 Adji Baskoro Dwi Nugroho
Adji Baskoro Dwi Nugroho- Department of Plant Science and Technology, Chung-Ang University, Anseong, Republic of Korea
Polycomb group proteins (PcG) play a crucial role in developmental programs in eukaryotic organisms, including plants. PcG-mediated gene repression is achieved by epigenetic histone modification on target chromatins. Loss of PcG components leads to severe developmental defects. CURLY LEAF (CLF), a PcG component in Arabidopsis, catalyzes the trimethylation of histone H3 on lysine 27 (H3K27me3), a repressive histone mark in numerous genes in Arabidopsis. In this study, we isolated a single homolog of Arabidopsis CLF, namely, BrCLF, in Brassica rapa ssp. trilocularis. Transcriptomic analysis revealed that BrCLF participated in B. rapa developmental processes, such as seed dormancy, leaf and flower organ development, and floral transition. BrCLF was also involved in stress signaling and stress-responsive metabolism, such as aliphatic and indolic glucosinolate metabolism in B. rapa. Epigenome analysis showed that H3K27me3 was substantially enriched in genes related to these developmental and stress-responsive processes. Thus, this study provided a basis for elucidating the molecular mechanism of the PcG-mediated regulation of development and stress responses in B. rapa.
Introduction
In eukaryotes, a zygote, a single diploid cell, is produced through the successful fertilization between egg and sperm cells. The zygote then undergoes successive cell divisions and differentiation and develops into multi-cellular tissues and organs with distinct functions and characteristics. Even though each cell has the same genomic contents, they differentiate into functionally specialized cells, indicating that well-systematized cell differentiation systems control differential transcriptional control in each cell lineage. However, studies have yet to determine how a single cell can delicately develop into distinctly specialized organs and tissues in multi-cellular organisms such as eukaryotes. Earlier genetic studies using Drosophila provided important information and identified polycomb group (PcG) genes, which are genes required for the proper development of the Drosophila body plan (Lewis, 1978).
PcG proteins are highly conserved in eukaryotes, including Drosophila, mammals, and plants, and they form large multimeric protein complexes, namely, polycomb repressive complexes (PRCs). PRCs are implicated in developmental programs and traditionally divided into PRC2 and PRC1 in eukaryotes (Margueron and Reinberg, 2011). In Drosophila, PRC2 has four core components, namely, Enhancer of Zeste (E(z)), Suppressor 12 of zeste (Su(z)12), Extra sex combs (ESC), and Nucleosome remodeling factor 55 (Nurf55) in Drosophila (Calonje, 2014). Each PRC2 component has a unique role; specifically, E(z) protein containing a SET domain catalyzes the deposition of a methyl group in histone H3 on lysine 27 (H3K27me3); ESC protein binds to H3K27me2/3 histone marks, thus stabilizing and enhancing the catalytic activity of E(z); Su(z)12 and Nurf55 are required for the nucleosome association of PRC2 to its target chromatin (Cao et al., 2002).
PRC2 components are well conserved in higher eukaryotes, including plants (Kim and Sung, 2014; Huang et al., 2019). Arabidopsis genome has three E(z) homologs: CURLY LEAF (CLF: At2g23380), SWINGER (SWN: At4g02020), and MEDEA (MEA, At1g02580); three homologs of Su(z)12, VERNALIZATION 2 (VRN2), EMBRYONIC FLOWRING 2 (EMF2), FERTILIZATION INDEPENDENT SEED 2 (FIS2); a homolog of ESC, FERTILIZATION-INDEPENDENT ENDOSPERM 1 (FIE1); and five homologs of Nurf55, MULTI-SUBUNIT SUPPRESSOR OF IRA 1–5 (MSI1–5) (Chanvivattana et al., 2004).
Three Arabidopsis homologs of E(z) (CLF, SWN, and MEA) encode histone methyltransferases involved in PRC2-mediated gene suppression by catalyzing H3K27me3 in target chromatins (Shu et al., 2020). While MEA is required for endosperm development in seeds, SWN plays a partially redundant role with CLF and MEA in development throughout the Arabidopsis lifespan (Godwin and Farrona, 2022). Nevertheless, CLF may play a major role in developmental programs in Arabidopsis because the null mutant of CLF displays severe defects in development, whereas the mutant of SWN does not show visible developmental defects compared with that of wild-type plants (Shu et al., 2020). The single CLF mutant, namely, clf, displays developmental defects in embryogenesis, seed dormancy, leaf development, floral transition, and floral organ development (Kim and Sung, 2014; Xiao and Wagner, 2015; Yan et al., 2020). For example, the expression of floral integrator genes, such as FLOWERING LOCUS T (FT) and SUPPRESSOR OF OVEREXPRESSION OF CO 1 (SOC1), was upregulated in the clf mutant, which is caused by the loss of the repressive histone mark H3K27me3 in FT and SOC1 chromatin (Jiang et al., 2008; Hou et al., 2014). In addition, the repression of seed dormancy genes, such as SOMNUS (SOM) and DELAY OF GERMINATION 1 (DOG1), is impaired in the clf mutant (Bouyer et al., 2011). Furthermore, the de-repression of homeotic genes, such as AGAMOUS (AG), AGAMOUS-LIKE 19 (AGL19), and SEPALATTA 3 (SEP3), in the clf mutant results in abnormal floral organ development (Shu et al., 2019). Therefore, loss of CLF, a catalytic subunit of PRC2, elicits detrimental effects on plant developments, thus showing an upward curly leaf, dwarf phenotype, premature floral transition, embryo lethality, reduced fertility, and deformed floral organs (Goodrich et al., 1997).
Several allelic mutants of B. rapa homologs of Arabidopsis CLF have been described for Chinese cabbage (Brassica rapa L. ssp. pekinensis, named as ebm1 and ebm3) and yellow sarson inbred line R-o-18 (B. rapa ssp. trilocularis, named braA.clf-1; Payá-Milans et al., 2019; Huang et al., 2020; Tan et al., 2021). braA.clf plants exhibited pleiotropic developmental changes which were resulted from the de-repression of developmental genes via the reduction of enrichment of H3K27me3 (Payá-Milans et al., 2019). For example, braA.clf plants have a smaller overall size, abnormal floral organs, and upward curling of leaves, resembling to the case of the Arabidopsis clf mutant (Goodrich et al., 1997). In addition, enrichment of H3K27me3 histone mark was substantially compromised in the braA.clf-1 mutant compared with that in wild-type plants (Payá-Milans et al., 2019). All of them are TILLING mutants developed via ethyl methanesulfonate (EMS) mutagenesis treatment. They commonly exhibit similar phenotypic defects such as upward curly leaves, reduced plant height, altered floral organs, and premature flowering phenotype observed in Arabidopsis. The enrichment of H3K27me3 histone mark is substantially compromised in the braA.clf-1 mutant compared with that in wild-type plants. Another EMS screening also isolated a mutant named as’tu8’ displayed a small malformation of shoot organs and reduced level of indole glucosinolates (GSLs). The TU8 was identified as an allele of TERMINAL FLOWER 2 (TFL2) (also referred to as LIKE-HETEROCHROMATIN 1 (LHP1)) that encodes a homeodomain protein functioning with PRC1 and PRC2 complexes (Ludwig-Müller et al., 1999).
Even though B. rapa CLF is required for several facets of developmental programs of B. rapa, the functional importance of BrCLF is poorly understood. In this study, a null mutant named brclf (same allele previously known as braA.clf-1) was isolated as a B. rapa CLF homolog of Arabidopsis CLF. Genome-wide transcriptomic, epigenomic, and metabolic analyses were performed to understand the functional significance of BrCLF in B. rapa. Thus, this study revealed that BrCLF-containing PRC epigenetically regulated developmental and stress response programs in B. rapa.
Materials and methods
Plant materials and growth conditions
Seeds of B. rapa yellow sarson (ssp. trilocularis) inbred line R-o-18 (Rusholme et al., 2007) and brclf mutant were sterilized with 5% sodium hypochlorite solution and then washed with distilled water. Sterilized seeds were plated on 1/2 Murashige and Skoog (MS) media and stored in a refrigerator at 4°C for 3 days for stratification. Two-week-old seedlings were transplanted to plastic pots containing vermiculite soil and watered with tap water. The plants were grown in a growth room under a long-day condition (16 h/8 h cycle) at 22°C with cool-white fluorescent illumination (120 mol m-2 s-1, FHF32SSEX-D fluorescent tube; Osram, South Korea). Phenotypic analysis was performed to compare ‘R-o-18’ and ‘brclf’ during vegetative and reproductive stages. A total of 8 plants were used from each ‘R-o-18’ and ‘brclf’ to measure hypocotyl, leaf, and rosette at 4-weeks after germination. Meanwhile, 5 plants were used in the measurement of plant height, petal, bud, and pistil at 2-weeks after bolting. The number of siliques and seeds was also quantified in this study at 2-3weeks after ‘R-o-18’ and ‘brclf’ flowering.
Characterization of brclf mutant
An EMS-induced TILLING mutant brclf (line number: ji32391-a) was purchased from RevGenUK (Stephenson et al., 2010). Genomic DNA from individual brclf mutants were extracted using DNeasy Plant mini kit (QIAGEN, Germany) and used for PCR amplification. Gene-specific primer pair (BrCLF_genoF and BrCLF_genoR) were used for genotyping of brclf mutant using T1000™ thermal cycler (Bio-rad, USA). Amplified PCR fragments (about 500bp) were run on 1% agarose gel and extracted from gel by using a QIAquick Gel extraction kit (QIAGEN, Germany) and then confirmed by the direct Sanger sequencing (Bionics Co., South Korea). Information on primer sequences is presented in Supplementary Table S1.
Quantitative RT-PCR expression analysis
Total RNAs from the whole seedling of 2-week-old B. rapa plants were purified using an RNeasy Plant mini kit in accordance with the manufacturer’s instructions (QIAGEN, Germany). Extracted RNAs were further treated with DNase I (New England Biolabs, USA) for 20 mins at 37°C to eliminate genomic DNA contamination. Purified RNAs were used to synthesize the complementary DNA synthesis using EasyScript RTase (TransGen Biotech, China). Quantitative RT-qPCR (qRT-PCR) reactions were performed using Sol™ 2X Real-Time PCR Smart mix under the following cycling conditions: 95°C for 10 mins followed by 45 cycles of 95°C for 20s, 60°C for 25 s, and 72°C for 35 s. BrPP2Aa (Bra012474) was used as the reference gene (Kim et al., 2022). qRT-PCR reactions were performed with three technical replicates by using the LineGene 9600 Plus (FQD-96A) Real-Time PCR Detection System (BioER, China). The detected quantification cycle (cq) values were examined using the 2–ΔCT method to calculate changes in gene expression. To ensure the reliability of quantitative analysis with standard deviation error bars. The primer sequences were designed on the basis of sequence information from the Brassica database (BRAD) (Chen et al., 2022) and summarized in the Supplementary Table S1.
Multiple sequence alignment and phylogenic analysis
The nucleotide and amino acid sequences of BrCLF were compared using the multiple sequence alignment webtool MultAlin (http://multalin.toulouse.inra.fr/multalin/) (Corpet, 1988). For phylogenic analysis, the amino acid sequences of AtCLF and AtCLF-like gene (SWN and MEA) in Arabidopsis genome were obtained from the TAIR database (http://www.arabidopsis.org). The sequence information of B. rapa homologs of CLF-like proteins were searched and downloaded from B. rapa genome database (http://brassicadb.cn/#/). The protein sequence information of Arabidopsis and B. rapa homologs were combined in fasta format and submitted into ClustalX (ver. 1.81) sequence alignment program.
RNA-seq library sequencing
Three biological replicates for 2-week-old seedlings of R-o-18 and brclf mutant were harvested and frozen in liquid nitrogen. Total RNAs were extracted using an RNeasy Plant mini kit (QIAGEN, Germany). The quantity and quality of RNA were checked with a 2100 Bioanalyzer (Agilent, USA), and only the samples with an RNA integrity number of >7 were used for library preparation. A library was constructed using TruSeq Stranded mRNA LT Sample Prep Kit (Illumina Inc., USA) in accordance with the manufacturer’s instructions. The constructed RNA-seq libraries were sequenced on a NovaSeq 6000 platform system (Macrogen Co., South Korea). Paired-end sequencing protocol was employed.
RNA-seq alignment and analysis
The quality of RNA-seq read counts was first evaluated using the FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). On the basis of FastQC results, raw reads were trimmed and quality-filtered before genome alignment by using Trimmomatic (ver0.36; Bolger et al., 2014). The trimmed and filtered reads with more than 90% portion (Q>30) were only used for mapping. The reference genome of B. rapa plants was obtained from Ensembl (https://plants.ensembl.org/info/website/ftp/index.html). Mapping was performed using the TopHat2 software with default parameters (Kim et al., 2013). Aligned reads were converted to digital counts using FeatureCounts (Liao et al., 2014) and were analyzed using edgeR (Robinson et al., 2010). Differentially expressed genes (DEGs) were identified on the basis of p < 0.05 and two-fold difference cut-off. A multi-dimensional scaling (MDS) plot and correlation heap were generated using R packages (ver. 3.6.0) (R Core Team, 2020) (https://www.R-project.org/). Venn diagram analysis was performed using VENNY (v2.1) webtool (https://bioinfogp.cnb.csic.es/tools/venny/). GO analysis was conducted using ShinyGO (ver 0.76) program (Ge et al., 2020). Aligned reads were visualized with the Integrative Genomics Viewer (IGV) program of Broad Institute (Thorvaldsdóttir et al., 2013). Heatmap analysis was performed using multi experiment viewer (MEV) program (ver 4.9.0).
