 Ning Mao
Ning Mao Wentai Shao
Wentai Shao Yuefeng Cai
Yuefeng Cai Xue Kong
Xue Kong Nanjing Ji
Nanjing Ji Xin Shen
Xin Shen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Mar. Sci. , 12 January 2024
Sec. Deep-Sea Environments and Ecology
Volume 10 - 2023 | https://doi.org/10.3389/fmars.2023.1269411
This article is part of the Research Topic From Cold Seeps to Hydrothermal Vents: Geology, Chemistry, Microbiology, and Ecology in Marine and Coastal Environments View all 23 articles
Barnacles have demonstrated adaptability to a range of habitats, spanning from shallow water to the deep sea. Given the harsh conditions present in hydrothermal vents, hydrothermal vent barnacles serve as the model organism for investigating the interplay between evolution and adaptability. In order to gain insights into barnacle adaptive characteristics, particularly within hydrothermal vents, we conducted a comprehensive analysis of the mitogenomes and transcriptome in a deep-sea barnacle (Neolepas marisindica), in comparison to its shallow-water related species. The mitogenomes with the same genetic skews and the non-synonymous/synonymous mutation ratios (Ka/Ks) of the mitogenomes indicate that the protein-coding genes (COIII, ND2, and ND6) of N. marisindica are under positive selection. Meanwhile, the functional annotation shows that distinctly positive selected orthologs in N. marisindica are predominately related to neural signal transduction, immunity, antiapoptotic, and energy metabolism. These results indicate that the mitogenomes and key genes found in transcriptomic analysis are under high-temperature and high-pressure conditions, and which may contribute N. marisindica to have evolved to adapt to the extreme hydrothermal vent environments. The findings shed light on the mitogenome and transcriptome of N. marisindica, which lays a foundation for the in-depth understanding of the adaptation mechanism of sessile invertebrates to the deep-sea environment.
The general characteristics of the deep-sea environment are high pressure, darkness, and a lack of oxygen. Moreover, the temperature is low and the food source is limited (Danovaro et al., 2014; Lyu et al., 2023). Despite the harsh deep-sea ecosystem, there are still many crustaceans living here, including Glyptelasma gigas (Annandale) (Gan et al., 2020b), Alvinocaris longirostris (Kikuchi & Ohta) (Hui et al., 2018a; Xin et al., 2021), and Hirondellea gigas (Birstein & Vinagradov) (Lan et al., 2017). Additionally, the hereditary characteristics and adaptive evolution of marine organisms may be influenced by these adverse factors. Consequently, a number of reserachers have explored the adaptive evolutionary traits of marine organisms within a specific ecological setting, elucidating the mechanisms by which these organisms adapt to their environment. For instance, the population genetic structure of Capitulum mitella (Linnaeus) was explored through single DNA fragments, such as mitochondrial DNA control regions, and a large amount of gene flow was found, indicating that populations living in different regions have significant genetic differentiation due to the differences in their genetic adaptability (Song and Yoon, 2013; Yoon et al., 2013).
In addition, the deep-sea hydrothermal region is the most anoxic and highly toxic marine ecosystem on earth, which plays an important role in mitochondrial division and biological function (Ki et al., 2009). Previous phylogenetic analysis based on fossil and mitochondrial genomes has shown that bathymodiolins (Bathymodiolus) gradually evolved characteristics that were adapted to their environment in the process of their origin and evolution (Lorion et al., 2013; Thubaut et al., 2013). However, how bathymodiolin mussels have adapted to deep-sea chemosynthetic environments over the long term, and the genetic bases for these adaptations are still unclear (Zheng et al., 2017). Moreover, there is a lack of molecular data, such as single genes or single proteins, that can be used to deduce the adaptive mechanisms of deep-sea marine organisms. Therefore, it is imperative to conduct an analysis of the crucial mitochondrial functional genes within the immunity metabolic pathway, as this would elucidate the underlying mechanism behind their adaptability to the hydrothermal vent environment.
It is universally acknowledged that the transcriptome can be used to determine the expression of genes at the level of messenger RNA (mRNA) and non-coding RNA transcription and explore the functional genes that are related to environmental adaptation or physiological functions (Wilhelm and Landry, 2009). Consequently, a transcriptomic analysis may also illustrate the adaptive mechanisms of deep-sea organisms in diverse environments. Transcriptome sequencing technology can be used to understand the response of various species to marine ecosystems at the mRNA level, and it has been widely used in researching deep-sea environmental adaptation mechanisms (Zhou et al., 2021; Sun et al., 2022; Yan et al., 2022). For instance, a transcriptomic analysis was conducted on A. longirostris from the Iheya North hydrothermal vents and a methane seep in the South China Sea, which found that various genes including cytochrome P450s and Rhodopsin were related to sulfur metabolism and detoxification, showing their different adaptability between the two extreme conditions (Hui et al., 2018a). Recently, researchers have made significant findings by conducting comparative genomic analysis on deep-sea anemone species. This investigation has led to the identification of multiple genes that exert influence on osmotic metabolism, membrane function, protein translation, and cytoskeleton functions, and which were adapted to the deep-sea environment (Feng et al., 2021). Furthermore, the deep-sea mussel Gigantidas platifrons (Hashimoto & Okutani) lived in vent and seep habitats with abundant toxic chemical substances, particularly hydrogen sulfide (H2S). To adapt to the harsh H2S-rich environments, its genes control oxidative phosphorylation and the mitochondrial sulfide oxidization pathway, playing important roles in the sulfide tolerance of the mussel (Sun et al., 2022).
Barnacles exhibit a wide widely distribution within the intertidal zone and attaching to marine organisms. Furthermore, certain barnacle have undergone evolutionary adaptations in diverse deep-sea habitats (Sha and Ren, 2015; Chan et al., 2021). Due to their extended attachment to the bottom and confinement to relatively narrow deep zones, deep-sea barnacle adults served as suitable model organisms for conducting research on evolutionary and ecological processes (Gan et al., 2020a). Notably, members of the Scalpellomorpha order have been discovered in hydrothermal vents and cold seeps, suggesting the presence of distinctive adaptive mechanisms enabling their survival in deep-sea chemosynthetic habitats (Gan et al., 2022). However, the difficulty associated with the collection of deep-sea Cirripedia have limited the current research on the barnacle transcriptome to mere transcriptome assembly and the mechanism of barnacle attachment (Al-Aqeel et al., 2016; Ryu et al., 2019; Yan et al., 2020). While studies on other organisms inhabiting extreme deep-sea environments have identified several potential mechanisms, the transcriptome of them remains unexplored. Moreover, few studies have investigated the molecular adaptive characteristics that assist with barnacles’ survival in deep-sea harsh chemosynthetic habitats. Due to the limited prior investigations concerning the transcriptome of barnacles, particularly with regard to the deepsea adaptation, our study attempted to address this research gap.
In this study, we collected the deep-sea and shallow-water barnacle individuals at Longqi hydrothermal field on the Southwest Indian Ridge and Guangxi Beibu Gulf, respectively. In order to investigate the adaptability of barnacles to the deep-sea environment, the analysis of positive selection pressure was conducted using the comparative transcriptome of these two individuals, as well as the 13 protein coding genes (PCGs) of mitogenomes from 11 barnacles. This research endeavor aimed to not only provide valuable genomic resources for deep-sea barnacles but also establish a foundation for comprehending their genetic strategies and adaptation mechanisms in the deep-sea habitat.
Samples of the deep-sea barnacles from a hydrothermal vent Longqi hydrothermal field on the Southwest Indian Ridge (49°64’E, 37°17’S) at a depth of 2,759 m in the Southwest Indian Ocean collected on board of “Explorer 1” scientific research ship of Institute of Deep-sea Science and Engineering (Chinese Academy of Sciences) by using “Deep-sea Warrior” manned submersible during the winter of 2018, we thank the crew for their help during the cruise. Samples of the shallow-water barnacles were obtained from the Guangxi Beibu Gulf (109° 12 ‘ E, 21° 04 ‘ N), depth of about 300 m. In contrast to deep-sea species, the depth of 300m can be considered relatively shallow. Therefore, we provisionally identified the two barnacle species as the deep water species Vulcanolepas fijiensis (Lee et al., 2019) and the shallow water Scalpellum stearnsi (Chan et al., 2010). As a result of the Chen et al. Commentary Paper (Chan et al., 2024) we became aware that our initial identification of V. fijiensis was incorrect. We have confirmed that the barnacle is actually Neolepas marisindica and have revised this paper to include the correct species indentation. After collection, they were immediately stored at -80°C.
According to the manufacturer’s directions, the TIANamp Marine Animal DNA Kit (TIANGEN Biotech, China) was used to extract DNA from the abdominal muscle. Then, 1% gel electrophoresis and ultraviolet spectroscopy (Eppendorf BioPhotometer D30, Eppendorf, Germany) were used to estimate and quantify the genomic DNA. Then, the DNA was randomly cut into 350 bp fragments, which were repaired at the end and amplified by a polymerase chain reaction to construct the sequencing library. Next, Sanger dideoxy sequencing (N. marisindica) was conducted on an ABI-3730XL (Thermo Fisher Scientific, USA), and high-throughput sequencing (S. stearnsi) was conducted on Illumina NovaSeq 6000 platform (TSINGKE Biotechnology Co, Ltd, China).
For each species of barnacle (N. marisindica and S. stearnsi), a random selection of three individuals was made, and all RNA was extracted using TRIzol reagent (Invitrogen, USA). The quality of the RNA was assessed through 1% agarose gel electrophoresis. The purity and concentration of the RNA were determined using a NanoPhotometer spectrophotometer (IMPLEN, Germany) and Qubit 2.0 Fluorimeter (Life Technologies, USA), respectively. The integrity of the RNA was evaluated using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, USA). The sequencing library was established according to the manufacturer’s instructions of the NEBNext Ultra RNA Library Prep Kit (NEB, USA). The mRNA was enriched using poly-T oligo-attached magnetic beads, and an Agilent Bioanalyzer 2100 system (Agilent Technologies) was used to evaluate the synthesis of the complementary DNA (cDNA), the construction of a pair-end library, and the quality of the library. Finally, the two different kinds of species’ cDNA libraries were clustered and sequenced using the Illumina HiSeq 2500 platform by NovoGene (Beijing, China).
The initial dataset of the mitochondrial genome consisted of 150 base pairs at the paired ends. Reads containing an adapter, reads with a mass score below 20 (Q< 20), and duplicated sequences were removed. A reference genome (Lepas anserifera) was used to retrieve the reads belonging to the mitochondrial genome for assembly. Then, de novo assembly was performed using SPAdes 3.13.0 (Bankevich et al., 2012), and the assembly results were corrected for mutual alignment to confirm the accuracy of the assembled genome sequence. The assembled mitochondrial genomes were subjected to analysis using the online MITOS software (http://mitos.bioinf.unileipzig.de) with default parameters. This software was utilized to predict the PCGs, transfer RNA (tRNA) genes, and ribosomal RNA (rRNA) genes. Manual corrections to the genes for the start/stop codons were performed in Sequin 15.10 (http://www.ncbi.nlm.nih.gov/Sequin/index.html) by referencing the mitochondrial genome. On this basis, BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) technology was used to compare the existing research data including the PCGs and rRNA to verify and correct the accuracy of the analysis results. The circular mitochondrial genome map was drawn using OGDRAW 1.3.1 (Greiner et al., 2019), and the complete mitogenomes of N. marisindica and S. stearnsi have been registered as MZ772032 and OP345466 on GenBank.
The biases for the codon usage of the mitogenome can regulate genes and reflect the evolutionary relationship between different species (Wei et al., 2014). Consequently, the usage of codons was analyzed for the PCGs and rRNA. Using the following formula, the inclination related to the nucleotide bases was calculated:
The low-quality reads and reads containing adaptors were removed from the raw reads using the internal software (ng_qc) with custom script of NovoGene, and the transcriptome clean reads were obtained. The de novo transcriptome assembly was performed using Trinity v2.5.1, with the minimum k-mer parameter coverage of 2, while default parameters were employed for other sections (Grabherr et al., 2011). The longest transcript of each transcription group was regarded as a unigene for the subsequent analysis. Next, BUSCO v1.22 was used to verify the quality of the assembly that was performed using the arthropoda_odb9 database (Simão et al., 2015). Additionally, all the unigenes were annotated by blasting the common databases, such as the National Center for Biotechnology Information non-redundant protein (Nr), Swiss-Prot, and euKaryotic Ortholog Group using DIAMOND v0.8.22 (Buchfink et al., 2015), and the NCBI nucleotide database using BLAST v2.2.28+ (Altschul et al., 1990). The Kyoto Encyclopedia of Genes and Genomes (KEGG) classification was performed using the KEGG Automatic Annotation Server (Moriya et al., 2007). The protein family (Pfam) alignments were put into practice using HMMER v3.0 (http://hmmer.org/) and were based on the results of Nr and Pfam, and Blast2GO v2.5 was used to classify the Gene Ontology (GO) (Götz et al., 2008). The Nr database was searched for unigenes with BLASTx, and then SOLAR was used to compare the link fragments (Rota-Stabelli et al., 2010), so all the open reading frames (ORF) of the unigenes were obtained and converted into amino acid sequences.
