
 Zhifei Wang
Zhifei Wang
- Molecular Neurobiology Section, National Institute of Mental Health, National Institutes of Health, Bethesda, MD, USA
The mood stabilizer lithium inhibits glycogen synthase kinase-3 (GSK-3) directly or indirectly by enhancing serine phosphorylation of both α and β isoforms. Lithium robustly protected primary brain neurons from glutamate-induced excitotoxicity; these actions were mimicked by other GSK-3 inhibitors or silencing/inhibiting GSK-3α and/or β isoforms. Lithium rapidly activated Akt to enhance GSK-3 serine phosphorylation and to block glutamate-induced Akt inactivation. Lithium also up-regulated Bcl-2 and suppressed glutamate-induced p53 and Bax. Induction of brain-derived neurotrophic factor (BDNF) was required for lithium’s neuroprotection to occur. BDNF promoter IV was activated by GSK-3 inhibition using lithium or other drugs, or through gene silencing/inactivation of either isoform. Further, lithium’s neuroprotective effects were associated with inhibition of NMDA receptor-mediated calcium influx and down-stream signaling. In rodent ischemic models, post-insult treatment with lithium decreased infarct volume, ameliorated neurological deficits, and improved functional recovery. Up-regulation of heat-shock protein 70 and Bcl-2 as well as down-regulation of p53 likely contributed to lithium’s protective effects. Delayed treatment with lithium improved functional MRI responses, which was accompanied by enhanced angiogenesis. Two GSK-3-regulated pro-angiogenic factors, matrix metalloproteinase-9 (MMP-9) and vascular endothelial growth factor were induced by lithium. Finally, lithium promoted migration of mesenchymal stem cells (MSCs) by up-regulation of MMP-9 through GSK-3β inhibition. Notably, transplantation of lithium-primed MSCs into ischemic rats enhanced MSC migration to the injured brain regions and improved the neurological performance. Several other GSK-3 inhibitors have also been reported to be beneficial in rodent ischemic models. Together, GSK-3 inhibition is a rational strategy to combat ischemic stroke and other excitotoxicity-related brain disorders.
Introduction
Glycogen synthase kinase-3 (GSK-3), an evolutionarily conserved ubiquitous serine–threonine kinase consisting of α and β isoforms, is a multifaceted protein with diverse cellular and neurophysiological functions. The main structural difference between GSK-3α and GSK-3β isoforms lies in the N- and C-terminal regions, while their sequences within the kinase domain are highly homologous. GSK-3 is considered to be constitutively active under non-stimulated basal conditions. A growing body of evidence indicates that GSK-3 is pro-apoptotic and that its dysfunction may be linked to the pathophysiology of mood disorders, schizophrenia, diabetes, and various neurological/neurodegenerative diseases, among others (for review, Meijer et al., 2004; Huang and Klein, 2006; Jope et al., 2007; Chiu and Chuang, 2010; Li and Jope, 2010). GSK-3 inhibition has attracted widespread attention as one of the critical therapeutic targets whereby lithium exerts its effects on mood stabilization, neurogenesis, neurotrophicity, neuroprotection, anti-inflammation, and others (for review, Rowe and Chuang, 2004; Rowe et al., 2007; Beurel et al., 2010). Pharmacological inhibition or gene knockout/knockdown of this kinase mimics the anti-depressant and anti-manic effects of lithium observed in rodent models (Gould et al., 2004; Kaidanovich-Beilin et al., 2004, 2009; O’Brien et al., 2004; Rosa et al., 2008; Omata et al., 2011). The activities of GSK-3 are negatively regulated by phosphorylation of GSK-3α at Ser21 and GSK-3β at Ser9. GSK-3 can be inhibited by lithium through direct binding to the ATP-dependent magnesium-sensitive catalytic site of the enzyme (Klein and Melton, 1996; Stambolic et al., 1996), and/or indirectly through enhanced serine phosphorylation of GSK-3 isoforms by multiple mechanisms (Figure 1). Lithium has been shown to enhance GSK-3 serine phosphorylation by activation of protein kinase A (PKA; Jope, 1999; Liang et al., 2008), or phosphatidylinositol 3-kinase (PI3-kinase)-dependent Akt (Chalecka-Franaszek and Chuang, 1999) and protein kinase C-α (Kirshenboim et al., 2004). It has also been reported that lithium can disrupt the β-arrestin-2–PP2A–Akt complex that dephosphorylates/inactivates Akt, thereby enhancing GSK-3 serine phosphorylation (Beaulieu et al., 2005). Moreover, it has been proposed that lithium can interrupt auto-regulation of GSK-3 via disinhibition of the inhibitory action of inhibitor-2 complex on protein phosphatase-1 (PP-1; Zhang et al., 2003). This article reviews the findings supporting the role of GSK-3 inhibition in mediating lithium’s neuroprotective effects against excitotoxicity in both cultured neurons and animal models of ischemic stroke. Potential down-stream mechanisms underlying lithium’s neuroprotection against excitotoxicity are also discussed.
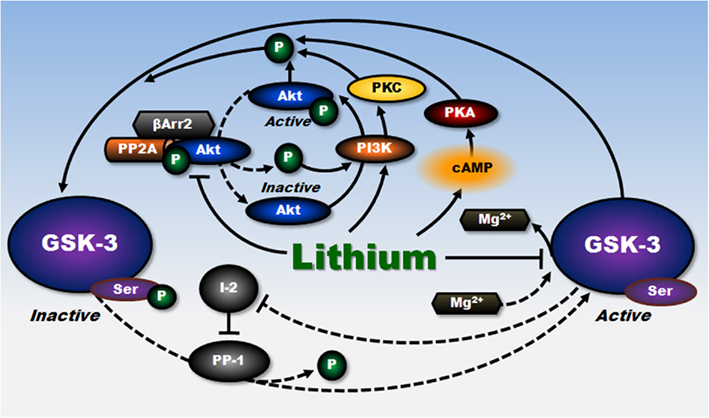
Figure 1. Inhibitory regulation of GSK-3 by lithium. Lithium negatively regulates the constitutively activated GSK-3 activity through multiple mechanisms. Lithium, a competitive inhibitor of magnesium, directly inhibits ATP–magnesium-dependent catalytic activity of GSK-3. The activity of GSK-3 is also reduced by phosphorylation at a specific serine residue. Lithium can indirectly increase this serine phosphorylation of GSK-3 through PI3-kinase-mediated phosphorylation/activation of Akt, PI3-kinase-mediated activation of PKC, and cAMP-dependent activation of PKA. Lithium can also increase the serine phosphorylation of GSK-3 by disrupting the β-arrestin-2 (βArr2)–PP2A–Akt complex that dephosphorylates and inactivates Akt. In addition, by disinhibiting the inhibitory action of inhibitor-2 (I-2) on protein phosphatase-l (PP-1) that dephosphorylates GSK-3 at serine residues, lithium’s direct inhibition of GSK-3 interrupts this auto-regulation of GSK-3 and further decreases GSK-3 activity. Lines with solid arrows represent stimulatory connections; lines with flattened ends represent inhibitory connections. Dashed lines represent pathways with reduced activity as a result of lithium treatment. I-2, inhibitor-2; PP-1, protein phosphatase-l. (Modified from Chiu and Chuang, 2010).
Regulation and Function of GSK-3 Isoforms
We have designed isoform-specific small interfering RNAs (siRNAs) to distinguish the functional and regulatory differences between the two GSK-3 isoforms in rat cerebral cortical neuronal cultures (Liang and Chuang, 2007). Transfection with siRNA for GSK-3α or GSK-3β or with dominant-negative mutants specific for either isoform produced almost complete protection against glutamate-induced, N-methyl-D-aspartate (NMDA) receptor-mediated excitotoxicity. The siRNA-induced neuroprotection was associated with enhanced N-terminal phosphorylation in both GSK-3 isoforms. Moreover, transfection with α or β isoform-specific dominant-negative mutants of GSK-3 mimicked lithium-induced neuroprotection against glutamate excitotoxicity. These results strongly suggest that both GSK-3α and β are involved in glutamate-induced neuronal death and that both isoforms are the initial targets of lithium-elicited neuroprotection.
GSK-3 has also been implicated in neuronal development, maturation/differentiation, and aging in the mammalian CNS (Spittaels et al., 2002; Kim et al., 2009; Sofola et al., 2010). Substrates phosphorylated by GSK-3 include metabolic, signaling, and structural proteins as well as transcription factors. It is known that inhibition of GSK-3 results in activation, and sometimes suppression, of an array of transcription factors (for review, Grimes and Jope, 2001; Jope and Roh, 2006; Chiu and Chuang, 2010). Among the long list of transcription factors regulated by GSK-3 are cyclic AMP response element binding protein (CREB), nuclear factor-κB (NF-κB), activating protein-1 (AP-1), heat-shock factor-1 (HSF-1), β-catenin, T-cell factor (Tcf)/lymphoid enhancer factor (Lef), and p53. Dysfunction of GSK-3-mediated phosphorylation of transcription factors is believed to relate with the pathophysiology of various pathological conditions (for review, Chiu and Chuang, 2010). We found that GSK-3α silencing activated cAMP response element (CRE)- and NF-κB-responsive transcription more robustly than GSK-3β silencing (Liang and Chuang, 2006). Our protein–DNA array further identified two novel GSK-3-regulated transcription factors, early growth response-1 (EGR-1) and Smad3/4, both of which play important roles in growth, differentiation, survival, and plasticity of brain cells (Harada et al., 2001; Derynck and Zhang, 2003; Lee and Kim, 2004; Droguett et al., 2010). Specifically, the binding activity of EGR-1 was down-regulated by siRNA for GSK-3α, but was up-regulated by siRNA for GSK-3β (Liang and Chuang, 2006). By using siRNAs or dominant-negative mutants specific to GSK-3 isoforms, inhibition of GSK-3α increased the transcriptional activity of Smad3/4, whereas inhibition of GSK-3β reduced it. The differential roles of GSK-3 isoforms are further supported by the opposite effects of GSK-3α and β siRNAs on the protein levels of plasminogen activator inhibitor type-1 (PAI-1), a Smad3/4-regulated gene product. These results demonstrate that selective silencing or inhibition of the two GSK-3 isoforms could produce different and sometimes opposite effects on the regulation of certain transcription factors including novel GSK-3 targets (Liang and Chuang, 2006).
