 Martina Rimoldi1Daniele Velardo1Simona Zanotti1Michela Ripolone1Roberto Del Bo2Patrizia Ciscato1Laura Napoli1Stefania Corti1,3†Giacomo Pietro Comi2,3†Dario Ronchi2,3*†
Martina Rimoldi1Daniele Velardo1Simona Zanotti1Michela Ripolone1Roberto Del Bo2Patrizia Ciscato1Laura Napoli1Stefania Corti1,3†Giacomo Pietro Comi2,3†Dario Ronchi2,3*†- 1Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Neuromuscular and Rare Disease Unit, Milan, Italy
- 2Dino Ferrari Center, Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy
- 3Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Neurology Unit, Milan, Italy
DNM2 encodes the dynamin-2 protein, a GTPase involved in clathrin-mediated endocytosis and other membrane trafficking pathways. The dynamin-2 protein is composed of several functional domains, including a GTPase domain, a middle domain, a pleckstrin homology (PH) domain, a GTPase effector domain (GED), and a proline-rich domain. Monoallelic variants in DNM2 are associated with Charcot–Marie–Tooth disease and a rare form of congenital centronuclear myopathy (CNM). Several DNM2 variants have been reported in patients with CNM, typically presenting with mild and slowly progressive symptoms. We report the case of a 47-year-old man with DNM2-related myopathy, who presented with progressive muscle weakness starting at the age of 40 years. Clinical exome sequencing revealed the presence of a heterozygous DNM2 variant c.1726G>A, p.(Glu576Lys). This variant, previously unreported, is located in the PH domain of the protein. Muscle biopsy findings showed several fibers with central nuclei, sometimes multiple. In addition, occasional centronucleated fibers showed a radial distribution of sarcoplasmic strands. This study expands the clinical and genetic repertoire of DNM2-related myopathy.
1 Introduction
The DNM2 gene encodes a protein called dynamin-2, a component of the dynamin family of GTPases, which plays a crucial role in various cellular processes, particularly in endocytosis and membrane trafficking (Antonny et al., 2016; Ferguson and De Camilli, 2012).
The DNM2 gene is located on the short arm of chromosome 19 (19p13.2). It spans approximately 59 kilobases and consists of 22 exons. The dynamin-2 protein includes a GTPase domain, a middle domain, a pleckstrin homology (PH), a GTPase effector domain (GED), and a proline-rich domain (Hayes et al., 2022).
Dynamin-2 plays a crucial role in clathrin-mediated endocytosis, facilitating the fission of clathrin-coated vesicles from the plasma membrane. Dynamin-2 is also involved in other membrane trafficking pathways, including receptor recycling, synaptic vesicle recycling, and the maintenance of organelle morphology (Puri et al., 2020).
Monoallelic variants in the DNM2 are associated with heterogeneous clinical presentations, including dominant intermediate Charcot–Marie–Tooth disease (CMTDIB, MIM 606482), dominant axonal Charcot–Marie–Tooth disease (CMT2M, MIM 606482), and a form of centronuclear myopathy (CNM, MIM 160150), characterized by muscle weakness. In DNM2-related CNM patients, muscle biopsy usually shows disorganization of myofibers and centralized nuclei (Jungbluth et al., 2008; Romero, 2010; Biancalana et al., 2018). Ultrastructural studies reveal disorganized transverse tubules and triads (Bitoun et al., 2005; Al-Qusairi e Laporte, 2011).
Pathogenic variants in DNM2 were associated with myopathic clinical presentations ranging from severe neonatal onset to adult-onset disorders with a milder phenotype (Böhm et al., 2012; Catteruccia et al., 2013; Casar-Borota et al., 2015; Hohendahl, Roux, and Galli, 2016; Biancalana et al., 2018; Vandersmissen et al., 2018; Westra et al., 2019; Reumers et al., 2021; Hayes et al., 2022). The prognosis of CNM can vary widely among affected individuals. Some patients may experience a relatively stable course with minimal progression of muscle weakness, while others may develop a more severe and rapidly progressive form of the disease. Regular follow-up and continuous monitoring of disease progression are essential to adapting patient management (Susman et al., 2010; Hayes et al., 2022).
The majority of the reported DNM2 variants result in amino acid changes in the PH and stalk domains.
In this report, we present the case of a 47-year-old man affected with DNM2-related myopathy associated with a novel missense variant in the PH domain of dynamin-2.
2 Methods
The study was approved by the institutional review board of the Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico. The patients provided written informed consent for all aspects of the study. Genomic DNA was extracted from peripheral blood samples taken from both the patient and his mother. The QIAsymphony automated nucleic acid extraction platform (QIAGEN) was utilized for this purpose.
Clinical exome sequencing was performed on the affected proband, starting with 100 ng of high-quality DNA. Agilent SureSelectXT Clinical Focused Exome library preparation and target enrichment kits were used. The libraries underwent paired-end sequencing on a NextSeq 2000 Illumina platform. The variants included in the generated VCF files were annotated based on the genome assembly of hg19 and classified using an internal analysis pipeline and the bioinformatic tool eVai Expert Variant Interpreter v2.7.
PCR amplification, followed by Sanger sequencing (using Thermo Fisher Scientific BigDye Terminator v3.1) on an ABI Prism 3130 automated DNA analyzer, was performed to validate the candidate variant in the DNM2 gene on both the proband and his mother.
