 Yanlin Huang†Chang Liu†Hongke DingYunan WangLihua YuFangfang GuoFake LiXiaomei ShiYan Zhang*Aihua Yin*
Yanlin Huang†Chang Liu†Hongke DingYunan WangLihua YuFangfang GuoFake LiXiaomei ShiYan Zhang*Aihua Yin*- Medical Genetic Center, Guangdong Women and Children Hospital, Guangzhou, Guangdong, China
Background: Prenatal diagnosis of fetal short long bones (SLBs) was reported to be associated with skeletal dysplasias, chromosomal abnormalities, and genetic syndromes. This study aims to identify the genetic causes for fetal short long bones, and retrospectively evaluate the additional diagnostic yield of exome sequencing (ES) for short long bones following the use of conventional genetic testing.
Methods: A cohort of ninety-four fetuses with sonographically identified short long bones was analyzed by trio-exome sequencing between January 2016 and June 2021. Fetuses with abnormal results of karyotype or chromosomal microarray analysis were excluded. Variants were interpreted based on ACMG/AMP guidelines. All diagnostic de novo variants were validated by Sanger sequencing.
Results: Of the 94 fetuses, 38 (40.4%) were found to carry causal genetic variants (pathogenic or likely pathogenic) in sixteen genes with 38 variants. Five fetuses (5.3%) had variant(s) of uncertain significance. Thirty-five cases (37.2%) were diagnosed as genetic skeletal dysplasias including 14 different diseases that were classified into 10 groups according to the Nosology and Classification of Genetic Skeletal Disorders. The most common disease in the cohort was achondroplasia (28.9%), followed by osteogenesis imperfecta (18.4%), thanatophoric dysplasia (10.5%), chondrogenesis (7.9%), and 3-M syndrome (5.3%). The diagnostic yield in fetuses with isolated short long bones was lower than the fetuses with non-isolated short long bones, but not reached statistical significance (27.3% vs. 44.4%; p = 0.151). Whereas, the rate in the fetuses with other skeletal abnormalities was significantly higher than those with non-skeletal abnormalities (59.4% vs. 32.5%, p = 0.023), and the diagnostic rate was significantly higher in femur length (FL) below -4SDs group compared with FL 2-4SDs below GA group (72.5% vs. 16.7%; p < 0.001). A long-term follow-up showed that outcomes for fetuses with FL 2-4SDs below GA were significantly better than those with FL below -4SDs. Additionally, fourteen (36.8%) novel short long bones-related variants were identified in the present study.
Conclusion: The findings suggest that in fetuses with short long bones routine genetic tests failed to determine the underlying causes, exome sequencing could add clinically relevant information that could assist the clinical management of pregnancies. Novel pathogenic variants identified may broaden the mutation spectrum for the disorders and contributes to clinical consultation and subsequent pregnancy examination.
1 Introduction
Routine fetal biometric evaluation includes femur diaphysis length measurement (Salomon et al., 2019). In cases where the femur length (FL) is 2 or more standard deviations (SDs) below the normal range, the guidelines recommend measuring the length of the other long bones and performing a thorough skeletal assessment (Beke et al., 2005; Speer et al., 2014; Rao and Platt, 2016; Tzadikevitch et al., 2021). Short long bones may be constitutional and/or attributed to race, ethnicity and familial tendency (Liu et al., 2019; Tzadikevitch et al., 2021). When no coexistence of other fetal abnormalities or abnormal uterine artery Doppler flows is demonstrated, a good pregnancy outcome may be expected (Zalel et al., 2002; Mohr-Sasson et al., 2020). Whereas, prenatal diagnosis of fetal SLBs was reported to be associated with skeletal dysplasias, chromosomal abnormalities and genetic disorders (Zalel et al., 2002; Papageorghiou et al., 2008; Kaijomaa et al., 2016; Mohr-Sasson et al., 2020). In some cases, it may be a sign of intrauterine growth restriction (FGR) and placental insufficiency, resulting in an increased risk for pregnancy complications, including pre-eclampsia, placental abruption and intrauterine fetal death (Weisz et al., 2008; Aviram et al., 2015; Kaijomaa et al., 2016; Tzadikevitch et al., 2021). According to the latest version of the Nosology, skeletal dysplasia comprises 461 different diseases that are classified into 42 groups based on their clinical, radiographic, and molecular phenotypes. Remarkably, 437 skeletal dysplasia-related genes are functionally diverse, involved in a broad range of cell biologic processes, and cause diseases by a variety of mutational mechanisms (Mortier et al., 2019). Pathogenic variants in FGFR3, COL1A1, COL1A2, and COL2A1 are common causes of isolated short stature or osteogenesis imperfecta. However, features of skeletal dysplasias such as epiphyseal stippling, bowing of the long bones, aberrant bone mineralization, and fractures are not always identified on antenatal scanning (Ramachandrappa et al., 2018; Victoria et al., 2018). Furthermore, disproportionate limb development may be a late sign as exemplified by achondroplasia where SLBs are rarely detected before the third trimester (Kurtz et al., 1986; Patel et al., 1995; Ramachandrappa et al., 2018). The complex etiology, combined with the highly variable and often overlapping presentations of skeletal dysplasias and SLBs, challenge the ability of traditional prenatal diagnosis.
The conventional molecular diagnosis strategies mainly involved karyotyping or chromosomal microarray analysis (CMA). Karyotyping has a diagnostic yield of about 13%–16% in fetuses with sonographically identified SLBs, and CMA provides an additional diagnostic yield of 6%–27% (Gaudry et al., 2012; Liu et al., 2019; Hu et al., 2021; Tzadikevitch et al., 2021). In the last decade, next-generation sequencing (NGS) has revolutionized the genetic testing of diseases with high genetic and allelic heterogeneity, such as skeletal dysplasias, allowing hundreds of genes to be screened simultaneously. Based on the statements released by the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), the Perinatal Quality Foundation (PQF), and the American College of Medical Genetics and Genomics (ACMG), NGS can be used with ultrasound anomalies when standard diagnostic genetic testing, such as CMA, failed to yield a definitive diagnosis (Lord et al., 2019; Tang et al., 2021). In addition, SLBs are mainly attributed to monogenic disorders, such as skeletal dysplasias (Mortier et al., 2019), and therefore NGS may be of importance in their prenatal evaluation. Especially, if a specific diagnosis is suspected or when FL is > 4 SDs below the mean, targeted capture sequencing or whole exome sequencing should be performed (Liu et al., 2019; Tang et al., 2021). Definitive molecular diagnosis can provide information about prognosis of the disease and treatment regimens, as well as the estimation of the likelihood of recurrence.
There is limited data in the literature regarding ES findings in fetuses with SLBs to evaluate its clinical value. In the present study, ES was performed to identify genetic causes for 94 fetuses with sonographically identified SLBs, and to evaluate the diagnostic yield of ES for fetal SLBs. Besides of genetic diagnosis, we also performed long-term follow-ups to assess the prognoses and outcomes in fetuses with SLBs.
2 Materials and methods
2.1 Ethics statement
This retrospective cohort study was conducted at Guangdong Women and Children Hospital. The study has been approved by our institutional review board and clinical research ethics committee. Written informed consents were obtained from all participants. Authors had access to information that could identify individual participants, and the information was anonymized prior to submission. All the procedures performed in the study were in accordance with the Declaration of Helsinki and as we previously described (Liu et al., 2022).
