 Yue Liu1Yaodong Lv2Tingwei Zhang1Tongtong Huang1Yating Lang1Qinghao Sheng1Yingxiao Liu1Zhijuan Kong1Ying Gao1Shangwei Lu1Meilin Yang1Yaqi Luan1Xining Wang1*†Zhimei Lv1*†
Yue Liu1Yaodong Lv2Tingwei Zhang1Tongtong Huang1Yating Lang1Qinghao Sheng1Yingxiao Liu1Zhijuan Kong1Ying Gao1Shangwei Lu1Meilin Yang1Yaqi Luan1Xining Wang1*†Zhimei Lv1*†- 1Department of Nephrology, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, China
- 2Department of Neurology, Yantai Yuhuangding Hospital, Shandong University, Yantai, China
Diabetic kidney disease (DKD) is the most common cause of end-stage renal disease and has gradually become a public health problem worldwide. DKD is increasingly recognized as a comprehensive inflammatory disease that is largely regulated by T cells. Given the pivotal role of T cells and T cells-producing cytokines in DKD, we summarized recent advances concerning T cells in the progression of type 2 diabetic nephropathy and provided a novel perspective of immune-related factors in diabetes. Specific emphasis is placed on the classification of T cells, process of T cell recruitment, function of T cells in the development of diabetic kidney damage, and potential treatments and therapeutic strategies involving T cells.
1 Introduction
Diabetic kidney disease (DKD) is a highly prevalent microvascular complication of diabetes that affects >50% of incident cases of diabetes mellitus (DM) and profoundly contributes to patient morbidity and mortality (1). Clinically, DKD is characterized by the presence of albuminuria and decreased estimated glomerular filtration. DKD is diagnosed based on glomerular basement membrane thickening, mesangial expansion, diffuse or nodular glomerulosclerosis, podocyte loss, and interstitial fibrosis on pathology and histology (2). Multiple mechanisms contribute to the outcome of DKD. Among them, nonimmune factors, metabolism, and hemodynamics are considered the most crucial causes of renal damage in patients with type 2 DM (T2DM) and DKD in traditional perceptions (3–5). Therefore, optimal control of hyperglycemia and intensive treatments for elevated blood pressure remain the current management strategies for patients with diabetes. However, current treatments are insufficient to prevent its progression in a large proportion of patients, and the prevalence of DKD is still increasing every year. The mechanisms leading to the development of renal dysfunction in diabetes are not fully understood; therefore, there is an urgent need to identify the pathogenesis and therapeutic approaches to DKD.
In comparison to merely considering DKD a non-immune metabolic disease induced by hyperglycemia, current studies emphasize that DKD is also an inflammatory disease (6, 7). The infiltration of immune cells, which is related to innate and adaptive immunity, may be involved in hyperglycemia-induced renal injury (8). In particular, the role of T-cells in the development of DKD has been confirmed (9). On the one hand, high glucose has been verified to induce T cells recruitment, activation, differentiation, and maturation, even the cytokine factor expression profiles of T cell (10). On the other hand, serum concentrations of chemokines and cytokines produced by T cells have been assessed in patients with diabetes, which are supposedly to predict the onset of diabetic complications.
Hence, we summarized the updated progress in the aspects of differentiation, recruitment, function of T cells, and their products in the DKD as well as the potential strategies for the treatment of DKD, hoping to provide insights for future research.
2 Classification and differentiation of T cells in diabetic kidney disease
T cells are involved in host defense and clearance of pathogens. In general, T cells are divided into two species according to their constitutive chains, called “conventional T cells” and “unconventional T cells,” which operate in utterly different ways to regulate and coordinate immune responses in the kidney.
Classically, T cells that express T-cell receptors (TCRs) with α- and β-chains are classified as conventional T cells; specifically, conventional T cells can be separated into CD8+ T cells and CD4+ T cells, and these cells recognize peptides presented by the major histocompatibility complex class II and I. Based on the specific function, the differentiated CD4+ T cell subsets were further distinguished into T-helper (Th) cells and regulatory T cells (Tregs).
According to previous research, unconventional T cells recognize antigens in the absence of classical restriction via the major histocompatibility complex and respond rapidly upon antigen encounters (11). In the kidneys, unconventional T cells include mucosal-associated invariant T (MAIT), natural killer T (NKT), and γδT cells (12).
In addition, tissue-resident memory (TRM) T-cells, the most abundant memory T-cell subset, have been identified as a class of T cells that reside in the kidney (13, 14). The different phenotypes and functions of TRM are derived from its position in various tissues (15). Due to the synergistic effect of the anatomical localization effector and memory phenotype, TRM T-cells located in the kidney are critically involved in DKDs.
2.1 Overview of T helper cells
Th cells are a cluster of highly plastic CD4+ T cells and are simultaneously important contributors to the autoimmunity and inflammation induced by DKD. Many modulatory mechanisms employed by Th cells contribute to the adjustment of renal tissue damage, such as by mediating the production of local cytokines. According to their cytokine and transcription factor expression profiles, Th cells are primarily grouped into Th1, Th2, Th3, Th9, Th17, Th22, T follicular helper (Tfh), and Tregs.
As a flock of plastic cells, Th cell subsets can acquire regulatory functions upon chronic stimulation in diabetes, opening a new perspective for the exploration of immunomodulatory mechanisms for diabetes (8, 16). Hence, the classification and differentiation of Th cells in diabetes and its renal complications are associated with their unique subsets, which are described in the following sections.
2.1.1 Th1
Since 1986, a groundbreaking study has elaborated the patterns of lymphokine activity production of Th1 and Th2 cells; Th1 expresses its signature cytokines such as interleukin (IL)-2, interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and transcription factor T-box (T-bet), among others (17–19), owing to which it participates in the activation of macrophage cell-mediated immunity and systematically regulates cellular function.
Notably, the level of Th1 can be mediated by multiple factors. For example, STAT4 and STAT1, members of the signal transducer and activator of transcription (STAT) family, are crucial for inducing differentiation and maintaining the Th1 cell phenotype (20, 21). T-bet also modifies the level of Th1 by activating STAT1 (22). In contrast, the cell immunoglobulin domain and mucin domain 3 are extensively considered suppressants of IFN-γ-producing T-cells (23).
Meanwhile, Th1 has been shown to respond to preceding and accompanying immunoreaction in DM (24). In clinical settings, Th1 cells dramatically increase in patients with type 2 diabetic nephropathy (T2DN), and the degree of proteinuria is positively correlated with aberrant cytokine production, such as IFN-γ and IL-2R (25). Creatinine clearance is also negatively correlated with plasma TNF-α and urinary MCP-1 levels.
2.1.2 Th2
Th2 and its produced IL-4, IL-5, IL-9, IL-10 and IL-13 are related to the pathogenesis of DKD (26, 27). Furthermore, the inherent link between Th1 and Th2 has been discussed in the immunopathogenesis of diabetes. In contrast, IL-10 and IL-4 produced by Th2 can dampen IFN-γ secretion and suppress Th1 cell activation (in the regulation of humoral immunity, among other processes (27, 28).
Contrastingly, the decrease in T-bet produced by Th1 corresponds with the increase in plasma IL-4 secreted by Th2, implying an imbalance between Th1 and Th2 (29). Hence, upregulating GATA-3 and IL-4 expression and downregulating T-bet and IFN-γ levels may provide a novel therapeutic method for type 1 diabetes (T1D) treatment in non-obese diabetic (NOD) mice (30). Intriguingly, GATA-3, a promoter of Th2 responses, was increased in diabetes (31, 32), while peritoneal dialysis may increase the frequency of Th2 cells during the treatment of DKD (33).
2.1.3 Th17
As discovered in 2005, Th17 secrete IL-17 as its signature cytokine (34). After Th17 cells receive active signaling, the JAK/STAT pathway directly culminates in activation of STAT3 of RORγT, resulting in the production of IL-17 (35). In contrast, IL-2-induced activation of STAT5 causes a decrease in ROR-γt and a transient downregulation of IL-17 (36). In addition to ROR-γt, the differentiation of Th17 cells can be directed by transforming growth factor (TGF)-β, IL-6, IL-1β, and IL-23 (37, 38). Interestingly, unique cytokines can induce different types of Th17 cells. For example, the proinflammatory subtype of Th17 cells is induced by TGF-β, whereas the less pathogenic subtype is promoted by IL-1β (36, 39). In peripheral blood lymphocytes from patients with diabetes, promoter activation was verified as the core principle of the change in IL-17 and its downstream signaling (40).
On the immune-mediated kidney disease, Th17 cells are likely to get upregulated in DKD, resulting in a general increase of IFN-γ and IL-17A in streptozotocin (STZ)-induced diabetes (41). A clinical study based on blood samples collected from 56 patients with nephropathy and 57 patients with diabetes revealed that patients carrying at least one allele of the IL-17A (rs2275913) gene polymorphism were vulnerable to DKDs (42). In a cross-sectional study, the level of serum IL-17 was also found to be lower in individuals with diabetes or renal lesions in Asian and Indian populations (43).
Furthermore, in terms of DKD treatment, IL-17A gradually demonstrates dose-dependent properties. As mentioned above, presence of IL-17A in individuals with diabetes and diabetic mouse models is an obvious characteristic, and serum and urinary levels of IL-17A in the former with advanced DKD confirms this finding; additionally, low doses of IL-17A have a noteworthy therapeutic effect on podocytes and tubular cells (44). The protective effect of IL-17 may also be dependent on its subsets, as low doses of IL-17A and IL-17F can prevent severe impairment of renal function at the beginning of the course of DKD; however, IL-17C or IL-17E do not show a similar effect (40).
With respect to the relative ratio of Th17 cells, interesting studies have demonstrated that the Th17/Treg ratio promotes inflammation and may hasten the development of diabetic complications. The increase in Th17 or decrease in Tregs may be a contributing factor to the deterioration of kidney function (45, 46). The Th17/Th1 response ratio is a potential contributor to β cell destruction and provides a novel biomarker for the rapid diagnosis of T1D preceding the clinical end. Moreover, similar investigations have been performed on the serum levels of relevant cytokines in patients with T2DM, and the Th1/Th2/Th17/Treg paradigm has been demonstrated to skew toward Th1 and Th17 (26).
2.1.4 Th3, Th9, and Th22
As the research has progressed, various types of Th cells have been discovered to be involved in diabetic complications. Characterized by high expression of TGF-β, Th3 has a negative correlation with DKD at the onset of the disease rather than in the prediabetic phase (47). Another subset, named Th9 cells, is designated as IL-9 producers. With the technical support of nanoscale flow cytometry, Semenchuk et al. found that IL-9 is inversely related to the quantification of urinary podocyte-derived extracellular vesicles (48). Additionally, Th22 participates in the regulation of DKD by producing IL-22 (49).
2.2 Tfh cells
Tfh cells are another distinctive Th subset of cells that require the synergistic action of IL-6 and IL-21 to drive differentiation (50, 51). Tfh cells are involved in diabetic syndrome, leading to elevated levels of CXCR5, ICOS, PDCD1, BCL6, and IL21 (52). Many subsets of Tfh cells, such as CXCR5+ PD1+ ICOS+ and CD4+ CXCR5+ PD-1+, are increased in children and adults with diabetes (53, 54). In particular, CD4+ CXCR5+ Tfh cells have been confirmed to manipulate the levels of estimated glomerular filtration rate (GFR), creatinine, urea, urinary protein, fasting and postprandial blood glucose, and hemoglobin A1c in patients with DKD (55).
2.3 Regulatory T cells
Analysis of gene polymorphisms revealed that FOXP3+ Tregs were reduced in patients at the onset of diabetes (56). The apoptosis of Tregs is affected by aberrant IL-2R signaling, leading to a decrease in FOXP3 persistence and impacting the establishment of tolerance (57). Therefore, a single infusion of autologous polyclonal Tregs and recombinant human low-dose IL-2 may be a novel treatment for diabetes (58).
In addition to suppressing T cells, NK cells, NKT cells, B cells, and dendritic cells in the adaptive immune responses, Treg cells play a fundamental role in the pathological development of DN, maintaining a dynamic equilibrium between inflammatory cytokines and anti-inflammatory cytokines (59–61). Treg cells can control phenotypic changes by increasing (IFN-γ, IL-2, and IL-17) and decreasing (IL-10, IL-35, and TGF-β) the levels of anti-inflammatory cytokines (62, 63). Generally, the population and function of Tregs has a peculiar effect on immunoregulation in patients with diabetes.
2.4 CD8+ T cells
Recent findings have reported that CD8+ T cells were increased in patients with diabetes and that suppressing CD8+ T cells may alleviate the pathological reaction of DKD (64). Furthermore, infiltration of CD8+ effector T cells is important for recruiting macrophages to ameliorate systemic insulin resistance in mice fed a high-fat diet (65). Interestingly, the proportion of CD8+ TRM cells was increased in DKD and further promoted podocyte injury and glomerulosclerosis, suggesting a pivotal role of CD8+ T cells in podocyte damage in insulin-resistant patients with DN (66).
2.5 NKT cells and γδ T Cells
NKT cells are another characteristic T-cell subset that links the innate and adaptive immune systems, and abnormalities in the frequency and activity of NKT cells may be attributed to the exacerbation of T1D (67). NKT cells play a fundamental role in various renal diseases involving abnormal metabolism. For instance, inappropriate overactivation of NKT cells can cause kidney damage via the TNF-α/Fas ligand pathway (68). In progressive non-alcoholic fatty liver disease, NKT cells also cause glomerular function and renal immunotoxicity (69). Furthermore, during chronic kidney disease (CKD) progression, the raise of CD3- CD56+ NK cells were observed in tubulointerstitial, and the frequency of CD3- CD56+ NK cells and CD3+ CD56+ NKT cells were also remarkably elevated in the peripheral blood of diabetic patients (70, 71). Simultaneously, NKT cells express IL-4, IFN-γ, natural-killer group 2 member D, and IL-17, thus inducing vascular injuries (72).
