C, N-fertilizer and tillage-crop residue management. Soil Till. Res. 159, 1–8. doi: 10.1016/j.still.2016.01.007
Delavaux, C. S., Smith-Ramesh, L. M., and Kuebbing, S. E. (2017). Beyond nutrients: a meta-analysis of the diverse effects of arbuscular mycorrhizal fungi on plants and soils. Ecology 98, 2111–2119. doi: 10.1002/ecy.1892
Dröge, J., Gregor, I., and Mchardy, A. C. (2015). Taxator-tk: precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics 31, 817–824. doi: 10.1093/bioinformatics/btu745
DuŽí, B., Frantál, B., and Rojo, M. S. (2017). The geography of urban agriculture: new trends and challenges. Morav. Geog. Rep. 25, 130–138. doi: 10.1515/mgr-2017-0012
DuŽí, B., Tóth, A., Bihunová, M., and Stojanov, R. (2014). Challenges of urban agriculture: highlights on the Czech and Slovak Republic specifics. Current Challenges of Central Europe. Soc. Environ. 185, 82–107. Available online at: http://www.ecoology.org/wp-content/uploads/Current-challenges-of-CE-09.pdf
Earle, S. G., Wu, C.-H., Charlesworth, J., Stoesser, N., Gordon, N. C., Walker, T. M., et al. (2016). Identifying lineage effects when controlling for population structure improves power in bacterial association studies. Nat. Microbiol. 1, 1–8. doi: 10.1038/nmicrobiol.2016.41
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14. doi: 10.1186/s13059-019-1832-y
Fadiji, A. E., and Babalola, O. O. (2020). Metagenomics methods for the study of plant-associated microbial communities: a review. J. Microbiol. Meth. 170, 1–13. doi: 10.1016/j.mimet.2020.105860
Farouk, F., El Shimy, R., Abdel-Motaleb, A., Essam, S., and Azzazy, H. M. (2020). Detection of Acinetobacter baumannii in fresh produce using modified magnetic nanoparticles and PCR. Anal. Biochem. 609, 113890. doi: 10.1016/j.ab.2020.113890
Fujii, K., Shibata, M., Kitajima, K., Ichie, T., Kitayama, K., and Turner, B. L. (2018). Plant-soil interactions maintain biodiversity and functions of tropical forest ecosystems. Ecol. Res. 33, 149–160. doi: 10.1007/s11284-017-1511-y
Garrido-Oter, R., Nakano, R. T., Dombrowski, N., Ma, K. W., Team, T. A., Mchardy, A. C., et al. (2018). Modular traits of the rhizobiales root microbiota and their evolutionary relationship with symbiotic rhizobia. Cell Host Microbe 24, 155–167.e5. doi: 10.1016/j.chom.2018.06.006
Ghosh, S., Ahmad, R., Gautam, V. K., and Khare, S. K. (2018). Cholesterol-oxidase-magnetic nanobioconjugates for the production of 4-cholesten-3-one and 4-cholesten-3, 7-dione. Biores. Technol. 254, 91–96. doi: 10.1016/j.biortech.2018.01.030
Goel, R., Suyal, D. C., Dash, B., and Soni, R. (2017). “Soil metagenomics: A tool for sustainable agriculture,” in Mining of Microbial Wealth and Metagenomics, eds V. C. Kalia, Y. Shouche, H. J. Purohit, and P. Rahi (New York, NY: Springer), 217–225.