Differential roles of GSK-3α and β have also been suggested by other investigators. For example, the disruption of GSK-3β in mice is embryonic lethal, despite the normal expression of GSK-3α, indicating that the presence of α isoform can not compensate for the loss of β isoform (Hoeflich et al., 2000). Transfection and siRNA studies suggested that GSK-3α inhibition decreased the processing of β-amyloid (Aβ) precursor protein to form Aβ1–40 and Aβ1–42, while GSK-3β appeared to have a lesser role (Phiel et al., 2003). In addition, GSK-3β, but not GSK-3α, is required for interferon-γ-induced activation of signal transducer and activator of transcription-3 (Beurel and Jope, 2009). Together, these findings underscore important similarities and differences between the roles of GSK-3 isoforms α and β in cell survival as well as transcription, and suggest that the development of isoform-specific inhibitors may be essential for therapeutic intervention of GSK-3-related neuropathological conditions.
We have also explored lithium’s effects on Smad3/4-dependent transcriptional activity and the underlying mechanisms. Smad3/4 is a down-stream mediator of the signaling pathway triggered by transforming growth factor-β (TGF-β), and plays a prominent role in regulating the expression of proteins involved in neuronal survival, differentiation, and synaptic plasticity (for review, Gomes et al., 2005). Treating cultured cortical neurons with therapeutically relevant concentrations of lithium significantly decreased Smad3/4-dependent transactivation and protein levels of PAI-1, a TGF-β-responsive Smad3/4-dependent gene product (Liang et al., 2008). Of particular relevance to the therapeutic efficacy of lithium, PAI-1 has been implicated in the etiology and progression of neurodegenerative diseases and mood disorders (for review, Pawlak et al., 2003; Liang et al., 2008).
Lithium’s effects on Smad3/4 likely result from cross-talk of signaling pathways between cAMP/PKA and PI3-kinase/Akt/GSK-3β. We have shown that lithium-induced Smad3/4 suppression involved GSK-3β inhibition through the activation of PKA and cell survival factor Akt followed by the phosphorylation of GSK-3β at Ser9 and CREB at Ser133 (Liang et al., 2008). CREB binding protein (CBP) and p300 are known to be co-activators of CREB. Our data further demonstrated that over-expression of p300, but not CBP, completely antagonized lithium-induced reduction of PAI-1 promoter activity. A series of experimental data support the notion that, in Smad3/4 signaling, the inhibitory effects of lithium are due to complex formation of activated CREB and p300, which results in limited interactions of p300 with the transcription factors/Smad complexes. This in turn prevents efficient Smad3/4-dependent transcription of Smad3/4-dependent genes such as PAI-1 and p21 (Figure 2).
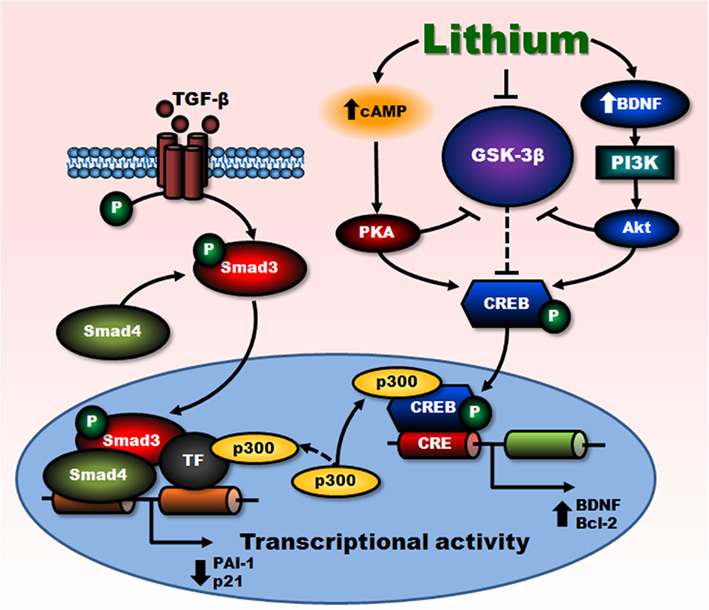
Figure 2. Negative regulation of Smad3/4-dependent transcription by lithium. Transcriptional activations triggered by stimulation of cell surface TGF-β and BDNF receptors are mediated by Smad3/4- and PI3-kinase/Akt-dependent pathways, respectively. Lithium treatment-induced inhibition of GSK-3β, directly and indirectly via cAMP-dependent activation of PKA as well as BDNF-stimulated activation of PI3-kinase/Akt pathways, potentiates BDNF-induced phosphorylation/activation of CREB. This in turn increases CRE-mediated transactivation and expression of survival factors such as BDNF and Bcl-2. Enhanced gene transcription triggered by BDNF, via sequestration of transcriptional co-activator p300, suppresses Smad3/4-dependent transactivation and subsequently decreases the expression of TGF-β-responsive genes, PAI-1, and p21. Lines with solid arrows represent stimulatory connections; lines with flattened ends represent inhibitory connections. Dashed lines represent pathways with reduced activity as a result of lithium treatment. CRE, cAMP response element. (Modified from Liang et al., 2008).
Involvement of GSK-3 and Other Molecules in Lithium-Elicited Neuroprotection Against Glutamate-Induced Excitotoxicity in Cellular Models
Lithium-induced neuroprotection against glutamate excitotoxicity was first noted in rodent primary neuronal cultures of cerebellar granule cells (CGCs), cerebral cortical neurons, and hippocampal neurons (Nonaka et al., 1998). This experimental paradigm was selected because glutamate-related excitotoxicity has been implicated in many neurodegenerative diseases including stroke (for review, Chuang, 2004; Chiu and Chuang, 2010). Our pioneering studies have shown that glutamate-induced, NMDA receptor-mediated excitotoxicity was robustly reduced by extended lithium chloride pretreatment (5–7 days) in cultured rat CGCs and cortical neurons, partly via inhibition of NMDA receptor-mediated calcium influx (Nonaka et al., 1998; Hashimoto et al., 2002a). Moreover, these effects of lithium were likely due to the attenuation of constitutive phosphorylation at Tyr1472 of the NR2B subunit of NMDA receptors, possibly as a result of inhibiting Src tyrosine kinase (Hashimoto et al., 2002a, 2003). Although glutamate-induced excitotoxicity in cultured cortical neurons was blocked by treatment with either lithium or MK-801 (an NMDA receptor antagonist), the Src kinase inhibitor SU6656 only partially diminished this toxicity (Hashimoto et al., 2003), suggesting that other components are involved. In CGCs, lithium-induced neuroprotection against glutamate excitotoxicity was associated with up-regulation of the anti-apoptotic protein Bcl-2, down-regulation of the pro-apoptotic proteins p53 and Bax, and suppressed release of cytochrome c from mitochondria (Chen and Chuang, 1999), whereas the involvement of GSK-3 in the regulation of NMDA signaling by lithium treatment is currently unclear and requires further investigations.
Cyclin-dependent kinase 5 (Cdk5) also regulates signaling mediated by NMDA receptors, either directly through phosphorylation of the NR2B subunit or indirectly through phosphorylation of PSD-95 (Morabito et al., 2004; Zhang et al., 2008). Cdk5 activity is primarily regulated by its co-activator p35. However, when it binds to p25 (the product of calpain-mediated cleavage of p35), Cdk5 becomes pro-apoptotic and its activity is dysregulated (Lee et al., 2000; Carmins et al., 2006). Accordingly, p25 accumulation was observed in neurons in response to glutamate or oxidative stress, and also in the brains of several animal models of neurodegenerative diseases. Sustained activation of Cdk5 in neurons has been implicated in many neurodegenerative diseases (Cruz and Tsai, 2004; Dhariwala and Rajadhyaksha, 2008). In cultured rat CGCs, lithium pretreatment prevented colchicine-induced apoptosis and associated increase in Cdk5 expression and fragmentation of p35 into p25 (Jorda et al., 2005). Additionally, pretreatment with lithium also attenuated intracellular calcium increase, calpain activity, Cdk5 activation, and cellular death in primary cultured hippocampal neurons and rat striatum following the treatment of 3-nitropropionic acid (Crespo-Biel et al., 2009), a succinate dehydrogenase inhibitor (for review, Brouillet et al., 1999). Therefore, lithium-induced inhibition of calpain and Cdk5 activation may also contribute to protection against glutamate excitotoxicity.