Determination of glucosinolate content through U-HPLC analysis
Glucosinolates were analyzed from 2-week-old R-o-18 and brclf mutant in accordance with a previously described protocol (Nugroho et al., 2021). They were extracted from whole fresh tissue samples with 70% aqueous methanol (methanol:water 70:30, v:v) at 70°C for 10 min. The extract was centrifuged at 3000 × g for 20 min, and the supernatant was transferred into a column containing Sephadex A-25 (Sigma-Aldrich Inc., USA). The column was reacted with 11.25 units of purified sulfatase (Sigma-Aldrich Inc., USA) at 37°C to allow desulfation for 12 h. Desulfo-glucosinolates (DS-GSLs) were eluted from the column with 1.5 ml of deionized water and evaporated using a speed vacuum. DS-GSLs were re-dissolved with 1 ml of HPLC water and filtered with 0.45 μm PVDF membrane (Biofact, Korea). They were chromatographically separated on a C18 reverse phase column (Zorbax XDB-C18, 4.6 × 250 mm2, 5 μm particle size, Agilent, USA) with a gradient system composed of water (Thermo Fisher Scientific, USA) and acetonitrile (Honeywell, USA) in the Dionex Ultimate 3000 ultra-high performance liquid chromatography (U-HPLC) systems (Thermo Fisher Scientific, USA). Samples (20 μL) of DS-GSLs were analyzed using a diode array detector at 229 nm. All DS-GSL peaks detected in this study were identified in accordance with a previous study (Nugroho et al., 2019). Each DS-GSL was independently quantified from three biological replicates based on sinigrin (Sigma-Aldrich Inc., USA) standard compounds (Brown et al., 2003). Data were presented as micromoles per gram dry weight (µmol/gr DW).
Analysis of H3K27me3 ChIP-seq dataset
The dataset of the genome-wide H3K27me3 profile (NCBI SRA number: PRJNA542357) in leaf and inflorescence tissues of R-o-18 inbred line (B. rapa ssp trilocularis) was downloaded. The reference genome of B. rapa was also downloaded from Ensembl Plants database (https://plants.ensembl.org/). The downloaded raw fastq files of H3K27me3 were initially quality-checked using the FastQC program (Andrews, 2010). On the basis of FastQC result, low-quality reads were trimmed and filtered using Trimmomatic (v0.36) (Bolger et al., 2014), and the filtered fastq files were used for mapping to B. rapa genome by using bowtie2 (Langmead and Salzberg, 2012). SAMtools (Li et al., 2009) were used to convert SAM files to BAM files. Then, duplicated reads were removed by Picard MarkDuplicates (v2.18.2.0; biotools:picard_tools; RRID : SCR_006525). Deduplicated reads were then used for peak calling on pooled replicates by using MACS2 (v2.1.1) for comparison between input DNA and ChIP samples. The distribution of H3K27me3 enrichment was visualized using the IGV (Thorvaldsdóttir et al., 2013).
Analysis of ChIP-qPCR
The 2-weeks seedlings of ‘R-o-18’ and ‘brclf’ were cross-linked with 1% formaldehyde solution under vacuum for 25 min and stop the reaction by addition of 0.1M glycine. The cross-linked seedlings were dried and grinded in liquid nitrogen. ChIP experiment was performed as previously reported. Ten micrograms of monoclonal antibody against H3K27me3 histone mark (ab6002, Abcam, United Kingdom) were treated for the individual ChIP sample. The immunoprecipitated and input DNAs were used for the qPCR analysis. ChIP-qPCR reactions were performed using Sol™ 2X Real-Time PCR Smart mix (SolGent, Korea) under the following cycling conditions: 95°C for 12 mins followed by 45 cycles of 95°C for 20s, 60°C for 25 s, and 72°C for 35 s. BrFUS3 (Bra032953) was used as the reference gene.
Statistical analysis
Data were statistically analyzed using a statistical software package (SAS; version 9.4; SAS Institute Inc., Cary, NC, USA). Statistical differences were calculated through one-way analysis of variance (ANOVA) and post-hoc Tukey’s test (p < 0.05). Data were presented as means ± standard deviation (SD) of three biological replicates.
Results
Phenotypic characterization of brclf mutant
In Arabidopsis, CURLY LEAF (CLF) plays a catalytic activity in PRC2 for gene suppression via the trimethylation of histone H3 at the K27 residue (H3K27me3) (Schubert et al., 2006). B. rapa genome has a single homolog of Arabidopsis CLF referred to as BrCLF (Bra032169) from the Brassica database (BRAD) (Payá-Milans et al., 2019; Supplementary Figure S1A). Because Arabidopsis CLF has two paralogs, namely, SWINGER (SWN) and MEDEA (MEA), in the genome, we searched homologs of these paralogs from the B. rapa genome database through BLAST search and phylogenic analysis. We found that B. rapa has one homolog of SWN (named BrSWN, Bra036300) and two homologs of MEA (BrMEA,a [Bra033334] and BrMEA,b [Bra032592]; Supplementary Figure S1B).
CLF plays a crucial role in developmental programs, including leaf development and floral organ development, in Arabidopsis (Goodrich et al., 1997). Thus, we aimed to obtain the loss-of-function mutant of BrCLF by searching an EMS-mutagenized TILLING mutant population in B. rapa ssp. trilocularis (hereafter referred to as brclf mutant). Point mutation (C to T) in the 1,843 bp region from the start codon of BrCLF was detected to produce a premature stop codon (Gln at the 615th residue from the start codon) in brclf mutant (Figure 1A; Supplementary Figure S1C). This mutation led to the loss of 292 amino acids in the C-terminal region, which has a catalytic SET domain (Supplementary Figure S1D). Non-functional brclf mutant exhibited severe developmental defects in vegetative and reproductive organs compared with those in the wild type (WT) R-o-18 plants. For example, the brclf mutant showed abnormal phenotypes such as upward curled and smaller leaves than the R-o-18 wild-type plant did (Figures 1B, C). The hypocotyl length, leaf width, rosette diameter of brclf mutant were significantly shorter than those of R-o-18 in 4-week-old vegetative-stage plant in our growth condition (Supplementary Figure S2A). At the reproductive stage, brclf was quite shorter than the wild-type plant (Figure 1D; Supplementary Figure S2B). Seed fertility was significantly reduced in brclf mutant than the wild-type plant (Figure 1E; Supplementary Figure S2B). A lower seed set in brclf mutant might cause by the deformed development of floral organs (Figure 1F). For instance, the pistil of brclf was extraordinarily elongated, which might substantially reduce the chance for pollination and successive fertilization between egg and sperm cells.

Figure 1 Phenotypic characterization between R-o-18 wild type and brclf mutant. (A) Identification of point mutation in the brclf coding sequence that generates a premature stop codon. The conversion of cytosine to thymine at 1,843 bp from the start codon was indicated with an upward red arrowhead. Glutamine (Q) amino acid at the 615th residue of BrCLF of R-o-18 wild type was converted to a stop codon (indicated with an asterisk) in the brclf mutant. (B) Comparison of leaf phenotypes between R-o-18 and brclf mutant. The brclf mutant exhibited a smaller and upward curly leaf phenotype than R-o-18 did. The red arrow indicates the upward curly leaf of the brclf mutant. (C) Whole plant phenotype of four-week-old R-o-18 and brclf mutant. The brclf mutant showed smaller rosette diameter and curly leaves (indicated with red color arrows) than the wild-type R-o-18 did. (D) Phenotypic comparison of R-o-18 and brclf at the flowering stage. Flowering occurred earlier in the brclf mutant than in R-o-18; furthermore, the height of the former was shorter than that of the latter. (E) Comparison of seed sets between R-o-18 and brclf mutant. brclf showed lower number of seeds and some aborted seeds (indicated with red color arrows) in a pod compared with R-o-18. (F) Comparison of flower organ between R-o-18 and brclf. brclf showed an abnormally elongated pistil compared with R-o-18 (indicated with red color arrow).
Transcriptomic change in brclf mutant
The brclf mutant exhibited severe developmental defects. Thus, we aimed to dissect the transcriptomic change between WT and brclf mutant by RNA-seq analysis. After low-quality reads were trimmed, high-quality paired reads (>Q30) were over 95% in all samples (Supplementary Table S2). They were used for alignment with the B. rapa reference genome downloaded from the Ensembl Plants (https://plants.ensembl.org/info/data/ftp/index.html). Correlation heatmap analyses showed a distinct clustering between R-o-18 and brclf samples (Supplementary Figure S3A), indicating that RNA-seq libraries were well constructed and sequenced. Differentially expressed genes (DEGs) between R-o-18 and brclf samples were isolated based on the pairwise sample comparisons using edgeR (Figure 2A; Supplementary Figure S3B). A total of 2,740 DEGs, including 1,844 upregulated genes and 896 downregulated genes in brclf mutant compared with wild type R-o-18, were detected as significant DEGs in pairwise sample comparisons (|log2|> 1, p < 0.05) (Supplementary Table S3).
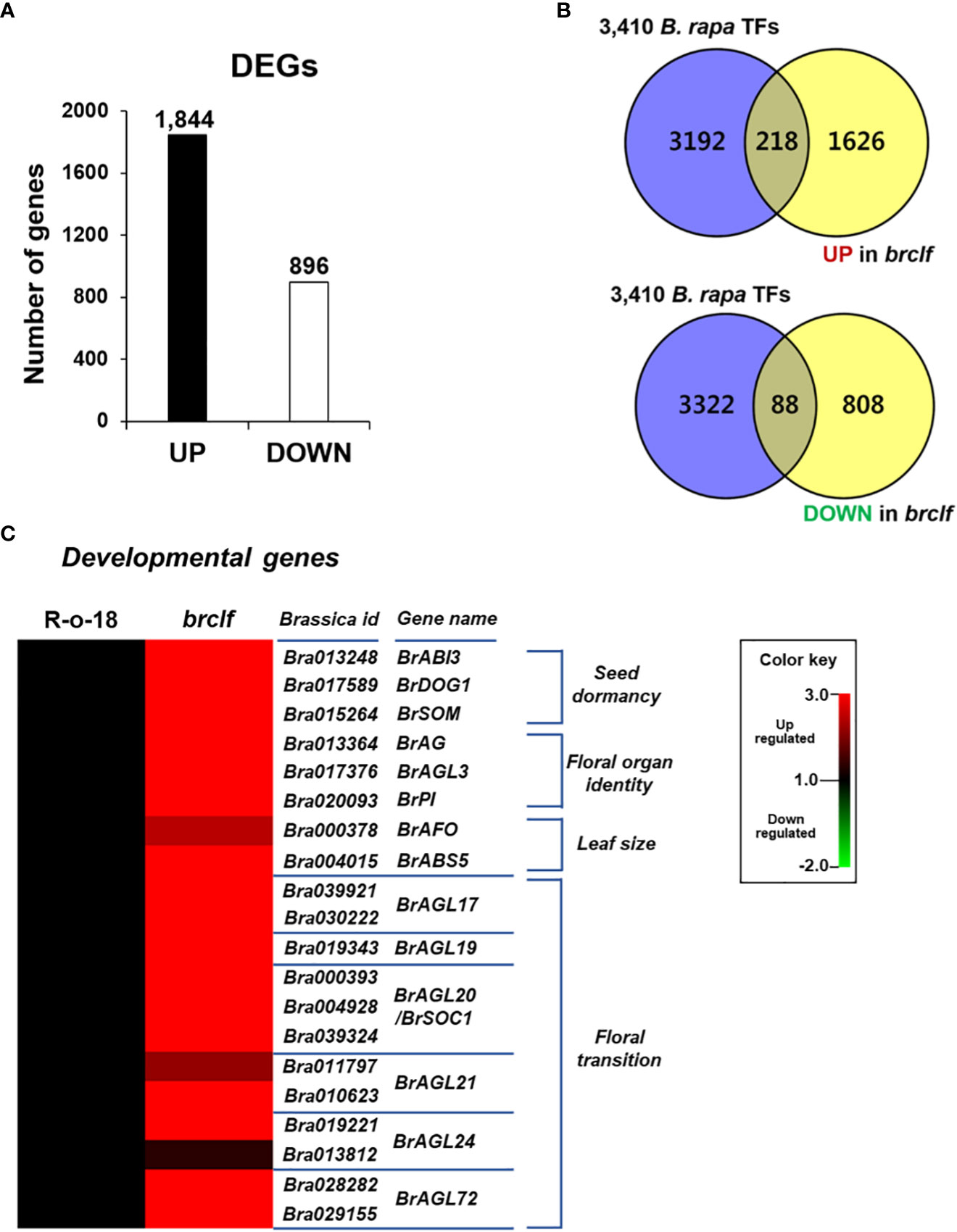
Figure 2 Identification of differentially expressed genes (DEGs) between “R-o-18-and brclf. (A) Number of DEGs between “R-o-18-and brclf. A total of 1,844 and 896 genes were respectively upregulated and downregulated in the brclf mutant compared with those in wild-type R-o-18. (B) Venn diagram showing the overlap between 3,540 TF genes and DEGs found in brclf in comparison with R-o-18 in B. rapa. A total of 218 and 88 TFs were upregulated and downregulated in brclf compared with those in R-o-18, respectively. (C) Heatmap showing normalized transcript levels of 20 TF genes related to developmental programs in B. rapa: 3 genes (BrABI3, BrSOM, BrDOG1) related to “seed dormancy,” 3 genes related to the “floral organ identity” (BrAG, BrAGL3, BrPI), 2 genes related to “leaf size” (BrAFO, BrABS5), and 12 genes related to the “floral transition” (BrAGL17.1/2, BrAGL19, BrAGL20.1/2/3, BrAGL21.1/2, BrAGL24.1/2, and BrAGL72.1/2).