Goodwin, S., Mcpherson, J. D., and Mccombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333. doi: 10.1038/nrg.2016.49
Grafton, R. Q., Daugbjerg, C., and Qureshi, M. E. (2015). Towards food security by 2050. Food Sec. 7, 179–183. doi: 10.1007/s12571-015-0445-x
Granjou, C., and Phillips, C. (2019). Living and labouring soils: metagenomic ecology and a new agricultural revolution? Biosocieties 14, 393–415. doi: 10.1057/s41292-018-0133-0
Guerra, A. B., Oliveira, J. S., Silva-Portela, R. C., Araújo, W., Carlos, A. C., Vasconcelos, A. T. R., et al. (2018). Metagenome enrichment approach used for selection of oil-degrading bacteria consortia for drill cutting residue bioremediation. Environ. Pollut. 235, 869–880. doi: 10.1016/j.envpol.2018.01.014
Gupta, N., Vats, S., and Bhargava, P. (2018). “Sustainable agriculture: role of metagenomics and metabolomics in exploring the soil microbiota,” in Silico Approach for Sustainable Agriculture, eds D. K. Choudhary, M. Kumar, R. Prasad, and V. Kumar (New York, NY: Springer), 183–199
Handelsman, J. (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 68, 669–685. doi: 10.1128/MMBR.68.4.669-685.2004
Handelsman, J., Rondon, M. R., Brady, S. F., Clardy, J., and Goodman, R. M. (1998). Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem. Biol. 5, R245–R249. doi: 10.1016/S1074-5521(98)90108-9
Head, I., Saunders, J., and Pickup, R. W. (1998). Microbial evolution, diversity, and ecology: a decade of ribosomal RNA analysis of uncultivated microorganisms. Microb. Ecol. 35, 1–21. doi: 10.1007/s002489900056
Hollister, E. B., Brooks, J. P., and Gentry, T. J. (2015). “Bioinformation and'omic approaches for characterization of environmental microorganisms,” in Environmental Microbiology, eds I. L. Pepper, C. P. Gerba, and T. J. Gentry (Cambridge; Boston, MA: Academic Press; Elsevier), 483–505.
Hu, A., Ju, F., Hou, L., Li, J., Yang, X., Wang, H., et al. (2017). Strong impact of anthropogenic contamination on the co-occurrence patterns of a riverine microbial community. Environ. Microbiol. 19, 4993–5009. doi: 10.1111/1462-2920.13942
Igiehon, N. O., and Babalola, O. O. (2018). Below-ground-above-ground plant-microbial interactions: focusing on soybean, rhizobacteria and mycorrhizal fungi. Open Microbiol. J. 12, 261. doi: 10.2174/1874285801812010261
Igiehon, N. O., and Babalola, O. O. (2019). Fungal bio-sorption potential of chromium in Norkrans liquid medium by shake flask technique. J. Basic Microbiol. 59, 62–73. doi: 10.1002/jobm.201800011
Igiehon, N. O., Babalola, O. O., and Aremu, B. R. (2019). Genomic insights into plant growth promoting rhizobia capable of enhancing soybean germination under drought stress. BMC Microbiol. 19, 159. doi: 10.1186/s12866-019-1536-1
Igiehon, N. O., Babalola, O. O., Cheseto, X., and Torto, B. (2021). Effects of rhizobia and arbuscular mycorrhizal fungi on yield, size distribution and fatty acid of soybean seeds grown under drought stress. Microbiol. Res. 242, 126640. doi: 10.1016/j.micres.2020.126640
Igiehon, O. N. (2015). Bioremediation potentials of Heterobasidion annosum 13.12 B and Resinicium bicolor in diesel oil contaminated soil microcosms. J. Appl. Sci. Environ. Manag. 19, 513–519. doi: 10.4314/jasem.v19i3.22
Jansson, J. K., and Hofmockel, K. S. (2018). The soil microbiome-from metagenomics to metaphenomics. Curr. Opin. Microbiol. 43, 162–168. doi: 10.1016/j.mib.2018.01.013
Johnson, J. S., Spakowicz, D. J., Hong, B. Y., Petersen, L. M., Demkowicz, P., Chen, L., et al. (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 10, 1–11. doi: 10.1038/s41467-019-13036-1
Kerkhof, L. J., Dillon, K. P., Häggblom, M. M., and Mcguinness, L. R. (2017). Profiling bacterial communities by MinION sequencing of ribosomal operons. Microbiome 5, 1–11. doi: 10.1186/s40168-017-0336-9
Khan, A. R., Pervez, M. T., Babar, M. E., Naveed, N., and Shoaib, M. (2018). A comprehensive study of de novo genome assemblers: current challenges and future prospective. Evolut. Bioinform. 14, 1176934318758650. doi: 10.1177/1176934318758650
