 Boyuan Huang
Boyuan Huang Jin Zhang2†
Jin Zhang2† Sisi Chen
Sisi Chen Hongbo Zhang
Hongbo Zhang- 1Department of Neurosurgery, Capital Medical University Electric Power Teaching Hospital/State Grid Beijing Electric Power Hospital, Beijing, China
- 2Department of Neurosurgery, The First Affiliated Hospital, Jinan University, Guangzhou, China
- 3Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, China
- 4Department of neurosurgery, Jiujiang Hospital of Traditional Chinese Medicine, Jiujiang, China
- 5Department of Neurosurgery, Shenzhen Key Laboratory of Neurosurgery, Shenzhen Second People’s Hospital, The First Affiliated Hospital of Shenzhen University, Shenzhen, China
- 6Department of Neurosurgery, The Second Affiliated Hospital of Nanchang University, Nanchang, China
Glioblastoma (GBM) is the most common and lethal malignant tumor of the central nervous system in adults. Conventional therapies, including surgery, radiotherapy, and chemotherapy, have limited success in ameliorating patient survival. The immunosuppressive tumor microenvironment, which is infiltrated by a variety of myeloid cells, has been considered a crucial obstacle to current treatment. Recently, immunotherapy, which has achieved great success in hematological malignancies and some solid cancers, has garnered extensive attention for the treatment of GBM. In this review, we will present evidence on the features and functions of different populations of myeloid cells, and on current clinical advances in immunotherapies for glioblastoma.
1 Introduction
Glioblastoma (GBM), accounting for more than 50% of diagnosed intracranial glioma and approximately 12% of all brain tumors, is the most common and lethal malignant tumor of the central nervous system (CNS) in adults (1–3). GBM is of a highly aggressive nature, with fast tumor growth, diffuse tumor invasiveness, and high levels of resistance to conventional therapies (4). It occurs frequently in people aged 50– 60 years and is more prevalent in men (1). GBM is traditionally classified into two groups: primary GBM arising de novo, of which 90% have a wild-type isocitrate dehydrogenase (IDH) profile, and secondary GBM, which develops from low-grade glioma (5). On the basis of a genomic profile, primary GBM can be further categorized into three subgroups: classical (CL) GBM, with abnormal expression of epidermal growth factor receptor (EGFR) and homozygous deletion of cyclin-dependent kinase inhibitor 2A (CDKN2A); proneural (PN) GBM, which is characterized by the amplification of platelet-derived growth factor receptor alpha (PDGFRα) and a tumor protein p53 (TP53) mutation; and mesenchymal (MES) GBM featuring co-mutated phosphatase and tensin homolog (PTEN) and TP53 tumor suppressor genes and the lack of the neurofibromatosis type 1 (NF1) gene function (5–8). It has been demonstrated that the MES subgroup showed the shortest median survival of 11.5 months, compared with 17 months for the PN subgroup and 14.7 months for the CL subgroup (6). Secondary GBM harbors IDH1 and IDH2 mutations, which can convert α-ketoglutarate (α-KG) to the oncometabolite 2-hydroxyglutarate (2-HG) to initiate tumorigenesis (6).
The current standard treatment for GBM, comprising a combination of maximal surgical resection, radiation, and chemotherapy, has limited success in improving the prognosis of patients, who are given only 15 months of median survival and a 5-year survival rate of less than 5% (9–11). Thus, opportunities and challenges remain in finding more efficient treatments for GBM. Currently, immunotherapy, which has obtained great success in treating hematopoietic malignancies, malignant melanoma, and some solid tumors, has garnered extensive attention for treating GBM (12). Current immunotherapy mainly focuses on the investigation of vaccine therapy, adoptive T-cell therapy, immune checkpoint inhibitors (ICIs) therapy, and oncolytic virus therapy. Unfortunately, immunotherapeutic successes in GBM are still lacking (13). Multifarious factors have attenuated the immunotherapeutic efficiency for GBM. It has been long recognized that the brain is an immune-privileged organ based on the restriction from the brain–blood barrier (BBB) (14). However, this concept has been challenged by the existence of functional lymphatic vessels and varied types of leukocytes in the CNS. Today, the brain is considered immunologically “distinct” rather than “privileged” (15). Moreover, a consensus has been reached on the idea that the immunosuppressive tumor microenvironment (TME) of GBM plays a crucial role in the resistance to current treatment. The GBM microenvironment is intensively infiltrated with a variety of myeloid cells, including bone marrow-derived macrophages (BMDMs), microglia, myeloid-derived suppressor cells (MDSCs), and neutrophils. Recent flow cytometric and single-cell analyses have revealed that glioma cells (40.5%) and myeloid cells (45%) constitute the tumor mass, both of which can secrete cytokines and metabolites to suppress tumor-infiltrating lymphocyte (TIL) function (16, 17). Thus, the lack of TILs and abundant immunosuppressive myeloid cells constitute a significant barrier against immunotherapy efficacy in GBM. A recent study that combined single-cell analysis and spatial transcriptomics identified reactive-hypoxia and reactive-immune regions in the GBM TME. These regions were found to be enriched with memory and exhausted T cells as well as myeloid cells, suggesting a local enhanced immunosuppression (18). Moreover, through analysis of cell-to-cell interactions among different cancer and immune cells, researchers found that bidirectional signaling between glioma cells and myeloid cells, and myeloid cells and T cells is much more abundant than signaling between T cells and glioma cells, indicating that the myeloid cells are major conduits of cell-to-cell interaction in the GBM microenvironment (19). Accordingly, investigating the features and functions of these myeloid cells as well as their interplay with cytotoxic lymphocytes could provide insight into the establishment of the TME, which could be applied to the development of immunotherapies against GBM. In this review, we will present evidence on the features and functions of different populations of myeloid cells, including those of BMDMs, microglia, MDSCs, and neutrophils. Moreover, we will also present evidence of current advances in immunotherapies for GBM, including vaccine therapy, adoptive T-cell therapy, ICIs therapy, and oncolytic virus therapy.
2 Myeloid cells in GBM
2.1 Microglia and macrophages in GBM
Tumor-associated macrophages (TAMs), which originate from bone marrow-derived monocytes, represent one of the most prominent populations in the tumor stroma. In GBM, the TAMs, also called glioma-associated macrophages/microglia (GAM), are composed of BMDMs and microglia, which constitute the majority of tumor-invading myeloid cells and approximately 30% of the tumor mass (20–22). Whereas microglia, derived from yolk sac progenitors, mainly reside in peritumoral regions in GBM, BMDMs, derived from hematopoietic stem cells (HSCs), are preferentially located in perivascular regions in GBM (23–25). Traditionally, microglia and BMDMs share the same marker of CD11b; however, microglia are distinguished by low levels of expression of CD45. Considering the phenomenon that activates microglia can rapidly up-regulate CD45 expression, two new markers are proposed to distinguish microglia from macrophages: the transmembrane protein 119 (TMEM119) and purinergic receptor P2RY12 (26–28). Typically, microglia and macrophages can be stimulated into two different polarizations in vitro: the pro-inflammatory M1 phenotype and the anti-inflammatory M2 phenotype. In cancer, this M1/M2 classification has also been used for decades to evaluate whether microglia and macrophages have anti-tumor (M1) or tumor-promoting (M2) characteristics (29, 30). However, compelling evidence suggests that this categorization does not apply to in vivo conditions because it only describes the extremes of a broader spectrum of functional states (31). This is supported by microarray analysis of GAMs isolated from GL261-implanted C57BL/6 mouse brains, which showed a different profile from the M1 and M2 phenotypes, including a mixture of M1- and M2-specific genes (32). Moreover, genetic analysis of TAMs from GBM patients demonstrated that these cells exhibited diverse immunological functions as a non-polarized M0 phenotype (33). However, a recent study has argued that GAM subtypes in vivo are not directly equivalent to in vitro-defined M0-, M1-, or M2-like macrophages (34). Recently, a single-cell analysis of GBM multiregionally and multidimensionally identified four molecular subtypes of microglia: MC1 (i-Mic), with high levels of expression of activated microglia markers and TNF, IL1B, and NFKBIZ; MC2 (h-Mic), with the highest level of expression of the homeostatic microglia marker CST3; MC6 (AP-Mic), with expression of both microglia and macrophage markers; and MC7 (a-Mic), identified by differential levels of expression of SPRY1, PYRY13, and microglia activation markers (19). Moreover, a further investigation of the relationship between the gene signature and patient survival showed that MC2 and MC7 were related to better survival, indicating an anti-tumorigenic role in the TME. Accordingly, a comprehensive and functional classification system is still needed to describe the diverse states of GAMs in GBM.
In the TME, the GAMs predominantly exhibit an anti-inflammatory M2 polarization and reduced phagocytic activity (35, 36). GAMs can be driven to the TME by a variety of chemokines, including CXCL12, CCL2, CX3CL1, MCP-1, MCP-3, glial cell-derived neurotrophic factor (GDNF), and osteopontin released by neoplastic cells (36–41). Moreover, GBM cells can induce GAM invasion by expressing the macrophage-stimulating factor CSF-1, which regulates macrophage proliferation and differentiation, and guides the macrophages to polarize toward the protumorigenic M2 phenotype (42). Recently, the secretion of periostin from glioma stem cells (GSCs) has also been demonstrated to support macrophage recruitment and transition into an M2 phenotype, and the deletion of POSTN in GSCs resulted in a dramatic reduction of GAM density and GBM growth, as well as an increased survival rate of mice bearing GSC-derived xenografts (43). Once infiltrated into the TME and induced by GBM into a protumoral phenotype, GAMs can promote the tumor cells’ growth and invasion through multifarious pathways. The GBM-associated microglia can secrete epidermal growth factor (EGF) to facilitate GBM cell migration and invasion through binding to the epidermal growth factor receptor (EGFR) on GBM cells (42). Moreover, the GAMs can release the tumor growth factor beta (TGF-β) 1 to bind the TGF-β receptor (TGFBR) 2 (TGFBR2) on GBM cells, which can promote tumor invasion (44). Furthermore, GBM-associated microglia can up-regulate matrix metallopeptidase (MMP14), which can cleave into an inactive form of MMP2 and thus facilitate the invasion of glioma cells into the brain parenchyma by metalloproteinase-mediated degradation of the extracellular matrix (45). Although microglia are the most powerful phagocytes in the TME, their phagocytic function is impaired by GBM cells through rendering microglia as an anti-inflammatory, antiphagocytic M2 phenotype (46). For example, GBM cells can secrete molecules, such as CSF1 and CSF2, to induce the shift of GAMs toward a protumoral phenotype and thus create a favorable TME for GBM growth (42, 46). Moreover, GBM cells can also inhibit microglial phagocytosis by overexpressing sialic acid-rich glycoproteins and up-regulating CD47, which often acts as an antiphagocytic protein in cancer cells (47–49).
Taken together, GAMs are plastic and heterogeneous in their profiles and functions. The recruitment of GAMs is regulated by multiple factors. Induced into a protumoral phenotype in the TME, GAMs play a critical role in promoting the proliferation, invasion, and function of GBM cells.
