 Lucija Kanjer1Klara Filek1Maja Mucko1Roksana Majewska2,3Romana Gračan1Adriana Trotta4Aliki Panagopoulou5Marialaura Corrente4Antonio Di Bello4Sunčica Bosak1*
Lucija Kanjer1Klara Filek1Maja Mucko1Roksana Majewska2,3Romana Gračan1Adriana Trotta4Aliki Panagopoulou5Marialaura Corrente4Antonio Di Bello4Sunčica Bosak1*- 1Department of Biology, Faculty of Science, University of Zagreb, Zagreb, Croatia
- 2Unit for Environmental Sciences and Management, Faculty of Natural and Agricultural Sciences, North-West University, Potchefstroom, South Africa
- 3Human Metabolomics, Department of Biochemistry, Faculty of Natural and Agricultural Sciences, North-West University, Potchefstroom, South Africa
- 4Department of Veterinary Medicine, University of Bari Aldo Moro, Bari, Italy
- 5ARCHELON, The Sea Turtle Protection Society of Greece, Athens, Greece
The loggerhead sea turtle is considered a keystone species with a major ecological role in Mediterranean marine environment. As is the case with other wild reptiles, their outer microbiome is rarely studied. Although there are several studies on sea turtle’s macro-epibionts and endo-microbiota, there has been little research on epibiotic microbiota associated with turtle skin and carapace. Therefore we aimed to provide the identification of combined epibiotic eukaryotic, bacterial and archaeal microbiota on Mediterranean loggerhead sea turtles. In this study, we sampled skins and carapaces of 26 loggerheads from the Mediterranean Sea during 2018 and 2019. To investigate the overall microbial diversity and composition, amplicon sequencing of 16S and 18S rRNA genes was performed. We found that the Mediterranean loggerhead sea turtle epibiotic microbiota is a reservoir of a vast variety of microbial species. Microbial communities mostly varied by different locations and seas, while within bacterial communities’ significant difference was observed between sampled body sites (carapace vs. skin). In terms of relative abundance, Proteobacteria and Bacteroidota were the most represented phyla within prokaryotes, while Alveolata and Stramenopiles thrived among eukaryotes. This study, besides providing a first survey of microbial eukaryotes on loggerheads via metabarcoding, identifies fine differences within both bacterial and eukaryotic microbial communities that seem to reflect the host anatomy and habitat. Multi-domain epi-microbiome surveys provide additional layers of information that are complementary with previous morphological studies and enable better understanding of the biology and ecology of these vulnerable marine reptiles.
Introduction
Microbial communities associated with the external surfaces of animals represent an important part of the animal microbiome. Animal integument is a physical barrier that protects animal’s internal environment while interacting with their external environment. In vertebrates, one of the most extensively studied epimicrobiomes is the human skin (Turnbaugh et al., 2007; Byrd et al., 2018). The epidermal microbes enhance the skin barrier performance by modulating innate immunity and developing adaptive immunity (Sanford and Gallo, 2013), therefore helping to battle skin pathogens (Belkaid and Segre, 2014; Belkaid and Tamoutounour, 2016). A shift in the host’s health can alter the composition and functions of the skin microbiota that can lead to various diseases (Sanford and Gallo, 2013). Similarly, the native microbial communities of the epidermis can be affected by sub-optimal environmental conditions, negatively influencing their protective properties (Scharschmidt and Fischbach, 2013; Byrd et al., 2018).
The skin and other external body surfaces (e.g., horns, carapaces, hair and other keratinous hard tissues) of vertebrates differ between taxonomic groups and provide fairly diversified habitats for various animal-associated microbes (Ross et al., 2019). Since both intrinsic (e.g., species, sex, age) and extrinsic (e.g., geographic location, biotic and abiotic environmental conditions, captivity affecting the natural behavior, and diet) factors shape the community composition of the epimicrobiome, differences between even closely related host species or individuals are to be expected (Ross et al., 2019; Woodhams et al., 2020). Nevertheless, a certain degree of phylosymbiosis, in which the microbiota mirrors the phylogeny of the host (Brooks et al., 2016), is also observed (Ross et al., 2018). This may be due to changes in host traits, or host microbial co-evolution. Apart from humans, much of the epimicrobiome research has focused on captive animals and pets as well as amphibians whose skin is more permeable and thus more susceptible to pollution and novel pathogens, potentially threatening the survival of entire populations and species (Ross et al., 2019). However, very little is known about the epimicrobiomes of reptiles, especially turtles, including terrestrial, freshwater, and marine species. Recently, there have been studies on external microbial communities of freshwater turtles (Trachemys scripta, Pseudemys concinna, and Emydura macquarii krefftii) that identified major microbial components of eukaryotic, bacterial and archaeal surface communities and showed that turtles’ microbiotas differ between body parts and between animals and their environment (McKnight et al., 2020; Parks et al., 2020). New knowledge about the functional and phylogenetic composition of epimicrobiomes of different species improves our understanding of the relationships between the host, its microbial flora, and the environment. Such advances in knowledge may contribute to a more efficient conservation of endangered and threatened macroorganisms. Skin microbiome research has a lot of potential in conservation biology of marine animals because of its accessibility and non-invasive sampling procedures. The potential of microbiome as bioindicator of ecosystem’s health has been recognized and effort is being put into the standardization of the methodology – e.g., from sampling to correct index calculations (Lau et al., 2015; Aylagas et al., 2017; Keeley et al., 2018; Cordier et al., 2019). It is possible that surface-associated microbiomes exhibit a stronger link with variations in the environment, while the internal microbial communities are more affected by the host’s intrinsic factors (Woodhams et al., 2020).
Although loggerheads are the most abundant sea turtle species in the Mediterranean Sea, they are threatened by coastal development, fishing bycatch, tourism, pollution and climate change (Casale et al., 2018). Skin and carapace of loggerheads provide habitats for a surprising variety of unique and taxonomically diverse macro-epibionts, including barnacles, amphipods and red algae (Hollenberg, 1971; Broderick et al., 2002; Frick and Pfaller, 2013). Some of these organisms require the sea turtle substratum to attach and thrive, and thus their survival is inextricably linked to the wellbeing and fitness of their hosts. The existing body of literature on loggerhead and other sea turtle microbiomes includes mainly studies investigating the internal microbiota, such as those living in the gut, cloaca, faces, and oral cavities (Abdelrhman et al., 2016; Arizza et al., 2019; Biagi et al., 2019; Scheelings et al., 2020a,b; Filek et al., 2021). The epimicrobiomes of sea turtles, in turn, have received far less attention. Recent years brought increased interest in micro-eukaryotic surface assemblages of sea turtles largely due to a series of projects exploring the diversity of sea turtle-associated diatoms (Majewska et al., 2015; Robinson et al., 2016; Rivera et al., 2018; Azari et al., 2020; Kanjer et al., 2020; Van de Vijver et al., 2020). Those studies identified a group of the diatom core taxa typical of sea turtles but also showed some biogeographic differences between diatom epizoic assemblages (Van de Vijver et al., 2020). Besides inventorial and ecological interest in diversity of epi-microbiome, there is a possible benefit for sea turtles’ health that could arise from these kinds of studies. For example, Fusarium spp. fungal infection of loggerhead eggs is considered a global threat (Bailey et al., 2018) and its detection on carapace and skin could be beneficial (Cafarchia et al., 2020). Further, evidence of antibiotic resistant bacteria found on sea turtles highlight the direct effect of antibiotic pollution in the seas (Pace et al., 2019; Alduina et al., 2020; Trotta et al., 2021). However, reports on bacterial, archaeal or micro-eukaryotic non-diatom communities associated with the skin and carapace of sea turtles are extremely scarce and include only a recent study by Blasi et al. (2022) that reported the composition of bacterial microbial community based on 16S rRNA gene profiling from the carapaces of three juvenile loggerheads from the Tyrrhenian Sea.
The aim of our study was to investigate the micro-eukaryotic, bacterial and archaeal diversity found on the external surfaces of loggerhead sea turtles from the Mediterranean Sea using the 18S and 16S rRNA genes amplicon sequencing approach, respectively. Furthermore, we aimed to describe both loggerhead skin and carapace microbial communities to allow for comparison between these two biochemically, micro-topographically, and physiologically different substrata. Detailed observation and statistical analyses of microbial assemblages’ taxonomic composition were addressed in accordance to our large and diverse loggerhead dataset. The roles of potential factors that could influence the microbial communities are considered and additional approaches in studies of this type are discussed.
