
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 13 May 2022
Sec. Human and Medical Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.878063
This article is part of the Research TopicFetal Phenotypes of Rare Diseases: Application and Evaluation of Prenatal Exome Sequencing and Pathogenesis Research of Rare DiseasesView all 5 articles
 Jingjing Liu1,2
Jingjing Liu1,2 Yuduo Wu1,2
Yuduo Wu1,2 Hairui Sun1,2
Hairui Sun1,2 Xiaowei Liu1,2Xiaoyan Gu1,2Ying Zhao1,2Ye Zhang1,2
Xiaowei Liu1,2Xiaoyan Gu1,2Ying Zhao1,2Ye Zhang1,2 Jiancheng Han1,2*
Jiancheng Han1,2* Yihua He1,2*
Yihua He1,2*Objective: This study aims to characterize the abnormal changes in placental DNA methylation associated with conotruncal heart defects (CTDs) and the level of methylation as epigenetic biomarkers for CTDs detection.
Methods: This was a prospective study involving 28 fetuses diagnosed with CTDs in the second trimester at Beijing Anzhen Hospital between September 2020 and June 2021. These cases were classified into four groups based on their subtypes. 12 normal fetuses were used as controls. Placental tissue was obtained after inducing labor in fetuses. To identify differential methylation sites (DMSs) and regions (DMRs) in cases vs. controls, an Infinium Human Methylation 850 k bead chip was used. Differential methylation was assessed by comparing the β-values for individual CpG loci. Based on the p-value (<0.05), the most discriminating CpG sites were identified. The area under the receiver-operating-characteristics curve (AUC) was used to determine the predictive accuracy of CpG loci with significant methylation changes for CTDs. The function of genes was assessed through KEGG enrichment analysis, Gene Ontology (GO) analysis, and KEGG pathway analysis.
Results: In comparison to the control group, the DNA methylation of the placental tissue is significantly different in fetuses with CTDs. We identified the most significantly different methylated loci and they demonstrated excellent individual predictive accuracy for CTDs detection with AUC >0.9 in cases compared with controls. HOXD9, CNN1, NOTCH1, and ECE1 were identified as CTDs-detection candidate genes.
Conclusion Our study established the abnormal changes in placental methylation associated with CTDs and potential epigenetic biomarkers for CTDs detection.
Conotruncal heart defects (CTDs) are a diverse subtype of congenital heart diseases (CHDs) that account for approximately 25–30% of all non-syndromic CHDs. Although fetuses with CTDs typically require catheter-based or surgical treatment in the early postnatal period, the mortality rate remains high. (Bouma and Mulder, 2017). The diagnosis of CTDs at an early stage is critical for postnatal outcomes. (Holland et al., 2015; Eckersley et al., 2016; Li et al., 2016). The detection rate of prenatal CTDs has increased as screening technology has improved. There is still a lack of accurate biomarkers that can be used in addition to echocardiography to improve the detection of CTDs.
Currently, there are known factors involved in the pathogenesis of CTDs, including the genetic factors associated with cardiac development genes such as GATA4, GATA6, TBX18, TBX20, NKX2-5, and SAMDS, as well as unique copy mutations and chromosomal aneuploidy such as trisomy 13, 18, or 21, which account for 20–30% of CHDs. (Pierpont et al., 2018).
The causes of CTDs are complex; current research indicates that environmental, genetic, and epigenetic factors all contribute to the development of malformations. (Blue et al., 2012). The pathogenesis is unknown in the majority of CTDs. Methylation induces a change in the three-dimensional structure of DNA and has been shown to suppress gene transcription in the past. DNA methylation modifications may be critical in the development of CTDs such as in Tetralogy of Fallot (TOF). (Marín-García, 2009; Csáky-Szunyogh et al., 2013; and Serra-Juhé et al., 2015).
The placenta and the heart are the two organs that develop most rapidly during pregnancy. According to their developmental connections, a phenomenon called the “placenta-heart axis” has been proposed. (Maslen, 2018). Cardiovascular malformations are associated with poor trophoblast invasion and reduced transfer of nutrients to the fetus as a result of placental abnormality. (Linask, 2013). Recently, the “placenta-heart axis” has gained a considerable research attention. Moreover, studies (Radhakrishna et al., 2019 and Bahado Singh et al., 2020) have shown that the placental DNA in CHD contains a specific methylation region that is closely related to the type and occurrence of CHD and may serve as a biomarker for its detection. The purpose of this study is to characterize the abnormal changes in placental DNA methylation associated with CTDs and to assess the utility of placental DNA methylation as an epigenetic biomarker for the prediction of CTDs.
Between September 2020 and June 2021, this prospective study enrolled 40 fetuses diagnosed with CTDs at Beijing Anzhen Hospital. Twelve fetuses were excluded due to chromosomal abnormalities. The medical Ethics Committee approved this study (note: GZR-2-006). Fetuses were classified according to the subtypes of CTDs which included TOF, double outlet right ventricle (DORV), transposition of the great artery (TGA), and pulmonary atresia with ventricular septum defect (PAVSD). All pregnant women consented to participate and signed an informed consent form. After inducing labor in fetuses, placental tissue was obtained. To ensure consistency in sampling, the placental tissue with the umbilical cord insertion part was chosen. Until DNA purification, the separated tissue blocks were stored in a refrigerator at minus 80°C. To avoid the effects of genetic malformations, Gene whole exon sequencing (GWES) analysis and copy number variation (CNV) detection were applied in all fetuses. We excluded fetuses with known or suspected genetic syndromes. As controls, 12 labor-induced fetuses with extracardiac anatomical abnormalities but no syndromic defects were used. Other obstetric complications were not noted.