2.2 Subject identification and enrollment
Between January 2016 and June 2021, a cohort of 97 couples consented to further ES with normal preliminary results of routine genetic tests. Apart from 2 couples who withdrew consent and one sample that was not of good enough quality for analysis, DNA samples from 94 eligible trios were therefore used for the analysis of the primary outcome (Figure 1). The sonographic criteria include the presence of short limb deformities in which fetal femur length and/or other long bones < −2 SD of our reference ranges at mid-trimester ultrasonography with/without other abnormalities (Papageorghiou et al., 2008). The traditional molecular diagnosis strategies include karyotyping and CMA. Fetuses with chromosomal aneuploidies or CNV abnormalities were excluded. The isolated-SLBs group includes fetuses with short long bones only, while the non-isolated-SLBs group includes fetuses with SLBs and another abnormality. To compare the degree of femur shortening, Z-score was calculated as (XGA − MGA)/SDGA, where XGA is the ultrasound measurement of the fetal femur, and MGA and SDGA are the mean value and standard deviation at the corresponding gestational age (GA) in weeks (Sananes et al., 2009). The clinical details of enrolled fetuses are summarized in Table 1 and Additional file 1 (Supplementary Table S1). The chorionic villi, amniotic fluid, umbilical cord blood or skin tissue of aborted fetuses, and peripheral blood of family members were collected by maternal-fetal medicine specialists at our center through the workup already being performed as part of the standard-of-care. Pretest counseling for prenatal ES was delivered in an intelligible fashion to every enrolled family by trained genetic professionals. Results were reported to the parents after a multidisciplinary team of clinical and laboratory geneticists, obstetricians, and genetic counselors reviewed all the variants related to the ultrasound anomalies.
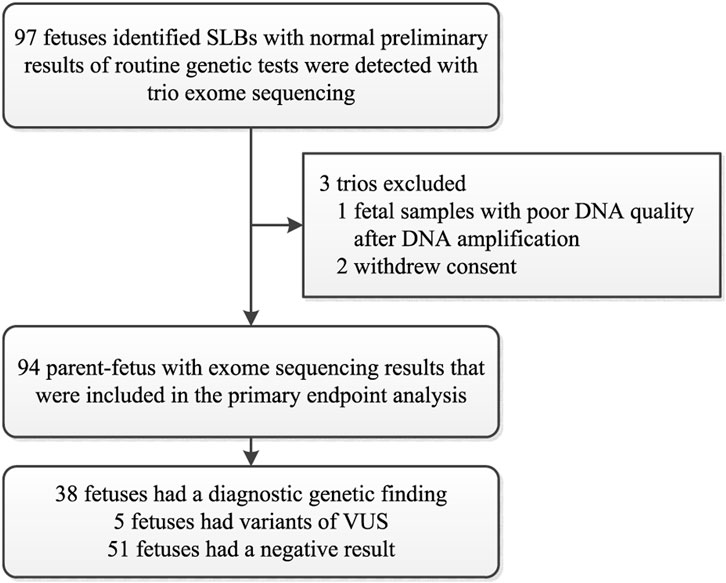
FIGURE 1. The flow diagram of the cohort and outcomes.
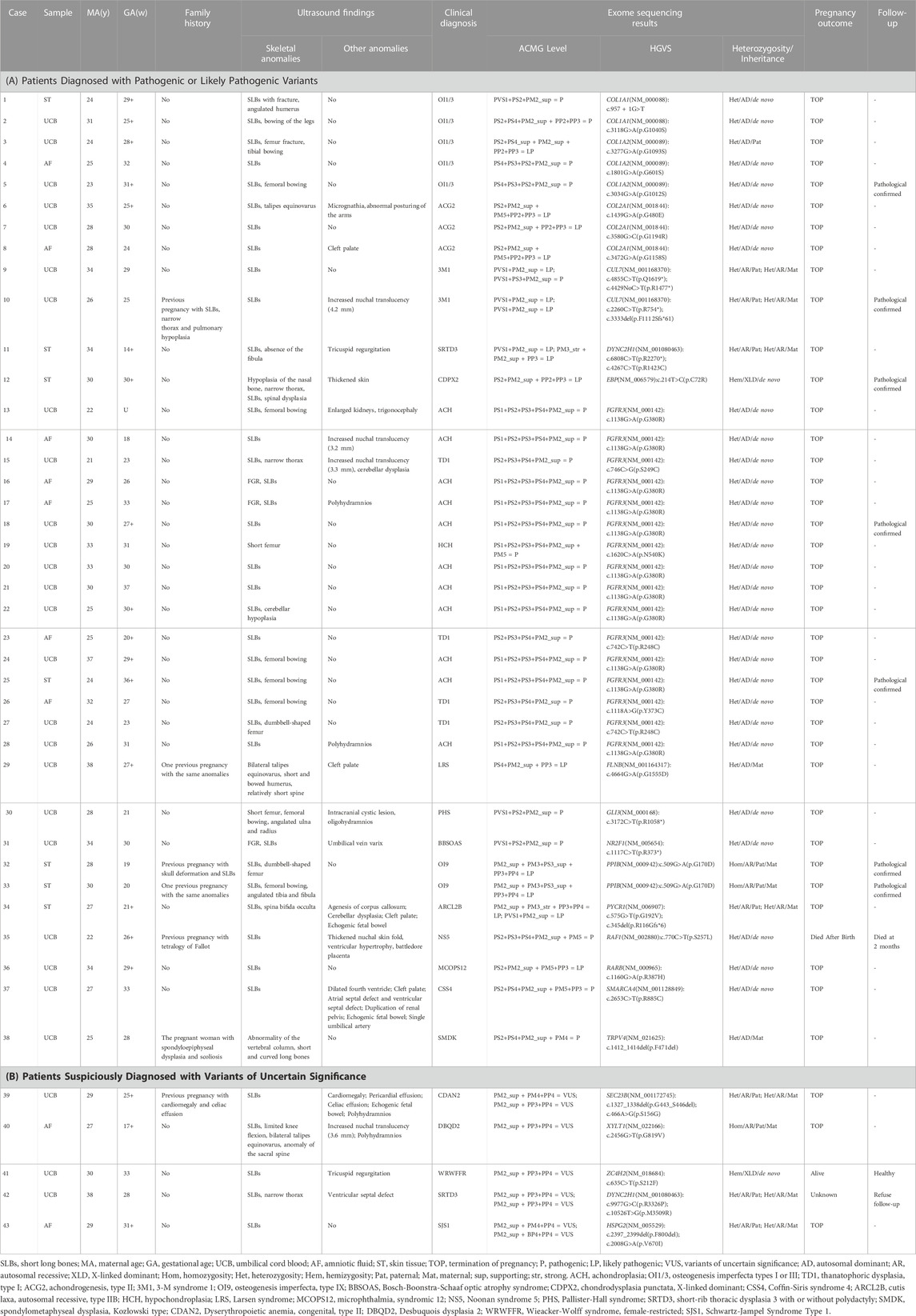
TABLE 1. Prenatal phenotype and genotype information of the genetically diagnosed fetuses in the cohort.
2.3 Exome sequencing
Ninety-four fetuses received genetics evaluations by using prenatal ES, as previously described (Zheng et al., 2018; Dai et al., 2019; Han et al., 2020; Zhang B et al., 2020; Liu et al., 2022). Genomic DNA was extracted by using the SolPure Blood DNA kit (Magen, Guangzhou, China) according to the manufacturer’s instructions and was fragmented by Q800R Sonicator (Qsonica, CT, United States). The paired-end libraries were prepared following the Illumina library preparation protocol. Custom-designed NimbleGen SeqCap probes (Roche NimbleGen, Madison, WI) were used for in-solution hybridization to enrich target whole-exome sequences for WES or target sequences included ∼5,000 genes potentially associated with known Mendelian genetic diseases for clinical exome sequencing (CES) (AmCare Genomic Laboratory, Guangzhou, China). Captured DNA samples were amplified by PCR with indexed primers and then sequenced on MGISEQ-2000RS, DNBSEQ-T7 sequencers (MGI, ShenZhen, China) or NovaSeq 6000 sequencers (Illumina, San Diego, CA) with pair-end 150 base pairs (bp) model following the manufacturer’s protocol. Low-quality reads (Phred score < Q20) were removed before demultiplexing. An overview of prenatal ES data analysis and interpretation process is summarized in Figure 2. Before bioinformatics analysis, data quality control would be conducted based on gender judgment, pedigree judgment, and data volume judgment. Raw fastq reads were filtered by using Fastp (v0.23) to remove low quality and adapter contaminated reads, leaving clean paired-end reads were aligned to the GRCh37/hg19 human reference sequence by NextGENe software (SoftGenetics, State College, PA). Then, NextGENe software was used for identifying single-nucleotide variants (SNVs) and small insertions/deletions (InDels) by using the recommended standard settings. Finally, variants were annotated by NextGENe software with multiple public database and in-house database (Figure 2).