BrCLF is required to regulate genes related to multiple developmental transitions in B. rapa L.
Because Arabidopsis CLF was reported to regulate thousands of genes related to various developmental processes, we examined how many transcription factors are transcriptionally affected in brclf in comparison to R-o-18 sample, we first searched the BRAD genome database and collected total 3,410 transcription factor genes in B. rapa genome. Among 3,410 TFs, total 218 and 88 TF genes were up- and downregulated in brclf compared with R-o-18, respectively (Figure 2B; Supplementary Table S4). Given that BrCLF-containing PRC2 exerts a repressive role in target gene transcription, the number of upregulated genes in brclf was possibly higher (2.5 times) than that of downregulated genes. The expression of several key TF genes involved in seed dormancy/germination, leaf development, floral organ identity, and floral transition were examined between R-o-18 and brclf. Twenty genes related to developmental processes were selected, and their transcript levels were compared between R-o-18 and brclf: 3 genes related to “seed dormancy” such as BrABI3 (B. rapa homolog of ABI3, Bra013248), BrDOG1 (B. rapa homolog of DOG1, Bra017589), BrSOM (B. rapa homolog of SOM, Bra015264), 3 genes associated with “floral organ identity,” BrAG (B. rapa homolog of AG, Bra013364), BrAGL3 (B. rapa homolog of AGL3, Bra017376), and BrPI (B. rapa homolog of PI, Bra020093), 2 genes related to “leaf size” (BrAFO, B. rapa homolog of AFO, Bra000378 and BrABS5, B. rapa homolog of ABS5, Bra004015), and 12 BrAGL genes related to the “floral transition” (BrAGL17, BrAGL19, BrAGL20/SOC1, BrAGL21, BrAGL24, and BrAGL72). All tested B. rapa developmental genes were upregulated in brclf in comparison with those of the R-o-18 wild type (Figure 2C). This result indicated that BrCLF (and its PRC2 complex) play a negative role in the expression of many key developmental TF genes in B. rapa plant. Quantitative RT-PCR (qRT-PCR) was used to determine the transcript levels of these TF genes between R-o-18 and brclf mutant and validate the RNA-seq results. In a consistency with RNA-seq result, qRT-PCR analysis showed that transcript levels of all tested developmental genes were substantially upregulated in brclf mutant compared with the wild type R-o-18 plant (Supplementary Figure S4). Altogether, our RNA-seq and qRT-PCR data confirmed that BrCLF play a repressive role in transcription of key developmental genes in B. rapa plant.
Gene ontology analysis of DEGs between R-o-18 and brclf
Besides developmental TF genes, thousands of genes were differentially expressed between R-o-18 and brclf. GO analysis was conducted using ShinyGO v. 0.76 (http://bioinformatics.sdstate.edu/go/) to further seize the biological role of BrCLF from the list of DEGs. GO analysis using the list of upregulated or downregulated genes displayed top 20 and top 9 GO categories, respectively (Figure 3). Unexpectedly, GO analysis using the list of upregulated genes exhibited that the top rankers in the top 20 categories were enriched with stress-related categories such as “hydrogen peroxide catabolic process” and “reactive oxygen species metabolic process” (Figure 3A). As expected, GO categories related to “regulation of transcription” were also shown in the top 20 categories, but they were in lower ranks. GO analysis was also conducted with the lists of downregulated genes (Figure 3B). GO analysis using list of downregulated genes showed the enrichment of GO categories related to metabolic processes such as “cellular response to blue light,” “xyloglucan metabolism,” “carbohydrate metabolism,” “response to abiotic stimulus,” and “response to oxygen-containing compound.” Based on the repressive role of PRC2 complex in gene regulation, it is likely that a majority of GO terms found with downregulated genes might not be the directly targeted functional categories. Results of GO analysis using DEGs suggested that BrCLF might play a regulatory role on the stress signaling genes involved in the “hydrogen peroxide catabolic process” and “reactive oxygen species metabolic process” in B. rapa. Based on this observation, we further analyzed the list of DEGs containing 1,844 up-regulated and 896 down-regulated genes. We collected 3,150 stress related genes from the Arabidopsis genome, which was obtained from the STIFDB2 (Stress-responsive Transcription Factor Database, ver. 2) website (http://caps.ncbs.res.in/stifdb2/index.html). Based on the sequence homolog search results between Arabidopsis and B. rapa, we found a total of 5,226 stress-responsive genes in B. rapa (Supplementary Table S5). Out of 5,225 stress-related genes, 301 genes were found in the 1,844 up-regulated genes in ‘brclf’ (Supplementary Figure S5; Table S6). It indicated that 16% (301/1,844) of stress-related genes were affected in the ‘brclf’ mutant.
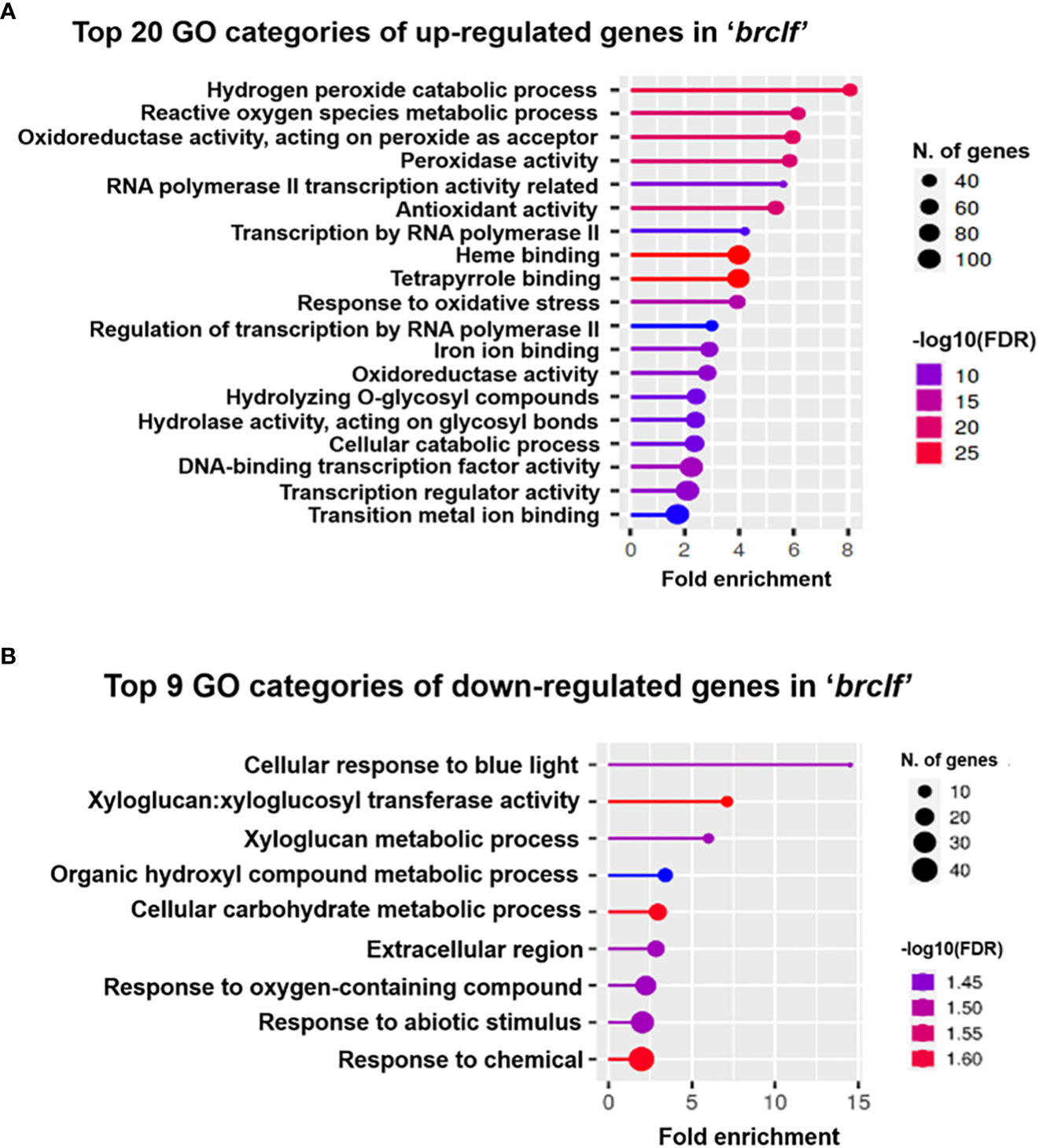
Figure 3 Gene ontology (GO) analysis of upregulated and downregulated genes in brclf mutant compared with those in R-o-18 wild-type plant. (A) Top 20 functional GO categories with 1,844 upregulated genes in the brclf mutant compared with those in the R-o-18 wild-type plant. (B) Top 9 functional GO categories with 896 downregulated genes in the brclf mutant compared with those in the R-o-18 wild-type plant.
BrCLF is involved in the regulation of plant stress signaling genes in B. rapa L.
Because GO enrichment suggested a possible role of BrCLF on stress signaling, we selected 13 genes related to “hydrogen peroxide catabolic process” and “reactive oxygen species metabolic process” response from B. rapa genome (Supplementary Table S7). It included B. rapa MYC2 (BrMYC2, Bra010178), B. rapa OCTADECANOID-RESPONSIVE ARABIDOPSIS AP2/ERF 59 (BrORA59, Bra010178), B. rapa NAC DOMAIN CONTAINING PROTEIN 55 (BrANAC55), B. rapa VEGETATIVE STORAGE PROTEIN 2 (BrVSP2, Bra020470), B. rapa MANAGANESE SUPEROXIDE DISMUTASE (BrSOD, Bra007239), B. rapa ASCORBATE PEROXIDASE (BrAPX, Bra023579), B. rapa GLTATHIONE S-TRANSFERASE 11 (BrGSTU11, Bra003970), B. rapa PEROXIDASE 1b (BrPER1b, Bra032241), B. rapa PEROXIDASE 1a (BrPER1a, Bra038102), B. rapa PLANT DEFENSIN 1.2b (BrPDF1.2b, Bra003970), B. rapa MAP KINASE 6a (BrMPK6a, Bra014528), B. rapa MAP KINASE 6b (BrMPK6b, Bra014527) homologs. The analysis of RNA-seq data showed that transcript levels of these 13 genes were commonly higher in brclf sample than those of R-o-18 sample (Figure 4A). The qRT-PCR analysis of these genes also confirmed the result of RNA-seq (Supplementary Figure S6). These results suggested that BrCLF is involved in the suppression of stress signaling genes in B. rapa.
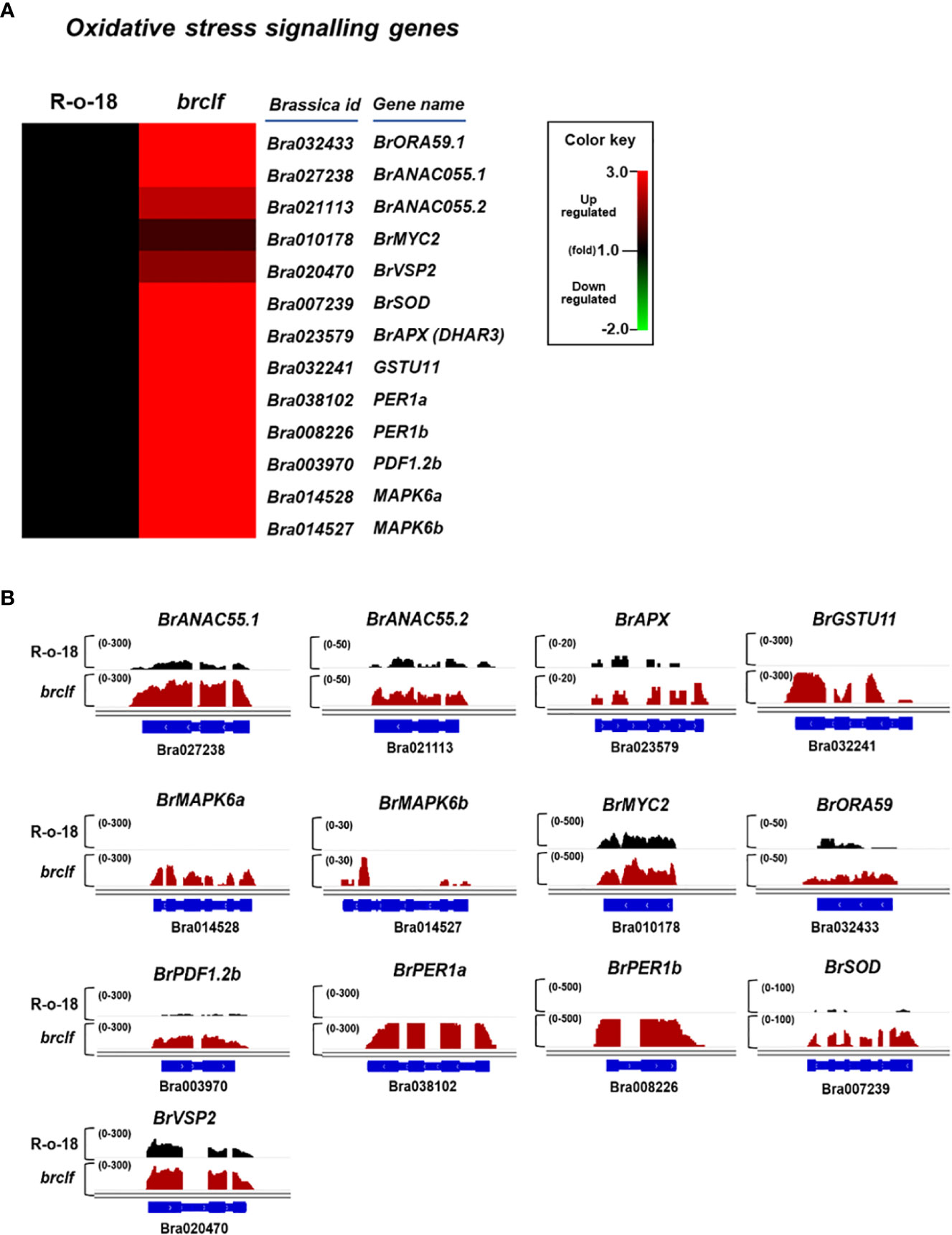
Figure 4 Expression of oxidative stress-related genes between R-o-18 and brclf. (A) Heatmap showing the normalized transcript levels of 13 oxidative stress-related genes in B. rapa. Transcript level of R-o-18 was set to value 1 and relative transcript level of each gene in brclf was presented. (B) Integrative genome browser (IGV) illustration on transcript levels of 13 stress signaling genes between ‘R-o-18’ and ‘brclf’. BrPP2Aa was used as the reference gene to confirm the normalization of RNA-seq reads between ‘‘R-o-18’ and ‘brclf’. Black colors and red colors indicates transcript reads mapped to individual genes in ‘R-o-18’ and ‘brclf’, respectively. Read coverage normalized by total number of mapped reads are indicated at the top left corner of each track in parentheses.