The subsets of yδ TCR+ cells, such as CD27- CD44hi and CD27+ CD44lo, have also been increased in prediabetic NOD mice; however, the knowledge of concrete mechanism of γδ T in DKDs has been limited until now (73).
2.6 Mucosal-associated invariant T cells
MAIT cells not only belong to a specialized subset of unconventional (non-major histocompatibility complex-restricted) T cells but have also emerged as key players in immunity and pathological inflammation. First, human MAIT cells express a semi-invariant TCRa chain (Va7.2, coupled with restricted Ja segments), coexpressed with high levels of the C-type lectin receptor CD161, which is beneficial to its presentation in human barrier sites such as the kidneys (74, 75). Moreover, MAIT cells were reported to sharply increase with a cytokine cocktail comprising IL-12, IL-15, and IL-18, which participates in the progression of chronic inflammation (76).
Furthermore, researchers have found that dysregulation of MAIT cells may influence the severity of insulin resistance. The frequency of MAIT has been shown to be influenced by BMI, and there is a positive correlation between MAIT and HbA1c levels, accompanied by an increase in CD25 and CD69 (77, 78). Another study by Harms et al. observed a significant increase in the CD27- MAIT cell subset and IL-17A in patients with T1DM, particularly in younger patients (77).
2.7 Tissue-resident memory T-cells
CD69+ CD103+ and CD69+ CD103- TRM cells have been identified as two primary subsets of TRM cells (15). CD69 binds to S1PR1 on the T cell membrane, restraining the migration of memory T cells from the blood to peripheral tissues (79). Therefore, a mass of TRM-T cells exists in the kidney rather than in the circulation. After encountering antigens in vivo and in vitro, native T cells rapidly produce effector T cells and swiftly migrate to lymphoid and non-lymphoid tissues, persisting through barrier tissues, such as the kidney (80, 81). Following inflammation resolution, antigen-specific effector T cells differentiate into diverse memory T cell subsets with distinct trafficking properties.
In the tubulointerstitium of DKD, a recent xCell analysis has identified immune cells, thus revealing significant changes, including activated Th2 cells, CD4+ T cells, CD8+ T cells, dendritic cells, conventional dendritic cells, M1 macrophages, and restrained Tregs (82). However, knowledge of T cell differentiation in DKD remains limited.
3 Recruitment and activation of T cells in diabetic kidney disease
As early as 2012, the aberrant recruitment and activation of T cells in DKD had been discussed (83). The results showed an increase in CD4+, CD8+, and CD20+ cells in the interstitium, indicating that aberrant intrarenal infiltration and recruitment of T cells are potential immunopathological mechanisms of diabetic kidney lesions. Immunohistochemical analysis also showed that a substantial proportion of juxtaglomerular apparatuses in patients with T1DM contained abundant T cells (84).
To exert their local effects on renal injury, circulating T cells must reach the site of inflammation. Typically, some T cells, such as Th cells, do not possess a residency status similar to that of other immune cells, such as kidney TRM-T cells.
A series of tissue-specific markers has been reported to activate T cells in the kidney. Once activated, T cells can expand their immunoreaction, inducing chemokine release and more widespread recruitment of T cells (85). However, little is known about the trafficking of T cells into the kidney under hyperglycemic conditions, and their migration patterns have been the subject of extensive studies (17). Hence, the methods for circulating T cell migration into kidney should be assessed in the next step of research.
3.1 Chemokines and its receptor
There is a positive feedback between chemokines and T cells in the inflammatory response and immune adjustment. In other words, chemokines facilitate the attraction of circulating T-cells and stimulate their infiltration into tissues. T cells also participate in the regulation of the pathophysiological progression of renal insufficiency by producing chemokines. In this section, we focus on the chemokines involved in the recruitment of T cells in DKD.
3.1.1 CXCL9-CXCR3
Multiple studies have shown that the urinary level of C-X-C motif chemokine ligand 9 (CXCL9) mRNA is significantly elevated and correlated with eGFR decline, which can be utilized to measure and stratify the risk of DKD (86, 87).
On the other side of the CXCL9-CXCR3 axis, C-X-C motif chemokine receptor 3 (CXCR3) is a well-known chemokine receptor predominantly expressed on the surface of Th1 polarized T cells and regulates the recruitment of Th1 cells (88). Moreover, CXCL9 and CXCR3 have been found to be influenced by advanced glycosylation end products (AGEs), implying that Th1 can be recruited under diabetic conditions (89).
3.1.2 CXCL10/CXCR3
In an exploratory study, the CXCL10/CXCR3 axis was observed in the autoimmune process in T1D. Serum levels of CXCL10, a well-known Th1 chemokine, are elevated in patients with T1D, suggesting that CXCL10 plays a critical role in predicting T1D (90). Most importantly, CXCL10 may be induced by IFN-γ, promoting T cell infiltration and accelerating beta cell destruction (91).
3.1.3 CXCR5
In individuals with DN, the increase in CD4+ CXCR5+ Tfh cells may significantly increase creatinine, urea, urinary protein levels, fasting blood glucose, postprandial blood glucose, and HbA1c and decrease estimated GFR (55). In the future, the increased number of CD4+ CXCR5+ PD-1+ Tfh cells in patients with DN may be a new target for intervention in DKD (47).
3.1.4 CX3CL1-CX3CR1
At an early stage of nephropathy, CX3CR1+ T cells are elevated and induce IL-17A production in renal impairment (92–94). In addition, the polarization of TH17 or Treg cells may be associated with an increase in CX3CR1 reporter gene expression in T cells (92). Several studies have shown that CX3CR1 and CX3CL1 are upregulated in the kidneys of patients with diabetes, accompanied by an increase in urea, creatinine, A/C ratio, HbA1C, and IgG; however, the concrete mechanism of CX3CL1-CX3CR1 recruiting T cells requires further exploration in DKD (93).
3.1.5 CCL5 (RANTES)- CCR5
CCL5 is a β-chemokine, which is also known as RANTES (regulated on activation, normal T cell expressed and secreted), and can function as a chemotactic factor for T cells and induce cellular activation of normal T cells (95) In inflammatory kidney diseases, constitutive RANTES expression facilitates the accumulation of CD4+ T cells in the kidney, while the administration of RANTES-neutralizing antibody is helpful in reducing the accumulation of T cells in the kidneys to a large degree. Moreover, RANTES-neutralizing antibodies can reduce the deposition of collagen in obstructed kidneys (96).
There is no doubt that CCR5 is a characteristic of Th1 lymphocytes and a critical chemokine receptor for trafficking of TH1 cells to the kidney (88, 97); however, the status of CCR5 in T2DM and microvascular complications remains controversial. The problems are mainly focused on the significant discrepancy in the allelic frequency of CCR5 between different ethnic groups. In Asian populations and people with T2DM, the CCR5 59029G/A polymorphism is significantly associated with an enhanced susceptibility to DN (98). Nevertheless, the CCR5 59029 A allele only has a convincing association with nephropathy in T2DM Malaysian Chinese population but is weakly associated with nephropathy in Malaysian Indian population (99). Additionally, in native Estonian patients with T2D, there was a lack of association between the CCR5-Δ32 mutation and DKDs (100). Hence, further research is needed to determine whether CCR5 is associated with DKD worldwide.
3.1.6 CCL2 (MCP-1)- CCR2
Chemokine ligand 2 (CCL2) binds to its receptor, C-C chemokine receptor 2 (CCR2), initiating the migration and infiltration of T cells and regulating tissue inflammation (101). A longitudinal analysis followed the fate of CCR2−/− T cells and observed that CCR2 regulates the immune response by modulating the effector/regulatory T ratio. Additionally, CCR2 deficiency in T cells decreases the levels of Th17 cells while promoting a program that induces the accumulation of Foxp3+ Tregs in vivo (102).
Recent studies have suggested that CCL2 (MCP-1) is a key chemokine involved in DN. In a blood sample analysis conducted in Iran, CCL2 was gradually elevated in patients with T1D with disease duration (103). Furthermore, the blockade of this pathway plays a protective role in insulin resistance, modulation of adipose tissue, restoration of renal function, and restraint of progressive fibrosis in hyperglycemic kidneys (104, 105). A phase Ia study targeting emapticap impeded the CCL2/CCR2 receptor axis and exerted beneficial effects on ACR and HbAlc in albuminuric T2D (106). Overall, the CCL2/CCR2 receptor axis is thought to be crucial for the progression of DKD.
3.1.7 Interleukins
T cells not only produce several members of the IL family but are also recruited by other immunocytes produced ILs, such as IL-18, IL-19, among others.
IL-18 is not mainly produced by T cells but plays an underlying pathophysiological role in the progression of T cell differentiation in DKD. IL-18 induces plasticity in established Th1 and Th2 cells (107–109). It also acts synergistically with IL-12 to increase the level of IFN-γ, a Th1 cytokine (110). In recent years, a cross-sectional study of patients with T2D showed that IL-18 levels were significantly boosted at a low eGFR and positively correlated with the development of DN and urinary albumin excretion (UAE) rate (111, 112).
Similarly, IL-19 were markedly positively correlated with Hs-CRP, cystatin C, and UAE in patients with DN (113). The reduction in IL-19 levels contributes to the suppression of T-cell responses and inhibition of the regulatory activity of CD4+ T cells, causing cell-mediated immunosuppression (114). Therefore, IL-19 may be another target for regulating T cell differentiation in DKD.
3.1.8 TGF-β
In renal inflammatory diseases, TGF-β has been demonstrated to orchestrate the differentiation of T cells, including Th17 and Foxp3+ Treg cells (96) Additionally, rats with hyperglycemia-induced microalbuminuria possess upregulated TGF-β and serum creatinine levels (115). Recently, the role of TGF-β in promoting the characterized T cell cytokines, IL-9 and IL-17, has become more widely accepted. TGF-β controls the secretion of both these cytokines, subsequently mediating fasting and postprandial glucose and HbAlc levels in patients with DN (116). Taken together, restraining TGF-β may be considered as an approach aimed at attenuating T1D in the immediate future.
3.2 Other factors that regulate T cells
Similar to chemokines, there are many other factors that facilitate the assembly and infiltration of T cells, such as C3a and its receptor, AGE, KIM-1, Chromogranin A, among others.
3.2.1 Complement C3a and its receptor
Emerging evidence suggests that the expression of C3a and C3aR is involved in DN pathogenesis (117). Compared with normal controls, C3aR was significantly increased in the renal specimens of patients with diabetes and wild-type (WT) diabetic mice. In vitro microarray profiling revealed the underlying mechanism that C3a plays a role in suppressing T-cell adaptive immunity by interfering with CD4+ and CD8+ T cell infiltration, and in an in vitro study, C3a was able to enhance differentiation of the T-cell lineage in inflammatory responses (118). Thus, C3aR may be a promising target for T cell recruitment and activation.
3.2.2 Advanced glycosylation end products
In peripheral blood T lymphocytes, the expression of AGE binding sites serves to target T cells to the AGE-rich renal tissues. With the increase and accumulation of AGE products and AGE-modified proteins, their binding to the AGE receptor on T cells is remarkably increased, promoting the synthesis and release of proinflammatory cytokines in diabetes (119).
3.2.3 KIM-1
KIM-1 is also known as T-cell lg mucin 1 (TIM-1) or hepatitis A virus cellular receptor 1 and has been reported as a transmembrane glycoprotein receptor on T cells (120). Recent studies have revealed elevations in KIM-1, suggesting that glycemic variations may increase the production of KIM-1 in CD8+ T cells in individuals with DKD, thereby increasing the risk of DKD (121). The elevations in circulating KIM-1 also increases the urinary KIM-1 in DN, verifying that KIM-1 can be a biomarker and a reliable predictor of diabetic kidney injury (122).
3.2.4 Chromogranin A
The β-cell secretory granule protein, also known as chromogranin A, is a new autoantigen in T1D. A recent study identified chromogranin A as a forceful inducer of the reacting CD4+ T cells in the pathogenic process of T1D in NOD mice (123). However, studies on the function of chromogranin A in diabetic vascular complications and DKD are still insufficient.
4 T cells regulate inflammation in diabetic kidney disease through inflammatory cytokines
In DKD, the inflammatory cytokines secreted by T cells can cause the epithelial-to-mesenchymal transition and the extracellular matrix accumulation (124). In this section, we have elaborated on the mechanisms by introducing, summarizing, and comparing the inflammatory mediators in DKD, which may prove useful in future researches.
4.1 IL-1β
Based on the different encoding genes, IL-1, a classical chemokine, is divided into IL-1α and IL-1β. Both can bind to the primary receptor point of distinction (IL-1RI), while only IL-1β is secreted by T cells and macrophages (125). In diabetic metabolic syndrome, high glucose and oxidative stress can induce IL-1 activation, which occurs earlier than the pathophysiological manifestations. IL-1β production may be related to TNFR-Fas-caspase-8-dependent pathway in CD4+ T cell-driven autoimmune pathology (126). Moreover, IL-1β was also identified to cause endothelial cell damage in resistance arteries and affect the NADPH oxidase activation (127, 128). In addition, studies have shown that repressing IL-1β and its receptor can reduce systemic inflammation in patients with T2DM (128, 129).