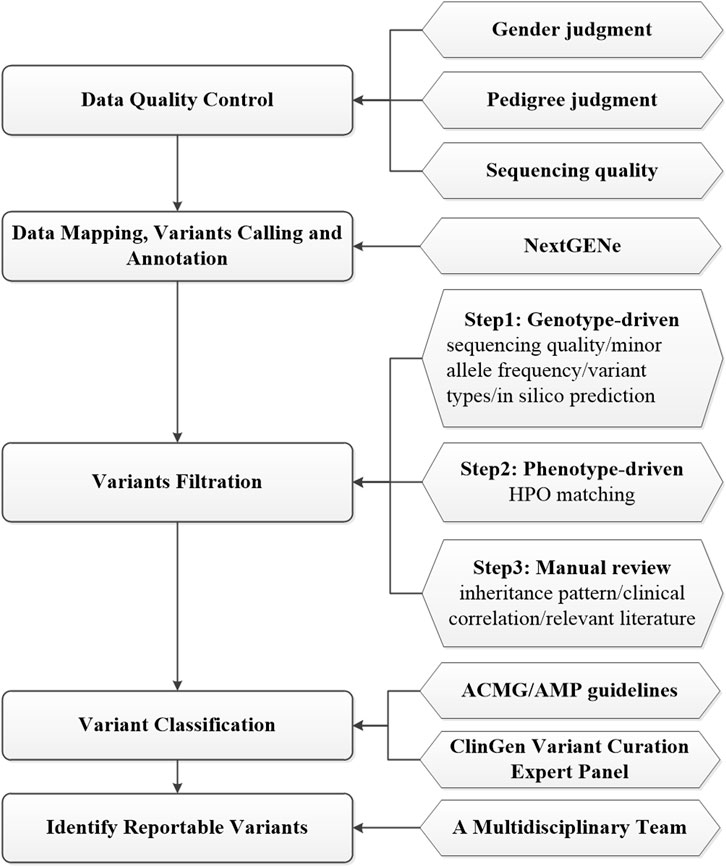
FIGURE 2. Data analysis process of exome sequencing.
2.4 Variants filtration and evaluation
Quality control for each sample included an average depth of >100X and >95% targeted region with at least 20X in the cohort. The annotated variants were filtrated in a stepwise model as shown in Figure 2. The first genotype-driven step prioritized the variants based on sequencing quality, minor allele frequency, variant types and in silico prediction without phenotypic data. All the annotated variants, excluding low quality ones (with a depth <10X or alternate allele proportion <0.20) were subject to downstream analysis. Variants with a minor allele frequency (MAF) > 0.5% in the Genome Aggregation Database (GnomAD, https://gnomad.broadinstitute.org/) were filtered out except for those in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), HGMD (http://www.hgmd.cf.ac.uk/) and ClinGen BA1 exception list (Ghosh R et al., 2018). Next, we mainly focused on genomic regions known or likely associated with the disease and potential protein-altering variants (e.g., missense, start loss, stop gain/loss, frameshift, in-frame insertion/deletion, or canonical splice-site) were retained. Variants were evaluated with the predictors included in the Varsome website (https://Varsome.com/), VarSEAK website (https://varseak.bio/), and SpliceAI software (Jaganathan et al., 2019). In the second phenotype-driven analysis step, the rare variants that potentially related to initial clinical indications for prenatal diagnosis was evaluated with the aid of HPO matching. In the third analysis step, the remaining variants were closely reviewed by trained genetic professionals based on inheritance pattern, clinical correlation and relevant literature. Search engines such as Mastermind (https://mastermind.genomenon.com/) and PubMed (https://pubmed.ncbi.nlm.nih.gov/) were used for retrieving published literature on variant-related case reports. The variant number in each trio after each screening step was shown in Additional file 2 (Supplementary Table S2). The information for all variants passing each screening step was shown in Additional file 3 (Supplementary Table S3). Candidate variants were classified according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines (Richards et al., 2015) and ClinGen variant curation expert panel guidelines (Abou et al., 2018; Brnich et al., 2019). A multidisciplinary team of clinical and laboratory geneticists, obstetricians and genetic counselors reviewed all the rare phenotype-related variants to identify the reportable variants. All diagnostic de novo variants were validated by Sanger sequencing. The primer sequences of de novo variants were shown in Additional file 4 (Supplementary Table S4).
2.5 Statistical analysis
Continuous data (maternal age, GA at initial diagnosis) are presented as mean (standard deviation) and categorical variables are presented as number (percentage). Comparison of diagnostic yield of ES between groups was conducted using the chi-square test or Fisher’s exact test. A two-sided p < 0.05 was considered statistically significant. All statistical analyses were performed using SPSS Statistics 21.0 (SPSS, Chicago, IL, United States).
3 Results
3.1 Clinical descriptions
A total of 94 parent-fetuses were enrolled in the current study. All of the enrolled fetuses underwent detailed ultrasound examinations during pregnancy, and the clinical manifestations were summarized in Supplementary Table S1 and Table 1. Maternal age ranged from 21 to 44 years (29.46 ± 4.48 years). GA at initial diagnosis ranged from 14+5 to 36+6 weeks (27.51 ± 4.38 weeks). Thirteen cases had a history of adverse pregnancy outcomes, and 9 of those were associated with skeletal dysplasias. Twenty-two fetuses had an ultrasonic diagnosis with isolated SLBs, 32 with SLBs combined with other skeletal abnormalities, and 40 with SLBs combined with non-skeletal abnormalities. Moreover, FL in 42.6% (40/94) of the fetuses were below -4SDs (Table 2).

TABLE 2. The yield of exome sequencing according to clinical parameters.
3.2 Diagnosis yields of Trio-ES
Of the 94 fetuses, 38 (40.4%) were found to carry causal genetic variants (pathogenic or likely pathogenic) in sixteen genes. Five (5.3%) fetuses had variant(s) of uncertain significance (VUS), thereby yielding a total detection rate of 45.7%. The diagnostic rate in fetuses with isolated SLBs was lower than those with non-isolated SLBs, but not reached statistical significance (27.3% vs. 44.4%; p = 0.151). While the yield of ES among the fetuses with other skeletal abnormalities was significantly higher than those with non-skeletal abnormalities (59.4% vs. 32.5%; p = 0.023). Moreover, the diagnostic rate was 58.7% among terminated fetuses, which was much higher than those in alive fetuses (3.6%, p < 0.001). Fetuses with Z-scores below −4SDs show a significantly higher diagnostic yield than the fetuses with Z-scores between −2SDs and −4SDs (72.5% vs. 16.7%; p < 0.001). However, we did not find any correlation between the yield of ES and family history, maternal age, sample type, or gestational week at initial diagnosis (Table 2).