2.2 Myeloid-derived suppressor cells in GBM
Myeloid-derived suppressor cells (MDSCs), presenting at very low frequencies of peripheral blood mononuclear cells, are a heterogeneous cluster of immature myeloid cells (IMCs) that can be differentiated into macrophages, dendritic cells (DCs), and granulocytes under physiological conditions (31, 50, 51). In pathological conditions such as GBM, the IMCs are driven to differentiate into MDSCs not only in the tumor bed but also in the peripheral blood (51). Human MDSCs are generally characterized by co-expressions of the myeloid differentiation markers, CD33 and CD11b, and with negative expression of markers of mature lymphoid and myeloid cells, such as human leukocyte antigen DR(HLA-DR) and major histocompatibility complex (MHC) class II molecule (52). In the human body, there are three major groups of MDSCs: early-stage MDSCs (eMDSCs), characterized by CD33+ and Lin–; granulocytic or polymorphonuclear MDSCs (PMN-MDSCs), defined as CD11b+Ly6G+Ly6Clow and CD33+CD15+CD14–HLA-DRlow/–; and monocytic MDSCs (M-MDSCs), featuring CD11b+CD33+CD14+ Ly6Chigh and Ly6G–D15–HLA-DRlow/–. Although PMN-MDSCs are morphologically and phenotypically analogous to neutrophils, M-MDSCs are similar to monocytes (52–54).
Increasing evidence demonstrates that MDSCs are elevated in the peripheral blood of GBM patients (55). In GBM patients, elevated MDSC accumulation at the time of recurrence predicts poor prognosis, whereas the reduction of MDSCs is correlated with extended survival (55). With respect to the subgroups of MDSCs, although both PMN- and M-MDSCs were found to be increased in the peripheral blood of GBM patients, only PMN-MDSCs were found to be increased in the TME of GBM patients (56). Moreover, the two subsets of MDSCs were found to expand and thus drive immune suppression in a sex-specific manner (57). For example, the proliferation of M-MDSCs was predominant in tumors from male GBM patients, whereas PMN-MDSCs were enriched in female GBM patients’ blood, with significant relation to poor prognosis (57). In addition, M-MDSCs have also been claimed to constitute the major population of MDSCs within the glioma microenvironment. In accordance with these findings, the improvement in survival induced by the depletion of M- and PMN- MDSCs in GBM mouse models was also displayed in a sex-specific manner (57).
In GBM, MDSCs play critical roles in the sustenance of tumor growth, invasion, vascularization, and immunosuppression (58). Several mechanisms have been demonstrated to affect the accumulation of MDSCs in the GBM TME and the promotion of their immunostimulatory phenotype, which enables them to exert their immunosuppressive effects. GBM cells can recruit MDSCs from the bone marrow through up-regulation of indoleamine 2,3-dioxygenase (IDO1) and CD200, as well as secretion of macrophage migration inhibitory factor (MIF) and CCL20 (59–61). Moreover, they can also overexpress hypoxia-inducible factor 1 alpha (HIF1α) and vascular endothelial growth factor (VEGF) to promote the recruitment of MDSCs by converting extracellular ATP to 50-AMP (62). After accumulation in the TME, the proliferation and functions of MDSCs are controlled by various factors, such as CCR2, IL-6, IL-10, IL-8, IL-12, TGF-β, M-CSF, galectin-1,GM-CSF, and INF-γ, which can stimulate JAK, STAT1, STAT3, STAT6, CCAAT/enhancer-binding protein (C/EBPs), S100A8, S100A9, and prostaglandin E2 (PGE2) in MDSCs to promote their proliferation (63–65).
MDSCs can contribute to immunosuppression by suppressing the anti-tumor activity of cytotoxic T cells, inhibiting the functions of natural killer (NK) cells, macrophages, and DCs, and introducing the accumulation of regulatory T cells (Tregs) and immune-suppressive regulatory B cells (Bregs) (66–69). In glioma rat models, MDSCs can produce NO, iNOS, ARG1, IDO, ROS, and peroxynitrite (PNT), and secrete anti-inflammatory cytokines such as IL-10 and TGF-β to suppress T-cell proliferation, induce T-cell apoptosis, and attenuate T-cell cytotoxic function (70–73). In addition, MDSCs can overexpress ICIs such as the program death 1 ligand (PD-L1), which binds to its receptor PD1 to cause T-cell exhaustion (74). In accordance with T cells, MDSCs can also inhibit the cytotoxicity of NK cells. For example, the TGF-β1 on MDSCs can attenuate cytotoxicity of NK cells by down-regulating the expression of INFγ and activating the receptor NKGD. Moreover, a subset of MDSCs in mice were found capable of suppressing perforin production of NK cells by regulating the Janus Kinase-Signal transducers and activators of transcription (JAK-STAT) pathway (67, 75). With respect to DCs, MDSCs-derived NO can suppress the antigen presentation from DCs to CD4+ T cells, which can be abrogated by treatment of MDSC with iNOS inhibitors (76). In addition, MDSC can suppress TLR-ligand-induced IL-12 production of DCs via IL-10 secretion and inhibit the T-cell stimulatory function of DCs (77).
Apart from the inhibition of T cells, NK cells, and DCs, MDSCs can also exert an immunosuppressive function by recruitment and regulation of Tregs, Bregs, and M2 macrophages. MDSCs can release cytokines such as IL-10, INFβ, and TGF-β to drive naive T cells to differentiate into Tregs, and chemokines such as CCL3, CCL4, and CCL5 through CCR5 to recruit Tregs into the TME (78, 79). In return, Tregs can affect the survival and/or the proliferation of MDSCs and their direct interactions can suppress the functions and promote the apoptosis of CD8+ T cells (80). In addition, MDSCs can recruit Bregs, which account for approximately 40% of the immune-infiltrating cells in GBMs, into the TME to exert their immunosuppressive functions. Bregs can inhibit CD8+T-cell cytotoxic functions by overexpressing PD-L1 through the uptake of MDSC-derived microvesicles containing PD-L1. In addition, Bregs can also up-regulate CD155 expression and secrete immunosuppressive cytokines IL-10 and TGF-β to suppress CD8+ T-cell cytotoxicity (81). With respect to macrophages, the presence of MDSCs in tumors is correlated with a declined level of the anti-inflammatory M1 macrophages and an elevated level of pro-tumor M2 macrophages (77). In addition, MDSC-derived IL-10 can inhibit macrophages from secreting IL-6 and TNF-α (82, 83). Furthermore, MDSCs can down-regulate the MHC class II expression on macrophages to suppress their antigen presentation functions (82, 83). Lastly, it was demonstrated that MDSCs themselves can differentiate into TAMs when faced with hypoxic conditions in the TME (84).
In summary, MDSCs play a crucial role in sustaining immunosuppression in the TME and their recruitment into the TME is regulated by GBM cells and other factors. MDSCs contribute to the immunosuppressive TME not only by suppressing the maturation, stimulation, and cytotoxic functions of T cells, DCs, and NK cells, but also by supporting the recruitment and inhibitory functions of Tregs, Bregs, and M2 macrophages. Further investigations will be carried out to explore the detailed mechanisms (Figure 1).
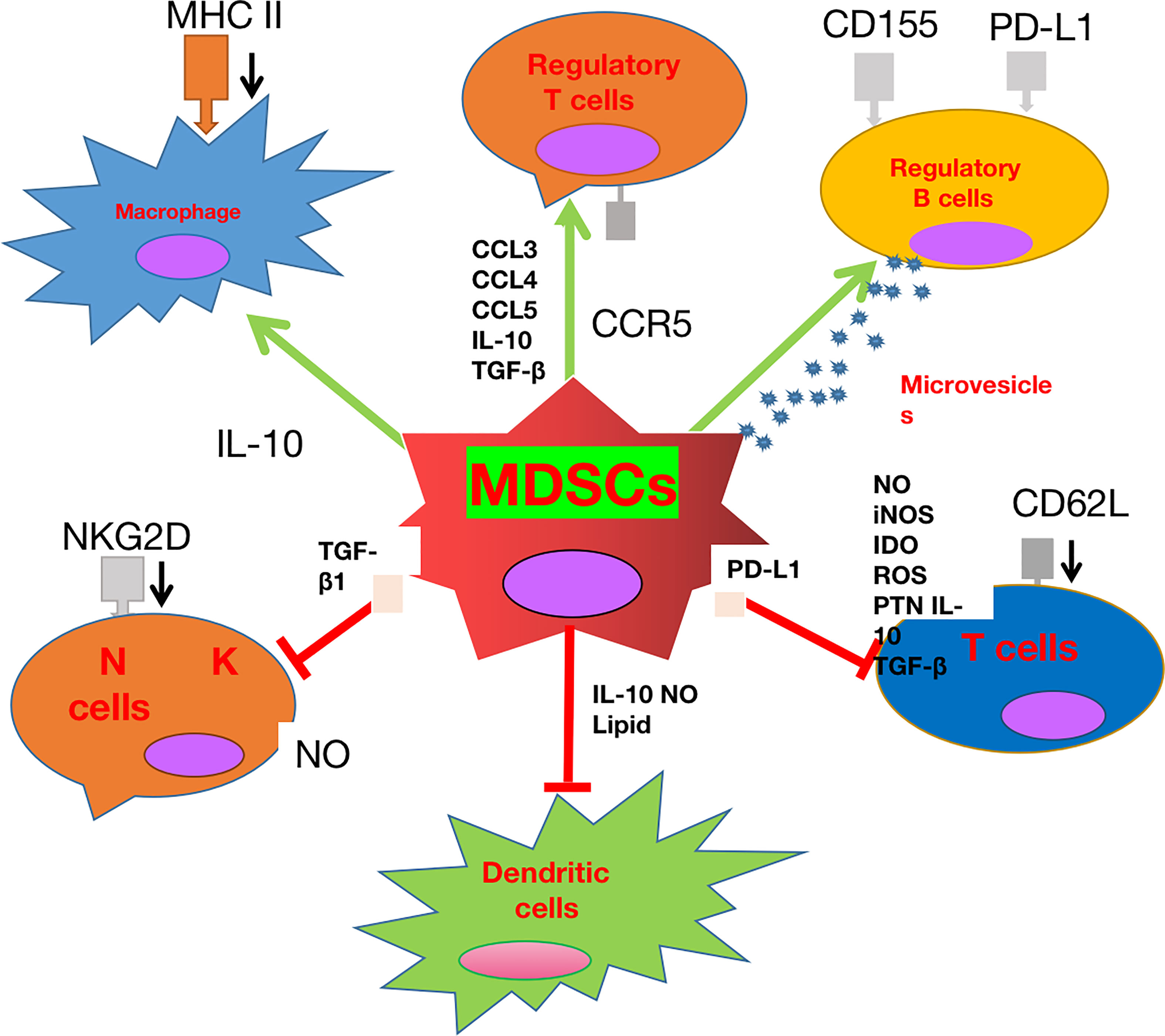
Figure 1 The role played by myeloid-derived suppressor cell (MDSC) glioblastoma (GBM). MDSCs can not only inhibit the cytotoxic effect and antigen presentation functions of T cells, natural killer (NK) cells, and dendritic cells (DCs), but also promote the recruitment and stimulation of immune-suppressive M2 macrophages, regulatory T cells (Tregs), and regulatory B cells (Bregs).