The target sequences for this analysis consisted of the 13 PCGs from N. marisindica and 10 other barnacle species, aligning using clustalW (codons) in MEGA v7.0. Manual removal of gap and stop codons was performed to ensure accurate alignment (Kumar et al., 2016). The resulting sequence alignment file was then utilized for the analysis of positive selection. A molecular phylogenetic tree was constructed using the maximum likelihood method in MEGA v7.0 to assess selective pressure. Subsequently, the reconstructed tree was subjected to EasyCodeML in the PAML software (Gao et al., 2019) to identify natural selection characteristics within the 13 PCGs. Among them, the 13 PCGs from N. marisindica were treated as the foreground branch, while PCGs from other barnacle species served as the background branch. Site models were employed to identify the individual sites of PCGs. Changes in the different sites of the 13 PCGs were explored by calculating the ω values (ω = Ka/Ks or dN/dS). For the positive selection sites (ω > 1), the Bayes Empirical Bayes (BEB) method was used to estimate the Bayesian posterior probabilities (Biswas and Akey, 2006). The likelihood Ratio Test (LRT) was applied to compare the hypothetical models with the corresponding original hypotheses, and the LRT P-value was determined using the Chi-square method. The sites of the PCGs with an LRT P-value< 0.05 or BEB > 95% were considered to be under evolution and positive selection pressure. The ProtParam program (http://www.expasy.ch/tools/protparam.html) software was utilized to compute the relative molecular weight and isoelectric points of the sequence, and SignalIP 4.1 (http://www.cbs.dtu.dk/services/SignalP/) was employed to predict the signal peptides. Additionally, the SMART program (http://smart.embl-heidelberg.de/) was utilized to predict the conserved functional domains of proteins (Letunic and Bork, 2021). On this basis, we matched the sites to the three-dimensional (3D) conformation of the PCGs to observe their role under positive selection more clearly. The 3D structure of the target proteins was simulated using the SWISS-MODEL (Waterhouse et al., 2018) server, which visualized the positive selection sites in the proteins.
Transcriptome sequence set from the two species (N. marisindica and S. stearnsi), internal reference (Amphibalanus Amphitrite) and external reference species (Eurytemora affinis) were utilized for the analysis of the orthologous genes and nonsynonymous (dN)/synonymous (dS) substitution rates. Subsequently, adaptive evolution was assessed by comparing the ω value (dN/dS). BLAST was employed to pair the orthologs with the best hit probabilities. Following this, sequence alignment was conducted using MUSCLE, and all gaps were eliminated. The ω numerical calculation for each ortholog was performed using PAML v4.48a. Genes with ω values exceeding 1 were considered as positively selected genes (PSGs). The PSGs were further subjected to GO and KEGG enrichment analysis using Omicshare CloudTools (http://www.omicshare.com/tools/), with a significance threshold set at a q-value less than 0.05.
The complete mitogenomes of N. marisindica and S. stearnsi are 16,960 bp and 15,462 bp in length, respectively. They each contain the general 13 PCGs (COI, COII, COIII, ATP8, ATP6, ND1, ND2, ND3, ND4, ND4L, ND5, ND6 and COB), 2 rRNA genes (16S and 12S rRNA), 22 tRNA genes, and a D-loop. With the exception of the nicotinamide adenine dinucleotide hydride (NADH) genes (ND5, ND4, ND4L, ND1) and eight tRNA genes (trnF, trnQ, trnH, trnP, trnC, trnY, trnL, trnV), the majority of genes are encoded by the heavy strand (H-strand). Furthermore, apart from a few gene overlaps, the majority of nucleotide gaps are observed between the tRNA. In addition, 16S and 12S are located between trnL1 (TAG), trnV (TAC) and trnW (TCA) respectively (Figure 1; Table 1).
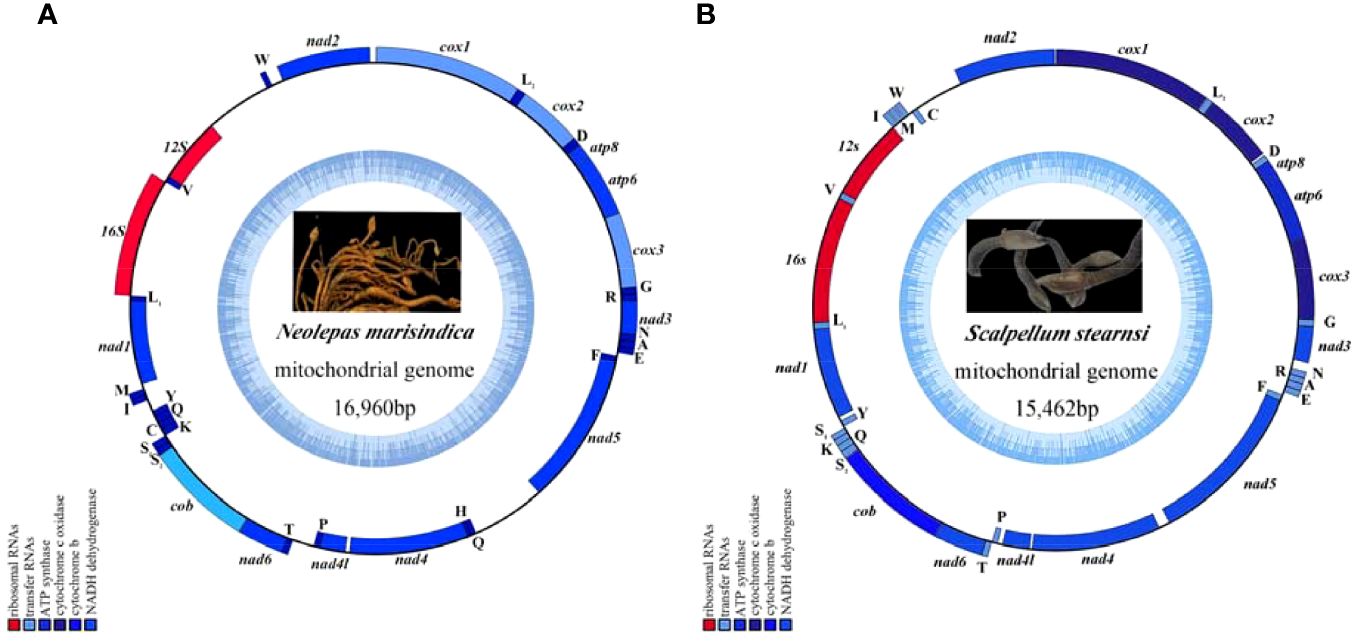
Figure 1. The mitochondrial genome map of deep-sea barnacle Neolepas marisindica (A) and shallow barnacle Scalpellum stearnsi (B) collected for this study, and photographs in the genome map are from preserved samples that were collected for this study, which were collected by Professor Haibin Zhang and kept in Institute of Deep-sea Science and Engineering (Chinese Academy of Sciences). The heavy strand is represented by the outer circle, while the light strand is represented by the inner circle; The tRNA and PCGs are denoted by the blue genes, whereas the 12S rRNA and 16S rRNA are indicated by the red genes.
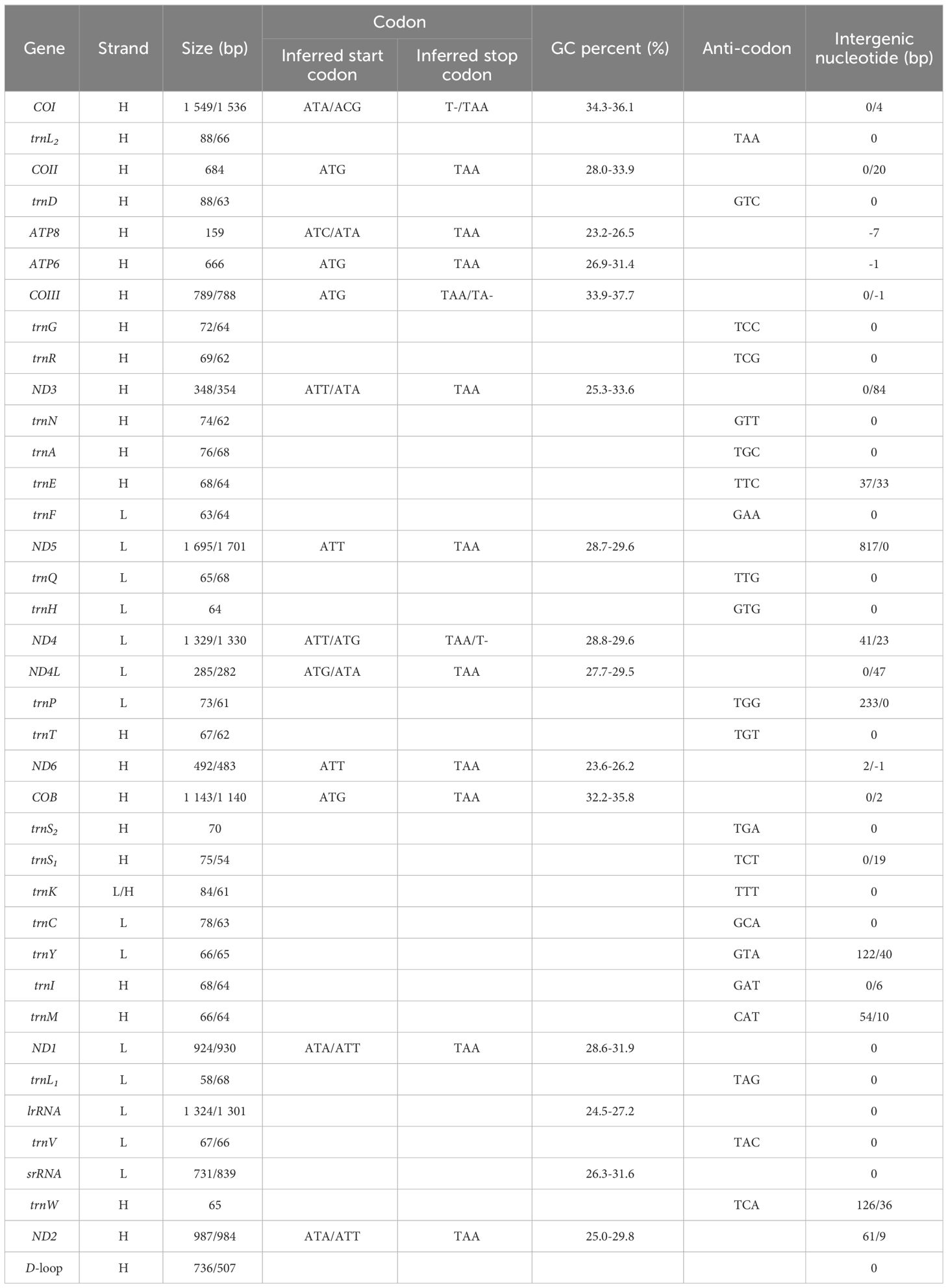
Table 1. Summary of the mitogenome features of Neolepas marisindica and Scalpellum stearnsi.
The nucleotide compositions (PCGs and rRNA) of N. marisindica and S. stearnsi are 20.9~39.8% A, 5.3~24.1% C, 32.9~48.3% T, and 3.1~24.2% G, with an AT content ranging from 62.4% to 76.8% (Supplementary Tables 1, 2). Their majority of the PCGs have a negative AT skew, except for ND1, ND4, ND4L, and ND5, which display a positive GC skew (Figure 2).