2.1 Histological and histochemical analysis
Tissue specimens were frozen in isopentane-cooled liquid nitrogen and processed according to the standard techniques. For histological analysis, 8-µm-thick cryosections were selected and processed for routine staining with hematoxylin and eosin (H&E), modified Gomori trichrome (MGT), myosin ATPase (pH 9.4-4.6-4.3), cytochrome c oxidase (COX), succinate dehydrogenase (SDH), phosphatase acid, NADH dehydrogenase, Oil Red O, and periodic acid–schiff (PAS).
2.2 Immunofluorescence
For immunofluorescence staining, 8-μm-thick muscle cryosections were fixed with acetone for 3 min, washed three times with PBS, and blocked with 1% BSA diluted in PBS for 30 min at RT. Muscle sections were incubated overnight at 4°C with one of the following antibodies: C5b-9 (1:50, mouse monoclonal, Dako, Glostrup, Denmark), αB-crystallin (1:100, rabbit polyclonal, Sigma), NH-Dystrophin (1:10 mouse monoclonal, Novocastra, Newcastle upon Tyne, United Kingdom), myotilin (1:10, mouse monoclonal, Novocastra), spectrin (1:100, rabbit polyclonal, Sigma), and p62 (1:100, rabbit polyclonal, Abcam, Cambridge, United Kingdom), lamp-2 (1:200, rabbit polyclonal, Sigma). Caveolin-3 (1:1,000, mouse monoclonal, BD Biosciences, Franklin Lakes, NJ, United States) and laminin-α2 (1:200, rabbit polyclonal, Thermo Fisher Scientific, Waltham, MA, United States) were used for membrane staining. Following three washes with PBS, sections were incubated with the secondary antibody goat anti-mouse Alexa-488 or goat anti-rabbit Alexa-596 (1:1,000; Thermo Fisher Scientific) for 2 hours at RT. Finally, after three washes with PBS, slides were mounted with the anti-fading reagent Fluoromount (Thermo Fisher Scientific).
2.3 Electron microscopy
For ultrastructural examination, a small part of the muscle sample was fixed in 2.5% glutaraldehyde at pH 7.4 (Electron Microscopy Sciences EMS, Hatfield, PA, United States) for 1 h at room temperature and overnight at 4°C; the specimens were washed in 0.1 M, pH 7.4 cacodylate buffer (EMS), and post-fixed for 1 h in 2% osmium tetroxide (EMS). Next, the specimens were dehydrated using a graded series of ethyl alcohol and embedded in Epon resin (EMS). Finally, ultrathin sections (80-nm-thick slices) were prepared using an Ultramicrotome PowerTome XL (RMC, Tucson, AZ, USA). The grids, stained with 0.5% lead citrate (EMS) and uranyl acetate replacement/methanol 1:1 (EMS), were examined using a Hitachi HT7800 Transmission Electron Microscope (Hitachi, Japan).
3 Case description and results
The patient is a 47-year-old man, first born from non-consanguineous healthy parents of Brazilian origin (Figure 1). Family history is negative for neuromuscular disorders.
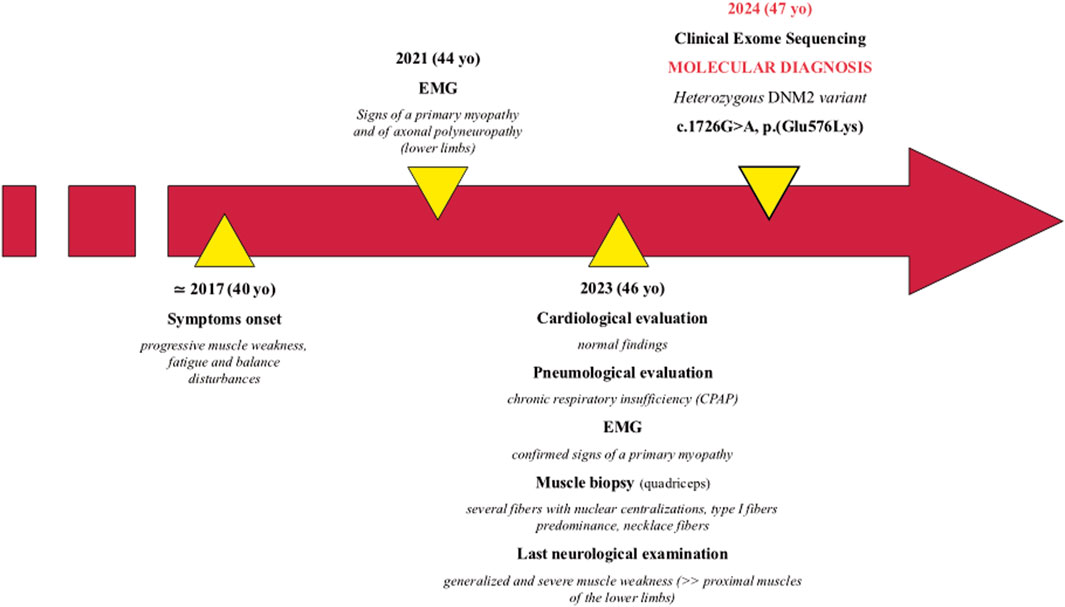
Figure 1. Patient’s timeline.
He presented with progressive muscle weakness, fatigue, and balance disturbances since his 40s, which were reported as difficulty in walking and climbing stairs. The symptoms mainly affected his lower limbs. During his last hospitalization at 46 years of age, the proband showed generalized and severe muscle weakness, with predominant involvement of the proximal muscles of the lower limbs. Needle electromyographic examination showed a myopathic pattern (motor unit action potentials of reduced duration and amplitude and early recruitment pattern) pointing toward a pathological process with a primary origin from the muscle. A nerve conduction study showed the presence of mild sensory–motor axonal polyneuropathy in the lower limbs in the absence of specific clinical symptoms.