3.3 Etiologic diagnosis
The etiologic diagnosis has been identified for 38 cases. These were linked to FGFR3 (16 cases), COL1A2 (3 cases), COL2A1 (3 cases), COL1A1 (2 cases), CUL7 (2 cases), and one case in each of the PPIB, EBP, FLNB, GLI3, NR2F1, PYCR1, RAF1, RARB, DYNC2H1, SMARCA4, and TRPV4 genes. 35 cases (37.2%) were diagnosed as genetic skeletal dysplasias including 14 different diseases that are classified into 10 groups according to the 2019 revision of the Nosology and Classification of Genetic Skeletal Disorders (Table 3). Three other cases were diagnosed with Noonan syndrome 5 (NS5), Microphthalmia, syndromic 12 (MCOPS12), and Bosch-Boonstra-Schaaf optic atrophy syndrome (BBSOAS) respectively. Among genetic skeletal dysplasias in our cohort, the most common disease is achondroplasia (28.9%), followed by osteogenesis imperfecta (18.4%), thanatophoric dysplasia (10.5%), chondrogenesis (7.9%), and 3-M syndrome (5.3%). One case in each of other skeletal dysplasias are shown in Table 3.
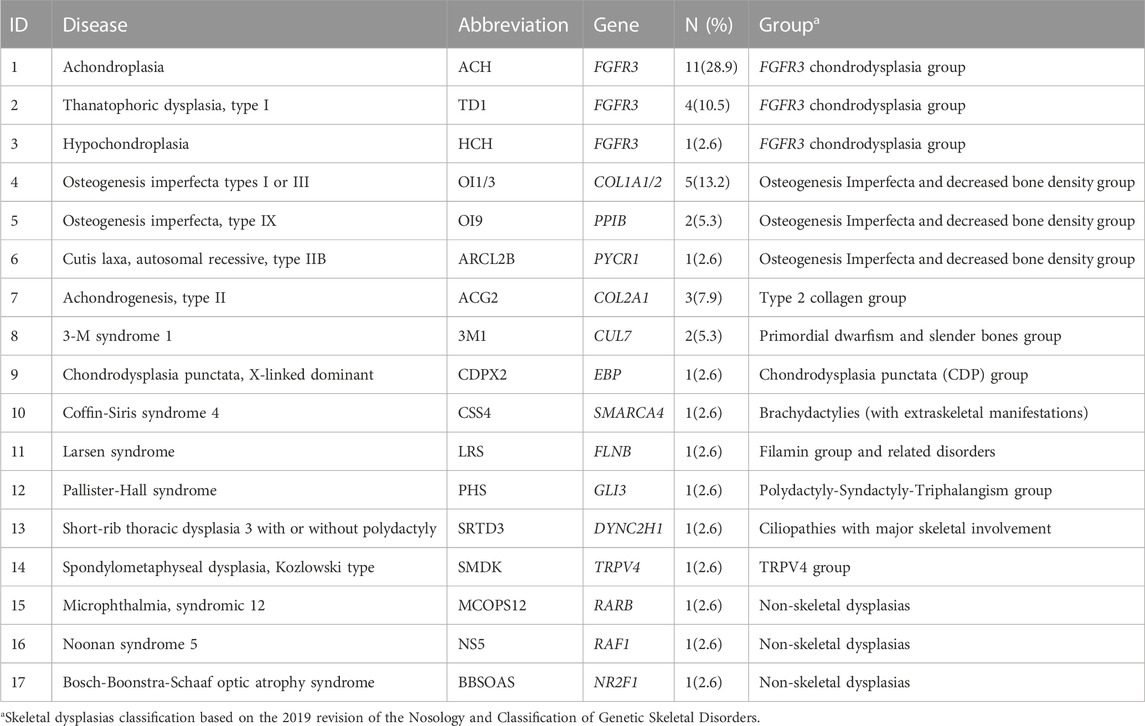
TABLE 3. Etiologic diagnosis and categories for 38 fetuses with genetic disorders.
3.4 Variant analysis
Of the 43 fetuses diagnosed by ES, 31 with an autosomal dominant (AD) inheritance pattern, 10 with an autosomal recessive (AR) pattern and 2 with an X-linked pattern. Thirty-eight variants were identified in 20 different genes, including 26 missense variants, 6 non-sense variants, 2 frameshift variants, 3 in-frame ins/del variants, and 1 affects splice-site. Additionally, 31 (72.1%) variants were identified to be de novo, 7 (16.3%) variants were compound heterozygous inherited from the parents, three (7.0%) were homozygous mutations inherited from the parents, and two (4.7%) arose as a result of maternal mutation. Furthermore, fourteen (36.8%) novel SLBs-related variants were identified and analyzed in the present study (Table 4).
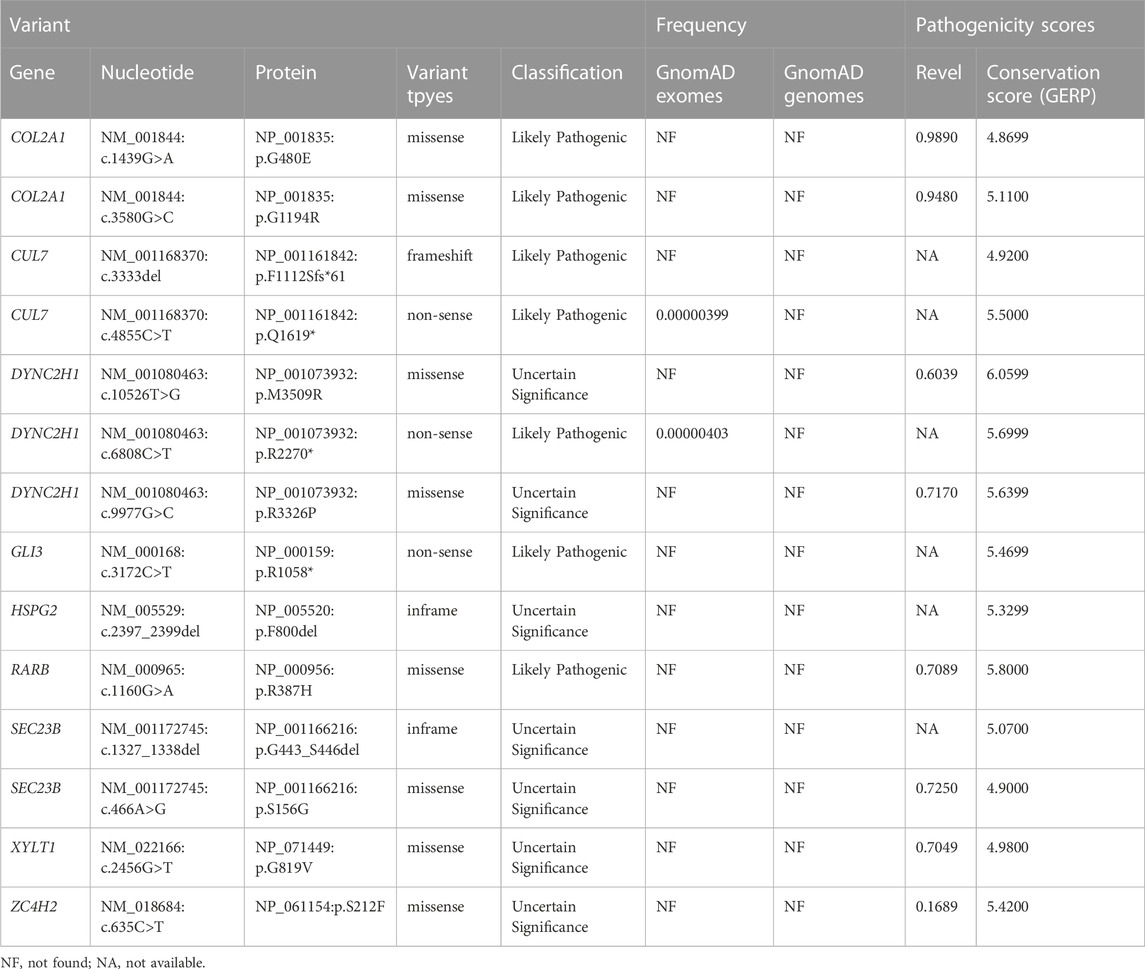
TABLE 4. Analysis of novel variants identified in this study.