QIAamp Mini Kit was used for the purification of DNA from placental tissues according to the manufacturer’s protocol. Genome-wide DNA methylation was determined using the Illumina Infinium Human Methylation 850K BeadChip (Illumina Inc., California, United States), which provides genome-wide coverage with >850000 CpG methylation sites per sample according to the manufacturer’s instructions. It is capable of producing high-quality data for DNA extracted from the samples. The genomic DNA from both the cases and the controls was subjected to bisulfite-conversion according to the manufacturers protocol using the Zymo EZ DNA Methylation kit (Zymo Research, Irvine, CA, United States). The fluorescence signals from the BeadArrays were measured using an Illumina ‘iScan’ and then analyzed using the Genome Studio software (Illumina, Inc.). The software assigned β-values to CpG probes based on the ratio of the methylated alleles (C) fluorescence signal to the sum of the methylated (C) and unmethylated (T) alleles’ fluorescence signals. Before performing detailed bioinformatics and statistical analysis, data preprocessing and quality control were performed, including examination of the background signal intensity in the affected cases and negative controls, the methylated and unmethylated signals, and the ratio of methylated and unmethylated signal intensities. The processing was carried out entirely according to the manufacturer’s protocol, and 99% of the CpG loci were unequivocally determined. The methylation status of all probes was denoted by the β-value, which is the ratio of the methylated probe intensity to the total probe intensity (sum of methylated and unmethylated probe intensities plus constant α, where α = 100). Between the two groups, the average β-values of the promoters were compared.
Locus quality control requires that detection p < 0.05 and Beadcount are not less than three in more than 95% of individuals. Meanwhile, loci located on X, Y chromosomes, and SNP should be removed. Individual quality control requires detection p < 0.05 in more than 95% loci. Based on the above quality control filtered data, BMIQ (Beta-mixture quantile normalization) was used to correct probe type bias to obtain the methylation level (β value) that could be used for different analyses.
We determined differential methylation by comparing the β-value of individual CpG loci in two groups. Based on the p-value (<0.05), the most discriminating CpG sites were identified. The R-package Limma was used to analyze the differential methylation sites, and the moderated T-statistics and empirical Bayes methods were used to test the significance of the differential sites in Limma. Bumphunters method was used to find differential methylation regions. The method first clustered according to site distance information, and then selected continuous candidate regions with consistent methylation levels in each cluster. Bumphunter constructed a linear statistical model according to candidate regions and filtered the regions with significant differences (p < 0.05). The methylation level, or β-value, of each CpG locus (one per gene), was used to calculate its predictive accuracy for CTDs and its subtype’s detection on an individual basis. The area under the receiver-operating characteristics curve was used to determine the predictive accuracy (AUC). Continuous variables are expressed as the mean ± standard deviation values. Normally distributed data were analyzed by using student’s t-test. A p value < 0.05 was considered statistically significant. The clinical data were statistically analyzed using the SPSS 22.0 software (IBM Corporation, Armonk, NY, United States).
KEGG Orthology Based Annotation System (KOBAS) software was used to analyze pathways, diseases, and functions at the same time. The whole analysis process consists of two steps. The first step is to map the input gene sets to the database genes and then annotate the pathways, diseases, and functions in which these gene sets are involved. In the second step, the results obtained in the previous step are compared with the background (usual genes in the whole genome, or all probes on the chip), and the statistically significant enriched pathways, diseases, or functions are mined. Statistical tests can be performed using the binomial test, chi-square test, Fisher’s exact test, or hypergeometric distribution test. Multi-hypothesis testing methods were used to adjust the p value. Genes associated with DMSs and DMRs were enriched in the Gene Ontology. Additionally, we identified overrepresented canonical pathways, biological processes, and molecular processes. We performed literature research to determine the known or plausible roles of differentially methylated genes in cardiac development.
Table 1 summarized the demographic and clinical characteristics of the cases and the controls and revealed no statistically significant differences. None of the participants experienced additional obstetric complications.

TABLE 1. Demographic and clinical data of cases and controls.
In comparison to the controls, we identified 670 DMSs corresponding to 401 genes in CTDs. 444 (66.3%) of all DMSs were hypomethylated, while 226 (33.7%) were hypermethylated. The heat map in Figure 1 illustrates the differentially methylated sites in CTDs and controls, as well as subtypes of CTDs. The AUC (95%CI) and β-value for each group’s prediction were calculated. Figure 2 depicts the two genes with the highest predictive accuracy for CTDs detection (AUC >0.90). Table 2 details the most variable methylation sites between groups and the highest predictive accuracy for CTDs and their subtypes detection.