Prior to changes in gene expression, lithium rapidly and transiently activated the cell survival PI3-kinase and its down-stream target, Akt-1, through phosphorylation at Ser473, thereby reversing glutamate-induced inactivation of this signaling pathway in CGCs (Chalecka-Franaszek and Chuang, 1999). Activated Akt is known to affect several anti-apoptotic targets including Bcl-2 associated death promoter (BAD), CREB, members of the forkhead family, and procaspase-9 (for review, Neri et al., 2002; Nicholson and Anderson, 2002; Huang and Reichardt, 2003). In addition, lithium also triggered Ser21 phosphorylation of the α isoform of GSK-3 (and hence resulted in inhibition), and this effect was prevented by a PI3-kinase inhibitor (Chalecka-Franaszek and Chuang, 1999). Another signaling pathway affected by lithium is the mitogen-activated protein (MAP) kinase pathway. One of the down-stream targets of MAP kinase is CREB, a transcription factor that is involved in learning and memory, and promotes the expression of Bcl-2 as well as brain-derived neurotrophic factor (BDNF; for review, Finkbeiner, 2000). In CGCs, toxic concentrations of glutamate-induced an NMDA receptor-dependent decrease in CREB phosphorylation at Ser133 and CREB-driven transcriptional activity (Kopnisky et al., 2003). Concurrent with its neuroprotective effects, long-term (but not acute) lithium treatment suppressed glutamate-induced dephosphorylation of CREB. We also found that glutamate rapidly activated c-Jun-N-terminal kinase (JNK) and p38 kinase in CGCs, resulting in a robust increase in AP-1 binding (Chen et al., 2003a). These two kinases are also activated by a variety of apoptotic insults (for review, Mielke and Herdegen, 2000), and AP-1 has been known to be activated by different stress factors as well. Experiments using lithium and curcumin, a selective AP-1 inhibitor, suggest that NMDA receptor-mediated apoptotic death requires concerted action of JNK and p38 to enhance AP-1 binding, and that lithium’s neuroprotection is mediated, at least in part, by suppressing the JNK and p38 kinase pathways.
As one of the major neurotrophins, BDNF is essential for cortical development, synaptic plasticity, and neural survival, and is likely a key mediator of the clinical efficacy of anti-depressants and anxiolytic drugs (for review, Woo and Lu, 2006). The notion that BDNF plays a key role in neuronal survival is supported by our observation that BDNF and neurotrophin-4 (NT-4), but not NT-3, completely protected immature CGCs from apoptosis induced by cytosine arabinoside (Leeds et al., 2005). It was first reported that chronic treatment with lithium increased the expression of BDNF in the rat brain (Fukumoto et al., 2001), and we have documented that BDNF protein levels were increased in cortical neurons following lithium treatment (Hashimoto et al., 2002b). We hypothesized that this BDNF up-regulation and subsequent activation of its receptor TrkB might play a critical role in mediating the neuroprotective effects of lithium. In confirmation of this hypothesis, we found that lithium’s neuroprotection against glutamate excitotoxicity was blocked by a TrkB inhibitor, K252a, or by a neutralizing antibody against BDNF, and was mimicked by exogenous BDNF in rat cortical neurons. In addition, lithium increased intracellular levels of BDNF and this was followed by activation of TrkB. Furthermore, lithium-induced neuroprotection was prevented in cortical neurons from heterozygous (+/−) or homozygous (−/−) BDNF knockout mice (Hashimoto et al., 2002b).
Rodent BDNF has a complex genomic structure that makes it an ideal target for multiple and complex regulation. It contains multiple 5′-untranslated exons and one protein-coding 3′-exon. Each untranslated exon is alternatively spliced to produce various species of BDNF mRNA. We found that treatment of rat cortical neurons with therapeutic concentrations of lithium (e.g., 1 mM) caused a significant increase in the levels of BDNF exon IV-containing mRNA, while levels of exon I, II, or VI-containing mRNA remained unchanged (Yasuda et al., 2009). It is known that exon IV-containing BDNF transcripts are expressed in response to KCl-induced depolarization in rat cortical neurons (Tao et al., 2002). This transcriptional activation requires utilization of the promoter region 80 bp up-stream from the transcription initiation site of exon IV-containing three calcium responsive elements (CaREs; Chen et al., 2003b). We generated various BDNF promoter IV deletion constructs to investigate whether lithium treatment causes an increase in BDNF promoter IV activity, and, if so, which region of promoter IV confers the sensitivity to this drug. We identified that the drug-induced up-regulation of exon IV-containing BDNF transcript was associated with a significant increase in the activity of BDNF promoter IV and total BDNF protein. To our surprise, the lithium-responsive element(s) in promoter IV resides in a region up-stream from the CaREs responsible for depolarization-induced BDNF induction (−170 to −704 bp). Moreover, activation of BDNF promoter IV occurred in cortical neurons depolarized with KCl and depletion of these three CaREs failed to abolish lithium-induced activation. Importantly, we found that lithium-induced activation of promoter IV was mimicked by pharmacological inhibitors of GSK-3 (SB216763, SB415286, inhibitor I, and inhibitor VII) or by transfection with specific siRNA for GSK-3α or GSK-3β. Additionally, their dominant-negative mutants also mimicked lithium-induced activation of promoter IV. These results demonstrate that GSK-3 is the initial target of lithium to selectively activate BDNF promoter IV and that BDNF induction by lithium involves a novel responsive region in promoter IV of the BDNF gene. Lithium-induced, GSK-3-dependent BDNF promoter IV activation could be a part of the molecular mechanisms underlying its neuroprotective effects and as such, possibly accounts for the therapeutic actions in bipolar patients.
It should be noted that in addition to lithium, other GSK-3 inhibitors have been shown to almost completely block glutamate-induced excitotoxicity in rat cortical neuronal cultures (Liang and Chuang, 2007). These include ATP-competitive inhibitors, SB216763 and SB415286, and ATP-non-competitive inhibitors, Inhibitor I and VII. As mentioned in the preceding section, glutamate-induced death of cortical neurons was mitigated by silencing of GSK-3α and/or β, or both isoforms, or inhibition of GSK-3 activity via transfection with dominant-negative mutants of GSK-3α/β isoforms (Liang and Chuang, 2007). Studies from other laboratories also supported the roles of GSK-3 inhibition in protecting neurons from glutamate neurotoxicity. For example, stimulation of NMDA receptors in cultured rat hippocampal or cortical neurons activated GSK-3 by PP-1-mediated serine dephosphorylation of GSK-3β (Szatmari et al., 2005). GSK-3 inhibition reduced the PP-1-mediated serine dephosphorylation of GSK-3 and CREB. Treatment of primary rat cortical neurons with α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA), lithium or SB216763 blocked glutamate-induced caspase-3 activation and excitotoxicity, and the protective effects of AMPA required PI3-kinase–Akt-dependent serine phosphorylation of GSK-3β (Nishimoto et al., 2008). Further, in organotypic cultures of chick embryo spinal cord, lithium prevented kainate-induced excitotoxic death of motoneurons by targeting GSK-3β, and this neuroprotection was associated with cytopathological changes (Caldero et al., 2010).
Neuroprotective Effects of Lithium in Preclinical Models of Ischemic Stroke
Stroke is the third leading cause of death in the United States and a major global cause of serious long-term disability in adults. Ischemic strokes represent approximately 87% of all cases, while the rest are hemorrhagic strokes (Roger et al., 2011). In addition to physical deficits, stroke victims also suffer from vascular depression and dementia, both of which are difficult to treat with conventional medicine. It is becoming clear that there is a substantial increase in extracellular glutamate in the brain following cerebral ischemia, and that a significant portion of ischemia-induced brain damage is mediated by over-stimulation of NMDA receptors. Shortly after ischemia, the interruption of cerebral blood flow depletes oxygen and glucose and subsequently prevents ATP production. Inadequate ATP supply will cause the malfunction of ATP-dependent ion pumps and alter the ion concentration gradient across the neuronal membranes. The resulting failure to transport glutamate leads to an accumulation of glutamate in the extracellular space and over-stimulates NMDA receptors, which leads to a toxic influx of calcium and in turn drives the activation of damaging calcium-mediated intracellular enzymes. This cascade of events ultimately results in mitochondrial failure, production of reactive oxygen species, neuroinflammation, and cell necrosis and apoptosis (Allen and Bayraktutan, 2009; Deb et al., 2010).
