 Sheetal Ambardar
Sheetal Ambardar Nancy Bhagat
Nancy Bhagat Jyoti Vakhlu
Jyoti Vakhlu Malali Gowda
Malali Gowda- 1National Center for Biological Sciences, Bangalore, India
- 2Centre for Functional Genomics and Bioinformatics, The University of Trans-Disciplinary Health Sciences and Technology, Bangalore, India
- 3School of Biotechnology, University of Jammu, Jammu, India
- 4Bengaluru Genomics Centre, Bangalore, India
Earlier plant growth promoting rhizo-bacteria (PGPRs) were isolated from the plants, by cultivation based techniques and the interaction was mostly thought to be bilateral. The routine bilateral study, with no information on the associated microbiome, could be one of the reasons for the limited success of PGPRs in the field conditions. Keeping in view the role of PGPRs in rhizo-bacteriome on the growth and production of plant, the present study was aimed at studying the diversity of the rhizo-bacteriome of saffron grown across three geographical locations namely Kashmir, Kishtwar and Bengaluru. Variation in the rhizo-bacteriome of saffron growing across 10 different sites from 3 geographical locations was studied using 16S rDNA amplicon metagenomic sequencing. 16 bacterial phyla, 261 genera and 73 bacterial species were cataloged from all the rhizosphere samples. Proteobacteria was a dominant phylum in all the rhizosphere samples. Rhizo-bacteriome of saffron grown in Kishtwar was found to be significantly different from the rhizo-bacteriome of saffron grown in Kashmir and Bengaluru. Interestingly, the rhizo-bacteriome of saffron grown in Bengaluru was very similar to the saffron grown in Kashmir, thereby indicating that the rhizo-bacteriome in saffron is “plant driven” as the corm sown in Bengaluru were from Kashmir. Despite variation in rhizo-bacteriome, core rhizo-bacteriome in saffron was identified that was represented by 53 genera and eight bacterial species belonging to 11 phyla irrespective of their geographical distribution. In addition, 21 PGPRs were reported for the first time from the saffron rhizosphere. The high yielding saffron field Wuyan was found to have the highest number of PGPRs; this indicates that the presence of PGPR is important for yield enhancement than diversity. The two PGPR Rhizobium leguminosarum and Luteibacter rhizovicinus were reported from all the locations except Kishtwar that had escaped isolation in our previous attempts using cultivation based techniques. It is being proposed instead of going for random isolation and screening for PGPRs from plant rhizosphere, an alternate strategy using metagenomic cataloging of the rhizo-bacteriome community and cultivation of the dominant PGPR should be undertaken. This strategy will help in the selection of dominant PGPRs, specific to the plant in question.
Introduction
Microbiomics is a fast-growing field in which collective genomes of microorganisms, of a given community (a microbiota) are investigated. Literature is replete, with the studies on the microbiomes of environmental samples such as soil and water (Gui-Feng et al., 2020), human gut and skin (Parello, 2020), plant rhizosphere, cormosphere and phyllosphere (Stone et al., 2018; Bhagat et al., 2021; Rüger et al., 2021). The plant microbiome is reported to be critical to host adaptation, productivity and health (Zhao et al., 2019; Trivedi et al., 2020). The plant microbiome acts as a reservoir of microbes, that directly influences the structure and composition of the plant, promotes plant growth, increases stress tolerance, mediates local patterns of nutrient cycling and can also be used as molecular markers (Bakker et al., 2013; Coats and Rumpho, 2014; Trivedi et al., 2020; Bhagat et al., 2021). Earlier microbiome associated with different plants would be studied mostly using cultivation based techniques. However, it is a fact now, that not more than 1% of bacteria can be cultivated by routine cultivation and 99% remained uncultivated (Steen et al., 2019). In order to study microbiomes associated with any niche, cultivation independent technique metagenomics, complements the cultivation based techniques. In metagenomics, genomes of the bacterial communities are extracted collectively and sequenced directly. Metagenomics has revolutionized the study of complex microbial communities, as it overcomes the limitation of cultivation based methods, as far as cataloging of bacteria present and functions performed by them, in any niche is concerned (Boughner and Singh, 2016; Alteio et al., 2020; Taş et al., 2021). Further, the attention has shifted from plant-microbe interaction to plant-microbiome interaction and the role of microbiome in plant growth promotion (Kour et al., 2019; Yadav et al., 2020). The rhizosphere microbiome's contribution to plants has been acknowledged by giving it the title of “plant's second genome” (Ofek-Lalzar et al., 2014; Yin et al., 2020). Plant growth promotion by Plant growth promoting bacteria (PGPBs) has been reported in various plants such as in rice (Chauhan et al., 2019), wheat (Çakmakçı et al., 2017), maize (Zerrouk et al., 2019), tomato (Cordero et al., 2018), soya bean (De Gregorio et al., 2017) and saffron (Ambardar and Vakhlu, 2013; Ambardar, 2014; Ambardar et al., 2014, 2016; Kour et al., 2018; Magotra et al., 2021).
Crocus sativus, commonly known as saffron, is the world's costliest spice with medicinal and cosmetic value (Zhao et al., 2019; Bhooma et al., 2020). 1 kg of saffron costs about 11,000 USD/Kg (Ambardar et al., 2014). The other reasons for the selection of the Crocus sativus, as a test organism, in addition to its economical status is, (i) reference microbiome of root and corm during various growth stages has been cataloged previously, (ii) PGPBs have been isolated and evaluated for their efficacy, (iii) and most importantly, it is reported to be a monogenetic herb world over with the uniform genotype, hence variation in rhizo-bacteriome and its effect on yield was the question worth asking. Variation in the quality and yield of saffron growing across different locations has been reported due to various factors such as epigenetic, climate change, soil characteristics and microbes associated with plants. It is a sterile triploid plant (2n = 3x = 24) and reproduces vegetatively by underground bulb-like, starch-storing organs, known as corms (Wani et al., 2018; Nemati et al., 2019). The herb has an interesting biannual life cycle that is characterized by three distinct growth stages, dormant stage from July to August, flowering stage from October to November and vegetative stage from January to May. The flowering stage lasts ~30 days a year. Each flower has three red trumpets like stigmas (2 to 3 cm long) which when dried, are commercialized as a saffron spice (Yasmin and Nehvi, 2013). The bacteriome and mycobiome of saffron rhizosphere were, for the first time, reported by our group (Ambardar et al., 2014, 2016). In addition, PGPBs were isolated and evaluated in the course of the study (Ambardar and Vakhlu, 2013; Ambardar, 2014; Kour et al., 2018; Magotra et al., 2021). Recently, the cormosphere bacteriome of saffron was compared across the geographical locations from India and Morocco and preliminary data suggests that the microbiome is location specific and simultaneously, there is a core microbiome that is common to all locations (Bhagat et al., 2021). Such reports of core and location specific microbiome have also been reported in other plants such as, rice (Eyre et al., 2019); coffee (Fulthorpe et al., 2020); switch grass (Grady et al., 2019); red sage (Chen et al., 2018), wheat (Kuzniar et al., 2020) etc.
Since saffron is reported to be a monogenetic plant and has the same genotype worldwide, the present study was undertaken to explore the variation in the rhizo-bacteriome associated with the plant grown across different geographical locations in India to evaluate the effect of geography on rhizo-bacteriome of the saffron. Rhizo-bacteriome associated with a saffron plant was compared and a core rhizo-bacteriome was identified using 16S rDNA gene targeted metagenomic analysis during the flowering stage in November 2016. In addition, an inventory of putative PGPRs present in the rhizo-bacteriome was made and compared across these locations and a correlation with the yield was attempted.
Materials and Methods
Sample Collection
Saffron rhizosphere samples were collected from 10 locations including eight saffron fields from Kashmir district (Wuyan, Patalbagh_upper range, Patalbagh_lower range, Alchibagh, Galandar, Chandhara, Hatiwara, Samboora); one from Berwar saffron field from Kishtwar, Jammu district and one laboratory experiment conducted in Bengaluru (Figure 1). The details of all the saffron fields, geographical coordinates (latitude and longitude), fertilizers usage, total production, and total area of cultivation have been tabulated in Table 1. Composite sampling was performed from all the fields wherein the Crocus sativus rhizosphere samples were collected from the three corners of five fields of each location and all the 15 samples per location were pooled together to form composite samples (Ambardar and Vakhlu, 2013; Ambardar, 2014). Rhizosphere samples were collected by uprooting the plants and vigorously shaking by hand. The soil that remained adheres to the roots after vigorous shaking was taken as rhizosphere soil. The roots were washed in normal saline (0.85% NaCl) that was taken further for metagenomic DNA extraction.
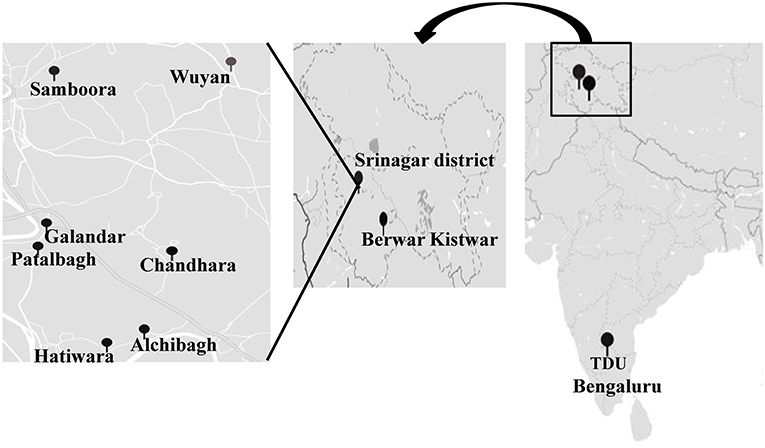
Figure 1. Geographic location of sample collection sites on the map of India. TDU (The University of Trans-Disciplinary Health Sciences and Technology), Bengaluru, (Berwar) Kishtwar and 8 locations with in Srinagar district (Wuyan, Patalbagh_upper range, Patalbagh_lower range, Alchibagh, Galandar, Chandhara, Hatiwara and Samboora).
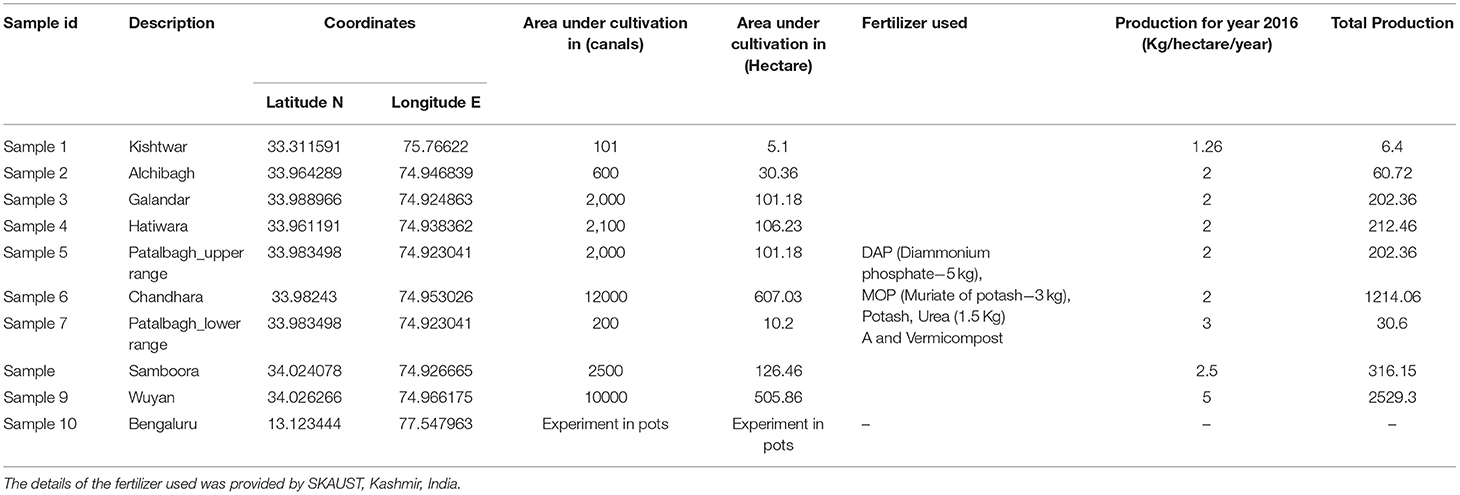
Table 1. Area under cultivation and production data of saffron from various saffron fields under study. Saffron production was maximum in the Wuyan fields and lowest in Kishtwar fields.
A separate experiment was conducted in which saffron corms were collected from Kashmir, Pampore during the dormant stage of the saffron life cycle (July, 2016). These corms were then planted in 10 pots (5 corms/pot) filled with the garden soil at Trans-Disciplinary University, Bengaluru (Karnataka). These pots were kept in open under natural conditions (temperature 20°C) for a period of 3 months (August–November 2016). The rhizosphere samples from 15 plants were collected randomly and pooled together to form a composite sample during the flowering stage similar to field samples. This experiment was performed to address the question whether the root microbiome is soil driven or plant driven. Root microbiome was harvested using same design and method as in case of field samples.