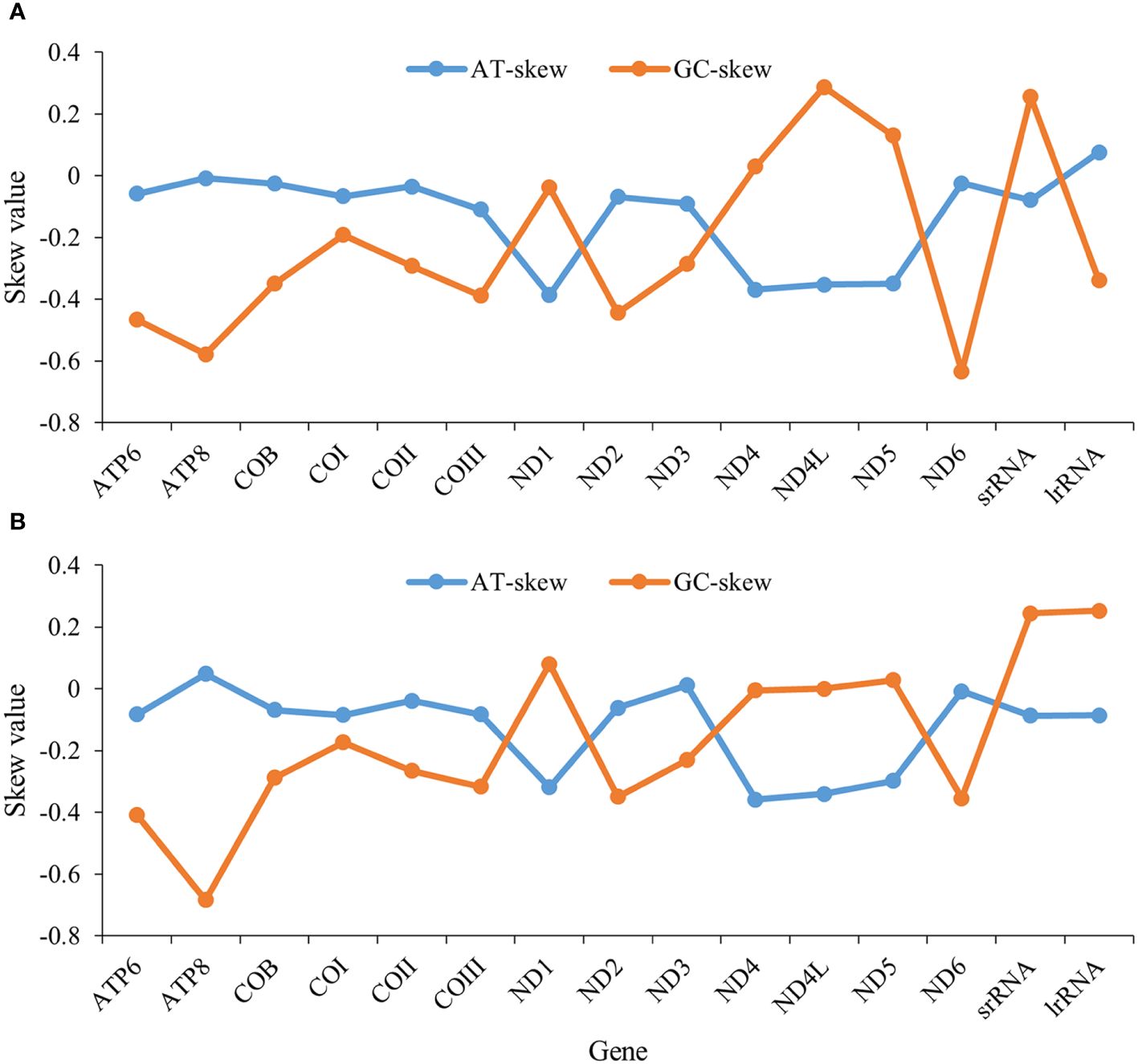
Figure 2. AT and GC skews in protein-coding genes of the mitochondrial genomes in deep-sea barnacle Neolepas marisindica (A) and shallow barnacle Scalpellum stearnsi (B). The horizontal and vertical coordinates represent gene name and skew value, respectively; The blue line represents the AT skew and the orange line represents the GC skew.
Except for COI, which is encoded by the ACG codon, the start codons of the PCGs predominantly follow canonical sequences (ATT/ATA/ATG). Additionally, within barnacle mitogenomes, three distinct types of stop codons are identified (T-, TA-, and TAA), with TAA being the most prevalent. Moreover, the T- and TA- stop codons only occur in COI, COIII, and ND4 of N. marisindica and S. stearnsi (Table 1).
The findings show that except for leucine and serine have six to eight codons, the other amino acids of the two examined species have two to four changeable codons (Figure 3). The codon usage analysis demonstrates that the following codon sequences appear with higher frequency: phenylalanine (UUU), isoleucine (AUU), and leucine (UUA). The codons with U or C in the third position occur more frequently than those with A or G. Different from the shallow-sea barnacle, the main amino acids in N. marisindica are asparagine and lysine (Figure 3).
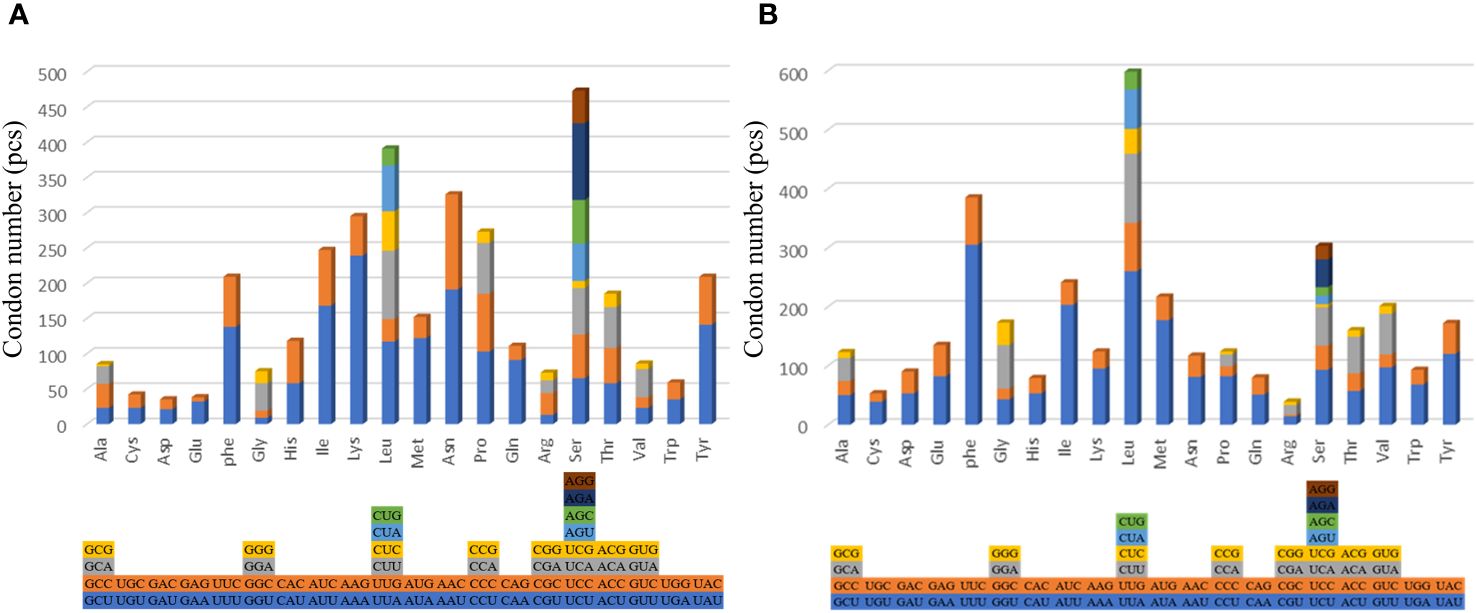
Figure 3. The codon usage of the protein-coding genes of the mitochondrial genomes of deep- sea barnacle Neolepas marisindica (A) and shallow barnacle Scalpellum stearnsi (B). The different colors represent different kinds of codons, and the different columns represent different amino acids.
A total of 47,593,764 and 47,752,536 raw reads are generated from S. stearnsi and N. marisindica, respectively. BUSCO assessment of assembly quality are showed in Supplementary Figures 1, 2. The complete single-copy and complete duplicated of the two origanisms are over 80%. Through the transcriptome assemblies, there are 98,686 unigenes identified with an N50 length of 916 bp for S. stearnsi, and 53,562 unigenes with an N50 length of 1,721 bp for N. marisindica. Among these unigenes, 38,666 (39.18%) and 20,628 (38.51%) are successfully annotated in S. stearnsi and N. marisindica, respectively (Table 2).
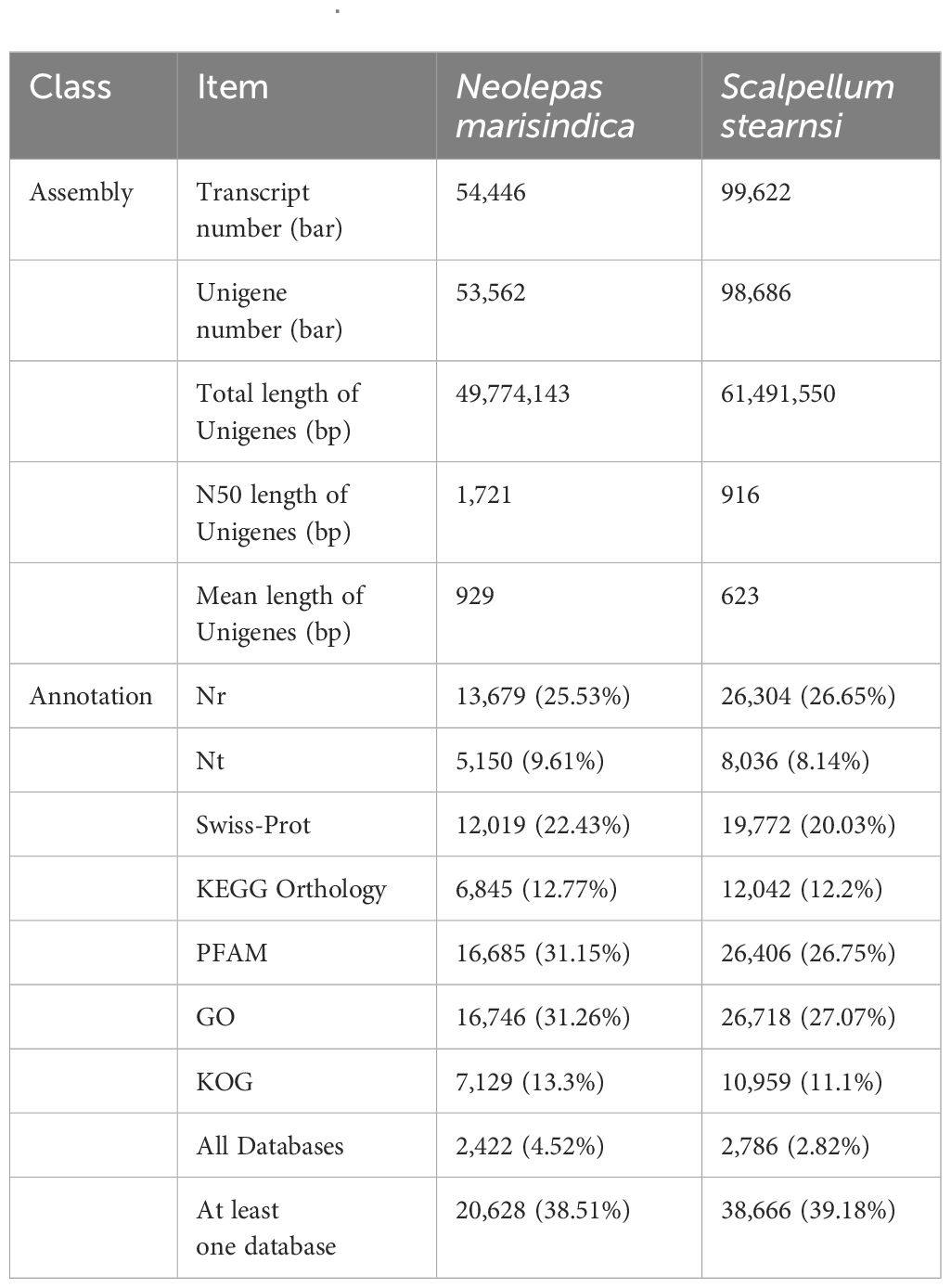
Table 2. Transcriptome library basic information of Scalpellum stearnsi and Neolepas marisindica.
The Gene Ontology functional cluster analysis indicates that 26,718 and 16,746 unigenes are successfully clustered in the three functional categories of GO (48 functional items). The major biological processes are the cellular, metabolic, and single-organism processes, with N. marisindica lacking the biological phase item and S. stearnsi lacking the rhythmic process item. Within the cellular components, the cell and cell part items are identified. The major molecular functions are binding and catalytic activity (Figure 4). In addition, The KEGG functional cluster analysis shows that 6,845 and 12,042 unigenes in N. marisindica and S. stearnsi are clustered into five functional groups, respectively, and more genes are annotated in the endocrine system, translation, and signal transduction pathways (Figure 5).