Kim, C., Wang, X., Lee, T. H., Jakob, K., Lee, G. J., and Paterson, A. H. (2014). Comparative analysis of Miscanthus and Saccharum reveals a shared whole-genome duplication but different evolutionary fates. Plant Cell 26, 2420–2429. doi: 10.1105/tpc.114.125583
Ksiazek, K., Tonietto, R., and Ascher, J. S. (2014). Ten bee species new to green roofs in the chicago area. Gt. Lakes Entomol. 47, 13, 1–17. Available online at: https://scholar.valpo.edu/tgle/vol47/iss1/13
Kulmatiski, A., Beard, K. H., Norton, J. M., Heavilin, J. E., Forero, L. E., and Grenzer, J. (2017). Live long and prosper: plant-soil feedback, lifespan, and landscape abundance covary. Ecology 98, 3063–3073. doi: 10.1002/ecy.2011
Kumar Awasthi, M., Ravindran, B., Sarsaiya, S., Chen, H., Wainaina, S., Singh, E., et al. (2020). Metagenomics for taxonomy profiling: tools and approaches. Bioengineered 11, 356–374. doi: 10.1080/21655979.2020.1736238
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547. doi: 10.1093/molbev/msy096
Langille, M. G., Zaneveld, J., Caporaso, J. G., Mcdonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Levy, M. A., and Lubell, M. N. (2018). Innovation, cooperation, and the structure of three regional sustainable agriculture networks in California. Reg. Environ. Change 18, 1235–1246. doi: 10.1007/s10113-017-1258-6
Li, X., Jousset, A., De Boer, W., Carrión, V. J., Zhang, T., Wang, X., et al. (2019). Legacy of land use history determines reprogramming of plant physiology by soil microbiome. ISME J. 13, 738–751. doi: 10.1038/s41396-018-0300-0
Li, Z., Feng, C., Luo, X., Yao, H., Zhang, D., and Zhang, T. (2018). Revealing the influence of microbiota on the quality of Pu-erh tea during fermentation process by shotgun metagenomic and metabolomic analysis. Food Microbiol. 76, 405–415. doi: 10.1016/j.fm.2018.07.001
Liu, P., Wang, X. H., Li, J. G., Qin, W., Xiao, C. Z., Zhao, X., et al. (2015). Pyrosequencing reveals fungal communities in the rhizosphere of Xinjiang jujube. Biomed. Res. Int. 2015, 1–8. doi: 10.1155/2015/972481
López-López, O., Cerdan, M. E., and Gonzalez Siso, M. I. (2014). New extremophilic lipases and esterases from metagenomics. Curr. Protein Pept. Sci. 15, 445–455. doi: 10.2174/1389203715666140228153801
Louca, S., Doebeli, M., and Parfrey, L. W. (2018). Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 6, 1–12. doi: 10.1186/s40168-018-0420-9
Lovell, S. T., and Taylor, J. R. (2013). Supplying urban ecosystem services through multifunctional green infrastructure in the United States. Landsc. Ecol. 28, 1447–1463. doi: 10.1007/s10980-013-9912-y
Lucas, M., Balb?n-Su?rez, A., Smalla, K., and Vetterlein, D. (2018). Root growth, function and rhizosphere microbiome analyses show local rather than systemic effects in apple plant response to replant disease soil. PLoS ONE. 13, e0204922. doi: 10.1371/journal.pone.0204922
Ma, C., Lo, P. K., Xu, J., Li, M., Jiang, Z., Li, G., et al. (2020). Molecular mechanisms underlying lignocellulose degradation and antibiotic resistance genes removal revealed via metagenomics analysis during different agricultural wastes composting. Biores. Technol. 314, 123731. doi: 10.1016/j.biortech.2020.123731
Macivor, J. S., and Ksiazek, K. (2015). “Invertebrates on green roofs,” in Green Roof Ecosystems, ed R. K. Sutton (New York, NY: Springer), 333–355.
Mahmoud, M., Zywicki, M., Twardowski, T., and Karlowski, W. M. (2019). Efficiency of PacBio long read correction by 2nd generation Illumina sequencing. Genomics 111, 43–49. doi: 10.1016/j.ygeno.2017.12.011
Makonde, H. M., Mwirichia, R., Osiemo, Z., Boga, H. I., and Klenk, H. P. (2015). 454 Pyrosequencing-based assessment of bacterial diversity and community structure in termite guts, mounds and surrounding soils. Springer Plus 4, 471. doi: 10.1186/s40064-015-1262-6
Manjula, A., and Narsimha, G. (2015). “XCYPF: A flexible and extensible framework for agricultural crop yield prediction,” in 2015 Institute of Electrical and Electronic Engineers 9th International Conference on Intelligent Systems and Control (Coimbatore: IEEE), 1–5.