Rhizosphere Metagenomic DNA Extraction
Metagenomic DNA was extracted from rhizosphere samples using a MoBio Power soil DNA extraction kit (MoBio Laboratories Inc. Carlsbad, CA, USA) following the manufacturer's instructions. The DNA quality and quantity were determined by using a Nano Drop device (Thermo Scientific, Wilmington, DE) and electrophoresis on 0.8% agarose gel, with 1 kb plus ladder as molecular weight marker.
16S rDNA Sequencing of the Extracted Rhizosphere Metagenome
A 16S rDNA sequencing library was constructed targeting the V3 and V4 hyper-variable regions of the 16S rDNA gene, according to the 16S rDNA metagenomic sequencing library preparation protocol (Illumina, San Diego, CA, USA). The PCR was performed with 12 ng template DNA and region-specific primers with Illumina index and sequencing adapters (forward_primer: 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTCGTCGGCAGCGT CAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′; reverse primer: 5′GTCTCGTG GGCTCGGAGATGTGTATAAGAGACAGGTCTCGTGGGCTCGGAGATGTGTATAAGAGAC AGGACTACHVGGGTATCTAATCC-3′) using KAPA HiFi Hot Start Ready Mix (KAPA Biosystems, Wilmington, MA, USA). The amplicons were purified using the Agencourt AMPure XP system (Beckman Coulter Genomics, Brea, CA, USA). Subsequently, purified PCR products were visualized after gel electrophoresis and quantified with a Qubit dsDNA HS Assay Kit (Thermo Scientific) on a Qubit 3.0 fluorometer. The PCR products of all the samples were pooled to 4 nM concentration, prior to sequencing, and were analyzed on an Agilent 2,200 Tape Station (Agilent Technologies, Santa Clara, CA, USA) for quantity and quality analysis. The pooled sample (4 nM) was denatured with 0.2 N NaOH, diluted further to 4 pM concentration, and combined with 20% (v/v) denatured 4 pM PhiX as control, following Illumina's guidelines. Samples were sequenced on the MiSeq sequencing platform (Illumina) using a 2 × 250 cycle V3 kit, following standard Illumina sequencing protocols.
Data Analysis
Raw data of 16S rDNA sequencing was analyzed for a quality check using the FastQc tool kit (Brown et al., 2017). The adapter and low quality reads (Q < 30 Phred score) were trimmed using the Cutadapt tool of Trimgalore (Jiang et al., 2014). The reads with phred score above 30 were selected for QIIME1 pipeline analysis (version 1.9.1). The QIIME 1 pipeline (Quantitative Insights into Microbial Ecology version 1.9.1) was used to process and filter multiplexed sequence reads. The sequencing reads were grouped into an operational taxonomic unit (OTUs) that were further clustered against GreenGenes_13_8 sequences to ascertain the taxonomical affiliation of bacteria (McDonald et al., 2012). Reads failing to hit the reference were subsequently clustered de novo at the 97% similarity level using the UCLUST greedy algorithm. Chimeric sequences were identified by the UCHIME algorithm of USEARCH61 and removed. OTU sequences were aligned using PYNAST. OTU taxonomy was determined using the Green Genes taxonomy database (Caporaso et al., 2010).
Statistical Analysis of Metagenomic Sequence Data
The bacterial community associated with each sample was compared for alpha and beta diversity analysis using Qiime 1 (Sinclair et al., 2015). For alpha diversity analysis, rarefaction curves and diversity indices like Chao1, Shannon, Simpson, phylogenetic diversity were calculated for estimating the richness and evenness in all the samples (Chao, 1987; Gotelli and Colwell, 2011). For rarefaction curves, the operational taxonomy units OTU table was rarified to an even depth of 1,00,000 sequences per sample, to avoid biases generated by the difference in sequencing depth. Rarefaction curves (97% identity) were plotted between the number of observed OTUs (cluster count) and the number of reads of the samples. In order to evaluate β diversity, PCOA plots, venn diagram and heat maps were constructed. In addition, a circos plot was generated for representing the core rhizo-bacteriome across all locations using the tool Circos Version 0.63-9 (Krzywinski et al., 2009).
Results
Ten rhizosphere samples were collected from 8 different saffron fields from Kashmir and 1 from Kishtwar (Jammu and Kashmir); and 1 from corms grown in pots at Trans-Disciplinary University, Bengaluru (Karnataka). The saffron fields were selected from different geographical locations having variations in total production of saffron as represented in Table 1. Wuyan in Kashmir had maximum saffron production and Kishtwar in Jammu had minimum production in 2016, the year of sample collection (Table 1). The corms were collected from Kashmir and sown in pots in Bengaluru to test the hypothesis that rhizo-bacteriome in saffron is plant driven, hence lacks production data, as flowering needs special climatic conditions. Rhizosphere samples were analyzed for bacterial diversity using 16S rDNA sequencing and the total number of 16S rDNA sequence reads per sample ranged between 51,918 and 97,484 (Table 2). The bacterial communities associated with all the rhizosphere samples were classified up to the phylum, genus and species level (Table 2).
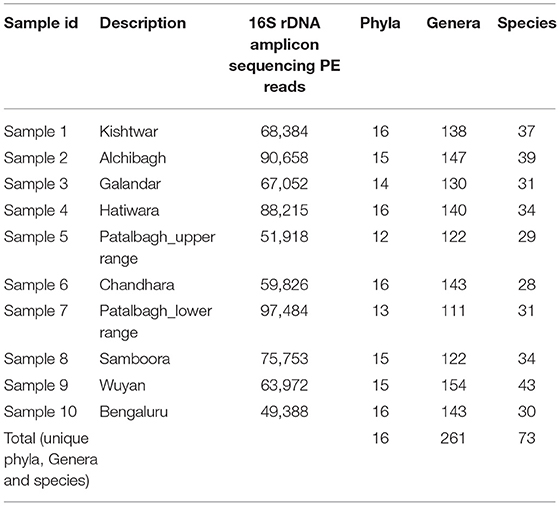
Table 2. 16S rDNA amplicon sequencing reads, total phyla, genera and species cataloged in saffron rhizosphere from various saffron fields under study.
Rhizo-Bacterial Diversity
Bacterial diversity associated with all the 10 rhizospheric samples was cataloged into 16 phyla, 261 genera and 73 species (Table 2). Out of 16 phyla, Proteobacteria was the dominant phyla in all the samples with higher relative abundance in Kashmir (avg 59.1%) followed by Bengaluru (49%) and Kishtwar (31.28%). However, Firmicutes and Planctomycetes were relatively abundant in Kishtwar (20, 10%) as compared to Kashmir (avg 4.9, 3.12%) and Bengaluru (5, 4%) respectively (Figure 2).
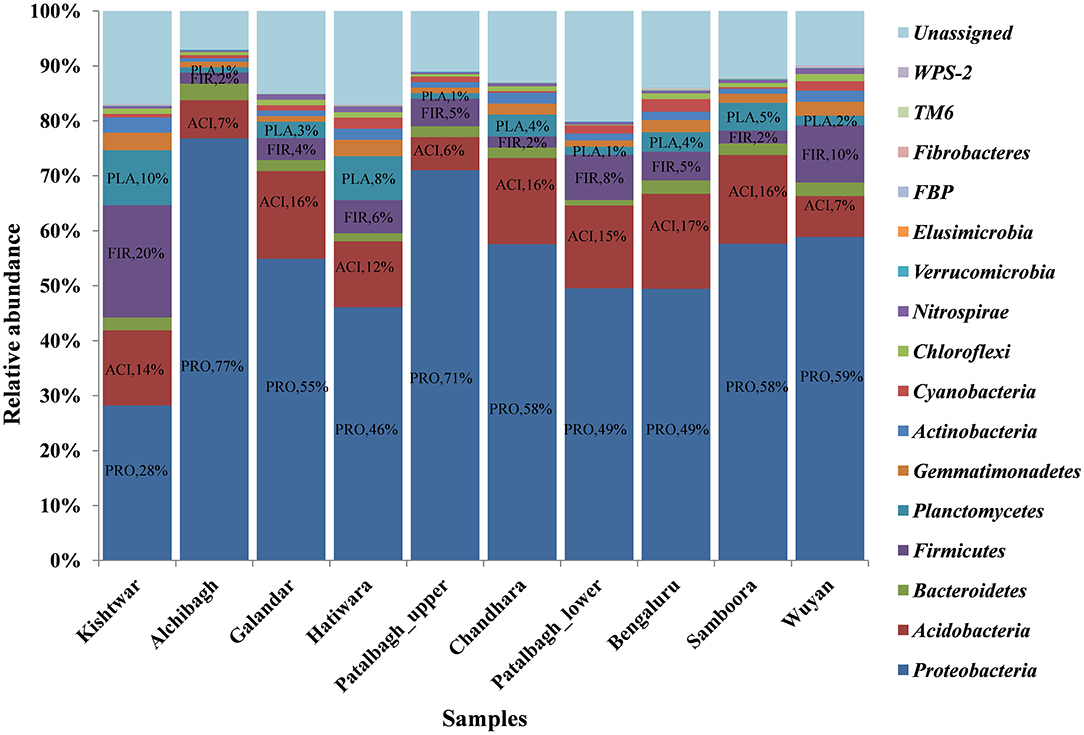
Figure 2. Comparison of rhizo-bacteriome of saffron from different geographical location at phylum level representing dominance of Proteobacteria in all 10 samples. The relative abundance of Proteobacteia is less in Kishtwar (Jammu) as compared to all the samples from Kashmir and Bengaluru whereas Firmicutes and Planctomyces are more in Kishtwar as compared to others. PRO, Proteobacteria; FIR, Firmicutes; ACI, Acidobacteria; PLA, Planctomycetes.
Out of total of 261 bacterial genera cataloged, the abundance of 4 bacterial genera namely Bacillus, Lysobacter, Rhodoplanes and Janthiobacterium were observed. Bacillus was dominant at four locations namely Kishtwar (54%), Hatiwara (24%), Patalbagh_lower range (22%) and Bengaluru (15%), (Figure 3). However, Bacillus was also found co-dominant with other genera at two locations namely Wuyan [Bacillus (16%) and Rhodoplane (17%)]; and Galandar [(Bacillus (12%) and Lysobacter (13%)]. The abundance of Bacillus was comparatively less in rest of the four locations i.e., Alchibagh (4%), Chandhara (7%), Samboora (8%), and Patalbagh_upper (11%). Janthiobacterium was dominant in Alchibagh (19%). Lysobacter was the most abundant bacterial genera in two locations namely Patalbagh_upper range (15%) and Samboora (18%), whereas Lysobacter (15%) shared dominance with Rhodoplanes (14%) in Chandhara (Figure 3).
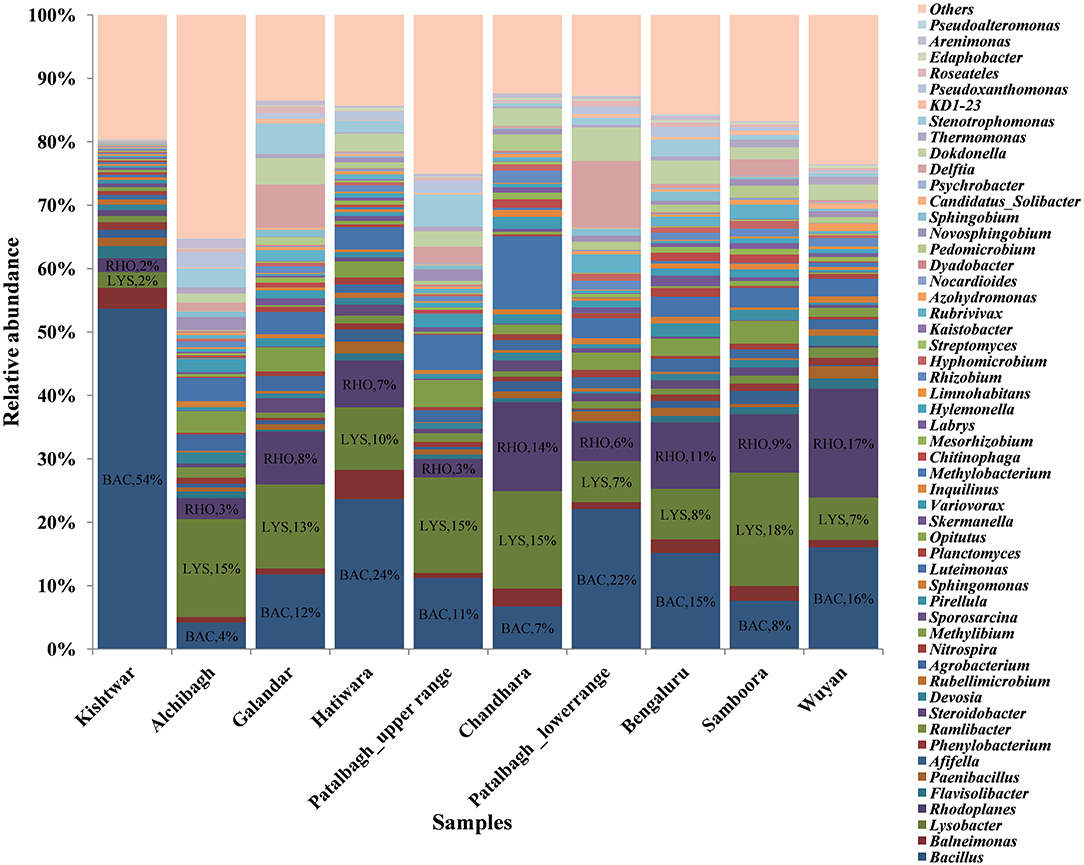
Figure 3. Comparison of bacterial genera in saffron rhizosphere from different geographical locations. The OTU cataloged up to genus level were normalized to 10,000. BAC, Bacillus; LYS, Lysobacter; RHO, Rhodoplane; JAN, Janthiobacterium were dominant genera in these samples. In this figure, Kishtwar sample is distinct from the Kashmir samples. The microbial communities of Kashmir and Bengaluru samples are similar as compared to Kishtwar sample.