2.3 Neutrophils in GBM
Neutrophils, as the first respondent against pathogens, are the most abundant circulating leukocytes in humans, with multiple functions including phagocytosis, secretion of reactive oxygen species and granules, and formation of neutrophil extracellular traps (85). Tumor-associated neutrophils (TANs) display two typical phenotypes of polarization, which can switch into one another during tumor progression: the anti-tumoral N1 polarization elicited mainly by IFN-β and the protumoral N2 polarization induced by G-CSF, TGF-β1, and IL-6 (86). The number of circulating and tumor-infiltrating neutrophils is correlated with glioma grade (87–89). In GBM, clinical data suggests that an elevated level of neutrophils is negatively correlated with patients’ prognosis (90, 91). It was found that the stimulation of neutrophils was correlated with elevated levels of IL-12 in GBM patients, which was considered an early sign of tumor progression (87). Moreover, GBM patients with high levels of stimulated neutrophils were found to have a worse prognosis than those with low levels (91). Accordingly, the stimulation of neutrophils also has a negative prognostic value for GBM patients. In addition, the neutrophil-to-lymphocyte ratio (NLR) in blood is positively related to glioma grade, and elevated baseline NLRs predict poor prognosis in GBM patients (88, 92–94).
Neutrophils can be recruited into the TME by various cytokines, such as IL8, MIF, and CXCL8, which in turn leads to aggressive tumor growth and therapeutic resistance in GBM (95–97). Once infiltrated into the TME, neutrophils can secrete elastase, which further facilitates neutrophil infiltration and promotes GBM cell invasion. For example, neutrophil infiltration can increase the expression of S100A4, which mediates the tumor progression with mesenchymal characteristics, favoring glioma invasion and resistance to anti-VEGF therapies (98). In addition to this, neutrophils can form neutrophil extracellular traps (NETs) to protect glioma cells and facilitate tumor progression (99). A recent study showed that radiation-induced senescence in GBM cells promoted the recruitment of Ly6G+ (TANs) through NFκB signaling (100, 101). However, these infiltrated Ly6G+ cells can in return support the conversion of GBM cells to glioma stem cells (GSCs) through dedifferentiation and nitrosative stress signaling (102). This mechanism was confirmed by the fact that Ly6G-neutralizing antibodies and NFκB inhibitors reduced GSCs and prolonged the survival of GBM-bearing mice (101). In addition, neutrophils can mediate tumor cell ferroptosis by inducing iron-dependent accumulation of lipid peroxides through transferring myeloperoxidase-containing granules into tumor cells. This ferroptosis induced by neutrophils was identified as a pro-tumorigenic role in GBM because of its promotion of tumor necrosis (103). Recently, results from CIBERSORT analyzing immune cell fractions among the molecular subtypes of GBM showed that mesenchymal GBM had a significantly elevated level of TANs compared with other subtypes, implying a potential role in the extreme immunosuppression of the TME and plausibly mediating immunotherapy resistance (104).
Taken together, circulating neutrophils and TANs are generally related to invasive tumor proliferation, tumor immunosuppression, and the poor prognosis of GBM. Further investigations are needed to explore the detailed mechanisms and establish a therapeutic strategy based on the neutrophils (Figure 2).
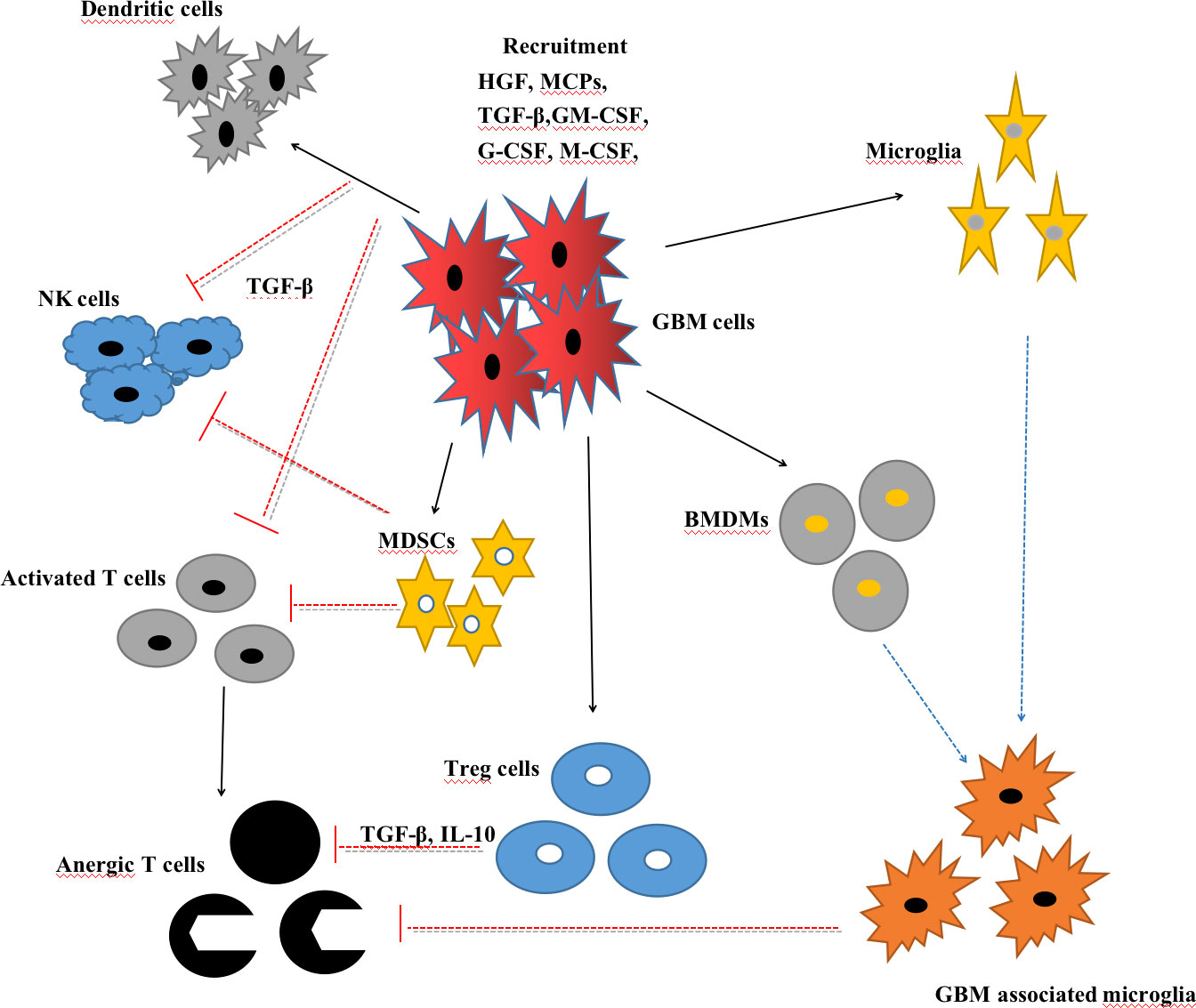
Figure 2 Interactions between glioblastoma (GBM) cells and myeloid cells, including microglia, GBM-associated microglia, bone marrow-derived macrophages (BMDMs), myeloid-derived suppressor cells (MDSCs), T cells, natural killer (NK) cells, and regulatory T cells (Tregs).
3 Current immunotherapy against GBM
The goal of immunotherapy is to harness the hosts’ innate and adaptive immune system by enhancing or suppressing immune responses to promote cancer eradication and overcome cancer’s immune resistance. Recently, immunotherapy has obtained great achievements in melanoma, renal cell carcinoma, Hodgkin’s lymphoma, and non-small cell lung cancer (NSCLC), in which conventional therapies have gained limited success (105–108). With the 2018 Nobel Prize in Medicine awarded to Tasuku Honjo and James Allison for their discovery of the inhibition of negative immune regulation in cancer therapy, immunotherapy has now taken the leading position in cancer therapies (12). In GBM, several immunotherapies including vaccine therapies, ICIs therapies, oncolytic virus therapy, and adoptive T-cell therapy have been investigated alone or combined with standard treatment in preclinical and clinical research.
3.1 Vaccine therapy
The vaccine therapy, which aims to enhance the adoptive immune response in the brain against GBM cells, generally involves two forms of therapies: peptide vaccines and DC vaccines. Peptide vaccines are generally designed to encompass tumor-specific antigens (TSAs) or tumor-associated antigens (TAAs) to induce a potent immunity. Currently, peptide vaccines that target a single GBM antigen include EGFRvIII, IDH1R132H, Wilms’ tumor 1 (WT1), and survivin. Results from phase II clinical trials have demonstrated that rindopepimut, an EGFRvIII-based vaccine, has a beneficial role in improving progression-free survival (PFS) and median overall survival (OS) in both primary and recurrent GBM patients (109, 110). However, no such therapeutic benefit of rindopepimut was observed in a randomized phase III clinical trial among newly diagnosed GBM (ndGBM) patients (111). Similar to EGFRvIII, the WT1-based vaccine has also shown benefits in improving GBM patients’ survival in a non-randomized trial (112). Recently, a novel vaccine named IMA950 that contains nine synthetic tumor-associated HLA-A2-restricted peptides (TUMAP), and thus can trigger the TUMAP-specific cytotoxic T cells, has shown therapeutic effect in improving the PFS and OS of ndGBM patients in phase I/II clinical trials (113, 114). Further evidence from phase III clinical trials is still lacking. GBM is notorious for its high heterogeneity and low mutational burden (115); thus, vaccines that target a single tumor antigen may theoretically lead to antigen escape. This was confirmed by the phenomenon in the EGFRvIII vaccine research, in which most patients experiencing recurrence had lost EGFRvIII expression (116). Therefore, alternative vaccine strategies that target multiple tumor neoantigens are needed. Heat shock protein (HSP) peptide complex 96 (HSPPC-96), a primary resident chaperone of the endoplasmic reticulum that can be internalized into antigen presenting cells (APCs) for efficient class I and II MHC-mediated presentation of tumor peptides, is a solution to this problem. In phase I and II clinical trials, HSSPC-96 vaccination showed a therapeutic effect in improving the OS in high-grade glioma and GBM patients (117). More evidence from phase III clinical trials is needed.
Dendritic cell (DC) vaccines are typically produced through the ex vivo generation of DCs harvested from patients and stimulated with either tumor antigens or mRNA-expressing MHC molecules, before being administered back to patients (118, 119). Similar to the peptide vaccine, the DC vaccines can also be loaded with a single tumor antigen or multiple tumor antigens. A WT1- and cytomegalovirus phosphoprotein 65 RNA (CMV pp65)-based DC vaccine is generated from a single tumor antigen, both of which have demonstrated efficiency in improving the PFS and OS of GBM patients in phase I clinical trials (120–123). On the other hand, DC vaccines loaded with multiple tumor antigens are, in theory, thought to induce more robust immune responses. ICT-107 is a DC vaccine loaded with class I peptides from a TAA highly expressed on gliomas and glioma stem cells (GSCs). It presented a survival benefit in a phase I clinical trial of 15 GBM patients, among whom six patients showed no evidence of tumor recurrence at a follow-up of over 40 months (124). Another α-type 1 polarized DC vaccine loaded with EphA2, IL13Ra2, YKL-40, and gp100 also showed survival benefits for recurrent glioma patients, with one patient achieving a sustained complete response (125). In addition, a novel DC vaccine named DCVax-L, which is loaded with tumor lysates, has shown survival benefits for GBM patients in a phase I/II clinical trial. Phase III clinical trial of DCVax-L in GBM patients is ongoing, and preliminary results indicate that it may improve patients’ OS (126).