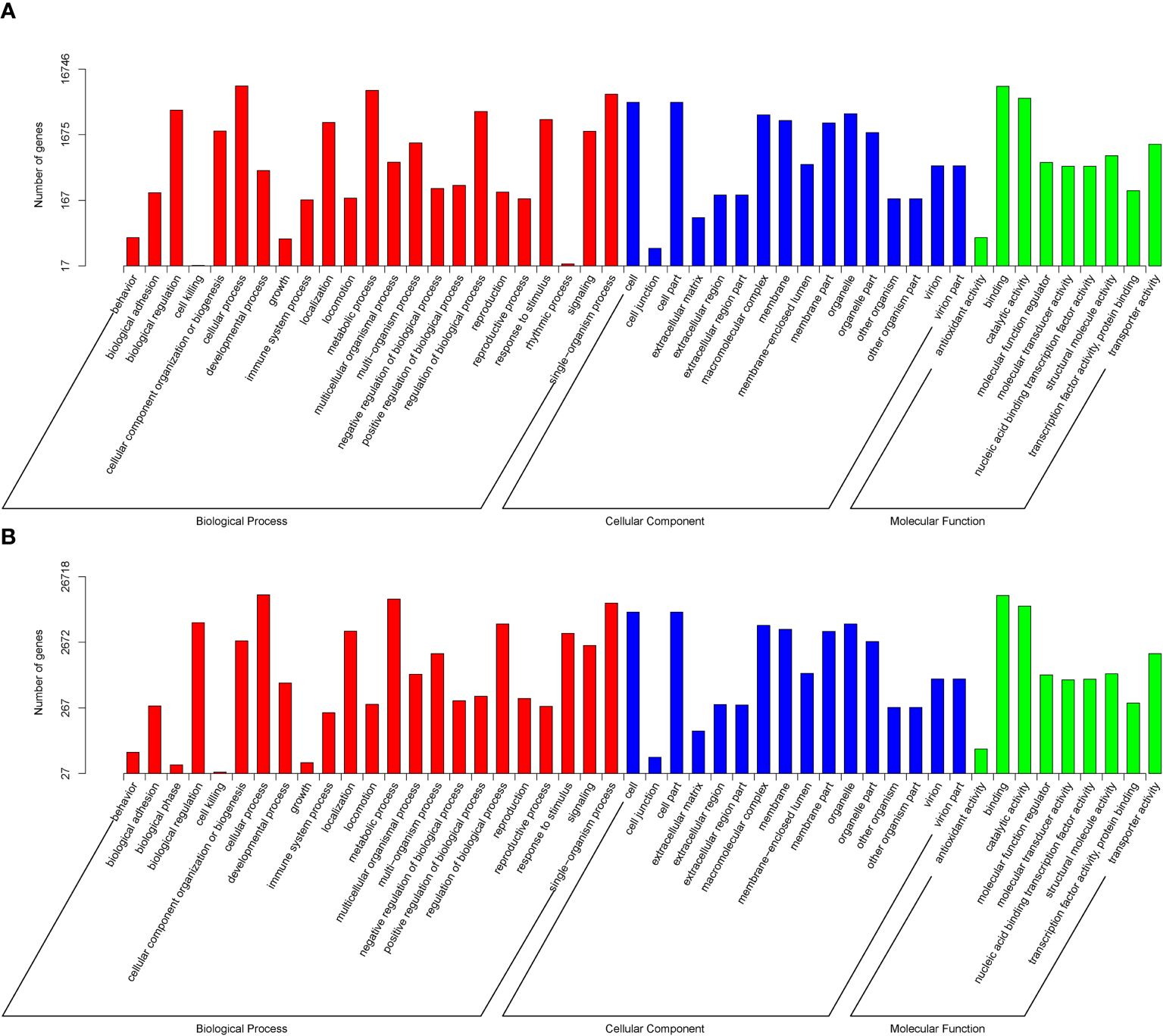
Figure 4. Gene Ontology function annotation of the transcriptomes of deep-sea barnacle Neolepas marisindica (A) and shallow barnacle Scalpellum stearnsi (B). The red terms represent Biological Process, the blue terms represent Cellular Composition, and the green terms represent Molecular Function.
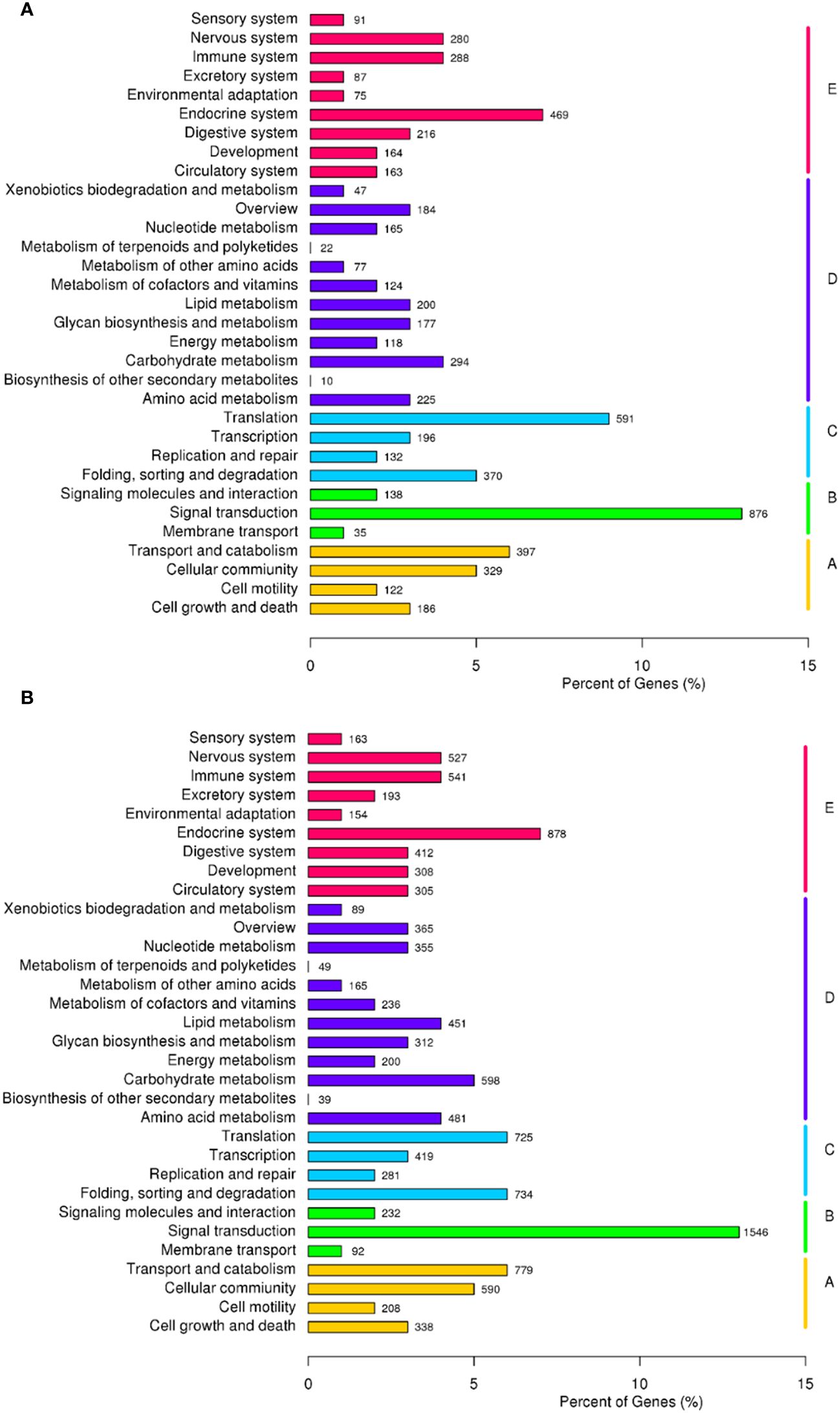
Figure 5. Classification of the Kyoto Encyclopedia of Genes and Genomes metabolic pathway of the transcriptomes of deep-sea barnacle Neolepas marisindica (A) and shallow barnacle Scalpellum stearnsi (B). Vertical lines represent five main pathways: red terms denote cellular processes, purple terms denote environmental information processing, blue terms denote genetic information processing, green terms denote metabolism, and yellow terms denote organismal systems.
From the complete mitochondrial genome of the 11 barnacle species, namely N. marisindica, Conchoderma hunteri (Owen), Lepas anserifera Linnaeus, L. australis Darwin, L. anatifera Linnaeus (Lepadidae), S. stearnsi, Arcoscalpellum epeeum Chan (Scalpellidae), Altiverruca navicula Hoek (Verrucidae), Capitulum mitella Linnaeus (Pollicipedidae), Glyptelasma annandalei Pilsbry (Poecilasmatidae), and Ibla cumingi Darwin (Iblidae) (Table 3), 13 PCGs were extracted for positive selection pressure analysis. The positive selection analysis yielded results indicating that the COIII and ND2 genes (ω > 1.0, P<0.05) of deep-sea species experience positive selection pressure. The three-dimensional structure model was utilized to identify and highlight specific sites within the genes that are under positive selection pressure, such as 22I, 29T, and 41E in the COIII gene, as well as 73F, 75S, and 76L in the ND2 gene (Table 4; Figure 6).
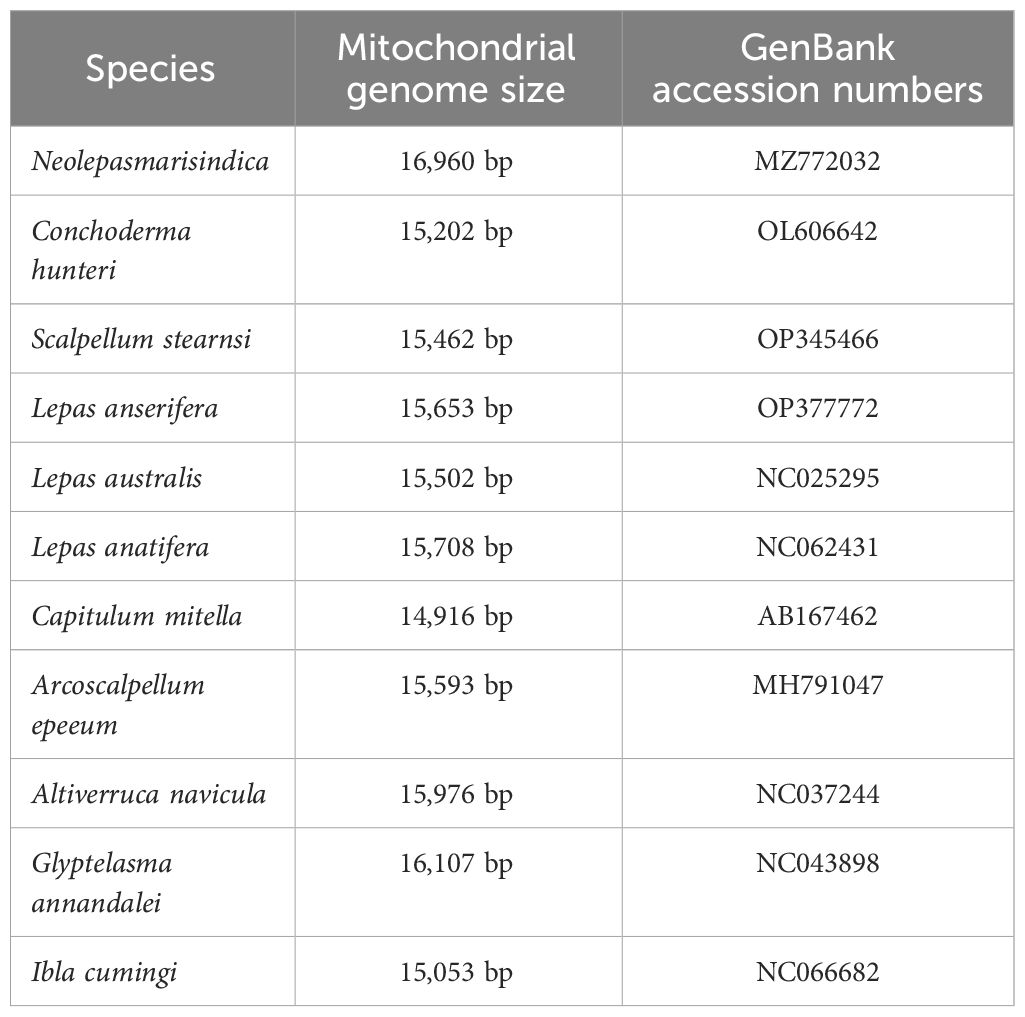
Table 3. Collection and mitogenome information of the specimens that were used for the positive selection analysis.