53 genera out of 261 bacterial genera were common in all the samples, are considered as the core rhizo-bacteriome that has been discussed in detail under core rhizo-bacteriome section. on comparing all the 10 samples, unique genera specific to each location were also identified. The maximum number of unique bacterial genera were identified in Wuyan (19) followed by Alchibagh (10), Galandar and Kishtwar (6 each), Chandhara (4), Patalbagh_lower range and Samboora (2 each), Patalbagh_upper range, Hatiwara and Bengaluru (1 each), (Figure 4).
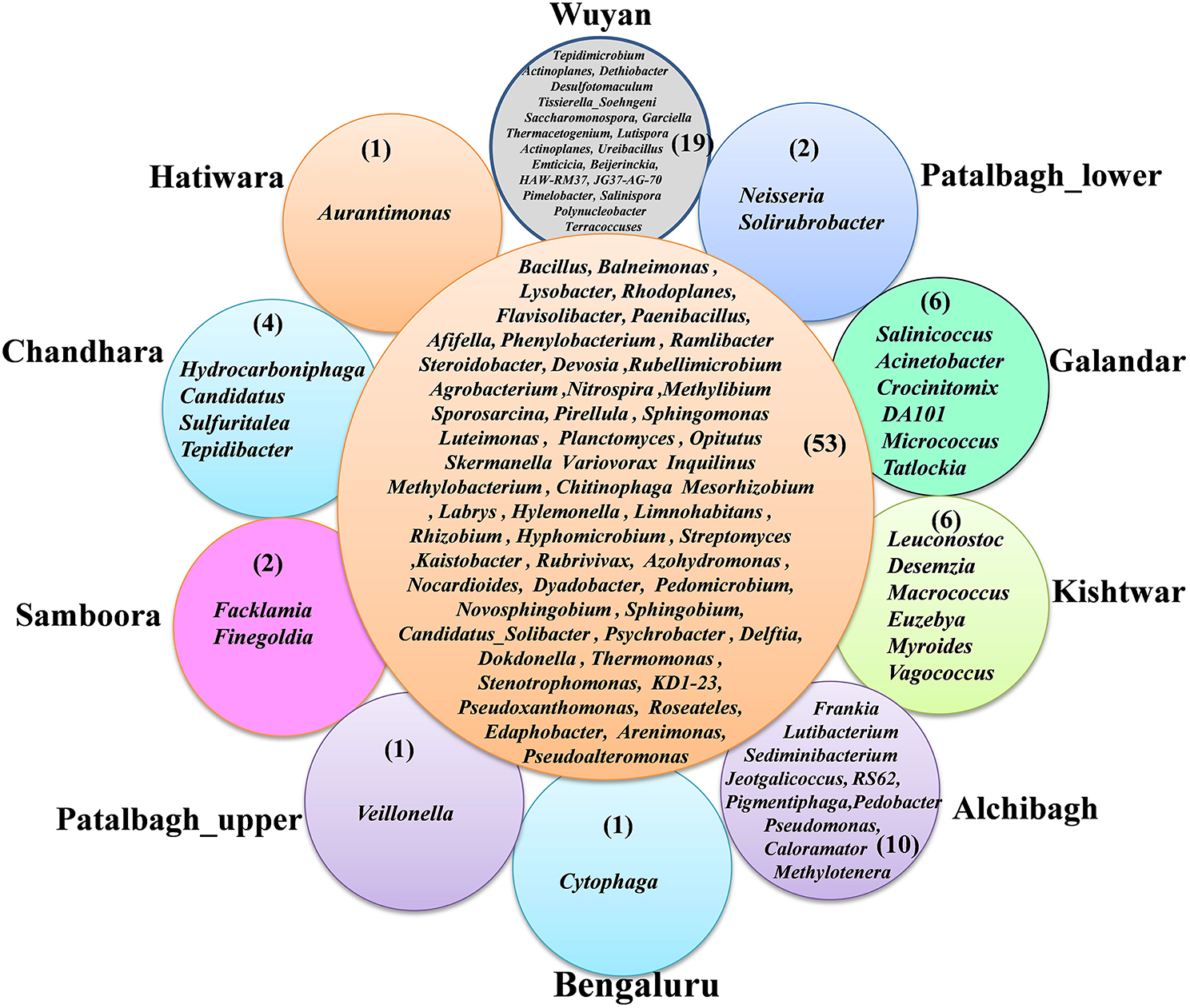
Figure 4. Radical Venn diagram representing the unique genera identified in 10 different locations. Wuyan-Kashmir has the maximum number (19) of unique genera followed by 10 in Alchibagh, 6 in Gullandar and Kishtwar, 4 in Chandhara, 2 in Samboora and Patalbagh_lower range and 1 genera in Hatiwara, Bengaluru, Patalbagh_upper range.
73 bacterial species were present in all the samples, out of which, 3 bacterial species i.e., Bacillus flexus, Lysobacter brunescens and Janthiobacterium lividum were dominant; but their abundance varied (Figure 5). Bacillus flexus was abundant in seven locations namely Kishtwar (49%), Patalbagh_lower range (38%), Hatiwara (35%), Bengaluru (31%), Galandar (28%), Wuyan (22%) and Patalbagh_upper range (20%). However, Janthiobacterium lividum (19%) was abundant in Alchibagh (Figure 5). Lysobacter brunescens was abundant in rest of the two locations namely Samboora (14%) and Chandhara (40%). Out of 73 bacterial species, 8 bacterial species were considered as the core rhizo-bacteriome (common in all the samples) that has been discussed in core rhizo-bacteriome section. In addition to core species, each sample has unique bacterial species that were specific to that particular location and absent in all other samples. Kishtwar had four unique bacterial species namely Bacillus marisflavi, Macrococcus caseolyticus, Myroides odoratimimus and Staphylococcus scuiri whereas two species were unique in Alchibagh namely Methylotenera mobilis and Sphingomonas suberifaciens and Galandar namely Paenibacillus barengoltzii and Paracoccus marcusii. Only one unique species was cataloged from Patalbagh_upper range (Vellonellas dispar), Wuyan (Paenibacillus ginsengarvi) and Samboora (Roseomonas mucosa). However, Bengaluru, Hatiwara, Chandhara and Patalbagh_lower range did not have any unique bacterial species (Figure 6).
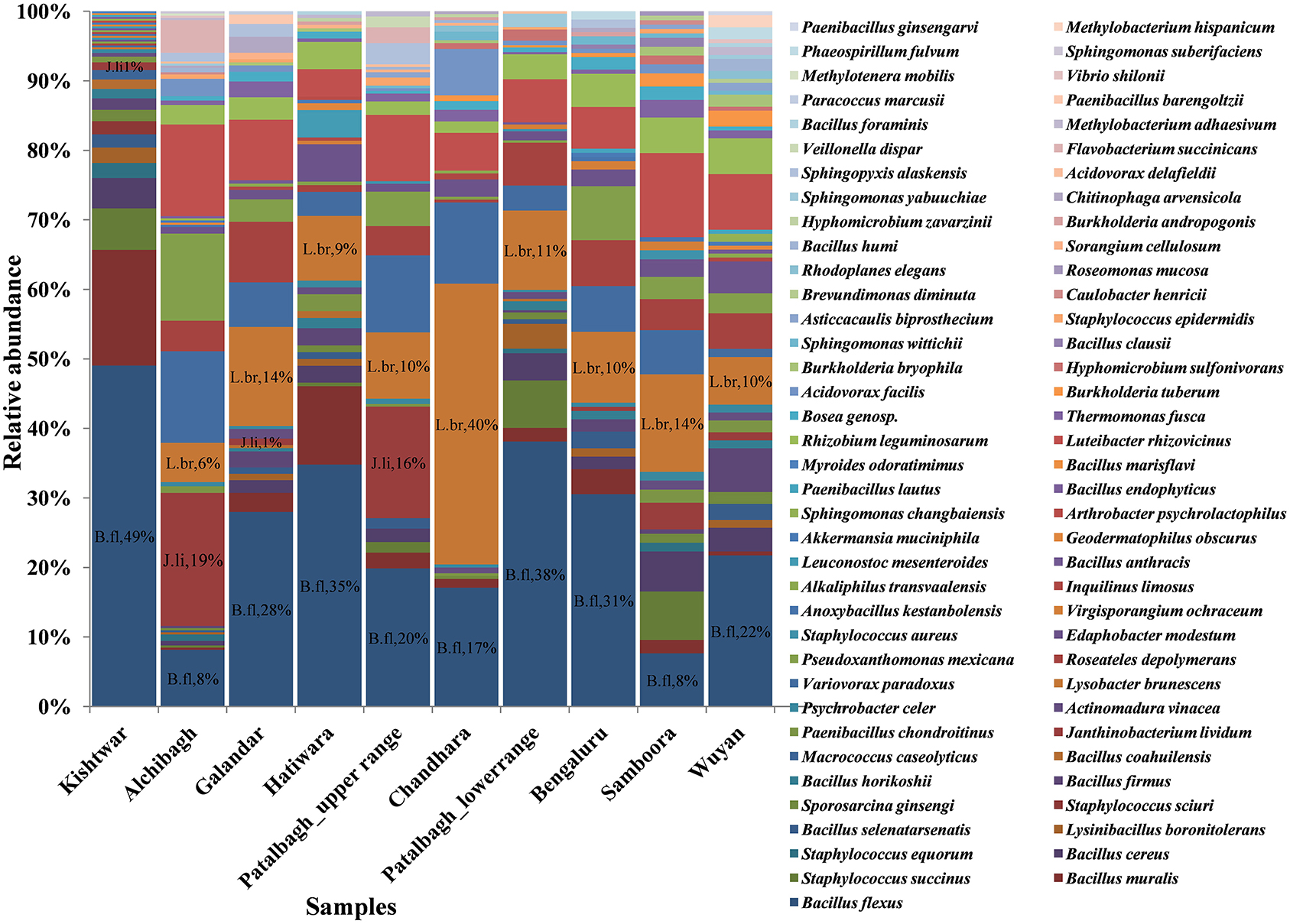
Figure 5. Comparison of bacterial species in saffron rhizosphere from different geographical locations. The OTU cataloged up to species level were normalized to 2,000. A total of 73 species were cataloged from all the samples wherein Bacillus flexus (B. fl), Lysobacter brunescens (L. br), Janthiobacterium lividum (J. li) were found dominant.