3.5 Follow-up assessments
Clinical follow-up assessments were able to be performed in 91 cases (96.8%). Of 38 diagnosed cases, 37 fetuses underwent termination of pregnancy (TOP), and one infant (Case 35) with genetic abnormality died 2 months after birth. Among the fetuses of TOP, seven fetuses were autopsied and pathologically confirmed (Table 1). In addition, for the 5 cases with variants of uncertain significance, three cases with a specific diagnosis suspected or fetal FL significantly short (−3.9 SDs) underwent TOP, one case (Case 41) was healthy without clinical defects at the age of 3 years, and the one case refused follow up. Of the 51 fetuses with no prenatally detected genetic abnormalities, 5 were of incomplete follow-up data and 46 had outcome data ascertained. Twenty-three of 51 fetuses with multi-system abnormalities or with FL significantly short but without identified genetic abnormalities underwent TOP. Sixteen cases (16/46, 34.8%) were healthy and of no short stature at the age of follow-up. Seven (7/46, 15.2%) cases were followed up with clinical manifestations such as hypertonia, hypospadias, bilateral ventriculomegaly, bilateral talipes equinovarus, aortic stenosis, etc., (Table 5). One case was stillbirth with maternal gestational hypertension. Furthermore, among the fetuses without genetic abnormalities, the rate of TOP was significantly lower in FL 2-4 SDs below GA group compared with FL below −4 SDs group (36.8% [14/38] vs. 75.0% [9/12], p = 0.021). 6 of 20 children in FL 2–4SDs below GA group were associated with clinical abnormalities at the age of follow-up, while 1 out of 3 children was found to have clinical abnormalities in FL below -4SDs group (Table 5).
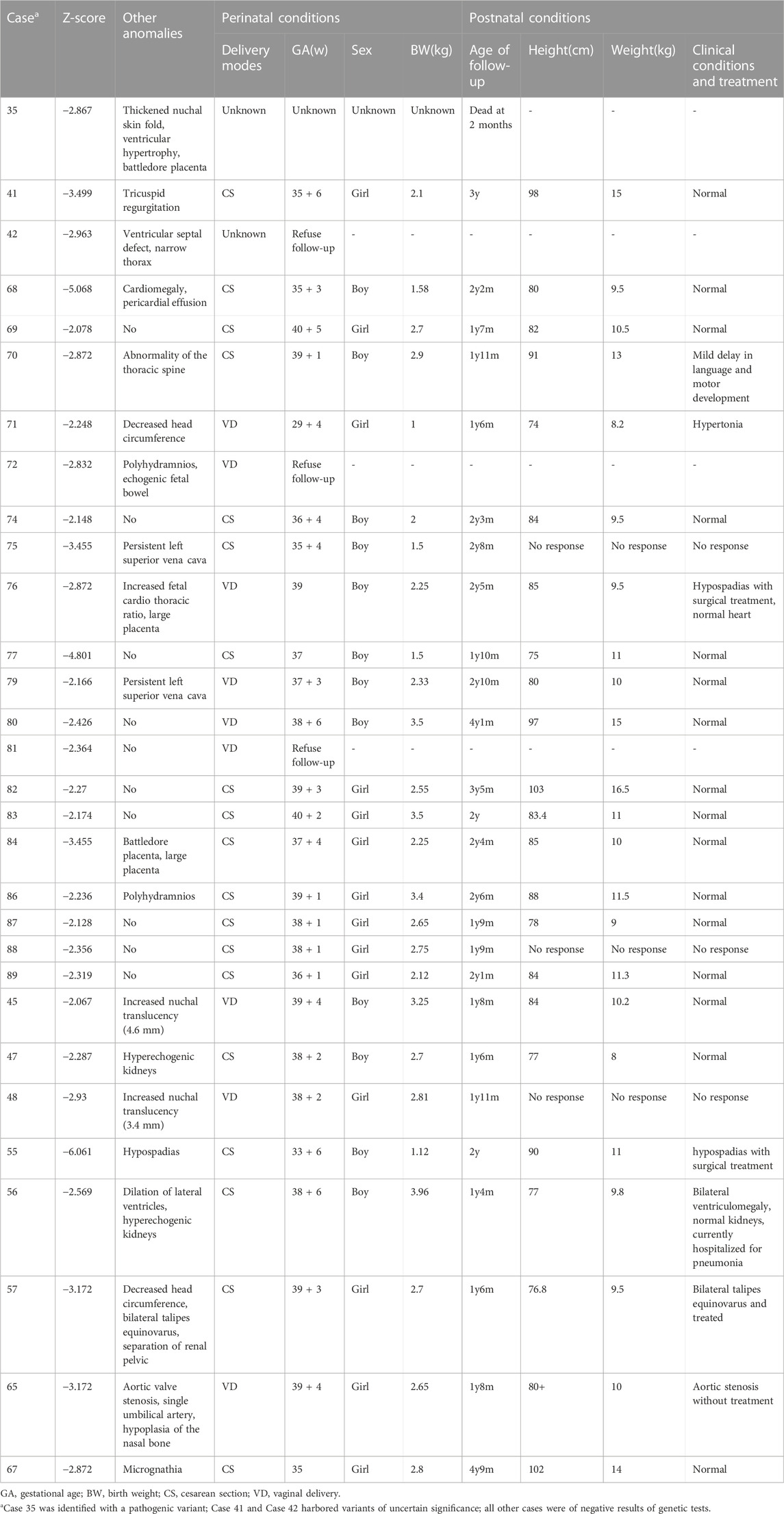
TABLE 5. Characteristics of clinically significant perinatal and postnatal conditions in 30 liveborns cases in whom follow up was ascertained.
4 Discussion
Fetal short long bones bring diagnostic challenges for clinicians. Although in most cases, it may be constitutional, it is also recognized as a marker of chromosomal abnormalities or skeletal dysplasias. The vast majority of skeletal dysplasias are associated with monogenic abnormalities (Snijders et al., 2000; Vermeer and Bekker, 2013; Li et al., 2021). We performed a cohort study to identify the genetic causes for fetal SLBs and retrospectively evaluate the additional diagnostic yield of ES after a negative result of CMA or karyotype analysis. Besides of genetic diagnosis, we also performed long-term follow-ups to assess the prognoses and outcomes in fetuses with SLBs.
Prenatal ES identified well-described genetic causes in 38 out of 94 cases (40.4%), and VUS in 5 cases, rendering a total diagnostic yield of 45.74%. Compared with our findings, three recent cohort studies reported a significantly higher diagnostic rate by prenatal ES, which was 80% (12 out of 15), 85% (11 out of 13), and 70% (21 out of 30), respectively (Liu et al., 2019; Peng et al., 2021; Tang et al., 2021). Many aspects could affect the detection rate of ES, such as the selected criteria of the study, the number of cases, proband-only or trio ES, and so on (Tang et al., 2021). The number of cases in previous studies were comparatively small (≤30 cases), and small study number may cause relatively high selected bias. In some previous studies, fetal growth restriction and some non-genetic causes were excluded, and may also bring in selected bias. It was reported that trio-ES had a higher diagnostic rate; fetuses with multiple anomalies also had a higher diagnostic rate, and when testing single structural anomalies, a particular organ system may have a higher diagnostic yield (Petrovski et al., 2019). In our study, ES yielded 27.3% (6 out of 22) in the isolated SLBs group and 44.4% (32 out of 72) in the non-isolated SLBs group. For fetuses with SLBs and other skeletal abnormalities (femur fracture, femoral bowing, spinal dysplasia, bilateral talipes equinovarus, et al.), the diagnostic rate was 59.4% (19 out of 32), and for fetuses with SLBs and non-skeletal abnormalities (increased nuchal translucency, tricuspid regurgitation, cerebellar dysplasia, polyhydramnios, et al.) the diagnostic rate was 32.5% (13 out of 40). The finding is consistent with previous studies, suggesting that fetuses with SLBs and other skeletal abnormalities were the most common associations with skeletal dysplasias (Papageorghiou et al., 2008). Therefore, when prenatal ultrasound scans show that the fetal femur is 2 or more SDs below the normal range, the length of the other long bones should be measured and a thorough skeletal assessment should be performed to determine whether there are other skeletal morphological abnormalities (Liu et al., 2019).