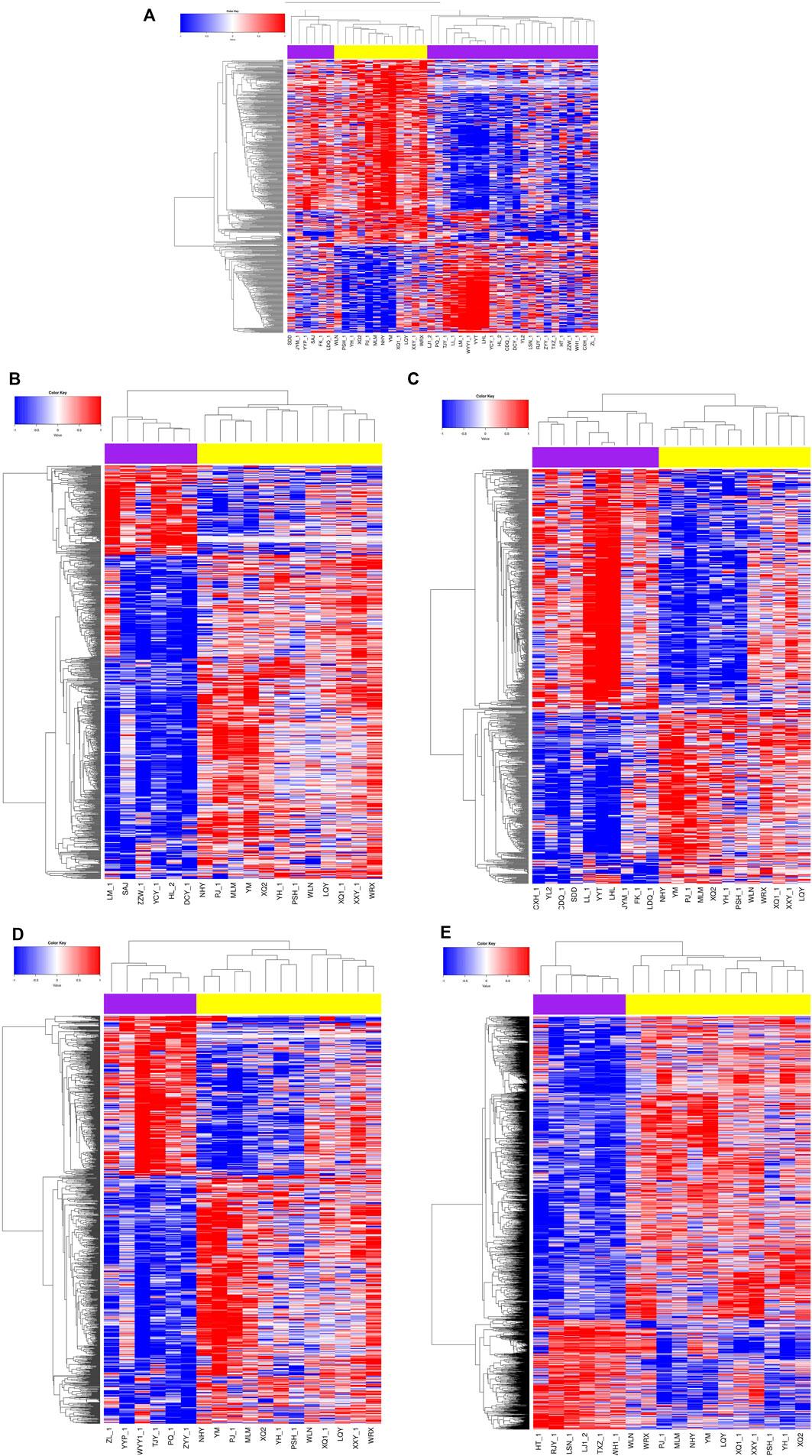
FIGURE 1. Heat map showing the differential methylation levels of CpG loci in placental DNA of cases compared with normal controls. Red represents hypermethylation, blue represents hypomethylation, the purple are the cases, the yellow are controls. (A) CTDs vs. controls; (B) TOF vs. controls; (C) DORV vs. controls; (D) TGA vs. controls; (E) PAVSD vs. controls.
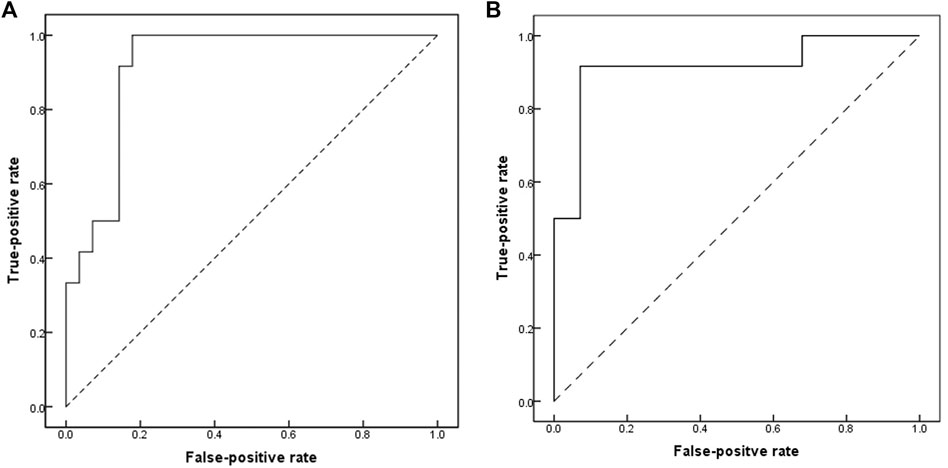
FIGURE 2. Receiver-operating-characteristics (ROC) curves showing accuracy of methylation profile of placental DNA CpG in prediction of CTDs. (A) cg04328562(CNN1); area under ROC curves (AUC) = 0.917 (95%CI, 0.831–1.0). (B) cg05167251 (chr2; HOXD9); area under ROC curves (AUC) = 0.917 (95%CI, 0.8–1.0).