BrCLF regulates genes related to stress-responsive “glucosinolate metabolism”
In plants, “oxidative stress,” “hydrogen peroxide metabolism” and “reactive oxygen species (ROS) responses are initial signaling events upon exposure to a diversity of stresses such as biotic (i.e., insect and pathogen attack) and abiotic stress (i.e., salinity, heat and drought) (Nazir et al., 2020). Stress-induced signaling cascade stimulates the accumulation of defensive secondary metabolites such as aliphatic and indolic glucosinolates (abiotic and biotic stress metabolites) (Kliebenstein, 2004; Hiruma et al., 2013; Meraj et al., 2020). Thus, we decided to examine whether BrCLF is involved in the regulation of genes related to these stress-responsive secondary metabolisms, glucosinolates. Glucosinolates (GSLs) are a group of sulfur-rich compounds present in Brassicaceae family crop plants including Brassica genus plants (i.e., B. rapa.) (Sonderby et al., 2010). We found total 104 GSL pathway genes (63 aliphatic and 41 indolic GSL genes) from the B. rapa genome (Supplementary Table S8). Among the list of 1,844 up-regulated genes, total 9 GSL pathway genes were significantly upregulated (fold change ≥2) in comparison to levels of ‘R-o-18’ (Supplementary Table S9). In addition, additional 18 GSL pathway genes were moderately up-regulated genes (fold change ≥1.5 & <2) in ‘brclf’ mutant compared with R-o-18. Thus, total 27 out of 104 genes (26%) were upregulated in brclf, whereas none GSL pathway genes was downregulated in ‘brclf’ in comparison to levels of R-o-18 (fold change cutoff ≥1.5).
GSLs biosynthetic genes were reported to be positively modulated by a group of R2-R3 type MYB transcription factors like BrMYB28, BrMYB29 for aliphatic GSLs and BrMYB34, BrMYB51, and BrMYB122 for indolic GSLs (Gigolashvili et al., 2007; Hirai et al., 2007; Sønderby et al., 2007; Malitsky et al., 2008; Gigolashvili et al., 2009). When we compared the transcript levels of these MYB-domain TF genes, BrMYB29 for aliphatic GSLs and BrMYB34s and BrMYB122s for indolic GSLs were substantially upregulated in brclf mutant in comparison to R-o-18 (Figure 5). It suggests that BrCLF play a negative role in the expression of BrMYB TFs genes regulating aliphatic and indolic GSL biosynthesis (Figures 6, 7). To confirm that BrCLF is involved in the GSL metabolism, we quantified endogenous two major types of GSLs, aliphatic and indolic GSLs. We successfully detected four aliphatic GSL compounds (PGT, GNA, GNP, and GBN) and four indolic GSL compounds (GBS, 4-MTGB, 4-HGB, and NGB) using U-HPLC analysis (Supplementary Figures S7A, B). Compared to levels of R-o-18, brclf mutant had significantly increased amounts of both aliphatic and indolic GSLs (Figure 8). PCA analysis displayed that GSL compounds of R-o-18 and brclf were clustered in different groups but closely associated each other (Supplementary Figure S7C). Further, the biplot picture which generated from the PCA data pointed out that loss of BrCLF affected GSL metabolism (Supplementary Figure S7D). Altogether, these data indicated that BrCLF (and its PRC2 complex) suppresses genes involved in the stress signaling cascade and downstream stress-responsive GSL metabolism in B. rapa plants.
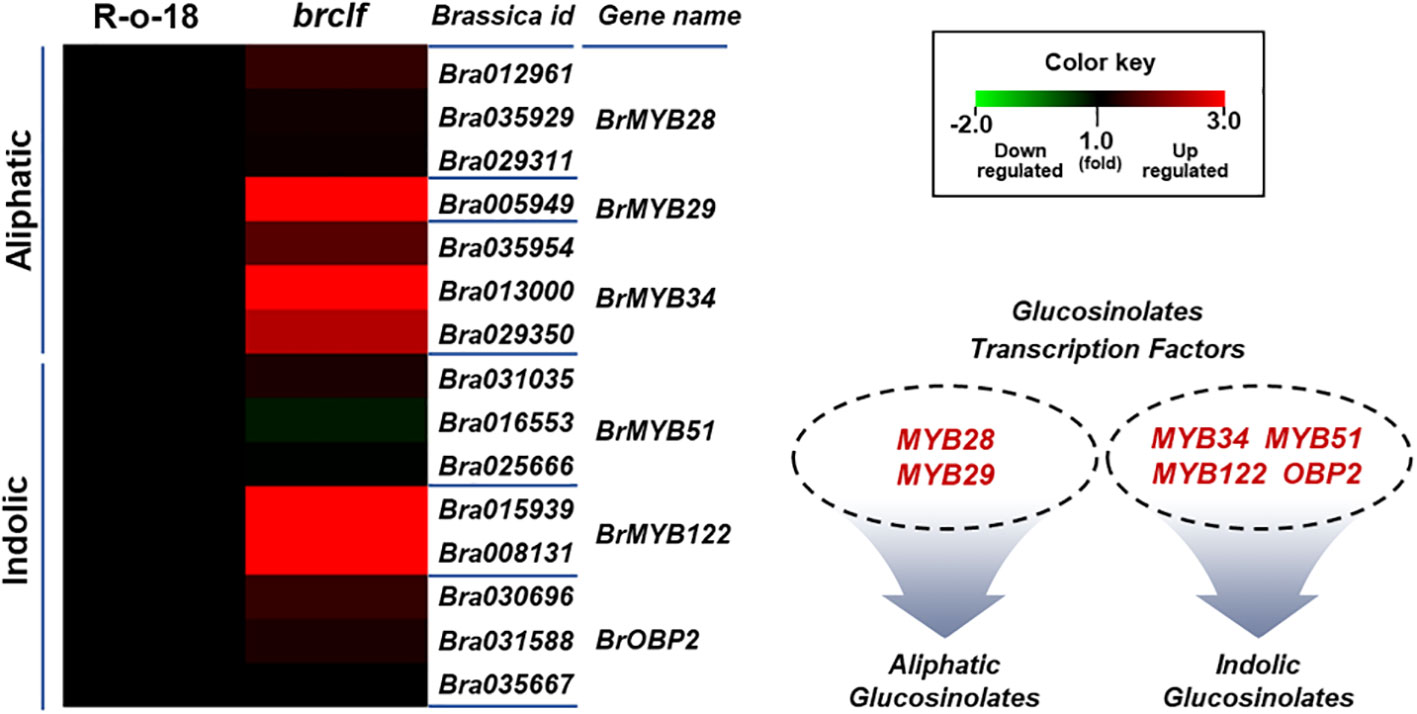
Figure 5 Expression levels of 15 TFs controlling aliphatic and indolic GSL biosynthesis genes between R-o-18 and brclf. Heatmap showing expression levels of 4 TFs (BrMYB28.1–3 and BrMY29) controlling aliphatic GSL biosynthesis genes and 11 TFs (BrMYB34.1–3, BrMYB51.1–3, BrMYB122.1–2, and BrOBP2.1–3) controlling indolic GSL biosynthesis gene factors between R-o-18 and brclf. Transcript level of R-o-18 was set to value 1, and relative transcript level of brclf was presented.

Figure 6 Expression levels of 59 aliphatic GSL biosynthesis genes between R-o-18 and brclf. (A) Heatmap showing expression levels of 59 aliphatic GSL biosynthesis genes between R-o-18 and brclf. Transcript level of R-o-18 was set to value 1, and relative transcript level of brclf was presented. (B) Diagram showing biosynthetic pathway for aliphatic GSL compounds. Genes responsible for each catalytic conversion were indicated with red letters.
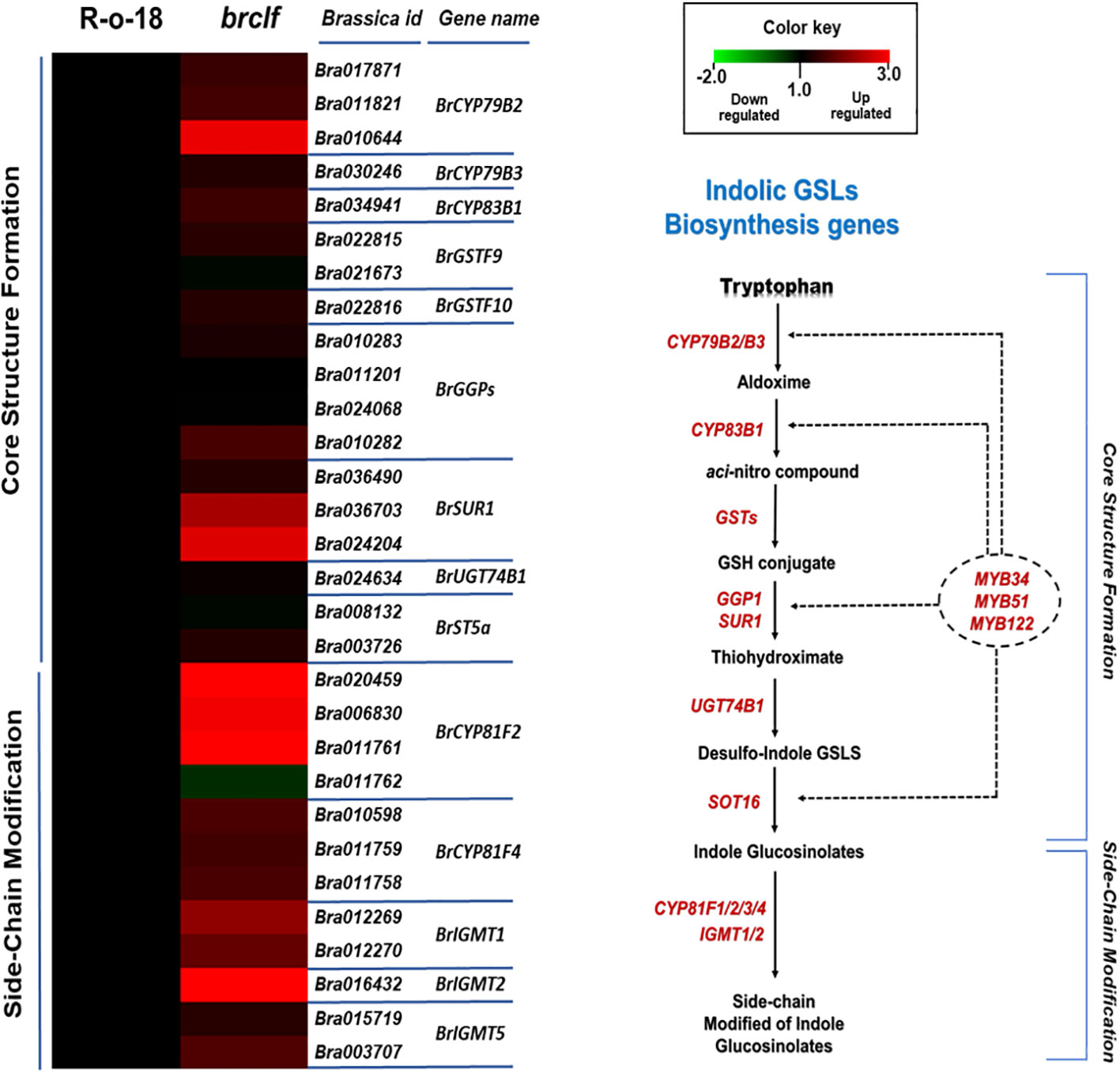
Figure 7 Expression levels of 40 indolic GSL biosynthesis genes between R-o-18 and brclf. (A) Heatmap showing the expression levels of 40 indolic GSL biosynthesis genes between R-o-18 and brclf. Transcript level of R-o-18 was set to value 1, and relative transcript level of brclf was presented. (B) Diagram showing biosynthetic pathway for indolic GSL compounds. Genes responsible for each catalytic conversion were indicated with red letters.