Materials and methods
Sampling
Twenty-six loggerhead sea turtles were sampled from four different Mediterranean areas: Adriatic (n = 14), Ionian (n = 9), Tyrrhenian (n = 1) and Aegean Sea (n = 2; Figure 1) following recommendations from Pinou et al. (2019). Two separate samples were collected from each turtle, one from the carapace and one from the skin (Supplementary Figure 1). Biofilm scrapings were taken using clean toothbrushes and/or a sterilized scalpel and were resuspended in 96% ethanol in sterile 50 ml conical tubes immediately after collection. Carapace samples were collected randomly from an entire carapace, whereas skin samples were taken from the animal head, neck, and flippers. All samples were stored at −20°C until further processing. One turtle (ID010) was sampled twice: immediately upon arrival to the rescue center and after approx. one year in rehabilitation. In total, 54 samples were collected from August 2018 until November 2019 (Table 1). Due to the heterogeneity of sampled turtles, we differentiate a turtle’s origin localities from “sampling locality” (Table 1 and Supplementary Table 1). Origin locality is the location where a turtle was found in the sea or on the beach and the sampling locality refers to the place where samples were obtained. Origin locality and sampling locality is identical for the turtles sampled where they were found but differs for the turtles that were being brought to rehabilitation centers. The turtles were sampled in three rescue centers (Marine Turtle Rescue Center Aquarium Pula and Blue World Institute Lošinj in Croatia, and The Archelon Sea Turtle Protection Society in Greece) and one veterinary clinic (The Sea Turtle Clinic, STC, Department of Veterinary Medicine, University of Bari “Aldo Moro” in Bari, Italy). The sea turtle status was designated as “wild” if the turtle was sampled immediately after capturing without being immersed into the rehabilitation pool, and “admitted” if the animal was admitted to a rehabilitation center and was immersed in the rehabilitation pool prior to sampling. Time between the turtle admission and the sampling of its biofilm spans between 1 and 10 days (except for ID010).
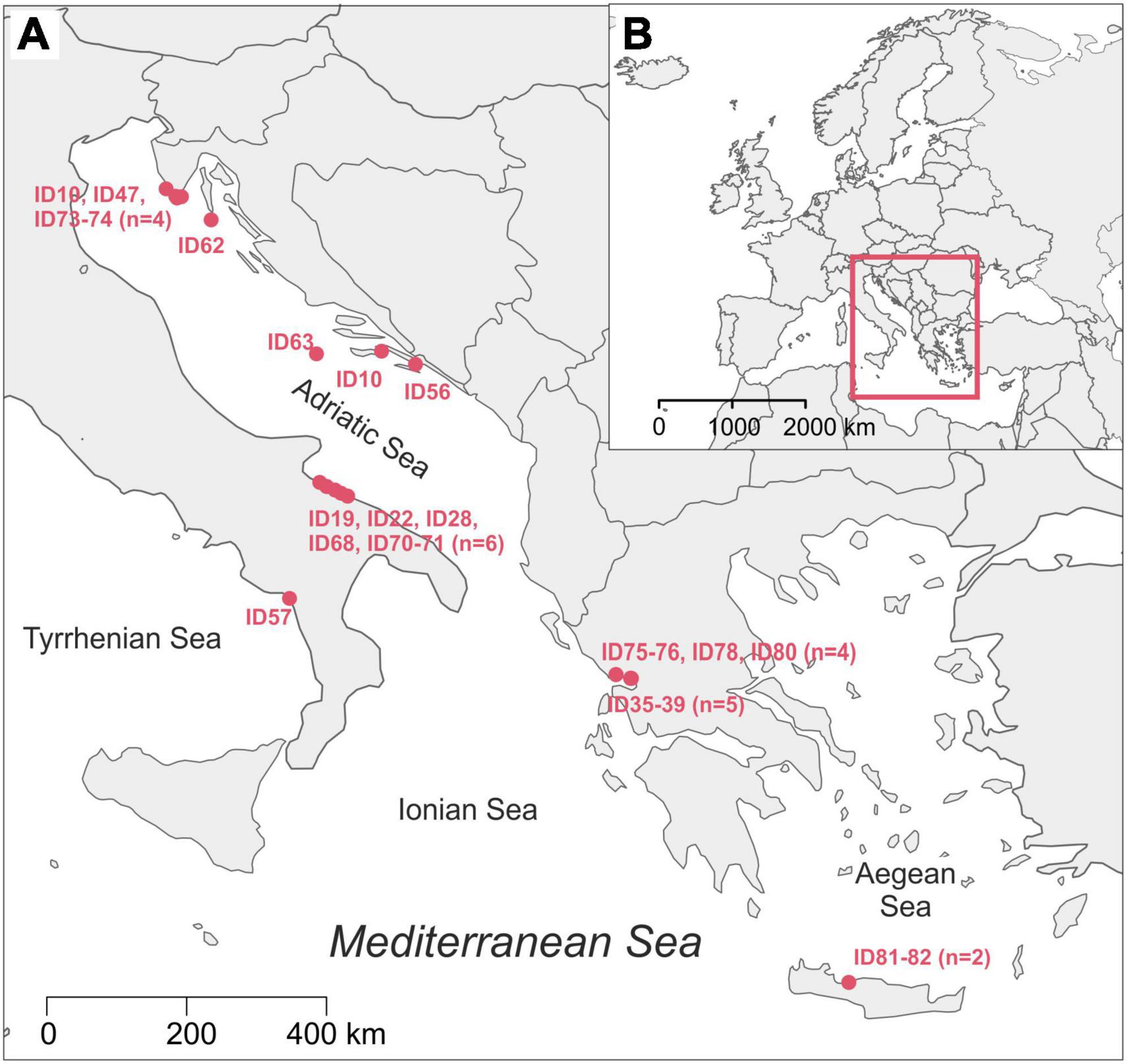
Figure 1. Map of the origin localities of sampled loggerhead turtles with indicated Turtle ID code (A); position of our study area in map of Europe (B). The map was made using R packages maps (Becker et al., 2021; RRID:SCR_019296) and mapdata (Becker et al., 2018).
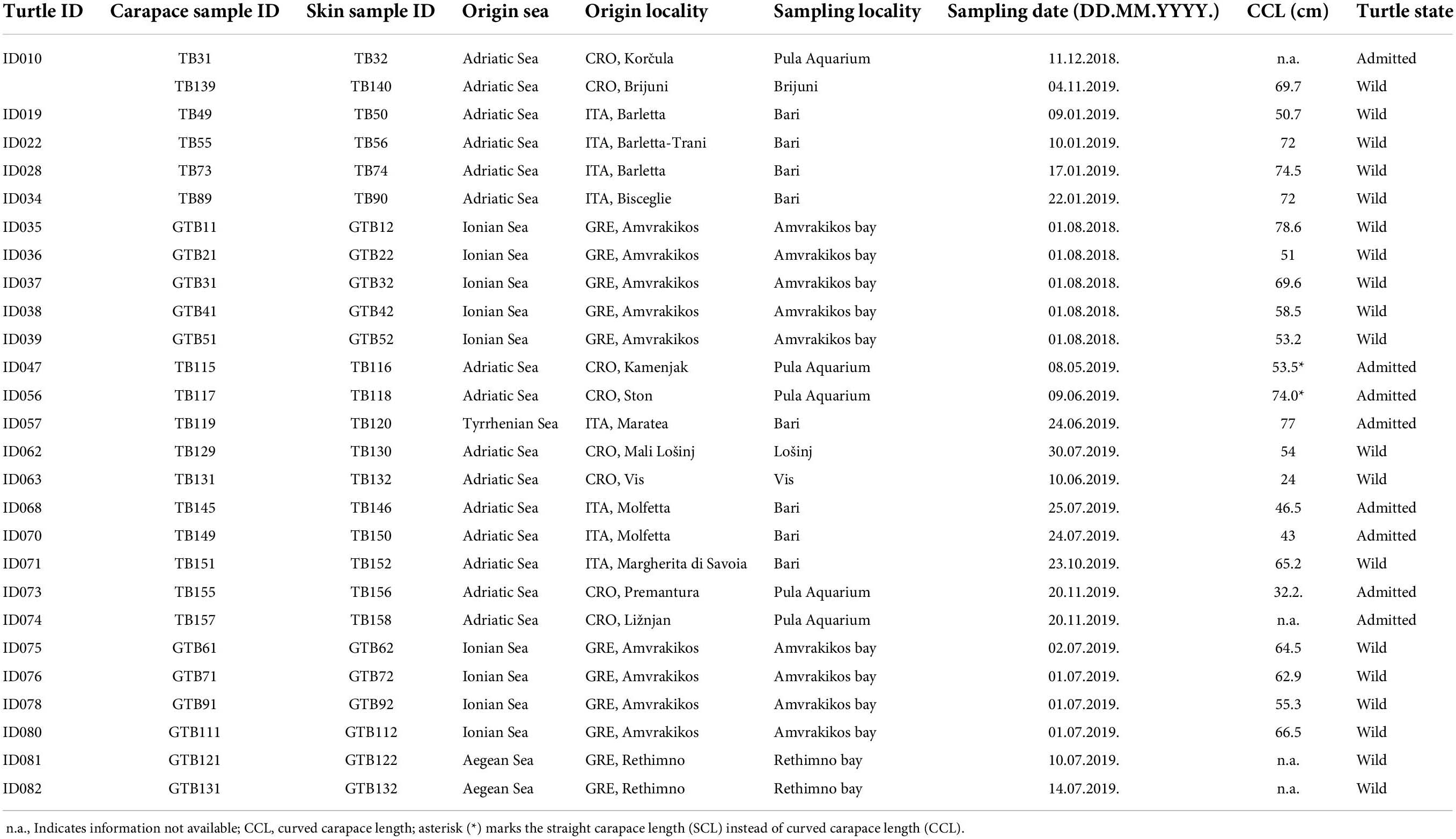
Table 1. Turtle and sample information.