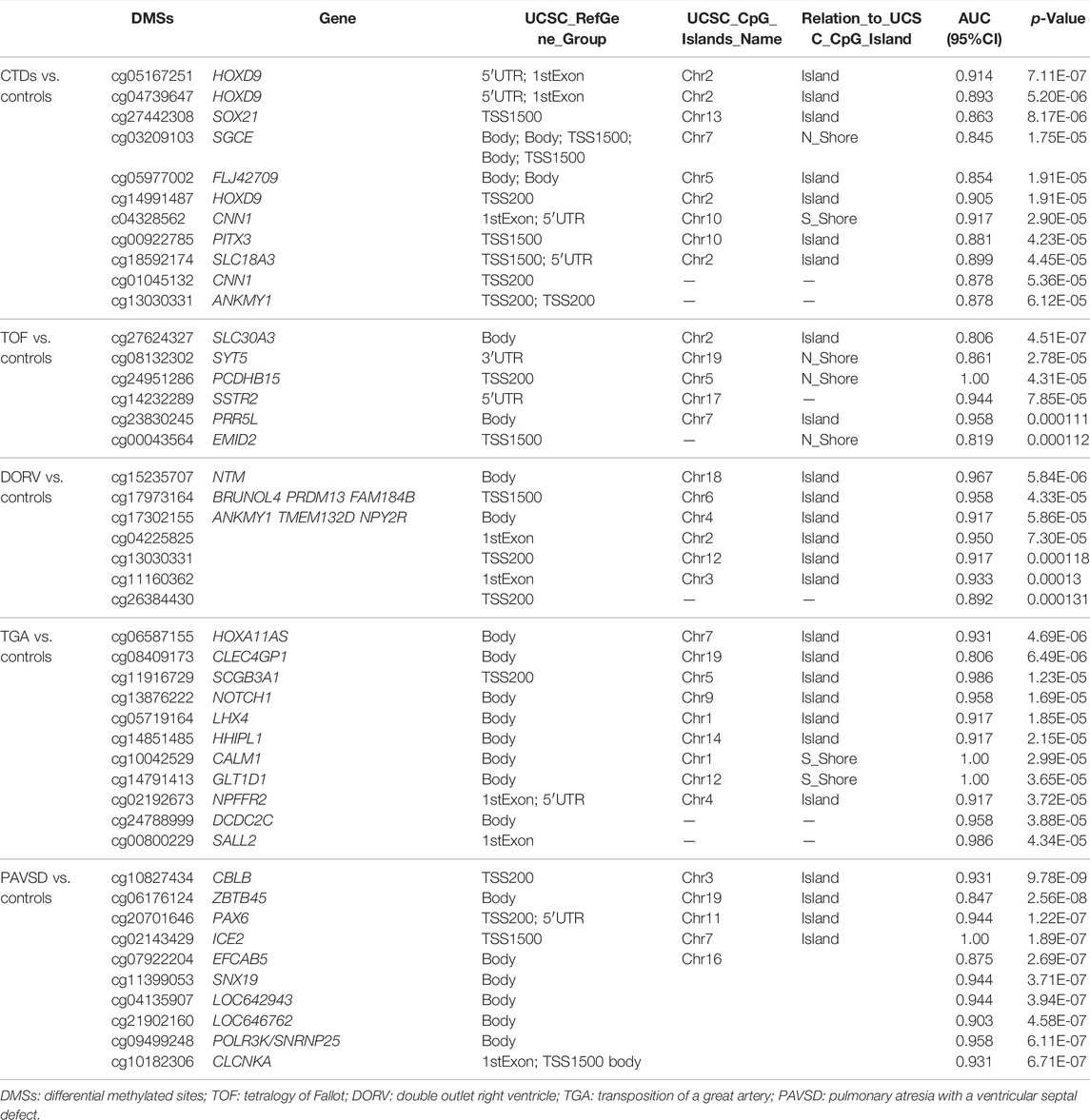
TABLE 2. The top DMSs between groups and AUC of epigenetic markers of each group.
Furthermore, when comparing CTDs and controls, we identified 29 DMRs. Three DMRs were hypermethylated and 26 were hypomethylated. The majority of the DMRs were located on the CpG island. In comparison to the controls, we identified 20, 6, 27, and 17 DMRs in TOF, DORV, TGA, and PAVSD, respectively. We discovered that the majority of DMRs in these cases were hypermethylated. Table 3 lists the gene-related sites and their locations. NR2E1 and ECE1 were identified in more than two groups of gene-related sites.
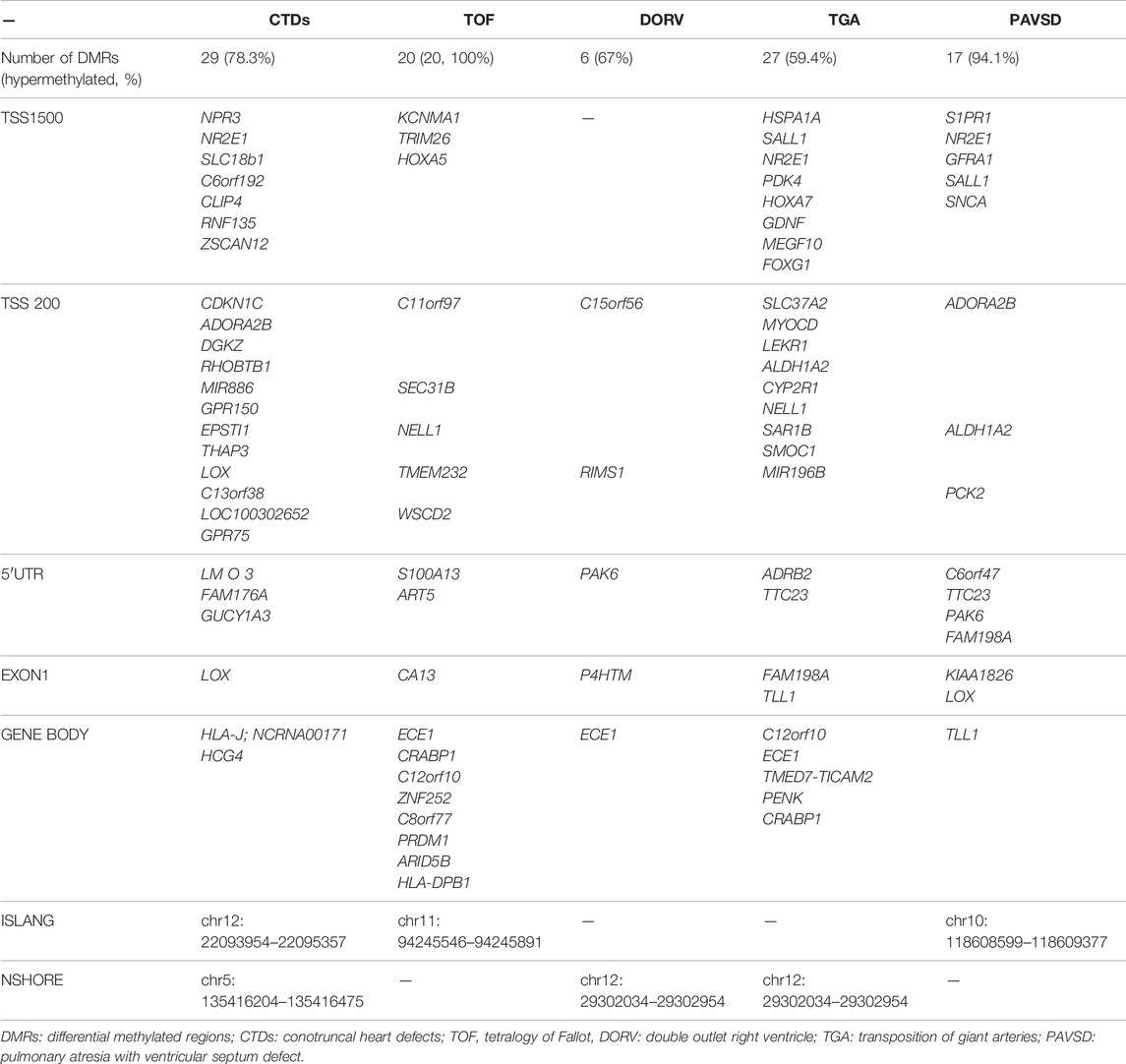
TABLE 3. Genes with differential methylation regions in each group.