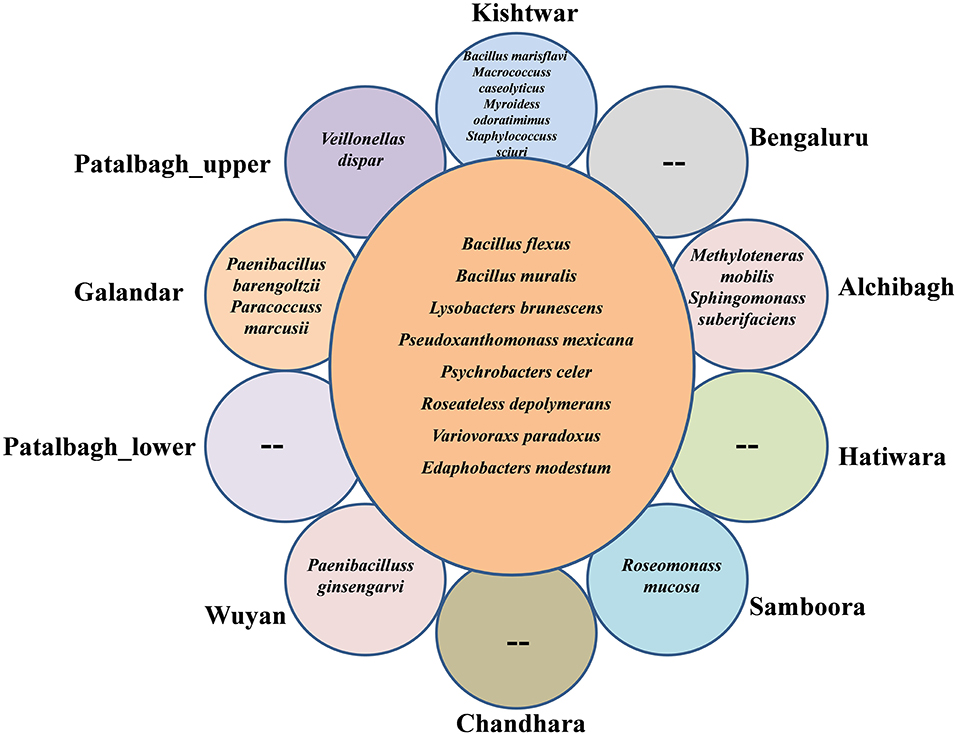
Figure 6. Radical Venn diagram representing the number of unique and common species identified in 10 different locations. 8 common species were identified in 10 samples. Kishtwar had maximum number (4) of unique bacterial species followed by 2 bacterial species each in Alchibagh and Galandar whereas only 1 bacterial species in Patalbagh_upper range, Wuyan and Samboora. Bengaluru, Hatiwara, Chandhara and patalbagh_lower range did not have any unique bacterial species.
Statistical Analysis of Rhizo-Bacterial Diversity
The bacterial diversity of each sample was also analyzed using the alpha diversity indices i.e., Chao1, Simpson, phylogenetic diversity and Shanon (Table 3), rarefaction curves (Figure 7), PCOA plots (Figure 8) generated by Qiime software. Rarefaction curves (97% identity) of rhizosphere samples did not reach plateau indicating bacterial diversity was well-represented but could increase on repetitive sampling (Figure 7). The bacterial community in saffron rhizosphere from Samboora field was diverse as compared to other sample, as depicted by higher number of different species (OTUs) in the rarefaction curve. This result was further complemented by diversity indices such as Chao1, phylogenetic diversity, Shannon and Simpson indexes were also higher in the case of Samboora (Table 3). Beta diversity analysis using PCOA plots was done to evaluate the significant variation in bacterial diversity among all the samples. Beta bacterial diversity of Kishtwar was significantly different from Kashmir and Bengaluru, as it does not cluster with the rest of the samples in PCOA plots (Figure 8).
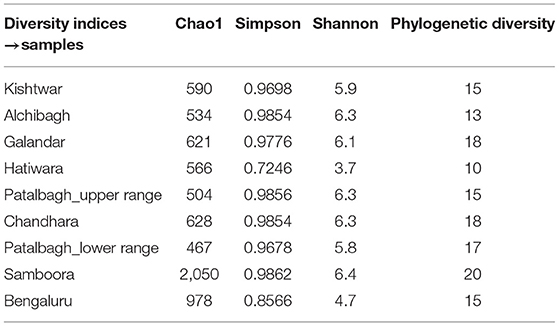
Table 3. Diversity indices were the maximum in Samboora representing maximum bacteria diversity and richness in rhizosphere of saffron growing in Samboora as compared to other samples.
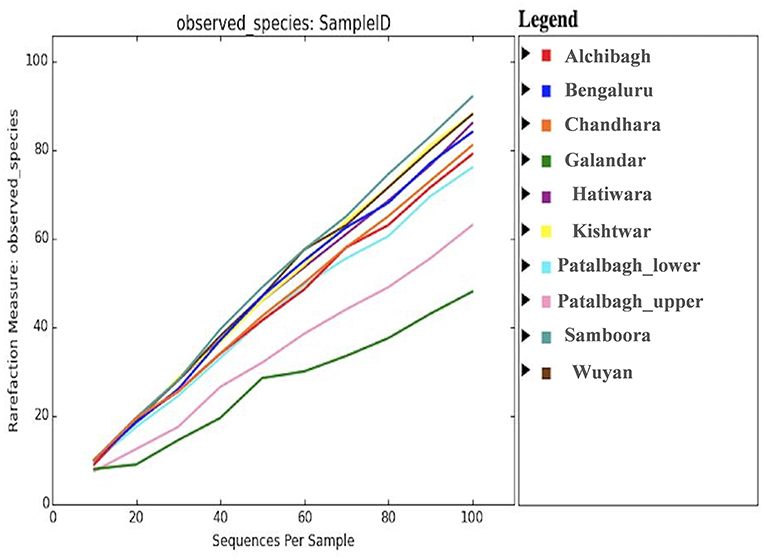
Figure 7. Rarefaction curve representing the bacterial diversity across 10 samples where in Samboora is more diverse and Galandar is least diverse as represented by the number of species observed as per plot.
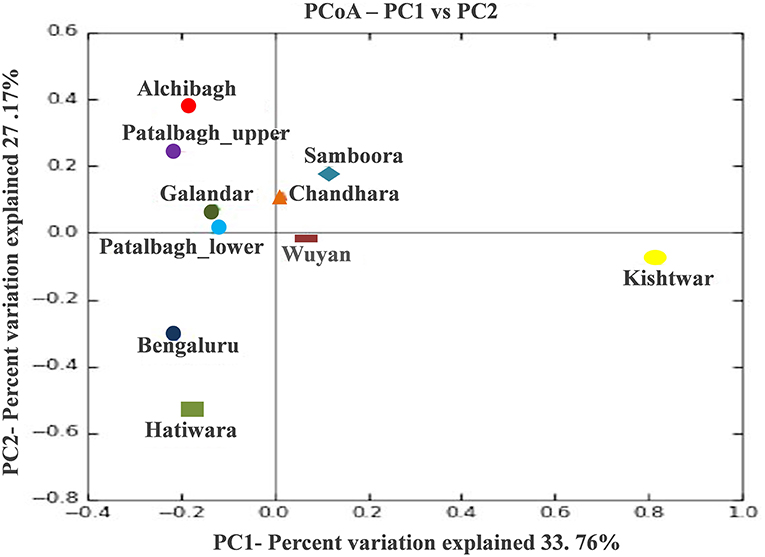
Figure 8. PCOA plots representing the bacterial diversity of Kishtwar is significantly different from rest of sample as it doesn't cluster with rest of the sample.
Core Rhizo-Bacteriome of Saffron
Despite the variation in the rhizobacterial community associated with saffron plant across different locations, the core rhizo-bacteriome that remains constant in the saffron growing across different geographical locations was identified. Core rhizo-bacteriome in saffron consisted of 11 phyla, out of total of 16 phyla i.e., Proteobacteria, Chloroflexi, Firmicutes, Acidobacteria, Cyanobacteria, Gemmatimonadetes, Bacteroidetes, Nitrosiprae, Planctomycetes, Verrucomicrobia and Actionobacteria. At genera level, it constituted 53 out of 261 genera and eight bacterial species out of 73 bacterial species. The bacterial species that constituted the core comprised of Bacillus flexus, Bacillus muralis, Edaphobacter modestum, Lysobacter brunescens, Pseudoxanthomonas mexicana, Psychrobacter celer, Roseateles depolymerans and Variovorax paradoxus. The core rhizo-bacteriome in saffron is also well-represented in heat maps at genus and species level and with circos plot (Figures 9–11).
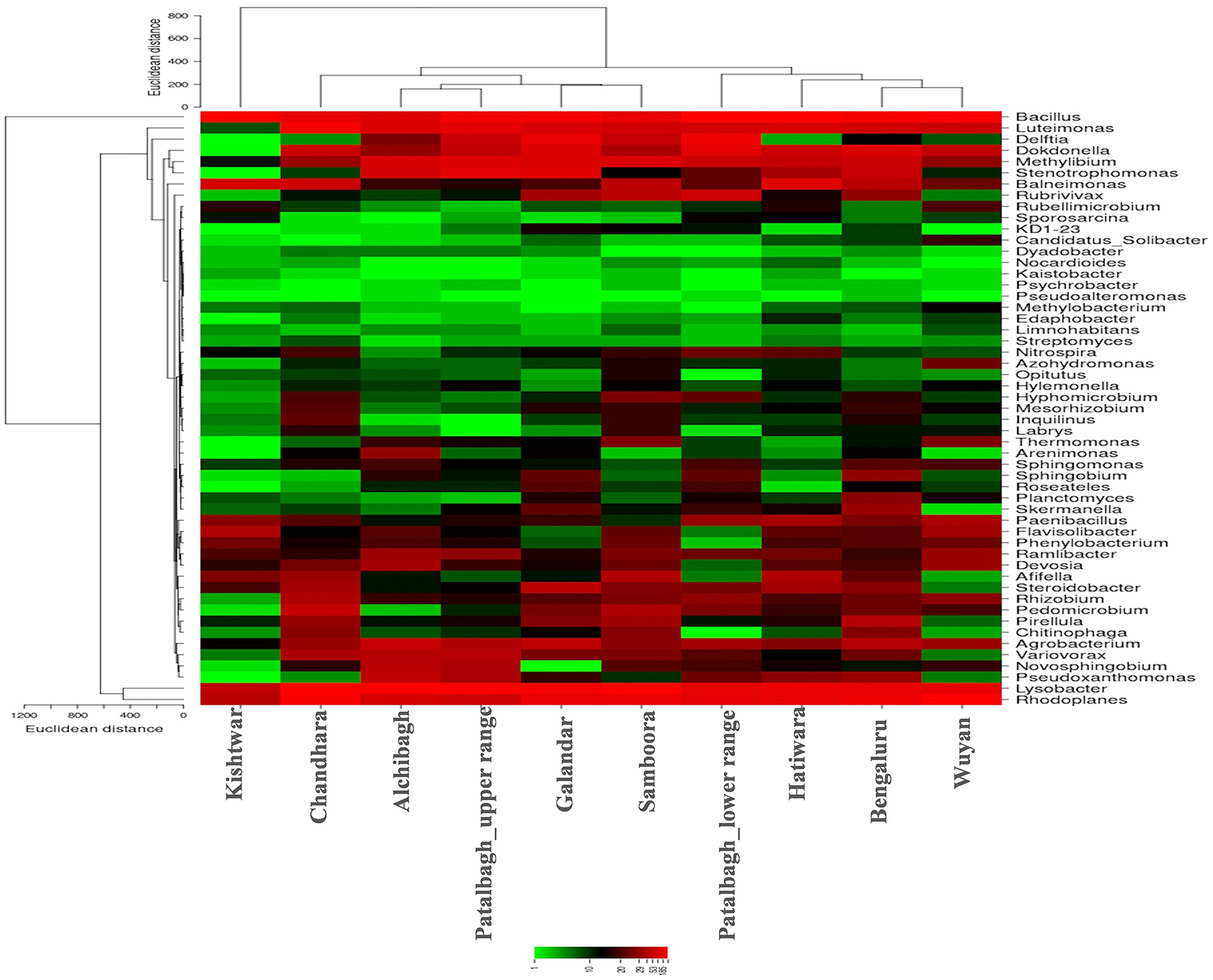
Figure 9. Heat map representing the relative abundance of core genera (53) in the Saffron rhizosphere from 10 different samples. Saffron rhizo-bacteriome in Kishtwar was different than rest of the samples as represented by phylogenetic tree comparing all the samples.