GSK-3β has been strongly implicated in the neuronal cell death caused by cerebral ischemic insult. One study in rats subjected to transient middle cerebral artery occlusion (MCAO) demonstrated a rapid increase in the expression of cytoplasmic and nuclear GSK-3β protein in ipsilateral lamina I, II, V, and VI in young rat brains, whereas in lamina V and VI in old rat brains (Sasaki et al., 2001). In addition, the distribution of GSK-3β was well correlated with TUNEL-staining. Although the phosphorylation status of GSK-3β was not mentioned, these findings implicate a role of GSK-3β in cerebral ischemic injury. It is well known that GSK-3β can be phosphorylated at serine and tyrosine residues in which Ser9 phosphorylation renders it inactive, while Tyr216 phosphorylation is necessary for its functional activity (Hughes et al., 1993). Discrepancies exist in the literature regarding changes of Ser9 and Tyr216 phosphorylation levels following cerebral ischemia. The phosphorylation levels of GSK-3β at Ser9 and Akt at Ser473 were reported to be markedly enhanced in the vulnerable hippocampal CA1 region, but not in the ischemia-resistant CA3 region in rats subjected to transient global cerebral ischemia, while there was no change in levels of Tyr216 phosphorylation or total GSK-3β (Endo et al., 2006). Levels of GSK-3β Ser9 phosphorylation were also increased shortly after permanent focal cerebral ischemia and decreased to basal levels or even lower 24 h after ischemic onset (Sasaki et al., 2006; Gao et al., 2008). However, it has also been reported that transient focal cerebral ischemia in rats caused an increase in GSK-3β Tyr216 phosphorylation in degenerating cortical neurons with no alteration in Ser9 phosphorylation (Bhat et al., 2000). This discrepancy may stem, in part, from the difference of ischemic severity and ischemic models across various studies. It appears that transient focal cerebral ischemia tends to activate GSK-3β and subsequently to induce apoptotic cell death. In contrast, GSK-3β is inactivated shortly after permanent focal cerebral ischemia or global cerebral ischemia, which in turn may promote survival of vulnerable neurons. A very recent study showed that following hypoxia–ischemia both GSK-3α and GSK-3β mediated the expression of a lethal protein, neuronal pentraxin 1 (Russell et al., 2011). In light of these findings, GSK-3 inhibition may provide neuroprotective effects against cerebral ischemia-induced injury. The beneficial effects of lithium in rodent cerebral ischemic models demonstrated by us and others support this notion.
In an initial study, long-term lithium pretreatment at therapeutically relevant doses decreased brain infarct volume, reduced apoptotic cell death and improved behavioral performance after permanent cerebral ischemia-induced by MCAO (Nonaka and Chuang, 1998; Xu et al., 2003). In a subsequent study, we demonstrated that subcutaneous injection of rats with lithium at therapeutic doses (e.g., 0.5 and 1.0 mEq/kg) after the onset of transient MCAO markedly decreased infarct volume, reduced TUNEL-positive DNA damage, and suppressed neurological deficits measured by sensory, motor, and reflex tests (Ren et al., 2003). The time window for these beneficial effects was at least 3 h after the onset of ischemia. Heat-shock protein 70 (HSP70), a well-established cytoprotective factor against apoptosis, was induced in the ischemic penumbra where neuronal recovery takes place. Post-insult treatment with lithium increased the DNA binding activity of HSF-1 to the heat-shock element, superinducing HSP70 which inhibits brain ischemia-induced apoptosis (Ren et al., 2003). Lithium-elicited GSK-3 inhibition is likely associated with HSF-1 activation and HSP70 induction (Bijur and Jope, 2000). Notably, post-insult lithium treatment mitigated apoptosis and brain damage by preventing GSK-3β and ERK dephosphorylation, suppressing calpain and caspase-3 activation, and inhibiting mitochondrial release of cytochrome c and apoptosis-inducing factor in a neonatal hypoxic–ischemic rat model (Li et al., 2010). These findings suggest that lithium-induced GSK-3 inhibition contributes to its anti-apoptotic effects under ischemic conditions.
In addition, it was found that lithium pretreatment largely suppressed ischemia-induced exploratory behavioral changes and memory impairments in gerbils after global cerebral ischemia (Bian et al., 2007). These behavioral benefits were associated with an increase in the number of viable cells and a decrease in apoptotic cells in the CA1 hippocampal area of ischemic gerbils. Moreover, lithium-induced neuroprotection in the ischemic brain was accompanied by down-regulation of pro-apoptotic p53 in the CA1, and up-regulation of anti-apoptotic Bcl-2 and HSP70, both of which are targets of GSK-3. It is likely that lithium protection against ischemia-induced injury involves multiple mechanisms. In the rat hippocampus, lithium was reported to inhibit ischemia-induced NMDA receptor hyperactivation by inhibiting NMDA subunit 2A tyrosine phosphorylation and its interactions with Src and Fyn through PSD-95 (Ma and Zhang, 2003). Lithium also attenuated hypoxia-induced serine dephosphorylation of GSK-3α and β in the mouse brain (Roh et al., 2005). Additionally, in organotypic cultures of rat hippocampus subjected to oxygen and glucose deprivation, lithium showed neuroprotection in conjunction with HSP27 activation (Cimarosti et al., 2001).
It is widely recognized that neuroinflammation plays a causative role in ischemic stroke injury. Post-ischemic inflammation is a dynamic process involving a complicated set of interactions between inflammatory cells and molecules (Iadecola and Alexander, 2001). A recent study documented the anti-inflammatory effects of lithium in a neonatal rat hypoxic–ischemic model. Post-insult lithium treatment significantly reduced total tissue loss following hypoxia–ischemia, and this beneficial effect of lithium was associated with inhibiting microglia activation and attenuating levels of pro-inflammatory cytokines or chemokines, such as interleukin-1β and chemokine ligand 2 (Li et al., 2011). One possible underlying mechanism is through HSP70 superinduction. HSP70 over-expression can inactivate the key inflammatory transcription factor NF-κB by stabilizing the NF-κB-IκB complex, and thereby preventing nuclear translocation of activated NF-κB subunits in a mouse MCAO model (Zheng et al., 2008).
Besides anti-inflammation, lithium also increased proliferation and differentiation of hippocampal neural progenitor cells in both non-ischemic and ischemic brains without altering the relative levels of neuronal and astrocytic differentiation, and this effect lasted at least 7 weeks after hypoxia–ischemia in neonatal rats (Li et al., 2011). In line with this finding, chronic lithium pretreatment was found to increase the generation and survival of newborn cells in the hippocampal dentate gyrus, and did not affect the neuronal or astrocytic differentiation of these newborn cells in a transient four-vessel occlusion model (Yan et al., 2007). ERK1/2 phosphorylation following ischemia was enhanced by lithium treatment, while ERK1/2 inhibitor U0126 prevented the effects of lithium in increasing BrdU-positive cells and improving spatial learning and memory (Yan et al., 2007). In fact, chronic lithium treatment has been demonstrated to increase activity in the MEK/ERK pathway in vivo, and lithium’s neuroprotection has been suggested to depend on the induction of this signaling pathway (Einat et al., 2003). In addition, it was reported that activation of ERK associates with and phosphorylates GSK-3β at the Thr43 residue, which primes this kinase for its subsequent phosphorylation at Ser9 by p90RSK, resulting in inactivation of GSK-3β and up-regulation of β-catenin (Ding et al., 2005). Therefore, lithium might affect GSK-3β phosphorylation through the MEK/ERK pathway, which in turn inhibits this kinase via RSK. GSK-3β is also negatively regulated by the Wnt/β-catenin signaling pathway and, accordingly, activating the canonical Wnt pathway has been shown to contribute to adult hippocampal neural progenitor cell proliferation triggered by lithium treatment (Wexler et al., 2008).
In a collaborative study, the neurohemodynamic aspects of recovery induced by delayed chronic lithium treatment were assessed using functional magnetic resonance imaging (MRI; Kim et al., 2008). Rats were subjected to transient MCAO and then injected with lithium (LiCl, 1 mEq/kg, s.c.) 12 h after the ischemic onset. This delayed lithium injection was followed by daily injections, and on day 15, an MRI scan was performed to monitor changes in blood oxygen level dependence (BOLD) and functional cerebral blood volume (fCBV) responses using electric stimulation of forelimbs. The mean activated volume ratio and total activation magnitude ratio between ipsilateral and contralateral cortices for both BOLD and fCBV were significantly higher in the lithium-treated than in the saline-treated rats. The lithium-induced increase in fCBV in the peri-infarct regions suggests a possible vascular transformation. Indeed, the size and distribution of immunohistochemical staining of CD31, a microvasculature marker, were enhanced by lithium treatment in the peri-infarct regions. Co-localized with CD31, the tissue staining of matrix metalloproteinase-9 (MMP-9) was also much more pronounced following lithium treatment, suggesting MMP-9-dependent neurovascular remodeling in the recovering brain area. Moreover, treatment of cultured rat brain endothelial cells with lithium in a follow-up study was also found to increase the protein levels of vascular endothelial growth factor (VEGF) via a mechanism involving the PI3-kinase and GSK-3 signaling pathways (Guo et al., 2009). Since VEGF has been linked to angiogenesis, neurogenesis, and neuroprotection (for review, Fan and Yang, 2007), VEGF over-expression may contribute to lithium’s ability to promote neurovascular remodeling and to induce functional recovery after ischemic stroke.