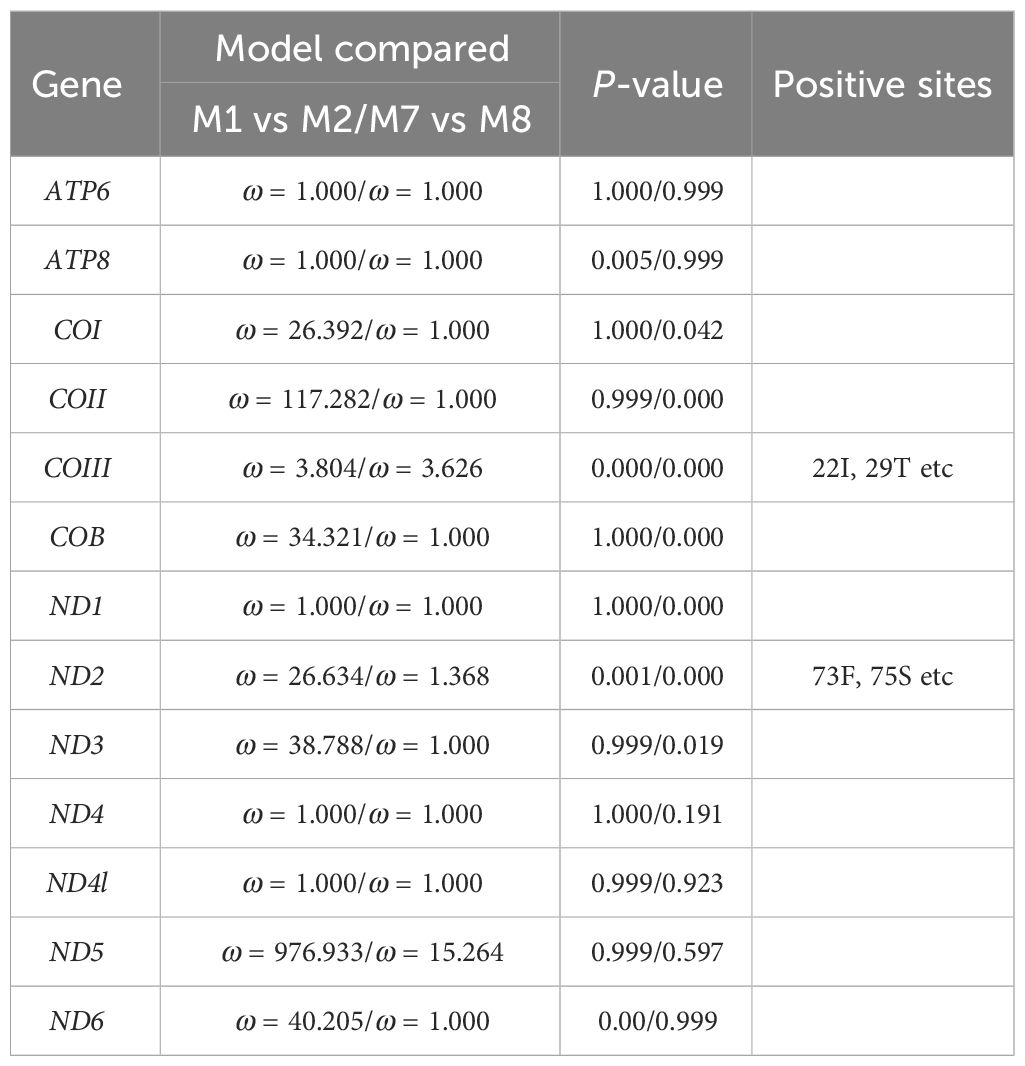
Table 4. Results of the selective pressure acting on the protein-coding genes of Neolepas marisindica.
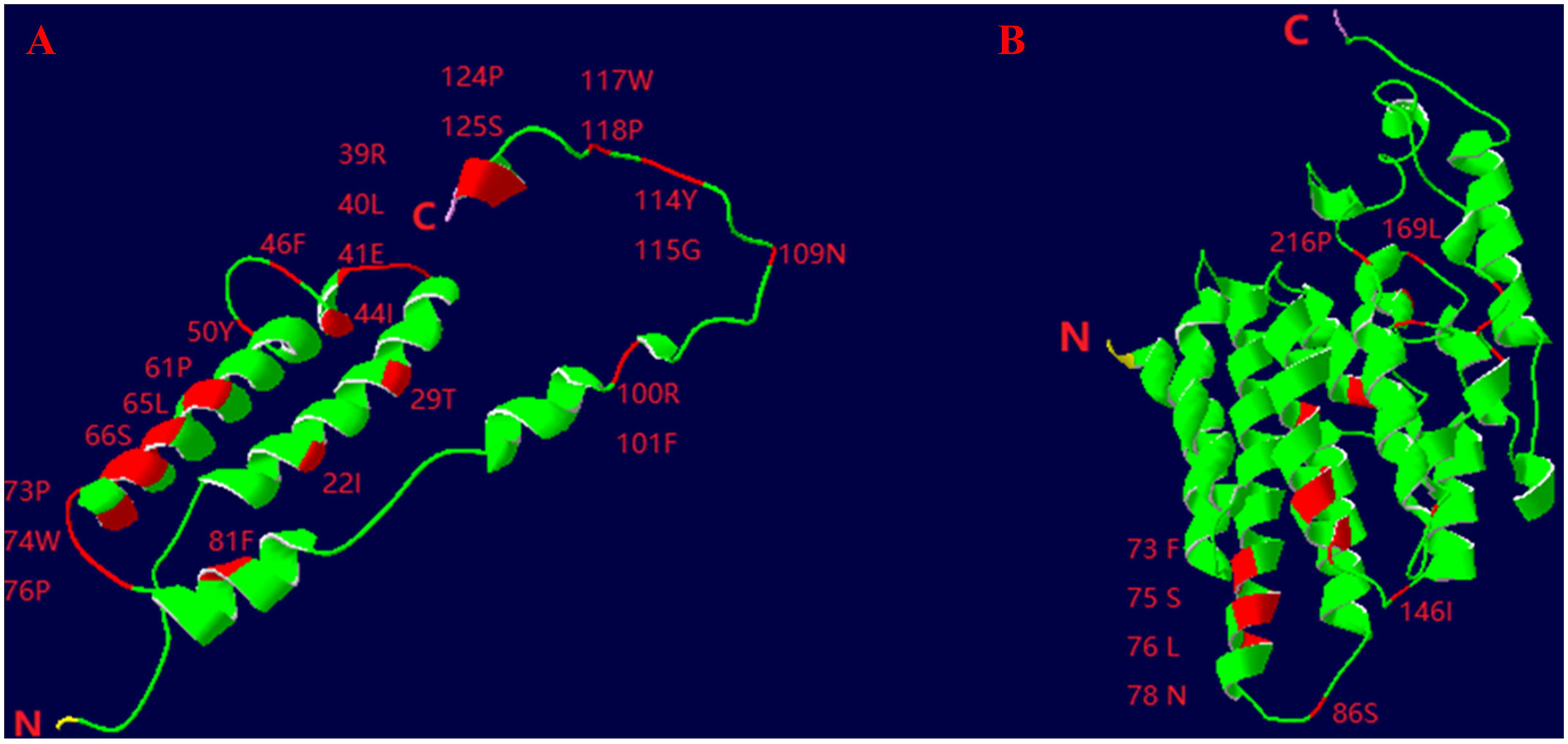
Figure 6. Three-dimensional structural models of the COIII (A) and ND2 (B) genes of deep- sea barnacle Neolepas marisindica. The red represents positive selection site; N represents amino terminus and (C) represents carboxy terminus.
Genes that are subjected to positive selection tend to respond to natural selection. We identified 148 pairwise orthologs within N. marisindica, S. stearnsi and two reference species in one-to-one correspondence to confirm the PSGs (Supplementary Table 3, Supplementary Figure 3) and performed an adaptive evolutionary analysis on them. The findings reveal the presence of five orthologs, namely chromosome 3 open reading frame 10 (C3ORF10), E74-like factor (ELF), ubiquitin-like protein ATG12 (ATG12), uracil phosphoribosyltransferase (UPRT), and small subunit ribosomal protein S21e (RPS21), all exhibiting ω values greater than 1.0. Notably, only RPS21 did not fall below the significance threshold of 0.05 (Table 5; Supplementary Figure 3). These PSGs are mainly enriched in the RIG-I-like receptor signaling, regulation of autophagy, apoptosis-fly, regulation of the actin cytoskeleton, pyrimidine metabolism, foxO signaling, and ribosome pathways (Figure 7; Table 5). It is predicted that the ORFs of the five genes under positive selection pressure are all complete. Except for C3ORF10, the amino acid domain positions of the other genes are predicted, there is no signal peptide, and the molecular weight range from 9.21 to 27.23 kDa. Additionally, the isoelectric point is relatively similar, ranging from 6.05 to 9.88 (Supplementary Table 4).
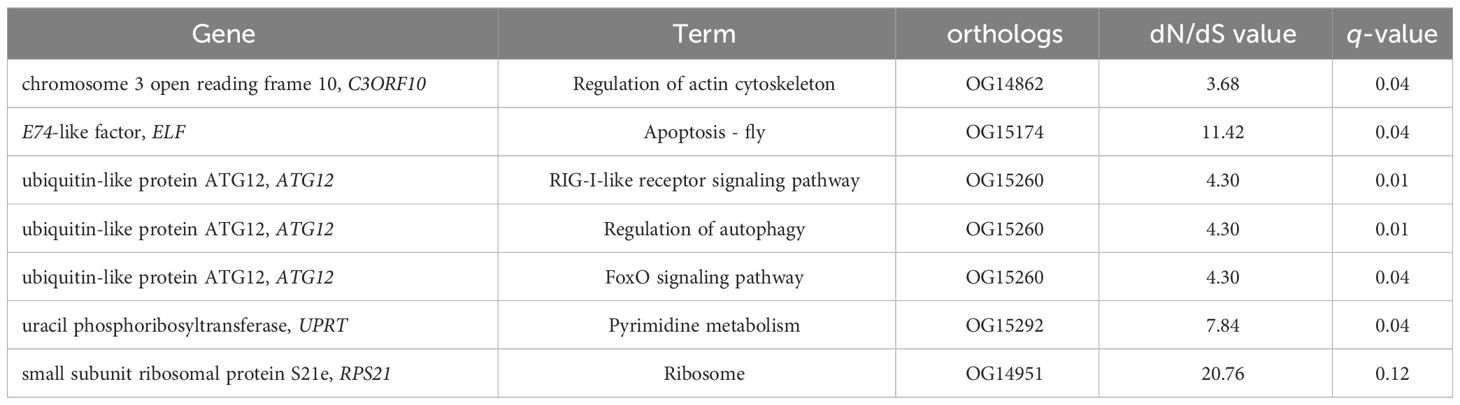
Table 5. Genes that were subject to positive selection pressure in Neolepas marisindica and Scalpellum stearnsi.