Currently, unassisted ambulation is possible, even though mild waddling gait is present. The patient is not able to stand up from a squatting position without external aids.
Cardiological evaluation performed at 46 years, including Holter electrocardiography and echocardiography, was normal.
A thoracic computed tomography scan showed subpleural interstitial thickening in the lower lobes bilaterally, without pleural effusions. The patient was thereby diagnosed with obstructive sleep apnea syndrome with chronic respiratory insufficiency, conditioning the introduction of continuous positive airway pressure (CPAP) therapy. Creatine kinase dosage tests returned normal results.
At 46 years, a quadriceps muscle biopsy was performed. Histological analysis revealed a primary muscular involvement and characteristic features associated with DNM2-related myopathy. MGT and H&E showed mild variability in fiber size and numerous fibers containing multiple centralized nuclei (Figures 2A, B), accumulating in rows when observed in longitudinal sections (Figure 2C). Occasionally, fibers showed a radial distribution of sarcoplasmic strands. Oil red O staining demonstrated an increase in the lipid content in some type I fibers (Figure 2D). NADH staining showed the presence of radial strand fiber, and semi-thin sections revealed the presence of necklace cytoplasmic bodies (Figures 2E, F). Type I fibers showed mild hypotrophy and were slightly more prevalent than type II fibers (58.3% versus 41.6%, respectively), with a moderate shift in the distribution pattern compared to that of the control quadriceps muscle (36% versus 64%, respectively).
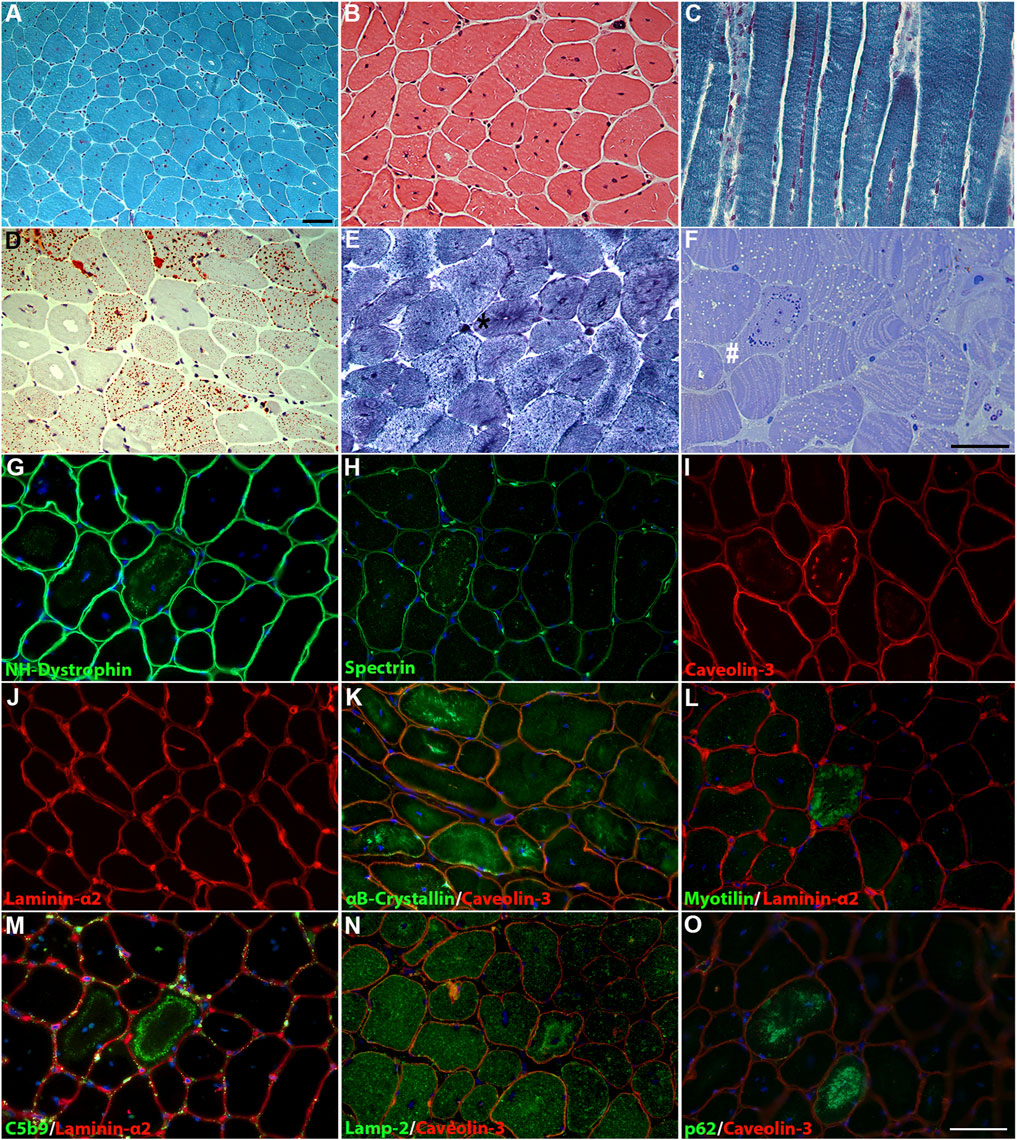
Figure 2. Histological findings in the patient’s muscle. (A) MGT and (B) H&E staining revealed fiber size variability and centronuclear fibers. (C) Longitudinal MGT section showed numerous nuclei accumulating in rows. (D) Oil red O staining revealed increased content of lipid droplets in numerous fibers. (E) NADH dehydrogenase staining showed a radial strand fiber (asterisk). (F) Semi-thin sections showed the presence of a “necklace fiber” (hashtag) and numerous fibers with an increase in lipid contents. Scale bar, 20 µm. Immunofluorescence staining showed positivity for the membrane proteins NH-dystrophin (G), spectrin (H), and caveolin-3 (I) associated with the cytoplasmic vacuolar structures, except for laminin-α2 (J). Altered distribution for αB-crystallin (K) in several fibers and increased deposition of myotilin (L) in necklace fibers. The cytoplasmic vacuolar structures showed marked positivity for C5b9 (M), lamp-2 (N), and p62 (O). DAPI for counterstaining of nuclei. Scale bar, 20 µm.