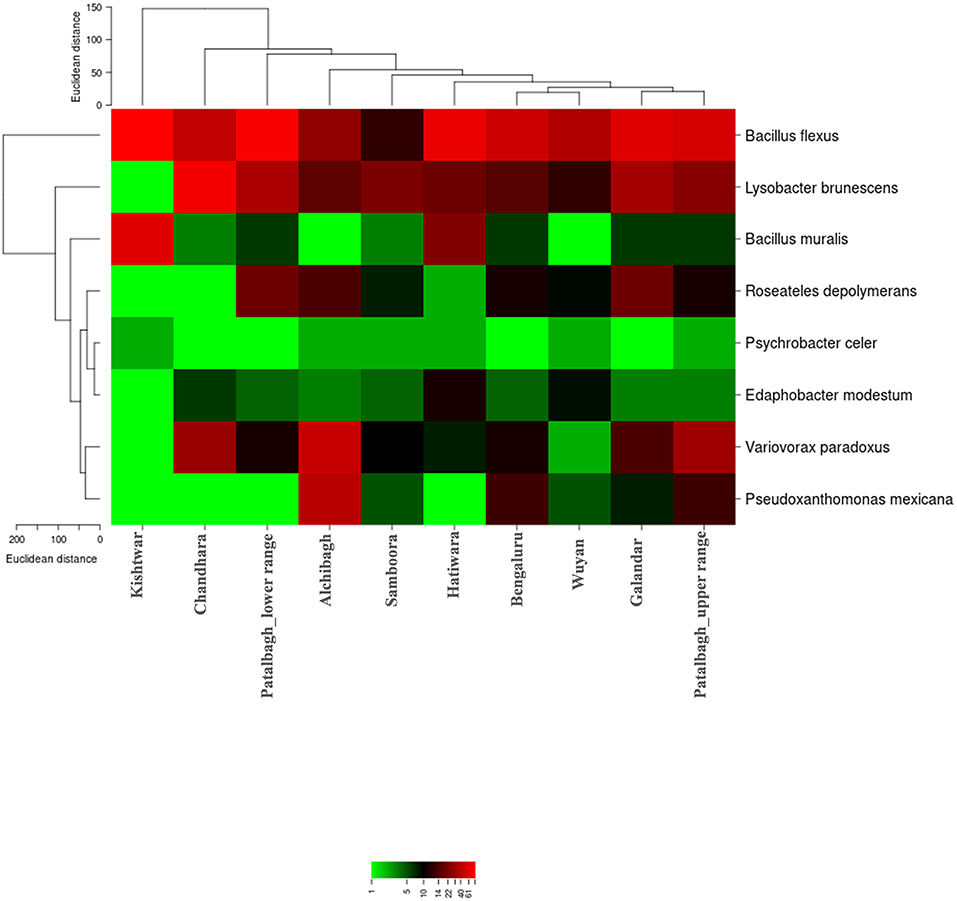
Figure 10. Heat map representing the relative abundance of core bacterial species (8) in the Saffron rhizosphere from 10 different samples. Saffron rhizo-bacteriome in Kishtwar was different than rest of the samples as represented by phylogenetic tree comparing all the samples.
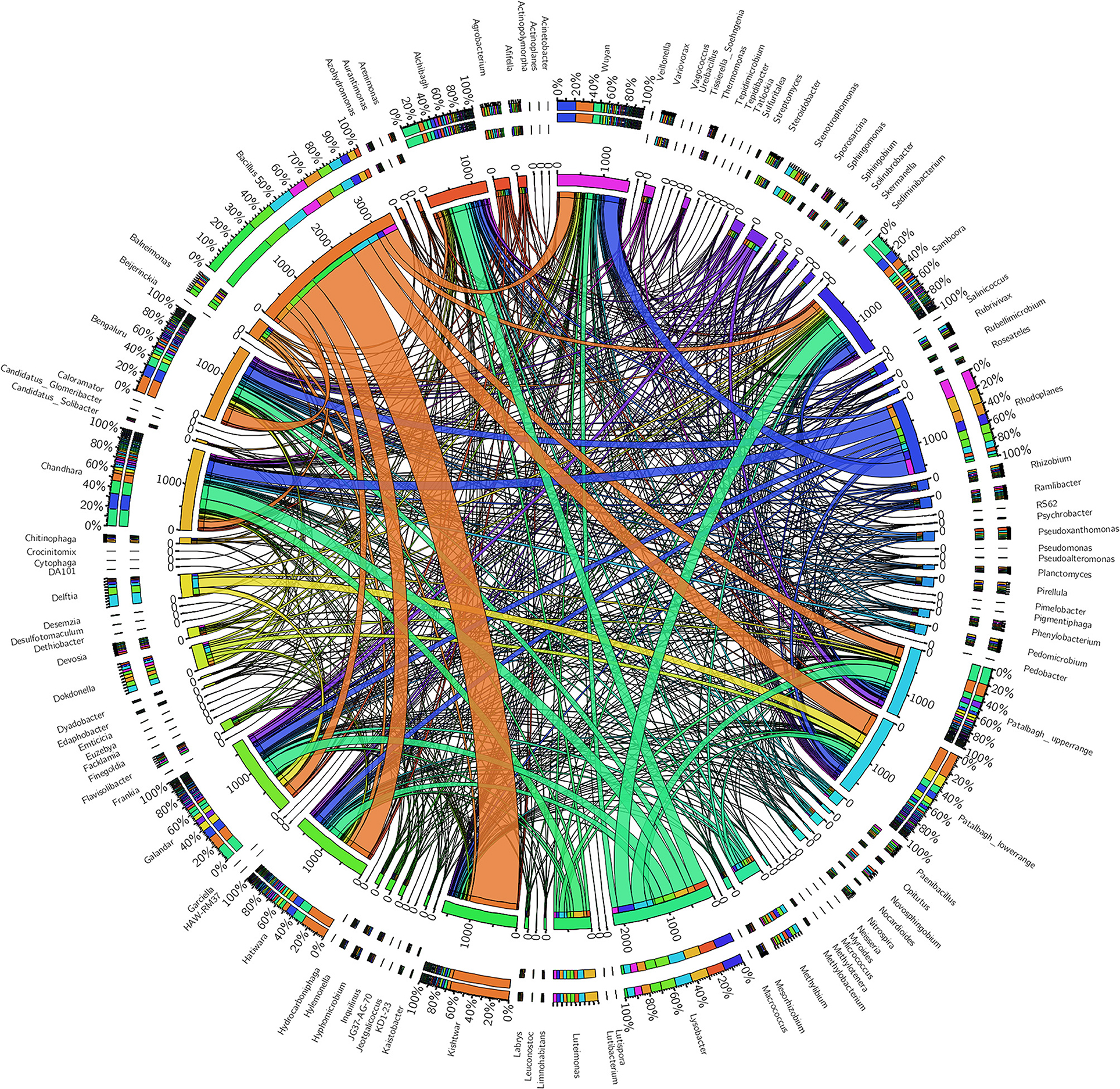
Figure 11. Circos plot representing the core rhizo-bacteriome and unique rhizobacteria in saffron grown across different geographical locations. Central line (ribbons) represents the abundance of each genus and connects genera to samples. The outer three rings are the stacked bars that represent the relative contribution of particular genera in every sample.
Discussion
Metagenomic approaches using NGS technologies can be used to explore the taxonomic and functional diversity of bacteria associated with the plants, thereby accelerating the research specific to the effect of plant bacteriome on plant health (Parvathi et al., 2019; Nathan et al., 2020). The present study was aimed at (i) exploring variation in the rhizo-bacteriome of the saffron grown across 10 sites from 3 geographical locations namely Kashmir, Kishtwar and Bengaluru, (ii) identify the core rhizo-bacteriome that persist in saffron irrespective of its geographical locations (if any), and (iii) presence of PGPRs in rhizo-bacteriome and its correlation with yield. The selected fields varied in the saffron production with Wuyan in Kashmir with the highest production i.e., 5 kg/hectare) whereas the fields in Kishtwar, Jammu with lowest production i.e., 2 kg/hectare in the year 2016 (Table 1).
Comparative Rhizo-Bacteriome Diversity
Out of 16 bacterial phyla, Proteobacteria was the most abundant phylum in all the samples (Figure 2). This is in accordance with a previously published report on saffron rhizosphere by our group (Ambardar and Vakhlu, 2013; Ambardar et al., 2014, 2016). Proteobacteria have been also reported to be the dominant phyla in the rhizosphere of other plants such as, Gossypium hirsutum, Artemisia argyi, Ageratum conyzoides, Erigeron annuus, Bidens biternata, Euphorbia hirta and Viola japonica (Qiao et al., 2017; Lei et al., 2019). Proteobacteria are known to regulate nutrients (carbon, nitrogen, sulfur) cycling in the environment that enhance plant growth (Rampelotto et al., 2013; Mukhtar et al., 2020). In our recent publication, Proteobacteria was also found to be the most abundant phylum in the cormosphere of saffron from Kashmir and Kishtwar, as well as in Morocco (Bhagat et al., 2021). On comparing all the samples, the rhizo-bacterial diversity of saffron growing in Kishtwar was found distinct from the rest of the samples as the relative abundance of Proteobacteria (PRO-31.28%) was comparatively less whereas the relative abundance of Firmicutes (FIR-20%) and Planctomycetes (PLA-10%) were more as compared to Kashmir (PRO-59.1%, FIR-4.9%, PLA-3.12%) and Bengaluru (PRO-49%, FIR-5%, PLA-4%), (Figure 2). This was further complemented by beta diversity analysis wherein rhizo-bacterial diversity of saffron grown in Kishtwar did not cluster with the rest of the samples based on the PCOA plots and heat maps at genus and species level (Figures 8–10) respectively. Similar to rhizo-bacteriome, cormo-bacteriome of Kishtwar was found significantly different than that of Kashmir and surprisingly, relatively similar to Morocco in our recent report (Bhagat et al., 2021). The significant variation in the rhizo-bacterial diversity of saffron growing in Kishtwar from rest of the samples can be preliminarily attributed to corms (plant) as Kishtwar is closer to Kashmir geographically than Morocco. Though saffron is reported to be a sterile monogenetic triploid plant, with no genetic variation and reproduces vegetatively by corms (Nemati et al., 2019), however, the variation in the yield and quality of saffron has been reported worldwide (Cardone et al., 2021). Climate and soil are thought to be two major abiotic factors (Cardone et al., 2019, 2020) and epigenetic influences have been reported to be one of the biotic factors to affect the quality and yield of saffron (Chen et al., 2021). In the preliminary observation, it seems that Cormo/rhizo-bacteriome has a role to play and is “plant driven” because rhizo-bacteriome of saffron grown in different climates and soil at Bengaluru was similar to that of Kashmir. The reason was the corms that were collected from Kashmir and grown in Bengaluru indicating that rhizo-bacteriome in saffron could be plant driven.
Comparison of rarefaction curve (97% identity) of all the samples indicated that the bacterial community in saffron rhizosphere from Samboora field in Kashmir was more diverse as compared to other samples, as the number of different species (OTUs) was higher here. This result was further complemented by diversity indices like Chao1, phylogenetic diversity, Shannon and Simpson indexes which were also higher in the case of Samboora (Table 3). However, higher bacterial diversity in the rhizosphere of saffron growing in Samboora field does not affect the saffron production positively as saffron yield in Samboora (2.5 kg/hectare) was less than that of Wuyan fields (5 kg/hectare) that had less diversity than Samboora.
The dominance of three bacterial species, Bacillus flexus, Lysobacter brunescens and Janthiobacterium lividium were observed in the rhizo-bacteriome of saffron grown in all the 10 locations (Figures 3, 5). Bacillus flexus was dominant in the rhizosphere of saffron grown in seven locations namely Kishtwar, Bengaluru, Wuyan, Galandar, Hatiwara, Patalbagh_upper range and Patalbagh_lower range. Lysobacter brunescens was dominant in Chandhara and Samboora; and Janthobacterium lividium in Alchibagh (Figure 5). Dominant bacterial genera identified in the saffron rhizosphere (Bacillus, Lysobacter, Janthobacterium and Rhodoplanes) have been reported from plant rhizospheres such as saffron (Ambardar and Vakhlu, 2013; Ambardar, 2014; Ambardar et al., 2014, 2016), green pepper (Liu et al., 2019), grapevine (Sacca et al., 2019) and oilseed rape (Gkarmiri et al., 2017), etc. However, only Bacillus flexus have been reported in Limoneum sinense and rice (Roy et al., 2020; Xiong et al., 2020) whereas Lysobacter brunescens and Janthobacterium lividium have not been reported from any plant so far.
In our previous studies, Pseudomonas was found dominant in the saffron rhizospheres by cloning based 16S rDNA metagenomic approach wherein full length 16S rDNA was amplified, TA cloned and sequenced using Sanger sequencing (Ambardar et al., 2014). Surprisingly, in the present study, Pseudomonas was only present in the saffron field from Alchibagh and absent in all other samples. The absence of Pseudomonas could be attributed to DNA extraction protocol and PCR primers and cloning approaches that were different from the present study. In a previous study, the metagenomic DNA was extracted manually using four protocols and pooled together and full length 16S rDNA primers followed by TA cloning were used to cataloge the diversity (Ambardar et al., 2014), whereas, in the present study, a commercial kit was used for DNA extraction followed by amplification of only V3-V4 region of 16S rDNA. In addition to this, the previous sampling was done in the year 2009 and present sampling was done after about 5 years in 2016, therefore climate change and floods in between in Kashmir can also be probable reasons, though this needs further investigation.
Various bacterial species, being reported in the present study, have been also reported by our group earlier by cultivation based method and were characterized in-vitro and in-vivo as well (Ambardar and Vakhlu, 2013; Ambardar et al., 2014; Kour et al., 2018; Bhagat et al., 2021). However, in the present study, six bacterial phyla, 212 bacterial genera and 70 bacterial species have been reported from the rhizosphere of saffron for the first time. Though, the rhizo-bacteria cataloged in the present study have been reported from other plants, but not reported from the cormosphere and rhizosphere saffron by our group earlier.