4.2 IL-2
IL-2 can be produced by Th and kidney-derived MAIT cells. The function of the IL-2/IL-2R in renal dysfunction has been discussed in early studies, which has indicated that serum soluble IL-2R (sIL-2R) levels increase with a decrease in creatinine clearance (130). In the autoimmune diabetic NOD mice, two separate research groups have revealed that deficiency in IL-2 production or the responsiveness of Tregs to IL-2 may be associated with the development of the immune response (131, 132). Given its crucial role in the expansion and function of Tregs, IL-2 has been used to regulate tissue damage and limit the immune response following infection (133). Low-dose IL-2 selectively induces CD4+ CD25+ FOXP3+ Tregs in patients with CKD, and these Tregs limit the levels of proinflammatory Th1 and Th17 cells (133). In other mouse models of autoimmune diseases, such as C57BL/6 mice, CD4+ CD25+ Tregs are also induced by recombinant IL-2, thus preventing the progression of diabetes (134). Hence, it would be interesting to explore the effect of IL-2 on new therapeutic schedules for patients with DKD.
4.3 IL-4
IL-4, partly produced by Th2 and NKT cells, can expand the proliferation of activated T and B cells and regulate the differentiation of Th1 and Th2 cells (135). The role of IL-4 in DM remains controversial. Data investigation of Filipino patients suggested that the risk of T1D was partly determined by specific polymorphisms. The variability in promoters, coding sequences, and specific combinations of genotypes indicated that IL-AR of IL-4 and IL-13 were significantly associated with susceptibility to T1DM (136). In contrast, no significant change in IL-4 plasma levels between patients with T2DN and those without nephropathy was observed in a study (25). The IL-4 rs2243250 polymorphism is irrelevant to DN in Slovenian patients with T2DM (137). As a result, the relationship between IL-4 and DN may depend on race, ancestry, geographical conditions, and national customs to some extent, which needs to be proven by more prospective evidence.
4.4 IL-6
IL-6 levels are significantly increased in the plasma of patients with DN patients than those with diabetes but without nephropathy (138). Furthermore, the IL-6-174 G allele was found to increase the occurrence rate of DN, confirming the correlation between IL-6 and DN (139). Similarly, a recent meta-analysis revealed the significance of different IL-6 polymorphisms in DN progression. The results showed that IL-6 rs1800795, rs1800796, and rs1800797 were associated with DN, whereas IL-6 rs2069837 and rs2069840 may be indifferent to the risk of renal complications in patients with T2DM (139).
Classically, IL-6 participates in the pathogenesis of DKD by various methods, including binding to the receptor IL-6R, sIL-6R trans-signaling pathway, and IL-6 autocrine signaling (140). IL-6 influences renal cells by relying on diverse signaling pathways. For instance, IL-6 facilitated mesangial expansion by infiltrating the mesangium, interstitium, and tubules, which has been observed in human renal biopsies (141). Second, the determination of samples from patients indicated that the width of the glioblastoma (GBM) was directly associated with fibrinogen and IL-6 levels in diabetic glomerulopathy (142). Moreover, the effects of IL-6 on diabetic renal injury may be due to increased insulin resistance and promotion of the inflammasome (143).
4.5 IL-9
IL-9 is mainly produced by a flock of T cells, such as Tregs and Th2 cells, and manipulates signaling pathways in renal immune diseases. For example, IL-9 protects against progressive glomerulosclerosis and tubulointerstitial fibrosis and regulates T cell-induced immune suppression in adriamycin-induced nephropathy and acute kidney injury (144, 145). Meanwhile, as characterized by T cell cytokines, IL-9 levels were evidently reduced in the diabetic group and positively correlated with the level of urea and microalbuminuria, which may be considered as an approach of T cells to address hyperglycemia damage (43).
4.6 IL-17
IL-17A can be produced by many types of CD4+αβ and γδ T cells, particularly Th17 cells (146). The IL-17 family is essential for the inflammatory response and includes six structurally related isoforms: IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F. However, only IL-17A and IL-17F have unique functions in DN, whereas IL-17C and IL-17E are indifferent to DN (44).
Clinically, the decline in IL-17 levels is synchronous with the progression of DKD and is correlated with declining GFR (43, 147). IL-17A has been proven to not only trigger inflammatory signaling pathways associated with NF-κB downstream but also regulate the viability of T cells (147). However, another study reported the opposite result, indicating that IL-17A may increase the infiltration of inflammatory cells in renal tissue and blood pressure in mice (148)
As a potential immunologic therapeutic target for DKD, studies have suggested that intrarenal IL-17A1 CD41 T cells can be suppressed by mycophenolate mofetil, which is beneficial for treating albuminuria and tubulointerstitial fibrosis (41). All-trans retinoic acid was used to retain the capacity of Tregs to secrete IL-17 during hyperglycemia, implying an important role of IL-17 in DKD (149).
4.7 IL-22
In addition to studies on DKD, IL-22, mainly produced by Th22, was found to be downregulated in patients with DKD. Further observations indicated that the mechanism of IL-22 participating in inflammatory processes of DKD is intricate and comprehensive. Chen et al. demonstrated that IL-22 induced AMPK/AKT signaling and PFBFK3 activity, alleviating the level of dysfunctional mitochondria and the accumulation of reactive oxygen species (150). In addition, IL-22 can ameliorate renal fibrosis and attenuate microalbuminuria in DKDs (150, 151).
4.8 IL-35
Anti-inflammatory cytokine IL-35 is expelled by Tregs, regulatory B cells, and tolerogenic antigen presenting cells. Tregs were reported to infiltrate renal tissues to maintain homeostasis of the immune system in patients with diabetes and use IL-35 to intervene in the development of DKD (63).
4.9 INF-γ
Several studies have reported that T cells can be stimulated by high glucose concentrations and expedite IFN-γ production (83, 130, 152). Under conditions of high glucose concentrations, IL-12 can stimulate CD4 cells to produce IFN-γ. AGE-modified proteins bind to the receptor for AGE and T cells, inducing the synthesis and release of IFN-γ and accelerating inflammation of renal tissues (130).
4.10 TNF-α
As a synthetic product of T cells, TNF-α may be used as an indicator for evaluating DKDs (153). Many clinical studies have found that TNF-α is increased in the plasma and urine of patients with diabetes, leading to a higher risk of mortality, more serious macroalbuminuria, sodium retention, and renal hypertrophy (154–157). Specifically, TNF-α participates in the pathophysiological reaction in DN via diverse pathways, including altering intraglomerular blood flow, reducing glomerular filtration, inducing cytotoxicity to renal cells, and producing local reactive oxygen species (158–160).
5 Summary of other functions of T cells in diabetic kidney disease
Pathologically, hyperglycemia stimulates T cells to produce chemokines and cytokines that not only participate in the promotion of inflammation and activation of macrophages and endothelial cells but also damage renal function through different mechanisms. First, these proinflammatory molecules highlight the role of T cells in the process of insulin resistance. Second, T cells mediate the glomerular filtration barrier through podocytes. Third, T cells contribute to extracellular matrix deposition and the differentiation and proliferation of myofibroblasts. Ultimately, T cells lead to proteinuria and the development of DKD (Figure 1).
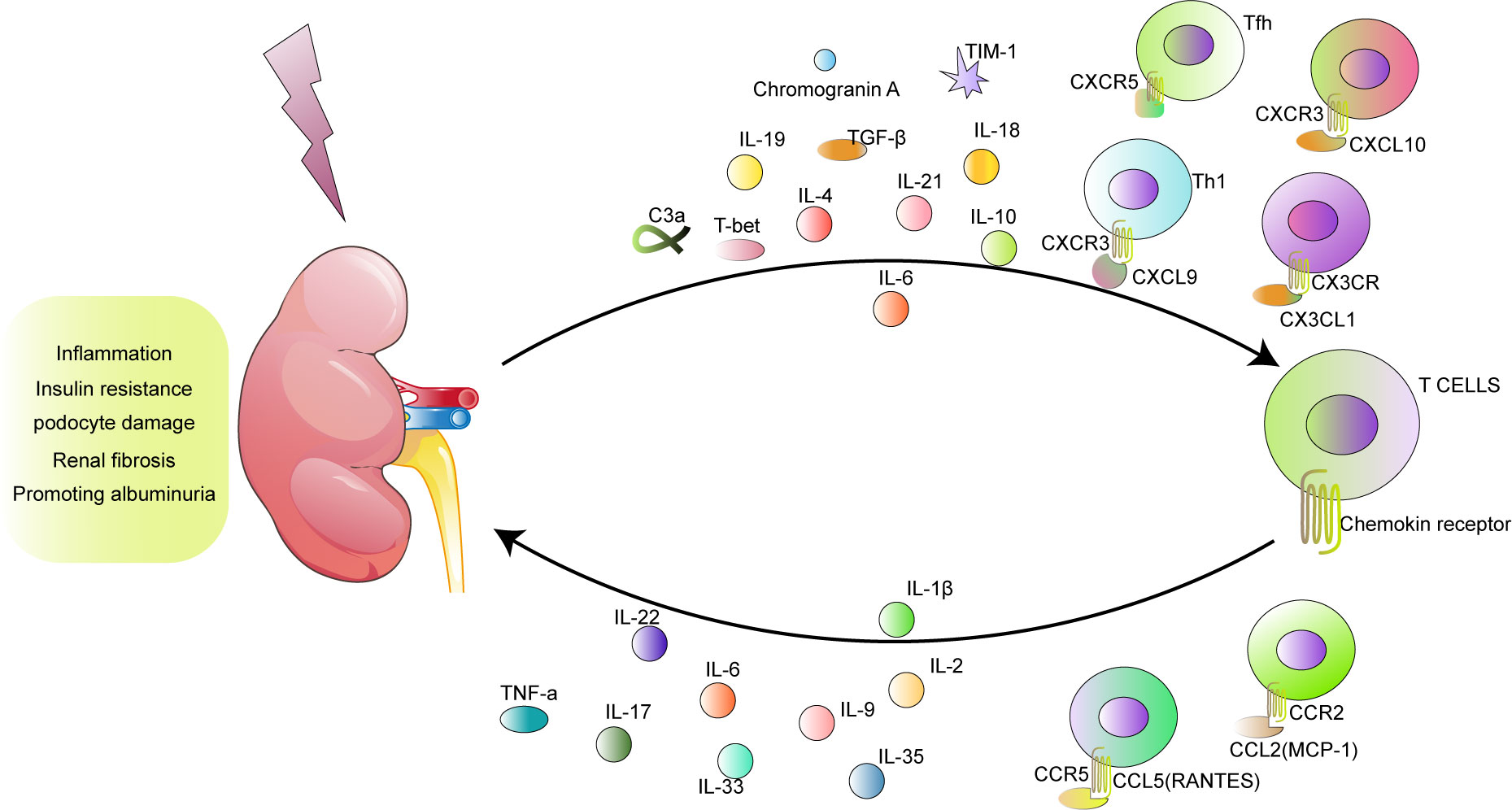
Figure 1 The model of T cells and their products participating in DKD.
5.1 Function of T cells in insulin resistance
Insulin resistance is often regarded as a strong marker of DKD and is characterized by hyperinsulinemia and reduced insulin action, affecting many classical insulin-regulated pathways in the kidney and vasculature (161). For instance, the lack of insulin resistance in the kidney has been verified as an inducer of sodium retention, resulting in salt-sensitive hypertension. Podocyte insulin sensitivity is critical for glomerular alterations and disorders in DKDs (162, 163). In recent years, T-cells have been reported to improve glucose tolerance, enhance insulin sensitivity, and reduce weight gain in mouse models (164). However, a summary of T cells in renal insulin resistance is currently insufficient.
The relationship between insulin resistance and T cells has been described, involving Tregs, CD8+ T cells, Th cells, and MAIT cells. The depletion of Tregs leads to enhanced insulin resistance and impaired insulin sensitivity accompanied by albuminuria and glomerular hyperfiltration (165). In contrast, insulin sensitivity in DKD can be significantly rescued by adoptive transfer of CD4+ FoxP3+ Tregs in a murine model, resulting in less diabetic kidney damage (166). In addition, CD4+T cells in visceral adipose tissue have also been demonstrated to regulate insulin resistance and control glucose homeostasis in diet-induced obesity progression. When Th1 statically overwhelms CD4+ FoxP3- Tregs, weight gain and insulin resistance are reversed (166).
Moreover, the depletion of CD8+ T cells has been reported to alleviate macrophage infiltration of CD8+ T cells. CD8 + T cells recruit macrophages to mediate insulin resistance and adipose tissue inflammation (65). Conversely, systemic insulin resistance is aggravated by adoptive transfer of CD8+ cells (167). In addition, Th1 and MAIT cells can regulate insulin resistance (77, 168).
As mentioned above, secretion and release of T cells produce proinflammatory cytokines that not only induce insulin resistance but also impair kidney function. IL-1, IL-6, IL-17, IL-33, GATA-3, and other proinflammatory cytokines also play important roles in renal insulin resistance. First, the blockade of IL-1 improves glycemia and β-cell secretory function; repression of the IL-6 receptor relieves diabetic renal injury and insulin resistance, and suppression of GATA-3 restores insulin sensitivity (129, 143, 169). In clinical investigations, TGF-β is positively correlated with insulin resistance markers, including fasting and postprandial glucose levels and HbA1c, whereas IL-17 is negatively associated with them (43). Additionally, studies have shown a serial decline in IL-33 levels in DN, resulting in an increased severity of insulin resistance and microalbuminuria (170). Overall, insulin resistance in DKD is closely associated with proinflammatory cytokines produced by T cells.
5.2 T cells and podocyte damage in DKD
Normal function and structural integrity of podocytes are essential for the occurrence of albuminuria and progression of diabetes (171). T cells and their production have been described as novel factors influencing podocytes in patients with diabetes (172). Therefore, podocytes may be regarded as an essential part of T cells that mediate pathological effects in DKD.