Immunofluorescence showed positivity for the membrane proteins NH-dystrophin, spectrin, and caveolin-3, indicating a membrane lining of these vacuoles, but positive staining for laminin-α2 was not observed (Figures 2G–J). Several fibers showed altered staining for αB-crystalline, with marked protein accumulation in numerous fibers (Figure 2K). The “necklace fibers” showed strong positivity for myotilin (Figure 2L). Strong positivity associated with these vacuolar structures was detected for C5b-9 (Figure 2M); in addition, lamp-2 (Figure 2N) and p62 (Figure 2O) positivity were detected in the same vacuoles, suggesting a lysosomal origin. Similar findings were previously reported by Rinnenthal et al. (2018) in two patients from two independent families carrying the DNM2 variant c.1106G>A, p.(Arg369Gln).
Ultrastructural analysis (Figure 3) revealed numerous central nuclei, a moderate increase in lipid droplets, and lipofuscin granules. Occasionally, altered mitochondria were observed, showing cristae rarefaction and matrix loss. In some fibers, round vacuoles surrounded by a membrane and containing a strongly electron-dense, homogeneous granular material were observed. A second type of inclusion containing less electron-dense material was rarely observed. These regions likely correspond to those observed in semi-thin sections, where some fibers exhibit vacuoles that were localized in a circular pattern, approximately two-thirds of the distance from the center of the fiber to the sarcolemma, appearing as necklace-like fibers. We did not observe any alterations in the T-tubules and triads.
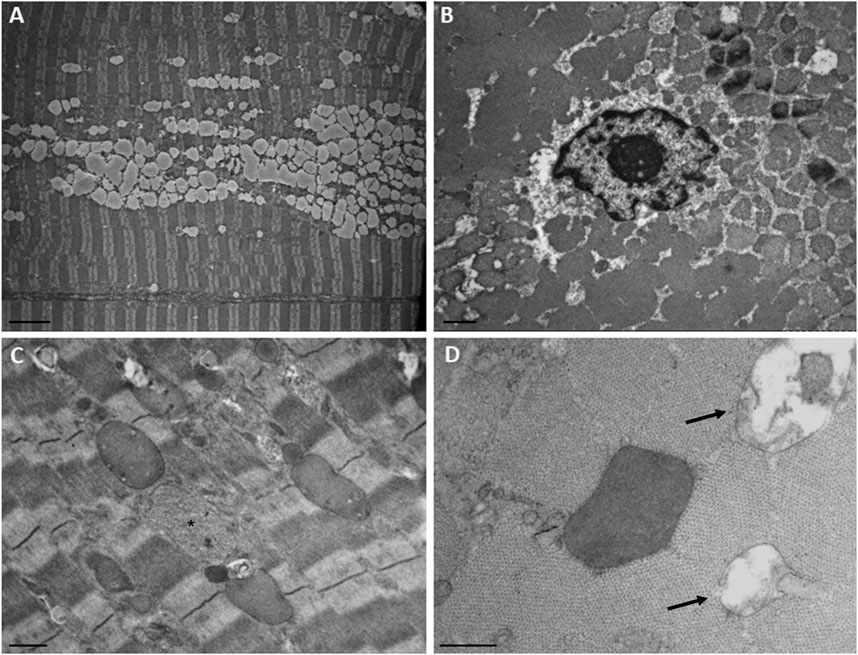
Figure 3. Ultrastructural findings in the patient’s muscle. (A) Numerous confluent lipid droplets in the center of the fiber. (B) Transverse section showed the nucleus in the center of the fiber. (C) Inclusions containing strongly electron-dense or finely granular material (asterisk). (D) Higher magnification of an electron-dense body and degraded mitochondria (arrows). Scale bar, A, 5 µm; B and C, 1 μm; D, 0.5 µm.
Clinical exome sequencing revealed the presence of the heterozygous chr19:10930710-G-A substitution (GrCh37) corresponding to the c.1726G>A, p.(Glu576Lys) variant in the DNM2 gene (NM_001005360.2). The variant was confirmed by Sanger sequencing in the proband, whereas it was absent in the patient’s healthy mother (Figures 4A, B). A DNA sample from the patient’s father was not available, preventing the conclusive determination of a de novo origin for the variant. This variant is absent in the reference databases, predicted to be pathogenic by computational prediction tools (Supplementary Table S2), and classified as a variant of uncertain significance (VUS) according to the ACMG criteria (Richards et al., 2015). The variant affects a codon that is conserved across species and in the human DNM1 and DNM3 homolog genes (Figure 4C). The p.(Glu576Lys) variant is located in the PH domain of the protein, like most of the other DNM2 variants that are reported as pathogenic (Figure 4D).