Core Rhizo-Bacteriome of Saffron
Comparative analysis of rhizo-bacteriome of saffron across different sites revealed the presence of various common bacterial phyla, genera and species representing the core rhizo-bacteriome of saffron. Core rhizo-bacteriome in saffron was represented by 53 bacterial genera and eight bacterial species namely Bacillus flexus, Bacillus muralis, Edaphobacter modestum, Lysobacter brunescens, Pseudoxanthomonas mexicana, Psychrobacter celer, Roseateles depolymerans, Variovorax paradoxus. These bacteria mostly belonged to 11 bacterial phyla namely Proteobacteria, Chloroflexi, Firmicutes, Acidobacteria, Cyanobacteria, Gemmatimonadetes, Bacteroidetes, Nitrosiprae, Planctomycetes and Actionobacteria. Core rhizo-bacteriome in saffron has been illustrated in the heat maps at genus & species level and with circos plot (Figures 9–11) respectively. Recently, core bacteriome associated with the corms of saffron were compared across different geographical locations i.e., Kashmir, Kishtwar and Morocco. 24 bacteria genera belonging to the phylum Proteobacteria, Actinobacteria, Bacteriodetes and Verrumicrobia made the core of cormo-bacteriome in saffron (Bhagat et al., 2021). Core rhizo-bacteriome in saffron represents 20.3% (53 out of 261) of the whole rhizo-bacteriome which was found comparatively less than the core cormo-bacteriome i.e., 32.8% of total cormo-bacteriome, however the number of bacterial genera constituting the core was more in rhizosphere (53 out of 261) as compared to cormosphere (24 out of 73), (Bhagat et al., 2021). Core microbiome of various plants have been studied such as wheat (Kuzniar et al., 2020), tomato (Cordero et al., 2020), Gymnadenia conopsea (Lin et al., 2020), coffee (Fulthorpe et al., 2020), switch grass (Grady et al., 2019), Phaseolus vulgaris (Pérez-Jaramillo et al., 2019), Oryza sativa (Eyre et al., 2019), Vineyards soil (Coller et al., 2019), Dalbergia spruceana (Skaltsas et al., 2019), and Salvia miltiorrhiza (Chen et al., 2018), etc. Core microbiome of 21 Salvia miltiorrhiza seeds represented 54% of the whole microbiome cataloged from seven different geographic origins (Chen et al., 2018). In the case of Phaseolus vulgaris rhizosphere, the core microbiome represented 25.9% of the total microbiome in native and agricultural soils (Pérez-Jaramillo et al., 2019). The core microbiome of 30 phylogenetically diverse angiosperm plants constitutes 40% of the whole microbiome (Fitzpatrick et al., 2018). Core rhizo-bacteriome in saffron was 20.3% of the total microbiome, which was comparatively less than Salvia miltiorrhiza, Phaseolus vulgaris and Angiosperm plants. In rice seeds, the core microbiome was enriched in Rhizobium, Pantoea, Sphingomonas, Methylobacterium, Xanthomonas, Paenibacillus, Alternaria, and Occultifur (Wang et al., 2020). Saffron's core rhizo-bacteriome was enriched with Bacillus, Rhizobium, Sphingomonas, Agrobacterium, and Methylobacterium which was similar to the core microbiome in rice.
Out of 53 core rhizosphere bacteria identified in the present study, 19 bacteria were also reported previously from the rhizosphere and cormosphere of saffron grown in Kashmir, by our group using culture dependent and independent approaches (Ambardar and Vakhlu, 2013; Ambardar, 2014; Ambardar et al., 2014, 2016; Kour et al., 2018; Bhagat et al., 2021). Interestingly, the core rhizo-bacteriome of saffron also harbored some common bacteria as in previously published core cormo-bacteriome. 11 bacterial genera namely Agrobacterium, Chitinophaga, Dyadobacter, Mesorhizobium, Methylobacterium, Nocardioides, Opitutus, Rhizobium, Sphingomonas, Streptomyces and Varivorax were common in both the underground organs of saffron that is corm (Bhagat et al., 2021) and roots. In addition, Agrobacterium, Bacillus, Delftia, Rhizobium, and Variovorax have also been reported from rhizosphere and cormosphere of saffron, grown in Morocco using cultivation dependent methods (Chamkhi et al., 2018). Though we could not capture the genera reported in Morocco cormo/rhizosphere by earlier used techniques but with the present technique they were captured and found to be present in Kashmir. However, 34 bacterial genera of the core rhizo-bacteriome of saffron grown in India were reported for the first time in the saffron, in the present study.
Eight core bacterial species identified in the present study was also reported from other plants such as Bacillus flexus from rice (Roy et al., 2020), Bacillus muralis from peanut (Jiang et al., 2016), Edaphobacter modestum from Magnolia grandiflora (Stone and Jackson, 2016), Pseudoxanthomonas mexicana from maize and peanut rhizosphere (Geng et al., 2018; Youseif, 2018), Roseateles depolymerans from ginger (Chen et al., 2014), Variovorax paradoxus from mustard roots (Belimov et al., 2005). However, Lysobacter brunescens and Psychrobacter celer has not been reported from any plant so far.
Plant Growth Promoting Bacteria From Saffron Rhizo-Bacteriome
64.1% of total core rhizo-bacteriome i.e., 34 out of 53 bacteria has been reported as PGPBs from saffron and other plants (Supplementary Table 1). While exploring the core rhizo-bacteriome for PGPR, it was found that 21 bacteria have been reported as PGPB for the first time from saffron in the present study as previously used methods failed to capture them. The core rhizo-bacteria that are being reported as PGPRs for the first in saffron are Azospirillum, Agromyces, Agrobacterium, Bradyrhizobium, Burkholderia, Dokdonella, Edaphobacter, Flavobacterium, Flavisolibacter, Frankia, Leuconostoc, Luteimonas, Lysobacter, Phenylobacterium, Pseudoxanthomonas, Mesorhizobium, Nocardia, Novosphingobium, Rhizobium, Rubellimicrobium, Variovorax etc.
In addition to core rhizo-bacteriome, saffron rhizo-bacteriomes of each location were enriched with some specific bacteria that were unique to that particular location. An inventory of unique PGPBs of all these bacteria across different locations was made based on the previous reports of PGPBs from saffron and other plants. The location specific unique PGPBs details have been summarized in Supplementary Table 2. Wuyan had the maximum number of unique genera i.e., 19 out of which 5 are reported as PGPB i.e., Actinoplanes, Beijerinckia, Desulfotomaculum Dethiobacter and Ureibacillus. In other locations unique number of genera was 4 in Chandhara, 2 in Samboora, 1 in Hatiwara and 1 in patalbagh_upper range but none of them have been reported as PGPB. Kishtwar had six unique genera and three genera namely Desemzia, Macrococcus and Myroides have been reported as PGPB (Supplementary Table 2). In addition, Alchibagh had 10 unique genera and four have been reported as PGPB. Galandar had six unique genera and three PGPB reported, Patalbagh_upper had two and Bengaluru has one unique genera, both locations have one PGPB reported (Supplementary Table 2).
In the present study, Bacillus flexus, Lysobacter brunescens and Janthiobacterium lividium were the dominant bacterial species identified in saffron rhizo-bacteriome. Bacillus genus dominant in the rhizosphere microbiome is predominantly used as plant growth promoting bacteria, due to their ability to colonize the roots rapidly, competitive colonization potential, production of various phytohormone and conversion of complex nutrients such as phosphorous and nitrogen into simple absorbable forms (Aloo et al., 2019; Enebe and Babalola, 2019; Hashem et al., 2019; Kashyap et al., 2019). Bacillus flexus KLBMP 4941 in Limoneum sinense (Xiong et al., 2020) and B. flexus U8 in rice (Roy et al., 2020) have been reported as PGPR. However, Lysobacter brunescens and Janthobacterium lividium have not been reported so far from any plant.
In addition to Bacillus flexus other bacterial species of core rhizo-bacteriome reported as PGPB are Bacillus muralis, Pseudoxanthomonas mexicana, Roseateles depolymerans and Variovorax paradoxus (Supplementary Table 3). Bacillus muralis strain HS4 has been reported as PGPR in peanut (Jiang et al., 2016). Pseudoxanthomonas Mexicana has been reported as PGPR from rhizosphere of maize and peanut (Geng et al., 2018; Youseif, 2018). Roseateles depolymerans has been isolated as an endophyte from the seedling stage of ginger and reported to have PGPR properties (Chen et al., 2014) and Variovorax paradoxus has been reported as PGPR from Indian mustard roots (Belimov et al., 2005). Lysobacter brunescens, Edaphobacter modestum and Psychrobacter celer have not been reported as PGPB so far (Supplementary Table 3).
Among different locations, Kishtwar had four unique species (Bacillus marisflavi, Macrococcus caseolyticus, Myroides odoratimimus and Staphylococcus scuiri) and all are reported as PGPB in different plant. In addition, Galandar from Kashmir had only one PGPB (Paracoccus marcusii) reported. All other locations have no unique species reported PGPB so far (Supplementary Table 4).
Altogether, 110 bacterial genera (out of total 261) and 21 bacterial species (out of total 73), cataloged in present study, have also been reported to be plant growth promoting bacteria from saffron and other plants (Supplementary Table 5). Plant growth promoting bacterial species cataloged in saffron are Acidovorax facilis, Bacillus cereus, Bacillus firmus, Bacillus flexus, Bacillus horikoshii, Bacillus marisflavi, Bacillus muralis, Brevundimonas diminuta, Burkholderia bryophila, Burkholderia tuberum, Luteibacter rhizovicinus, Macrococcus caseolyticus, Myroides odoratimimus, Paracoccus marcusii, Pseudoxanthomonas mexicana, Rhizobium leguminosarum, Roseateles depolymerans, Staphylococcuss epidermidis, Staphylococcus equorum, Staphylococcus sciuri, Staphylococcus succinus and Variovorax paradoxus.
On comparison of PGPB from each sample, Wuyan had maximum number of PGPBs (76 out of 154) followed by Galandar (75 out of 132), Alchibagh (73 out of 147), Chandhara (72 out of 143), Hatiwara (73 out of 140), Kishtwar (66 out of 139), Bengaluru (63 out of 143), Samboora (62 out of 124), Patalbagh_lower (61 out of 111) whereas Patalbagh_upper (60 out of 122) with the minimum number of PGPBs (Supplementary Table 6). The highest number of PGPB in the Wuyan sample could be correlated to the yield as the production in Wuyan is two times as compared to other fields. In the previous report by our group, PGP Bacillus amyloliquefaciens W2 for growth promotion of saffron and corm rot inhibition were isolated from the Wuyan by cultivation based method (Gupta and Vakhlu, 2015). However, in present study a well-reported PGPR Rhizobium leguminosarum and Luteibacter rhizovicinus was relatively abundant in all the nine locations, including Wuyan and completely absent in Kishtwar. Rhizobium leguminosarum possess various PGP characteristics such as phosphorous solubilization, siderophore, IAA, HCN production and nitrogen fixation which enhance the growth of host plants in pea plant, lettuce and carrot (Mishra et al., 2009; Tank and Saraf, 2010; Flores-Félix et al., 2013; Gopalakrishnan et al., 2015). Luteibacter rhizovicinus MIMR1 have been reported as plant growth promoting bacteria that promotes root development in barley (Guglielmetti et al., 2013). Absence of Rhizobium leguminosarum and Luteibacter rhizovicinus may be attributed to the low production status of Kishtwar as compared to Kashmir, but the real confirmation will come only after their evaluation in-vivo. The presence of unique location specific PGPB at places other than Wuyan, with a lower yield than Wuyan indicates that the potential of PGP bacteria varies and maybe all the PGPB are not as effective as that of Wuyan fields. The PGPB cataloge in the present and previous studies in saffron by metagenomic approaches will be captured by culture based technique and evaluated for PGP potential in the future.
Conclusion
The present study investigated the variation in rhizo-bacteriome of saffron grown across different locations i.e., Kashmir, Kishtwar and Bengaluru in India. The rhizo-bacteriome of saffron grown in Kishtwar was found significantly different from the saffron grown in Kashmir and Bengaluru. Interestingly, rhizo-bacteriome in saffron seems to be “plant driven” though in our earlier report we have suggested the bacteriome of corm is location specific. This need to be further confirmed as for cormo-bacteriome corms were directly taken from the fields and analyzed whereas in the present study, corms from Kashmir were grown in the pots with garden soil from Bengaluru and analyzed. Despite growing in non-native soil, the similarity of Bengaluru rhizo-bacteriome to Kashmir rhizo-bacteriome indicates that it is plant driven. This needs to be studied extensively further by analyzing rhizo-bacteriome of saffron roots developed from corms from Kishtwar and Kashmir, cultivated in Bengaluru in non-native soil. Further, the total PGPBs and unique PGPBs were highest in the Wuyan field that has maximum production indicating, a correlation between number of PGPBs and production. However, unique and common PGPB were cataloged across geographical locations in the present study, in the future they will be cultivated by media engineering (for those that are difficult to cultivate) and evaluated in-vivo to estimate their actual efficacy.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA687631.