Ample evidence supports the therapeutic potential of mesenchymal stem cells (MSCs) in several human diseases including stroke. However, it is increasingly recognized that the effectiveness of MSC transplantation is limited by their poor migration toward disease target sites such as ischemic brain regions. In a recent study, we investigated whether treatment of MSCs with lithium and another mood stabilizing drug, valproic acid (VPA), would enhance cell migration (Tsai et al., 2010). We found that treatment of MSCs with lithium (2.5 mM for 1 day) selectively elevated the transcript and protein levels of MMP-9 and its enzymatic activity. These effects were mimicked by pharmacological inhibition or gene silencing of GSK-3β. Lithium treatment also potentiated stromal cell-derived factor-1α (SDF-1α)-dependent MSC migration across the extracellular matrix, which was suppressed by two MMP-9 inhibitors, doxycycline and GM6001. Short-term (3 h) exposure of MSCs to a relatively high concentration (2.5 mM) of VPA markedly increased the transcript and protein levels of CXC chemokine receptor 4 (CXCR4). VPA-induced CXCR4 expression required its ability to inhibit histone deacetylases (HDACs), including the HDAC1 isoform, and involved histone hyperacetylation at the CXCR4 gene promoter. VPA treatment enhanced SDF-1α-mediated MSC migration, which was completely blocked by AMD3100, a CXCR4 antagonist. Notably, combining lithium and VPA treatment further increased MSC migration, and the additive enhancement of migration was completely blocked by the co-presence of AMD3100 and GM6001. Our results suggest that lithium and VPA stimulate MSC migration through distinct targets and mediators: GSK-3β–MMP-9 and HDAC–CXCR4, respectively (Tsai et al., 2010).
In a follow-up in vivo study, MSCs were primed with lithium and/or VPA and then injected into the tail vein of transient MCAO rats 24 h after ischemic onset. Priming with lithium or VPA increased the number of MSCs homing to the cerebral infarcted regions such as the cortex and striatum 2 weeks after transplantation, and co-priming with lithium and VPA further enhanced this migratory effect (Tsai et al., 2011). MCAO rats receiving lithium- and/or VPA-primed MSCs showed improved functional recovery, reduced infarct volume, and enhanced angiogenesis in the infarcted penumbra regions. These beneficial effects of lithium and VPA priming were reversed by pharmacological inhibition of MMP-9 and CXCR4, respectively, suggesting that these effects were likely mediated by lithium-induced MMP-9 up-regulation and VPA-induced CXCR4 over-expression. Together, these findings raise the potential utility of using MSCs primed with inhibitors of GSK-3 and HDAC to enhance the migration and homing capacity for transplantation into stroke victims.
In addition to lithium, other pharmacological GSK-3 inhibitors have been shown to exert neuroprotective effects against cerebral ischemia by various groups. A specific GSK-3β inhibitor, Chir025, was demonstrated to protect cultured hippocampal neurons from glutamate excitotoxicity and to attenuate death of cortical neurons following oxygen–glucose deprivation, an in vitro model of cerebral ischemia (Kelly et al., 2004). Moreover, Chir025 reduced infarct size in focal cerebral ischemic rats, but did not affect TUNEL-positive neurons or caspase-3/9 activities, although Bcl-2 expression was increased. GSK-3 enzymatic activity was markedly elevated after transient MCAO in rats, and this GSK-3 activation was blocked by jugular vein injection of GSK-3 inhibitor VIII (Koh et al., 2008). Pre- or post- (up to 2 h) MCAO injection with inhibitor VIII also reduced blood glucose levels, infarct size, caspase-3 activity, and water content in the ipsilateral brain hemisphere. Furthermore, ischemia-induced inflammation-related signals such as COX-2 over-expression and neutrophil infiltration were alleviated by this GSK-3 inhibitor. Prophylactic or therapeutic administration of a GSK-3β inhibitor TDZD-8 reduced infarct volume and cerebral injury in the rat hippocampus after transient ischemia (Collino et al., 2008). This was accompanied by suppression of ischemia-induced oxidative stress, apoptosis, and neuroinflammation. Delayed treatment with Compound I, a GSK-3β and Cdk inhibitor, decreased TUNEL-positive cells in the ipsilateral hippocampus and striatum of adult (but not juvenile) mice subjected to hypoxic–ischemic injury (Cowper-Smith et al., 2008). These neuroprotective effects of Compound I were associated with long-lasting functional recovery. Finally, GSK-3 inhibition by SB216763 counteracted oxygen–glucose deprivation-induced mitochondrial biogenesis impairment and reduced mitochondrial reactive oxygen species generation in primary cortical neurons (Valerio et al., 2011). When systematically administrated to permanent MCAO mice, SB216763 decreased infarct volume and restored the loss of mitochondrial DNA, thus supporting a novel role of GSK-3 inhibitors in stimulating the renewal of functional mitochondria following ischemic stroke.
Conclusion
A growing body of evidence supports that lithium, a mood stabilizer used to treat bipolar disorder, has neuroprotective properties in both cellular and in vivo experimental settings. One of the major targets of lithium is GSK-3, a serine/threonine kinase implicated in the pathogenesis of diverse CNS disorders. Lithium inhibits GSK-3 activity by direct binding to the enzyme or indirectly by enhancing serine phosphorylation of both α and β isoforms through multiple mechanisms. Lithium has been used as a prototype drug to seek evidence for the involvement of GSK-3 inhibition in lithium-induced protection against excitotoxicity in cultured neurons and animal models of cerebral ischemic stroke. Lithium at therapeutically relevant concentrations robustly protected primary brain neurons from glutamate-induced, NMDA receptor-mediated excitotoxicity. The neuroprotective effects of lithium were associated with GSK-3 inhibition, and were mimicked by other pharmacological GSK-3 inhibitors, by silencing GSK-3α and/or β isoforms, or by expression of isoform-specific dominant-negative mutants. These results support the roles of GSK-3 inhibition in lithium-elicited protection against excitotoxicity. Lithium rapidly activated the cell survival PI3-kinase–Akt signaling pathway to enhance GSK-3 serine phosphorylation and to block glutamate-induced Akt inactivation as well as apoptosis. Lithium also caused an increase in the expression of cytoprotective Bcl-2 and suppressed glutamate-induced up-regulation of pro-apoptotic p53 and Bax, resulting in blocking cytochrome c release from mitochondria. Induction of BDNF and activation of the BDNF–TrkB signaling were prerequisite for lithium’s neuroprotection. BDNF promoter IV was selectively activated by GSK-3 inhibition using lithium or other drugs or through gene silencing/inactivation of either isoform. This effect on promoter IV resulted in BDNF transcriptional activation and protein up-regulation. However, there is a gap in the understanding of how GSK-3 inhibition causes an increase in BDNF promoter activity. In addition, lithium’s neuroprotective effects were associated with inhibition of NMDA receptor-mediated calcium influx and suppression of p38/JNK and AP-1 activation, thus reducing apoptosis. This effect appears to stem from inhibition of Src/Fyn kinase to suppress NR2B Tyr1472 phosphorylation of the receptor. It remains to be explored as to whether this lithium-induced action on NMDA receptors is related to GSK-3 inhibition. It should be noted that lithium has other direct targets such as inositol phosphatases. The potential roles of these other targets in mediating the neuroprotective effects of this drug also deserve future investigation.
It is well known that glutamate overflow and NMDA receptor hyper-stimulation are early events following cerebral ischemia. In rodent ischemic models, pre- or post-insult treatment with therapeutic doses of lithium decreased infarct volume, caspase-3 activity and apoptotic cells in the injured brain. Importantly, lithium administration ameliorated neurological deficits, and improved functional recovery. The beneficial time window of lithium is at least 3 h after the ischemic onset. Up-regulation of HSP70 and Bcl-2 as well as down-regulation of p53 likely contributed to the protective effects of lithium in the ischemic conditions, thus supporting similar underlying neuroprotective mechanisms in the excitotoxic cellular models and animal models of ischemic stroke. Limited data suggested that lithium might also display anti-inflammatory effects by inhibiting ischemia-induced microglia activation and pro-inflammatory factors release. Delayed and chronic injections of lithium improved functional MRI responses such as increases in BOLD and fCBV. The improved fCBV was concurrent with enhanced angiogenesis and neurovascular remodeling. Indeed, lithium was found to induce two pro-angiogenic factors, MMP-9 and VEGF in a GSK-3-dependent manner. Lithium has also been reported to stimulate ERK1/2 activity and to enhance proliferation of hippocampal neural progenitor cells and memory performance after ischemia. Finally, lithium promoted migration of MSCs in vitro by up-regulation of MMP-9 through GSK-3β inhibition and this migratory effect was potentiated by co-treatment with VPA, another mood stabilizer. Notably, transplantation of lithium–VPA co-primed MSCs into ischemic rats markedly increased MSC migration to the injured brain regions, decreased infarct size and improved the neurological performance. Lithium-induced stem cell migration, neurogenesis, and angiogenesis all likely contribute to functional recovery. Figure 3 illustrates proposed molecular events leading to lithium-induced beneficial effects following cerebral ischemia. It should be noted that several other GSK-3 inhibitors have also been reported to exert beneficial effects in rodent ischemic models and their actions were accompanied by suppression of ischemia-increased GSK-3 activity. Accordingly, GSK-3 inhibitors have therapeutic potential to treat stroke and other excitotoxicity-related neurodegenerative diseases. Lithium has been used in bipolar patients over 60 years and its clinical profiles are well understood. Therefore, lithium is a prime candidate for use in clinical trials of new therapies for stroke victims.