Marchesi, J. R., and Ravel, J. (2015). The vocabulary of microbiome research: a proposal. Microbiome 3, 1–3. doi: 10.1186/s40168-015-0094-5
Markowitz, V. M., Chen, I. M. A., Chu, K., Szeto, E., Palaniappan, K., Grechkin, Y., et al. (2012). IMG/M: the integrated metagenome data management and comparative analysis system. Nucleic Acids Res. 40, D123–D129. doi: 10.1093/nar/gkr975
Maropola, M. K. A., Ramond, J. B., and Trindade, M. (2015). Impact of metagenomic DNA extraction procedures on the identifiable endophytic bacterial diversity in Sorghum bicolor (L. Moench). J. Microbiol. Meth. 112, 104–117. doi: 10.1016/j.mimet.2015.03.012
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12. doi: 10.14806/ej.17.1.200
Martínez-Porchas, M., and Vargas-Albores, F. (2017). Microbial metagenomics in aquaculture: a potential tool for a deeper insight into the activity. Rev. Aquacult. 9, 42–56. doi: 10.1111/raq.12102
Mcguire, K. L., Payne, S. G., Orazi, G., and Palmer, M. I. (2015). “Bacteria and fungi in green roof ecosystems.,” in Green Roof Ecosystems, ed R. K. Sutton (Cham; New York, NY: Springer), 175–191.
Medvedev, P., Georgiou, K., Myers, G., and Brudno, M. (2007). “Computability of models for sequence assembly,” in International Workshop on Algorithms in Bioinformatics (New York, NY: Springer), 289–301
Mok, H.-F., Williamson, V. G., Grove, J. R., Burry, K., Barker, S. F., and Hamilton, A. J. (2014). Strawberry fields forever? Urban agriculture in developed countries: a review. Agron. Sustain. Dev. 34, 21–43. doi: 10.1007/s13593-013-0156-7
Murphy, C. J., Baggs, E. M., Morley, N., Wall, D. P., and Paterson, E. (2015). Rhizosphere priming can promote mobilisation of N-rich compounds from soil organic matter. Soil Biol. Biochem. 81, 236–243. doi: 10.1016/j.soilbio.2014.11.027
Nehls, T., Jiang, Y., Dennehy, C., Zhan, X., and Beesley, L. (2015). “From waste to value: urban agriculture enables cycling of resources in cities,” in Urban Agriculture Europe, eds F. Lohrberg, L. Licka, L. Scazzosi, and A. Timpe (Berlin: Jovis), 170–173. Available online at: https://scholar.google.com/scholar?lookup=0&q=Nehls,+T.,+Jiang,+Y.,+Dennehy,+C.,+Zhan,+X.,+and+Beesley,+L.+(2015).+From+waste+to+value:+urban+agriculture+enables+cycling+of+resources+in+cities.+In:+Urban+Agriculture+Europe+(Lohrberg,+F.,+Licka,+L.,+Scazzosi,+L.+%26+Timpe,+A.,+eds).+JOVIS,+Berlin,+German&hl=en&as_sdt=2007
Ngara, T. R., and Zhang, H. (2018). Recent advances in function-based metagenomic screening. Genom. Proteom. Bioinform. 16, 405–415. doi: 10.1016/j.gpb.2018.01.002
O'brien, P. A., Webster, N. S., Miller, D. J., and Bourne, D. G. (2019). Host-microbe coevolution: applying evidence from model systems to complex marine invertebrate holobionts. MBio 10, e02241–e02218. doi: 10.1128/mBio.02241-18
Oulas, A., Pavloudi, C., Polymenakou, P., Pavlopoulos, G. A., Papanikolaou, N., Kotoulas, G., et al. (2015). Metagenomics: tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform. Biol. Insights 9, 75–88. doi: 10.4137/BBI.S12462
Peng, W., Li, X., Song, J., Jiang, W., Liu, Y., and Fan, W. (2018). Bioremediation of cadmium-and zinc-contaminated soil using Rhodobacter sphaeroides. Chemosphere 197, 33–41. doi: 10.1016/j.chemosphere.2018.01.017
Philippot, L., Raaijmakers, J. M., Lemanceau, P., and Van Der Putten, W. H. (2013). Going back to the roots: the microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 11, 789–799. doi: 10.1038/nrmicro3109
Pinu, F. R., Villas-Boas, S. G., and Aggio, R. (2017). Analysis of intracellular metabolites from microorganisms: quenching and extraction protocols. Metabolites 7, 53. doi: 10.3390/metabo7040053
Plakias, A. C. (2016). The Farm on the Roof: What Brooklyn Grange Taught us About Entrepreneurship, Community, and Growing a Sustainable Business. Englang: Penguin Publishers.