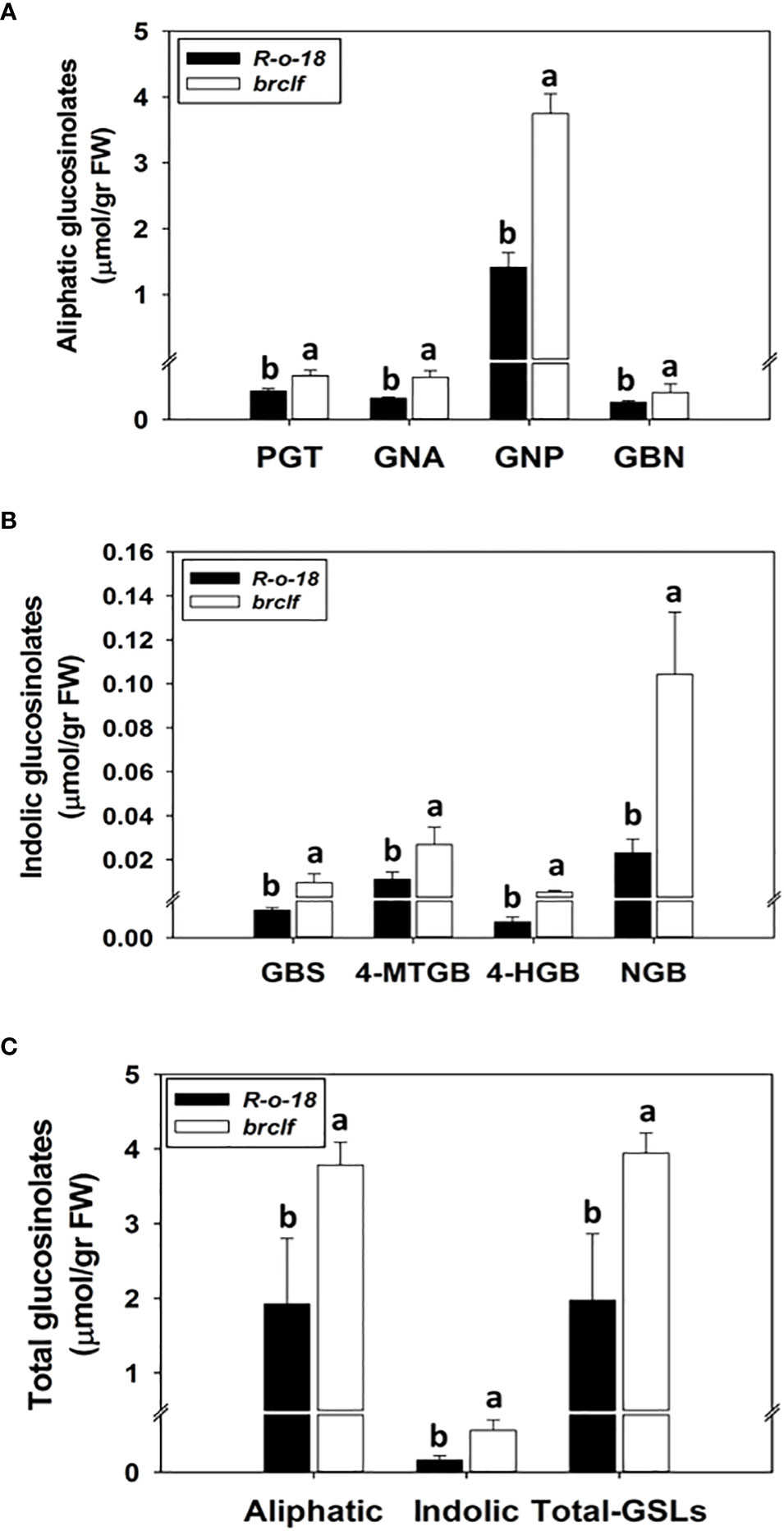
Figure 8 Comparison of the amounts of aliphatic and indolic GSL compounds between R-o-18 and brclf mutant. (A) Comparison of the amounts of four aliphatic GSL compounds between R-o-18 and brclf mutant. PGT: progoitrin, GNA: gluconapoleiferin, GNP: gluconapin, GBN: glucobrassicanapin. (B) Comparison of amounts of four indolic GSL compounds between R-o-18 and brclf mutant. GBS: glucobrassicin, 4-MTGB: 4-Methoxyglucobrassicin, 4-HGB: 4-hydroxyglucobrassicin, NGB: neoglucobrassicin. (A–C) One-way ANOVA with Tukey’s post-hoc test was applied to calculate statistical differences (p<0.05), and data were expressed as means ± standard deviation (SD). Statistical difference was indicated with different letters above each bar.
H3K27me3 was enriched in “developmental genes,” “stress signaling” and stress-responsive “GSLs pathway genes”
Arabidopsis CLF-containing PRC2 complex play a suppressive role on target genes via deposit of H3K27me3 (Bouyer et al., 2011). Recently, genome-wide H3K27me3 profiles in leaf and flower tissues of R-o-18 inbred line (B. rapa ssp. trilocularis) were published (Payá-Milans et al., 2019). Thus, enrichment of H3K27me3 on developmental genes, stress signaling genes, and stress-responsive metabolic genes were analyzed in both “leaf” and “inflorescence”. In case of developmental genes, all tested 20 genes (100%) were markedly enriched with H3K27me3 in both leaf and flower tissues (Supplementary Figure S8). To validate this, we performed ChIP assay using H3K27me3 antibody between R-o-18 and brclf mutant. ChIP-qPCR analysis was performed on three developmental genes related to seed dormancy (BrABI3-Bra013248), leaf size (BrABS5-Bra004015), and floral organ identity (BrAG-Bra013364). All tested genes exhibited that enrichment of H3K27me3 was evidently compromised in the brclf mutant compared to R-o-18. (Figure 9). This result clearly explained why RNA-seq and qRT-PCR analyses of these developmental genes showed higher transcript levels in brclf mutants than R-o-18. It indicated that BrCLF regulates the expression and H3K27me3 marking of those genes, probably through direct deposition of the mark at these loci. A total of 13 “stress signaling” genes were also checked whether they have H3K27me3 enrichment on their genomic regions. 10 genes out of 13 (77%) stress-signaling genes were densely enriched with H3K27me3 (Supplementary Figure S9; Supplementary Table S10). Because stress signaling genes play a crucial role in the early stage of defense system, it is reasoned that BrCLF-containing PRC2 complex might contribute to suppress and maintain basal level expression of stress signal genes in normal condition prior to experience of challenging stress condition.
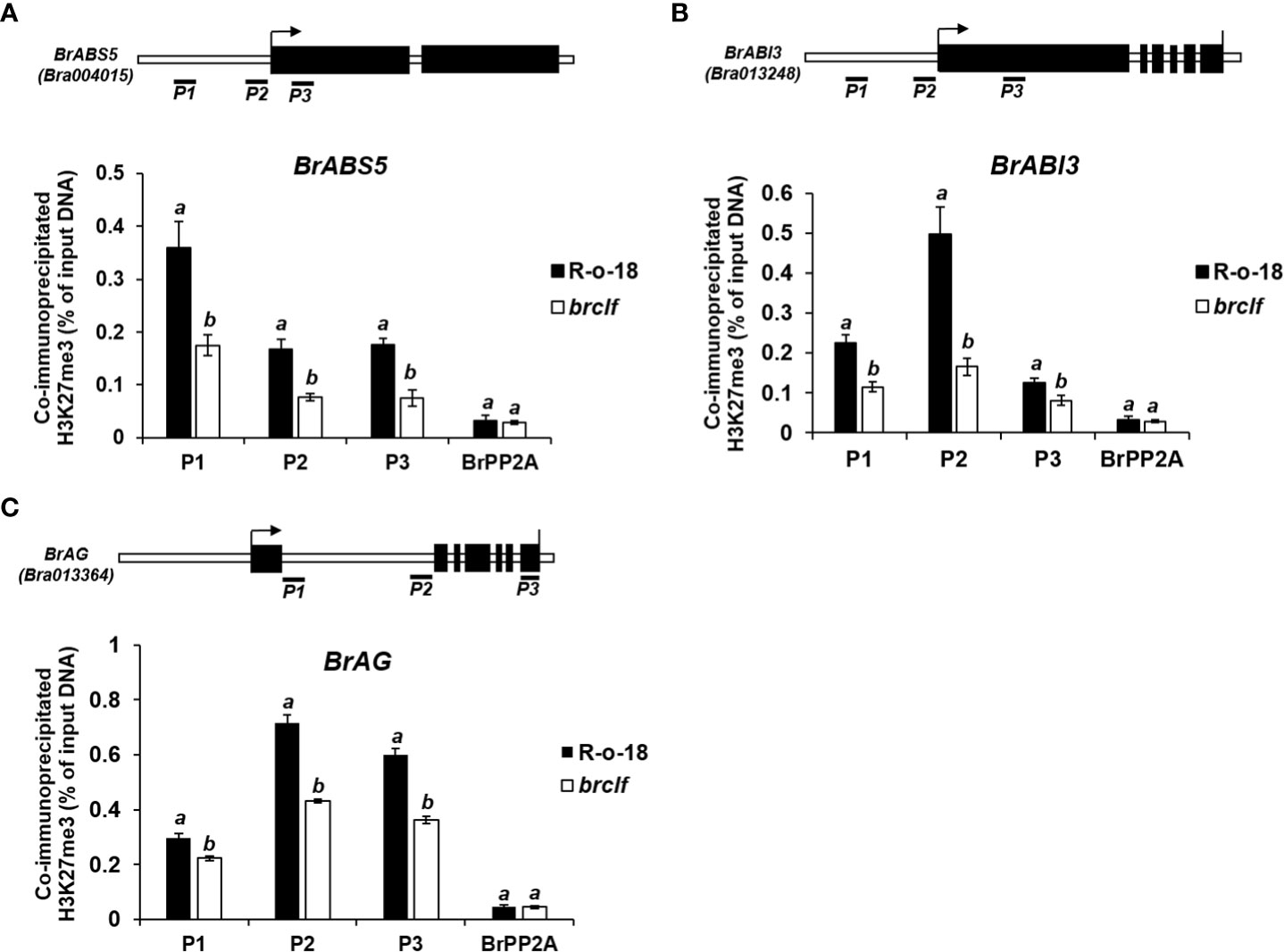
Figure 9 ChIP-qPCR analysis of 3 developmental genes between R-o-18 and brclf mutant. The enrichment level of H3K27me3 in developmental genes related to (A) leaf size (BrABS5-Bra004015) (B) seed dormancy (BrABI3-Bra013248), (C) floral organ identity (BrAG-Bra-013364) were calculated between R-o-18 and brclf mutant. All tested genes exhibited substantially reduced levels of H3K27me3 in brclf mutant compared with R-o-18 plant. Enrichment level was calculated as % of input DNA. BrPP2A (Bra012474) was used as a negative control (H3K27me3-unmarked gene) which was used as a housekeeping reference gene (Kim et al., 2022; Supplementary Figure S13). BrFUS3 (Bra032953) was used as a positive control, H3K27me3-enriched gene (Supplementary Figure S13). Mean and standard deviation (SD) of three biological replicates were calculated and presented. Statistical analysis was performed with the one-way analysis of variance (ANOVA) and post-hoc Tukey’s test (p < 0.05).
In case of stress-responsive metabolites, aliphatic and indolic GSLs, we checked the presence of H3K27me3 in 104 pathway genes, 15 TF genes (4 genes for aliphatic and 11 genes for indolic pathway), and 89 GSL biosynthesis genes comprising 59 aliphatic and 30 indolic pathway genes. Interestingly, fourteen genes out of 15 TF genes (93%) except BrMYB34.3 were enriched with H3K27me3 in either leaf or flower (Supplementary Figure S10; Supplementary Table S11). To quantify relative enrichment of H3K27me3 in the GSL TFs, ChIP-qPCR analysis was applied for both of aliphatic and indolic GSL TFs which includes three TFs involved in aliphatic GSL (BrMYB28.1, BrMYB28.2, and BrMYB28.3) and five TFs for indolic GSL (Br BrMYB51.3, BrMYB122.1, BrOBP2.1, BrOBP2.2, BrOBP2.3). The result showed that ‘brclf’ contained low level of H3K27me3 compared to levels of R-o-18 (Figure 10). Thus, it is confirmed that BrCLF, a component of B. rapa PRC2 complex is required for H3K27me3 enrichment for the suppression of target genes.