Firstly, CD28/B7 and cytotoxic T lymphocyte-associated antigen-4 (CTLA4) are critical for Th cells and podocytes. With regard to T-cell proliferation, differentiation, and survival, costimulatory molecules composed of CD28, B7-1 (CD80), and B7-2 (CD86) have been reported to play crucial roles (173). As a novel biomarker for podocyte damage, B7-1 is upregulated in podocytes under high glucose conditions. After activation, the CD28/B7-1 pathway mediates circulating T cells to aggravate podocyte damage (174, 175). Moreover, CTLA4 is a negative regulator of T cell activation, and genetic polymorphisms in CD28/B7/CTLA4 are related to susceptibility to T2DM (176). However, the fact that B7-1 was not inducible in podocytes in patients with DKD is contradictory; therefore, further investigation is required (177).
Researchers have also discovered that IL-17A is a characteristic proinflammatory cytokine in the serum and urine of patients with diabetes, and CD40 expression was observed to be increased in podocytes with DN (38, 147). The synergistic action of IL-17 and CD40L regulates the inflammatory response and mediates remodeling of glomerular sclerosis in DN.
Furthermore, podocyte damage is affected by TNF. Albuminuria is partly attributed to TNF-induced ABCA1 deficiency in podocytes. Studies have indicated that TNF is sufficient to cause free cholesterol-dependent podocyte injury through an NFATC1/ABCA1 dependent mechanism (155).
Podocyte apoptosis is triggered by CD8+ TRM cells. In db/db mice, the relative proportion of CD8+ TRM cells is remarkably increased under pathological conditions, and renal CD8+ TRM cells have cytotoxic effects on podocytes and enhance podocyte apoptosis (66).
5.3 T cells and renal fibrosis in DKD
Pathologically, fibrosis is one of the most fundamental characteristic mechanisms in the onset and progression of DKD, and renal T-cell infiltration is helpful for fibrosis. Therefore, hyperglycemia stimulates T cells and T cell-derived products, including IL-1, IL-6, IL-17, and IL-22, which are of central importance in progressive fibrosis in DKD (66).
Primarily, IL-1β induces proximal tubule damage and fibrosis in renal tubule interstitials (178). One study showed that IL-1β participates in the dysregulation of glycolysis and matrix activation, leading to tubulointerstitial fibrosis (147). In contrast, another report on CKD described the relationship between IL-1β and fibrosis initiation and progression (178).
Second, IL-6 trans-signaling may be a crucial factor in the development of renal fibrosis, thus influencing the width of the GBM in the pathogenesis of diabetic glomerulopathy (142, 179). Simultaneously, targeting IL-6 trans-signaling, Fc-gp130, could be a novel therapeutic strategy for renal fibrosis.
Moreover, IL-17 suppresses fibrosis via the STAT-3 and WAP domain protein pathways in models of T1D and T2D, and tubulointerstitial fibrosis can be rescued by suppressing intrarenal IL-17A1 CD41 T cells (41, 44). Furthermore, through the NLRP3/caspase-1/IL-1β pathway, IL-22 can reverse the overexpression of fibronectin, collagen IV, and extracellular matrix in mouse renal glomerular mesangial cells, thereby ameliorating renal fibrosis and proteinuria excretion in DN (150, 180).
Additionally, sphingosine 1-phosphate receptor 1 activation in T cells leads to fibrosis in normoglycemic conditions but exacerbates fibrosis in a model of STZ-induced diabetic cardiomyopathy (181).
5.4 T cells and albuminuria in DKD
5.4.1 The quantity of T cells and albuminuria
To elaborate the internal relationship between T cells and albuminuria in DN, preliminary exploration was performed. Under STZ stimulation, only Rag1(+/+) mice, which have mature T lymphocytes, had glomerular immunoglobulin deposition. However, Rag1(-/-) mice, which lack kidney infiltration with T cells, were protected from albuminuria (172). Additionally, the degree of albuminuria is regulated by the number of T cells infiltrating the kidneys of DKD animals, and abatacept ameliorates DKD by blocking systemic T-cell activation (182).
However, there are also a series of contradictory reports. Absolute and percent T-lymphocytes were found to be relatively lower in patients with nephrotic proteinuria and long-standing insulin-dependent diabetes (183). In contrast, Another study showed that T cell-positive patients had a shorter duration of diabetes and lower albumin excretion rates than T cell-negative patients (84). Hence, the exploration and summary of the relationship between T cells and proteinuria in DN are significant.
5.4.2 Types of T cells associated with proteinuria
In general, circulating CD8+ T cells and Tregs are considered the main types of T cells that are associated with albuminuria in DN. A cross-sectional study showed that the percentage of circulating CD8+ T cells was correlated with albuminuria in T2DM, indicating that systemic inhibition of T lymphocytes provides a new therapeutic direction for albuminuria in DKD (184). In addition, FoxP3+ Tregs exert a protective effect in the kidneys of diabetic mice, although it reduces glomerular hyperfiltration and albuminuria. Moreover, depletion of Tregs with anti-CD25 antibodies can accelerate the progression of albuminuria (165).
5.4.3 Product of T cells and albuminuria
T cells regulate albuminuria through cytokines including IL-6, IL-9, TNF-α, IL-22, IL-33, and IL-233.
IL-6, associated with higher albuminuria, has been reported in db/db mice and patients with diabetes (143, 185). The IL-6 receptor antibody (tocilizumab) can reduce proteinuria and glomerular mesangial matrix accumulation. Furthermore, the levels of IL-9 and TNF-α are positively correlated with the levels of urea and microalbuminuria (43, 185, 186). Albuminuria may be caused by TNF-α via alterations in the glomerular capillary wall and an increase in albumin permeability.
In addition, studies on IL-22 support the hypothesis that cytokines drive proteinuria. IL-22 can alleviate mesangial matrix expansion and proteinuria in mice (151, 172). IL-33 also represses microalbuminuria in DKDs (170, 187). Intriguingly, the increase in IL-33 levels in DN is only associated with diabetes but not with kidney injury (188). Therefore, the exact role of IL-33 in DKD remains controversial.
Notably, a novel cytokine (named “IL-233”) possesses the activities of both IL-2 and IL-33 and protects against type-2 DN by promoting T-regulatory cells. Treatment with IL233 reduces hyperglycemia, plasma glycated proteins, and albuminuria, protecting mice from T2DN (189).
6 Promising novel therapies targeting T cells in DKD
Until now, the standard management strategy for DKD has prioritized strict glucose control and blood pressure with RAAS blockade. However, the therapeutic means are limited to stopping or reversing the progression of DN. Therefore, new drugs targeting the pathological mechanisms of DKD, such as T cells and their products, have drawn increasing attention (Table 1).
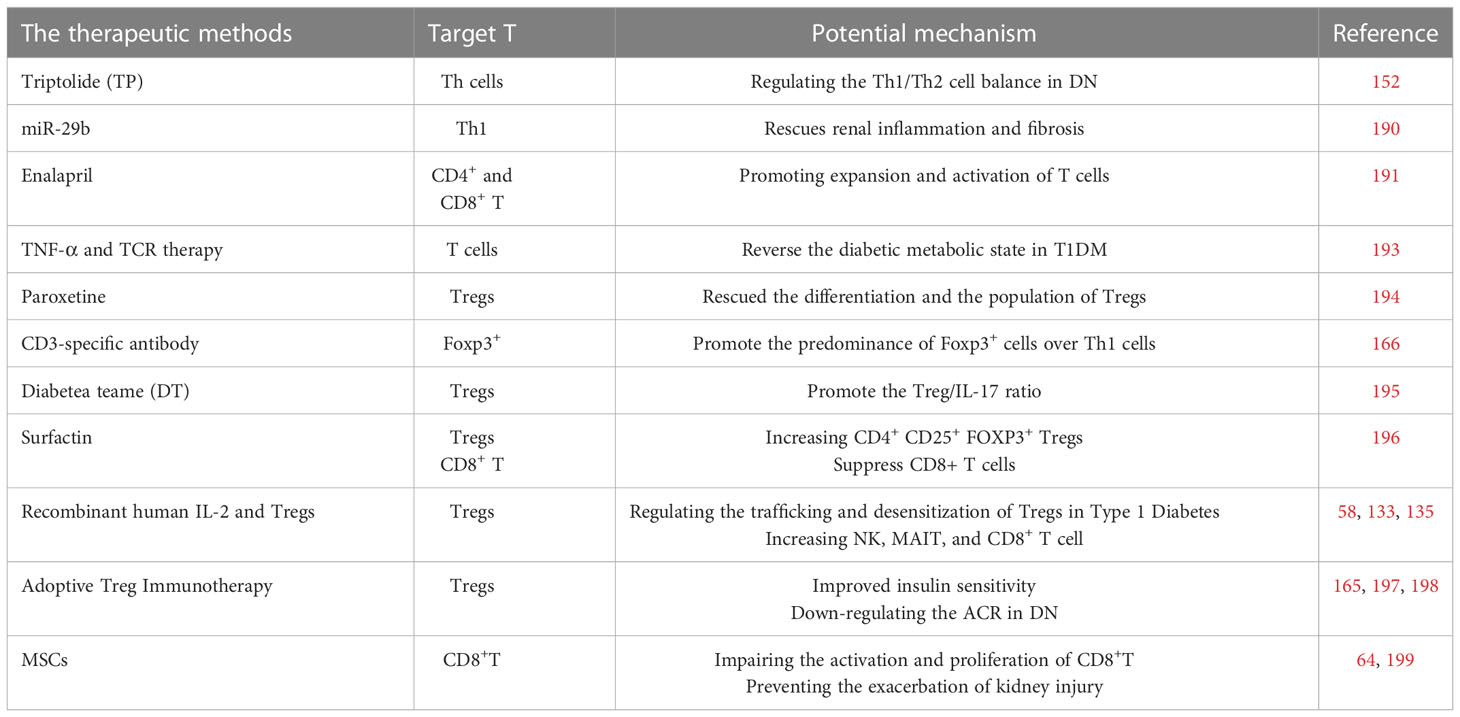
Table 1 The therapeutic methods targeting to T cells in DKD.
6.1 Traditional drug therapy with T cells
First, there are some drugs that target the Th cells. Similar to triptolide, a well-known drug for DN, influences Th lymphocyte cells in rat models of DN by regulating the Th1/Th2 cell balance. DN is associated with the upregulation of Th1 cells and downregulation of Th2 cells; however, triptolide can alter this ratio in high-fat diets and STZ-induced rats (152). Concurrently, animal experiments have shown that miR-29b is a novel therapeutic agent for treating T2D that effectively rescues renal inflammation and fibrosis by inhibiting T-bet/Th1-mediated immune response (190).
Second, the expansion and activation of CD4+ and CD8+ T cells can be enhanced by Enalapril and γ-aminobutyric acid receptors in DKDs (191, 192).
In addition, combining anti-TNF-α therapy and the T-cell-specific antibody anti-TCR can reverse the diabetic metabolic state in a model of human T1D (193).
6.2 Tregs-targeted drugs
Researchers have discovered that administering drugs targeting Tregs can be beneficial in diabetic diseases. For instance, Paroxetine, a G protein-coupled receptor kinase 2 inhibitor, has been approved to rescue Treg differentiation and restore the population of circulating Tregs in vitro and in vivo (194). In obese WT and ob/ob (leptin-deficient) mice, a CD3-specific antibody or its F(ab’)2 fragment can promote the predominance of Foxp3+ cells over Th1 cells (166).
Ethnopharmacological relevance Diabetea teame was verified to promote the Treg/IL-17 ratio in clinical settings, suggesting the protective effect of DT against diabetes-related complications in the long term (195). In addition, Surfactin, a bacillus-produced natural immunomodulator, could increase CD4+ CD25+ FOXP3+ Tregs while simultaneously suppressing T cell proliferation and downregulating the activated CD8+ T cells (196).
6.3 Recombinant human IL-2 and Tregs
IL-2 plays an essential role in the expansion of Tregs and can reduce tissue damage by limiting immune response. A single ultra-low dose of Aldesleukin (proleukin; recombinant human IL-2) has been demonstrated to regulate early altered trafficking and desensitization of Tregs in T1D (133). Simultaneously, low expression of mlL-2 also prevents the progression of diabetes by regulating Tregs in islets (135). Furthermore, combining low doses of IL-2 with exogenously administered Tregs leads to an increase in the number of Tregs, NK cells, mucosal associated invariant T cells, and clonal CD8+ T cells (58).
6.4 Adoptive Treg immunotherapy
Recently, expanded Tregs have been used to treat deficits in the number and suppressive activity of Tregs in immune-related diseases. Two separate research groups have explored adoptive Treg immunotherapy and demonstrated its safety, tolerance, and efficacy in patients with DM (197, 198). Bluestone et al. reported a phase 1 trial of adoptively transferred self-derived Tregs to repair or replace Tregs in patients with T1D. Simultaneously, adoptive transfer of CD4+ FoxP3+ Tregs significantly improved insulin sensitivity and decreased the albumin-to-creatinine ratio in DN (165).
6.5 Mesenchymal stem cells
In the last decade, MSCs have been widely used to treat DN. Intriguingly, MSC-CM pretreatment reduced CD8+ T cell priming and proliferation capacities in the kidneys of DN rats (64). Furthermore, MSC transplantation not only impairs the activation and proliferation of CD8+ T cells but also prevents the exacerbation of kidney injury, providing a new insight into the treatment of DN (199).
7 Conclusion
With the increase in the number of patients suffering from DKD, exploration of the function of T cells in DKD is increasingly important. After circulating T cells are recruited into the renal tissue or T cells are amplificated, differentiated, and activated in the kidney, T cells play protective or pathogenic roles through multiple pathways, including influencing insulin resistance, mediating podocyte damage, participating in fibrosis, and regulating proteinuria (Table 2 and Figure 1).