3.2 Adoptive T-cell therapy
The primary forms of adoptive T-cell therapy can be generally classified as T-cell receptor (TCR) treatment, tumor-infiltrating lymphocytes (TILs), and chimeric antigen receptor T (CAR-T) cells. TCR treatment is the first successful application of an adoptive T-cell therapy, which utilizes autologous T cells transduced with human TCR to target a melanoma antigen recognized by T cells 1 (MART-1) in order to treat metastatic melanoma patients (127). However, no achievement has been obtained in clinical trials based on TCR-T cell treatment. In addition, the administration of autologous TILs has induced regressions in metastatic melanoma patients (128); however, such powerful cytotoxicity was not seen in GBM patients (129). In fact, the application of TILs requires highly accessible and immunogenic tumor cells. Melanoma can meet sufficient isolation and expansion of TILs from their respective tumor samples, but cannot (128) when it comes to GBM characterized by a high heterogeneity and a low mutational burden. The limited progress made in TCR and TILs against GBM has necessitated the development of CAR-T cell therapy.
Genetically engineered T cells express chimeric antigen receptors (CARs), which consist of an intracellular T-cell signaling domain with one or more single-chain variable fragments (scFvs) and an extracellular antigen recognition domain to target specific neoplastic cells (130, 131). The advantages of CAR-T cells lie in not only the recognition of specific antigens and stimulation without MHC limitation, but also their capability of generating an anti-tumor immune microenvironment featuring decreased levels of anti-inflammatory factors and increased levels of pro-inflammatory factors (132, 133). Recently, CD19-specific CAR-T cell therapy has shown excellent therapeutic safety and efficacy in hematological malignancies and solid cancers, and is therefore preferred in clinical treatment (134–139). Owing to these advances in other kinds of malignancies, CAR-T cell therapy is of extreme interest in GBM treatment and can be applied intravenously, intracranially, or intralesionally (140). So far, results from clinical trials of GBM patients are available for CAR-T cells targeting three antigens: EGFRvIII, IL-13 receptor alpha 2(IL-13Rα2), and human epidermal growth factor receptor 2 (HER2) (141–144). EGFRvIII is supposed to enhance GBM proliferation, angiogenesis, and invasiveness (145), and IL-13Rα2 is associated with GBM invasiveness and poor prognosis; both are GBM-specific antigens (146, 147). HER2 is a GBM-associated antigen that has been identified as an independent unfavorable
prognostic indicator for GBM patients (148). CAR-T cells that target these three antigens showed feasibility, safety, and potential efficiency in early clinical trials against GBM (141–144). However, CAR-T cell therapy is still faced with several intractable obstacles to the treatment of GBM. First, the non-uniform antigen expression resulting from the high heterogeneity of GBM will lead to antigen escape and tumor recurrence. For example, a recurrent GBM (rGBM) patient obtained tumor intracranial and spinal regression after intracranial administration of IL-13Rα2-targeted CAR-T cells, but IL-13Rα2-negative tumors resulted in a subsequent relapse (147). Second, CAR-T cell proliferation and persistence in vitro and CAR-T exhaustion in the TME is also an intractable problem that requires maximal sustenance of CAR-T cells. Last, the CAR-T cells’ administration may trigger some resistant mechanisms, including up-regulation of immunosuppressive factors (e.g., IDO1, PD-L1) and recruitment of immunosuppressive cells (e.g., Tregs, MDSCs) (140, 149). These obstacles require CAR-T cell therapy to target multiple antigens or to be combined with other therapies. Hegde et al. successfully created bivalent CAR-T cells (targeting HER2 and IL-13Rα2) and trivalent CAR-T cells (targeting HER2, IL-13Rα2, and EphA2), both of which showed more efficacy in preclinical studies than monovalent CAR-T cells (150–152). Notably, trivalent CAR-T cells can capture nearly 100% of tumor cells and exhibit superior anti-tumor efficacy in a murine model (151). That is, to deal with the immunosuppressive TME, researchers have exploited the advantages of an oncolytic virus that has been demonstrated to enhance anti-tumor immunity and regulate the TME to improve CAR-T cells for solid tumor therapy (152, 153). Recently, researchers have successfully promoted de novo CD19-CAR-T cells to target solid tumor cells by tumor-selective delivery of oncolytic viruses encoded a truncated CD19 protein (154).
3.3 Immune checkpoint inhibitors therapy
Immune checkpoint inhibitors (ICIs) are antibodies that block the endogenous negative regulatory pathways mediated by immune checkpoints to reduce their inhibiting effects on T-cell stimulation, proliferation, and function. Currently, ICIs that target cytotoxic T lymphocyte-associated antigen 4 (CTLA4) or programmed cell death protein 1 and its ligand (PD-1 and PD-L1) have been approved by the US Food and Drug Administration (FDA) for clinical application owing to their notable successes against melanoma and other solid malignancies (155–159). For the treatment of GBM, results from preclinical studies are encouraging; however, clinical success is still lacking (160–162). Recently, three phase III clinical trials (CheckMate 143, 498, and 548) investigated nivolumab, a PD-1 monoclonal antibody, alone or combined with another therapy for the treatment of rGBM or ndGBM patients. All these trials have declared a failure to meet the primary end point of OS, although some results await publication (163, 164).
Multifarious factors attenuate the efficiency of the PD-1 inhibitor in treating GBM patients, including extensive tumor heterogeneity, low tumor mutation burden (TMB), T-cell dysfunction and exhaustion, and DNA mismatch repair (dMMR) system status (165–167). The extensive tumor heterogeneity will lead to a heterogeneous expression of PD-1 in GBM, which is negatively correlated with patient prognosis (168). Recent studies show that tumors with high TMB are more responsive to ICIs (169, 170). In GBM, high TMB usually arises late after the extended application of chemotherapy. This suggests that patients with recurrent malignancies who have accepted chemotherapy may be more responsive to ICIs, and TMB should be regarded as a biomarker to select patients who are more likely to benefit from ICIs. Accordingly, the FDA has identified a high level of TMB [≥ 10 mutations per megabase (mut/mb)] as one of the inclusion criteria for choosing patients with malignancies for ICIs therapy, including those with gliomas (171). Clinical data show that only a subgroup of GBM patients has benefited from ICIs therapy and obtained prolonged survival (172). For example, in a phase II clinical trial, 35 rGBM patients accepted pembrolizumab (PD-1 monoclonal antibody) as neoadjuvant or adjuvant-only therapy. Patients accepting neoadjuvant pembrolizumab showed a statistically significant increase in OS and PFS compared with those in the adjuvant group. It was also demonstrated that neoadjuvant ICIs therapy was related to a tumor-specific up-regulation of the IFN-g-responsive gene signature and a declined cell cycle-related gene signature in the tumor (173). Moreover, another factor that limits the efficacy of ICIs therapy is T-cell dysfunction and exhaustion, which has long been identified as a hallmark of GBM (174–176). Apart from the PD-1/PD-L1 pathway, GBM is characterized by the up-regulation of multiple alternative immune checkpoints such as T-cell immunoglobulin and mucin domain 3 (TIM-3), B7 homolog 3 (B7-H3), indoleamine 2,3-dioxygenase-1 (IDO1), and lymphocyte activation gene 3 (LAG3), which usually mediate T transition into a dysfunctional, exhausted status (177–179). From this perspective, multitargeted inhibitors or combined ICIs therapies may theoretically be more likely to avoid T-cell dysfunction and exhaustion. Recently, a preclinical study has highlighted a possible combination of PD1, LAG3, and TIM3 blockade in a glioma murine model (180). Furthermore, antibodies targeting TIM-3 and LAG3 either alone or combined with anti-PD1 blockade are being tested among GBM patients clinically (NCT02658981, NCT02817633). In addition, the DNA mismatch repair (dMMR) system status, which is responsible for correcting DNA mismatches during replication to maintain DNA stability, is also associated with the efficacy of ICIs therapy (181). It has been demonstrated that GBM with dMMR deficiency is correlated with high TMB and overexpressed neoantigens, which are usually identified as “immunoresponsive” (182). Clinically, high-grade glioma patients without dMMR expression showed statistically longer OS and PFS after pembrolizumab administration than those with weak dMMR expression, indicating that patients without dMMR expression may benefit from pembrolizumab monotherapy (183) (Table 1).
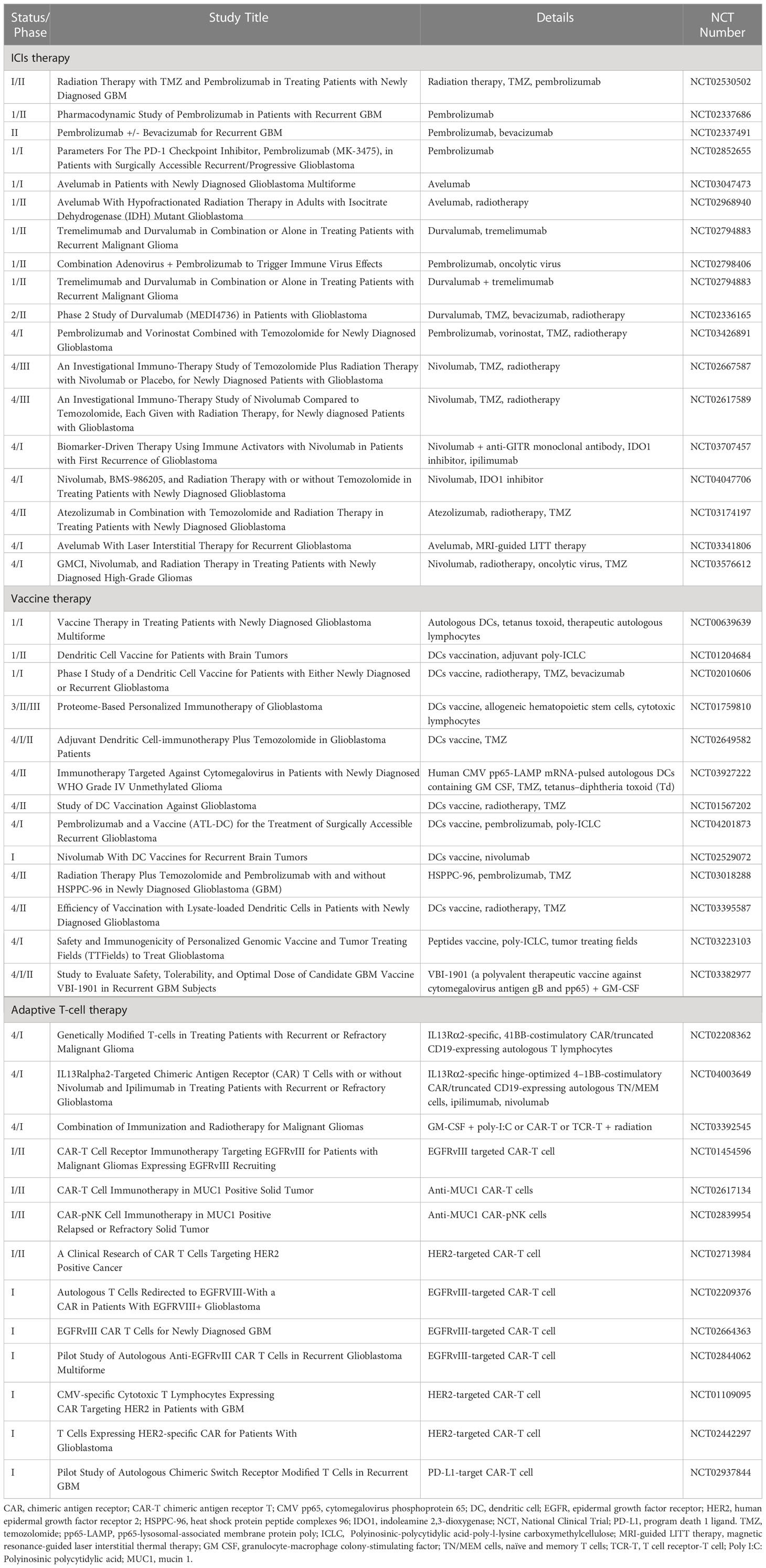
Table 1 Ongoing clinical trials involving immune checkpoint inhibitors (ICIs), vaccines, and adaptive T cells. .