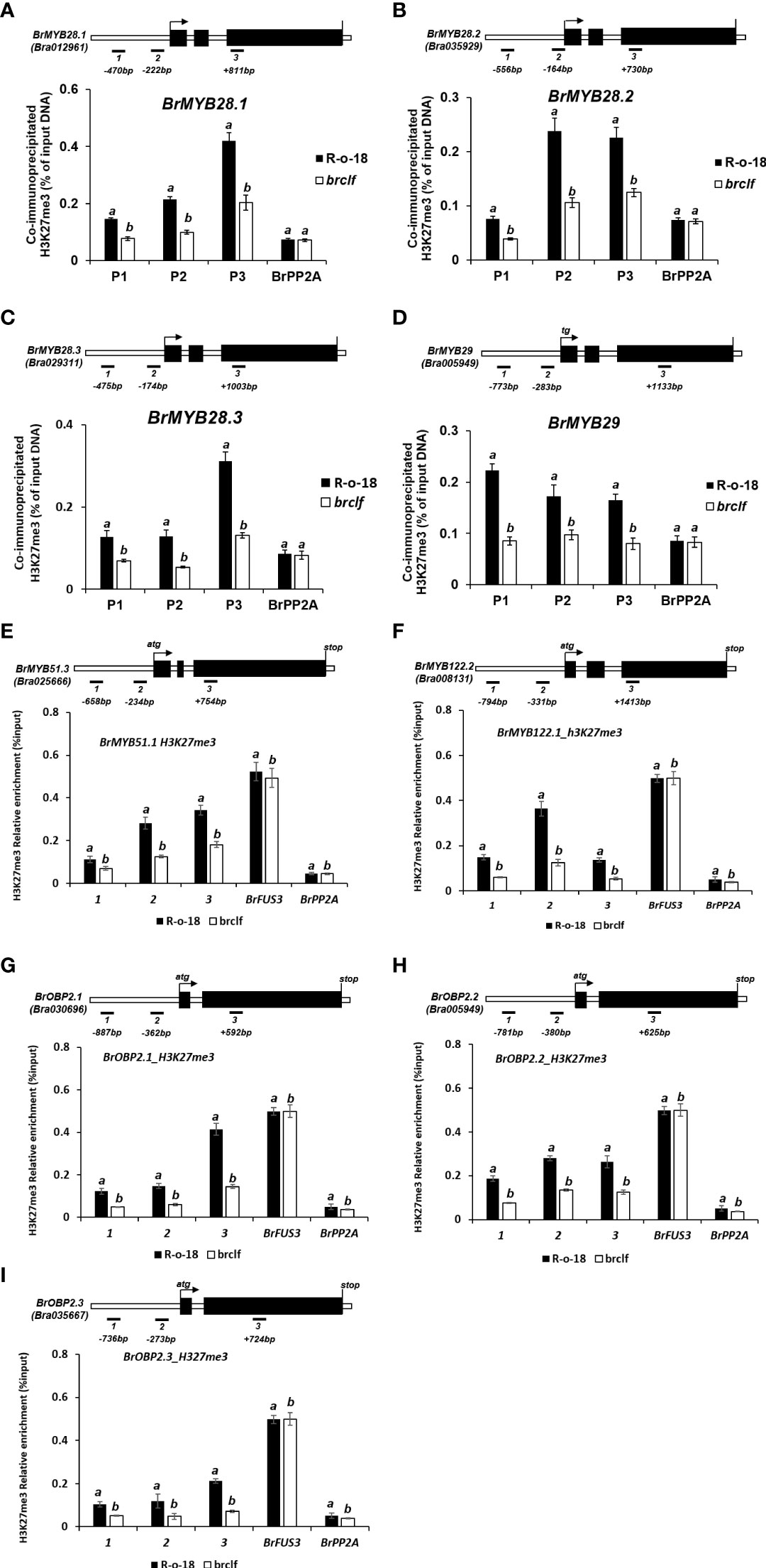
Figure 10 ChIP-qPCR analysis of 9 GSL TFs genes between R-o-18 and brclf mutant. The enrichment level of H3K27me3 in TF genes involved in the regulation of aliphatic GSL [BrMYB28.1 (A), BrMYB28.2 (B), BrMYB28.3 (C), and BrMYB29 (D)] and Indolic GSL [BrMYB51.3 (E), BrMYB122.2 (F), BrOBP2.1 (G), BrOBP2.2 (H), BrOBP2.3 (I)]. All tested genes exhibited substantially reduced levels of H3K27me3 in brclf mutant compared with R-o-18 plant. Enrichment level was calculated as % of input DNA. BrPP2A (Bra012474) was used as a negative control (H3K27me3-unmarked gene) which was used as a housekeeping reference gene (Kim et al., 2022; Supplementary Figure S13). BrFUS3 (Bra032953) was used as a positive control, H3K27me3-enriched gene (Supplementary Figure S13). Mean and standard deviation (SD) of three biological replicates were calculated and presented. Statistical analysis was performed with the one-way analysis of variance (ANOVA) and post-hoc Tukey’s test (p < 0.05).
We also checked the presence of H3K27me3 in 59 aliphatic and 30 indolic glucosinolates biosynthesis genes. Of the total 59 aliphatic pathway genes, 32 genes (54%) were enriched with H3K27me3 in their genomic regions in either leaf or flower tissue (Supplementary Figure S11; Table S11). In case of 30 indolic GSL pathway genes, 18 (60%) genes were accumulated with H3K27me3 in their genomic regions in either leaf or flower tissue (Supplementary Figure S12; Table S11). These results indicated that many TF genes and biosynthetic genes (to a lesser degree) related to aliphatic and indolic GSLs were under the epigenetic control mediated by BrCLF-containing PRC2 complex in B. rapa. Thus, our study combining RNA-seq, H3K27me3 ChIP-qPCR dataset, and metabolic analysis between R-o-18 and brclf demonstrated that BrCLF-containing PRC2 complex modulate expression of key developmental genes (i.e., floral transition, seed dormancy, floral organ development) and genes related to stress signaling and stress-responsive secondary metabolisms (aliphatic and indolic glucosinolates) in B. rapa plants.
Discussion
Plants are constantly exposed to biotic (e.g., insect, fungi, and bacterial attacks) and abiotic stresses (e.g., drought, heat, salinity) during their lifetime. As sessile organisms with limited resources, they need to execute growth-defense tradeoffs, which are well-balanced optimization between growth and defense programs (Huot et al., 2014). Both programs are costly and critical for their survival, adaptation, and successful reproduction. Upon challenging stress, plants rapidly respond and use a defense mode by initiating a complex network of signaling cascades, which trigger genome-wide transcriptomic changes to build a multi-layered defense system against threatening stressors. However, molecular mechanisms underlying growth and defense tradeoffs remain poorly elucidated. In this study, we found that genes involved in crucial developmental transitions were highly enriched with an H3K27me3 histone mark, highlighting the importance of epigenetic control in developmental programs in B. rapa (Figure 9; Supplementary Figure S8). In addition, we revealed that high proportions of stress signaling genes and glucosinolate (aliphatic and indolic) biosynthesis genes were densely enriched with H3K27me3, indicating that these stress responsive genes were suppressed by a BrCLF-containing PRC2 complex in B. rapa (Figure 10; Supplementary Figures S9–S12). It is worthy to note that recent transcriptomic analysis of Arabidopsis found that CLF regulate the genes involved in the aliphatic and indolic GSLs pathways including side-chain elongation, core structure formation, and secondary modification steps of Arabidopsis model plant (Tyler et al., 2019). Thus, it is likely that CLF and BrCLF-mediated H3K27me3 might commonly suppress stress-responsive genes in the absence of challenging stress to benefit plant developmental growth and simultaneously save unessential costs on defense programs.
PRCs play an essential role in diverse developmental programs in many eukaryotes (Margueron and Reinberg, 2011). In an Arabidopsis model plant, CLF and SWN, which are PRC2 components, encode H3K27me3 methyltransferases. Thus, CLF/SWN-containing PRC2 catalyzes the trimethylation of H3K27, which is a histone mark for gene inactivation (Kim and Sung, 2014). Numerous genes related to developmental transition processes, such as seed germination, juvenile-to-adult phase transition, leaf and floral organ development, and floral transition, are enriched with H3K27me3 in Arabidopsis (Bouyer et al., 2011). In addition to the role of PRCs in developmental gene regulation, the functional role of PRCs in the regulation of stress-related pathway genes in Arabidopsis model plant has been described. For instance, a recent study reported that Arabidopsis CLF, a PRC2 component, is positively required for the expression of ORA59, a stress-responsive AP2/ERF transcription factor. The reduced ORA59 expression in a clf mutant results in at least a partial defect in leaf immunity because of the low expression of defense-related genes such as PDF1.2a (Kosaka et al., 2020; Singkaravanit-Ogawa et al., 2021). On the contrary, in our study using B. rapa, ORA59 was upregulated in the brclf mutant compared with that in the R-o-18 wild type; furthermore, H3K27me3 was densely enriched in the ORA59 chromatin region (Figure 4B; Supplementary Figure S9). These data indicated that BrCLF played a negative role in the ORA59 expression by depositing H3K27me3 in B. rapa. Therefore, even though Arabidopsis and B. rapa belong to Brassicaceae, B. rapa homologs probably evolved to acquire a different and divergent role from Arabidopsis homologs.
LHP1/TFL2 is a PRC1 component that physically recognizes and binds to H3K27me3; then, it subsequently carries other PRC1 components to stably silence target genes (Turck et al., 2007; Veluchamy et al., 2016). LHP1 is required for the repression of stress signaling genes, such as MYC2 and NAC domain protein, ANAC55 (NAC DOMAIN CONTAINING PROTEIN 55), which are downstream target genes of ORA59 and involved in the jasmonate (JA) and ethylene (ET) signaling pathway of immunity in Arabidopsis (Ramirez-Prado et al., 2019). H3K27me3 and LHP1 are highly enriched in the MYC2 and ANAC55 region, indicating that LHP1-containing PRC1 suppresses this defense signaling factors in Arabidopsis. Consequently, MYC2 and ANAC55 are upregulated in the loss-of-function LHP1 because of the reduced enrichment of the H3K27me3 mark. In the lhp1 mutant, aphid resistance, drought stress tolerance, and abscisic acid (ABA) sensitivity are increased. In this study, we identified one B. rapa MYC2 homolog (named BrMYC2) and two B. rapa ANAC55 homologs (named BrANAC55.1 and BrANAC55.2) from the B. rapa genome (Supplementary Table S7). These homologs were upregulated in brclf mutant compared with R-o-18 (Figure 4). Regarding the enrichment of H3K27me3, BrANAC55.2 (Bra021113) was evidently enriched with H3K27me3, indicating that it is the direct target of BrCLF-containing PRC2 complex in B. rapa. Meanwhile, BrMYC2 (Bra010178) and BrANAC55.1 (Bra027238) were not enriched with H3K27me3 indicating that they might be not the direct target of PRC2. Nevertheless, their expressions were higher in the brclf mutant than R-o-18, possibly because of the upregulated upstream transcription factors like BrORA59. It need further investigation that BrORA59 is direct positive regulator of BrMYC2 and BrANAC55s mediated defense signaling pathway in B. rapa.
In Arabidopsis, MYC2 activates glucosinolate biosynthesis in signaling mediated by a defense hormone, namely, jasmonic acid (JA) (Pangesti et al., 2016). B. rapa has only a single homolog of MYC2 in B. rapa, (BrMYC2). This study showed that BrMYC2 is not enriched with H3K27me3, indicating that BrMYC2 might not be the direct target of PRC2 (Supplementary Table S10). Nonetheless, the BrMYC2 expression was higher in brclf than in R-o-18 (Figure 4; Supplementary Table S7). Therefore, the upstream regulators of BrMYC2 in the brclf mutant were influenced, affecting BrMYC2 expression. Alternatively, JA-related biosynthesis genes were upregulated in BrCLF mutation; as a result, the increased amount of endogenous JA enhanced the BrMYC2 expression in the brclf mutant.
In Arabidopsis, CLF and its closest paralog, SWN, functioned as H3K27me3 methyltransferase in genes related to many facets of developmental programs (Supplementary Figure S1B). The null mutant of CLF exhibited serious developmental defects, such as small and curly leaves, deformed floral organs, and early flowering, indicating that CLF are implicated in Arabidopsis developmental programs. Meanwhile, swn, loss-of-function SWN, does not exhibit an abnormal phenotype compared with wild-type plants (Shu et al., 2020). Double mutants of CLF and SWN display more severe developmental defects than the single mutant of CLF does, indicating that CLF and SWN play at least partly redundant roles in developmental programs in Arabidopsis. We noticed that the percentage of upregulated genes in brclf (35 genes/104 total, 34%) was lower than that of the GSL pathway genes enriched with H3K27me3 (64 genes/104 total, 62%) (Supplementary Tables S8, S11). Regarding this discrepancy, we reasoned that some genes not affected in brclf might be redundantly suppressed by BrSWN; thus, they were enriched with H3K27me3, but their expression was not significantly affected. Therefore, the development of a BrSWN mutant might further clarify the functional redundancy between BrCLF and BrSWN in B. rapa.
MEA, another paralog of CLF and SWN, also encodes an H3K27me3 methyltransferase and is uniquely expressed in the early embryo of Arabidopsis (Arnaud and Feil, 2006). It serves as a catalytic unit of PRC2 to negatively regulate seed development in the absence of fertilization (Haun et al., 2007). Even though MEA acts as an H3K27me3 transferase in genomic imprinting for proper embryo development, it likely plays another role in plant immune system (Roy et al., 2018). Its transcription is triggered when plants are inoculated with pathogens such as Pseudomonas syringae pv. Tomato (Pst); this process also has a positive role in Pst-AvrRpt2 carrier-mediated pathogenesis (Roy et al., 2018). An induced MEA acts to suppress the plant defense gene RESISTANCE TO P. SYRINGAE 2 (RPS2), thus weakening plant immunity against pathogen attack. Therefore, the functional role of MEA and CLF in stress response might be divergently evolved in Arabidopsis although they have a similar repressive role in developmental genes by depositing H3K27me3. In this study, B. rapa CLF homolog was determined, and the importance of BrCLF in the transcriptional regulation of genes related to development and stress-responsive genes (i.e., glucosinolates); however, further studies on other BrCLF paralogs, BrSWN, BrMEA.a, and BrMEA.b should be conducted to clarify functional similarity and diversity in response to different types of abiotic and biotic stresses in B. rapa.