Moreover, diverse degree of femoral shortening may confer different associated genetic risks (Li et al., 2021). Of the 94 fetuses that underwent ES, 54 fetuses had Z-scores between −2 SDs and −4 SDs, and 40 fetuses had Z-scores below −4 SDs. The diagnostic rate was 16.7% (9 out of 54) in fetuses with Z-scores between −2 SDs and −4 SDs, and 72.50% (29 out of 40) in fetuses with Z-scores below −4 SDs. In the present study, the degree of femoral shortening is correlated with the rate of adverse pregnancy outcomes. In fetuses with FL below −4 SDs, the abnormal pregnancy outcomes were significantly higher. Therefore, prenatal ES can be used as an effective clinical diagnostic platform for fetuses with short long bones, especially in those with multiple congenital anomalies or severely short limbs.
In the present study, 38 variants affecting 20 genes including FGFR3, CUL7, COL1A2, COL2A1, COL1A1, DYNC2H1, PPIB, PYCR1, EBP, HSPG2, FLNB, GLI3, NR2F1, RAF1, RARB, SEC23B, SMARCA4, TRPV4, XYLT1, and ZC4H2 were found to be associated with monogenic disorders. Of 38 diagnosed cases, 92.1% (35/38) were diagnosed as genetic skeletal dysplasias, including 14 different diseases that are classified into 10 groups according to the 2019 revision of the Nosology and Classification of Genetic Skeletal Disorders (Mortier et al., 2019). Among the fetuses with genetic skeletal dysplasias, 42.1% (16/38) contained variants in FGFR3, which were categorized into FGFR3 chondrodysplasia group based on the Nosology and Classification of Genetic Skeletal Disorders. Moreover, 13.2% (5/38) of fetuses contained variants in COL1A1/COL1A2, 5.3% (2/38) contained variants in PPIB, and one fetus contained variants in PYCR1. These genes were categorized into the group of osteogenesis imperfecta and decreased bone density. In addition, 7.9% (3/38) of the fetuses contained variants in COL2A1 that were diagnosed as achondrogenesis, type II. 5.3% (2/38) of the fetuses harbored variants in CUL7 diagnosed with 3-M syndrome 1, which would lead to primordial dwarfism. These findings are in line with the high genetic variability of fetal SLBs, and also highlights that pathogenic variants in FGFR3 and collagen genes are the most common genetic causes for SLBs, as well as for skeletal dysplasias.
Additionally, fourteen variants identified in this study were novel. Novel pathogenic variants identified may broaden the mutation spectrum for the disorders and contributes to clinical consultation and subsequent pregnancy examination.
Fibroblast growth factor receptor 3 (FGFR3) is one of four distinct membrane-spanning tyrosine kinases that serve as high-affinity receptors for a number of fibroblast growth factors and play essential roles in skeletal development (Ornitz et al., 2015; Peng et al., 2021). Variants in FGFR3 have been associated with at least 10 different skeletal disorders (Mortier et al., 2019; Peng et al., 2021). In the present study, 11 cases (Case 13–14, Case 16–18, Case 20–22, Case 24–25, Case 28) had a hotspot mutation, c.1138G>A in FGFR3, that would lead to achondroplasia (MIM_100,800) (Hafner et al., 2006; Natacci et al., 2008). Two cases (Case 23, Case 27) carried an identical variant c.742C>T in FGFR3 gene, which has been detected in different populations (Chen et al., 2001; Tonni et al., 2010; Tang et al., 2021). The variant c.746C>G and c.1118A>G in FGFR3 were identified in one case each (Case 15, Case 26). Variants c.742C>T, c.746C>G, and c.1118A>G have been associated with thanatophoric dysplasia, type I (MIM_187,600) (Brodie et al., 1999). Case 19 harbored a variant c.1620C>A (p.N540K) in the TK-1 domain of FGFR3, which is a common cause of hypochondroplasia (MIM_146,000) (Mortier et al., 2019).
Mutations in the type I collagen coding genes (COL1A1 and COL1A2) affecting collagen quantity or structure count for approximately 85% of osteogenesis imperfecta (OI) cases (Marini et al., 2017; Tournis and Dede, 2018; Peng et al., 2021). The helical domains contain Gly-X-Y triplets where glycine substitutions are the most frequent cause of OI (Marini et al., 2017; Peng et al., 2021). In five cases of the present study, variant c.3118G>A, COL1A2 c.3277G>A, c.3034G>A and c.1801G>A in COL1A1 were identified, causing a glycine in the helical domain of type 1 collagen was substituted. These variants may disrupt type 1 collagen functions, and therefore lead to skeletal dysplasias. Besides, Case 1 harbored a variant c.957 + 1G>T in COL1A1, that was predicted to disrupt splicing by SpliceAI. COL2A1 gene encodes type II collagen that plays a role in the regulation of intramembranous and endochondral ossification. Heterozygous COL2A1 variants are associated with a spectrum of dwarfism and skeletal malformation diseases (Zhang R et al., 2020). In the present study, 2 novel likely pathogenic mutations (c.1439G>A and c.3580 G>C) and 1 known likely pathogenic mutation (c. 3472G>A) were identified in COL2A1 (Table 1). These findings further indicate the high genetic variability of fetal SLBs and also highlight the importance of molecular diagnosis for fetuses with SLBs.
In the present study, we performed a long-term follow-up for up to 5 years after birth. The pregnancy outcomes for fetuses with FL 2- 4 SDs below GA were significantly better than those with FL below −4 SDs. Consistent with previous studies, the severe femoral shortening was the main reason for TOP in our cohort with fetal SLBs (Li et al., 2021). Fifty-six cases in the present study did not have a definitive molecular diagnosis, and 12 of them (21.4%) with Z-scores below −4SDs. The negative ES results could be attributed to limited phenotypes or the limitations of ES (Tang et al., 2021). For prenatal ES, several aspects could affect the detection rate, especially incomplete prenatal phenotypes (Aarabi et al., 2018; Tang et al., 2021). Meanwhile, many aspects should be considered, such as ethical concerns, analysis of variants of unknown significance, and secondary findings (Best et al., 2018; Monaghan et al., 2020; Tang et al., 2021). In the future, with the development of fetal-specific phenotype-genotype database, and increased studies on the molecular mechanisms of genes associated with SLBs, the uncertainty in cases will become less frequent (Aarabi et al., 2018; Tang et al., 2021). Furthermore, the causative variants may reside in the non-coding-regulatory or deep-intronic regions, and may not be detected by ES, but could be identified by whole genome sequencing in the future (Tang et al., 2021). Additionally, our study has some limitations. For instance, this is a single-center study with a relatively small case number and selected population. Prospective multicenter studies with large sample sizes are needed to obtain more reliable data (Li et al., 2021). In some cases, telephone follow-up may induce recall bias (Peng et al., 2021).
5 Conclusion
Prenatal exome sequencing analysis facilitates genetic diagnosis and improves the management of pregnancies with fetal short long bones detected by ultrasonography especially in those with multiple congenital anomalies or severely short limbs. Additionally, novel pathogenic variants identified may broaden the mutation spectrum for the disorders and contributes to clinical consultation and subsequent pregnancy examination.