Between CTDs and controls, disease ontology analysis, go enrichment analysis, and pathway analysis of the DMRs were performed. Figure 3 summarizes the top 30 items. Following our cases, disease ontology revealed that the DMRs of CTDs were associated with congenital developmental disorders, vascular diseases, and cardiovascular diseases. CTDs were associated with a variety of pathways, including vascular smooth muscle contraction, Notch signaling, glycerolipid metabolism, long-term depression, renin secretion, PPAR signaling, gap junctions, salivary secretion, circadian entrainment, and endocrine resistance. CNN1 was found to be involved in smooth muscle contraction; NOTCH1 was found to be involved in the NOTCH signaling pathway.
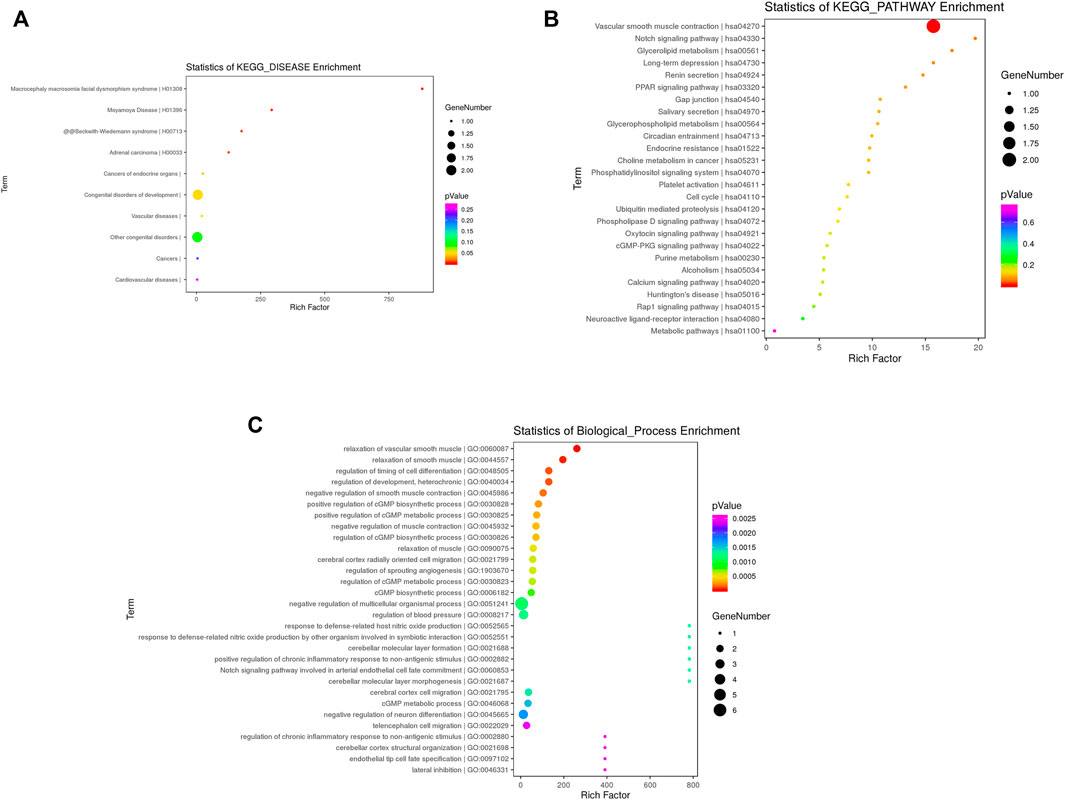
FIGURE 3. Functional enrichment analysis of differential methylation regions annotated genes in CTDs vs. controls KEGG enrichment analysis in disease (A) and pathway (B); (C) Top 30 terms of biological process (BP).
Even with today’s highly developed diagnostic methods, early detection and classification of CHD remain a significant clinical challenge. Epigenetic biomarkers of CHD are of great clinical interest and the predictive value of DNA methylation changes in CHD has been demonstrated in numerous studies. Since the placenta and the heart develop in the same way, the placenta may be an ideal tissue for both understanding the mechanism of CHD and developing biomarkers for it. Several studies (Fantasia et al., 2018; Fantasia et al., 2019; and Binder et al., 2020) have demonstrated placental dysfunction in the absence of impaired placental perfusion in pregnancies with isolated CHD, reinforcing the preceding position. Additionally, the placenta is available during the first to third trimesters of pregnancy.