3.4 Oncolytic virotherapy
Oncolytic virus (OV) therapy is a novel and promising therapeutic remedy in
the treatment of various solid tumors, including GBM. It can be divided into two broad categories: replication-competent viruses, which function as viral vectors to introduce specific genes, such as tumor suppressor genes, suicide genes, or immunostimulatory genes, into tumor cells to trigger an anti-tumor response; and selectively replication-competent viruses, which infect tumor cells and replicate until the cell lyses, and then infect and lyse neighboring cells (1–3, 184–186). Thus, the anti-tumor effect introduced by OVs occurs through two mechanisms: the direct killing of tumor cells and the subsequent stimulation of innate and adaptive immune responses. After the direct tumor lysis and apoptosis introduced by OVs, TAAs, pathogen-associated molecular patterns (PAMPs), and damage-associated molecular patterns (DAMPs) are released from the disrupted tumor cells. Both PAMPs and DAMPs can not only trigger innate immunity by stimulating pattern recognition receptors but also improve antigen cross-presentation and adaptive immune responses (4, 5, 187, 188). Therefore, OVs are also thought to be capable of turning the TME from immunologically “cold” to “hot”.
Oncolytic virus (OV) therapy has been investigated for the treatment of malignancies for decades. In 2015, the FDA approved the application of talimogene laherparepvec (T-VEC), a genetically modified Herpes Simplex Virus (HSV), to treat advanced melanoma as the first OV therapy in the USA (6, 189). GBM virotherapy clinical trials started in 1991 (7, 190). Since then, multifarious genetically engineered OV candidates have been investigated to treat GBM both in preclinical and clinical research. These include parvovirus, oncolytic herpes simplex virus (oHSV1716, oHSVG207), conditionally replicating adenovirus (ONYX-015, DNX-2401, DNX-2440), measles paramyxovirus (MV-CEA, MV expressing IL-13), oncolytic polio/rhinovirus recombinant (PVSRIPO), and retroviral replicating vector (Toca 511). Among these, DNX-2401, PVSRIPO, and Toca 511 are the most promising, having shown complete durable responses when intratumorally administered in GBM patients (8–10, 191–193). However, these trials failed to meet primary end points. Recently, the novel strategy of inserting immunomodulatory genes into the viral genome to conduct OVs to express immunomodulatory transgenes has been evaluated in several preclinical trials to treat gliomas. These include OVs with an expression of IL-12, IL-15, ICIs, tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), immune stimulators (GM-CSF, OX40 ligand), tumor suppressors (PTEN, P53), E-cadherin, and FMS-like tyrosine kinase 3 ligand (Flt3L) (11–20, 152, 194–202). Some have demonstrated safety and efficacy in preclinical trials, but others still require further investigation.
Although OV therapy has drawn concentrations, powerful clinical effectiveness in treating GBM is still lacking. The first obstacle is the limited efficiency of OV delivery. Several factors impact its systemic delivery, including complement factor neutralization, off-target dispersion risk, virus reduction during hepatic metabolism, and limitation from the BBB. To address these problems, mesenchymal stem cells (MSCs) with tumor-tropic and immune-evasive abilities are introduced as carriers for the virus delivery. Not only can MSCs shield OVs from host immunity, but they also alleviate the systemic sequestration of viruses and off-target toxicities. A recent study has successfully conducted MSCs to migrate and deliver therapeutic viruses to distant glioma cells (21, 203). That is, most trials concerning GBM adopted intracranial or intratumoral administration of OVs to overcome the BBB. However, it is also intractable for the second obstacle introduced by the immunosuppressive TME. Recent studies have demonstrated that GSCs and glioma-associated mesenchymal stem cells in the TME are likely to generate resistance to OV therapy (22, 23, 204, 205). Moreover, it has been demonstrated that TAMs can restrict the replication and spread of oncolytic viruses (24, 206). Furthermore, MDSCs can induce B-cell-mediated immunosuppression, which may limit the effective response to OVs with the expression of immunomodulatory transgenes (25, 81). Accordingly, a better-designed delivery and a better understanding of the TME should be pursued to optimize OV therapy against GBM.
4 Conclusion
Myeloid cells, including BMDMs, microglia, MDSCs, and neutrophils, play a crucial role in maintaining the immunosuppressive TME in GBM. Numerous factors impact the recruitment, stimulation, and function of myeloid cells in the TME, which constitutes a significant challenge for therapy efficacy. Currently, immunotherapies predominantly focus on the investigation of vaccines, CAR-T cells, ICIs, and OVs therapies. However, successful advances in a clinical environment are still lacking. Moreover, single immunotherapy is always insufficient in GBM and leads to the formation of a “cold tumor”, because multifarious factors challenge immunotherapy, including the immunosuppressive TME, the high tumor heterogeneity, the low TMB, the dMMR status, persistence and delivery of the vaccines, and CAR-T cells. Thus, a better understanding of the functions of myeloid cells in the TME and their interactions with GBM cells should be pursued in the future to optimize therapeutic strategy. Recently, combined therapy of OVs with ICIs in GBM demonstrated an improved efficiency over OV therapy alone (207). Similar results have also been obtained from the combined therapy of OVs with CAR-T cell therapy compared with CAR-T cells alone (154). Thus, this combination should be further investigated in order to be applied in clinical environments. Accordingly, immunotherapy for GBM requires integrated efforts to reduce and deactivate immunosuppressive myeloid cells, improve the TME suppression, promote CAR-T cell function by equipping multi-antigens targets, and reinforce T-cell effector function with the blockade of immune checkpoints. These contributions will promote the development of an optimal personalized therapeutic strategy for GBM patients.
Author contributions
BH and JZ wrote the initial draft. Figures were prepared by WZ. HZ prepared the final version. ZZ recommended a structure for the review and substantially advanced the draft. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82260578 to HZ).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Committee of Brain Tumor Registry of Japan. Brain tumor registry of Japan (2005–2008). Neurol Med Chir. (2017) 57(Suppl. 1):9–102. doi: 10.2176/nmc.sup.2017-0001
2. Crocetti E, Trama A, Stiller C, Caldarella A, Soffietti R, Jaal J, et al. RARECARE working group. epidemiology of glial and non-glial brain tumours in EuropeEpidemiology of glial and non-glial brain tumours in Europe. Eur J Cancer. (2012) 48(10):1532–42. doi: 10.1016/j.ejca.2011.12.013
3. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS, et al. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2011-2015. Neuro Oncol (2018) 20(suppl_4):iv1–iv86. doi: 10.1093/neuonc/noy131
4. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA Methylation-based classification of central nervous system tumours. Nature (2018) 555(7697):469–74. doi: 10.1038/nature26000
5. Clark O, Yen K, Mellinghoff IK. Molecular pathways: Isocitrate dehydrogenase mutations in cancer. Clin Cancer Res (2016) 22(8):1837–42. doi: 10.1158/1078-0432.CCR-13-1333
6. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell (2018) 33(1):152. doi: 10.1016/j.ccell.2017.12.012
7. Ozawa T, Riester M, Cheng Y-K, Huse JT, Squatrito M, Helmy K, et al. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell (2014) 26(2):288–300. doi: 10.1016/j.ccr.2014.06.005
8. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455:1061–8. doi: 10.1038/nature07385
9. Davis ME. Epidemiology and overview of gliomas. Semin Oncol Nurs (2018) 34(5):420–9. doi: 10.1016/j.soncn.2018.10.001
10. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med (2005) 352:987–96. doi: 10.1056/NEJMoa043330
11. Cohen MH, Shen YL, Keegan P, Pazdur R. FDA Drug approval summary: Bevacizumab (Avastin®) as treatment of recurrent glioblastoma multiforme. Oncologist (2012) 17:1482–2. doi: 10.1634/theoncologist.2009-0121erratum
12. Ballas ZK. The 2018 Nobel prize in physiology or medicine: An exemplar of bench to bedside in immunology. J Allergy Clin Immunol (2018) 142(6):1752–3. doi: 10.1016/j.jaci.2018.10.021
13. Huang B, Li X, Li Y, Zhang J, Zong Z, Zhang H. Current immunotherapies for glioblastoma multiforme. Front Immunol (2021) 11:603911. doi: 10.3389/fimmu.2020.603911
14. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12(9):623–35. doi: 10.1038/nri3265
15. John H S, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. (2020) 20(1):12–25. doi: 10.1038/s41568-019-0224-7
16. Landry AP, Balas M, Alli S, Spears J, Zador Z. Distinct regional ontogeny and activation of tumor associated macrophages in human glioblastoma. Sci Rep (2020) 10:19542. doi: 10.1038/s41598-020-76657-3
17. Fu W, Wang W, Li H, Jiao Y, Huo R, Yan Z, et al. Single-cell atlas reveals complexity of the immunosuppressive microenvironment of initial and recurrent glioblastoma. Front Immunol (2020) 11:835. doi: 10.3389/fimmu.2020.00835
18. Ravi VM, Will P, Kueckelhaus J, Sun N, Joseph K, Salié H, et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell (2022) 40(6):639–655.e13. doi: 10.1016/j.ccell.2022.05.009
19. Abdelfattah N, Kumar P, Wang C, Leu J-S, Flynn WF, Gao R, et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat Commun (2022) 13(1):767. doi: 10.1038/s41467-022-28372-y
20. Cassetta L, Pollard JW. Tumor-associated macrophages. Curr Biol (2020) 30(6):R246–8. doi: 10.1016/j.cub.2020.01.031
21. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science (2014) 344(6186):921–5. doi: 10.1126/science.1252510
22. Chen X, Zhang L, Zhang IY, Liang J, Wang H, Ouyang M, et al. RAGE expression in tumor-associated macrophages promotes angiogenesis in glioma. Cancer Res (2014) 74(24):7285–97. doi: 10.1158/0008-5472.CAN-14-1240
23. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res (2017) 77(9):2266–78. doi: 10.1158/0008-5472.CAN-16-2310