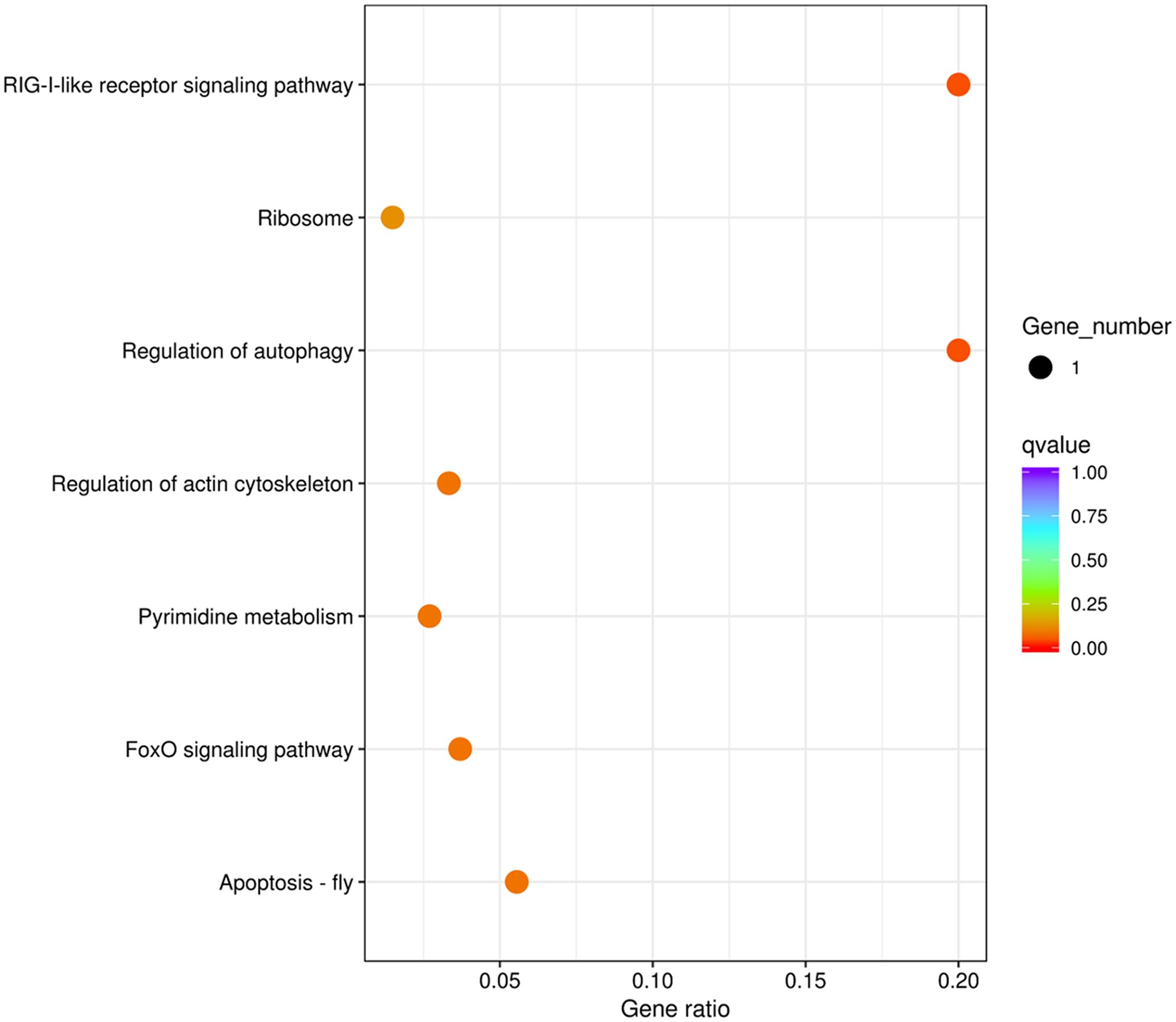
Figure 7. Enrichment of the Kyoto Encyclopedia of Genes and Genomes metabolic pathway of the positive selection genes in deep-sea barnacle Vulcanolepas fijiensis compared with shallow barnacle Scalpellum stearnsi. The circle size represents the number of genes, and the color from cool to warm represents the smaller q value.
Previous research has identified that certain barnacles exhibit similar arrangement structures within their mitogenomes (Shen et al., 2015). We also find similar genetic structures in the mitogenomes of the deep-sea and shallow barnacles. The AT content in the complete mitogenomes of both organisms, exceeded the GC content, aligning with the base composition observed in the majority of invertebrates (Liu et al., 2018; Wang et al., 2021; Sato et al., 2023). More negative AT skews are observed in ND1, ND4, ND4L, and ND5 genes of the two barnacles (Figure 2). This phenomenon might be attributed to the deep-sea habitat of these organisms, where DNA replication and transcription processes are subject to the mutations and the natural selection resulting in a bias towards specific nucleotides (Sahyoun et al., 2014; Yu et al., 2019). It is speculated that the different genetic structures of these four genes might be closely related to the pressure of natural selection in the extreme habitats. However, how nucleotide bias affects the initiation and orientation of gene replication and what selective pressures induced nucleotide skews need further study. Furthermore, the amino acid usage patterns observed in N. marisindica are similar to those of the shallow barnacle S. stearnsi (Figure 3), which provides more evidence for the barnacle nucleotide composition and gene skew results. Generally, more than 95% of energy comes from the oxidative phosphorylation of mitochondria (Auger et al., 2021). In the abyssal zone, the dark and absence of autotrophic primary producers pose constraints on the availability of sustenance (Gattuso et al., 2006). The observed nucleotide compositional biases between N. marisindica and S. stearnsi indicated that, in their mitogenomes, comparable alterations occurred during the evolutionary progression which might be a means of adapting to the extreme conditions and nutritional scarcity of the deep-sea environment.
Mitochondria serve as a vital pathway for aerobic respiration within cells. To cope with the extreme environment, selection pressure would have been put on the mitogenome, and non-synonymous mutations are regarded as an adaptive substitution to meet the needs of a new environment (Srivastava et al., 2020). The results of our study indicate that the mitogenomes in N. marisindica experienced significant selective pressure, particularly the COIII and ND2 genes, which have specific functional protein sites (Table 4; Figure 6). There is a prevalent belief that functional proteins contain positive selection sites, which serve as a foundation for adaptive responses to alterations in the environment (Rausher and Huang, 2016). COIII is one of the core members of cytochrome C oxidase, a key rate-limiting enzyme at the end of the mitochondrial respiratory chain, which produces ATP and changes oxygen into water for cell metabolism (Timón-Gómez et al., 2018). At the level of mitochondrial transcription and translation, during the respiratory chain embedding in the inner mitochondria, the respiratory supercomplex factor protein family acts in response to biological hypoxia by binding to COIII to adapt to environmental and metabolic changes (Lobo-Jarne and Ugalde, 2018). Through bioinformatics analysis, several adaptive residues potentially presented in COIII and ND2 of alvinocaridid shrimp (Alvinocarididae) inhabiting the deep-sea hydrothermal vent environment also have been identified by researchers. These proteins played an important role in regulating mitogenome intracellular energy metabolism and contributed to the adaptation of organisms to the hydrothermal vent environment (Hui et al., 2018b). Consequently, we speculate that the PCGs help N. marisindica to resist multiple pressures when adapting to the deep-sea environment.
The NADH dehydrogenases are the largest enzyme complex in the mitochondrial electron transport chain; thus, mutations in these subunits may disturb the efficiency of the electron transport process (Da Fonseca et al., 2008). The NADH dehydrogenase genes of deep-sea anemones exhibited novel genetic structures, which were evidence of adaptation to the deep-sea environment (Zhang et al., 2017). Likewise, the hydrothermal vent environment may potentially exert potential selection pressure on NADH dehydrogenase genes from mitogenomes in N. marisindica. This pressure may influence the modulation of mitochondrial complexes and electron transport efficiency and hence, adapting to cold and hypoxic conditions (Luo et al., 2012). Given the crucial role of mitochondria in aerobic respiration and their indispensability for metabolism and energy production, they likely aid the deep-water barnacle in surmounting the constraints imposed by hypoxia, cold and food availability. Additionally, hydrothermal vents are also associated with hydrogen sulfide (H2S), and the primary cytotoxic effect of H2S is to interrupt the mitochondrial respiratory chain by directly inhibiting cytochrome C oxidase (Cooper and Brown, 2008). Therefore, we infer that the mitogenomes of N. marisindica are subjected to multiple selection pressures which led to adaptation to the hydrothermal vent environment under extreme deep-sea conditions.
Since they cannot swim in the ocean, barnacles are constrained to inhabit a comparatively restricted narrow region, rendering the process of acclimating to the deep-sea milieu arduous. The Southwest Indian Ridge (SWIR) is a typical ultraslow-spreading oceanic ridge, and the Longqi hydrothermal field is the first confirmed active field on the SWIR. Due to the general lack of deep-sea sediments in this hydrothermal zone, which can provide good channels for hydrothermal circulation and spillage, there are a large number of sulfides (Liang et al., 2023). The extreme environment impacts on population vicariance and dispersal, both significant biological processes. However, few researches have focused on the biodiversity of this region (Zhou et al., 2018). Therefore, our research endeavors to fill this gap by examining the effects of N. marisindica and S. stearnsi in the marine habitat, thereby enhancing our understanding of the adaptations in the harsh deep-sea environment.
ATG12, a core autophagy protein. The interaction between ATG12 and its active complex can form autophagosome. Autophagy is an evolutionarily conserved catabolic process that generally promotes cell survival by eliminating intracellular pathogens and removing damaged organelles, playing a pivotal role in cellular homeostasis and adaptation to adverse environments (Chen et al., 2014). During autophagy, ATG12 binds the E2 ubiquitin conjugating enzyme to form sulfolipid intermediate, which further mediate the binding of phosphatidylethanolamine to LC3 to promote autophagy formation (Yuan et al., 2022). Although the regulation of this process remains incompletely understood, it has been found to induce the formation of the autophagosome membrane and type I interferons (IFNs). The process have been shown to play a crucial role in the antiviral immunity of Larimichthys crocea (Richardson) (Wei et al., 2021), Epinephelus coioides (Hamilton) (Li et al., 2019), and Ctenopharyngodon idella (Valenciennes in Cuvier and Valenciennes) (Chu et al., 2019), shedding light on the potential involvement of ATG12 in antiviral immunity by stimulating both autophagy and type I interferon response. Consequently, it is hypothesized that ATG12 may eliminate intracellular pathogens and damaged celles within N. marisindica by regulating autophagy homeostasis, which have contributed to its adaptation of harsh deep-sea environments. Furthermore, previous studies have demonstrated the involvement of ATG12 in various cell biological processes such as viability, apoptosis, and autophagy (Wei et al., 2019). Meanwhile, ATG12 has been found to directly modulate the mitochondrial apoptotic pathway by binding and inactivating prosurvival Bcl-2 family members (Rubinstein et al., 2011). In contrast to barnacles residing in shallow waters, the survival of N. marisindica exposed to diverse toxic substances, including H2S. This exposure may have potentially facilitated the evolution of more effective immune responses and antiapoptotic metabolism (Cooper and Brown, 2008; Liang et al., 2023).
In Addition, the ELF (OG15174) and UPRT (OG15292) genes are also positively selected from the enriched pathways of apoptosis-fly and pyrimidine metabolism in N. marisindica, and they are both involved in adaptive immune responses. ELF, a pleiotropic transcription factor that modulates disease susceptibility and mediates several biotic and abiotic stress processes, playing a significant role in the host innate immune defense system (Suico et al., 2017). pH, temperature, and salinity can affect the immune response of crustaceans. ELF was involved in regulatory processes in organisms through synergistic or retrograde interactions, maintaining osmotic pressure homeostasis (Zhang et al., 2019). Previous researches has demonstrated that ELF was significantly expressed in the hepatopancreas and gill tissue of Penaeus monodon Fabricius under salt stress, showing that ELF was involved in the immune stress process of organisms by regulating immune responses and the development of immune-related cells (Si et al., 2022). Simultaneously, UPRT maintains the immunocompetent and regulates the sensitivity of organisms to harmful cells (Koyama et al., 2000; Hasegawa et al., 2013). UPRT is a pyrimidine salvage enzyme. It can convert 5-flurouracil to a toxic compound, which kills the deleterious cells but subsequent conversion to other metabolites enhances its anti-apoptotic effect, by irreversibly inhibiting thymidylate synthase, DNA synthesis and the processing of rRNA and mRNA (Gopinath and Ghosh, 2008). Thus, it is plausible that these PSGs can potentially contribute to maintaining the immune system of N. marisindica, enabling it to withstand the adverse conditions of the deep sea, including low temperatures and toxic substances. Furthermore, high hydrostatic pressure and low temperatures have caused the inhibition of ligand binding and ion channel function which in turn affected signal transduction (Shinozaki and Yamaguchi-Shinozaki, 2000; Siebenaller, 2000). C3ORF10 has been proposed to regulate actin dynamics, and its expression played a role in regulating actin dynamics during the process of neurite outgrowth (Stradal and Scita, 2006). C3ORF10 is a member of the SCAR/WAVE complex, which is the effector of Rac1 to regulate actin dynamics, regulating Arp2/3-dependent actin polymerization and the number of axonal collaterals and dendrites (Wang et al., 2013). Consequently, the genes that have undergone positive selection and involved energy utilization, immune response, and signal transduction metabolism, might enhance the ability of N. marisindica to effectively employ various diverse adaptive strategies within the extreme conditions of the deep-sea environment. N. marisindica also exhibits enrichment of several significant pathways involving genes subject to positive selection, including the apoptosis-fly, regulation of actin cytoskeleton, pyrimidine metabolism, and ribosome pathways. Functionally, the ribosome pathway (K02971) influences almost every aspect of cell and organismal biology including maintaining cell homeostasis in response to changing environmental conditions and acute stress (Ni and Buszczak, 2023). Moreover, impairment of translation can result in the activation of the ribotoxic stress response, implying that ribosomes serve as a signal for the metabolic regulation of stress response (Snieckute et al., 2022), and the inhibition of ribosomal activity hinders the progression of the cell cycle, thereby regulating the adaptive stress response (Stoneley et al., 2022). Consequently, the presence of genes enriched in the ribosomal pathway indicates that the organisms have experienced environmental stress in their habitat.