DNA analysis
The DNA isolation and sequencing were performed in two batches, in 2019 (20 samples from ID10, ID19-39) and in 2020 (34 samples from ID10, ID47-82). The DNA was extracted from 0.25 g of an ethanol-free sample in duplicates using the DNeasy PowerSoil kit (QIAGEN, RRID:SCR_008539). The extraction protocol followed the manufacturer’s guidelines with several modifications (as described below). Samples were transferred into the PowerBead tubes and incubated in a sonicator at 50°C at 35 kHz for 15 min. The incubation times for C1, C2, and C3 solutions were extended (30 min at 65°C for C1 and 15 min at 4°C), and bead-beating was replaced with horizontal vortexing on IKA VXR basic Vibrax shaker (10 min at maximum speed of 2,200 rpm). The DNA was eluted with 50 μl of DNase-free molecular grade water (incubated at room temperature for 2 min). The quantity and purity of extracted DNA were measured by NanoDrop ND-1000 V3.8 spectrophotometer (Thermo Fisher). The extracted DNA samples were sent for 2 × 250 bp paired-end sequencing (Illumina MiSeq System, RRID:SCR_016379) of the 16S rRNA gene V4 region by 515F (5′-GTGYCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACNVGGGTWTCTAAT-3′) primers (Apprill et al., 2015; Parada et al., 2016), and the 18S rRNA gene V4 region by eukV4F (5′-CCAGCASCYGCGGTAATTCC-3′) and zigeukV4R (5′-ACTTTCGTTCTTGATYRATGA-3′) primers (Stoeck et al., 2010; Piredda et al., 2017) at Molecular Research MrDNA (Shallowater, TX, United States).
Sequence data processing and analysis
Sequences obtained from MrDNA were processed by FASTqProcessor (MrDNA), and all non-biological sequences were removed prior to exporting the data in QIIME2-readable format (“EMP protocol” multiplexed paired-end fastq format). The sequences were then imported to the QIIME2 (RRID:SCR_021258) environment, versions 2020.6 for 16S and 2021.4 for 18S (Bolyen et al., 2019). Demultiplexing of sequences was done by q2-demux plugin. DADA2 (q2-dada2 plugin) was used for sequence denoising (Callahan et al., 2016). A 18S rRNA sequences were truncated at 220 bp for forward and reverse sequences. Sequence alignment was performed with MAFFT (Katoh et al., 2002) and a phylogenetic tree was constructed with fasttree2 using q2-phylogeny (Price et al., 2010). Taxonomy was assigned to amplicon sequence variants (ASVs) via q2-feature-classifier (Bokulich et al., 2018) classify-sklearn naïve Bayes taxonomy classifier against the SILVA v.138 (99% 505F-806R nb classifier) (Quast et al., 2013) and PR2 4.13.0 (Guillou et al., 2013; del Campo et al., 2018) databases for 16S and 18S datasets, respectively. Prior to downstream analyses, mitochondria and chloroplast sequences were filtered from the 16S dataset, and metazoan and macroalgal sequences were filtered from the 18S dataset. For alpha and beta diversity analyses, we rarefied the 16S dataset to the sampling depth of 34 000 and the 18S dataset to 10 000 based on rarefaction curves (q2-diversity plugin). We calculated two alpha diversity indices via q2-diversity: observed ASVs (features) and Faith’s phylogenetic diversity (PD) index (Faith, 1992) for both 16S and 18S datasets, and made visualizations using boxplots. Beta diversity was estimated using three distance matrices via q2-diversity: Bray-Curtis, weighted UniFrac (Lozupone et al., 2007) and robust Aitchison’s distance (Aitchison and Shen, 1980; Aitchison and Ho, 1989; Martino et al., 2019). Principal Coordinates Analysis (PCoA) plots for Bray-Curtis and weighted UniFrac distance were produced using the q2-diversity plugin, and Robust Aitchison Principal Components Analysis (rPCA) was performed on non-rarefied data via DEICODE plugin (Martino et al., 2019). To compare the 16S and 18S datasets, we plotted the first principal coordinate (PC1) of each dataset’s robust Aitchison’s distance and performed the Procrustes analysis via q2-diversity. The permutational multivariate analysis of variance (PERMANOVA) with the q2-diversity plugin was used to test for significant differences between sample groups. Turtles from the Aegean and Tyrrhenian Seas were excluded from PERMANOVA calculations for “Origin Sea” due to the low number of samples in these two groups. For PERMANOVA statistic, turtles from Adriatic Sea were divided on East Adriatic (Croatian samples) and West Adriatic (Italian samples). PERMANOVA tested factor “Season” was obtained as following: samples obtained in spring and summer are put into “warm” category, while samples from autumn and winter are put into “cold” category. Data visualizations were made using ggplot2 (RRID:SCR_014601) (Wickham, 2016), phyloseq (RRID:SCR_013080) (McMurdie and Holmes, 2013), vegan (RRID:SCR_011950) (Oksanen et al., 2020), and pheatmap (RRID:SCR_016418) (Kolde, 2019) within R Studio (R Project for Statistical Computing, RRID:SCR_001905). The relative abundance of different groups of samples was calculated as a sum of the ASV count of selected taxon and then divided by the total sequence number in that sample group. Community composition was summarized by heatmaps (Figure 2) produced based on centered log-ratio (clr) transformed data from ASV counts.

Figure 2. Heatmaps of surface microbiota of loggerhead sea turtles’ skin and carapace samples for 16S (A) and 18S dataset (B). Each column represents individual sample. Samples are color-coded by categories (Origin sea, Body site and Sampling locality) in the first three rows of each heatmap.
Results
High throughput sequencing of 54 samples yielded 6,242,910 high quality 16S and 1,675,191 18S sequences. Median frequency per sample was 102,920.0 (min. 34,669.0; max. 257,399.0) for 16S while median frequency per sample for 18S was 20,048.5 (min. 1,634.0; max. 123,842.0). Sequences obtained for 16S and 18S were denoised to 17,636 ASVs and 1,917 ASVs, respectively (Supplementary Tables 2–4).
Community composition
Bacterial and archaeal microbiota
The microbial community showed the dominance of bacterial over archaeal taxa. The most abundant bacterial phylum in all samples was Proteobacteria, followed by Bacteriodota, Bdellovibrionota and Cyanobacteria (Figures 2, 3). Classes Gammaproteobacteria, Alphaproteobacteria and Bacteroidia dominated in all samples (Figure 2). Family Rhodobacteraceae was more abundant overall than any other bacterial family, followed by Moraxellaceae and Pseudoalteromonadaceae (Supplementary Table 5).
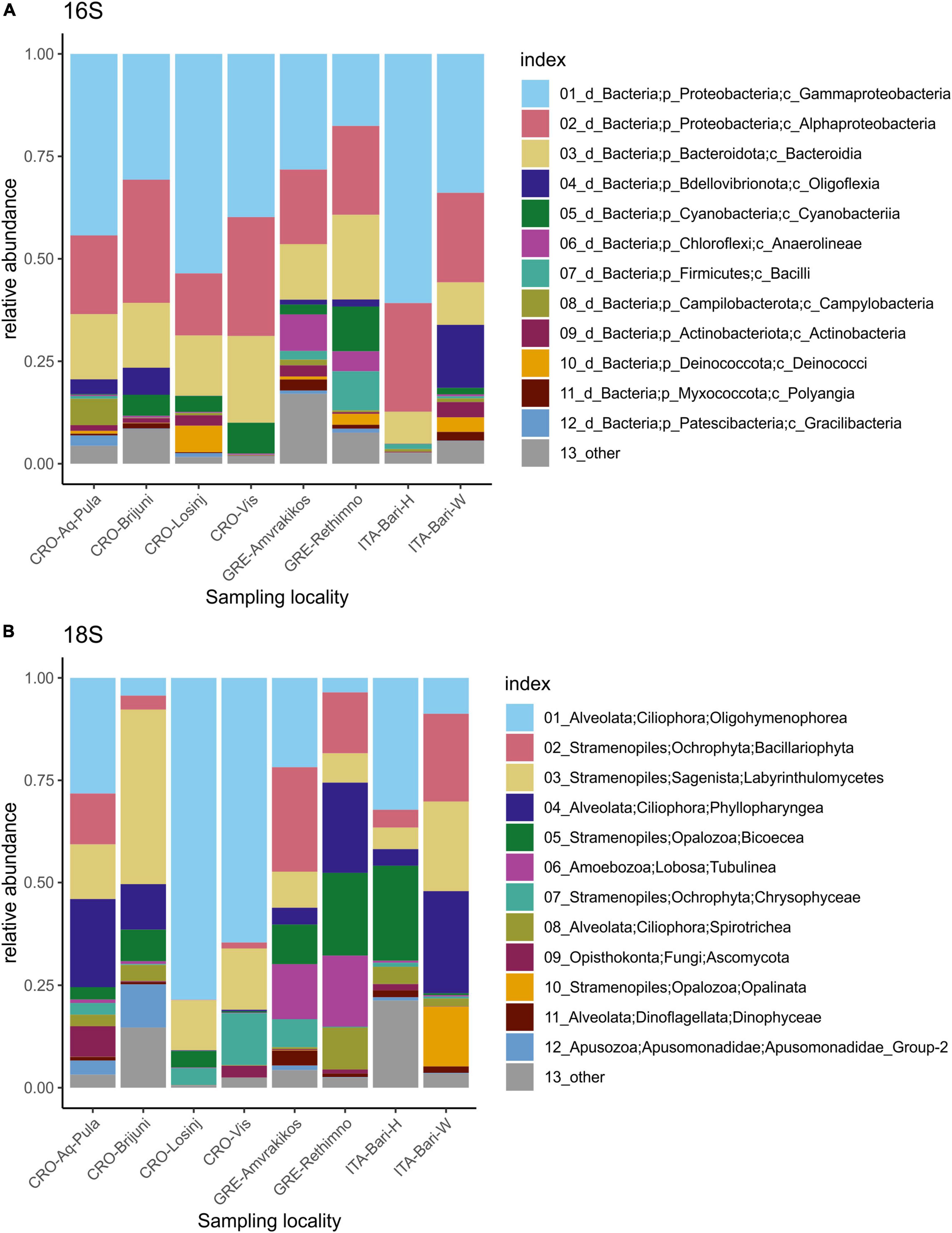
Figure 3. Relative abundances of 12 most abundant microbial taxa present on loggerhead sea turtles from the same sampling locality 16S dataset (A), 18S dataset (B).