24. Pinton L, Masetto E, Vettore M, Solito S, Magri S, D’Andolfi M, et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J Immunother Cancer. (2019) 7(1):58. doi: 10.1186/s40425-019-0536-x
25. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci (2014) 15(5):300–12. doi: 10.1038/nrn3722
26. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep (2016) 17(9):2445–59. doi: 10.1016/j.celrep.2016.10.052
27. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. (2016) 113(12):E1738–46. doi: 10.1073/pnas.1525528113
28. Woolf Z, Swanson MEV, Smyth LC, Mee EW, Schweder P, Heppner P, et al. Single-cell image analysis reveals a protective role for microglia in glioblastoma. Neurooncol Adv (2021) 3(1):vdab031. doi: 10.1093/noajnl/vdab031
29. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill. AM. Pillars article: M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol (2017) 199(7):2194–201. doi: 10.4049/jimmunol.1701141
30. Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. (2009) 210(1-2):3–12. doi: 10.1016/j.jneuroim.2009.02.003
31. Ransohoff. RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci (2016) 19(8):987–91. doi: 10.1038/nn.4338
32. Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express gpnmb and Spp1. PloS One (2015) 10(2):e0116644. doi: 10.1371/journal.pone.0116644
33. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight (2016) 1(2):e85841. doi: 10.1172/jci.insight.85841
34. Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol (2017) 18(1):234. doi: 10.1186/s13059-017-1362-4
35. Kees T, Lohr J, Noack J, Mora R, Gdynia G, Tödt G, et al. Microglia isolated from patients with glioma gain antitumor activities on poly (I:C) stimulation. Neuro Oncol (2012) 14(1):64–78. doi: 10.1093/neuonc/nor182
36. Hambardzumyan D, Gutmann DH, Kettenmann. H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci (2016) 19(1):20–7. doi: 10.1038/nn.4185
37. Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M, et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest. (2019) 129(1):137–49. doi: 10.1172/JCI121266
38. Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, Yong VW. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis (2012) 33(2):312–9. doi: 10.1093/carcin/bgr289
39. Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S, et al. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann Neurol (2003) 54(3):388–92. doi: 10.1002/ana.10679
40. Okada M, Saio M, Kito Y, Ohe N, Yano H, Yoshimura S, et al. Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int J Oncol (2009) 34(6):1621–7. doi: 10.3892/ijo_00000292
41. Feng X, Szulzewsky F, Yerevanian A, Chen Z, Heinzmann D, Rasmussen RD, et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget (2015) 6(17):15077–94. doi: 10.18632/oncotarget.3730
42. Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med (2012) 18(1):519–27. doi: 10.2119/molmed.2011.00217
43. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol (2015) 17(2):170–82. doi: 10.1038/ncb3090
44. Ye X-z, Xu S-l, Xin Y-h, Yu S-c, Ping Y-f, Chen L, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J Immunol (2012) 189(1):444–53. doi: 10.4049/jimmunol.1103248
45. Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A. (2009) 106(30):12530–5. doi: 10.1073/pnas.0804273106
46. Li W, Graeber MB. The molecular profile of microglia under the influence of glioma. Neuro Oncol (2012) 14(8):958–78. doi: 10.1093/neuonc/nos116
47. Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA (2012) 109(17):6662–7. doi: 10.1073/pnas.1121623109
48. Büll C, Stoel MA, Brok MHd, Adema. GJ. Sialic acids sweeten a tumor’s life. Cancer Res (2014) 74(12):3199–204. doi: 10.1158/0008-5472.CAN-14-0728
49. Veillon L, Fakih C, Abou-El-Hassan H, Kobeissy F, Mechref Y. Glycosylation changes in brain cancer. ACS Chem Neurosci (2018) 9(1):51–72. doi: 10.1021/acschemneuro.7b00271
50. Kamran N, Kadiyala P, Saxena M, Candolfi M, Li Y, Moreno-Ayala MA, et al. Immunosuppressive myeloid cells’ blockade in the glioma microenvironment enhances the efficacy of immune-stimulatory gene therapy. Mol Ther (2017) 25(1):232–48. doi: 10.1016/j.ymthe.2016.10.003
51. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
52. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19(2):108–19. doi: 10.1038/s41590-017-0022-x
53. Gieryng A, Kaminska. B. Myeloid-derived suppressor cells in gliomas. Contemp Oncol (Pozn) (2016) 20(5):345–51. doi: 10.5114/wo.2016.64592
54. Condamine T, Dominguez GA, Youn J-I, Kossenkov AV, Mony S, Alicea-Torres. K. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol (2016) 1(2):aaf8943. doi: 10.1126/sciimmunol.aaf8943
55. Alban TJ, Alvarado AG, Sorensen MD, Bayik D, Volovetz J, Serbinowski E, et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight (2018) 3(21):e122264. doi: 10.1172/jci.insight.122264
56. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Petersen-Baltussen HMJM, Laan Mt, et al. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J Neuropathol Exp Neurol (2015) 74(5):390–400. doi: 10.1097/NEN.0000000000000183
57. Bayik D, Zhou Y, Park C, Hong C, Vail D, Silver DJ, et al. Myeloid-derived suppressor cell subsets drive glioblastoma growth in a sex-specific manner. Cancer Discovery (2020) 10(8):1210–25. doi: 10.1158/2159-8290.CD-19-1355
58. Ho IAW, Shim. WSN. Contribution of the microenvironmental niche to glioblastoma heterogeneity. BioMed Res Int (2017) 2017:9634172. doi: 10.1155/2017/9634172
59. Moertel CL, Xia J, LaRue R, Waldron NN, Andersen BM, Prins RM, et al. CD200 in CNS tumor-induced immunosuppression: the role for CD200 pathway blockade in targeted immunotherapy. J Immunother Cancer. (2014) 2(1):46. doi: 10.1186/s40425-014-0046-9
60. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res (2016) 76(19):5671–82. doi: 10.1158/0008-5472.CAN-16-0144
61. Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infiltrating T cells increase IDO1 expression in glioblastoma and contribute to decreased patient survival. Clin Cancer Res (2017) 23(21):6650–60. doi: 10.1158/1078-0432.CCR-17-0120
62. Chiu DK-C, Tse AP-W, Xu IM-J, Cui JD, Lai RK-H, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun (2017) 8(1):517. doi: 10.1038/s41467-017-00530-7
63. Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res (2005) 65(20):9525–35. doi: 10.1158/0008-5472.CAN-05-0529
64. Ding AS, Routkevitch D, Jackson C, Lim. M. Targeting myeloid cells in combination treatments for glioma and other tumors. Front Immunol (2019) 10:1715. doi: 10.3389/fimmu.2019.01715
65. Ji XY, Ma. J-W, Dong. J. Myeloid-derived suppressor cells and nonresolving inflammatory cells in glioma microenvironment: Molecular mechanisms and therapeutic strategies. Glioma (2018) 1:2–8. doi: 10.4103/glioma.glioma_2_17
66. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer. (2014) 134(12):2853–64. doi: 10.1002/ijc.28622
67. Li H, Han Y, Guo Q, Zhang M, Cao. X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol (2009) 182(1):240–9. doi: 10.4049/jimmunol.182.1.240
68. Ugolini A, Tyurin VA, Tyurina YY, Tcyganov EN, Donthireddy L, Kagan VE, et al. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight (2020) 5(15):e138581. doi: 10.1172/jci.insight.138581
69. Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi: 10.1158/0008-5472.CAN-05-1299
70. Mondanelli G, Ugel S, Grohmann U, Bronte. V. The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr Opin Pharmacol (2017) 35:30–9. doi: 10.1016/j.coph.2017.05.002
71. Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy. F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood (2011) 117(24):6532–41. doi: 10.1182/blood-2010-11-317321
72. Hart KM, Byrne KT, Molloy MJ, Usherwood EM, Berwin. B. IL-10 immunomodulation of myeloid cells regulates a murine model of ovarian cancer. Front Immunol (2011) 2:29. doi: 10.3389/fimmu.2011.00029
73. Feng S, Cheng X, Zhang L, Lu X, Chaudhary S, Teng R, et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc Natl Acad Sci U S A. (2018) 115(40):10094–9. doi: 10.1073/pnas.1800695115
74. Law AMK, Valdes-Mora F, Gallego-Ortega. D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells (2020) 9(3):561. doi: 10.3390/cells9030561
75. Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, et al. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood (2007) 109(10):4336–42. doi: 10.1182/blood-2006-09-046201
76. Markowitz J, Wang J, Vangundy Z, You J, Yildiz V, Yu L, et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4 + T cells in cancer and measurement of STAT1 nitration. Sci Rep (2017) 7(1):15424. doi: 10.1038/s41598-017-14970-0
77. Hu C-E, Gan J, Zhang R-D, Cheng Y-R, Huang Guang-Jian. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol (2011) 46(2):156–64. doi: 10.3109/00365521.2010.516450
78. Jarnicki AG, Lysaght J, Todryk S, Mills. KHG. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol (2006) 177(2):896–904. doi: 10.4049/jimmunol.177.2.896
79. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol (2012) 189(12):5602–11. doi: 10.4049/jimmunol.1201018
80. Siret C, Collignon A, Silvy F, Robert S, Cheyrol T, André P, et al. Deciphering the crosstalk between myeloid-derived suppressor cells and regulatory T cells in pancreatic ductal adenocarcinoma. Front Immunol (2020) 10:3070. doi: 10.3389/fimmu.2019.03070
81. Lee-Chang C, Rashidi A, Miska J, Zhang P, Pituch KC, Hou D, et al. Myeloid-derived suppressive cells promote b cell-mediated immunosuppression via transfer of PD-L1 in glioblastoma. Cancer Immunol Res (2019) 7(12):1928–43. doi: 10.1158/2326-6066.CIR-19-0240
82. Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, Ostrand-Rosenberg. S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukoc Biol (2014) 96(6):1109–18. doi: 10.1189/jlb.3A0414-210R
83. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg. S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol (2007) 179(2):977–83. doi: 10.4049/jimmunol.179.2.977
84. Tcyganov E, Mastio J, Chen E, Gabrilovich. DI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol (2018) 51:76–82. doi: 10.1016/j.coi.2018.03.009
85. Nicolás-Ávila JÁ, Adrover JM, Hidalgo. A. Neutrophils in homeostasis, immunity, and cancer. Immunity (2017) 46(1):15–28. doi: 10.1016/j.immuni.2016.12.012
86. Shaul ME, Fridlender. ZG. Cancer-related circulating and tumor-associated neutrophils - subtypes, sources and function. FEBS J (2018) 285(23):4316–42. doi: 10.1111/febs.14524
87. Zadora P, Dabrowski W, Czarko K, Smolen A, Kotlinska-Hasiec E, Wiorkowski K, et al. Preoperative neutrophil-lymphocyte count ratio helps predict the grade of glial tumor -a pilot study. Neurol Neurochir Pol (2015) 49(1):41–4. doi: 10.1016/j.pjnns.2014.12.006