This study is the first to sequence the transcriptome of a deep-sea barnacle N. marisindica and compared it with a shallow-water related species. The presence of positive selection in the mitogenome of N. marisindica suggested that this species probably evolved efficient adaption to cope with the challenges posed by the extreme deep-sea conditions, including limited oxygen and food availability, as well as a high concentration of H2S in the hydrothermal vent environment. Moreover, the identified PSGs from the enriched KEGG pathways are involved in crucial biological processes such as neural signal transduction, immunity, and antiapoptotic metabolism. Nevertheless, it should be noted that the findings presented in this study are preliminary, and additional research is needed to fully understand the evolutionary and adaptive processes of PSGs in the hydrothermal vent environment. Nonetheless, this study provided valuable genomic resources and insighted into the mechanisms of N. marisindica and S. stearnsi, serving as a foundation for further investigations into the evolution and adaptation of invertebrates within deep-sea ecosystems.
The Neolepas marisindica and Scalpellum stearnsi datasets generated during the current study are available in the National Center for Biotechnology Information (NCBI) databases in GenBank repository at the following links: https://www.ncbi.nlm.nih.gov/nuccore/MZ772032 and https://www.ncbi.nlm.nih.gov/nuccore/OP345466.
NM: Data curation, Formal analysis, Methodology, Software, Writing – original draft. WS: Data curation, Formal analysis, Methodology, Software, Writing – original draft. YC: Conceptualization, Funding acquisition, Project administration, Resources, Writing – review & editing. XK: Methodology, Project administration, Writing – review & editing. NJ: Methodology, Project administration, Writing – review & editing. XS: Conceptualization, Funding acquisition, Project administration, Resources, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Natural Science Foundation of China (#42376139), the Jiangsu Provincial Basic Research Program (Natural Science Foundation) Distinguished Youth Fund Project (BK20190048), Postgraduate Research & Practice Innovation Program of Jiangsu Ocean University (KYCX2023-69), Jiangsu Agriculture Science and Technology Innovation Fund (JASTIF) [CX (22) 2032], and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
We would like to thank the reviewers and Chan et al. for their detailed comments about misidentification. We agree with the commentary paper of Chan et al. and have changed all reference to Vulcanolepas fijiensis to Neolepas marisindica in our corrected paper. We would also like to thank Professor Haibin Zhang from Institute of Deep-sea Science and Engineering (Chinese Academy of Sciences) for providing the samples and pictures of the deepsea barnacle.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1269411/full#supplementary-material
Al-Aqeel S., Ryu T., Zhang H. M., Chandramouli K. H., Ravasi T. (2016). Transcriptome and proteome studies reveal candidate attachment genes during the development of the barnacle Amphibalanus amphitrite. Front. Mar. Sci. 3. doi: 10.3389/fmars.2016.00171
Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Auger C., Vinaik R., Appanna V. D., Jeschke M. G. (2021). Beyond mitochondria: Alternative energy-producing pathways from all strata of life. Metabolism 118. doi: 10.3389/fmars.2016.00171
Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Biswas S., Akey J. M. (2006). Genomic insights into positive selection. Trends. Genet. 22, 437–446. doi: 10.1016/j.tig.2006.06.005
Buchfink B., Xie C., Huson D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Chan B. K., Prabowo R. E., Lee K. S. (2010). North West Pacific deep-sea barnacles (Cirripedia, Thoracica) collected by the Taiwan expeditions, with descriptions of two new species. Zootaxa 2405, 1–47. doi: 10.11646/zootaxa.2405.1.1
Chan B. K., Dreyer N., Gale A. S., Glenner H., Ewers-Saucedo C., Pérez-Losada M., et al. (2021). The evolutionary diversity of barnacles, with an updated classification of fossil and living forms. Zool. J. Linn Soc 193, 789–846. doi: 10.1093/zoolinnean/zlaa160
Chan B. K., Watanabe H. K., Chen C. (2024). Commentary: Comparative omics analysis of a new deep-sea barnacle species (Cirripedia, Scalpellomorpha) and shallow-water barnacle species provides insights into deep-sea adaptation. Front. Mar. Sci 11, 1374419. doi: 10.3389/fmars.2024.1374419
Chen Z. H., Cao J. F., Zhou J. S., Liu H., Che L. Q., Mizumura K., et al. (2014). Interaction of caveolin-1 with ATG12-ATG5 system suppresses autophagy in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 306, L1016–L1025. doi: 10.1152/ajplung.00268.2013
Chu P. F., He L. B., Yang C., Zeng W. C., Huang R., Liao L. J., et al. (2019). Grass carp ATG5 and ATG12 promote autophagy but down-regulate the transcriptional expression levels of IFN-I signaling pathway. Fish. Shellfish. Immunol. 92, 600–611. doi: 10.1016/j.fsi.2019.06.014
Cooper C. E., Brown G. C. (2008). The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 40, 533–539. doi: 10.1007/s10863-008-9166-6
Da Fonseca R. R., Johnson W. E., O’brien S. J., Ramos M. J., Antunes A. (2008). The adaptive evolution of the mammalian mitochondrial genome. BMC. Genomics 9, 119. doi: 10.1186/1471-2164-9-119
Danovaro R., Snelgrove P. V., Tyler P. (2014). Challenging the paradigms of deep-sea ecology. Trends. Ecol. Evol. 29, 465–475. doi: 10.1016/j.tree.2014.06.002
Feng C. G., Liu R. Y., Xu W. J., Zhou Y., Zhu C. L., Liu J., et al. (2021). The genome of a new anemone species (Actiniaria: Hormathiidae) provides insights into deep-sea adaptation. Deep. Sea. Res. 1. Oceanogr. Res. Pap. 170, 103492. doi: 10.1016/j.dsr.2021.103492
Gan Z. B., Jones D. S., Liu X. M., Sui J. X., Dong D., Li X. Z. (2022). Phylogeny and adaptative evolution to chemosynthetic habitat in barnacle (Cirripedia: Thoracica) revealed by mitogenomes. Front. Mar. Sci. 9. doi: 10.3389/fmars.2022.964114
Gan Z. B., Xu P., Li X. Z., Wang C. S. (2020a). Integrative taxonomy reveals two new species of stalked barnacle (Cirripedia, thoracica) from seamounts of the western pacific with a review of barnacles distributed in seamounts worldwide. Front. Mar. Sci. 7. doi: 10.3389/fmars.2020.582225
Gan Z. B., Yuan J. B., Liu X. M., Dong D., Li F. H., Li X. Z. (2020b). Comparative transcriptomic analysis of deep-and shallow-water barnacle species (Cirripedia, Poecilasmatidae) provides insights into deep-sea adaptation of sessile crustaceans. BMC. Genomics 21, 1–13. doi: 10.1186/s12864-020-6642-9
Gao F. L., Chen C. J., Arab D. A., Du Z. G., He Y. H., Ho S. Y. (2019). EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 9, 3891–3898. doi: 10.1002/ece3.5015
Gattuso J. P., Gentili B., Duarte C. M., Kleypas J., Middelburg J. J., Antoine D. (2006). Light availability in the coastal ocean: impact on the distribution of benthic photosynthetic organisms and their contribution to primary production. Biogeosciences 3, 489–513. doi: 10.5194/bg-3-489-2006
Gopinath P., Ghosh S. S. (2008). Implication of functional activity for determining therapeutic efficacy of suicide genes in vitro. Biotechnol. Lett. 30, 1913–1921. doi: 10.1007/s10529-008-9787-1
Götz S., García-Gómez J. M., Terol J., Williams T. D., Nagaraj S. H., Nueda M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic. Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Greiner S., Lehwark P., Bock R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic. Acids Res. 47, W59–W64. doi: 10.1101/545509
Hasegawa N., Abei M., Yokoyama K. K., Fukuda K., Seo E., Kawashima R., et al. (2013). Cyclophosphamide enhances antitumor efficacy of oncolytic adenovirus expressing uracil phosphoribosyltransferase (UPRT) in immunocompetent Syrian hamsters. Int. J. Cancer. 133, 1479–1488. doi: 10.1002/ijc.28132
Hui M., Cheng J., Sha Z. L. (2018a). Adaptation to the deep-sea hydrothermal vents and cold seeps: insights from the transcriptomes of Alvinocaris longirostris in both environments. Deep. Sea. Res. 1. Oceanogr. Res. Pap. 135, 23–33. doi: 10.1016/j.dsr.2018.03.014
Hui M., Wang M. X., Sha Z. L. (2018b). The complete mitochondrial genome of the alvinocaridid shrimp Shinkaicaris leurokolos (Decapoda, Caridea): Insight into the mitochondrial genetic basis of deep-sea hydrothermal vent adaptation in the shrimp. Comp. Biochem. Physiol. Part. D. Genomics Proteomics. 25, 42–52. doi: 10.1016/j.cbd.2017.11.002
Ki J. S., Dahms H. U., Hwang J. S., Lee J. S. (2009). The complete mitogenome of the hydrothermal vent crab Xenograpsus testudinatus (Decapoda, Brachyura) and comparison with brachyuran crabs. Comp. Biochem. Physiol. Part. D. Genomics Proteomics. 4, 290–299. doi: 10.1016/j.cbd.2009.07.002
Koyama F., Sawada H., Fujii H., Hamada H., Hirao T., Ueno M., et al. (2000). Adenoviral-mediated transfer of Escherichia coli uracil phosphoribosyltransferase (UPRT) gene to modulate the sensitivity of the human colon cancer cells to 5-fluorouracil. Eur. J. Cancer. 36, 2403–2410. doi: 10.1016/S0959-8049(00)00338-5
Kumar S., Stecher G., Tamura K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lan Y., Sun J., Tian R., Bartlett D. H., Li R. S., Wong Y. H., et al. (2017). Molecular adaptation in the world’s deepest-living animal: Insights from transcriptome sequencing of the hadal amphipod Hirondellea gigas. Mol. Ecol. 26, 3732–3743. doi: 10.1111/mec.14149
Lee W. K., Mi Kang H., Chan B. K., Ju S. J., Kim S. J. (2019). Complete mitochondrial genome of the hydrothermal vent stalked barnacle Vulcanolepas fijiensis (Cirripedia, Scalpelliforms, Eolepadidae). Mitochondrial DNA B 4(2), 2725–2726. doi: 10.1080/23802359.2019.1644564
Letunic L., Bork P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic. Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li C., Yu Y. P., Zhang X., Wei J. G., Qin Q. W. (2019). Grouper Atg12 negatively regulates the antiviral immune response against Singapore grouper iridovirus (SGIV) infection. Fish. Shellfish. Immunol. 93, 702–710. doi: 10.1016/j.fsi.2019.08.037
Liang J., Tao C. H., Zheng Y., Zhang G. Y., Su C., Yang W. F., et al. (2023). Geology context, vent morphology, and sulfide paragenesis of the Longqi-1 modern seafloor hydrothermal system on the ultraslow-spreading Southwest Indian ridge. Deep. Sea. Res. 1. Oceanogr. Res. Pap. 194, 103962. doi: 10.1016/j.dsr.2023.103962
Liu J., Liu H. L., Zhang H. B. (2018). Phylogeny and evolutionary radiation of the marine mussels (Bivalvia: Mytilidae) based on mitochondrial and nuclear genes. Mol. Phylogenet. Evol. 126, 233–240. doi: 10.1016/j.ympev.2018.04.019
Lobo-Jarne T., Ugalde C. (2018). Respiratory chain supercomplexes: structures, function and biogenesis. Semin. Cell. Dev. Biol. 76, 179–190. doi: 10.1016/j.semcdb.2017.07.021
Lorion J., Kiel S., Faure B., Kawato M., Ho S. Y., Marshall B., et al. (2013). Adaptive radiation of chemosymbiotic deep-sea mussels. Proc. R. Soc Lond. B. Biol. Sci. 280, 20131243. doi: 10.1098/rspb.2013.1243
Luo Y., Chen Y., Liu F., Gao Y. (2012). Mitochondrial genome of Tibetan wild ass (Equus kiang) reveals substitutions in NADH which may reflect evolutionary adaptation to cold and hypoxic conditions. Asia. Life. Sci. 21, 1–11.