There were five core features (ASVs) identified in 100% of samples in the 16S dataset, four belonging to class Gammaproteobacteria and one to Oligoflexia (Supplementary Table 6). These features are classified as an uncultured bacterium from order Oligoflexales, genus Pseudoalteromonas, an unidentified ASV from class Gammaproteobacteria, an uncultured bacterium from family Sedimenticolaceae, and an uncultured bacterium from family Saccharospirillaceae. In carapace samples, additional five core features were identified in 100% of the samples classified as genus Vibrio (Gammaproteobacteria), BD1-7 clade (family Spongiibacteraceae, Gammaproteobacteria), an uncultured bacterium from family Arcobacteraceae (Campylobacteria), genus Deinococcus (Deinococci), and genus Halarcobacter (Campylobacteria). In skin samples, additional seven core features were identified and classified as an uncultured bacterium from family Nannocystaceae (Polyangia, Myxococcota), family Rhodobacteraceae (Alphaproteobacteria), an uncultured bacterium from genus Psychrobacter (Gammaproteobacteria), an uncultured bacterium from genus Ahniella (Gammaproteobacteria), genus Tenacibaculum (Bacteroidia), family Stappiaceae (Alphaproteobacteria), and genus Poseidonibacter (Campylobacteria).
Within the 16S dataset, Cyanobacteria were the most abundant photoautotrophic prokaryotes, with Phormidesmiaceae and Paraspirulinaceae being the most abundant families (Figure 4). Phormidesmiaceae and Xenococcaceae dominated in carapace samples, whereas Paraspirulinaceae and Phormidesmiaceae were most abundant in skin samples. Many of the detected cyanobacterial sequences remained unclassified (Figure 4A, pink bars). On average, Cyanobacteria comprised 3% of all ASV sequences. In individual samples, this group accounted for 0.01–19.77% of all sequences (Figure 4B).
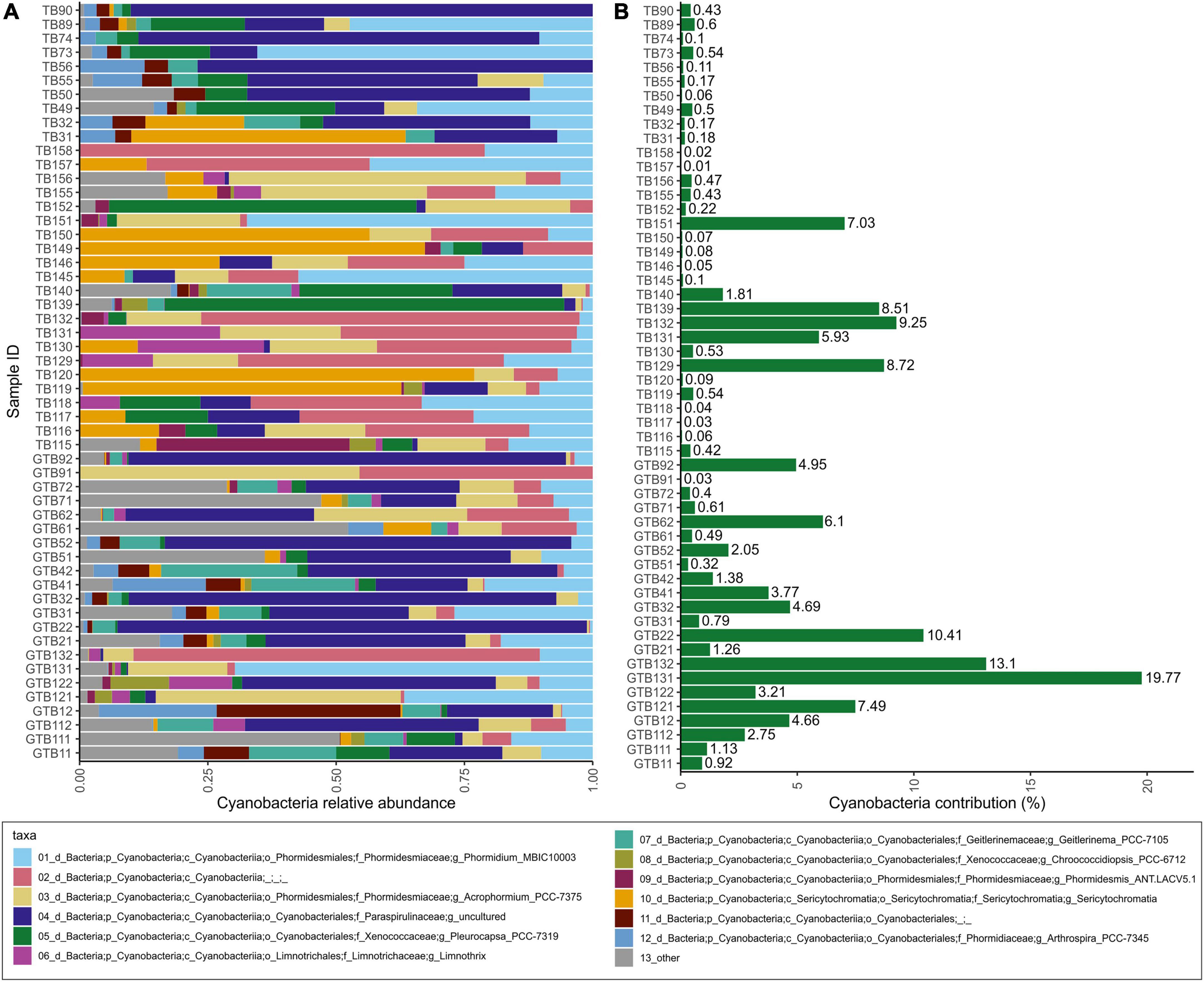
Figure 4. Relative abundances within the cyanobacterial group (A); contribution of Cyanobacteria to total bacterial community within a sample based on relative abundance (B).
Eukaryotic microbiota
The most abundant supergroups of micro-eukaryotes in the dataset were Alveolata and Stramenopiles. The dominant classes included Oligohymenophorea, Bacillariophyta, Labyrinthulomycetes, and Phyllopharyngea. The class Opalinata was highly prevalent in samples TB73 and TB74 (turtle ID28). Samples TB119 (carapace) and TB120 (skin) (from turtle ID57, the only animal sampled in the Tyrrhenian Sea) were dominated by Biocoeca. A high abundance of Ascomycota (Fungi) was recorded in the skin sample TB156 (Adriatic Sea), while Chrysophyceae were particularly abundant in the carapace sample GTB61 (Ionian Sea).
One core feature, belonging to the genus Zoothamnium (Oligohymenophorea, Ciliophora), was identified in all samples within the 18S dataset. An additional core feature of the carapace samples was found to be Nitzschia communis (Bacillariophyta). No additional core features were shared by all skin samples (Supplementary Table 6).
At the genus level (level 7 in the PR2 database), apart from the above-mentioned Zoothamnium, a taxon assigned to the level of “Raphid-pennate” group (Bacillariophyta) was found in all biofilm samples. In all carapace samples, Nitzschia (Bacillariophyta) and Labyrinthula (Labyrinthulomycetes) were identified as additional core genera. In 95% of all biofilm samples, the following core features were identified at the genus level: “Raphid-pennate” group (Bacillariophyta), Zoothamnium (Oligohymenophorea, Ciliophora), Nitzschia (Bacillariophyta) and Labyrinthula (Labyrinthulomycetes). In 95% of carapace samples, an additional core genus, Caecitellus (Opalozoa), was found. Four additional core genera were detected in 95% of skin samples: Thraustochytrium (Labyrinthulomycetes), Uronema (Oligohymenophorea), Labyrinthulaceae X (Labyrinthulomycetes), and Fistulifera (Bacillariophyta).