Data availability statement
The datasets presented in this study have been uploaded in online repositories. The names of the repository/repositories and accession number(s) can be found below: Genome Sequence Archive (Genomics, Proteomics, and Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2021), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA003147).
Ethics statement
The studies involving human participants were reviewed and approved by Medical Ethics Committee of Guangdong Women and Children Hospital (IRB reference number: 202001209). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YH, CL, YZ, and AY conceived and designed the study. YH, CL, HD, YW, LY, FG, FL, and XS performed the genetic diagnosis. YH analyzed and interpreted data. YH and CL drafted the manuscript, and YZ and AY revised it. All authors read and approved the final manuscript.
Funding
This study was supported by the Medical Science and Technology Research Project of Guangdong Province (Grant No. B2021258) and the Guangzhou Basic and Applied Basic Research Funding Project (Grant Nos. 202102080358, 202102080015, and 202102080263). The funding did not play role in the design of the study, collection, analysis, interpretation of data, or in writing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1032346/full#supplementary-material
Abbreviations
SLBs, short long bones; SDs, standard deviations; CMA, chromosomal microarray analysis; NGS, next-generation sequencing; FL, femur length; MAF, minor allele frequency; CNV, Copy number variations; AD, autosomal dominant; AR, autosomal recessive; VUS, Variants of uncertain clinical significance.
References
Aarabi, M., Sniezek, O., Jiang, H., Saller, D. N., Bellissimo, D., Yatsenko, S. A., et al. (2018). Importance of complete phenotyping in prenatal whole exome sequencing. Hum. Genet. 137, 175–181. doi:10.1007/s00439-017-1860-1
Abou, T. A., Pesaran, T., Distefano, M. T., Oza, A., Rehm, H. L., Biesecker, L. G., et al. (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524. doi:10.1002/humu.23626
Aviram, A., Bardin, R., Wiznitzer, A., Yogev, Y., and Hadar, E. (2015). Midtrimester isolated short femur length as a predictor of adverse pregnancy outcome. Fetal Diagn Ther. 38, 205–211. doi:10.1159/000375446
Beke, A., Papp, C., Toth-Pal, E., Mezei, G., Joo, J. G., Csaba, A., et al. (2005). Trisomies and other chromosome abnormalities detected after positive sonographic findings. J. Reprod. Med. 50, 675–691.
Best, S., Wou, K., Vora, N., Van der Veyver, I. B., Wapner, R., and Chitty, L. S. (2018). Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat. Diagn 38, 10–19. doi:10.1002/pd.5102
Brnich, S. E., Abou, T. A., Couch, F. J., Cutting, G. R., Greenblatt, M. S., Heinen, C. D., et al. (2019). Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 12, 3. doi:10.1186/s13073-019-0690-2
Brodie, S. G., Kitoh, H., Lachman, R. S., Nolasco, L. M., Mekikian, P. B., and Wilcox, W. R. (1999). Platyspondylic lethal skeletal dysplasia, San Diego type, is caused by FGFR3 mutations. Am. J. Med. Genet. 84, 476–480. doi:10.1002/(sici)1096-8628(19990611)84:5<476:aid-ajmg12>3.0.co;2-x
Chen, C. P., Chern, S. R., Wang, W., and Wang, T. Y. (2001). Second-trimester molecular diagnosis of a heterozygous 742-> T (R248C) mutation in the FGFR3 gene in a thanatophoric dysplasia variant following suspicious ultrasound findings. Ultrasound Obstet. Gynecol. 17, 272–273. doi:10.1046/j.1469-0705.2001.00377.x
Dai, Y., Liang, S., Dong, X., Zhao, Y., Ren, H., Guan, Y., et al. (2019). Whole exome sequencing identified a novel DAG1 mutation in a patient with rare, mild and late age of onset muscular dystrophy-dystroglycanopathy. J. Cell. Mol. Med. 23, 811–818. doi:10.1111/jcmm.13979
Gaudry, P., Esvan, M., Meynard, J. B., Lebbar, A., Choiset, A., Lewin, F., et al. (2012). Amniocentesis performed for karyotyping after identified ultrasonographic abnormalities: What to expect? Fetal Diagn Ther. 31, 55–62. doi:10.1159/000330373
Ghosh, R., Harrison, S. M., Rehm, H. L., Plon, S. E., Biesecker, L. G., et al. (2018). Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum. Mutat. 39 (11), 1525–1530. doi:10.1002/humu.23642
Hafner, C., van Oers, J. M., Vogt, T., Landthaler, M., Stoehr, R., Blaszyk, H., et al. (2006). Mosaicism of activating FGFR3 mutations in human skin causes epidermal nevi. J. Clin. Invest. 116, 2201–2207. doi:10.1172/JCI28163
Han, P., Wei, G., Cai, K., Xiang, X., Deng, W. P., Li, Y. B., et al. (2020). Identification and functional characterization of mutations in LPL gene causing severe hypertriglyceridaemia and acute pancreatitis. J. Cell. Mol. Med. 24, 1286–1299. doi:10.1111/jcmm.14768
Hu, T., Tian, T., Zhang, Z., Wang, J., Hu, R., Xiao, L., et al. (2021). Prenatal chromosomal microarray analysis in 2466 fetuses with ultrasonographic soft markers: A prospective cohort study. Am. J. Obstet. Gynecol. 224, 516.e1–516.e16. doi:10.1016/j.ajog.2020.10.039
Jaganathan, K., Kyriazopoulou, P. S., Mcrae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., et al. (2019). Predicting splicing from primary sequence with deep learning. Cell. 176, 535–548.e24. doi:10.1016/j.cell.2018.12.015
Kaijomaa, M., Ulander, V. M., Ryynanen, M., and Stefanovic, V. (2016). Risk of adverse outcomes in euploid pregnancies with isolated short fetal femur and humerus on second-trimester sonography. J. Ultrasound Med. 35, 2675–2680. doi:10.7863/ultra.16.01086
Kurtz, A. B., Filly, R. A., Wapner, R. J., Golbus, M. S., Rifkin, M. R., Callen, P. W., et al. (1986). In utero analysis of heterozygous achondroplasia: Variable time of onset as detected by femur length measurements. J. Ultrasound Med. 5, 137–140. doi:10.7863/jum.1986.5.3.137
Li, Q., Zhang, Z., Wang, J., Zhang, H., Zhu, H., Lai, Y., et al. (2021). Prenatal diagnosis of genetic aberrations in fetuses with short femur detected by ultrasound: A prospective cohort study. Prenat. Diagn 41, 1153–1163. doi:10.1002/pd.6006
Liu, J., Huang, L., He, Z., Lin, S., Wang, Y., and Luo, Y. (2019). Clinical value of genetic analysis in prenatal diagnosis of short femur. Mol. Genet. Genomic Med. 7, e978. doi:10.1002/mgg3.978
Liu, C., Huang, Y., Zhang, Y., Ding, H., Yu, L., Wang, A., et al. (2022). Next-generation sequencing facilitates genetic diagnosis and improves the management of patients with hearing loss in clinical practice. Int. J. Pediatr. Otorhinolaryngol. 161, 111258. doi:10.1016/j.ijporl.2022.111258
Lord, J., Mcmullan, D. J., Eberhardt, R. Y., Rinck, G., Hamilton, S. J., Quinlan-Jones, E., et al. (2019). Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): A cohort study. Lancet 393, 747–757. doi:10.1016/S0140-6736(18)31940-8