Lyu L., Fang K., Zhu Z., Li J., Chen Y., Wang L., et al. (2023). Bioaccumulation of emerging persistent organic pollutants in the deep-sea cold seep ecosystems: Evidence from chlorinated paraffin. J. Hazard. Mater. 445, 130472. doi: 10.1016/j.jhazmat.2022.130472
Moriya Y., Itoh M., Okuda S., Yoshizawa A. C., Kanehisa M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic. Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Ni C., Buszczak M. (2023). The homeostatic regulation of ribosome biogenesis. Cell. Dev. Biol. 136, 13–26. doi: 10.1016/j.semcdb.2022.03.043
Rausher M. D., Huang J. (2016). Prolonged adaptive evolution of a defensive gene in the solanaceae. Mol. Biol. Evol. 33, 143–151. doi: 10.1093/molbev/msv205
Rota-Stabelli O., Kayal E., Gleeson D., Daub J., Boore J. L., Telford M. J., et al. (2010). Ecdysozoan mitogenomics: evidence for a common origin of the legged invertebrates, the Panarthropoda. Genome. Biol. Evol. 2, 425–440. doi: 10.1093/gbe/evq030
Rubinstein A. D., Eisenstein M., Ber Y., Bialik S., Kimchi A. (2011). The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell. 44, 698–709. doi: 10.1016/j.molcel.2011.10.014
Ryu T., Woo S., Lee N. (2019). The first reference transcriptome assembly of the stalked barnacle, Neolepas marisindica, from the Onnuri Vent Field on the Central Indian Ridge. Mar. Genomics 48, 100679. doi: 10.1016/j.margen.2019.04.004
Sahyoun A. H., Bernt M., Stadler P. F., Tout K. (2014). GC skew and mitochondrial origins of replication. Mitochondrion 17, 56–66. doi: 10.1016/j.mito.2014.05.009
Sato C., Nendai N., Nagata N., Okuzaki Y., Ikeda H., Minamiya Y., et al. (2023). Origin and diversification of pheretimoid megascolecid earthworms in the Japanese Archipelago as revealed by mitogenomic phylogenetics. Mol. Phylogenet. Evol. 182, 107735. doi: 10.1016/j.ympev.2023.107735
Sha Z. L., Ren X. Q. (2015). A new species of the genus Arcoscalpellum (Cirripedia, Thoracica, Scalpellidae) from deep waters in the South China Sea. Chin. J. Oceanol. Limnol. 33, 732–734. doi: 10.1007/s00343-015-4164-0
Shen X., Tsang L. M., Chu K. H., Achituv Y., Chan B. K. K. (2015). Mitochondrial genome of the intertidal acorn barnacle Tetraclita serrata Darwin 1854 (Crustacea: Sessilia): gene order comparison and phylogenetic consideration within Sessilia. Mar. Genomics 22, 63–69. doi: 10.1016/j.margen.2015.04.004
Shinozaki K., Yamaguchi-Shinozaki K. (2000). Molecular responses to dehydration and low temperature: differences and cross-talk between two stress signaling pathways. Curr. Opin. Plant Biol. 3, 217–223. doi: 10.1016/S1369-5266(00)80068-0
Si M. R., Li Y. D., Jiang S. G., Yang Q. B., Jiang S., Yang L. S., et al. (2022). Identification of multifunctionality of the PmE74 gene and development of SNPs associated with low salt tolerance in Penaeus monodon. Fish. Shellfish. Immunol. 128, 7–18. doi: 10.1016/j.fsi.2022.07.010
Siebenaller J. (2000). The effects of hydrostatic pressure on signal transduction in brain membranes of deep-sea fishes of the genus Coryphaenoides. Fish. Physiol. Biochem. 23, 99–106. doi: 10.1023/A:1007830722804
Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Snieckute G., Genzor A. V., Vind A. C., Ryder L., Stoneley M., Chamois S., et al. (2022). Ribosome stalling is a signal for metabolic regulation by the ribotoxic stress response. Cell. Metab. 34, 2036–2046. doi: 10.1016/j.cmet.2022.10.011
Song Y. J., Yoon J. M. (2013). Genetic differences of three Pollicipes mitella populations identified by PCR analysis. Dev. Reprod. 17, 199. doi: 10.12717/DR.2013.17.3.199
Srivastava A., Barth E., Ermolaeva M. A., Guenther M., Frahm C., Marz M., et al. (2020). Tissue-specific gene expression changes are associated with aging in mice. Genomics Proteomics. Bioinf. 18, 430–442. doi: 10.1016/j.gpb.2020.12.001
Stoneley M., Harvey R. F., Mulroney T. E., Mordue R., Jukes-Jones R., Cain K., et al. (2022). Unresolved stalled ribosome complexes restrict cell-cycle progression after genotoxic stress. Mol. Cell. 82, 1557–1572. doi: 10.1016/j.molcel.2022.01.019
Stradal T. E., Scita G. (2006). Protein complexes regulating Arp2/3-mediated actin assembly. Curr. Opin. Cell. Biol. 18, 4–10. doi: 10.1016/j.ceb.2005.12.003
Suico M. A., Shuto T., Kai H. (2017). Roles and regulations of the ETS transcription factor ELF4/MEF. J. Mol. Cell Biol. 9, 168–177. doi: 10.1093/jmcb/mjw051
Sun Y., Wang M. X., Zhong Z. S., Chen H., Wang H., Zhou L., et al. (2022). Adaption to hydrogen sulfide-rich environments: Strategies for active detoxification in deep-sea symbiotic mussels, Gigantidas platifrons. Sci. Total. Environ. 804, 150054. doi: 10.1016/j.scitotenv.2021.150054
Thubaut J., Puillandre N., Faure B., Cruaud C., Samadi S. (2013). The contrasted evolutionary fates of deep-sea chemosynthetic mussels (Bivalvia, Bathymodiolinae). Ecol. Evol. 3, 4748–4766. doi: 10.1002/ece3.749
Timón-Gómez A., Nývltová E., Abriata L. A., Vila A. J., Hosler J., Barrientos A. (2018). Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell. Dev. Biol. 76, 163–178. doi: 10.1016/j.semcdb.2017.08.055
Wang P. P., Mao Y., Su Y. Q., Wang J. (2021). Comparative analysis of transcriptomic data shows the effects of multiple evolutionary selection processes on codon usage in Marsupenaeus japonicus and Marsupenaeus pulchricaudatus. BMC. Genomics 22, 1–14. doi: 10.1186/s12864-021-08106-y
Wang J. L., Tong C. W., Chang W. T., Huang A. M. (2013). Novel genes FAM134C, C3orf10 and ENOX1 are regulated by NRF-1 and differentially regulate neurite outgrowth in neuroblastoma cells and hippocampal neurons. Gene 529, 7–15. doi: 10.1016/j.gene.2013.08.006
Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic. Acids Res. 46, W296–W303. doi: 10.1093/nar/gky427
Wei L., He J., Jia X., Qi Q., Liang Z. S., Zheng H., et al. (2014). Analysis of codon usage bias of mitochondrial genome in Bombyx mori and its relation to evolution. BMC. Evol. Biol. 14, 262. doi: 10.1186/s12862-014-0262-4
Wei H. M., Hu J., Pu J., Tang Q. L., Li W. C., Ma R. H., et al. (2019). Long noncoding RNA HAGLROS promotes cell proliferation, inhibits apoptosis and enhances autophagy via regulating miR-5095/ATG12 axis in hepatocellular carcinoma cells. Int. Immunopharmacol. 73, 72–80. doi: 10.1016/j.intimp.2019.04.049
Wei Z. Y., Wen Q., Li W. R., Yuan X. Q., Fu Q. L., Cui Z. W., et al. (2021). ATG12 is involved in the antiviral immune response in large yellow croaker (Larimichthys crocea). Fish. Shellfish. Immunol. 119, 262–271. doi: 10.1016/j.fsi.2021.10.015
Wilhelm B. T., Landry J. R. (2009). RNA-Seq—quantitative measurement of expression through massively parallel RNA-sequencing. Methods 48, 249–257. doi: 10.1016/j.ymeth.2009.03.016
Xin Q., Hui M., Li C. L., Sha Z. L. (2021). Eyes of differing colors in Alvinocaris longirostris from deep-sea chemosynthetic ecosystems: genetic and molecular evidence of its formation mechanism. J. Oceanol. Limnol. 39, 282–296. doi: 10.1007/s00343-020-9312-5
Yan G. Y., Lan Y., Sun J., Xu T., Wei T., Qian P. Y. (2022). Comparative transcriptomic analysis of in situ and onboard fixed deep-sea limpets reveals sample preparation-related differences. iScience 25, 104092. doi: 10.1016/j.isci.2022.104092
Yan G. Y., Sun J., Wang Z. S., Qian P. Y., He L. S. (2020). Insights into the synthesis, secretion and curing of barnacle cyprid adhesive via transcriptomic and proteomic analyses of the cement gland. Mar. Drugs 18, 186. doi: 10.3390/md18040186
Yoon M., Jung J. Y., Kim D. S. (2013). Genetic diversity and gene flow patterns in Pollicipes mitella in Korea inferred from mitochondrial DNA sequence analysis. Fish. Aquatic. Sci. 16, 243–251. doi: 10.5657/FAS.2013.0243
Yu P., Zhou L., Zhou X. Y., Yang W. T., Zhang J., Zhang X. J., et al. (2019). Unusual AT-skew of Sinorhodeus microlepis mitogenome provides new insights into mitogenome features and phylogenetic implications of bitterling fishes. Int. J. Biol. Macromol. 129, 339–350. doi: 10.1016/j.ijbiomac.2019.01.200
Yuan Y., Huang Y., Jian J., Cai S., Yang S. (2022). Autophagy genes of ATG5-ATG12 complex in response to exogenous stimulations in Litopenaeus vannamei. Isr. J. Aquacult-Bamid. 74, 17. doi: 10.46989/001c.55792
Zhang C., Zhang Q., Pang Y., Song X., Zhou N., Wang J. (2019). The protective effects of melatonin on oxidative damage and the immune system of the Chinese mitten crab (Eriocheir sinensis) exposed to deltamethrin. Sci. Total Environ. 653, 1426–1434. doi: 10.1016/j.scitotenv.2018.11.063
Zhang B., Zhang Y. H., Wang X., Zhang H. X., Lin Q. (2017). The mitochondrial genome of a sea anemone Bolocera sp. exhibits novel genetic structures potentially involved in adaptation to the deep-sea environment. Ecol. Evol. 7, 4951–4962. doi: 10.1002/ece3.3067
Zheng P., Wang M. X., Li C. L., Sun X. Q., Wang X. C., Sun Y., et al. (2017). Insights into deep-sea adaptations and host–symbiont interactions: A comparative transcriptome study on Bathymodiolus mussels and their coastal relatives. Mol. Ecol. 26, 5133–5148. doi: 10.1111/mec.14160
Zhou L., Li M. N., Zhong Z. S., Chen H., Wang X. C., Wang M. X., et al. (2021). Biochemical and metabolic responses of the deep-sea mussel Bathymodiolus platifrons to cadmium and copper exposure. Aquat. Toxicol. 236, 105845. doi: 10.1016/j.aquatox.2021.105845
Zhou Y. D., Zhang D. S., Zhang R. Y., Liu Z. S., Tao C. H., Lu B., et al. (2018). Characterization of vent fauna at three hydrothermal vent fields on the Southwest Indian Ridge: implications for biogeography and interannual dynamics on ultraslow-spreading ridges. Deep. Sea. Res. 1. Oceanogr. Res. Pap. 137, 1–12. doi: 10.1016/j.dsr.2018.05.001
Keywords: barnacle, Neolepas marisindica, deep-sea adaptation, mitogenome, transcriptome
Citation: Mao N, Shao W, Cai Y, Kong X, Ji N and Shen X (2024) Comparative omics analysis of a deep-sea barnacle species (Cirripedia, Scalpellomorpha) and shallow-water barnacle species provides insights into deep-sea adaptation. Front. Mar. Sci. 10:1269411. doi: 10.3389/fmars.2023.1269411
Received: 30 July 2023; Accepted: 28 December 2023;
Published: 12 January 2024.
Edited by:
Andrew R Thurber, Oregon State University, United StatesReviewed by:
Guoyong Yan, Hong Kong University of Science and Technology, ChinaCopyright © 2024 Mao, Shao, Cai, Kong, Ji and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuefeng Cai, eXVlZmVuZ2NhaUBqb3UuZWR1LmNu; Xin Shen, c2hlbnRoaW5AMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.