Alpha diversity
Alpha diversity indices for community richness (observed ASVs) and diversity [Faith’s Phylogenetic Diversity (PD) index] were highly variable and ranged from 127 to 2,833 (richness) and from 12.68 to 135.65 (diversity) for the bacterial community (Figures 5A,B). Bacterial communities from turtles in different seas and from different body sites (“Origin Sea” and “Body Site” categories as shown in Table 1) showed significant differences (Kruskal-Wallis H test, p < 0.05). Within the 16S dataset, carapace samples showed higher median values of richness [1.032; interquartile range (IQR) = 750.0] and diversity (52.23; IQR = 40.11) than skin samples (richness 811; IQR = 386.5 and diversity 46.47; IQR = 20.64). The highest median values of bacterial Faith’s PD were observed for samples from the Ionian Sea (75.36; IQR = 23.37), followed by the Aegean (59.39; IQR = 16.10) and Adriatic Seas (42.17; IQR = 18.92). The lowest Faith’s PD values were recorded for the hospitalized turtle ID057 from the Tyrrhenian Sea (20.69; IQR = 1.77).
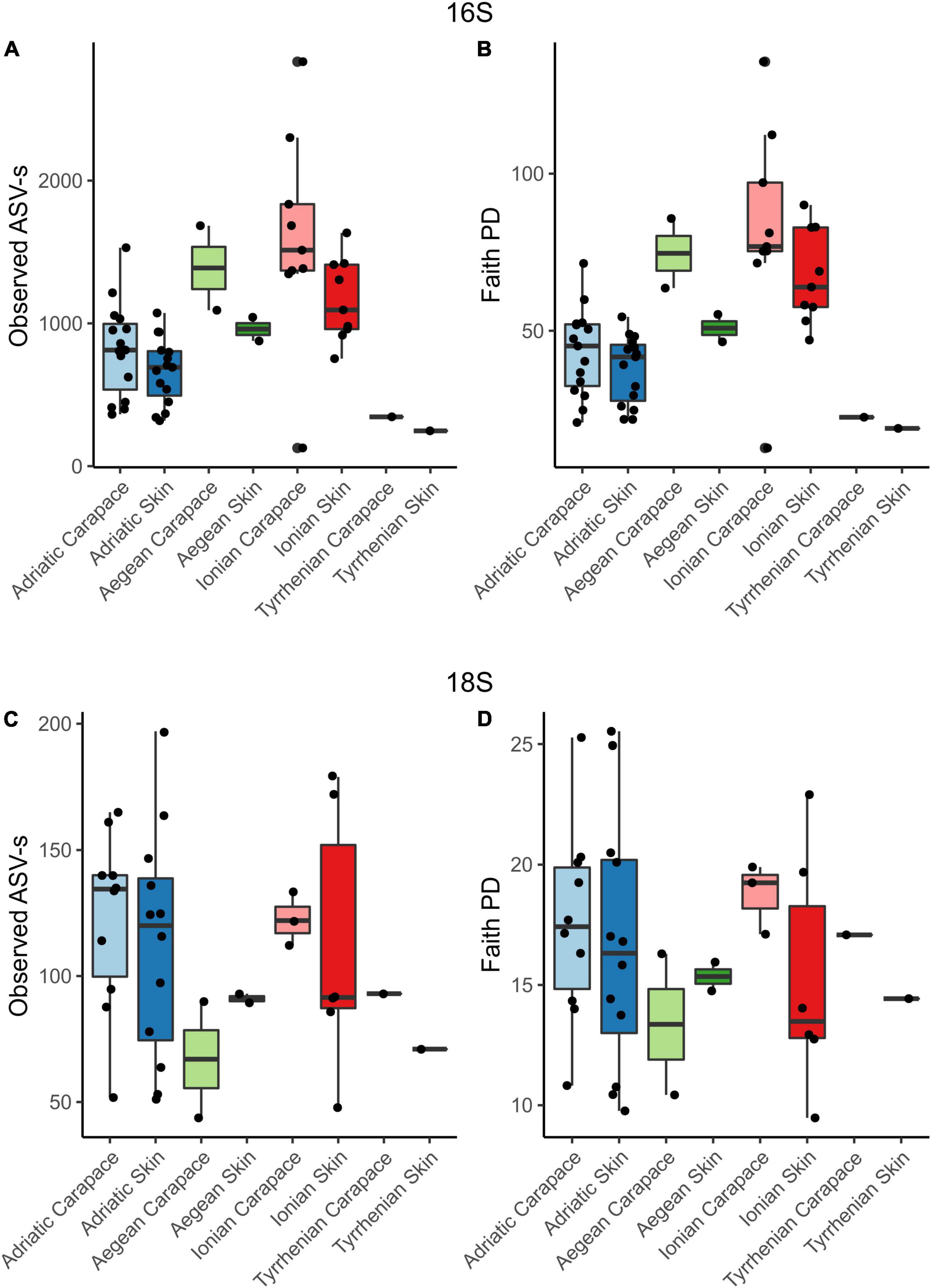
Figure 5. Observed ASV richness and Faith’s phylogenetic diversity index for 16S and 18S datasets for skin and carapace microbiome of loggerhead sea turtles sampled at four locations in the Mediterranean. Bar colors are paired and represent locations: blues, Adriatic; greens, Aegean; reds, Ionian; and no color, Tyrrhenian.
Microbial eukaryotes’ community ASVs richness ranged from 44 to 197, and diversity values ranged from 9.47 to 25.53 (Figures 5C,D) which is considerably lower comparing to the prokaryotes. The highest median value of micro-eukaryotic ASV richness was observed for the Adriatic Sea (124.5; IQR = 50.25), followed by Ionian Sea (112; IQR = 42.00), Aegean Sea (89.5; IQR = 13.00), and Tyrrhenian Sea (82; IQR = 11.00). The highest median value of micro-eukaryotic Faith’s PD was observed for Ionian Sea (17.11; IQR = 6.75), followed by Adriatic Sea (16.91; IQR = 6.00), Tyrrhenian Sea (15.76; IQR = 1.33) and Aegean Sea (15.35; IQR = 2.36), similar to the bacterial communities. Carapace microbial eukaryotes showed higher median values of ASVs richness (118; IQR = 44) and diversity (17.13; IQR = 3.61) than skin samples (richness 93; IQR = 58 and diversity 14.75; IQR = 6.75); however, no significant differences between the different seas or body sites were observed.
Beta diversity
Principal Components Analyses of robust Aitchison distance (rPCA) indicate groupings based on sampling locality and body site for prokaryotes (Figure 6A) and eukaryotes (Figure 6B). For the 16S dataset (Figure 6A) we can observe groupings based on sampled body site (carapace on the right and skin on the left) and sampling locality. ASVs that drive those groupings belong to uncultured Oligoflexales, Nannocystales, Rhodobacteraceae, Saccharospirillaceae, Pseudoalteromonas, and Vibrio. PERMANOVA results (Table 2) show that there is a significant difference (p < 0.05) between the bacterial and archaeal communities of skin and carapace body sites the seas of origin (only Adriatic and Ionian), turtle state (wild vs. admitted), and sampling season (warm vs. cold). The only non-significant value was detected between body site groups for unweighted UniFrac. The highest pseudo-F values for all distance matrices were observed between “Origin Sea” categories.