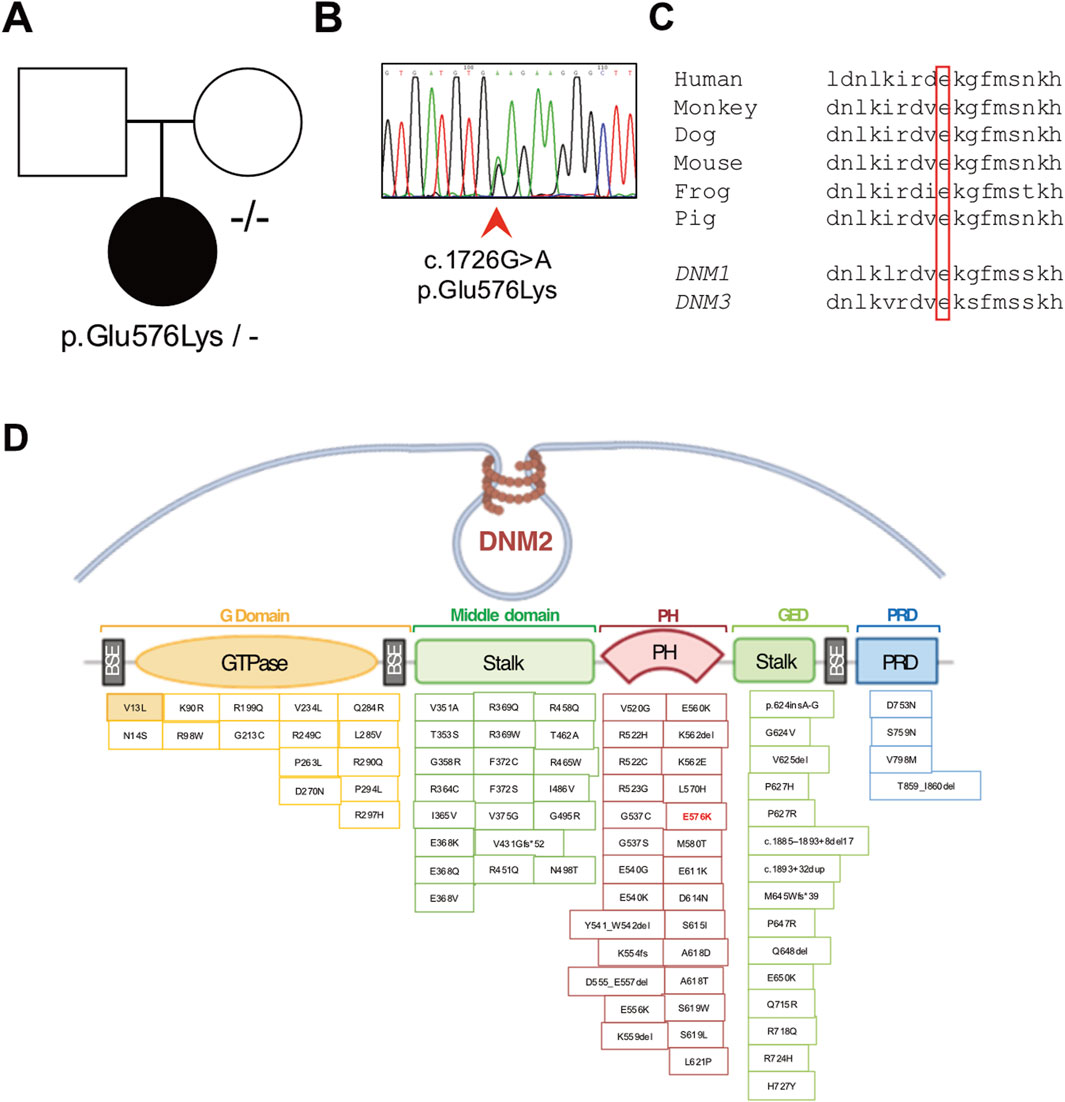
Figure 4. Molecular studies. (A) Pedigree of the family (affected member is represented by a black symbol) and (B) electropherogram showing the presence of the novel heterozygous c.1726G>A p.(Glu576Lys) variant detected in the proband’s genomic DNA. (C) Conservation of the affected amino acidic positions across species and among human homolog genes DNM1 and DNM3. (D) Schematic representation of the DNM2 gene containing the identified pathogenic variations. The p.(Glu576Lys) change is indicated in red (E576K).