We identified several potential methylation markers for the prediction of CTDs in our study. The best predictive performance (AUC >0.90) was achieved by HOXD9 and CNN1. To gain a better understanding of the function of HOXD9 and CNN1, we investigated the human protein atlas (Uhlén et al., 2015) database. HOXD9 is not restricted to the placenta or the heart but is expressed in multicellular organisms, with a high level of expression in the endometrium. According to the latest published data, it plays a critical role in the promotion of carcinoma (Zhu et al., 2019 and Wen et al., 2020) and arterial inflammation (Souilhol et al., 2020). However, the role of HOXD9 in the development of the heart and placenta remains unknown. CNN1 is expressed in the cytoplasm of smooth muscle and myoepithelial cells with a high degree of specificity. Additionally, CNN1 encodes a protein that plays a role in the regulation and modulation of smooth muscle contraction. It also plays a role in the development of cardiomyopathy (Lu et al., 2014), placental vascular development, and tissue morphology (de Barros Mucci et al., 20052020). Further studies will be critical to elucidate its role in the pathogenesis of CHD.
Additionally, we identified methylation predictors in subtypes of CTDs, including TOF, DORV, TGA, and PAVSD, as shown in Table 2. The intriguing thing is that the genes with the highest prediction for each group are distinct, although these cases are classified as CHD in anatomy. The most frequently studied difference between these cases is the methylation changes in TOF. Bahado-Singh. R et al. reported that methylation changes in ARHGAP22, CDK5, TRIM27, and IER3 in the placenta are excellent individual predictive markers of TOF, a finding that was not replicated in our study. Different gestation weeks and tissue preservation in the cases may be the primary reasons. PCDHB15, SSTR2, and PRR5L were identified in TOF in our study. PCDHB15 has a low tissue specificity and its specific functions are unknown, but it is highly likely to involve in the establishment and function of specific cellcell neural connections (Lee and Lee, 2008). SSTR2 expression in the heart from day 14–15 with a peak at day 17 was discovered in a study (Ludvigsen et al., 2015) of mouse embryos. This coincides with the development of the heart and the placenta. It is not, however, specified in these two organs. There is insufficient evidence to suggest that it plays a role in the development of cardiovascular disease. The PPR5L protein is associated with the mTORC2 complex, which regulates cellular processes such as survival and organization of the cytoskeleton. (Pearce et al., 2007). It acts as a substrate-specific regulator of the mTORC2 complex, preventing, for example, the specific phosphorylation of PKCs and eventually controlling the cell migration. (Gan et al., 2012)mTORC2 is required for cardiomyocyte differentiation from embryonic stem cells (Zheng et al., 2017), postnatal heart growth, and heart function maintenance in mice (Zhao et al., 2014). Nevertheless, the molecular mechanism by which PPR5L functions in CHD is unknown and requires further investigation. NTM, BROUNL4, PRDM13, FAM184B, ANKMY1, and TMEM132D were identified as DMSs genes in the DORV group. Both of them were not tissue-specified and had unknown functions during heart development. Among these TGA group DMSs, NOTCH1, and NPFFR2 have been associated with heart development. Some studies (High et al., 2009 and Koenig et al., 2016) established the role of endothelial NOTCH1 in the proper development of semilunar valves, cardiac outflow tract remodeling, and aortic arch artery remodeling. Transposition of the great arteries is associated with abnormal outflow tract development. A study (Kerstjens-Frederikse et al., 2016) found that cardiovascular malformation caused by NOTCH1 mutation does not keep left. The phenotypic spectrum included CTDs, which was consistent with our findings. NOTCH1 was recently identified as a major candidate gene for TOF in a whole-exome sequencing study conducted by (Page et al., 2019) While the highest concentration of NPFF receptor 2 (NPFFR2) mRNA is found in the placenta, a recent study (Zhu et al., 2020) demonstrated that NPFF acts via NPFFR2 to increase HCG-β production and promotes GCM1-dependent expression of syncytin one and two in cytotrophoblasts. Trophoblast cells are critical in the formation of placental blood vessels. In the PAVSD group, several genes were identified as the predictive makers, but none of them were specified as expressed in the placenta or heart in the human protein atlas. As discussed previously, methylation changes in placental tissue may serve as predictive biomarkers for CHD, even when differentiating between different subtypes.
In comparison to the controls, our study identified 29, 20, 6, 27, and 17 DMRs in CTDs, TOF, DORV, TGA, and PAVSD, respectively. The majority of them had been hypermethylated. Among the genes with differential methylation regions (Table 3), NR2E1 located at transpositional start site (TSS) 1500 and ECE1 located within the gene body were found in more than two groups. NR2E1 mRNA has been detected in a variety of organs, including the placenta, but its precise function is unknown. Endothelin-converting enzyme-1 (ECE1) is required for the development of a subset of neural crest lineages, including cardiogenesis, and a case-control study (Wang et al., 2012) suggested that ECE1 polymorphisms may contribute to susceptibility to sporadic CHD in the Chinese population, particularly in TOF and peri membranous ventricular septal defect (pmVSD). In our study, we also discovered differential methylation of ECE1 in TOF. As a result, additional research is required to elucidate the mechanisms. Consequently, the subtypes of CTDs may share some common differential methylation genes. However, distinct subgroups may have distinct differential methylation genes that influenced the development of the heart. Due to the interaction of all of these genes, distinct subtypes of CTDs exist.
Our study had some limitations. To begin, only a few cases were included, especially in the subtype group. Additional research is required in a large number of cases. Second, one should consider the effect of gestational age on methylation. Additionally, we did not examine the levels of gene expression (mRNA or protein), which is the ultimate determinant of the biological consequence of DNA methylation. Moreover, our specimens were all derived from fetuses undergoing labor induction, so we could not observe their efficacy in predicting pregnancy outcome or even post-birth prognosis. These are the things we need to improve next.
In conclusion, our study established that abnormal changes in placental methylation are associated with CTDs and may be predictive of CTDs. Numerous altered genes are already known or suspected to play a role in cardiovascular or placental development, additional research is planned to corroborate our findings.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by The Ethics Committee of Beijing Anzhen Hospital, Capital Medical University (IORG NO: GZR-2-006). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
JL, JH and YH conceived of the presented idea; JL and YW performed the experiment; JL and HS contributed to data analysis; JL took the lead in writing the manuscript; all authors discussed the results and commented on the manuscript.