Pölling, B., Mergenthaler, M., and Lorleberg, W. (2016). Professional urban agriculture and its characteristic business models in Metropolis Ruhr, Germany. Land Use Policy 58, 366–379. doi: 10.1016/j.landusepol.2016.05.036
Pölling, B., Sroka, W., and Mergenthaler, M. (2017). Success of urban farming's city-adjustments and business models-Findings from a survey among farmers in Ruhr Metropolis, Germany. Land Use Policy 69, 372–385. doi: 10.1016/j.landusepol.2017.09.034
Priya, G., Lau, N. S., Furusawa, G., Dinesh, B., Foong, S. Y., and Amirul, A. A. A. (2018). Metagenomic insights into the phylogenetic and functional profiles of soil microbiome from a managed mangrove in Malaysia. Agri. Gene 9, 5–15. doi: 10.1016/j.aggene.2018.07.001
Prosser, J. I. (2015). Dispersing misconceptions and identifying opportunities for the use of'omics' in soil microbial ecology. Nat. Rev. Microbiol. 13, 439–446. doi: 10.1038/nrmicro3468
Rabiee, M., Sayyari, E., and Mirarab, S. (2019). Multi-allele species reconstruction using astral. Mol. Phylogenet. Evol. 130, 286–296. doi: 10.1016/j.ympev.2018.10.033
Raymann, K. (2014). Reconstructing the evolutionary relationships between Archaea and Eukaryotes: a phylogenomic approach. Universit? Pierre et Marie Curie-Paris VI, Paris, France
Revillini, D., Gehring, C. A., and Johnson, N. C. (2016). The role of locally adapted mycorrhizas and rhizobacteria in plant-soil feedback systems. Funct. Ecol. 30, 1086–1098. doi: 10.1111/1365-2435.12668
Ruiz, O. N., Brown, L. M., Radwan, O., Bowen, L. L., Gunasekera, T. S., Mueller, S. S., et al. (2021). Metagenomic characterization reveals complex association of soil hydrocarbon-degrading bacteria. Int. Biodeter. Biodegr. 157, 105161. doi: 10.1016/j.ibiod.2020.105161
Russo, A., Escobedo, F. J., Cirella, G. T., and Zerbe, S. (2017). Edible green infrastructure: an approach and review of provisioning ecosystem services and disservices in urban environments. Agric. Ecosyst. Environ. 242, 53–66. doi: 10.1016/j.agee.2017.03.026
Sabale, S. N., Suryawanshi, P. P., and Krishnaraj, P. (2019). Soil Metagenomics: Concepts and Applications. Metagenomics-Basics, Methods and Applications. London: IntechOpen.
Salem, M. A., Perez De Souza, L., Serag, A., Fernie, A. R., Farag, M. A., Ezzat, S. M., et al. (2020). Metabolomics in the context of plant natural products research: from sample preparation to metabolite analysis. Metabolites 10, 37. doi: 10.3390/metabo10010037
Santamaria, M., Fosso, B., Licciulli, F., Balech, B., Larini, I., Grillo, G., et al. (2018). ITSoneDB: a comprehensive collection of eukaryotic ribosomal RNA Internal Transcribed Spacer 1 (ITS1) sequences. Nucleic Acids Res. 46, D127–D132. doi: 10.1093/nar/gkx855
Schwarz, K., Cutts, B. B., London, J. K., and Cadenasso, M. L. (2016). Growing gardens in shrinking cities: a solution to the soil lead problem? Sustainability 8, 141. doi: 10.3390/su8020141
Sergaki, C., Lagunas, B., Lidbury, I., Gifford, M. L., and Schäfer, P. (2018). Challenges and approaches in microbiome research: from fundamental to applied. Front. Plant Sci. 9, 1–12. doi: 10.3389/fpls.2018.01205
Sessitsch, A., Hardoim, P., Döring, J., Weilharter, A., Krause, A., Woyke, T., et al. (2012). Functional characteristics of an endophyte community colonizing rice roots as revealed by metagenomic analysis. Mol. Plant Microbe Interact. 25, 28–36. doi: 10.1094/MPMI-08-11-0204
Sharma, B., Sarkar, A., Singh, P., and Singh, R. (2017). Agricultural utilization of biosolids: a review on potential effects on soil and plant grown. J. Waste Manag. 64, 117–132. doi: 10.1016/j.wasman.2017.03.002