4 Discussion and conclusion
Despite the wide clinical variability, patients affected with DNM2-related congenital CNM often exhibit late-childhood, early adolescence, or adulthood-onset forms, although there are instances of DNM2-related CNM presenting as severe congenital myopathy. Common symptoms include slowly progressive skeletal muscle weakness, mainly involving the distal and axial muscles (Fischer et al., 2006). However, the involvement of the facial and bulbar muscles might impair the capacity of swallowing and speaking (bulbar weakness), while restrictive ventilatory impairment, with mild to severe forced vital capacity reduction, may require nocturnal non-invasive ventilation. Therefore, DNM2-related pathology is insidious and potentially life-threatening, even in patients presenting a late-onset, slowly progressing disease course (Bitoun et al., 2009; Catteruccia et al., 2013; Biancalana et al., 2018; Hayes et al., 2022; Ogasawara and Nishino, 2023). In addition to these “myopathic” findings, recent studies have also reported a mixed phenotype, with a coexistence of neuropathic signs, in terms of axonal peripheral nerve involvement evidenced at nerve conduction velocity (NCV) studies (Reumers et al., 2021). These findings highlight the partial clinical overlap between DNM2-related CNM and Charcot–Marie–Tooth neuropathy (Echaniz-Laguna et al., 2007). Regardless of the age at which symptoms appear, this condition progressively worsens, impacting independent ambulation: most of the patients perceive their condition as progressively deteriorating and require walking assistance during adulthood, if not earlier (Hayes et al., 2022).
Pathologically, DNM2-related centronuclear myopathy is usually characterized by centralized nuclei, predominance of type 1 muscle fibers, and radial distribution of sarcoplasmic strands (RSS) (Jeub et al., 2008; Romero, 2010; Park et al., 2014; Abath Neto et al., 2015). Nonetheless, in instances of severe cases, most of the fibers exhibit centrally located hyperintense responses surrounded by a faint peripheral halo, without typical RSS. This closely resembles the observations seen in individuals afflicted with XLMTM, an X-linked myotubular myopathy stemming from mutations in the MTM1 (myotubularin 1) gene. In a single study, authors also reported evidence of necklace fibers at muscle biopsy, potentially suggesting a shared pathogenic pathway in centronuclear myopathies caused by both DNM2 and MTM1 gene mutations (Casar-Borota et al., 2015).
The patient’s muscle biopsy displayed several fibers with nuclear centralization, a characteristic feature in DNM2-mutated subjects, along with slight type I fiber predominance and the presence of necklace fibers (Romero, 2010).
The patient we are reporting presented with progressive skeletal muscle weakness, predominantly affecting the proximal muscles, sparing the facial and bulbar muscles. Disease onset was in his first adulthood, and the course is currently slowly progressive. He also required assisted ventilation from the age of 46 years old. He did not display ptosis, eye movement disorders, speech difficulties, or orofacial malformations, which have long been considered typical features of DNM2-related clinical presentation. However, recently published case series now acknowledge the existence of a wide phenotypic spectrum of DNM2 myopathy. These additional manifestations are not consistently observed in all the patients in contrast to muscle weakness, fatigue, and exercise intolerance, which are described in almost every DNM2-mutated myopathic patient (Hayes et al., 2022; Reumers et al., 2021).
Since the first report (Bitoun et al., 2005), 76 different variants have been reported in DNM2, with half of them described as causative for CNM (Supplementary Table S1). In a single case, authors reported a patient affected with CNM carrying compound heterozygous DNM2 mutations: despite his genotype, the subject displayed milder clinical manifestations than subjects carrying biallelic variants in DNM2, which are usually associated with the “lethal congenital contracture syndrome” (MIM #615368) (Chen et al., 2015). The majority of the reported pathogenic DNM2 variants consist of missense changes situated within the PH and stalk domains. Recent studies (Böhm et al., 2012; Fujise et al., 2022; Hayes et al., 2022) have demonstrated that patients with mutations located at the autoinhibitory interface between the PH and middle domains, particularly p.(Glu368Lys), p.(Phe372Ser), p.(Ala618Asp), and p.(Ser619Leu), tend to exhibit more severe phenotypes characterized by earlier onset and faster progression of motor signs and respiratory difficulties. All these disease-causing substitutions are proposed to release autoinhibition of dynamin-2 and promote protein oligomerization, thus providing support for the “gain-of-function” mechanism advanced for DNM2-related CNM (Ogasawara and Nishino, 2023), based on their specific locations.
The variant detected in the patient we are reporting (p.Glu576Lys) affects a highly conserved position and is absent from population databases. However, the lack of functional experiments addressing the DNM2 transcript and protein stability or the evaluation of DNM2 GTPase activity prevents us from drawing a conclusion about the pathogenicity of this variant that should be considered of uncertain significance.
Currently, there is no specific treatment available for DNM2-related CNM. Management is primarily focused on symptom alleviation and improving quality of life. This may involve a multidisciplinary approach, including physiotherapy to maintain muscle strength and mobility, occupational therapy to assist with activities of daily living, and the use of assistive devices as needed. However, promising therapies are being developed for this debilitating disease (Hayes et al., 2022). Recent studies using a mouse model of DNM2-related centronuclear myopathy have shown that molecular strategies aiming to reduce mutant dynamin-2 levels through the systemic delivery of viral vectors or the intramuscular administration of antisense oligonucleotides can improve the phenotype (Buono et al., 2018; de Carvalho Neves et al., 2023; Trochet et al., 2022). Based on these promising findings, a clinical trial using DYN101, an antisense oligonucleotide targeting DNM2 pre-mRNA, was started in 2020 but was recently stopped because of liver enzyme elevations and low platelet counts in treated patients.