This work was supported by Beijing Lab for Cardiovascular Precision Medicine, Beijing, China (PXM 2020_014226_000017_00377132_FCG) and Beijing Advanced Innovation Center for Big Data-based Precision Medicine, Capital Medical University, Beijing, China (PXM 2021_014226_000026). Beijing municipal change form medical research institutes pilots reform project (grant number 2021-07) and National Natural Science Foundation of China (NO. 8217020250).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bahado-Singh, R., Vishweswaraiah, S., Mishra, N. K., Guda, C., and Radhakrishna, U. (2020). Placental DNA Methylation Changes in Detection of Tetralogy of Fallot. Ultrasound Obstet. Gynecoljun 55 (6), 768–775. doi:10.1002/uog.20292
Binder, J., Carta, S., Carvalho, J. S., Kalafat, E., Khalil, A., and Thilaganathan, B. (2020). Evidence for Uteroplacental Malperfusion in Fetuses with Major Congenital Heart Defects. PloS one 15 (2), e0226741. doi:10.1371/journal.pone.0226741
Blue, G. M., Kirk, E. P., Sholler, G. F., Harvey, R. P., and Winlaw, D. S. (2012). Congenital Heart Disease: Current Knowledge about Causes and Inheritance. Med. J. Aust. 197 (3), 155–159. doi:10.5694/mja12.10811
Bouma, B. J., and Mulder, B. J. M. (2017). Changing Landscape of Congenital Heart Disease. Circ. Res. 120 (6), 908–922. doi:10.1161/circresaha.116.309302
Csáky-Szunyogh, M., Vereczkey, A., Kósa, Z., Gerencsér, B., and Czeizel, A. E. (2013). Risk and Protective Factors in the Origin of Conotruncal Defects of Heart-Aa Population-Based Case-Control Study. Am. J. Med. Genet. A. 161a (10), 2444–2452. doi:10.1002/ajmg.a.36118
de Barros Mucci, D., Kusinski, L. C., Wilsmore, P., Loche, E., Pantaleão, L. C., Ashmore, T. J., et al. (20052020). Impact of Maternal Obesity on Placental Transcriptome and Morphology Associated with Fetal Growth Restriction in Mice. Int. J. Obes. (Lond)05 44 (5), 1087–1096. doi:10.1038/s41366-020-0561-3
Eckersley, L., Sadler, L., Parry, E., Finucane, K., and Gentles, T. L. (2016). Timing of Diagnosis Affects Mortality in Critical Congenital Heart Disease. Arch. Dis. Child. 101 (6), 516–520. doi:10.1136/archdischild-2014-307691
Fantasia, I., Kasapoglu, D., Kasapoglu, T., Syngelaki, A., Akolekar, R., and Nicolaides, K. H. (2018). Fetal Major Cardiac Defects and Placental Dysfunction at 11-13 Weeks' Gestation. Ultrasound Obstet. Gynecol. 51 (2), 194–198. doi:10.1002/uog.18839
Fantasia, I., Andrade, W., Syngelaki, A., Akolekar, R., and Nicolaides, K. H. (2019). Impaired Placental Perfusion and Major Fetal Cardiac Defects. Ultrasound Obstet. Gynecoljan 53 (1), 68–72. doi:10.1002/uog.20149
Gan, X., Wang, J., Wang, C., Sommer, E., Kozasa, T., Srinivasula, S., et al. (2012). PRR5L Degradation Promotes mTORC2-Mediated PKC-δ Phosphorylation and Cell Migration Downstream of Gα12. Nat. Cel Biol 14 (7), 686–696. doi:10.1038/ncb2507
High, F. A., Jain, R., Stoller, J. Z., Antonucci, N. B., Lu, M. M., Loomes, K. M., et al. (2009). Murine Jagged1/Notch Signaling in the Second Heart Field Orchestrates Fgf8 Expression and Tissue-Tissue Interactions during Outflow Tract Development. J. Clin. Invest. 119 (7), 1986–1996. doi:10.1172/JCI38922
Holland, B. J., Myers, J. A., and Woods, C. R. (2015). Prenatal Diagnosis of Critical Congenital Heart Disease Reduces Risk of Death from Cardiovascular Compromise Prior to Planned Neonatal Cardiac Surgery: a Meta-Analysis. Ultrasound Obstet. Gynecoljun 45 (6), 631–638. doi:10.1002/uog.14882
Kerstjens-Frederikse, W. S., van de Laar, I. M. B. H., Vos, Y. J., Verhagen, J. M. A., Berger, R. M. F., Lichtenbelt, K. D., et al. (2016). Cardiovascular Malformations Caused by NOTCH1 Mutations Do Not Keep Left: Data on 428 Probands with Left-Sided CHD and Their Families. Genet. Med. 18 (9), 914–923. doi:10.1038/gim.2015.193
Koenig, S. N., Bosse, K., Majumdar, U., Bonachea, E. M., Radtke, F., and Garg, V. (2016). Endothelial Notch1 Is Required for Proper Development of the Semilunar Valves and Cardiac Outflow Tract. J. Am. Heart Assoc. 5 (4). doi:10.1161/JAHA.115.003075
Lee, B., and Lee, D. (2008). DAhunter: a Web-Based Server that Identifies Homologous Proteins by Comparing Domain Architecture. Nucleic Acids Res. 36 (Web Server issue), W60–W64. doi:10.1093/nar/gkn172
Li, Y.-F., Zhou, K.-Y., Fang, J., Wang, C., Hua, Y.-M., and Mu, D.-Z. (2016). Efficacy of Prenatal Diagnosis of Major Congenital Heart Disease on Perinatal Management and Perioperative Mortality: a Meta-Analysis. World J. Pediatr. 12 (3), 298–307. doi:10.1007/s12519-016-0016-z