88. Mason M, Maurice C, McNamara MG, Tieu MT, Lwin Z, Millar B-A, et al. Neutrophil-lymphocyte ratio dynamics during concurrent chemo-radiotherapy for glioblastoma is an independent predictor for overall survival. J Neurooncol. (2017) 132(3):463–71. doi: 10.1007/s11060-017-2395-y
89. Massara M, Persico P, Bonavita O, Poeta VM, Locati M, Simonelli M, et al. Neutrophils in gliomas. Front Immunol (2017) 8:1349. doi: 10.3389/fimmu.2017.01349
90. Schernberg A, Nivet A, Dhermain F, Ammari S, Escande A, Pallud J, et al. Neutrophilia as a biomarker for overall survival in newly diagnosed high-grade glioma patients undergoing chemoradiation. Clin Transl Radiat Oncol (2018) 10:47–52. doi: 10.1016/j.ctro.2018.04.002
91. Rahbar A, Cederarv M, Wolmer-Solberg N, Tammik C, Stragliotto G, Peredo I, et al. Enhanced neutrophil activity is associated with shorter time to tumor progression in glioblastoma patients. Oncoimmunology (2015) 5(2):e1075693. doi: 10.1080/2162402X.2015.1075693
92. Bambury RM, Teo MY, Power DG, Yusuf A, Murray S, Battley JE, et al. The association of pre-treatment neutrophil to lymphocyte ratio with overall survival in patients with glioblastoma multiforme. J Neurooncol. (2013) 114(1):149–54. doi: 10.1007/s11060-013-1164-9
93. Gan Y, Zhou X, Niu X, Li J, Wang T, Zhang H, et al. Neutrophil/Lymphocyte ratio is an independent prognostic factor in elderly patients with high-grade gliomas. World Neurosurg (2019) 127:e261-e267. doi: 10.1016/j.wneu.2019.03.085
94. Deng Q, He B, Liu X, Yue J, Ying H, Pan Y, et al. Prognostic value of pre-operative inflammatory response biomarkers in gastric cancer patients and the construction of a predictive model. J Transl Med (2015) 13:66. doi: 10.1186/s12967-015-0409-0
95. Hor W-S, Huang W-L, Lin Y-S, Yang. B-C. Cross-talk between tumor cells and neutrophils through the fas (APO-1, CD95)/FasL system: human glioma cells enhance cell viability and stimulate cytokine production in neutrophils. J Leukoc Biol (2003) 73(3):363–8. doi: 10.1189/jlb.0702375
96. Chio CC, Wang YS, Chen YL, Lin SJ, Yang BC. Down-regulation of fas-l in glioma cells by ribozyme reduces cell apoptosis, tumour-infiltrating cells, and liver damage but accelerates tumour formation in nude mice. Br J Cancer. (2001) 85(8):1185–92. doi: 10.1054/bjoc.2001.2055
97. McGranahan T, Li G, Nagpal S. History and current state of immunotherapy in glioma and brain metastasis. Ther Adv Med Oncol (2017) 9(5):347–68. doi: 10.1177/1758834017693750
98. Liang J, Piao Y, Holmes L, Fuller GN, Henry V, Tiao N, et al. Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res (2014) 20(1):187–98. doi: 10.1158/1078-0432.CCR-13-1279
99. Manda-Handzlik A, Demkow. U. The brain entangled: The contribution of neutrophil extracellular traps to the diseases of the central nervous system. Cells (2019) 8(12):1477. doi: 10.3390/cells8121477
100. Lee PY, Wang J-X, Parisini E, Dascher CC, Nigrovic. PA. Ly6 family proteins in neutrophil biology. J Leukoc Biol (2013) 94(4):585–94. doi: 10.1189/jlb.0113014
101. Jeon H-Y, Ham SW, Kim J-K, Jin X, Lee SY, Shin YJ, et al. Ly6G + inflammatory cells enable the conversion of cancer cells to cancer stem cells in an irradiated glioblastoma model. Cell Death Differ (2019) 26(10):2139–56. doi: 10.1038/s41418-019-0282-0
102. Palumbo P, Lombardi F, Siragusa G, Dehcordi SR, Luzzi S, Cimini A, et al. Involvement of NOS2 activity on human glioma cell growth, clonogenic potential, and neurosphere generation. Int J Mol Sci (2018) 19(9):2801. doi: 10.3390/ijms19092801
103. Yee PP, Wei Y, Kim S-Y, Lu T, Chih SY, Lawson C, et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun (2020) 11(1):5424. doi: 10.1038/s41467-020-19193-y
104. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell (2017) 32(1):42–56.e6. doi: 10.1016/j.ccell.2017.06.003
105. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi: 10.1056/NEJMoa1302369
106. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory hodgkin’s lymphoma. N Engl J Med (2015) 372(4):311–9. doi: 10.1056/NEJMoa1411087
107. Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, et al. Nivolumab for metastatic renal cell carcinoma: Results of a randomized phase II trial. J Clin Oncol (2015) 33(13):1430–7. doi: 10.1200/JCO.2014.59.0703
108. Rizvi NA, Mazières J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol (2015) 16(3):257–65. doi: 10.1016/S1470-2045(15)70054-9
109. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol (2015) 17:854–61. doi: 10.1093/neuonc/nou348
110. Reardon DA, Schuster J, Tran DD, Fink KL, Nabors LB, Li G, et al. ReACT: overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. J Clin Oncol (2015) 33(uppl):2009. doi: 10.1227/01.neu.0000467069.86811.3f
111. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII expressing glioblastoma (ACT IV): a randomized, double-blind, international phase 3 trial. Lancet Oncol (2017) 18:1373–785. doi: 10.1016/S1470-2045(17)30517-X
112. Oji Y, Hashimoto N, Tsuboi A, Murakami Y, Iwai M, Kagawa N, et al. Association of WT1 IgG antibody against WT1 peptide with prolonged survival in glioblastoma multiforme patients vaccinated with WT1 peptide. Int J Cancer. (2016) 139(6):1391–401. doi: 10.1002/ijc.30182
113. Rampling R, Peoples S, Mulholland PJ, James A, Al-Salihi O, Twelves CJ, et al. A cancer research UK first time in human phase I trial of IMA950 (Novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin Cancer Res (2016) 22(19):4776–85. doi: 10.1158/1078-0432.CCR-16-0506
114. Migliorini D, Dutoit V, Allard M, Grandjean Hallez N, Marinari E, Widmer V, et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro Oncol (2019) 21(7):923–33. doi: 10.1093/neuonc/noz040
115. Ott M, Prins RM, Heimberger. AB. The immune landscape of common CNS malignancies: implications for immunotherapy. Nat Rev Clin Oncol (2021) 18(11):729–744. doi: 10.1038/s41571-021-00518-9
116. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol (2010) 28(31):4722–9. doi: 10.1200/JCO.2010.28.6963
117. Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, et al. Individual patient-specifc immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res (2013) 19:205–14. doi: 10.1158/1078-0432.CCR-11-3358
118. Filley AC, Dey M. Dendritic cell based vaccination strategy: an evolving paradigm. J Neurooncol (2017) 133:223–35. doi: 10.1007/s11060-017-2446-4
119. Lynes J, Sanchez V, Dominah G, Nwankwo A, Nduom E. Current options and future directions in immune therapy for glioblastoma. Front Oncol (2018) 8:578. doi: 10.3389/fonc.2018.00578
120. Sakai K, Shimodaira S, Maejima S, Udagawa N, Sano K, Higuchi Y, et al. Dendritic cell-based immunotherapy targeting wilms’ tumor 1 in patients with recurrent malignant glioma. J Neurosurg (2015) 123(4):989–97. doi: 10.3171/2015.1.JNS141554
121. Nair SK, Sampson JH, Mitchell DA. Immunological targeting of cytomegalovirus for glioblastoma therapy. OncoImmunology (2014) 3:e29289. doi: 10.4161/onci.29289
122. Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature (2015) 519(7543):366–9. doi: 10.1038/nature14320
123. Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res (2017) 23(8):1898–909. doi: 10.1158/1078-0432.CCR-16-2057
124. Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuño MA, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother (2013) 62(1):125–35. doi: 10.1007/s00262-012-1319-0
125. Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol (2011) 29(3):330–6. doi: 10.1200/JCO.2010.30.7744
126. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med (2018) 16(1):142. doi: 10.1186/s12967-018-1507-6
127. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314:5. doi: 10.1126/science.1129003
128. Rosenberg SA, Dudley ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci U S A. (2004) 101 Suppl 2(Suppl 2):14639–45. doi: 10.1073/pnas.0405730101
129. Quattrocchi KB, Miller CH, Cush S, Bernard SA, Dull ST, Smith M, et al. Pilot study of local autologous tumor infiltrating lymphocytes for the treatment of recurrent malignant gliomas. Clin Trial J Neurooncol (1999) 45(2):141–57. doi: 10.1023/a:1006293606710
130. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609
131. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery (2013) 3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548
132. van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of secondgeneration chimeric antigen receptors. Nat Rev Drug Discovery (2015) 14(7):499–509. doi: 10.1038/nrd4597
133. Srivastava S, Riddell SR. Engineering CAR-T cells: design concepts. Trends Immunol (2015) 36(8):494–502. doi: 10.1016/j.it.2015.06.004
134. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
135. Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med (2015) 373:1040–7. doi: 10.1056/NEJMoa1504542
136. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult b cell ALL patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/JCI85309
137. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
138. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 results of ZUMA-1: A multicenter study of KTE-C19 anti-CD19 CAR t cell therapy in refractory aggressive lymphoma. Mol Ther (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
139. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large b-cell lymphoma and indolent b-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J ClinOncol (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
140. Salinas RD, Durgin JS, O’Rourke. DM. Potential of glioblastoma-targeted chimeric antigen receptor (CAR) T-cell therapy. CNS Drugs (2020) 34(2):127–45. doi: 10.1007/s40263-019-00687-3
141. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIIIdirected CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
142. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ra2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res (2015) 21(18):4062–72. doi: 10.1158/1078-0432.CCR-15-0428
143. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specifc chimeric antigen receptormodifed virus-specifc T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
144. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
145. Yang J, Yan J, Liu B. Targeting EGFRvIII for glioblastoma multiforme. Cancer Lett (2017) 403:224–30. doi: 10.1016/j.canlet.2017.06.024
146. Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res (1999) 5:985–90.
147. Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC, et al. Glioma IL13Ra2 is associated with mesenchymal signature gene expression and poor patient prognosis. PloS One (2013) 8(10):e77769. doi: 10.1371/journal.pone.0077769
148. Koka V, Potti A, Forseen SE, Pervez H, Fraiman GN, Koch M, et al. Role of her-2/neu overexpression and clinical determinants of early mortality in glioblastoma multiforme. Am J Clin Oncol (2003) 26:332–5. doi: 10.1097/01.COC.0000020922.66984.E7
149. Medikonda R, Dunn G, Rahman M, Fecci P, Lim. M. A review of glioblastoma immunotherapy. J Neurooncol. (2021) 151(1):41–53. doi: 10.1007/s11060-020-03448-1
150. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ra2 mitigate tumor antigen escape. J Clin Invest (2016) 126:3036–52. doi: 10.1172/JCI83416
151. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol (2018) 20:506–18. doi: 10.1093/neuonc/nox182
152. Jiang H, Rivera-Molina Y, Gomez-Manzano C, Clise-Dwyer K, Bover L, Vence LM, et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res (2017) 77(14):3894–907. doi: 10.1158/0008-5472.CAN-17-0468
153. Liu S, Liu F, Zhao M, Zhang. J. Antitumor efficacy of oncolytic herpes virus type 1 armed with GM-CSF in murine uveal melanoma xenografts. Cancer Manag Res (2020) 12:11803–12. doi: 10.2147/CMAR.S274605
154. Park AK, Fong Y, Kim S-I, Yang J, Murad JP, Lu J, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Transl Med (2020) 12(559):eaaz1863. doi: 10.1126/scitranslmed.aaz1863
155. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372(26):2521–32. doi: 10.1056/NEJMoa1503093
156. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
157. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) 373(19):1803–13. doi: 10.1056/NEJMoa1510665
158. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
159. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med (2015) 372(21):2018–28. doi: 10.1056/NEJMoa1501824
160. Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res (2016) 4(2):124–35. doi: 10.1158/2326-6066.CIR-15-0151
161. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res (2014) 20(20):5290–301. doi: 10.1158/1078-0432.CCR-14-0514
162. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol (2018) 15(7):422–42. doi: 10.1038/s41571-018-0003-5
163. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol (2020) 6(7):1003–1010. doi: 10.1001/jamaoncol.2020.1024
164. Bristol-Myers Squibb Provides update on phase 3 opdivo. (nivolumab) CheckMate -548 trial in patients with newly diagnosed MGMT-methylated glioblastoma multiforme. US National Library of Medicine. ClinicalTrials.gov. (2015).
165. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, et al. Programmed death ligand 1 expression and tumor-infltrating lymphocytes in glioblastoma. Neuro Oncol (2015) 17(8):1064–75. doi: 10.1093/neuonc/nou307
166. Antonios JP, Soto H, Everson RG, Moughon D, Orpilla JR, Shin NP, et al. Immunosuppressive tumor-infltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro Oncol (2017) 19(6):796–807. doi: 10.1093/neuonc/now287
167. Maxwell R, Jackson CM, Lim M. Clinical trials investigating immune checkpoint blockade in glioblastoma. Curr Treat Options Oncol (2017) 18(8):51. doi: 10.1007/s11864-017-0492-y
168. Nduom EK, Wei J, Yaghi NK, Huang N, Kong L-Y, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol (2016) 18(2):195–205. doi: 10.1093/neuonc/nov172
169. McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (2016) 351(6280):1463–9. doi: 10.1126/science.aaf1490
170. Joshi K, de Massy MR, Ismail M, Reading JL, Uddin I, Woolston A, et al. Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat Med (2019) 25(10):1549–59. doi: 10.1038/s41591-019-0592-2
171. Khasraw M, Walsh KM, Heimberger AB, Ashley DM. What is the burden of proof for tumor mutational burden in gliomas? Neuro Oncol (2020) 23(1):17–22. doi: 10.1093/neuonc/noaa256
172. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med (2019) 25(3):477–86. doi: 10.1038/s41591-018-0337-7
173. Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn GP, Lim M, et al. Phase 2 study to evaluate safety and efcacy of MEDI4736 (durvalumab [DUR]) in glioblastoma (GBM) patients: an update. J Clin Oncol (2017) 35(15_Suppl):2042.
174. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res (2018) 24(17):4175–86. doi: 10.1158/1078-0432.CCR-17-1846
175. Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med (2018) 379(8):722–30. doi: 10.1056/NEJMoa1805453
176. Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE, et al. T-Cell dysfunction in glioblastoma: Applying a new framework. Clin Cancer Res (2018) 24(16):3792–802. doi: 10.1158/1078-0432.CCR-18-0047
177. Kanzaki M, Wada J, Sugiyama K, Nakatsuka A, Teshigawara S, Murakami K, et al. Galectin-9 and T cell immunoglobulin mucin-3 pathway is a therapeutic target for type 1 diabetes. Endocrinology (2012) 153(2):612–20. doi: 10.1210/en.2011-1579
178. Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res (2017) 77(24):6795–811. doi: 10.1158/0008-5472.CAN-17-2285
179. Wang Z, Wang Z, Zhang C, Liu X, Li G, Liu S, et al. Genetic and clinical characterization of B7-H3 (CD276) expression and epigenetic regulation in diffuse brain glioma. Cancer Sci (2018) 109(9):2697–705. doi: 10.1111/cas.13744
180. Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res (2017) 23(1):124–36. doi: 10.1158/1078-0432.CCR-15-1535
181. Jiricny. J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol (2006) 7(5):335–46. doi: 10.1038/nrm1907
182. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol (2016) 34(19):2206–11. doi: 10.1200/JCO.2016.66.6552
183. Giuseppe Lombardi MC, Simonelli M, Fassan M, Padovan M, Persico P, Bellu L, et al. Pembrolizumab (Pem) in recurrent high-grade glioma (HGG) patients (PTS) with mismatch repair defciency (MMRd): an observational study. Am Soc Clin Oncol (2019), 2043:2043–2043. doi: 10.1200/JCO.2019.37.15_suppl.2043
184. Estevez-Ordonez D, Chagoya G, Salehani A, Atchley TJ, Laskay NMB, Parr MS, et al. Immunovirotherapy for the treatment of glioblastoma and other malignant gliomas. Neurosurg Clin N Am (2021) 32:265–81. doi: 10.1016/j.nec.2020.12.008
185. Stavrakaki E, Dirven CM, Lamfers ML. Personalizing oncolytic virotherapy for glioblastoma: in search of biomarkers for response. Cancers (2021) 13:614. doi: 10.3390/cancers13040614
186. Russell SJ, Barber GN. Oncolytic viruses as antigen-agnostic cancer vaccines. Cancer Cell (2018) 33(4):599–605. doi: 10.1016/j.ccell.2018.03.011
187. Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. (2013) 12(1):1–16. doi: 10.1186/1476-4598-12-103
188. Chiocca EA, Rabkin SD, Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res (2014) 2(4):295–300. doi: 10.1158/2326-6066.CIR-14-0015
189. Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol (2012) 30(7):658–70. doi: 10.1038/nbt.2287
190. Andtbacka RHII, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol (2015) 33(25):2780–8. doi: 10.1200/JCO.2014.58.3377
191. Cloughesy TF, Landolfi J, Vogelbaum MA, Ostertag D, Elder JB, Bloomfield S, et al. Durable complete responses in some recurrent highgrade glioma patients treated with toca 511 + toca FC. Neuro Oncol (2018) 20:1383–92. doi: 10.1093/neuonc/noy075
192. Desjardins A, Gromeier M, 2nd JEH, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
193. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol (2018) 36:1419–27. doi: 10.1200/JCO.2017.75.8219
194. Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell (2017) 32:253–267, e255. doi: 10.1016/j.ccell.2017.07.006
195. Oh E, Hong J, Kwon OJ, Yun CO. A hypoxia- and telomeraseresponsive oncolytic adenovirus expressing secretable trimeric TRAIL triggers tumour-specific apoptosis and promotes viral dispersion in TRAIL-resistant glioblastoma. Sci Rep (2018) 8:1420. doi: 10.1038/s41598-018-19300-6
196. Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, et al. Superagonist IL-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with PD-1 blockade. Mol Ther (2018) 26:2476–86. doi: 10.1016/j.ymthe.2018.07.013
197. Passaro C, Alayo Q, De Laura I, McNulty J, Grauwet K, Ito H, et al. Arming an oncolytic herpes simplex virus type 1 with a single-chain fragment variable antibody against PD-1 for experimental glioblastoma therapy. Clin Cancer Res (2019) 25:290–9. doi: 10.1158/1078-0432.CCR-18-2311
198. Lun X, Chan J, Zhou H, Sun B, Kelly JJP, Stechishin OO, et al. Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol Ther (2010) 18:1927–36. doi: 10.1038/mt.2010.183
199. Cheney IW, Johnson DE, Vaillancourt MT, Avanzini J, Morimoto A, Demers GW, et al. Suppression of tumorigenicity of glioblastoma cells by adenovirus-mediated MMAC1/PTEN gene transfer. Cancer Res (1998) 58:2331–4.
200. Fan X, Lu H, Cui Y, Hou X, Huang C, Liu G, et al. Overexpression of p53 delivered using recombinant NDV induces apoptosis in glioma cells by regulating the apoptotic signaling pathway. Exp Ther Med (2018) 15:4522–30. doi: 10.3892/etm.2018.5935
201. Fan X, Lu H, Cui Y, Hou X, Huang C, Liu G. An oncolytic herpesvirus expressing e-cadherin improves survival in mouse models of glioblastoma. Nat Biotechnol (2018) 37:45–54. doi: 10.1038/nbt.4302
202. Ali S, Curtin JF, Zirger JM, Xiong W, King GD, Barcia C, et al. Inflammatory and anti-glioma effects of an adenovirus expressing human soluble fms-like tyrosine kinase 3 ligand (hsFlt3L): Treatment with hsFlt3L inhibits intracranial glioma progression. Mol Ther (2004) 10:1071–84. doi: 10.1016/j.ymthe.2004.08.025
203. Zhang Q, Xiang W, Yi D, Xue B, Wen W, Abdelmaksoud A, et al. Current status and potential challenges of mesenchymal stem cell-based therapy for malignant gliomas. Stem Cell Res Ther (2018) 9:228. doi: 10.1186/s13287-018-0977-z
204. Zhang Q, Yi D-Y, Xue B-Z, Wen W-W, Lu Y-P, Abdelmaksou A, et al. CD90 determined two subpopulations of glioma-associated mesenchymal stem cells with different roles in tumour progression. Cell Death Dis (2018) 9:1101. doi: 10.1038/s41419-018-1140-6
205. Audia A, Conroy S, Glass R, Bhat KPL. The impact of the tumor microenvironment on the properties of glioma stem-like cells. Front Oncol (2017) 7:143. doi: 10.3389/fonc.2017.00143
206. Denton NL, Chen CY, Scott TR, Cripe TP. Tumor-associated macrophages in oncolytic virotherapy: Friend or foe? Biomedicines (2016) 4:1–9. doi: 10.3390/biomedicines4030013
Keywords: myeloid cells, immunosuppressive, tumor microenvironment, glioblastoma, immunotherapies
Citation: Huang B, Zhang J, Zong W, Chen S, Zong Z, Zeng X and Zhang H (2023) Myeloidcells in the immunosuppressive microenvironment in glioblastoma: The characteristics and therapeutic strategies. Front. Immunol. 14:994698. doi: 10.3389/fimmu.2023.994698
Received: 15 July 2022; Accepted: 31 January 2023;
Published: 27 February 2023.
Edited by:
Junxia Zhang, Nanjing Medical University, ChinaReviewed by:
Serena Pellegatta, Carlo Besta Neurological Institute Foundation (IRCCS), ItalyYa-Jui Lin, Linkou Chang Gung Memorial Hospital, Taiwan
Copyright © 2023 Huang, Zhang, Zong, Chen, Zong, Zeng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhitao Zong, eGlhbmdmYW56aGJAMTYzLmNvbQ==; Xiaojun Zeng, enhqMjg5Njk5QDE2My5jb20=; Hongbo Zhang, aG9uZ2Jvemhhbmc5OUAxNjMuY29t
†These authors have contributed equally to this work