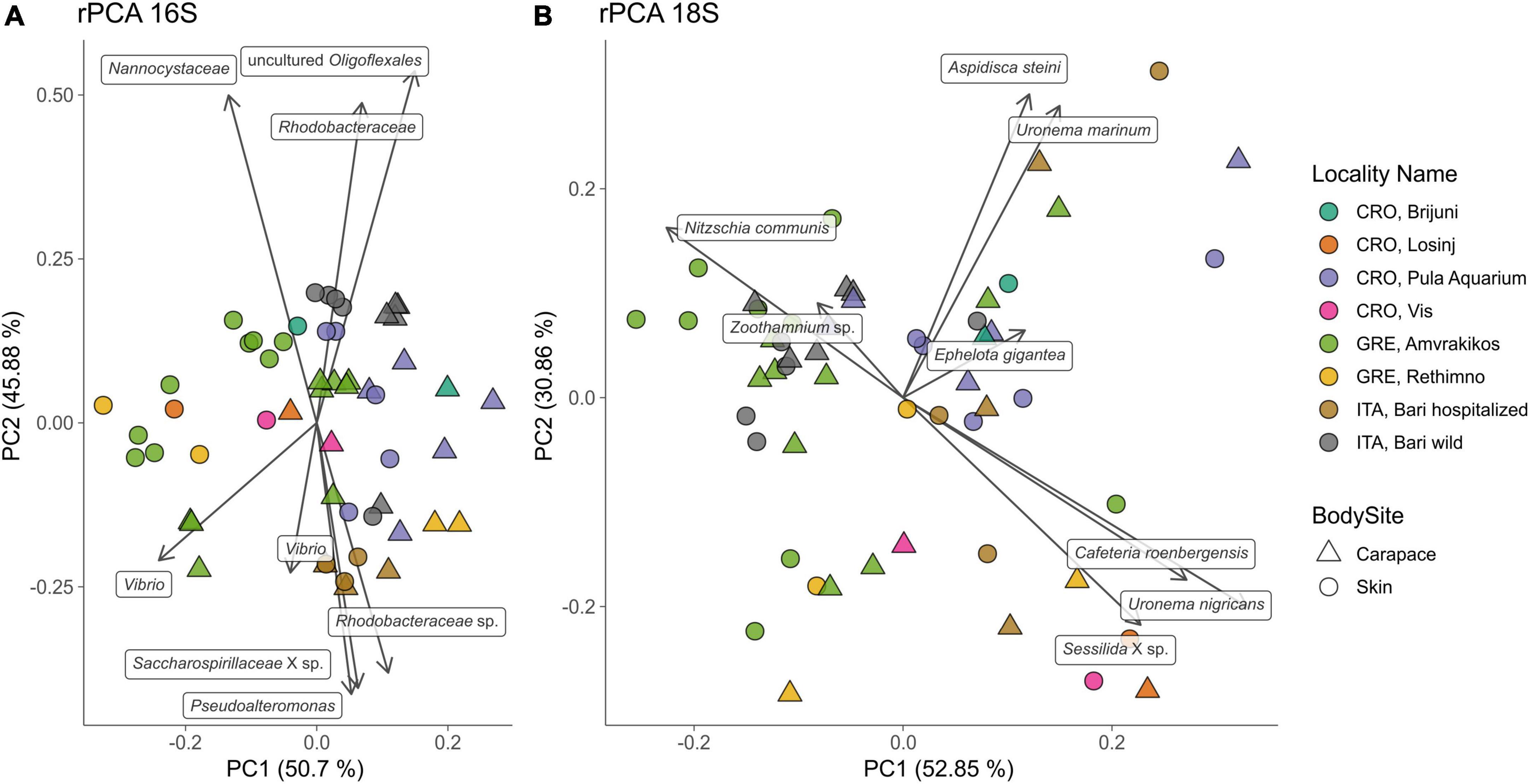
Figure 6. Principal component analysis (PCA) biplot of robust Aitchison distance for bacterial and archaeal (16S, A) and eukaryotic diversity (18S, B); arrows indicate individual highly ranked ASVs that contribute to the displayed positions of the samples; lowest taxonomic assignment of each ASV is written in textboxes at the end of each arrow. Sampling locality are indicated by color, body sites are indicated by shape.
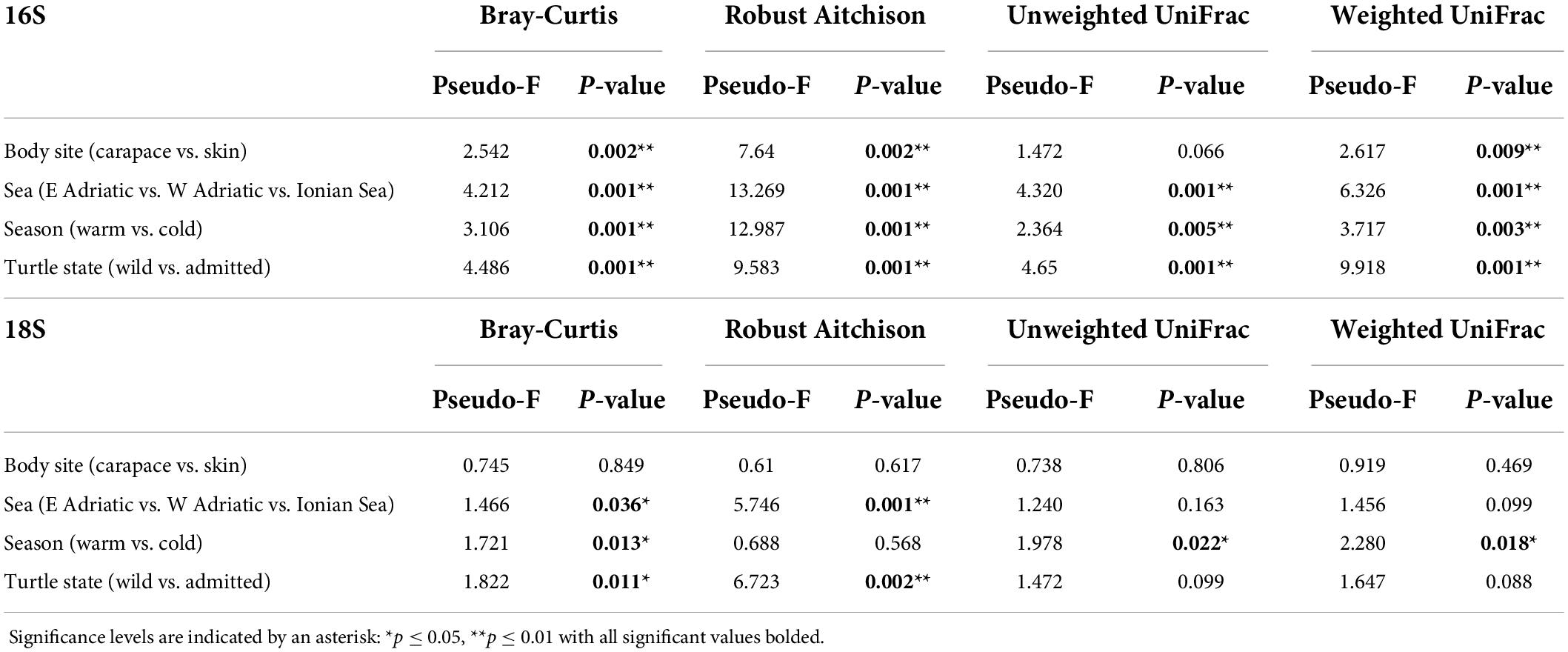
Table 2. Permutational multivariate analysis of variance (PERMANOVA) for Bray-Curtis, Robust Aitchison, unweighted and weighted UniFrac distance metrics.
For the 18S dataset (Figure 6B) we cannot observe clear groupings based on body site but there is an indication of samples grouping based on sampling locality. ASVs that drive the sample distribution for micro-eukaryotic communities (Figure 6B) belong to Ciliophora (Zoothamnium sp., Sessilida, Uronema marinum, Uronema nigricans, Ephelota gigantea, Aspidiscida steini), Nitzschia communis (Bacillariophyta) and Cafeteria roenbergensis (Bicoecea). PERMANOVA of eukaryotic communities showed no significant differences between sampled body sites. Significant differences between origin seas and turtle states were observed for Robust Aitchison and Bray-Curtis distances. According to all but one distance metrics tested, PERMANOVA showed a significant difference between sampling seasons (Table 2). To gain insight into the whole epi-microbiome (bacterial, archaeal and eukaryotic) we combined the principal components (PC1s) of the rPCA for 16S and 18S datasets where clear groupings based on sampling locality can be distinguished (Figure 7). To compare and detect any congruence between the bacterial and eukaryotic communities the rPCA ordinates of both datasets were compared by the Procrustes analysis (Supplementary Figure 2) which showed the bacterial, archaeal and eukaryotic dataset congruence is low (m2 = 0.93095, p = 0.043). Additional PCoA and rPCA ordinations are performed in order to visualize grouping of samples based on categories “Sampling locality” (Supplementary Figure 3), “Origin sea” (Supplementary Figures 4, 5), “Season” (Supplementary Figures 6, 7), and “Turtle state” (Supplementary Figures 8, 9).