Linask, K. K. (2013). The Heart-Placenta axis in the First Month of Pregnancy: Induction and Prevention of Cardiovascular Birth Defects. J. Pregnancy 2013, 320413. doi:10.1155/2013/320413
Lu, D., Zhang, L., Bao, D., Lu, Y., Zhang, X., Liu, N., et al. (2014). Calponin1 Inhibits Dilated Cardiomyopathy Development in Mice through the εPKC Pathway. Int. J. Cardiol. 173 (2), 146–153. doi:10.1016/j.ijcard.2014.02.032
Ludvigsen, E., Carlsson, C., Tiensuu Janson, E., Sandler, S., and Stridsberg, M. (2015). Somatostatin Receptor 1-5; Expression Profiles during Rat Development. Upsala J. Med. Sci. 120 (3), 157–168. doi:10.3109/03009734.2015.1035413
Marín-García, J. (2009). Advances in Molecular Genetics of Congenital Heart Disease. Revista espanola de cardiologia 62 (3), 242–245. doi:10.1016/s1885-5857(09)71552-x
Maslen, C. L. (2018). Recent Advances in Placenta-Heart Interactions. Front. Physiol. 9, 735. doi:10.3389/fphys.2018.00735
Page, D. J., Miossec, M. J., Williams, S. G., Monaghan, R. M., Fotiou, E., Cordell, H. J., et al. (2019). Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circ. Res. 124 (4), 553–563. doi:10.1161/CIRCRESAHA.118.313250
Pearce, L. R., Huang, X., Boudeau, J., Pawłowski, R., Wullschleger, S., Deak, M., et al. (2007). Identification of Protor as a Novel Rictor-Binding Component of mTOR Complex-2. Biochem. J. 405 (3), 513–522. doi:10.1042/BJ20070540
Pierpont, M. E., Brueckner, M., Chung, W. K., Garg, V., Lacro, R. V., McGuire, A. L., et al. (2018). Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement from the American Heart Association. Circulation 138 (21), e653–e711. doi:10.1161/CIR.0000000000000606
Radhakrishna, U., Albayrak, S., Zafra, R., Baraa, A., Vishweswaraiah, S., Veerappa, A. M., et al. (2019). Placental Epigenetics for Evaluation of Fetal Congenital Heart Defects: Ventricular Septal Defect (VSD). PloS one 14 (3), e0200229. doi:10.1371/journal.pone.0200229
Serra-Juhé, C., Cuscó, I., Homs, A., Flores, R., Torán, N., and Pérez-Jurado, L. A. (2015). DNA Methylation Abnormalities in Congenital Heart Disease. Epigenetics 10 (2), 167–177. doi:10.1080/15592294.2014.998536
Souilhol, C., Gauci, I., Feng, S., Tardajos Ayllon, B., Mahmoud, M., Canham, L., et al. (2020). Homeobox B9 Integrates Bone Morphogenic Protein 4 with Inflammation at Atheroprone Sites. Cardiovasc. Res. 116 (7), 1300–1310. doi:10.1093/cvr/cvz235
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-Based Map of the Human Proteome. Science (New York, N.Y.). 347 (6220), 1260419. Jan 23. doi:10.1126/science.1260419
Wang, Y., Liu, Y., Peng, W., Wang, M., Sun, J., Zhang, Z., et al. (2012). ECE1Polymorphisms May Contribute to the Susceptibility of Sporadic Congenital Heart Disease in a Chinese Population. DNA Cel. Biol. 31 (8), 1425–1430. doi:10.1089/dna.2012.1626
Wen, D., Wang, L., Tan, S., Tang, R., Xie, W., Liu, S., et al. (2020). doi:10.23736/s0031-0808.20.03911-7HOXD9 Aggravates the Development of Cervical Cancer by Transcriptionally Activating HMCN1Panminerva Med.
Zhao, X., Lu, S., Nie, J., Hu, X., Luo, W., Wu, X., et al. (2014). Phosphoinositide-dependent Kinase 1 and mTORC2 Synergistically Maintain Postnatal Heart Growth and Heart Function in Mice. Mol. Cel Biol 34 (11), 1966–1975. doi:10.1128/mcb.00144-14
Zheng, B., Wang, J., Tang, L., Shi, J., and Zhu, D. (2017). mTORC1 and mTORC2 Play Different Roles in Regulating Cardiomyocyte Differentiation from Embryonic Stem Cells. Int. J. Dev. Biol. 61 (1-2), 65–72. doi:10.1387/ijdb.160207dz
Zhu, H., Dai, W., Li, J., Xiang, L., Wu, X., Tang, W., et al. (2019). HOXD9 Promotes the Growth, Invasion and Metastasis of Gastric Cancer Cells by Transcriptional Activation of RUFY3. J. Exp. Clin. Cancer Res. 38 (1), 412. doi:10.1186/s13046-019-1399-1
Keywords: placenta, methylation, biomarkers, epigenetics, conotruncal heart defects
Citation: Liu J, Wu Y, Sun H, Liu X, Gu X, Zhao Y, Zhang Y, Han J and He Y (2022) Placental DNA Methylation Abnormalities in Prenatal Conotruncal Heart Defects. Front. Genet. 13:878063. doi: 10.3389/fgene.2022.878063
Received: 17 February 2022; Accepted: 11 April 2022;
Published: 13 May 2022.
Edited by:
Brian Hon-Yin Chung, The University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Christopher Chun Yu Mak, The University of Hong Kong, Hong Kong SAR, ChinaCopyright © 2022 Liu, Wu, Sun, Liu, Gu, Zhao, Zhang, Han and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiancheng Han, aGFuX2pjMTk3N0Bob3RtYWlsLmNvbQ==; Yihua He, aGV5aWh1YWVjaG9AaG90bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.