Gene activation and repression are highly correlated with the contents of histone modifications in an individual genomic region. For example, active histone marks such as H3K4me3, H3K36me3, and H3ac are correlated with gene activation, whereas H3K27me3, H3K9me3, and deacetylated histone H3/H4 are highly related to gene repression (Kouzarides, 2007; Ramirez-Prado et al., 2018). In this context, stress signaling and stress-responsive metabolic genes might undergo dynamic histone modification before and after stress. However, further studies have yet to clarify whether the activation of stress-responsive genes upon exposure to stress requires the removal of accumulated H3K27me3 in their chromatin regions prior to activation; studies have also yet to determine if the H3K27me3 mark is constantly retained and if the activation of these stress-responsive genes can be independently achieved by stress-triggered transcriptional activators (i.e., MYC2). One recent study using Arabidopsis investigated the genome-wide profiles of different histone modifications before and after wounding stress (Rymen et al., 2019). This study suggested that enriched profiles of H3K27me3 were not considerably affected; instead, histone acetylation or active marks evidently accumulated in stress-related genes. To our best knowledge, however, studies have yet to determine whether stress signaling and stress-responsive metabolic genes in B. rapa undergoes dynamic histone modifications upon exposure to stress, similar to the case of Arabidopsis. Therefore, further genome-wide epigenetic profiling of different (active and repressive) histone modifications might provide further insights into the epigenetic regulation of stress response in B. rapa.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, GSE214271.
Author contributions
D-HK planned this study. AN and SK prepared all materials and performed genetic and molecular experiments. AN, SK, and D-HK performed bioinformatic analysis. AN and SK performed HPLC analysis. and D-HK wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the National Research Foundation (NRF) grant (No. 2021R1A5A1047822), a grant from the BioGreen 21 Agri-Tech Innovation Program of the Rural Development Administration, Republic of Korea (No. PJ01566203), and a grant from the New Breeding Technologies Development Program (No. PJ01652702) of the Rural Development Administration, Republic of Korea to D-HK.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1079218/full#supplementary-material
Supplementary Figure 1 | Identification of loss of function mutant of BrCLF, brclf. (A) Multiple alignment of amino acid sequences of Arabidopsis CLF (AtCLF) clade and B. rapa homologous genes, (named BrCLF, BrSWN, BrMEA.a, and BrMEA.b). Catalytic SET domain for H3K27 (H3K27me3) trimethylation was indicated with a green box. (B) Phylogenic analysis of Arabidopsis CLF (AtCLF) clade and B. rapa homologous proteins (BrCLF, BrSWN, BrMEA.a, and BrMEA.b). AtCLF has a single B. rapa homolog, BrCLF (Bra032169, indicated with red letters). (C) Sanger sequencing validation of point mutation in 11th exon of BrCLF coding sequence in brclf mutant. Nucleotide at 1,843bp from the start codon of BrCLF was converted from C to T which generate a premature stop codon in brclf mutant. (D) Comparison of the full amino acid sequence of BrCLF of R-o-18 and brclf mutant. Glutamine (Gln) residue at 615th was converted to a stop codon (indicated with asterisk), generating the truncated form of BrCLF missing the H3K27me3 catalytic SET domain in the C-terminal region.
Supplementary Figure 2 | Measurement of phenotypic differences between R-o-18 wild type and brclf mutant at (A) vegetative and (B) reproductive stages. (A) Measurement of hypocotyl length, leaf length, leaf width, and rosette diameter at 4-week-old seedling R-o-18 and brclf. (B) Measurement of plant height, petal length, number of siliques per plant, bud length, pistil length and number of seeds per a pod between R-o-18 and brclf. (A–B) One-way ANOVA with Tukey’s post-hoc test was applied to calculate statistical differences (p < 0.05), and data were expressed as means ± standard deviation (SD). Statistical difference was indicated with different letters above each bar. Phenotypic measurement for vegetative organs including hypocotyl, leaf, rosette, and plant height was analyzed from 8 individual plants (n=8). Meanwhile, the measurement of petal, siliques, bud, pistil, and number of seeds were obtained from 5 individual plants (n=5).
Supplementary Figure 3 | Quality check of RNA-seq reads. (A) Correlation heatmap analysis showing distinct clustering between R-o-18 and brclf samples. Three biological replicates of R-o-18 and brclf were used for RNA-seq library construction. (B) MA plots of differentially expressed genes between R-o-18 and brclf. X-axis indicates the average expression and logCPM (log2 transformed expression value), and the Y-axis indicates the value of log2-transformed fold change. Red dots indicate differentially expressed genes from R-o-18 and brclf samples. Black dots indicate non-DEGs.
Supplementary Figure 4 | qRT-PCR analysis of 12 developmental genes between R-o-18 and brclf mutant. Transcript levels of developmental genes related to (A) seed dormancy, (B) leaf size, (C) floral organ identity, and (D) floral transition were substantially upregulated in brclf mutant compared with those in the wild type R-o-18 plant. Total RNAs extracted from 2-week-old seedlings were used for qRT-PCR analysis. Mean and standard deviation (SD) of three biological replicates were calculated and presented. One-way ANOVA analysis with Tukey post-hoc test was applied to determine statistical significance (p < 0.05).
Supplementary Figure 5 | Venn diagram between the list of B. rapa 5,122 stress-related genes and 1,844 upregulated genes in brclf compared to R-o-18. Total 301 stress-related genes were found in the 1,844-upregulated genes in the brclf mutant. VENNY (ver. 2.1) was used to generate a Venn diagram ().
Supplementary Figure 6 | Quantification of transcript levels of oxidative stress-related genes between R-o-18 and brclf via qRT-PCR. Mean and standard deviation (SD) of three biological replicates were calculated and presented. One-way ANOVA analysis with Tukey’s post-hoc test was applied to determine statistical significance (p < 0.05).
Supplementary Figure 7 | Profiles of glucosinolate (GSL) compounds detected from R-o-18 and brclf seedling plants through HPLC analysis. HPLC analysis detected total 8 GSL compounds (4 aliphatic GSL compounds and 4 indolic GSL compounds) were detected from both (A) R-o-18 and (B) brclf. Principal component analysis (PCA) scores (C) and biplot (D) of GSL distribution between R-o-18 and brclf. Green dots and red triangle denote R-o-18 and brclf, respectively. Principal component analysis (PCA) and biplot for the GSL content of R-o-18 and brclf was performed using Metaboanalyst 5.0 (). PGT: progoitrin, GNA: gluconapoleiferin, GNP: gluconapin, GBN: glucobrassicanapin. GBS: glucobrassicin, 4-MTGB: 4-Methoxyglucobrassicin, 4-HGB: 4-hydroxyglucobrassicin, NGB: neoglucobrassicin. Three biological replicates for R-o-18 and brclf were used for HPLC analysis.
Supplementary Figure 8 | Integrative genome browser (IGV) illustration showing enrichment of H327me3 on 20 developmental transitions in leaf and flower tissues. Normalized enrichment of H3K27me3 on 20 developmental genes related to (A) seed dormancy, (B) floral organ development, (C) leaf development, and (D) floral transition in B. rapa from leaf and flower tissues compared with the normalized input signal levels. All the 20 tested developmental genes were enriched with H3K27me3. Black tracks indicate the normalized input DNA intensity; green and orange tracks refer to the normalized α-H3K27me3-immunoprecipitated DNA intensity of leaf and flower tissues, respectively. ChIP-seq read intensity in the Y-axis is indicated with a bracket on the left upper part of each track.
Supplementary Figure 9 | Integrative genome browser (IGV) illustration showing enrichment of H327me3 on stress signaling related genes in leaf and flower tissues. Normalized enrichment of H3K27me3 histone mark on 13 oxidative stress signaling genes from leaf and flower tissues compared with normalized input signal levels. Of the 13 stress signaling genes, 10 (77%) were enriched with H3K27me3. Black tracks indicate normalized input DNA intensity; green and orange tracks indicate the normalized α-H3K27me3-immunoprecipitated DNA intensity of leaf and flower tissues, respectively. ChIP-seq read intensity in the Y-axis is indicated with a bracket on the left upper part of each track.
Supplementary Figure 10 | IGV illustration showing H327me3 enrichment on transcription factor genes controlling aliphatic and indolic GSL biosynthesis in leaf and flower tissues. (A) Normalized enrichment of H3K27me3 histone mark on 4 BrMYB TF genes controlling aliphatic GSL biosynthesis from both leaf and flower tissues compared with levels of the normalized input signal. (B) Normalized enrichment of H3K27me3 histone mark on 11 TF genes controlling indolic GSL biosynthesis from both leaf and flower tissues compared with levels of the normalized input signal. (A–B) Black color tracks indicate normalized input DNA intensity, green and orange color tracks indicate the normalized α-H3K27me3-immunoprecipitated DNA intensity of leaf and flower tissues, respectively. ChIP-seq read intensity of Y-axis is indicated with bracket on left upper part of each track.
Supplementary Figure 11 | Integrative genome browser (IGV) illustration showing H327me3 enrichment in 59 aliphatic glucosinolate (GSL) biosynthesis genes in leaf and flower tissues. Normalized enrichment of H3K27me3 histone mark in genes related to the aliphatic GSL biosynthesis from leaf and flower tissues compared with levels of the normalized input signal. (A) Enrichment of H3K27me3 in 19 genes related to side-chain elongation process of aliphatic GSLs pathway. (B) Enrichment of H3K27me3 in 17 genes related to core structure formation process of aliphatic GSLs pathway. (C) Enrichment of H3K27me3 in 16 genes related to secondary modification process of aliphatic GSL pathway. (A–C) Black tracks indicate normalized input DNA intensity; green and orange tracks indicate the normalized α-H3K27me3-immunoprecipitated DNA intensity of leaf and flower tissues, respectively. ChIP-seq read intensity of Y-axis is indicated with a bracket on the left upper part of each track.
Supplementary Figure 12 | Integrative genome browser (IGV) illustration showing H327me3 enrichment in 40 indolic glucosinolate (GSL) biosynthesis genes in leaf and flower tissues. Normalized enrichment of H3K27me3 histone mark in genes related to indolic GSL biosynthesis from both leaf and flower tissues compared with levels of the normalized input signal. (A) Enrichment of H3K27me3 in 10 genes related to core structure formation process of indolic GSLs pathway. (B) Enrichment of H3K27me3 in 12 genes related to secondary modification process of indolic GSLs pathway. (C) Enrichment of H3K27me3 on 7 genes commonly involved in both aliphatic and indolic GSL pathways. (A–C) Black tracks indicate normalized input DNA intensity; green and orange tracks indicate the normalized α-H3K27me3-immunoprecipitated DNA intensity of leaf and flower tissues, respectively. ChIP-seq read intensity of Y-axis is indicated with a bracket on the left upper part of each track.
Supplementary Figure 13 | Verification of H3K27me3-unmarked BrPP2A and H3K27me3-marked BrFUS3 in B. rapa. (A) IGV genome browser visualization showing the transcript levels of BrPP2A (Bra012474) between R-o-18 and brclf mutant. Transcription of BrPP2A was not affected in brclf in comparison to level of R-o-18. Three biological replicates from RNA-seq read counts were indicated with black color (R-o-18) and brown color (brclf). (B) IGV genome browser visualization showing the enriched level of H3K27me3 at the genomic locus of BrPP2A (Bra012474) in either leaf or flower tissue in comparison to input DNA. Upper diagram presents a primer location used for ChIP-qPCR analysis. A housekeeping reference gene, BrPP2A was not enriched with H3K27me3. ChIP-seq read intensity of Y-axis is indicated with a bracket on the left upper part of each track. (C) Result of ChIP-qPCR on H3K27me3 enrichment between R-o-18 and brclf mutant at BrFUS3 (Bra032953) genomic locus. Genomic region of BrFUS3 was enriched with H3K27me3 in comparison to input DNA. Upper diagram presents a primer location used for ChIP-qPCR analysis. One-way ANOVA analysis with Tukey’s post-hoc test was applied to determine statistical significance (p < 0.05).