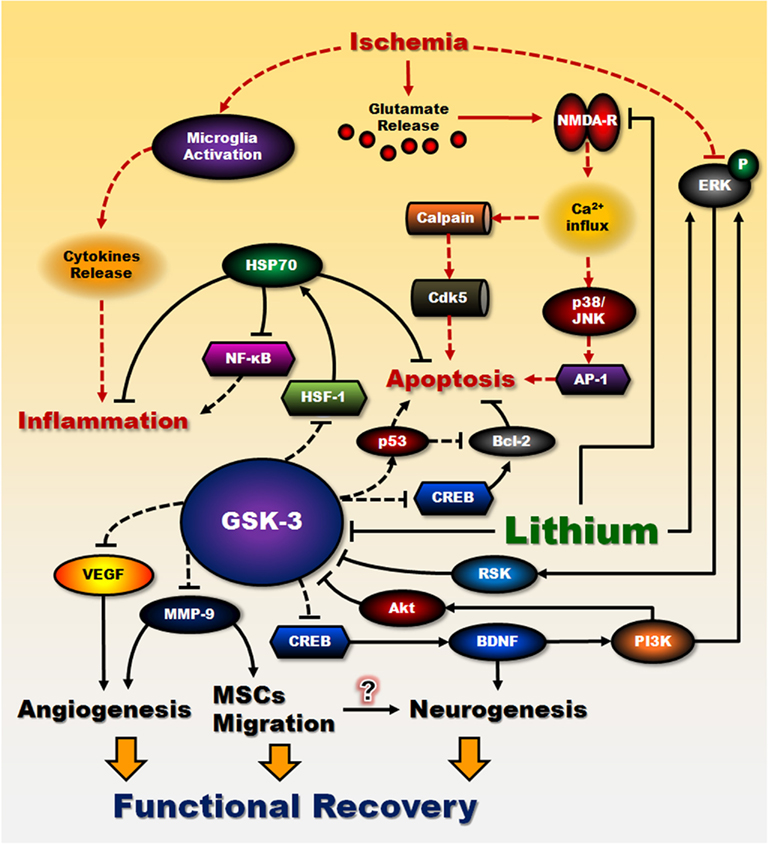
Figure 3. Proposed lithium’s neuroprotective effects against cerebral ischemia. The neuroprotective effects of lithium against cerebral ischemia are proposed to result from its interactions with cell survival and apoptotic machinery. A significant portion of brain damage following cerebral ischemia is caused by an increase in extracellular glutamate and subsequent over-stimulation of NMDA receptor-mediated toxic increase in intracellular calcium. This signaling pathway plays a critical role in mediating glutamate-induced caspase activation and apoptosis. Lithium at therapeutically relevant concentrations inhibits NMDA receptor-mediated calcium influx, which in turn decreases subsequent activation of JNK, p38 kinase, and transcription factor AP-1. Inhibition of intracellular calcium increase also attenuates the activity of calpain and calpain-mediated activation of pro-apoptotic Cdk5/p25 kinase. On the other hand, lithium can directly and indirectly reduce the activity of constitutively activated GSK-3 by multiple mechanisms, leading to disinhibition of several transcription factors, such as CREB and HSF-1, and resulting in induction of major cytoprotective proteins such as BDNF, VEGF, MMP-9, HSP70, and Bcl-2. A decrease in GSK-3 activity further reduces the activity of pro-apoptotic protein p53 and its downregulating effect on Bcl-2. BDNF, via activating its cell surface receptor and the down-stream ERK and PI3-kinase/Akt pathways, induces neuroprotective effects in part by inhibiting GSK-3 and stimulating CREB. Induction of BDNF is an early and essential step for neuroprotection and is involved in lithium-induced neurogenesis. In addition, superinduction of HSP70 by lithium treatment not only inhibits brain ischemia-induced apoptosis, but also contributes to the anti-inflammatory effects of lithium through inactivation of NF-κB. Counteraction of GSK-3 inhibition of VEGF and MMP-9 by lithium enhances angiogenesis and neurovascular remodeling. MMP-9 is also a key molecule involved in potentiating MSCs migration by lithium. Improvement in transplanted MSCs migration toward ischemic sites might increase neurogenesis as well. Taken together, these effects of lithium in reducing apoptosis, suppressing inflammation, enhancing angiogenesis and neurogenesis, contribute to behavioral improvement and functional recovery after ischemia. Lines with solid arrows represent stimulatory connections; lines with flattened ends represent inhibitory connections. Dashed lines represent pathways with reduced activity as a result of lithium treatment. NMDA-R, NMDA receptor.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Mental Health (NIMH), National Institutes of Health, and the Hsu family gift fund. The authors would like to thank Dr. Elizabeth Sherman, Peter Leeds, and Fairouz Chibane of the NIMH for their editorial assistance.
References
Allen, C. L., and Bayraktutan, U. (2009). Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 4, 461–470.
Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273.
Beurel, E., and Jope, R. S. (2009). Glycogen synthase kinase-3 promotes the synergistic action of interferon-gamma on lipopolysaccharide-induced IL-6 production in RAW264.7 cells. Cell Signal. 21, 978–985.
Beurel, E., Michalek, S. M., and Jope, R. S. (2010). Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 31, 24–31.
Bhat, R. V., Shanley, J., Correll, M. P., Fieles, W. E., Keith, R. A., Scott, C. W., and Lee, C. M. (2000). Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 97, 11074–11079.
Bian, Q., Shi, T., Chuang, D. M., and Qian, Y. (2007). Lithium reduces ischemia-induced hippocampal CA1 damage and behavioral deficits in gerbils. Brain Res. 1184, 270–276.
Bijur, G. N., and Jope, R. S. (2000). Opposing actions of phosphatidylinositol 3-kinase and glycogen synthase kinase-3beta in the regulation of HSF-1 activity. J. Neurochem. 75, 2401–2408.
Brouillet, E., Conde, F., Beal, M. F., and Hantraye, P. (1999). Replicating Huntington’s disease phenotype in experimental animals. Prog. Neurobiol. 59, 427–468.
Caldero, J., Brunet, N., Tarabal, O., Piedrafita, L., Hereu, M., Ayala, V., and Esquerda, J. E. (2010). Lithium prevents excitotoxic cell death of motoneurons in organotypic slice cultures of spinal cord. Neuroscience 165, 1353–1369.
Carmins, A., Verdaguer, E., Folch, J., Canudas, A. M., and Pallas, M. (2006). The rold of CDK5/P25 formation/inhibition in neurodegeneration. Drug News Perspect. 19, 453–460.
Chalecka-Franaszek, E., and Chuang, D. M. (1999). Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc. Natl. Acad. Sci. U.S.A. 96, 8745–8750.
Chen, R. W., and Chuang, D. M. (1999). Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity. J. Biol. Chem. 274, 6039–6042.
Chen, R. W., Qin, Z. H., Ren, M., Kanai, H., Chalecka-Franaszek, E., Leeds, P., and Chuang, D. M. (2003a). Regulation of c-Jun N-terminal kinase, p38 kinase and AP-1 DNA binding in cultured brain neurons: roles in glutamate excitotoxicity and lithium neuroprotection. J. Neurochem. 84, 566–575.
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C., Jaenisch, R., and Greenberg, M. E. (2003b). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889.
Chiu, C. T., and Chuang, D. M. (2010). Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol. Ther. 128, 281–304.
Chuang, D. M. (2004). Lithium protection from glutamate excitotoxicity: therapeutic implications. Clin. Neurosci. Res. 4, 243–252.
Cimarosti, H., Rodnight, R., Tavares, A., Paiva, R., Valentim, L., Rocha, E., and Salbego, C. (2001). An investigation of the neuroprotective effect of lithium in organotypic slice cultures of rat hippocampus exposed to oxygen and glucose deprivation. Neurosci. Lett. 315, 33–36.
Collino, M., Thiemermann, C., Mastrocola, R., Gallicchio, M., Benetti, E., Miglio, G., Castiglia, S., Danni, O., Murch, O., Dianzani, C., Aragno, M., and Fantozzi, R. (2008). Treatment with the glycogen synthase kinase-3beta inhibitor, TDZD-8, affects transient cerebral ischemia/reperfusion injury in the rat hippocampus. Shock 30, 299–307.
Cowper-Smith, C. D., Anger, G. J., Magal, E., Norman, M. H., and Robertson, G. S. (2008). Delayed administration of a potent cyclin dependent kinase and glycogen synthase kinase 3 beta inhibitor produces long-term neuroprotection in a hypoxia-ischemia model of brain injury. Neuroscience 155, 864–875.
Crespo-Biel, N., Camins, A., Pallas, M., and Canudas, A. M. (2009). Evidence of calpain/cdk5 pathway inhibition by lithium in 3-nitropropionic acid toxicity in vivo and in vitro. Neuropharmacology 56, 422–428.
Cruz, J. C., and Tsai, L. H. (2004). A Jekyll and Hyde kinase: roles for Cdk5 in brain development and disease. Curr. Opin. Neurobiol. 14, 390–394.
Deb, P., Sharma, S., and Hassan, K. M. (2010). Pathophysiologic mechanisms of acute ischemic stroke: an overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology 17, 197–218.
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584.
Dhariwala, F. A., and Rajadhyaksha, M. S. (2008). An unusual member of the Cdk family: Cdk5. Cell. Mol. Neurobiol. 28, 351–369.