Sieber, C. M., Probst, A. J., Sharrar, A., Thomas, B. C., Hess, M., Tringe, S. G., et al. (2018). Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843. doi: 10.1038/s41564-018-0171-1
Simon Rojo, M., Moratalla, A. Z., Alonso, N. M., and Jimenez, V. H. (2014). Pathways towards the integration of periurban agrarian ecosystems into the spatial planning system. Ecol. Process. 3, 1–16. doi: 10.1186/s13717-014-0013-x
Singh, T., Gangil, B., Patnaik, A., Biswas, D., and Fekete, G. (2019). Agriculture waste reinforced cornstarch-based biocomposites: effect of rice husk/walnut shell on physicomechanical, biodegradable and thermal properties. Mater. Res. Express 6, 1–11. doi: 10.1088/2053-1591/aafe45
Smith-Ramesh, L. M., and Reynolds, H. L. (2017). The next frontier of plant-soil feedback research: unraveling context dependence across biotic and abiotic gradients. J. Veg. Sci. 28, 484–494. doi: 10.1111/jvs.12519
Soni, R., and Goel, R. (2011). nifH homologs from soil metagenome. Ekologija 57. doi: 10.6001/ekologija.v57i3.1914
Suárez, J., and Stencel, A. (2020). A part-dependent account of biological individuality: why holobionts are individuals and ecosystems simultaneously. Biol. Rev. 95, 1308–1324. doi: 10.1111/brv.12610
Sun, K. (2020). Ktrim: an extra-fast and accurate adapter-and quality-trimmer for sequencing data. Bioinformatics 36, 3561–3562. doi: 10.1093/bioinformatics/btaa171
Tamames, J., and Puente-Sánchez, F. (2019). SqueezeMeta, a highly portable, fully automatic metagenomic analysis pipeline. Front. Microbiol. 9, 3349. doi: 10.3389/fmicb.2018.03349
Tan, B., Ng, C. M., Nshimyimana, J. P., Loh, L. L., Gin, K. Y. H., and Thompson, J. R. (2015). Next-generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Front. Microbiol. 6, 1027. doi: 10.3389/fmicb.2015.01027
Tan, W., Wang, J., Bai, W., Qi, J., and Chen, W. (2020). Soil bacterial diversity correlates with precipitation and soil pH in long-term maize cropping systems. Scient. Rep. 10, 1–12. doi: 10.1038/s41598-020-62919-7
Teste, F. P., Kardol, P., Turner, B. L., Wardle, D. A., Zemunik, G., Renton, M., and Laliberté, E. (2017). Plant-soil feedback and the maintenance of diversity in Mediterranean-climate shrublands. Science 355, 173–176. doi: 10.1126/science.aai8291
Thomas, F., Hehemann, J.-H., Rebuffet, E., Czjzek, M., and Michel, G. (2011). Environmental and gut bacteroidetes: the food connection. Front. Microbiol. 2, 93. doi: 10.3389/fmicb.2011.00093
Trivedi, P., Mattupalli, C., Eversole, K., and Leach, J. E. (2021). Enabling sustainable agriculture through understanding and enhancement of microbiomes. New Phytol. 230, 2129–2147. doi: 10.1111/nph.17319
Turner, T. R., James, E. K., and Poole, P. S. (2013). The plant microbiome. Genome Biol. 14, 1–10. doi: 10.1186/gb-2013-14-6-209
Van Niftrik, L., Geerts, W. J., Van Donselaar, E. G., Humbel, B. M., Webb, R. I., Fuerst, J. A., et al. (2008). Linking ultrastructure and function in four genera of anaerobic ammonium-oxidizing bacteria: cell plan, glycogen storage, and localization of cytochrome c proteins. J. Bacteriol. 190, 708–717. doi: 10.1128/JB.01449-07
Van Veenhuizen, R., and Danso, G. (2007). Profitability and Sustainability of Urban and Periurban Agriculture. Rome: Food and Agriculture Organization. p. 1–95
Vargas-Albores, F., Martínez-Córdova, L. R., Martínez-Porchas, M., Calderón, K., and Lago-Lestón, A. (2019). Functional metagenomics: a tool to gain knowledge for agronomic and veterinary sciences. Biotechnol. Genet. Eng. Rev. 35, 69–91. doi: 10.1080/02648725.2018.1513230