Further research is needed to explore new avenues for advancing the understanding and therapeutic development for this rare genetic muscle disorder. The identification of pathogenic DNM2 variants in clinically affected subjects through NGS-based sequencing is crucial for accelerating patient and potential inclusion in future clinical trials.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Comitato Etico Milano Area 2 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
MR: data curation, writing–original draft, and writing–review and editing. DV: data curation and writing–review and editing. SZ: data curation and writing–review and editing. MR: data curation and writing–review and editing. RD: data curation and writing–review and editing. PC: data curation and writing–review and editing. LN: data curation and writing–review and editing. SC: supervision and writing–review and editing. GC: supervision and writing–review and editing. DR: conceptualization, data curation, supervision, writing–original draft, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was (partially) funded by the Italian Ministry of Health—Current research IRCCS Ca’ Granda Ospedale Maggiore Policlinico and by SEQMD project (IRCCS Cà Granda Ospedale Maggiore Policlinico, PI: GC).
Acknowledgments
This work was promoted within the European reference network (ERN) for rare neuromuscular diseases. The authors thank the Associazione Centro Dino Ferrari for its support. The PNC “Hub Life Science-Diagnostica Avanzata (HLS-DA), PNC-E3-2022-23683266– CUP: C43C22001630001” is funded by the Italian Ministry of Health. The support of the Italian Ministry of Education and Research (MUR) “Dipartimenti di Eccellenza Program 2023–2027,” Dept of Pathophysiology and Transplantation, University of Milan, to DR, SC, and GPC is gratefully acknowledged. The authors acknowledge support from the University of Milan through the APC initiative.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1559773/full#supplementary-material
References
Abath Neto, O., Martins Cde, A., Carvalho, M., Chadi, G., Seitz, K. W., Oliveira, A. S., et al. (2015). DNM2 mutations in a cohort of sporadic patients with centronuclear myopathy. Genet. Mol. Biol. 38 (2), 147–151. doi:10.1590/S1415-4757382220140238
Al-Qusairi, L., and Laporte, J. (2011). T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet. Muscle 1 (1), 26. doi:10.1186/2044-5040-1-26
Antonny, B., Burd, C., De Camilli, P., Chen, E., Daumke, O., Faelber, K., et al. (2016). Membrane fission by dynamin: what we know and what we need to know. EMBO J. 35 (21), 2270–2284. doi:10.15252/embj.201694613
Biancalana, V., Romero, N. B., Thuestad, I. J., Ignatius, J., Kataja, J., Gardberg, M., et al. (2018). Some DNM2 mutations cause extremely severe congenital myopathy and phenocopy myotubular myopathy. Acta Neuropathol. Commun. 6 (1), 93. doi:10.1186/s40478-018-0593-2
Bitoun, M., Durieux, A. C., Prudhon, B., Bevilacqua, J. A., Herledan, A., Sakanyan, V., et al. (2009). Dynamin 2 mutations associated with human diseases impair clathrin-mediated receptor endocytosis. Hum. Mutat. 30 (10), 1419–1427. doi:10.1002/humu.21086
Bitoun, M., Maugenre, S., Jeannet, P. Y., Lacène, E., Ferrer, X., Laforêt, P., et al. (2005). Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet. 37 (11), 1207–1209. doi:10.1038/ng1657
Böhm, J., Biancalana, V., Dechene, E. T., Bitoun, M., Pierson, C. R., Schaefer, E., et al. (2012). Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum. Mutat. 33 (6), 949–959. doi:10.1002/humu.22067
Buono, S., Ross, J. A., Tasfaout, H., Levy, Y., Kretz, C., Tayefeh, L., et al. (2018). Reducing dynamin 2 (DNM2) rescues DNM2-related dominant centronuclear myopathy. Proc. Natl. Acad. Sci. U. S. A. 115 (43), 11066–11071. doi:10.1073/pnas.1808170115
Casar-Borota, O., Jacobsson, J., Libelius, R., Oldfors, C. H., Malfatti, E., Romero, N. B., et al. (2015). A novel dynamin-2 gene mutation associated with a late-onset centronuclear myopathy with necklace fibres. Neuromuscul. Disord. 25 (4), 345–348. doi:10.1016/j.nmd.2015.01.001
Catteruccia, M., Fattori, F., Codemo, V., Ruggiero, L., Maggi, L., Tasca, G., et al. (2013). Centronuclear myopathy related to dynamin 2 mutations: clinical, morphological, muscle imaging and genetic features of an Italian cohort. Neuromuscul. Disord. 23 (3), 229–238. doi:10.1016/j.nmd.2012.12.009
Chen, T., Pu, C., Wang, Q., Liu, J., Mao, Y., and Shi, Q. (2015). Clinical, pathological, and genetic features of dynamin-2-related centronuclear myopathy in China. Neurol. Sci. 36 (5), 735–741. doi:10.1007/s10072-014-2028-6
de Carvalho Neves, J., Moschovaki-Filippidou, F., Böhm, J., and Laporte, e J. (2023). DNM2 levels normalization improves muscle phenotypes of a novel mouse model for moderate centronuclear myopathy. Mol. Ther. Nucleic Acids 33, 321–334. doi:10.1016/j.omtn.2023.07.003