References
Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Arnaud, P., Feil, R. (2006). MEDEA takes control of its own imprinting. Cell 124 (3), 468–470. doi: 10.1016/j.cell.2006.01.020
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bouyer, D., Roudier, F., Heese, M., Andersen, E.D., Gey, D., Nowack, M.K., et al. (2011). Polycomb repressive complex 2 controls the embryo-to-Seedling phase transition. PloS Genet. 7, e1002014. doi: 10.1371/journal.pgen.1002014
Brown, P. D., Tokuhisa, J. G., Reichelt, M., Gershenzon, J. (2003). Variation of glucosinolate accumulation among different organs and developmental stages of Arabidopsis thaliana. Phytochemistry 62, 471–481. doi: 10.1016/S0031-9422(02)00549-6
Calonje, M. (2014). PRC1 marks the difference in plant PcG repression. Mol. Plant 7, 459–471. doi: 10.1093/mp/sst150
Cao, R., Wang, L., Wang, H., Xia, L., Erdjument-Bromage, H., Tempst, P., et al. (2002). Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043. doi: 10.1126/science.1076997
Chanvivattana, Y., Bishopp, A., Schubert, D., Stock, C., Moon, Y. H., Sung, Z. R., et al. (2004). Interaction of polycomb-group proteins controlling flowering in Arabidopsis. Development 131, 5263–5276. doi: 10.1242/dev.01400
Chen, H., Wang, T., He, X., Cai, X., Lin, R., Liang, J., et al. (2022). BRAD V3.0: an upgraded brassicaceae database. Nucleic Acids Res. 50, D1432–D1441. doi: 10.1093/nar/gkab1057
Corpet, F. (1988). Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890. doi: 10.1093/nar/16.22.10881
Ge, S. X., Jung, D., Jung, D., Yao, R. (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. doi: 10.1093/bioinformatics/btz931
Gigolashvili, T., Berger, B., Mock, H. P., Müller, C., Weisshaar, B., Flügge, U. I. (2007). The transcription factor HIG1/MYB51 regulates indolic glucosinolate biosynthesis in Arabidopsis thaliana. Plant J. 50, 886–901. doi: 10.1111/j.1365-313X.2007.03099.x
Gigolashvili, T., Yatusevich, R., Rollwitz, I., Humphry, M., Gershenzon, J., Flügge, U. I. (2009). The plastidic bile acid transporter 5 is required for the biosynthesis of methionine-derived glucosinolates in Arabidopsis thaliana. Plant Cell 21, 1813–1829. doi: 10.1105/tpc.109.066399
Godwin, J., Farrona, S. (2022). The importance of networking: Plant polycomb repressive complex 2 and its interactors. Epigenomes 2022 6, 8. doi: 10.3390/epigenomes6010008
Goodrich, J., Puangsomlee, P., Martin, M., Long, D., Meyerowitz, E. M., Coupland, G. (1997). A polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nat. 386, 44–51. doi: 10.1038/386044a0
Haun, W. J., Laoueillé-Duprat, S., O’Connell, M. J., Spillane, C., Grossniklaus, U., Phillips, A. R., et al. (2007). Genomic imprinting, methylation and molecular evolution of maize enhancer of zeste (Mez) homologs. Plant J. 49, 325–337. doi: 10.1111/j.1365-313X.2006.02965.x
Hirai, M. Y., Sugiyama, K., Sawada, Y., Tohge, T., Obayashi, T., Suzuki, A., et al. (2007). Omics-based identification of Arabidopsis myb transcription factors regulating aliphatic glucosinolate biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 104, 6478–6483. doi: 10.1073/pnas.0611629104
Hiruma, K., Fukunaga, S., Bednarek, P., Piślewska-Bednarek, M., Watanabe, S., Narusaka, Y., et al. (2013). Glutathione and tryptophan metabolism are required for Arabidopsis immunity during the hypersensitive response to hemibiotrophs. Proc. Natl. Acad. Sci. U. S. A. 110, 9589–9594. doi: 10.1073/pnas.1305745110
Hou, X., Zhou, J., Liu, C., Liu, L., Shen, L., Yu, H. (2014). Nuclear factor y-mediated H3K27me3 demethylation of the SOC1 locus orchestrates flowering responses of Arabidopsis. Nat. Commun. 5, 1–14. doi: 10.1038/ncomms5601
Huang, S., Hou, L., Fu, W., Liu, Z., Li, C., Li, X., et al. (2020). An insertion mutation in Bra032169 encoding a histone methyltransferase is responsible for early bolting in Chinese cabbage (Brassica rapa l. ssp. pekinensis). Front. Plant Sci. 11, 547. doi: 10.3389/fpls.2020.00547
Huang, Y., Jiang, L., Liu, B. Y., Tan, C. F., Chen, D. H., Shen, W. H., et al. (2019). Evolution and conservation of polycomb repressive complex 1 core components and putative associated factors in the green lineage. BMC Genomics 20, 1–18. doi: 10.1186/s12864-019-5905-9
Huot, B., Yao, J., Montgomery, B. L., He, S. Y. (2014). Growth–defense tradeoffs in plants: A balancing act to optimize fitness. Mol. Plant 7, 1267–1287. doi: 10.1093/mp/ssu049
Jiang, D., Wang, Y., Wang, Y., He, Y. (2008). Repression of FLOWERING LOCUS c and FLOWERING LOCUS T by the Arabidopsis polycomb repressive complex 2 components. PloS One 3, e3404. doi: 10.1371/journal.pone.0003404
Kim, S., Kim, J. A., Kang, H., Kim, D. H. (2022). A premature stop codon in BrFLC2 transcript results in early flowering in oilseed-type brassica rapa plants. Plant Mol. Biol. 108, 241–255. doi: 10.1007/s11103-021-01231-y
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., Salzberg, S. L. (2013). TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, 1–13. doi: 10.1186/gb-2013-14-4-r36
Kim, D. H., Sung, S. (2014). Polycomb-mediated gene silencing in Arabidopsis thaliana. Mol. Cells 37, 841. doi: 10.14348/molcells.2014.0249
Kliebenstein, D. J. (2004). Secondary metabolites and plant/environment interactions: A view through Arabidopsis thaliana tinged glasses. Plant Cell Environ. 27, 675–684. doi: 10.1111/j.1365-3040.2004.01180.x
Kosaka, A., Suemoto, H., Singkaravanit-Ogawa, S., Takano, Y. (2020). Plant defensin expression triggered by fungal pathogen invasion depends on EDR1 protein kinase and ORA59 transcription factor in Arabidopsis thaliana. Plant Sig. Behav. 15 (12), 1823120. doi: 10.1080/15592324.2020.1823120
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lewis, E. B. (1978). A gene complex controlling segmentation in drosophila. Nat. 276, 565–570. doi: 10.1038/276565a0
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liao, Y., Smyth, G. K., Shi, W. (2014). featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Ludwig-Müller, J., Pieper., K., Ruppel, M., Cohen, J. D., Epstein, E., Kiddle, G., et al. (1999). Indole glucosinolate and auxin biosynthesis in arabidopsis thaliana (L.) heynh. glucosinolate mutants and the development of clubroot disease. Planta. 208 (3), 409–419. doi: 10.1007/s004250050576
Malitsky, S., Blum, E., Less, H., Venger, I., Elbaz, M., Morin, S., et al. (2008). The transcript and metabolite networks affected by the two clades of Arabidopsis glucosinolate biosynthesis regulators. Plant Physiol. 148, 2021–2049. doi: 10.1104/pp.108.124784
Margueron, R., Reinberg, D. (2011). The polycomb complex PRC2 and its mark in life. Nature 469, 343. doi: 10.1038/nature09784
Meraj, T. A., Fu, J., Raza, M. A., Zhu, C., Shen, Q., Xu, D., et al. (2020). Transcriptional factors regulate plant stress responses through mediating secondary metabolism. Genes 11, 346. doi: 10.3390/genes11040346
Nazir, F., Fariduddin, Q., Khan, T. A. (2020). Hydrogen peroxide as a signalling molecule in plants and its crosstalk with other plant growth regulators under heavy metal stress. Chemosphere 252, 126486. doi: 10.1016/j.chemosphere.2020.126486
Nugroho, A. B. D., Han, N. R., Pervitasari, A. N., Kim, D. H., Kim, J. (2019). Differential expression of major genes involved in the biosynthesis of aliphatic glucosinolates in intergeneric baemoochae (Brassicaceae) and its parents during development. Plant Mol. Biol. 102, 171–184. doi: 10.1007/s11103-019-00939-2
Nugroho, A. B. D., Lee, S. W., Pervitasari, A. N., Moon, H., Choi, D., Kim, J., et al. (2021). Transcriptomic and metabolic analyses revealed the modulatory effect of vernalization on glucosinolate metabolism in radish (Raphanus sativus l.). Sci. Rep. 11, 1–15. doi: 10.1038/s41598-021-03557-5
Pangesti, N., Reichelt, M., van de Mortel, J. E., Kapsomenou, E., Gershenzon, J., Loon, J. J. A., et al. (2016). Jasmonic acid and ethylene signaling pathways regulate glucosinolate levels in plants during rhizobacteria-induced systemic resistance against a leaf-chewing herbivore. J. Chem. Ecol. 42, 1212–1225. doi: 10.1007/s10886-016-0787-7
Payá-Milans, M., Poza-Viejo, L., Martín-Uriz, P. S., Lara-Astiaso, D., Wilkinson, M. D., Crevillén, P. (2019). Genome-wide analysis of the H3K27me3 epigenome and transcriptome in brassica rapa. Gigascience 8, 1–13. doi: 10.1093/gigascience/giz147
Ramirez-Prado, J. S., Latrasse, D., Rodriguez-Granados, N. Y., et al. (2019). The polycomb protein LHP1 regulates Arabidopsis thaliana stress responses through the repression of the MYC2-dependent branch of immunity. Plant J. 100, 1118–1131. doi: 10.1111/tpj.14502
Ramirez-Prado, J. S., Piquerez, S. J. M., Bendahmane, A., Hirt, H., Raynaud, C., Benhamed, M. (2018). Modify the histone to win the battle: Chromatin dynamics in plant–pathogen interactions. Front. Plant Sci. 9, 355. doi: 10.3389/fpls.2018.00355
R Core Team (2020). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing).
Robinson, M. D., McCarthy, D. J., Smyth, G. K. (2010). edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Roy, S., Gupta, P., Rajabhoj, M. P., Maruthachalam, R., Nandi, A. K. (2018). The polycomb-group repressor MEDEA attenuates pathogen defense. Plant Physiol. 177, 1728–1742. doi: 10.1104/pp.17.01579
Rusholme, R. L., Higgins, E. E., Walsh, J. A., Lydiate, D. J. (2007). Genetic control of broad-spectrum resistance to turnip mosaic virus in brassica rapa (Chinese cabbage). J. Gen. Virol. 88 (Pt 11), 3177–3186. doi: 10.1099/vir.0.83194-0
Rymen, B., Kawamura, A., Lambolez, A., Inagaki, S., Takebayashi, A., Iwase, A., et al. (2019). Histone acetylation orchestrates wound-induced transcriptional activation and cellular reprogramming in Arabidopsis. Commun. Biol. 2, 1–15. doi: 10.1038/s42003-019-0646-5
Sønderby, I. E., Hansen, B. G., Bjarnholt, N., Ticconi, C., Halkier, B. A., Kliebenstein, D. J. (2007). A systems biology approach identifies a R2R3 MYB gene subfamily with distinct and overlapping functions in regulation of aliphatic glucosinolates. PloS One 2, e1322. doi: 10.1371/journal.pone.0001322
Schubert, D., Primavesi, L., Bishopp, A., Roberts, G., Doonan, J., Jenuwein, T., et al. (2006). Silencing by plant polycomb-group genes requires dispersed trimethylation of histone H3 at lysine 27. EMBO J. 25, 4638–4649. doi: 10.1038/sj.emboj.7601311
Shu, J., Chen, C., Li, C., Cui, Y. (2020). The complexity of PRC2 catalysts CLF and SWN in plants. Biochem. Soc Trans. 48, 2779–2789. doi: 10.1042/BST20200660
Shu, J., Chen, C., Thapa, R. K., Bian, S., Nguyen, V., Yu, K., et al. (2019). Genome-wide occupancy of histone H3K27 methyltransferases CURLY LEAF and SWINGER in Arabidopsis seedlings. Plant Direct 3, e00100. doi: 10.1002/pld3.100
Singkaravanit-Ogawa, S., Kosaka, A., Kitakura, S., Uchida, K., Nishiuchi, T., Ono, E., et al. (2021). Arabidopsis CURLY LEAF functions in leaf immunity against fungal pathogens by concomitantly repressing SEPALLATA3 and activating ORA59. Plant J. 108, 1005–1019. doi: 10.1111/tpj.15488
Sonderby, I. E., Burow, M., Rowe, H. C., Kliebenstein, D. J., Halkier, B. A. (2010). A complex interplay of three R2R3 MYB transcription factors determines the profile of aliphatic glucosinolates in Arabidopsis. Plant Physiol. 153, 348–363. doi: 10.1104/pp.109.149286
Stephenson, P., Baker, D., Girin, T., Perez, A., Amoah, S., King, G.J, et al. A rich TILLING resource for studying gene function in Brassica rapa. BMC Plant Biol 10, (62) (2010). doi: 10.1186/1471-2229-10-62
Tan, C., Ren, J., Wang, L., Ye, X., Fu, W., Zhang, J., et al. (2021). A single amino acid residue substitution in BraA04g017190.3C, a histone methyltransferase, results in premature bolting in Chinese cabbage (Brassica rapa l. ssp. pekinensis). BMC Plant Biol. 21, 1–15. doi: 10.1186/s12870-021-03153-9
Thorvaldsdóttir, H., Robinson, J. T., Mesirov, J. P. (2013). Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Turck, F., Roudier, F., Farrona, S., Martin-Magniette, M.-L., Guillaume El Buisine, N., et al. (2007). Arabidopsis TFL2/LHP1 specifically associates with genes marked by trimethylation of histone H3 lysine 27. PloS Genet. 3, e86. doi: 10.1371/journal.pgen.0030086
Tyler, L., Miller, M. J., Fletcher, J. C. (2019). The trithorax group factor ULTRAPETALA1 regulates developmental as well as biotic and abiotic stress response genes in arabidopsis. G3 (Bethesda) 9 (12), 4029–4043. doi: 10.1534/g3.119.400559
Veluchamy, A., Jégu, T., Ariel, F., Latrasse, D., Mariappan, K.G., Kim, S.-K., et al. (2016). LHP1 regulates H3K27me3 spreading and shapes the three-dimensional conformation of the Arabidopsis genome. PloS One 11, e0158936. doi: 10.1371/journal.pone.0158936
Xiao, J., Wagner, D. (2015). Polycomb repression in the regulation of growth and development in Arabidopsis. curr. Opin. Plant Biol. 23, 15–24. doi: 10.1016/j.pbi.2014.10.003
Keywords: polycomb, H3K27me3, curly leaf, stress response, glucosinolates, Brassica rapa
Citation: Nugroho ABD, Kim S, Lee SW and Kim D-H (2023) Transcriptomic and epigenomic analyses revealed that polycomb repressive complex 2 regulates not only developmental but also stress responsive metabolism in Brassica rapa. Front. Plant Sci. 14:1079218. doi: 10.3389/fpls.2023.1079218
Received: 25 October 2022; Accepted: 06 February 2023;
Published: 20 February 2023.
Edited by:
Cristel C. Carles, Université Grenoble Alpes, FranceReviewed by:
Clara Richet-bourbousse, INSERM U1024 Institut de biologie de l’Ecole Normale Supérieure, FranceAikaterini Symeonidi, Helmholtz Association of German Research Centres (HZ), Germany
Copyright © 2023 Nugroho, Kim, Lee and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong-Hwan Kim, dhkim92@cau.ac.kr