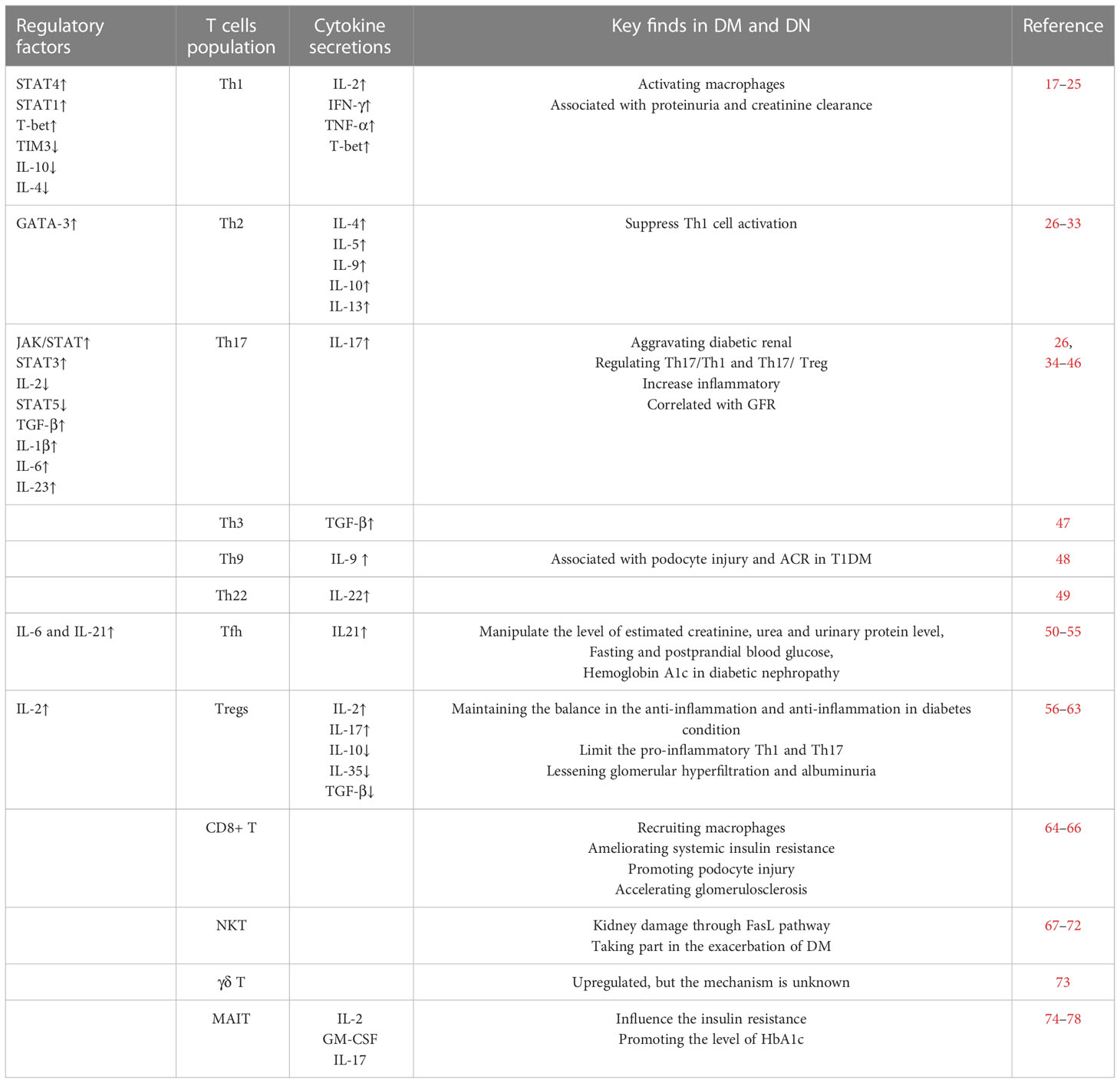
Table 2 The fundamental function of T cells in DM and DN.
As Table 1 showed, based on the significant functions of T cells and cytokines, the application of T cell-associated therapies in DKD has been attempted and preliminary achievements have been made. Promising studies on T cell biology will unquestionably contribute to a more profound understanding of DKDs, highlighting the need to identify new therapeutic approaches.
Author contributions
YLi and YLv conceived and drafted the review article. TZ, TH and YLa created the model figure. QS and YLi prepared the tables. ZK, YG, SL were involved in the compilation of the references. MY and YLu revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by National Natural Science Foundation of China (Grant: 81873615, 82070744, 81770723, 81400732), Academic promotion programme of Shandong First Medical University (NO: 2019QL022) and Taishan Scholars Program (NO: ts201712090, tsqn201812138).
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
DKD, Diabetic kidney disease; MHC, Major histocompatibility complex; MAIT, Mucosal-associated invariant T cells; NKT, Natural killer T cells; Trm, Tissue-resident memory T-cells; TFH, T follicular helper cells; IFN-γ, Interferon-γ; TNF-α, Tumor necrosis factor-α; STAT, Signal transducer and activator of transcription; TIM3, Immunoglobulin domain and mucin domain 3; Treg, Regulatory T cell; FasL, NF-α/Fas ligand; S1PR1, Sphingosine 1-phosphate receptor 1; HbAlc, Glycated hemoglobin; CXCL, C-X-C motif chemokine ligand; CXCR, C-X-C motif chemokine receptor; CCL2, Chemokine ligand 2; CCR, Receptor C-C chemokine receptor; AGE, Advanced glycosylation end; TIM-1, T-cell immunoglobulin and mucin-containing molecule-1; KIM-1, kidney injury molecule-1; tolAPCs, Tolerogenic antigen presenting cells; IDDM, Insulin-dependent diabetic; TP, Triptolide; TCR, T-cell-specific antibody anti-T-cell receptor; GRK2, G protein coupled receptor kinase 2; DT, Diabetea teame.
References
1. Chen Y, Lee K, Ni Z, He JC. Diabetic kidney disease: Challenges, advances, and opportunities. Kidney Dis (Basel Switzerland) (2020) 6(4):215–25. doi: 10.1159/000506634
2. Zheng Z, Zheng FJ. Immune cells and inflammation in diabetic nephropathy. Jodr (2016) 2016. doi: 10.1155/2016/1841690
3. Anders HJ, Huber TB, Isermann B, Schiffer M. CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nat Rev Nephrol (2018) 14(6):361–77. doi: 10.1038/s41581-018-0001-y
4. Dronavalli S, Duka I, Bakris GL. The pathogenesis of diabetic nephropathy. Nat Clin Pract Endocrinol Metab (2008) 4(8):444–52. doi: 10.1038/ncpendmet0894
5. Bondar IA, Klimontov VV. [Immune inflammatory mechanisms in the development of diabetic nephropathy]. Probl Endokrinol (Mosk) (2007) 53(2):34–40. doi: 10.14341/probl200753234-40
6. Alsaad KO, Edrees B, Rahim KA, Alanazi A, Ahmad M, Aloudah N. Collagenofibrotic (Collagen type III) glomerulopathy in association with diabetic nephropathy. Saudi J Kidney Dis Transpl (2017) 28(4):898–905. Available at: https://www.sjkdt.org/text.asp?2017/28/4/898/211324.
7. Harjutsalo V, Groop P-H. Epidemiology and risk factors for diabetic kidney disease. Adv Chronic Kidney Dis (2014) 21(3):260–6. doi: 10.1053/j.ackd.2014.03.009
8. Yang X, Mou S. Role of immune cells in diabetic kidney disease. Curr Gene Ther (2017) 17(6):424–33. doi: 10.2174/1566523218666180214100351
9. Wu CC, Sytwu HK, Lu KC, Lin YF. Role of T cells in type 2 diabetic nephropathy. Exp Diabetes Res (2011) 2011:514738. doi: 10.1155/2011/514738
10. Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol (2015) 6:1. doi: 10.3389/fimmu.2015.00001
11. Constantinides MG, Belkaid Y. Early-life imprinting of unconventional T cells and tissue homeostasis. Science (2021) 374(6573):eabf0095. doi: 10.1126/science.abf0095
12. Kaminski H, Couzi L, Eberl M. Unconventional T cells and kidney disease. Nat Rev Nephrol (2021) 17(12):795–813. doi: 10.1038/s41581-021-00466-8
13. Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell (2015) 161(4):737–49. doi: 10.1016/j.cell.2015.03.031
14. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol (2012) 188(10):4866–75. doi: 10.4049/jimmunol.1200402
15. Woon HG, Braun A, Li J, Smith C, Edwards J, Sierro F, et al. Compartmentalization of total and virus-specific tissue-resident memory CD8+ T cells in human lymphoid organs. PloS Pathog (2016) 12(8):e1005799. doi: 10.1371/journal.ppat.1005799
16. Soukou S, Huber S, Krebs CF. T Cell plasticity in renal autoimmune disease. Cell Tissue Res (2021) 385(2):323–33. doi: 10.1007/s00441-021-03466-z
17. Galkina E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol (2006) 17(2):368–77. doi: 10.1681/ASN.2005080859
18. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100(6):655–69. doi: 10.1016/S0092-8674(00)80702-3
19. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. i. definition according to profiles of lymphokine activities and secreted proteins. J Immunol (1986) 136(7):2348–57.
20. Tipping PG, Kitching AR. Glomerulonephritis, Th1 and Th2: what’s new? Clin Exp Immunol (2005) 142(2):207–15. doi: 10.1111/j.1365-2249.2005.02842.x
21. Yang R, Mele F, Worley L, Langlais D, Rosain J, Benhsaien I, et al. Human T-bet governs innate and innate-like adaptive IFN-γ immunity against mycobacteria. Cell (2020) 183(7):1826–47.e31. doi: 10.1016/j.cell.2020.10.046
22. Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-Bet is a STAT1-induced regulator of IL-12R expression in naïve CD4+ T cells. Nat Immunol (2002) 3(6):549–57. doi: 10.1038/ni794
23. Lu C, Chen H, Wang C, Yang F, Li J, Liu H, et al. An emerging role of TIM3 expression on T cells in chronic kidney inflammation. Front Immunol (2021) 12:798683. doi: 10.3389/fimmu.2021.798683
24. van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev (2011) 91(1):79–118. doi: 10.1152/physrev.00003.2010
25. Wu CC, Chen JS, Lu KC, Chen CC, Lin SH, Chu P, et al. Aberrant cytokines/chemokines production correlate with proteinuria in patients with overt diabetic nephropathy. Clin Chim Acta (2010) 411(9-10):700–4. doi: 10.1016/j.cca.2010.01.036
26. Zhang C, Xiao C, Wang P, Xu W, Zhang A, Li Q, et al. The alteration of Th1/Th2/Th17/Treg paradigm in patients with type 2 diabetes mellitus: Relationship with diabetic nephropathy. Hum Immunol (2014) 75(4):289–96. doi: 10.1016/j.humimm.2014.02.007
27. Mosmann TR, Moore KW. The role of IL-10 in crossregulation of TH1 and TH2 responses. Immunol Today (1991) 12(3):A49–53. doi: 10.1016/S0167-5699(05)80015-5
28. Fathy SA, Mohamed MR, Ali MAM, El-Helaly AE, Alattar AT. Influence of IL-6, IL-10, IFN-γ and TNF-α genetic variants on susceptibility to diabetic kidney disease in type 2 diabetes mellitus patients. Biomarkers (2019) 24(1):43–55. doi: 10.1080/1354750X.2018.1501761
29. Vaseghi H, Sanati MH, Jadali Z. T-Helper cell type-1 transcription factor T-bet is down-regulated in type 1 diabetes. Iran J Allergy Asthma Immunol (2016) 15(5):386–93. Available at: https://ijaai.tums.ac.ir/index.php/ijaai/article/view/734.
30. Tang A, Li C, Chen Z, Li T. Anti-CD20 monoclonal antibody combined with adenovirus vector-mediated IL-10 regulates spleen CD4+/CD8+ T cells and T-bet/GATA-3 expression in NOD mice. Mol Med Rep (2017) 16(4):3974–82. doi: 10.3892/mmr.2017.7111
31. Al-Jaber H, Al-Mansoori L, Elrayess MA. GATA-3 as a potential therapeutic target for insulin resistance and type 2 diabetes mellitus. Curr Diabetes Rev (2021) 17(2):169–79. doi: 10.2174/1573399816666200705210417
32. Luopajärvi K, Skarsvik S, Ilonen J, Akerblom HK, Vaarala O. Reduced CCR4, interleukin-13 and GATA-3 up-regulation in response to type 2 cytokines of cord blood T lymphocytes in infants at genetic risk of type 1 diabetes. Immunology (2007) 121(2):189–96. doi: 10.1111/j.1365-2567.2007.02557.x
33. Zamauskaite A, Yaqoob MM, Madrigal JA, Cohen SB. The frequency of Th2 type cells increases with time on peritoneal dialysis in patients with diabetic nephropathy. Eur Cytokine Netw (1999) 10(2):219–26. Available at: https://www.jle.com/fr/revues/ecn/e-docs/the_frequency_of_th2_type_cells_increases_with_time_on_peritoneal_dialysis_in_patients_with_diabetic_nephropathy._90189/article.phtml.
34. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi: 10.1038/ni1261
35. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
36. Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature (2012) 484(7395):514–8. doi: 10.1038/nature10957
37. Lee JY, Hall JA, Kroehling L, Wu L, Najar T, Nguyen HH, et al. Serum amyloid a proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell (2020) 183(7):2036–9. doi: 10.1016/j.cell.2020.12.008
38. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol (2007) 8(12):1390–7. doi: 10.1038/ni1539
39. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature (2010) 467(7318):967–71. doi: 10.1038/nature09447
40. Kumar P, Natarajan K, Shanmugam N. High glucose driven expression of pro-inflammatory cytokine and chemokine genes in lymphocytes: molecular mechanisms of IL-17 family gene expression. Cell Signal (2014) 26(3):528–39. doi: 10.1016/j.cellsig.2013.11.031
41. Kim SM, Lee SH, Lee A, Kim DJ, Kim YG, Kim SY, et al. Targeting T helper 17 by mycophenolate mofetil attenuates diabetic nephropathy progression. Transl Res (2015) 166(4):375–83. doi: 10.1016/j.trsl.2015.04.013
42. Nazarian A, Hejazian SM, Ahmadian E, Vahed SZ, Haghi M, Mobasseri M, et al. IL-17A rs2275913 gene polymorphism in patients with diabetic nephropathy. Immunopathol Per (2022), 1–5. doi: 10.34172/ipp.2022.29320
43. Vasanthakumar R, Mohan V, Anand G, Deepa M, Babu S, Aravindhan V. Serum IL-9, IL-17, and TGF-β levels in subjects with diabetic kidney disease (CURES-134). Cytokine (2015) 72(1):109–12. doi: 10.1016/j.cyto.2014.10.009
44. Mohamed R, Jayakumar C, Chen F, Fulton D, Stepp D, Gansevoort RT, et al. Low-dose IL-17 therapy prevents and reverses diabetic nephropathy, metabolic syndrome, and associated organ fibrosis. J Am Soc Nephrol (2016) 27(3):745–65. doi: 10.1681/ASN.2014111136
45. Ryba-Stanisławowska M, Skrzypkowska M, Myśliwiec M, Myśliwska J. Loss of the balance between CD4+ Foxp3+ regulatory T cells and CD4+ IL17A+ Th17 cells in patients with type 1 diabetes. JHi (2013) 74(6):701–7. doi: 10.1016/j.humimm.2013.01.024
46. Zhu X, Li S, Zhang Q, Zhu D, Xu Y, Zhang P, et al. Correlation of increased Th17/Treg cell ratio with endoplasmic reticulum stress in chronic kidney disease. Med (Baltimore) (2018) 97(20):e10748. doi: 10.1097/MD.0000000000010748
47. Ryden A, Stechova K, Durilova M, Faresjö M. Switch from a dominant Th1-associated immune profile during the pre-diabetic phase in favour of a temporary increase of a Th3-associated and inflammatory immune profile at the onset of type 1 diabetes. Diabetes Metab Res Rev (2009) 25(4):335–43. doi: 10.1002/dmrr.958
48. Semenchuk J, Sullivan K, Moineddin R, Mahmud F, Dart A, Wicklow B, et al. Urinary interleukin-9 in youth with type 1 diabetes mellitus. Acta Diabetol (2022) 59(7):939–47. doi: 10.1007/s00592-022-01873-4
49. Jia L, Wu C. The biology and functions of Th22 cells. Adv Exp Med Biol (2014) 841:209–30. doi: 10.1007/978-94-017-9487-9_8
50. Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, et al. IL-21 and IL-6 are critical for different aspects of b cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PloS One (2011) 6(3):e17739. doi: 10.1371/journal.pone.0017739
51. Ferreira RC, Simons HZ, Thompson WS, Cutler AJ, Dopico XC, Smyth DJ, et al. IL-21 production by CD4+ effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia (2015) 58(4):781–90. doi: 10.1007/s00125-015-3509-8
52. Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE, et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest (2015) 125(1):292–303. doi: 10.1172/JCI76238
53. Viisanen T, Ihantola EL, Näntö-Salonen K, Hyöty H, Nurminen N, Selvenius J, et al. Circulating CXCR5+PD-1+ICOS+ follicular T helper cells are increased close to the diagnosis of type 1 diabetes in children with multiple autoantibodies. Diabetes (2017) 66(2):437–47. doi: 10.2337/db16-0714
54. Zhang N, Tai J, Qu Z, Zhang Z, Zhao S, He J, et al. Increased CD4(+)CXCR5(+)T follicular helper cells in diabetic nephropathy. Autoimmunity (2016) 49(6):405–13. doi: 10.1080/08916934.2016.1196677
55. Abdelhameed SZ, Zahra MK, Mohamed WS, Okasha K. POS-361 CD4 AND CXCR5 in patients with diabetic nephropathy. JKIR (2021) 6(4):S157. doi: 10.1016/j.ekir.2021.03.378
56. Iwase K, Shimada A, Kawai T, Okubo Y, Kanazawa Y, Irie J, et al. FOXP3/Scurfin gene polymorphism is associated with adult onset type 1 diabetes in Japanese, especially in women and slowly progressive-type patients. Autoimmunity (2009) 42(2):159–67. doi: 10.1080/08916930802488258
57. Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes (2010) 59(2):407–15. doi: 10.2337/db09-0694
58. Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, et al. The effect of low-dose IL-2 and treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight (2021) 6(18). doi: 10.1172/jci.insight.147474
59. Ralainirina N, Poli A, Michel T, Poos L, Andrès E, Hentges F, et al. Control of NK cell functions by CD4+CD25+ regulatory T cells. J Leukoc Biol (2007) 81(1):144–53. doi: 10.1189/jlb.0606409
60. Ye C, Low BE, Wiles MV, Brusko TM, Serreze DV, Driver JP. CD70 inversely regulates regulatory T cells and invariant NKT cells and modulates type 1 diabetes in NOD mice. J Immunol (2020) 205(7):1763–77. doi: 10.4049/jimmunol.2000148
61. Ly D, Mi QS, Hussain S, Delovitch TL. Protection from type 1 diabetes by invariant NK T cells requires the activity of CD4+CD25+ regulatory T cells. J Immunol (2006) 177(6):3695–704. doi: 10.4049/jimmunol.177.6.3695
62. McClymont SA, Putnam AL, Lee MR, Esensten JH, Liu W, Hulme MA, et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J Immunol (2011) 186(7):3918–26. doi: 10.4049/jimmunol.1003099
63. Singh K, Kadesjö E, Lindroos J, Hjort M, Lundberg M, Espes D, et al. Interleukin-35 administration counteracts established murine type 1 diabetes–possible involvement of regulatory T cells. Sci Rep (2015) 5:12633. doi: 10.1038/srep12633
64. Zhang F, Wang C, Wen X, Chen Y, Mao R, Cui D, et al. Mesenchymal stem cells alleviate rat diabetic nephropathy by suppressing CD103(+) DCs-mediated CD8(+) T cell responses. J Cell Mol Med (2020) 24(10):5817–31. doi: 10.1111/jcmm.15250
65. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med (2009) 15(8):914–20. doi: 10.1038/nm.1964
66. Li L, Tang W, Zhang Y, Jia M, Wang L, Li Q, et al. Targeting tissue-resident memory CD8(+) T cells in the kidney is a potential therapeutic strategy to ameliorate podocyte injury and glomerulosclerosis. Mol Ther (2022) 30(8):2746–59. doi: 10.1016/j.ymthe.2022.04.024
67. Rodacki M, Milech A, de Oliveira JE. NK cells and type 1 diabetes. Clin Dev Immunol (2006) 13(2-4):101–7. doi: 10.1080/17402520600877182
68. Turner JE, Becker M, Mittrücker HW, Panzer U. Tissue-resident lymphocytes in the kidney. J Am Soc Nephrol (2018) 29(2):389–99. doi: 10.1681/ASN.2017060599
69. Alhasson F, Dattaroy D, Das S, Chandrashekaran V, Seth RK, Schnellmann RG, et al. NKT cell modulates NAFLD potentiation of metabolic oxidative stress-induced mesangial cell activation and proximal tubular toxicity. Am J Physiol Renal Physiol (2016) 310(1):F85–f101. doi: 10.1152/ajprenal.00243.2015
70. Law BM, Wilkinson R, Wang X, Kildey K, Lindner M, Rist MJ, et al. Interferon-γ production by tubulointerstitial human CD56bright natural killer cells contributes to renal fibrosis and chronic kidney disease progression. Kidney Int (2017) 92(1):79–88. doi: 10.1016/j.kint.2017.02.006
71. Lv X, Gao Y, Dong T, Yang L. Role of natural killer T (NKT) cells in type II diabetes-induced vascular injuries. Med Sci Monit (2018) 24:8322–32. doi: 10.12659/MSM.912446
72. Zhou T, Hu Z, Yang S, Sun L, Yu Z, Wang G. Role of adaptive and innate immunity in type 2 diabetes mellitus. J Diabetes Res (2018) 2018:7457269. doi: 10.1155/2018/7457269
73. Markle JG, Mortin-Toth S, Wong AS, Geng L, Hayday A, Danska JS. γδ T cells are essential effectors of type 1 diabetes in the nonobese diabetic mouse model. J Immunol (2013) 190(11):5392–401. doi: 10.4049/jimmunol.1203502
74. Tilloy F, Treiner E, Park SH, Garcia C, Lemonnier F, de la Salle H, et al. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med (1999) 189(12):1907–21. doi: 10.1084/jem.189.12.1907
75. Terpstra ML, Remmerswaal EBM, van der Bom-Baylon ND, Sinnige MJ, Kers J, van Aalderen MC, et al. Tissue-resident mucosal-associated invariant T (MAIT) cells in the human kidney represent a functionally distinct subset. Eur J Immunol (2020) 50(11):1783–97. doi: 10.1002/eji.202048644
76. Law BMP, Wilkinson R, Wang X, Kildey K, Giuliani K, Beagley KW, et al. Human tissue-resident mucosal-associated invariant T (MAIT) cells in renal fibrosis and CKD. J Am Soc Nephrol (2019) 30(7):1322–35. doi: 10.1681/ASN.2018101064
77. Harms RZ, Lorenzo KM, Corley KP, Cabrera MS, Sarvetnick NE. Altered CD161 bright CD8+ mucosal associated invariant T (MAIT)-like cell dynamics and increased differentiation states among juvenile type 1 diabetics. PloS One (2015) 10(1):e0117335. doi: 10.1371/journal.pone.0117335
78. Touch S, Clément K, André S. T Cell populations and functions are altered in human obesity and type 2 diabetes. Curr Diabetes Rep (2017) 17(9):81. doi: 10.1007/s11892-017-0900-5
79. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol (2015) 194(5):2059–63. doi: 10.4049/jimmunol.1402256
80. Carbone FR, Mackay LK, Heath WR, Gebhardt T. Distinct resident and recirculating memory T cell subsets in non-lymphoid tissues. Curr Opin Immunol (2013) 25(3):329–33. doi: 10.1016/j.coi.2013.05.007
81. Beura LK, Masopust D. SnapShot: resident memory T cells. Cell (2014) 157(6):1488–.e1. doi: 10.1016/j.cell.2014.05.026
82. Jia Y, Xu H, Yu Q, Tan L, Xiong Z. Identification and verification of vascular cell adhesion protein 1 as an immune-related hub gene associated with the tubulointerstitial injury in diabetic kidney disease. Bioengineered (2021) 12(1):6655–73. doi: 10.1080/21655979.2021.1976540
83. Moon JY, Jeong KH, Lee TW, Ihm CG, Lim SJ, Lee SH. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am J Nephrol (2012) 35(2):164–74. doi: 10.1159/000334928
84. Moriya R, Manivel JC, Mauer M. Juxtaglomerular apparatus T-cell infiltration affects glomerular structure in type 1 diabetic patients. Diabetologia (2004) 47(1):82–8. doi: 10.1007/s00125-003-1253-y
85. Wu CC, Sytwu HK, Lin YF. Cytokines in diabetic nephropathy. Adv Clin Chem (2012) 56:55–74. doi: 10.1016/B978-0-12-394317-0.00014-5
86. Higurashi M, Ohya Y, Joh K, Muraguchi M, Nishimura M, Terawaki H, et al. Increased urinary levels of CXCL5, CXCL8 and CXCL9 in patients with type 2 diabetic nephropathy. J Diabetes Complications (2009) 23(3):178–84. doi: 10.1016/j.jdiacomp.2007.12.001
87. Wang G, Lai FM, Chow KM, Kwan BC, Pang WF, Luk CC, et al. Urinary mRNA levels of ELR-negative CXC chemokine ligand and extracellular matrix in diabetic nephropathy. Diabetes Metab Res Rev (2015) 31(7):699–706. doi: 10.1002/dmrr.2654
88. Turner JE, Steinmetz OM, Stahl RA, Panzer U. Targeting of Th1-associated chemokine receptors CXCR3 and CCR5 as therapeutic strategy for inflammatory diseases. Mini Rev Med Chem (2007) 7(11):1089–96. doi: 10.2174/138955707782331768
89. Yu J, Wu H, Liu ZY, Zhu Q, Shan C, Zhang KQ. Advanced glycation end products induce the apoptosis of and inflammation in mouse podocytes through CXCL9-mediated JAK2/STAT3 pathway activation. Int J Mol Med (2017) 40(4):1185–93. doi: 10.3892/ijmm.2017.3098
90. Corrado A, Ferrari SM, Ferri C, Ferrannini E, Antonelli A, Fallahi P. Type 1 diabetes and (C-X-C motif) ligand (CXCL) 10 chemokine. Clin Ter (2014) 165(2):e181–5. doi: 10.7471/CT.2014.1706
91. Bender C, Christen S, Scholich K, Bayer M, Pfeilschifter JM, Hintermann E, et al. Islet-expressed CXCL10 promotes autoimmune destruction of islet isografts in mice with type 1 diabetes. Diabetes (2017) 66(1):113–26. doi: 10.2337/db16-0547
92. Zhuang Q, Cheng K, Ming Y. CX3CL1/CX3CR1 axis, as the therapeutic potential in renal diseases: Friend or foe? Curr Gene Ther (2017) 17(6):442–52. doi: 10.2174/1566523218666180214092536
93. Kikuchi Y, Ikee R, Hemmi N, Hyodo N, Saigusa T, Namikoshi T, et al. Fractalkine and its receptor, CX3CR1, upregulation in streptozotocin-induced diabetic kidneys. Nephron Exp Nephrol (2004) 97(1):e17–25. doi: 10.1159/000077594
94. Yadav AK, Lal A, Jha V. Association of circulating fractalkine (CX3CL1) and CX3CR1(+)CD4(+) T cells with common carotid artery intima-media thickness in patients with chronic kidney disease. J Atheroscler Thromb (2011) 18(11):958–65. doi: 10.5551/jat.8722
95. Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature (1990) 347(6294):669–71. doi: 10.1038/347669a0
96. Peng X, Xiao Z, Zhang J, Li Y, Dong Y, Du J. IL-17A produced by both γδ T and Th17 cells promotes renal fibrosis via RANTES-mediated leukocyte infiltration after renal obstruction. J Pathol (2015) 235(1):79–89. doi: 10.1002/path.4430
97. Loetscher P, Uguccioni M, Bordoli L, Baggiolini M, Moser B, Chizzolini C, et al. CCR5 is characteristic of Th1 lymphocytes. Nature (1998) 391(6665):344–5. doi: 10.1038/34814
98. Cao M, Tian Z, Zhang L, Liu R, Guan Q, Jiang J. Effects of CCR5 59029G/A polymorphism on the risk to diabetic nephropathy. Oncotarget (2017) 8(63):106926–34. doi: 10.18632/oncotarget.22148
99. Yahya MJ, Ismail PB, Nordin NB, Akim ABM, Yusuf W, Adam NLB, et al. Association of CCL2, CCR5, ELMO1, and IL8 polymorphism with diabetic nephropathy in Malaysian type 2 diabetic patients. Int J Chronic Dis (2019) 2019:2053015. doi: 10.1155/2019/2053015
100. Kalev I, Mikelsaar AV, Beckman L, Tasa G, Pärlist P. High frequency of the HIV-1 protective CCR5 delta32 deletion in native estonians. Eur J Epidemiol (2000) 16(12):1107–9. doi: 10.1023/A:1010829816334
101. Carr MW, Roth SJ, Luther E, Rose SS, Springer TA. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci U.S.A. (1994) 91(9):3652–6. doi: 10.1073/pnas.91.9.3652
102. Bakos E, Thaiss CA, Kramer MP, Cohen S, Radomir L, Orr I, et al. CCR2 regulates the immune response by modulating the interconversion and function of effector and regulatory T cells. J Immunol (2017) 198(12):4659–71. doi: 10.4049/jimmunol.1601458
103. Jamali Z, Nazari M, Khoramdelazad H, Hakimizadeh E, Mahmoodi M, Karimabad MN, et al. Expression of CC chemokines CCL2, CCL5, and CCL11 is associated with duration of disease and complications in type-1 diabetes: A study on Iranian diabetic patients. Clin Lab (2013) 59(9-10):993–1001. doi: 10.7754/Clin.Lab.2012.120810
104. Kang YS, Lee MH, Song HK, Ko GJ, Kwon OS, Lim TK, et al. CCR2 antagonism improves insulin resistance, lipid metabolism, and diabetic nephropathy in type 2 diabetic mice. Kidney Int (2010) 78(9):883–94. doi: 10.1038/ki.2010.263
105. Kitagawa K, Wada T, Furuichi K, Hashimoto H, Ishiwata Y, Asano M, et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol (2004) 165(1):237–46. doi: 10.1016/S0002-9440(10)63292-0
106. Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z, et al. C-c motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant (2017) 32(2):307–15. doi: 10.1093/ndt/gfv459
107. Wu N, Chen D, Sun H, Tan J, Zhang Y, Zhang T, et al. MAP3K2 augments Th1 cell differentiation via IL-18 to promote T cell-mediated colitis. Sci China Life Sci (2021) 64(3):389–403. doi: 10.1007/s11427-020-1720-9
108. Hayashi N, Yoshimoto T, Izuhara K, Matsui K, Tanaka T, Nakanishi K. T Helper 1 cells stimulated with ovalbumin and IL-18 induce airway hyperresponsiveness and lung fibrosis by IFN-gamma and IL-13 production. Proc Natl Acad Sci U.S.A. (2007) 104(37):14765–70. doi: 10.1073/pnas.0706378104