Marini, J. C., Forlino, A., Bachinger, H. P., Bishop, N. J., Byers, P. H., Paepe, A., et al. (2017). Osteogenesis imperfecta. Nat. Rev. Dis. Prim. 3, 17052. doi:10.1038/nrdp.2017.52
Mohr-Sasson, A., Toussia-Cohen, S., Shapira, M., Perlman, S., Achiron, R., and Gilboa, Y. (2020). Long-term follow-up on fetuses with isolated sonographic finding of short long bones: A cohort study. Arch. Gynecol. Obstet. 301, 459–463. doi:10.1007/s00404-019-05421-4
Monaghan, K. G., Leach, N. T., Pekarek, D., Prasad, P., and Rose, N. C. (2020). The use of fetal exome sequencing in prenatal diagnosis: A points to consider document of the American College of medical genetics and genomics (ACMG). Genet. Med. 22, 675–680. doi:10.1038/s41436-019-0731-7
Mortier, G. R., Cohn, D. H., Cormier-Daire, V., Hall, C., Krakow, D., Mundlos, S., et al. (2019). Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 179, 2393–2419. doi:10.1002/ajmg.a.61366
Natacci, F., Baffico, M., Cavallari, U., Bedeschi, M. F., Mura, I., Paffoni, A., et al. (2008). Germline mosaicism in achondroplasia detected in sperm DNA of the father of three affected sibs. Am. J. Med. Genet. A 146A, 784–786. doi:10.1002/ajmg.a.32228
Ornitz, D. M., and Marie, P. J. (2015). Fibroblast growth factor signaling in skeletal development and disease. Genes. Dev. 29, 1463–1486. doi:10.1101/gad.266551.115
Papageorghiou, A. T., Fratelli, N., Leslie, K., Bhide, A., and Thilaganathan, B. (2008). Outcome of fetuses with antenatally diagnosed short femur. Ultrasound Obstet. Gynecol. 31, 507–511. doi:10.1002/uog.5265
Patel, M. D., and Filly, R. A. (1995). Homozygous achondroplasia: US distinction between homozygous, heterozygous, and unaffected fetuses in the second trimester. Radiology 196, 541–545. doi:10.1148/radiology.196.2.7617874
Peng, Y., Yang, S., Huang, X., Pang, J., Liu, J., Hu, J., et al. (2021). Whole exome sequencing analysis in fetal skeletal dysplasia detected by ultrasonography: An analysis of 38 cases. Front. Genet. 12, 728544. doi:10.3389/fgene.2021.728544
Petrovski, S., Aggarwal, V., Giordano, J. L., Stosic, M., Wou, K., Bier, L., et al. (2019). Whole-exome sequencing in the evaluation of fetal structural anomalies: A prospective cohort study. Lancet 393, 758–767. doi:10.1016/S0140-6736(18)32042-7
Ramachandrappa, S., Kulkarni, A., Gandhi, H., Ellis, C., Hutt, R., Roberts, L., et al. (2018). SHOX haploinsufficiency presenting with isolated short long bones in the second and third trimester. Eur. J. Hum. Genet. 26, 350–358. doi:10.1038/s41431-017-0080-4
Rao, R., and Platt, L. D. (2016). Ultrasound screening: Status of markers and efficacy of screening for structural abnormalities. Semin. Perinatol. 40, 67–78. doi:10.1053/j.semperi.2015.11.009
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Salomon, L. J., Alfirevic, Z., Da, S. C. F., Deter, R. L., Figueras, F., Ghi, T., et al. (2019). ISUOG practice guidelines: Ultrasound assessment of fetal biometry and growth. Ultrasound Obstet. Gynecol. 53, 715–723. doi:10.1002/uog.20272
Sananes, N., Guigue, V., Kohler, M., Bouffet, N., Cancellier, M., Hornecker, F., et al. (2009). Use of Z-scores to select a fetal biometric reference curve. Ultrasound Obstet. Gynecol. 34, 404–409. doi:10.1002/uog.6439
Snijders, R. J., Platt, L. D., Greene, N., Carlson, D., Krakow, D., Gregory, K., et al. (2000). Femur length and trisomy 21: Impact of gestational age on screening efficiency. Ultrasound Obstet. Gynecol. 16, 142–145. doi:10.1046/j.1469-0705.2000.00198.x
Speer, P. D., Canavan, T., Simhan, H. N., and Hill, L. M. (2014). Prenatal midtrimester fetal long bone measurements and the prediction of small-for-gestational-age fetuses at term. Am. J. Perinatol. 31, 231–236. doi:10.1055/s-0033-1345260
Tang, H., Zhang, Q., Xiang, J., Yin, L., Wang, J., and Wang, T. (2021). Whole exome sequencing aids the diagnosis of fetal skeletal dysplasia. Front. Genet. 12, 599863. doi:10.3389/fgene.2021.599863
Tonni, G., Azzoni, D., Ventura, A., Ferrari, B., Felice, C. D., and Baldi, M. (2010). Thanatophoric dysplasia type I associated with increased nuchal translucency in the first trimester: Early prenatal diagnosis using combined ultrasonography and molecular biology. Fetal Pediatr. Pathol. 29, 314–322. doi:10.3109/15513811003796938
Tournis, S., and Dede, A. D. (2018). Osteogenesis imperfecta - a clinical update. Metabolism 80, 27–37. doi:10.1016/j.metabol.2017.06.001
Tzadikevitch, G. K., Singer, A., Maya, I., Sagi-Dain, L., Khayat, M., Ben-Shachar, S., et al. (2021). Chromosomal microarray should be performed for cases of fetal short long bones detected prenatally. Arch. Gynecol. Obstet. 303, 85–92. doi:10.1007/s00404-020-05729-6
Vermeer, N., and Bekker, M. N. (2013). Association of isolated short fetal femur with intrauterine growth restriction. Prenat. Diagn 33, 365–370. doi:10.1002/pd.4068
Victoria, T., Zhu, X., Lachman, R., Epelman, M., Oliver, E. R., Adzick, N. S., et al. (2018). What is new in prenatal skeletal dysplasias? AJR Am. J. Roentgenol. 210, 1022–1033. doi:10.2214/AJR.17.19337
Weisz, B., David, A. L., Chitty, L., Peebles, D., Pandya, P., Patel, P., et al. (2008). Association of isolated short femur in the mid-trimester fetus with perinatal outcome. Ultrasound Obstet. Gynecol. 31, 512–516. doi:10.1002/uog.5349
Zalel, Y., Lehavi, O., Schiff, E., Shalmon, B., Cohen, S., Schulman, A., et al. (2002). Shortened fetal long bones: A possible in utero manifestation of placental function. Prenat. Diagn 22, 553–557. doi:10.1002/pd.364
Zhang, B., Zhang, Y., Wu, N., Li, J., Liu, H., and Wang, J. (2020). Integrated analysis of COL2A1 variant data and classification of type II collagenopathies. Clin. Genet. 97, 383–395. doi:10.1111/cge.13680
Zhang, R., Chen, S., Han, P., Chen, F., Kuang, S., Meng, Z., et al. (2020). Whole exome sequencing identified a homozygous novel variant in CEP290 gene causes Meckel syndrome. J. Cell. Mol. Med. 24, 1906–1916. doi:10.1111/jcmm.14887
Keywords: fetal short long bones, skeletal dysplasia, exome sequencing, ultrasound examination, prenatal diagnosis
Citation: Huang Y, Liu C, Ding H, Wang Y, Yu L, Guo F, Li F, Shi X, Zhang Y and Yin A (2023) Exome sequencing in fetuses with short long bones detected by ultrasonography: A retrospective cohort study. Front. Genet. 14:1032346. doi: 10.3389/fgene.2023.1032346
Received: 30 August 2022; Accepted: 16 February 2023;
Published: 27 February 2023.
Edited by:
Sigve Nakken, Oslo University Hospital, NorwayReviewed by:
Li Zhuo, Central South University, ChinaSantasree Banerjee, Beijing Genomics Institute (BGI), China
Copyright © 2023 Huang, Liu, Ding, Wang, Yu, Guo, Li, Shi, Zhang and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aihua Yin, yinaihua0131@163.com; Yan Zhang, zhangyan1981_2003@aliyun.com
†These authors have contributed equally to this work