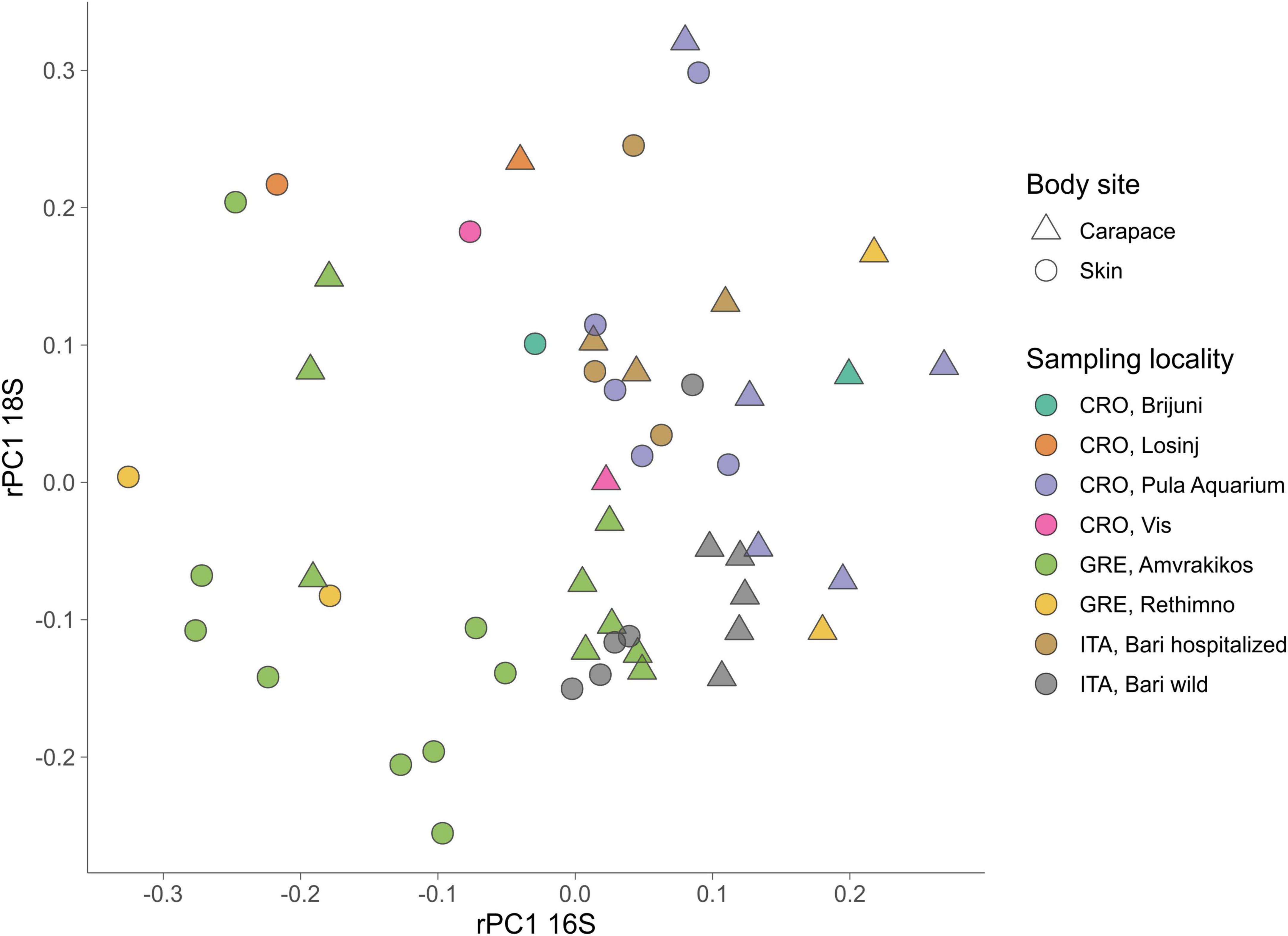
Figure 7. Combination plot of the first principal component (PC1) of PCAs based on robust Aitchison distance matrix for 16S (x-axis) and 18S (y-axis). Sampling locality are indicated by color, body sites are indicated by shape.
Discussion
In this study we provide insights into the epi-microbiota of loggerhead sea turtles using a combined 16S and 18S metabarcoding approach. Our results show that overall bacterial microbiota is dominated by a few classes of bacteria (Gammaproteobacteria, Alphaproteobacteria and Bacteroidia) and that the communities may differ depending on multiple extrinsic and intrinsic factors, which has been previously described in studies on other aquatic animals (as reviewed in Apprill, 2017). On the other hand, in spite of eukaryotic microbiota showing high heterogeneity, core taxa such as Oligohymenophorea, Bacillariophyta, Labyrinthulomycetes, and Phyllopharyngea were commonly present in the majority of samples. Despite the sampled turtles coming from different locations in the Mediterranean Sea, varying in age and health conditions, and being sampled in different seasons forming a diverse dataset, it is clear that several of the tested factors influenced their surface microbial community composition. Bacterial communities seem to be affected by the locality of origin, body site, turtle state, and sampling season while the eukaryotic microbiota followed a similar pattern, although to a lesser extent, and without detected differences between body sites.
The highest microbial diversity was observed on Ionian turtles from the lagoonal complex of the Amvrakikos Gulf, that is one of the most important and productive lagoonal complexes in Greece (Rees et al., 2013). The lagoonal shallow coastal aquatic systems, with a maximum depth of 65 m, are separated from the sea by sediment barriers and connected to it through channels, often characterized by salinity fluctuations and development of low dissolved oxygen conditions (Kapsimalis et al., 2005; Ferentinos et al., 2010). While the Amvrakikos Gulf offers a rich neritic foraging ground for subadult and adult loggerheads (Rees et al., 2013), the second locality with highest diversity is the Rethimno bay in northern Crete (Greece) that is an important nesting site for adult females (Margaritoulis and Rees, 2011). The Tyrrhenian Sea has shorter continental shelf than the Adriatic Sea (Cognetti et al., 2000) and possibly lower availability of the rich benthic environment as a source of microbes which could colonize the loggerhead’s body. That could explain lower diversity and richness of turtle-associated microbial communities from the Tyrrhenian Sea.
Bacterial diversity and richness of carapace samples was consistently higher than those of skin samples which could be explained by the large and rigid surface of the carapace covered by keratinous scutes that could allow for easier attachment and colonization of diverse microbes. Compared to the carapace, the skin of the neck and flippers (sampled in this study) is prone to higher mechanical disturbance caused by the turtle’s movements. Parks et al. (2020) reported a higher diversity and richness of microbial communities on the freshwater turtles’ carapace in comparison to the plastron, and provide the movement of the turtles as one of the possible explanations. Furthermore, Blasi et al. (2022) reported significant differences between microbial communities of differently positioned carapace scutes. The difference in bacterial community composition of anterior and posterior scutes of the sea turtle carapace might have been caused by different abiotic (hydrodynamics or sun exposure) and biotic factors (uneven distribution of macroorganisms across the carapace) affecting those areas (Blasi et al., 2022). The epi-microbiota of three juvenile loggerheads from the Tyrrhenian Sea harbored Firmicutes and Proteobacteria as the most prevalent phyla (Blasi et al., 2022). Contrastingly, in our dataset Proteobacteria were found to be the most abundant while Firmicutes were not among the highly abundant phyla. The most abundant bacterial family was Rhodobacteraceae which is known to be widely distributed in marine benthic habitats (Pohlner et al., 2019). Although the metabolic diversity within Rhodobacteraceae is great, they are mainly aerobic photo- and chemoheterotrophs, and purple non-sulfur bacteria that are known for anaerobic photosynthesis (Pujalte et al., 2014). Interestingly, we observed uncultured members of Psychrobacter and Tenacibaculum genera on all of the skin samples which were also reported as a part of the core microbiome on the humpback whales (Bierlich et al., 2018). This raises a question about Psychrobacter and Tenacibaculum genera members’ dependence on animal skin metabolites, possibly making them mutualistic or commensal to marine animals. It is worth mentioning that some species of Tenacibaculum are known as pathogens on fish skin (Nowlan et al., 2020), however, we cannot be certain of the exact ecological role on the turtle skin without further research.
The micro-eukaryotic taxa that were dominant in the majority of samples, ciliates Oligohymenophorea and Phyllopharyngea (Alveolata), are mainly free-living heterotrophs that could possibly graze on the other microbes colonizing the turtles’ surfaces. Commonly found Stramenopiles were mostly represented by diatoms (Bacillariophyta) and Labyrinthulomycetes (marine fungus-like organisms that produce filamentous webs for nutrient absorption). Diatoms are photosynthesizing microalgae with characteristic silica shells (Round et al., 1990) known for being among the first colonizers of submerged surfaces including marine vertebrates (Hooper et al., 2019). Our results show that diatoms are one of the major micro-eukaryotic groups present on sea turtles’ bodies and the most dominant phototrophs in those communities. Contrary to bacterial community, differences between carapace and skin community were not detected for micro-eukaryotes. This is also not in congruence with the morphological study on diatoms from loggerheads of where they reported higher diversity and richness of carapace than in skin diatom community. Common epiphytic and epipelic diatom genera were found in abundance on carapace while putatively epizoic taxa were dominating in skin diatom samples (Van de Vijver et al., 2020).
Light availability on the sea turtle surfaces enables the development of phototrophic microbes that cannot be found as a part of the endozoic microbiome. Moreover, unlike endozoic microbial communities which are dependent on nutrient inflow from the host, epizoic communities are probably dependent mostly on the nutrients available in the surrounding environment and from the primary producers in those communities. The microalgae and cyanobacteria, i.e., main phototrophic taxa in the epizoic biofilms are usually firmly attached and embedded in thick extracellular organic matrix. However, protozoans and metazoan grazers successfully adapted to feed on biofilm-dwelling microalgae and cyanobacteria that forms a strong trophic intra-biofilm link between primary and secondary producers (Weitere et al., 2018). The most studied primary producers associated with sea turtles are diatoms (Majewska et al., 2015). Recent morphology-based studies on turtle-associated diatoms revealed that they are highly abundant, diverse, and that there are several putative obligate epizoic diatom taxa (Robinson et al., 2016; Rivera et al., 2018; Van de Vijver et al., 2020). Besides diatoms, photoautotrophic Chrysophyceae and Dinophyceae were detected in noticeable abundances, and Chrysophyte stomatocysts of unknown species were previously reported on the sea turtle carapace (Pang et al., 2021). Additionally, Labyrinthulomycetes that were the third most abundant taxon in our samples are known to be mainly decomposers or, rarely, parasitic (Tsui et al., 2009) with recently emphasized importance in carbon sequestration (Bai et al., 2021). Either turtle- or microbe-derived particulate carbon (photoautotrophs or heterotrophs) could provide Labyrinthulomycetes with significant amounts of energy sources leading to their high relative abundance across samples.