Ding, Q., Xia, W., Liu, J. C., Yang, J. Y., Lee, D. F., Xia, J., Bartholomeusz, G., Li, Y., Pan, Y., Li, Z., Bargou, R. C., Qin, J., Lai, C. C., Tsai, F. J., Tsai, C. H., and Hung, M. C. (2005). Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 19, 159–170.
Droguett, R., Cabello-Verrugio, C., Santander, C., and Brandan, E. (2010). TGF-beta receptors, in a Smad-independent manner, are required for terminal skeletal muscle differentiation. Exp. Cell Res. 316, 2487–2503.
Einat, H., Yuan, P., Gould, T. D., Li, J., Du, J., Zhang, L., Manji, H. K., and Chen, G. (2003). The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J. Neurosci. 23, 7311–7316.
Endo, H., Nito, C., Kamada, H., Nishi, T., and Chan, P. H. (2006). Activation of the Akt/GSK3beta signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 26, 1479–1489.
Fan, Y., and Yang, G. Y. (2007). Therapeutic angiogenesis for brain ischemia: a brief review. J. Neuroimmune Pharmacol. 2, 284–289.
Fukumoto, T., Morinobu, S., Okamoto, Y., Kagaya, A., and Yamawaki, S. (2001). Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology (Berl.) 158, 100–106.
Gao, X., Zhang, H., Takahashi, T., Hsieh, J., Liao, J., Steinberg, G. K., and Zhao, H. (2008). The Akt signaling pathway contributes to postconditioning’s protection against stroke; the protection is associated with the MAPK and PKC pathways. J. Neurochem. 105, 943–955.
Gomes, F. C., Sousa Vde, O., and Romao, L. (2005). Emerging roles for TGF-beta1 in nervous system development. Int. J. Dev. Neurosci. 23, 413–424.
Gould, T. D., Einat, H., Bhat, R., and Manji, H. K. (2004). AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int. J. Neuropsychopharmacol. 7, 387–390.
Grimes, C. A., and Jope, R. S. (2001). The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 65, 391–426.
Guo, S., Arai, K., Stins, M. F., Chuang, D. M., and Lo, E. H. (2009). Lithium upregulates vascular endothelial growth factor in brain endothelial cells and astrocytes. Stroke 40, 652–655.
Harada, T., Morooka, T., Ogawa, S., and Nishida, E. (2001). ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat. Cell Biol. 3, 453–459.
Hashimoto, R., Fujimaki, K., Jeong, M. R., Christ, L., and Chuang, D. M. (2003). Lithium-induced inhibition of Src tyrosine kinase in rat cerebral cortical neurons: a role in neuroprotection against N-methyl-D-aspartate receptor-mediated excitotoxicity. FEBS Lett. 538, 145–148.
Hashimoto, R., Hough, C., Nakazawa, T., Yamamoto, T., and Chuang, D. M. (2002a). Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J. Neurochem. 80, 589–597.
Hashimoto, R., Takei, N., Shimazu, K., Christ, L., Lu, B., and Chuang, D. M. (2002b). Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 43, 1173–1179.
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90.
Huang, E. J., and Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642.
Huang, H. C., and Klein, P. S. (2006). Multiple roles for glycogen synthase kinase-3 as a drug target in Alzheimer’s disease. Curr. Drug Targets 7, 1389–1397.
Hughes, K., Nikolakaki, E., Plyte, S. E., Totty, N. F., and Woodgett, J. R. (1993). Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 12, 803–808.
Iadecola, C., and Alexander, M. (2001). Cerebral ischemia and inflammation. Curr. Opin. Neurol. 14, 89–94.
Jope, R. S. (1999). Anti-bipolar therapy: mechanism of action of lithium. Mol. Psychiatry 4, 117–128.
Jope, R. S., and Roh, M. S. (2006). Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr. Drug Targets 7, 1421–1434.
Jope, R. S., Yuskaitis, C. J., and Beurel, E. (2007). Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem. Res. 32, 577–595.
Jorda, E. G., Verdaguer, E., Canudas, A. M., Jimenez, A., Garcia de Arriba, S., Allgaier, C., Pallas, M., and Camins, A. (2005). Implication of cyclin-dependent kinase 5 in the neuroprotective properties of lithium. Neuroscience 134, 1001–1011.
Kaidanovich-Beilin, O., Lipina, T. V., Takao, K., van Eede, M., Hattori, S., Laliberte, C., Khan, M., Okamoto, K., Chambers, J. W., Fletcher, P. J., MacAulay, K., Doble, B. W., Henkelman, M., Miyakawa, T., Roder, J., and Woodgett, J. R. (2009). Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain 2, 35.
Kaidanovich-Beilin, O., Milman, A., Weizman, A., Pick, C. G., and Eldar-Finkelman, H. (2004). Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol. Psychiatry 55, 781–784.
Kelly, S., Zhao, H., Hua Sun, G., Cheng, D., Qiao, Y., Luo, J., Martin, K., Steinberg, G. K., Harrison, S. D., and Yenari, M. A. (2004). Glycogen synthase kinase 3beta inhibitor Chir025 reduces neuronal death resulting from oxygen-glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp. Neurol. 188, 378–386.
Kim, W. Y., Wang, X., Wu, Y., Doble, B. W., Patel, S., Woodgett, J. R., and Snider, W. D. (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12, 1390–1397.
Kim, Y. R., van Meer, M. P., Tejima, E., Murata, Y., Mandeville, J. B., Dai, G., Chuang, D. M., Rosen, B. R., and Lo, E. H. (2008). Functional MRI of delayed chronic lithium treatment in rat focal cerebral ischemia. Stroke 39, 439–447.
Kirshenboim, N., Plotkin, B., Shlomo, S. B., Kaidanovich-Beilin, O., and Eldar-Finkelman, H. (2004). Lithium-mediated phosphorylation of glycogen synthase kinase-3beta involves PI3 kinase-dependent activation of protein kinase C-alpha. J. Mol. Neurosci. 24, 237–245.
Klein, P. S., and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459.
Koh, S. H., Yoo, A. R., Chang, D. I., Hwang, S. J., and Kim, S. H. (2008). Inhibition of GSK-3 reduces infarct volume and improves neurobehavioral functions. Biochem. Biophys. Res. Commun. 371, 894–899.
Kopnisky, K. L., Chalecka-Franaszek, E., Gonzalez-Zulueta, M., and Chuang, D. M. (2003). Chronic lithium treatment antagonizes glutamate-induced decrease of phosphorylated CREB in neurons via reducing protein phosphatase 1 and increasing MEK activities. Neuroscience 116, 425–435.
Lee, J. H., and Kim, K. T. (2004). Induction of cyclin-dependent kinase 5 and its activator p35 through the extracellular-signal-regulated kinase and protein kinase A pathways during retinoic-acid mediated neuronal differentiation in human neuroblastoma SK-N-BE(2)C cells. J. Neurochem. 91, 634–647.
Lee, M. S., Kwon, Y. T., Li, M., Peng, J., Friedlander, R. M., and Tsai, L. H. (2000). Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405, 360–364.
Leeds, P., Leng, Y., Chalecka-Franaszek, E., and Chuang, D. M. (2005). Neurotrophins protect against cytosine arabinoside-induced apoptosis of immature rat cerebellar neurons. Neurochem. Int. 46, 61–72.
Li, H., Li, Q., Du, X., Sun, Y., Wang, X., Kroemer, G., Blomgren, K., and Zhu, C. (2011). Lithium-mediated long-term neuroprotection in neonatal rat hypoxia-ischemia is associated with antiinflammatory effects and enhanced proliferation and survival of neural stem/progenitor cells. J. Cereb. Blood. Flow. Metab. doi: 10.1038/jcbfm.2011.75. [Epub ahead of print].
Li, Q., Li, H., Roughton, K., Wang, X., Kroemer, G., Blomgren, K., and Zhu, C. (2010). Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell Death Dis. 1, e56.
Li, X., and Jope, R. S. (2010). Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology 35, 2143–2154.
Liang, M. H., and Chuang, D. M. (2006). Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J. Biol. Chem. 281, 30479–30484.
Liang, M. H., and Chuang, D. M. (2007). Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J. Biol. Chem. 282, 3904–3917.
Liang, M. H., Wendland, J. R., and Chuang, D. M. (2008). Lithium inhibits Smad3/4 transactivation via increased CREB activity induced by enhanced PKA and AKT signaling. Mol. Cell. Neurosci. 37, 440–453.
Ma, J., and Zhang, G. Y. (2003). Lithium reduced N-methyl-D-aspartate receptor subunit 2A tyrosine phosphorylation and its interactions with Src and Fyn mediated by PSD-95 in rat hippocampus following cerebral ischemia. Neurosci. Lett. 348, 185–189.
Meijer, L., Flajolet, M., and Greengard, P. (2004). Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 25, 471–480.
Mielke, K., and Herdegen, T. (2000). JNK and p38 stresskinases–degenerative effectors of signal-transduction-cascades in the nervous system. Prog. Neurobiol. 61, 45–60.
Morabito, M. A., Sheng, M., and Tsai, L. H. (2004). Cyclin-dependent kinase 5 phosphorylates the N-terminal domain of the postsynaptic density protein PSD-95 in neurons. J. Neurosci. 24, 865–876.