Wang, C. Y., Zhou, X., Guo, D., Zhao, J. H., Yan, L., Feng, G. Z., et al. (2019). Soil pH is the primary factor driving the distribution and function of microorganisms in farmland soils in northeastern China. Ann. Microbiol. 69, 1461–1473. doi: 10.1007/s13213-019-01529-9
Williams, N. S., Lundholm, J., and Scott Macivor, J. (2014). Do green roofs help urban biodiversity conservation? J. Appl. Ecol. 51, 1643–1649. doi: 10.1111/1365-2664.12333
Winand, R., Bogaerts, B., Hoffman, S., Lefevre, L., Delvoye, M., Van Braekel, J., et al. (2019). Targeting the 16s rRNA gene for bacterial identification in complex mixed samples: comparative evaluation of second (illumina) and third (oxford nanopore technologies) generation sequencing technologies. Int. J. Mol. Sci. 21, 298. doi: 10.3390/ijms21010298
Wongkiew, S., Polprasert, C., Koottatep, T., Limpiyakorn, T., Surendra, K., and Khanal, S. K. (2022). Chicken manure-based bioponics: effects of acetic acid supplementation on nitrogen and phosphorus recoveries and microbial communities. J. Waste Manag. 137, 264–274. doi: 10.1016/j.wasman.2021.11.023
Wu, L., Chen, J., Xiao, Z., Zhu, X., Wang, J., Wu, H., et al. (2018a). Barcoded pyrosequencing reveals a shift in the bacterial community in the rhizosphere and rhizoplane of Rehmannia glutinosa under consecutive monoculture. Int. J. Mol. Sci. 19, 850. doi: 10.3390/ijms19030850
Wu, L., Wang, J., Wu, H., Chen, J., Xiao, Z., Qin, X., et al. (2018b). Comparative metagenomic analysis of rhizosphere microbial community composition and functional potentials under Rehmannia glutinosa consecutive monoculture. Int. J. Mol. Sci. 19, 2394. doi: 10.3390/ijms19082394
Xu, L., Naylor, D., Dong, Z., Simmons, T., Pierroz, G., Hixson, K. K., et al. (2018). Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. 115, E4284–E4293. doi: 10.1073/pnas.1717308115
Young, A. D., and Gillung, J. P. (2020). Phylogenomics-principles, opportunities and pitfalls of big-data phylogenetics. Syst. Entomol. 45, 225–247. doi: 10.1111/syen.12406
Yu, J., Blom, J., Glaeser, S., Jaenicke, S., Juhre, T., Rupp, O., et al. (2017). A review of bioinformatics platforms for comparative genomics. Recent developments of the EDGAR 2.0 platform and its utility for taxonomic and phylogenetic studies. J. Biotechnol. 261, 2–9. doi: 10.1016/j.jbiotec.2017.07.010
Zarik, L., Meddich, A., Hijri, M., Hafidi, M., Ouhammou, A., Ouahmane, L., et al. (2016). Use of arbuscular mycorrhizal fungi to improve the drought tolerance of Cupressus atlantica G. C.R. Biol. 339, 185–196. doi: 10.1016/j.crvi.2016.04.009
Zhang, L., Chen, F., Zeng, Z., Xu, M., Sun, F., Yang, L., et al. (2021). Advances in metagenomics and its application in environmental microorganisms. Front. Microbiol. 12, 766364–766364. doi: 10.3389/fmicb.2021.766364
Keywords: bioinformatics, micro-ecological niche, omic techniques, rhizosphere, shotgun metagenomic, sustainable food production
Citation: Nwachukwu BC and Babalola OO (2022) Metagenomics: A Tool for Exploring Key Microbiome With the Potentials for Improving Sustainable Agriculture. Front. Sustain. Food Syst. 6:886987. doi: 10.3389/fsufs.2022.886987
Received: 01 March 2022; Accepted: 05 May 2022;
Published: 17 June 2022.
Edited by:
Everlon Cid Rigobelo, São Paulo State University, BrazilReviewed by:
Nicolas Desoignies, HEPH Condorcet, BelgiumLuis Gabriel Cueva-Yesquén, State University of Campinas, Brazil
Tanvir Kaur, Eternal University, India
Copyright © 2022 Nwachukwu and Babalola. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olubukola Oluranti Babalola, b2x1YnVrb2xhLmJhYmFsb2xhQG53dS5hYy56YQ==