Echaniz-Laguna, A., Nicot, A. S., Carré, S., Franques, J., Tranchant, C., Dondaine, N., et al. (2007). Subtle central and peripheral nervous system abnormalities in a family with centronuclear myopathy and a novel dynamin 2 gene mutation. Neuromuscul. Disord. 17 (11-12), 955–959. doi:10.1016/j.nmd.2007.06.467
Ferguson, S. M., and De Camilli, P. (2012). Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 13 (2), 75–88. doi:10.1038/nrm3266
Fischer, D., Herasse, M., Bitoun, M., Barragán-Campos, H. M., Chiras, J., Laforêt, P., et al. (2006). Characterization of the muscle involvement in dynamin 2-related centronuclear myopathy. Brain 129 (Pt 6), 1463–1469. doi:10.1093/brain/awl071
Fujise, K., Okubo, M., Abe, T., Yamada, H., Takei, K., Nishino, I., et al. (2022). Imaging-based evaluation of pathogenicity by novel DNM2 variants associated with centronuclear myopathy. Hum. Mutat. 43 (2), 169–179. doi:10.1002/humu.24307
Hayes, L. H., Perdomini, M., Aykanat, A., Genetti, C. A., Paterson, H. L., Cowling, B. S., et al. (2022). Phenotypic spectrum of DNM2-related centronuclear myopathy. Neurol. Genet. 8 (6), e200027. doi:10.1212/NXG.0000000000200027
Hohendahl, A., Roux, A., and Galli, V. (2016). Structural insights into the centronuclear myopathy associated functions of BIN1 and dynamin 2. J. Struct. Biol. 196 (1), 37–47. doi:10.1016/j.jsb.2016.06.015
Jeub, M., Bitoun, M., Guicheney, P., Kappes-Horn, K., Strach, K., Druschky, K. F., et al. (2008). Dynamin 2-related centronuclear myopathy: clinical, histological and genetic aspects of further patients and review of the literature. Clin. Neuropathol. 27 (6), 430–438. doi:10.5414/npp27430
Jungbluth, H., Wallgren-Pettersson, C., and Laporte, J. (2008). Centronuclear (myotubular) myopathy. Orphanet J. Rare Dis. 3, 26. doi:10.1186/1750-1172-3-26
Ogasawara, M., and Nishino, I. (2023). A review of major causative genes in congenital myopathies. J. Hum. Genet. 68 (3), 215–225. doi:10.1038/s10038-022-01045-w
Park, Y. E., Choi, Y. C., Bae, J. S., Lee, C. H., Kim, H. S., Shin, J. H., et al. (2014). Clinical and pathological features of Korean patients with DNM2-related centronuclear myopathy. J. Clin. Neurol. 10 (1), 24–31. doi:10.3988/jcn.2014.10.1.24
Puri, C., Manni, M. M., Vicinanza, M., Hilcenko, C., Zhu, Y., Runwal, G., et al. (2020). A DNM2 centronuclear myopathy mutation reveals a link between recycling endosome scission and autophagy. Dev. Cell 53 (2), 154–168.e6. doi:10.1016/j.devcel.2020.03.018
Reumers, S. F. I., Erasmus, C. E., Bouman, K., Pennings, M., Schouten, M., Kusters, B., et al. (2021). Clinical, genetic, and histological features of centronuclear myopathy in The Netherlands. Clin. Genet. 100 (6), 692–702. doi:10.1111/cge.14054
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Rinnenthal, J. L., Dittmayer, C., Irlbacher, K., Wacker, I., Schröder, R., Goebel, H.-H., et al. (2018). New variant of necklace fibres display peculiar lysosomal structures and mitophagy. Neuromuscul. Disord. NMD 28 (10), 846–856. doi:10.1016/j.nmd.2018.06.010
Romero, N. B. (2010). Centronuclear myopathies: a widening concept. Neuromuscul. Disord. 20 (4), 223–228. doi:10.1016/j.nmd.2010.01.014
Susman, R. D., Quijano-Roy, S., Yang, N., Webster, R., Clarke, N. F., Dowling, J., et al. (2010). Expanding the clinical, pathological and MRI phenotype of DNM2-related centronuclear myopathy. Neuromuscul. Disord. 20 (4), 229–237. doi:10.1016/j.nmd.2010.02.016
Trochet, D., Prudhon, B., Mekzine, L., Lemaitre, M., Beuvin, M., Julien, L., et al. (2022). Benefits of therapy by dynamin-2-mutant-specific silencing are maintained with time in a mouse model of dominant centronuclear myopathy. Mol. Ther. Nucleic Acids 27, 1179–1190. doi:10.1016/j.omtn.2022.02.009
Vandersmissen, I., Biancalana, V., Servais, L., Dowling, J. J., Vander Stichele, G., Van Rooijen, S., et al. (2018). An integrated modelling methodology for estimating the prevalence of centronuclear myopathy. Neuromuscul. Disord. 28 (9), 766–777. doi:10.1016/j.nmd.2018.06.012
Keywords: DNM2, centronuclear myopathy, myopathy, dynamin-2, neuromuscular disease
Citation: Rimoldi M, Velardo D, Zanotti S, Ripolone M, Del Bo R, Ciscato P, Napoli L, Corti S, Comi GP and Ronchi D (2025) A novel DNM2 variant associated with centronuclear myopathy: a case report. Front. Genet. 16:1559773. doi: 10.3389/fgene.2025.1559773
Received: 20 January 2025; Accepted: 17 March 2025;
Published: 07 April 2025.
Edited by:
Mara Marongiu, National Research Council (CNR), ItalyReviewed by:
Jorge Alfredo Bevilacqua, University of Chile, ChileYa-Wen Liu, National Taiwan University, Taiwan
Copyright © 2025 Rimoldi, Velardo, Zanotti, Ripolone, Del Bo, Ciscato, Napoli, Corti, Comi and Ronchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dario Ronchi, dario.ronchi@unimi.it
‡ORCID: Stefania Corti, Dario Ronchi, orcid.org/0000-0001-5425-969X; Giacomo Pietro Comi, https://orcid.org/0000-0002-1383-5248https://orcid.org/0000-0002-6093-9816