Neri, L. M., Borgatti, P., Capitani, S., and Martelli, A. M. (2002). The nuclear phosphoinositide 3-kinase/AKT pathway: a new second messenger system. Biochim. Biophys. Acta 1584, 73–80.
Nicholson, K. M., and Anderson, N. G. (2002). The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 14, 381–395.
Nishimoto, T., Kihara, T., Akaike, A., Niidome, T., and Sugimoto, H. (2008). alpha-Amino-3-hydroxy-5-methyl-4-isoxazole propionate attenuates glutamate-induced caspase-3 cleavage via regulation of glycogen synthase kinase 3beta. J. Neurosci. Res. 86, 1096–1105.
Nonaka, S., and Chuang, D. M. (1998). Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport 9, 2081–2084.
Nonaka, S., Hough, C. J., and Chuang, D. M. (1998). Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc. Natl. Acad. Sci. U.S.A. 95, 2642–2647.
O’Brien, W. T., Harper, A. D., Jove, F., Woodgett, J. R., Maretto, S., Piccolo, S., and Klein, P. S. (2004). Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 24, 6791–6798.
Omata, N., Chiu, C. T., Moya, P. R., Leng, Y., Wang, Z., Hunsberger, J. G., Leeds, P., and Chuang, D. M. (2011). Lentivirally mediated GSK-3beta silencing in the hippocampal dentate gyrus induces antidepressant-like effects in stressed mice. Int. J. Neuropsychopharmacol. 14, 711–717.
Pawlak, R., Magarinos, A. M., Melchor, J., McEwen, B., and Strickland, S. (2003). Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat. Neurosci. 6, 168–174.
Phiel, C. J., Wilson, C. A., Lee, V. M., and Klein, P. S. (2003). GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423, 435–439.
Ren, M., Senatorov, V. V., Chen, R. W., and Chuang, D. M. (2003). Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc. Natl. Acad. Sci. U.S.A. 100, 6210–6215.
Roger, V. L., Go, A. S., Lloyd-Jones, D. M., Adams, R. J., Berry, J. D., Brown, T. M., Carnethon, M. R., Dai, S., de Simone, G., Ford, E. S., Fox, C. S., Fullerton, H. J., Gillespie, C., Greenlund, K. J., Hailpern, S. M., Heit, J. A., Ho, P. M., Howard, V. J., Kissela, B. M., Kittner, S. J., Lackland, D. T., Lichtman, J. H., Lisabeth, L. D., Makuc, D. M., Marcus, G. M., Marelli, A., Matchar, D. B., McDermott, M. M., Meigs, J. B., Moy, C. S., Mozaffarian, D., Mussolino, M. E., Nichol, G., Paynter, N. P., Rosamond, W. D., Sorlie, P. D., Stafford, R. S., Turan, T. N., Turner, M. B., Wong, N. D., and Wylie-Rosett, J. (2011). Heart disease and stroke statistics – 2011 update: a report from the American Heart Association. Circulation 123, e18–e209.
Roh, M. S., Eom, T. Y., Zmijewska, A. A., De Sarno, P., Roth, K. A., and Jope, R. S. (2005). Hypoxia activates glycogen synthase kinase-3 in mouse brain in vivo: protection by mood stabilizers and imipramine. Biol. Psychiatry 57, 278–286.
Rosa, A. O., Kaster, M. P., Binfare, R. W., Morales, S., Martin-Aparicio, E., Navarro-Rico, M. L., Martinez, A., Medina, M., Garcia, A. G., Lopez, M. G., and Rodrigues, A. L. (2008). Antidepressant-like effect of the novel thiadiazolidinone NP031115 in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 1549–1556.
Rowe, M. K., and Chuang, D. M. (2004). Lithium neuroprotection: molecular mechanisms and clinical implications. Expert Rev. Mol. Med. 6, 1–18.
Rowe, M. K., Wiest, C., and Chuang, D. M. (2007). GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci. Biobehav. Rev. 31, 920–931.
Russell, J. C., Kishimoto, K. O’Driscoll, C., and Hossain, M. A. (2011). Neuronal pentraxin 1 induction in hypoxic-ischemic neuronal death is regulated via a glycogen synthase kinase-3alpha/beta dependent mechanism. Cell Signal. 23, 673–682.
Sasaki, C., Hayashi, T., Zhang, W. R., Warita, H., Manabe, Y., Sakai, K., and Abe, K. (2001). Different expression of glycogen synthase kinase-3beta between young and old rat brains after transient middle cerebral artery occlusion. Neurol. Res. 23, 588–592.
Sasaki, T., Han, F., Shioda, N., Moriguchi, S., Kasahara, J., Ishiguro, K., and Fukunaga, K. (2006). Lithium-induced activation of Akt and CaM kinase II contributes to its neuroprotective action in a rat microsphere embolism model. Brain Res. 1108, 98–106.
Sofola, O., Kerr, F., Rogers, I., Killick, R., Augustin, H., Gandy, C., Allen, M. J., Hardy, J., Lovestone, S., and Partridge, L. (2010). Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer’s disease. PLoS Genet. 6, e1001087. doi: 10.1371/journal.pgen.1001087
Spittaels, K., Van den Haute, C., Van Dorpe, J., Terwel, D., Vandezande, K., Lasrado, R., Bruynseels, K., Irizarry, M., Verhoye, M., Van Lint, J., Vandenheede, J. R., Ashton, D., Mercken, M., Loos, R., Hyman, B., Van der Linden, A., Geerts, H., and Van Leuven, F. (2002). Neonatal neuronal overexpression of glycogen synthase kinase-3 beta reduces brain size in transgenic mice. Neuroscience 113, 797–808.
Stambolic, V., Ruel, L., and Woodgett, J. R. (1996). Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6, 1664–1668.
Szatmari, E., Habas, A., Yang, P., Zheng, J. J., Hagg, T., and Hetman, M. (2005). A positive feedback loop between glycogen synthase kinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J. Biol. Chem. 280, 37526–37535.
Tao, X., West, A. E., Chen, W. G., Corfas, G., and Greenberg, M. E. (2002). A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron 33, 383–395.
Tsai, L. K., Leng, Y., Wang, Z., Leeds, P., and Chuang, D. M. (2010). The mood stabilizers valproic acid and lithium enhance mesenchymal stem cell migration via distinct mechanisms. Neuropsychopharmacology 35, 2225–2237.
Tsai, L. K., Wang, Z., Munasinghe, J., Leng, Y., Leeds, P., and Chuang, D. M. (2011). Mesenchymal stem cells primed with valproate and lithium robustly migrate to infarcted regions and facilitate recovery in a stroke Model. Stroke (in press).
Valerio, A., Bertolotti, P., Delbarba, A., Perego, C., Dossena, M., Ragni, M., Spano, P., Carruba, M. O., De Simoni, M. G., and Nisoli, E. (2011). Glycogen synthase kinase-3 inhibition reduces ischemic cerebral damage, restores impaired mitochondrial biogenesis and prevents ROS production. J. Neurochem. 116, 1148–1159.
Wexler, E. M., Geschwind, D. H., and Palmer, T. D. (2008). Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol. Psychiatry 13, 285–292.
Woo, N. H., and Lu, B. (2006). Regulation of cortical interneurons by neurotrophins: from development to cognitive disorders. Neuroscientist 12, 43–56.
Xu, J., Culman, J., Blume, A., Brecht, S., and Gohlke, P. (2003). Chronic treatment with a low dose of lithium protects the brain against ischemic injury by reducing apoptotic death. Stroke 34, 1287–1292.
Yan, X. B., Hou, H. L., Wu, L. M., Liu, J., and Zhou, J. N. (2007). Lithium regulates hippocampal neurogenesis by ERK pathway and facilitates recovery of spatial learning and memory in rats after transient global cerebral ischemia. Neuropharmacology 53, 487–495.
Yasuda, S., Liang, M. H., Marinova, Z., Yahyavi, A., and Chuang, D. M. (2009). The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry 14, 51–59.
Zhang, F., Phiel, C. J., Spece, L., Gurvich, N., and Klein, P. S. (2003). Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J. Biol. Chem. 278, 33067–33077.
Zhang, S., Edelmann, L., Liu, J., Crandall, J. E., and Morabito, M. A. (2008). Cdk5 regulates the phosphorylation of tyrosine 1472 NR2B and the surface expression of NMDA receptors. J. Neurosci. 28, 415–424.
Keywords: lithium, glycogen synthase kinase-3, excitotoxicity, cerebral ischemia, mesenchymal stem cells
Citation: Chuang D-M, Wang Z and Chiu C-T (2011) GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front. Mol. Neurosci. 4:15. doi: 10.3389/fnmol.2011.00015
Received: 28 June 2011; Accepted: 24 July 2011;
Published online: 09 August 2011.
Edited by:
Richard Scott Jope, University of Alabama at Birmingham, USAReviewed by:
Richard Scott Jope, University of Alabama at Birmingham, USAXiaohua Li, University of Alabama at Birmingham, USA
Copyright: © 2011 Chuang, Wang and Chiu. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: De-Maw Chuang, Molecular Neurobiology Section, National Institute of Mental Health, National Institutes of Health, Building 10, Room 3D38, 10 Center Drive, MSC 1363, Bethesda, MD 20892-1363, USA. e-mail:chuang@mail.nih.gov