Author Contributions
SA, JV, and MG have designed the experiments. SA conducted the experiments, analyzed the sequencing data, and experiment regarding growth of saffron plants in Bengaluru. SA and NB wrote the manuscript, analyzed the taxonomy data. MG conceptualized and supervised the study. JV guided in collection of samples from Kashmir and Kishtwar. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Authors are grateful to Mr. Chuni Lal Bhat, Former Chief Agriculture officer, Pulwama, Kashmir for his guidance in sample collection from saffron fields in Kashmir. Authors are also thankful to Mr. Gulzar Mohd Mir, Saffron Development Assistant, Kishtwar for his help in sample collection from Kistwar fields. SA acknowledges DST-SERB National Post-Doctoral Fellowship (PDF/2015/000712) for fellowship and funding the research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fsufs.2021.644230/full#supplementary-material
References
Aloo, B. N., Makumba, B. A., and Mbega, E. R. (2019). The potential of bacilli rhizobacteria for sustainable crop production and environmental sustainability. Microb. Res. 219, 26–39. doi: 10.1016/j.micres.2018.10.011
Alteio, L. V., Schulz, F., Seshadri, R., Varghese, N., Rodriguez-Reillo, W., Ryan, E., et al. (2020). Complementary metagenomic approaches improve reconstruction of microbial diversity in a forest soil. mSystems 5, 768–719. doi: 10.1128/mSystems.00768-19
Ambardar, S. (2014). Mining Microbial Diversity of Rhizosphere of Crocus sativus by Metagenomic Approach. University of Jammu.
Ambardar, S., Sangwan, N., Manjula, A., Rajendhran, J., Gunasekaran, P., Lal, R., et al. (2014). Identification of bacteria associated with underground parts of Crocus sativus by 16S rRNA gene targeted metagenomic approach. World J. Microbiol. Biotechnol. 30, 2701–2709. doi: 10.1007/s11274-014-1694-0
Ambardar, S., Singh, H. R., Gowda, M., and Vakhlu, J. (2016). Comparative metagenomics reveal phylum level temporal and spatial changes in mycobiome of belowground parts of Crocus sativus. PLoS ONE. 11:e0163300. doi: 10.1371/journal.pone.0163300
Ambardar, S., and Vakhlu, J. (2013). Plant growth promoting bacteria from Crocus sativus rhizosphere. World J. Microbiol. Biotechnol. 29, 2271–2279. doi: 10.1007/s11274-013-1393-2
Bakker, M. G., Otto-Hanson, L., Lange, A. J., Bradeen, J. M., and Kinkel, L. L. (2013). Plant monocultures produce more antagonistic soil Streptomyces communities than high-diversity plant communities. Soil Biol. Biochem. 65, 304–312. doi: 10.1016/j.soilbio.2013.06.007
Belimov, A. A., Hontzeas, N., Safronova, V. I., Demchinskaya, S. V., Piluzza, G., Bullitta, S., et al. (2005). Cadmium-tolerant plant growth-promoting bacteria associated with the roots of Indian mustard (Brassica juncea L. Czern.). Soil Biol. Biochem. 37, 241–250. doi: 10.1016/j.soilbio.2004.07.033
Bhagat, N., Sharma, S., Ambardar, S., Raj, S., Trakroo, D., Horacek, M., et al. (2021). Microbiome fingerprint as biomarker for geographical origin and heredity in Crocus sativus: a feasibility study. Front. Sustain. Food Syst. 5:688393. doi: 10.3389/fsufs.2021.688393
Bhooma, V., Nagasathiya, K., Vairamani, M. and Parani, M. (2020). Identification of synthetic dyes magenta III (new fuchsin) and rhodamine Bas common adulterants in commercial saffron. Food Chem. 309:125793. doi: 10.1016/j.foodchem.2019.125793
Boughner, L. A., and Singh, P. (2016). Microbial ecology: where are we now? Postdoc J. 4:3. doi: 10.14304/SURYA.JPR.V4N11.2
Brown, J., Pirrung, M., and McCue, L. A. (2017). FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139. doi: 10.1093/bioinformatics/btx373
Çakmakçı, R., Turan, M., Kıtır, N., Güneş, A., Nikerel, E., Özdemir, B. S., et al. (2017). The Role of Soil Beneficial Bacteria in Wheat Production: A Review. Wheat Improvement, Management and Utilization. Rijeka: Intech, 115–149.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cardone, L., Candido, V., Castronuovo, D., Perniola, M., and Cicco, N. (2021). Comparing annual and biennial crop cycle on the growth, yield and quality of saffron using three corm dimensions. Sci. Hortic. 288:110393. doi: 10.1016/j.scienta.2021.110393
Cardone, L., Castronuovo, D., Perniola, M., Cicco, N., and Candido, V. (2019). Evaluation of corm origin and climatic conditions on saffron (Crocus sativus L.) yield and quality. J. Sci. Food Agric. 99, 5858–5869. doi: 10.1002/jsfa.9860
Cardone, L., Castronuovo, D., Perniola, M., Scrano, L., Cicco, N., and Candido, V. (2020). The influence of soil physical and chemical properties on saffron (Crocus sativus L.) growth, yield and quality. Agronomy 10:1154. doi: 10.3390/agronomy10081154
Chamkhi, I., Sbabou, L., and Aurag, J. (2018). Endophytic fungi isolated from Crocus sativus L. (saffron) as a source of bioactive secondary metabolites. Pharmacogn. Mag. 10, 1143-1148. doi: 10.5530/pj.2018.6.195
Chao, A. (1987). Estimating the population size for capture-recapture data with unequal catchability. Biometrics 43, 783–791. doi: 10.2307/2531532
Chauhan, P. S., Lata, C., Tiwari, S., Chauhan, A. S., Mishra, S. K., Agrawal, L., et al. (2019). Transcriptional alterations reveal Bacillus amyloliquefaciens-rice cooperation under salt stress. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-019-48309-8
Chen, H., Wu, H., Yan, B., Zhao, H., Liu, F., Zhang, H., et al. (2018). Core microbiome of medicinal plant Salvia miltiorrhiza seed: a rich reservoir of beneficial microbes for secondary metabolism? Int. J. Mol. Sci. 19:672. doi: 10.3390/ijms19030672
Chen, J., Zhou, G., Dong, Y., Qian, X., Li, J., Xu, X., et al. (2021). Screening of key proteins affecting floral initiation of saffron under cold stress using iTRAQ-based proteomics. Front. Plant Sci. 12:708. doi: 10.3389/fpls.2021.644934
Chen, T., Chen, Z., Ma, G. H., Du, B. H., Shen, B., Ding, Y. Q., et al. (2014). Diversity and potential application of endophytic bacteria in ginger. Genet. Mol. Res. 13, 4918–4931. doi: 10.4238/2014.July.4.6
Coats, V. C., and Rumpho, M. E. (2014). The rhizosphere microbiota of plant invaders: an overview of recent advances in the microbiomics of invasive plants. Front. Microbiol. 5:368. doi: 10.3389/fmicb.2014.00368
Coller, E., Cestaro, A., Zanzotti, R., Bertoldi, D., Pindo, M., Larger, S., et al. (2019). Microbiome of vineyard soils is shaped by geography and management. Microbiome 7, 1–15. doi: 10.1186/s40168-019-0758-7
Cordero, I., Balaguer, L., Rincón, A., and Pueyo, J. J. (2018). Inoculation of tomato plants with selected PGPR represents a feasible alternative to chemical fertilization under salt stress. J. Plant Nutr. Soil Sci. 181, 694–703. doi: 10.1002/jpln.201700480
Cordero, J., de Freitas, J. R., and Germida, J. J. (2020). Bacterial microbiome associated with the rhizosphere and root interior of crops in Saskatchewan, Canada. Can. J. Microbiol. 66, 71–85. doi: 10.1139/cjm-2019-0330
De Gregorio, P. R., Michavila, G., Ricciardi Muller, L., de Souza Borges, C., Pomares, M. F., Saccol de, Sá,. E. L., et al. (2017). Beneficial rhizobacteria immobilized in nanofibers for potential application as soybean seed bioinoculants. PLoS ONE 12:e0176930. doi: 10.1371/journal.pone.0176930
Enebe, M. C., and Babalola, O. O. (2019). The impact of microbes in the orchestration of plants' resistance to biotic stress: a disease management approach. Appl. Microbiol. Biotechnol. 103, 9-25. doi: 10.1007/s00253-018-9433-3
Eyre, A. W., Wang, M., Oh, Y., and Dean, R. A. (2019). Identification and characterization of the core rice seed microbiome. Phytobiomes J. 3, 148–157. doi: 10.1094/PBIOMES-01-19-0009-R
Fitzpatrick, C. R., Copeland, J., Wang, P. W., Guttman, D. S., Kotanen, P. M., and Johnson, M. T. (2018). Assembly and ecological function of the root microbiome across angiosperm plant species. Proc. Nat. Acad. Sci. U.S.A. 115, 1157–1165. doi: 10.1073/pnas.1717617115
Flores-Félix, J. D., Menéndez, E., Rivera, L. P., Marcos-García, M., Martínez-Hidalgo, P., Mateos, P. F., et al. (2013). Use of Rhizobium leguminosarum as a potential biofertilizer for Lactuca sativa and Daucus carota crops. J. Plant Nutr. Soil Sci. 176, 876–882. doi: 10.1002/jpln.201300116
Fulthorpe, R., Martin, A. R., and Isaac, M. E. (2020). Root endophytes of coffee (Coffea arabica): variation across climatic gradients and relationships with functional traits. Phytobiomes J. 4, 27–39. doi: 10.1094/PBIOMES-04-19-0021-R
Geng, L. L., Shao, G. X., Raymond, B., Wang, M. L., Sun, X. X., Shu, C. L., et al. (2018). Subterranean infestation by Holotrichia parallela larvae is associated with changes in the peanut (Arachis hypogaea L.) rhizosphere microbiome. Microbiol. Res. 211, 13–20. doi: 10.1016/j.micres.2018.02.008
Gkarmiri, K., Mahmood, S., Ekblad, A., Alström, S., Högberg, N., and Finlay, R. (2017). Identifying the active microbiome associated with roots and rhizosphere soil of oilseed rape. Appl. Environ. Microbiol. 83:e01938-17. doi: 10.1128/AEM.01938-17
Gopalakrishnan, S., Sathya, A., Vijayabharathi, R., Varshney, R. K., Gowda, C. L., and Krishnamurthy, L. (2015). Plant growth promoting rhizobia: challenges and opportunities. 3 Biotech 5, 355–377. doi: 10.1007/s13205-014-0241-x
Gotelli, N. J., and Colwell, R. K. (2011). Estimating species richness. In: Biological Diversity: Frontiers in Measurement and Assessment. Oxford University Press, UK. 12, 39–54. Available online at: https://www.google.co.in/books/edition/Biological_Diversity/7mwVDAAAQBAJ?hl=en&gbpv=0
Grady, K. L., Sorensen, J. W., Stopnisek, N., Guittar, J., and Shade, A. (2019). Assembly and seasonality of core phyllosphere microbiota on perennial biofuel crops. Nat. Commun. 10, 1–10. doi: 10.1038/s41467-019-11974-4
Guglielmetti, S., Basilico, R., Taverniti, V., Arioli, S., Piagnani, C., and Bernacchi, A. (2013). Luteibacter rhizovicinus MIMR1 promotes root development in barley (Hordeum vulgare L.) under laboratory conditions. World J. Microbiol. Biotechnol. 29, 2025–2032. doi: 10.1007/s11274-013-1365-6
Gui-Feng, G. A. O., and Hai-Yan., C. H. U. (2020). Techniques and methods of microbiomics and their applications. Chin. J. Plant Ecol. 44:395. doi: 10.17521/cjpe.2019.0222
Gupta, R., and Vakhlu, J. (2015). Native Bacillus amyloliquefaciens W2 as a potential biocontrol for Fusarium oxysporum R1 causing corm rot of Crocus sativus. Eur. J. Plant Pathol. 143, 123–131. doi: 10.1007/s10658-015-0670-3
Hashem, A., Tabassum, B., and Abd_Allah, E. F. (2019). Bacillus subtilis: a plant-growth promoting rhizobacterium that also impacts biotic stress. Saudi. J. Biol. Sci. 26, 1291-1297. doi: 10.1016/j.sjbs.2019.05.004
Jiang, H., Lei, R., Ding, S. W., and Zhu, S. (2014). Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 15:182. doi: 10.1186/1471-2105-15-182