109. Nakanishi K. [Regulation of Th1 and Th2 immune responses by IL-18]. Kekkaku (2002) 77(2):87–93.
110. Hirooka Y, Nozaki Y. Interleukin-18 in inflammatory kidney disease. Front Med (Lausanne) (2021) 8:639103. doi: 10.3389/fmed.2021.639103
111. Al-Rubeaan K, Nawaz SS, Youssef AM, Al Ghonaim M, Siddiqui K. IL-18, VCAM-1 and p-selectin as early biomarkers in normoalbuminuric type 2 diabetes patients. biomark Med (2019) 13(6):467–78. doi: 10.2217/bmm-2018-0359
112. Yaribeygi H, Atkin SL, Sahebkar A. Interleukin-18 and diabetic nephropathy: A review. J Cell Physiol (2019) 234(5):5674–82. doi: 10.1002/jcp.27427
113. Li L, Jiang XG, Hu JY, Yu ZQ, Xu JY, Liu F, et al. The association between interleukin-19 concentration and diabetic nephropathy. BMC Nephrol (2017) 18(1):65. doi: 10.1186/s12882-017-0488-7
114. Yeh CH, Cheng BC, Hsu CC, Chen HW, Wang JJ, Chang MS, et al. Induced interleukin-19 contributes to cell-mediated immunosuppression in patients undergoing coronary artery bypass grafting with cardiopulmonary bypass. Ann Thorac Surg (2011) 92(4):1252–9. doi: 10.1016/j.athoracsur.2011.04.061
115. Habib HA, Heeba GH, Khalifa MMA. Comparative effects of incretin-based therapy on early-onset diabetic nephropathy in rats: Role of TNF-α, TGF-β and c-caspase-3. Life Sci (2021) 278:119624. doi: 10.1016/j.lfs.2021.119624
116. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14(5):500–8. doi: 10.1038/ni.2556
117. Morigi M, Perico L, Corna D, Locatelli M, Cassis P, Carminati CE, et al. C3a receptor blockade protects podocytes from injury in diabetic nephropathy. JCI Insight (2020) 5(5). doi: 10.1172/jci.insight.131849
118. Li XQ, Chang DY, Chen M, Zhao MH. Deficiency of C3a receptor attenuates the development of diabetic nephropathy. BMJ Open Diabetes Res Care (2019) 7(1):e000817. doi: 10.1136/bmjdrc-2019-000817
119. Berg TJ, Bangstad HJ, Torjesen PA, Osterby R, Bucala R, Hanssen KF. Advanced glycation end products in serum predict changes in the kidney morphology of patients with insulin-dependent diabetes mellitus. Metabolism (1997) 46(6):661–5. doi: 10.1016/S0026-0495(97)90010-X
120. Mariat C, Degauque N, Balasubramanian S, Kenny J, DeKruyff RH, Umetsu DT, et al. Tim-1 signaling substitutes for conventional signal 1 and requires costimulation to induce T cell proliferation. J Immunol (2009) 182(3):1379–85. doi: 10.4049/jimmunol.182.3.1379
121. Forbes JM, McCarthy DA, Kassianos AJ, Baskerville T, Fotheringham AK, Giuliani KTK, et al. T-Cell expression and release of kidney injury molecule-1 in response to glucose variations initiates kidney injury in early diabetes. Diabetes (2021) 70(8):1754–66. doi: 10.2337/db20-1081
122. Nowak N, Skupien J, Niewczas MA, Yamanouchi M, Major M, Croall S, et al. Increased plasma kidney injury molecule-1 suggests early progressive renal decline in non-proteinuric patients with type 1 diabetes. Kidney Int (2016) 89(2):459–67. doi: 10.1038/ki.2015.314
123. Baker RL, Bradley B, Wiles TA, Lindsay RS, Barbour G, Delong T, et al. Cutting edge: Nonobese diabetic mice deficient in chromogranin a are protected from autoimmune diabetes. J Immunol (2016) 196(1):39–43. doi: 10.4049/jimmunol.1501190
124. Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol (2011) 7(12):684–96. doi: 10.1038/nrneph.2011.149
125. Herder C, Dalmas E, Böni-Schnetzler M, Donath MY. The IL-1 pathway in type 2 diabetes and cardiovascular complications. Trends Endocrinol Metab (2015) 26(10):551–63. doi: 10.1016/j.tem.2015.08.001
126. Jain A, Irizarry-Caro RA, McDaniel MM, Chawla AS, Carroll KR, Overcast GR, et al. T Cells instruct myeloid cells to produce inflammasome-independent IL-1β and cause autoimmunity. Nat Immunol (2020) 21(1):65–74. doi: 10.1038/s41590-019-0559-y
127. Vila E, Salaices M. Cytokines and vascular reactivity in resistance arteries. Am J Physiol Heart Circ Physiol (2005) 288(3):H1016–21. doi: 10.1152/ajpheart.00779.2004
128. Vallejo S, Palacios E, Romacho T, Villalobos L, Peiró C, Sánchez-Ferrer CF. The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovasc Diabetol (2014) 13:158. doi: 10.1186/s12933-014-0158-z
129. Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med (2007) 356(15):1517–26. doi: 10.1056/NEJMoa065213
130. Imani F, Horii Y, Suthanthiran M, Skolnik E, Makita Z, Sharma V, et al. Advanced glycosylation endproduct-specific receptors on human and rat T-lymphocytes mediate synthesis of interferon gamma: role in tissue remodeling. J Exp Med (1993) 178(6):2165–72. doi: 10.1084/jem.178.6.2165
131. Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med (2010) 207(9):1871–8. doi: 10.1084/jem.20100209
132. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity (2008) 28(5):687–97. doi: 10.1016/j.immuni.2008.03.016
133. Todd JA, Evangelou M, Cutler AJ, Pekalski ML, Walker NM, Stevens HE, et al. Regulatory T cell responses in participants with type 1 diabetes after a single dose of interleukin-2: a non-randomised, open label, adaptive dose-finding trial. PLoS Med (2016) 13(10):e1002139. doi: 10.1371/journal.pmed.1002139
134. Flores R, Zhou L, Robbins P. Expression of IL-2 in β cells by AAV8 gene transfer in pre-diabetic NOD mice prevents diabetes through activation of FoxP3-positive regulatory T cells. JGt (2014) 21(8):715–22. doi: 10.1038/gt.2014.45
135. Seder RA, Paul WE, Davis MM, Fazekas de St Groth B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. JTJoem (1992) 176(4):1091–8. doi: 10.1084/jem.176.4.1091
136. Bugawan TL, Mirel DB, Valdes AM, Panelo A, Pozzilli P, Erlich HA. Association and interaction of the IL4R, IL4, and IL13 loci with type 1 diabetes among filipinos. JTAJoHG (2003) 72(6):1505–14. doi: 10.1086/375655
137. Qiu LJ, Ni J, Cen H, Wen PF, Zhang M, Liang Y, et al. Relationship between the IL-4 gene promoter-590C/T (rs2243250) polymorphism and susceptibility to autoimmune diseases: A meta-analysis. J Eur Acad Dermatol Venereol (2015) 29(1):48–55. doi: 10.1111/jdv.12435
138. Mahadevan P, Larkins R, Fraser J, Fosang A, Dunlop M. Increased hyaluronan production in the glomeruli from diabetic rats: a link between glucose-induced prostaglandin production and reduced sulphated proteoglycan. JD (1995) 38(3):298–305. doi: 10.1007/BF00400634
139. Chen G, Chen H, Ren S, Xia M, Zhu J, Liu Y, et al. Aberrant DNA methylation of mTOR pathway genes promotes inflammatory activation of immune cells in diabetic kidney disease. Kidney Int (2019) 96(2):409–20. doi: 10.1016/j.kint.2019.02.020
140. Feigerlová E, Battaglia-Hsu S-F. IL-6 signaling in diabetic nephropathy: From pathophysiology to therapeutic perspectives. JC (2017) 37:57–65. doi: 10.1016/j.cytogfr.2017.03.003
141. Francois ME, Little JP. Effectiveness and safety of high-intensity interval training in patients with type 2 diabetes. JDsapotADA (2015) 28(1):39. doi: 10.2337/diaspect.28.1.39
142. Dalla Vestra M, Mussap M, Gallina P, Bruseghin M, Cernigoi AM, Saller A, et al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol (2005) 16(3 suppl 1):S78–82. doi: 10.1681/asn.2004110961
143. Wu R, Liu X, Yin J, Wu H, Cai X, Wang N, et al. IL-6 receptor blockade ameliorates diabetic nephropathy via inhibiting inflammasome in mice. Metabolism (2018) 83:18–24. doi: 10.1016/j.metabol.2018.01.002
144. Xiong T, Attar M, Gnirck A-C, Wunderlich M, Becker M, Rickassel C, et al. Interleukin-9 protects from early podocyte injury and progressive glomerulosclerosis in adriamycin-induced nephropathy. Kidney Int (2020) 98(3):615–29. doi: 10.1016/j.kint.2020.04.036
145. Eller K, Wolf D, Huber JM, Metz M, Mayer G, McKenzie AN, et al. IL-9 production by regulatory T cells recruits mast cells that are essential for regulatory T cell-induced immune suppression. J Immunol (2011) 186(1):83–91. doi: 10.4049/jimmunol.1001183
146. Cortvrindt C, Speeckaert R, Moerman A, Delanghe JR, Speeckaert MM. The role of interleukin-17A in the pathogenesis of kidney diseases. JP (2017) 49(3):247–58. doi: 10.1016/j.pathol.2017.01.003
147. Kuo H-L, Huang C-C, Lin T-Y, Lin C-Y. IL-17 and CD40 ligand synergistically stimulate the chronicity of diabetic nephropathy. JNDT (2018) 33(2):248–56. doi: 10.1093/ndt/gfw397
148. Orejudo M, Rodrigues-Diez RR, Rodrigues-Diez R, Garcia-Redondo A, Santos-Sánchez L, Rández-Garbayo J, et al. Interleukin 17A participates in renal inflammation associated to experimental and human hypertension. Front Pharmacol (2019) 10:1015. doi: 10.3389/fphar.2019.01015
149. Scottà C, Esposito M, Fazekasova H, Fanelli G, Edozie FC, Ali N, et al. Differential effects of rapamycin and retinoic acid on expansion, stability and suppressive qualities of human CD4(+)CD25(+)FOXP3(+) T regulatory cell subpopulations. Haematologica (2013) 98(8):1291–9. doi: 10.3324/haematol.2012.074088
150. Chen W, Shen Y, Fan J, Zeng X, Zhang X, Luan J, et al. IL-22-mediated renal metabolic reprogramming via PFKFB3 to treat kidney injury. Clin Transl Med (2021) 11(2):e324. doi: 10.1002/ctm2.324
151. Liu Y, Zhao L-s. Effect of interlukin-22 antibody on diabetic nephropathy via inhibition on Snail1 expression. JCJoP (2018), 939–44. Available at: https://pesquisa.bvsalud.org/portal/resource/pt/wpr-701220.