Jiang, Y., Li, P., Wu, Y., Liu, Y., Wang, Q., and Han, Y. (2016). A new isolated indole-3-acetic acid producing rhizobacteria Bacillus muralis HS4 from sandy soil: characterization and growth enhancement on peanuts. J. Comput. Theor. Nanosci. 13, 5171–5177. doi: 10.1166/jctn.2016.5399
Kashyap, B. K., Solanki, M. K., Pandey, A. K., Prabha, S., Kumar, P., and Kumari, B. (2019). “Bacillus as plant growth promoting rhizobacteria (PGPR): a promising green agriculture technology,” in Plant Health Under Biotic Stress, eds R. Ansari, and I. Mahmood (Gateway East: Springer), 219-236. doi: 10.1007/978-981-13-6040-4_11
Kour, D., Rana, K. L., Yadav, N., Yadav, A. N., Kumar, A., Meena, V. S., et al. (2019). “Rhizospheric microbiomes: Biodiversity, Mechanisms of Plant Growth Promotion, and Biotechnological Applications for Sustainable Agriculture,” In: Kumar A., Meena V. (eds): Plant Growth Promoting Rhizobacteria for Agricultural Sustainability Springer, Singapore, 19-65. doi: 10.1007/978-981-13-7553-8_2
Kour, R., Ambardar, S., and Vakhlu, J. (2018). Plant growth promoting bacteria associated with corm of Crocus sativus during three growth stages. Lett. Appl. Microbiol. 67, 458–464. doi: 10.1111/lam.13042
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kuzniar, A., Włodarczyk, K., Grzadziel, J., Goraj, W., Gałazka, A., and Wolińska, A. (2020). Culture-independent analysis of an endophytic core microbiome in two species of wheat: Triticum aestivum L. (cv. “Hondia”) and the first report of microbiota in Triticum spelta L. (cv. “Rokosz”). Syst. Appl. Microbiol. 43:126025. doi: 10.1016/j.syapm.2019.126025
Lei, S., Xu, X., Cheng, Z., Xiong, J., Ma, R., Zhang, L., et al. (2019). Analysis of the community composition and bacterial diversity of the rhizosphere microbiome across different plant taxa. Microbiol. Open 8:e00762. doi: 10.1002/mbo3.762
Lin, M., Xiong, H., Xiang, X., Zhou, Z., Liang, L., and Mei, Z. (2020). The Effect of Plant Geographical location and developmental stage on root-associated microbiomes of Gymnadenia conopsea. Front. Microbial. 11:1257. doi: 10.3389/fmicb.2020.01257
Liu, Y., Qiao, J., Liu, Y., Liang, X., Zhou, Y., and Liu, J. (2019). Characterization of Lysobacter capsici strain NF87-2 and its biocontrol activities against phytopathogens. Eur. J. Plant Pathol. 155, 859–869. doi: 10.1007/s10658-019-01817-9
Magotra, S., Bhagat, N., Ambardar, S., Ali, T., Hurek, B. R., Hurek, T., et al. (2021). Field evaluation of PGP Bacillus sp. strain D5 native to Crocus sativus, in traditional and non traditional areas, and mining of PGP genes from its genome. Sci. Rep. 11, 1-16 doi: 10.1038/s41598-021-84585-z
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2012). An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. doi: 10.1038/ismej.2011.139
Mishra, P. K., Mishra, S., Selvakumar, G., Bisht, J. K., Kundu, S., and Gupta, H. S. (2009). Coinoculation of Bacillus thuringeinsis-KR1 with Rhizobium leguminosarum enhances plant growth and nodulation of pea (Pisum sativum L.) and lentil (Lens culinaris L.). World J. Microbiol. Biotechnol. 25, 753–761. doi: 10.1007/s11274-009-9963-z
Mukhtar, S., Hirsch, A. M., Khan, N., Malik, K. A., Humm, E. A., Pellegrini, M., et al. (2020). Impact of soil salinity on the cowpea nodule-microbiome and the isolation of halotolerant PGPR strains to promote plant growth under salinity stress. Phytobiomes J. 4, 364–374. doi: 10.1094/PBIOMES-09-19-0057-R
Nathan, V. K., Vijayan, J., and Ammini, P. (2020). Comparison of bacterial diversity from two mangrove ecosystems from India through metagenomic sequencing. Reg. Stud. Mar. Sci. 35:101184. doi: 10.1016/j.rsma.2020.101184
Nemati, Z., Harpke, D., Gemicioglu, A., Kerndorff, H., and Blattner, F. R. (2019). Saffron (Crocus sativus) is an autotriploid that evolved in Attica (Greece) from wild Crocus cartwrightianus. Mol. Phylogenetics Evol. 136, 14–20. doi: 10.1016/j.ympev.2019.03.022
Ofek-Lalzar, M., Sela, N., Goldman-Voronov, M., Green, S. J., Hadar, Y., and Minz, D. (2014). Niche and host-associated functional signatures of the root surface microbiome. Nat. Commun. 5, 1–9. doi: 10.1038/ncomms5950
Parello, C. S. (2020). “Microbiomics.” In: Stephen T. Sonis and Allessandro Villa (eds): Translational Systems Medicine and Oral Disease (Cambridge, MA: Academic Press), 137–162. doi: 10.1016/B978-0-12-813762-8.00006-2
Parvathi, A., Jasna, V., Aswathy, V. K., Aparna, S., Vinod Kumar, N., and Jyothibabu, R. (2019). Bacterial metataxonomic sequence analysis reveals dominance of Wolbachia sp. in the deep sea sediment of the Bay of Bengal, Indian Ocean. Genomics 112, 1030-1041. doi: 10.1016/j.ygeno.2019.06.019
Pérez-Jaramillo, J. E., de Hollander, M., Ramírez, C. A., Mendes, R., Raaijmakers, J. M., and Carrión, V. J. (2019). Deciphering rhizosphere microbiome assembly of wild and modern common bean (Phaseolus vulgaris) in native and agricultural soils from Colombia. Microbiome 7, 1–16. doi: 10.1186/s40168-019-0727-1
Qiao, Q., Wang, F., Zhang, J., Chen, Y., Zhang, C., Liu, G., et al. (2017). The variation in the rhizosphere microbiome of cotton with soil type, genotype and developmental stage. Sci. Rep. 7, 1–10. doi: 10.1038/s41598-017-04213-7
Rampelotto, P. H., de Siqueira Ferreira, A., Barboza, A. D. M., and Roesch, L. F. W. (2013). Changes in diversity, abundance, and structure of soil bacterial communities in Brazilian Savanna under different land use systems. Microb. Ecol. 66, 593–607. doi: 10.1007/s00248-013-0235-y
Roy, P., Chatterjee, S., Saha, N. C., and Gantait, V. V. (2020). Characterization of a starch hydrolysing Bacillus flexus U8 isolated from rhizospheric soil of the paddy plants. Proc. Natl. Acad. Sci. India: Sect. B Biol. Sci. 90, 1075-1081. doi: 10.1007/s40011-020-01176-0
Rüger, L., Feng, K., Dumack, K., Freudenthal, J., Chen, Y., Sun, R., et al. (2021). Assembly patterns of the rhizosphere microbiome along the longitudinal root axis of maize (Zea mays L.). Front. Microbiol. 12:237. doi: 10.3389/fmicb.2021.614501
Sacca, M. L., Manici, L. M., Caputo, F., and Frisullo, S. (2019). Changes in rhizosphere bacterial communities associated with tree decline: grapevine esca syndrome case study. Can. J. Microbiol. 65, 930–943. doi: 10.1139/cjm-2019-0384
Sinclair, L., Osman, O. A., Bertilsson, S., and Eiler, A. (2015). Microbial community composition and diversity via 16S rRNA gene amplicons: evaluating the illumina platform. PLoS ONE 10:e0116955. doi: 10.1371/journal.pone.0116955
Skaltsas, D. N., Badotti, F., Vaz, A. B. M., da Silva, F. F., Gazis, R., Wurdack, K., et al. (2019). Exploration of stem endophytic communities revealed developmental stage as one of the drivers of fungal endophytic community assemblages in two Amazonian hardwood genera. Sci. Rep. 9, 1–14. doi: 10.1038/s41598-019-48943-2
Steen, A. D., Crits-Christoph, A., Carini, P., DeAngelis, K. M., Fierer, N., Lloyd, K. G., et al. (2019). High proportions of bacteria and archaea across most biomes remain uncultured. ISME J. 13, 3126–3130. doi: 10.1038/s41396-019-0484-y
Stone, B. W., and Jackson, C. R. (2016). Biogeographic patterns between bacterial phyllosphere communities of the Southern Magnolia (Magnolia grandiflora) in a small forest. Microb. Ecol. 71, 954–961. doi: 10.1007/s00248-016-0738-4
Stone, B. W., Weingarten, E. A., and Jackson, C. R. (2018). The role of the phyllosphere microbiome in plant health and function. Annu. Rev. Plant Biol. 533–556. doi: 10.1002/9781119312994.apr0614
Tank, N., and Saraf, M. (2010). Salinity-resistant plant growth promoting rhizobacteria ameliorates sodium chloride stress on tomato plants. J. Plant Interact. 5, 51–58. doi: 10.1080/17429140903125848
Taş, N., de Jong, A. E., Li, Y., Trubl, G., Xue, Y., and Dove, N. C. (2021). Metagenomic tools in microbial ecology research. Curr. Opin. Biotechnol. 67, 184–191. doi: 10.1016/j.copbio.2021.01.019
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T., and Singh, B. K. (2020). Plant–microbiome interactions: from community assembly to plant health. Nat. Rev. Microbiol. 18, 607–621. doi: 10.1038/s41579-020-0412-1
Wang, M., Eyre, A. W., Thon, M. R., Oh, Y., and Dean, R. A. (2020). Dynamic changes in the microbiome of rice during shoot and root growth derived from seeds. Front. Micrbiol. 11:2183. doi: 10.3389/fmicb.2020.559728
Wani, Z. A., Ahmad, T., Nalli, Y., Ali, A., Singh, A. P., Vishwakarma, R. A., et al. (2018). Porostereum sp., associated with saffron (Crocus sativus L.), is a latent pathogen capable of producing phytotoxic chlorinated aromatic compounds. Curr. Microbiol. 75, 880–887. doi: 10.1007/s00284-018-1461-9
Xiong, Y. W., Li, X. W., Wang, T. T., Gong, Y., Zhang, C. M., Xing, K., et al. (2020). Root exudates-driven rhizosphere recruitment of the plant growth-promoting rhizobacterium Bacillus flexus KLBMP 4941 and its growth-promoting effect on the coastal halophyte Limonium sinense under salt stress. Ecotoxicol. Environ. Saf. 194:110374. doi: 10.1016/j.ecoenv.2020.110374
Yadav, A. N., Rastegari, A. A., Yadav, N., and Kour, D. (Eds.). (2020). Advances in Plant Microbiome and Sustainable Agriculture: Functional Annotation and Future Challenges, Vol. 20. Basingstoke: Springer Nature.
Yasmin, S., and Nehvi, F. A. (2013). Saffron as a valuable spice: a comprehensive review. Afr. J. Agric. Res. 8, 234–242. doi: 10.5897/AJAR12.1955
Yin, L., Liu, B., Wang, H., Zhang, Y., Wang, S., Jiang, F., et al. (2020). The rhizosphere microbiome of Mikania micrantha provides insight into adaptation and invasion. Front. Microbiol. 11:1462. doi: 10.3389/fmicb.2020.01462
Youseif, S. H. (2018). Genetic diversity of plant growth promoting rhizobacteria and their effects on the growth of maize plants under greenhouse conditions. Ann. Agri. Sci. 63, 25–35. doi: 10.1016/j.aoas.2018.04.002
Zerrouk, I. Z., Rahmoune, B., Khelifi, L., Mounir, K., Baluska, F., and Ludwig-Müller, J. (2019). Algerian Sahara PGPR confers maize root tolerance to salt and aluminum toxicity via ACC deaminase and IAA. Acta Physiol. Plant. 41:91. doi: 10.1007/s11738-019-2881-2
Keywords: PGPR–plant growth-promoting rhizobacteria, Crocus sativus, rhizo-bacteriome, core microbiome, 16S rDNA
Citation: Ambardar S, Bhagat N, Vakhlu J and Gowda M (2021) Diversity of Rhizo-Bacteriome of Crocus sativus Grown at Various Geographical Locations and Cataloging of Putative PGPRs. Front. Sustain. Food Syst. 5:644230. doi: 10.3389/fsufs.2021.644230
Received: 20 December 2020; Accepted: 08 October 2021;
Published: 03 November 2021.
Edited by:
Everlon Cid Rigobelo, São Paulo State University, BrazilReviewed by:
Aejaz Ahmad Dar, Sher-e-Kashmir University of Agricultural Sciences and Technology of Jammu, IndiaNoemi Carla Baron Cozentino, São Paulo State University, Brazil
Copyright © 2021 Ambardar, Bhagat, Vakhlu and Gowda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheetal Ambardar, c2hlZXRhbGFtYmFyZGFyQGdtYWlsLmNvbQ==