152. Guo H, Pan C, Chang B, Wu X, Guo J, Zhou Y, et al. Triptolide improves diabetic nephropathy by regulating Th cell balance and macrophage infiltration in rat models of diabetic nephropathy. Exp Clin Endocrinol Diabetes (2016) 124(6):389–98. doi: 10.1055/s-0042-106083
153. Sun L, Kanwar YS. Relevance of TNF-α in the context of other inflammatory cytokines in the progression of diabetic nephropathy. Kidney Int (2015) 88(4):662–5. doi: 10.1038/ki.2015.250
154. Navarro JF, Mora C, Muros M, García J. Urinary tumour necrosis factor-alpha excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol Dial Transplant (2006) 21(12):3428–34. doi: 10.1093/ndt/gfl469
155. Pedigo CE, Ducasa GM, Leclercq F, Sloan A, Mitrofanova A, Hashmi T, et al. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J Clin Invest (2016) 126(9):3336–50. doi: 10.1172/JCI85939
156. Pestana RM, Domingueti CP, Duarte RC, Fóscolo RB, Reis JS, Rodrigues AM, et al. Cytokines profile and its correlation with endothelial damage and oxidative stress in patients with type 1 diabetes mellitus and nephropathy. Immunol Res (2016) 64(4):951–60. doi: 10.1007/s12026-016-8806-x
157. DiPetrillo K, Gesek FA. Pentoxifylline ameliorates renal tumor necrosis factor expression, sodium retention, and renal hypertrophy in diabetic rats. Am J Nephrol (2004) 24(3):352–9. doi: 10.1159/000079121
158. Baud L, Perez J, Friedlander G, Ardaillou R. Tumor necrosis factor stimulates prostaglandin production and cyclic AMP levels in rat cultured mesangial cells. FEBS Lett (1988) 239(1):50–4. doi: 10.1016/0014-5793(88)80543-X
159. Bertani T, Abbate M, Zoja C, Corna D, Perico N, Ghezzi P, et al. Tumor necrosis factor induces glomerular damage in the rabbit. Am J Pathol (1989) 134(2):419–30.
160. Koike N, Takamura T, Kaneko S. Induction of reactive oxygen species from isolated rat glomeruli by protein kinase c activation and TNF-alpha stimulation, and effects of a phosphodiesterase inhibitor. Life Sci (2007) 80(18):1721–8. doi: 10.1016/j.lfs.2007.02.001
161. Artunc F, Schleicher E, Weigert C, Fritsche A, Stefan N, Häring HU. The impact of insulin resistance on the kidney and vasculature. Nat Rev Nephrol (2016) 12(12):721–37. doi: 10.1038/nrneph.2016.145
162. Zheng Y, Yamada H, Sakamoto K, Horita S, Kunimi M, Endo Y, et al. Roles of insulin receptor substrates in insulin-induced stimulation of renal proximal bicarbonate absorption. J Am Soc Nephrol (2005) 16(8):2288–95. doi: 10.1681/ASN.2005020193
163. Welsh GI, Coward RJ. Podocytes, glucose and insulin. Curr Opin Nephrol Hypertens (2010) 19(4):379–84. doi: 10.1097/MNH.0b013e32833ad5e4
164. Sell H, Habich C, Eckel J. Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol (2012) 8(12):709–16. doi: 10.1038/nrendo.2012.114
165. Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes (2011) 60(11):2954–62. doi: 10.2337/db11-0358
166. Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med (2009) 15(8):921–9. doi: 10.1038/nm.2001
167. Harford KA, Reynolds CM, McGillicuddy FC, Roche HM. Fats, inflammation and insulin resistance: insights to the role of macrophage and T-cell accumulation in adipose tissue. Proc Nutr Soc (2011) 70(4):408–17. doi: 10.1017/S0029665111000565
168. Zeyda M, Huber J, Prager G, Stulnig TM. Inflammation correlates with markers of T-cell subsets including regulatory T cells in adipose tissue from obese patients. Obes (Silver Spring) (2011) 19(4):743–8. doi: 10.1038/oby.2010.123
169. Al-Mansoori L, Al-Jaber H, Madani AY, Mazloum NA, Agouni A, Ramanjaneya M, et al. Suppression of GATA-3 increases adipogenesis, reduces inflammation and improves insulin sensitivity in 3T3L-1 preadipocytes. Cell Signal (2020) 75:109735. doi: 10.1016/j.cellsig.2020.109735
170. Anand G, Vasanthakumar R, Mohan V, Babu S, Aravindhan V. Increased IL-12 and decreased IL-33 serum levels are associated with increased Th1 and suppressed Th2 cytokine profile in patients with diabetic nephropathy (CURES-134). JIjoc Pathol e (2014) 7(11):8008.
171. Wolf G, Ziyadeh FN. Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron Physiol (2007) 106(2):26–31. doi: 10.1159/000101797
172. Lim AK, Ma FY, Nikolic-Paterson DJ, Kitching AR, Thomas MC, Tesch GH. Lymphocytes promote albuminuria, but not renal dysfunction or histological damage in a mouse model of diabetic renal injury. Diabetologia (2010) 53(8):1772–82. doi: 10.1007/s00125-010-1757-1
173. Nagai S, Azuma M. The CD28-B7 family of Co-signaling molecules. Adv Exp Med Biol (2019) 1189:25–51. doi: 10.1007/978-981-32-9717-3_2
174. Goldwich A, Burkard M, Olke M, Daniel C, Amann K, Hugo C, et al. Podocytes are nonhematopoietic professional antigen-presenting cells. J Am Soc Nephrol (2013) 24(6):906–16. doi: 10.1681/ASN.2012020133
175. Novelli R, Benigni A, Remuzzi G. The role of B7-1 in proteinuria of glomerular origin. Nat Rev Nephrol (2018) 14(9):589–96. doi: 10.1038/s41581-018-0037-z
176. Li Y, Jin L, Yan J, Zhang H, Zhang R, Hu C. CD28 genetic variants increase susceptibility to diabetic kidney disease in Chinese patients with type 2 diabetes: A cross-sectional case control study. Mediators Inflammation (2021) 2021:5521050. doi: 10.1155/2021/5521050
177. Gagliardini E, Novelli R, Corna D, Zoja C, Ruggiero B, Benigni A, et al. B7-1 is not induced in podocytes of human and experimental diabetic nephropathy. J Am Soc Nephrol (2016) 27(4):999–1005. doi: 10.1681/ASN.2015030266
178. Otto G. IL-1β switches on kidney fibrosis. Nat Rev Nephrol (2018) 14(8):475. doi: 10.1038/s41581-018-0026-2
179. Chen B, Wu M, Zang C, Li Y, Xu Z. Association between IL-6 polymorphisms and diabetic nephropathy risk: a meta-analysis. JTAjotms (2019) 358(5):363–73. doi: 10.1016/j.amjms.2019.07.011
180. Wu M, Yang Z, Zhang C, Shi Y, Han W, Song S, et al. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism (2021) 118:154748. doi: 10.1016/j.metabol.2021.154748
181. Abdullah CS, Jin ZQ. Targeted deletion of T-cell S1P receptor 1 ameliorates cardiac fibrosis in streptozotocin-induced diabetic mice. FASEB J (2018) 32(10):5426–35. doi: 10.1096/fj.201800231R
182. Chang TT, Kuchroo VK, Sharpe AH. Role of the B7-CD28/CTLA-4 pathway in autoimmune disease. Curr Dir Autoimmun (2002) 5:113–30. doi: 10.1159/000060550
183. Bending JJ, Lobo-Yeo A, Vergani D, Viberti GC. Proteinuria and activated T-lymphocytes in diabetic nephropathy. Diabetes (1988) 37(5):507–11. doi: 10.2337/diab.37.5.507
184. Lei L, Mao Y, Meng D, Zhang X, Cui L, Huo Y, et al. Percentage of circulating CD8+ T lymphocytes is associated with albuminuria in type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes (2014) 122(1):27–30. doi: 10.1055/s-0033-1358666
185. Lee BT, Ahmed FA, Hamm LL, Teran FJ, Chen CS, Liu Y, et al. Association of c-reactive protein, tumor necrosis factor-alpha, and interleukin-6 with chronic kidney disease. BMC Nephrol (2015) 16:77. doi: 10.1186/s12882-015-0068-7
186. McCarthy ET, Sharma R, Sharma M, Li JZ, Ge XL, Dileepan KN, et al. TNF-alpha increases albumin permeability of isolated rat glomeruli through the generation of superoxide. J Am Soc Nephrol (1998) 9(3):433–8. doi: 10.1681/ASN.V93433
187. Onk D, Onk OA, Turkmen K, Erol HS, Ayazoglu TA, Keles ON, et al. Melatonin attenuates contrast-induced nephropathy in diabetic rats: The role of interleukin-33 and oxidative stress. Mediators Inflamm (2016) 2016:9050828. doi: 10.1155/2016/9050828
188. Caner S, Usluoğulları CA, Balkan F, Büyükcam F, Kaya C, Saçıkara M, et al. Is IL-33 useful to detect early stage of renal failure? Ren Fail (2014) 36(1):78–80. doi: 10.3109/0886022X.2013.832313
189. Sabapathy V, Stremska ME, Mohammad S, Corey RL, Sharma PR, Sharma R. Novel immunomodulatory cytokine regulates inflammation, diabetes, and obesity to protect from diabetic nephropathy. Front Pharmacol (2019) 10:572. doi: 10.3389/fphar.2019.00572
190. Chen HY, Zhong X, Huang XR, Meng XM, You Y, Chung AC, et al. MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol Ther (2014) 22(4):842–53. doi: 10.1038/mt.2013.235
191. Cucak H, Nielsen Fink L, Højgaard Pedersen M, Rosendahl A. Enalapril treatment increases T cell number and promotes polarization towards M1-like macrophages locally in diabetic nephropathy. Int Immunopharmacol (2015) 25(1):30–42. doi: 10.1016/j.intimp.2015.01.003
192. Tian J, Dang H, O’Laco KA, Song M, Tiu BC, Gilles S, et al. Homotaurine treatment enhances CD4(+) and CD8(+) regulatory T cell responses and synergizes with low-dose anti-CD3 to enhance diabetes remission in type 1 diabetic mice. Immunohorizons (2019) 3(10):498–510. doi: 10.4049/immunohorizons.1900019
193. Jörns A, Ertekin ÜG, Arndt T, Terbish T, Wedekind D, Lenzen S. TNF-α antibody therapy in combination with the T-Cell-Specific antibody anti-TCR reverses the diabetic metabolic state in the LEW. 1AR1-iddm Rat. Diabetes (2015) 64(8):2880–91. doi: 10.2337/db14-1866
194. Han Y, Lai J, Tao J, Tai Y, Zhou W, Guo P, et al. Sustaining circulating regulatory T cell subset contributes to the therapeutic effect of paroxetine on mice with diabetic cardiomyopathy. Circ J (2020) 84(9):1587–98. doi: 10.1253/circj.CJ-19-1182
195. Mahmoud F, Al-Ozairi E, Haines D, Novotny L, Dashti A, Ibrahim B, et al. Effect of diabetea tea ™ consumption on inflammatory cytokines and metabolic biomarkers in type 2 diabetes patients. J Ethnopharmacol (2016) 194:1069–77. doi: 10.1016/j.jep.2016.10.073
196. Gao Z, Zhao X, Yang T, Shang J, Shang L, Mai H, et al. Immunomodulation therapy of diabetes by oral administration of a surfactin lipopeptide in NOD mice. Vaccine (2014) 32(50):6812–9. doi: 10.1016/j.vaccine.2014.08.082
197. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127- regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care (2012) 35(9):1817–20. doi: 10.2337/dc12-0038
198. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
Keywords: diabetic kidney disease, T cells, cytokines, immunologic function, differentiation, recruitment, therapeutic methods
Citation: Liu Y, Lv Y, Zhang T, Huang T, Lang Y, Sheng Q, Liu Y, Kong Z, Gao Y, Lu S, Yang M, Luan Y, Wang X and Lv Z (2023) T cells and their products in diabetic kidney disease. Front. Immunol. 14:1084448. doi: 10.3389/fimmu.2023.1084448
Received: 30 October 2022; Accepted: 02 January 2023;
Published: 26 January 2023.
Edited by:
Xuefeng Wang, Soochow University, ChinaReviewed by:
Bernhard Fidelis Maier, Purdue University Indianapolis, United StatesMaria J. Stangou, Aristotle University of Thessaloniki, Greece
Copyright © 2023 Liu, Lv, Zhang, Huang, Lang, Sheng, Liu, Kong, Gao, Lu, Yang, Luan, Wang and Lv. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xining Wang, xningwang@163.com; Zhimei Lv, sdlvzhimei@163.com
†These authors have contributed equally to this work and share last authorship