Bacterial photoautotrophic communities are dominated by Cyanobacteria with the most common in our samples being filamentous cyanobacteria like Phormidium and Leptolyngbya (Acrophorium), both known for cyanotoxin production (Frazão et al., 2010; McAllister et al., 2016; Li et al., 2019). It has been observed that cyanobacterial toxic compounds can interfere with composition and function of animal intestinal microbiome (Duperron et al., 2019; Li et al., 2019; Sehnal et al., 2021). Blasi et al. (2022) also highlighted the presence of Cyanobacteria on anterior scutes, specifically families Pseudanabenaceae and Rivulariaceae. However, Phormidesmiaceae, Paraspirulinaceae and Xenococcaceae were prevalent in our dataset. All reported cyanobacterial genera in our study are commonly found in marine benthic habitats forming colonies and cyanobacterial mats (Komárek et al., 2014). Sea turtles seem to provide additional surfaces for cyanobacterial colonization and could act as a highly mobile reservoir with unknown implications for the host’s health and effects on the environment.
It should be noted, however, that observed significant differences in multiple groupings of microbial communities in this study could be explained by overlapping metadata categories (e.g., wild animals being sampled mostly in Greece and during summer months) that could not be controlled for within our study design due to the unpredictability/stochasticity of opportunistic sampling. Additionally, reference databases play an important role in investigating microbial eukaryotes, as we cannot grasp the full diversity of micro-eukaryotes through metabarcoding alone because of a lack of sequenced representatives and eukaryotes often being overlooked as a part of microbial communities (Lind and Pollard, 2021 and references therein). In our study, a major portion of the cyanobacterial ASVs could not be properly identified via metabarcoding as the current version of SILVA reference database taxonomy is based on Bergey’s Manual of Systematic Bacteriology (Boone et al., 2001) in which cyanobacterial taxonomy higher than genus is not defined. Therefore, SILVA and Genome Taxonomy Database (GTDB) (Quast et al., 2013) proposed their own names for some taxa based on 16S rRNA phylogeny that is not in agreement with the currently valid cyanobacterial taxonomy in the CyanoDB database (Komárek et al., 2014). As microbial eukaryotes and cyanobacteria are an important part of microbial communities associated with Mediterranean loggerhead sea turtles, further efforts in their characterization are needed to reconcile multiple taxonomy databases and better understand the turtle-associated taxa and their possible effects on the host.
The Mediterranean loggerheads are widely distributed large hard-shelled top predators, and a highly migratory species which occupies different marine habitats at different life stages. Their major ecological role in bioturbation, energy flow, trophic status, mineral cycling, soil dynamics and connectivity between habitats makes it a keystone species in Mediterranean marine environment (Casale et al., 2018). This research brings us one step closer to much needed understanding of the complexity of microbial communities associated with loggerheads and wild animals in general. We show in this study that microbial communities of loggerhead sea turtles are rich and highly diverse with reservoirs of microbial taxa potentially important both for turtles’ and the ecosystem’s state. Moreover, DNA-based surveys focusing on epizoic bacterial, archaeal and eukaryotic microbiota could prove to be a valuable addition to non-invasive methods for monitoring the status of endangered marine species and their environment.
Data availability statement
The amplicon sequence data are deposited in the European Nucleotide Archive (ENA) under accession numbers PRJEB51458 for 16S and PRJEB51472 for 18S.
Ethics statement
Sampling was performed in accordance with the 1975 Declaration of Helsinki, as revised in 2013 and the applicable national laws. The sampling at the Sea Turtle Clinic (Bari, Italy) was conducted with the permission of the Department of Veterinary Medicine Animal Ethic Committee (Authorization # 4/19), while sampling in Croatia was done in accordance with the authorization of the Marine Turtle Rescue Centre by the Ministry of Environment and Energy of the Republic of Croatia. Sampling activities in Greece were carried out with permission from the Hellenic Ministry of Agriculture and Environment.
Author contributions
SB, RG, and RM designed the study. LK, KF, AT, MC, AD, AP, and SB collected the samples. KF and MM carried out the laboratory work. LK and KF conducted the bioinformatics, statistical analyses, and data visualization and interpretation. LK wrote the first draft of the manuscript. SB conceived project and obtained the funding. All authors revised the manuscript and approved the final version of the manuscript.
Funding
This study was supported by the Croatian Science Foundation under the project number UIP-2017-05-5635 (Loggerhead sea turtle (Caretta caretta) microbiome: Insight into endozoic and epizoic communities – TurtleBIOME). The work of doctoral student KF has been fully supported by the “Young Researchers’ Career Development Project – Training of Doctoral Students” of the Croatian Science Foundation funded by the European Union from the European Social Fund.
Acknowledgments
We are thankful to Milena Mičić, Karin Gobić Medica, and the rest of the staff from the Marine Turtle Rescue Center (Aquarium Pula) as well as Mateja Zekan and Draško Holcer from Blue World Institute for sample collection and to Hrvoje Višić for help with laboratory work and DNA isolations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.907368/full#supplementary-material
Supplementary Table 1 | Metadata table adapted for input in QIIME2, with information about samples and sampled sea turtles.
Supplementary Table 2 | Sequencing results (raw counts) and taxonomy assignments of 16S dataset per ASV for all samples in this study.
Supplementary Table 3 | Sequencing results (raw counts) and taxonomy assignments of 18S dataset per ASV for all samples in this study.
Supplementary Table 4 | Sequencing results (raw counts) and taxonomy assignments of filtered cyanobacterial sequences of 16S dataset per ASV for all samples in this study.
Supplementary Table 5 | Relative abundance and absolute sequence count of bacterial and archaeal families found on skin and carapace of loggerhead sea turtles.
Supplementary Table 6 | Core features of loggerhead sea turtles from 16S to 18S amplicon sequencing dataset.
References
Abdelrhman, K. F. A., Bacci, G., Mancusi, C., Mengoni, A., Serena, F., and Ugolini, A. (2016). A first insight into the gut microbiota of the Sea Turtle Caretta caretta. Front. Microbiol. 7:1060. doi: 10.3389/FMICB.2016.01060
Aitchison, J., and Ho, C. H. (1989). The multivariate poisson-log normal distribution. Biometrika 76, 643–653. doi: 10.1093/biomet/76.4.643
Aitchison, J., and Shen, S. M. (1980). Logistic-normal distributions: some properties and uses. Biometrika 67, 261–272. doi: 10.2307/2335470
Alduina, R., Gambino, D., Presentato, A., Gentile, A., Sucato, A., Savoca, D., et al. (2020). Is Caretta caretta a carrier of antibiotic resistance in the Mediterranean Sea? Antibiotics 9:116. doi: 10.3390/ANTIBIOTICS9030116
Apprill, A. (2017). Marine animal microbiomes: toward understanding host–microbiome interactions in a changing ocean. Front. Mar. Sci. 4:222. doi: 10.3389/FMARS.2017.00222
Apprill, A., Mcnally, S., Parsons, R., and Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137. doi: 10.3354/AME01753
Arizza, V., Vecchioni, L., Caracappa, S., Sciurba, G., Berlinghieri, F., Gentile, A., et al. (2019). New insights into the gut microbiome in loggerhead sea turtles Caretta caretta stranded on the Mediterranean coast. PLoS One 14:e0220329. doi: 10.1371/JOURNAL.PONE.0220329
Aylagas, E., Borja, Á, Tangherlini, M., Dell’Anno, A., Corinaldesi, C., Michell, C. T., et al. (2017). A bacterial community-based index to assess the ecological status of estuarine and coastal environments. Mar. Pollut. Bull. 114, 679–688. doi: 10.1016/J.MARPOLBUL.2016.10.050
Azari, M., Farjad, Y., Nasrolahi, A., De Stefano, M., Ehsanpour, M., Dobrestov, S., et al. (2020). Diatoms on sea turtles and floating debris in the Persian Gulf (Western Asia). Phycologia 59, 292–304. doi: 10.1080/00318884.2020.1752533
Becker, R. A., Wilks, A. R., and Brownrigg, R. (2018). mapdata: Extra Map Databases. Available online at: https://CRAN.R-project.org/package=mapdata (accessed July 21, 2022).
Becker, R. A., Wilks, A. R., Brownrigg, R., Minka, T., and Deckmyn, A. (2021). maps: Draw Geographical Maps. Available online at: https://CRAN.R-project.org/package=maps (accessed July 21, 2022).
Bai, M., Xie, N., He, Y., Li, J., Collier, J. L., Hunt, D. E., et al. (2021). Vertical community patterns of Labyrinthulomycetes protists reveal their potential importance in the oceanic biological pump. Environ. Microbiol. 24, 1703–1713. doi: 10.1111/1462-2920.15709
Bailey, J. B., Lamb, M., Walker, M., Weed, C., and Craven, K. S. (2018). Detection of potential fungal pathogens Fusarium falciforme and F. keratoplasticum in unhatched loggerhead turtle eggs using a molecular approach. Endanger. Species Res. 36, 111–119. doi: 10.3354/ESR00895
Belkaid, Y., and Segre, J. (2014). Dialogue between skin microbiota and immunity. Science 346, 954–959. doi: 10.1126/SCIENCE.1260144
Belkaid, Y., and Tamoutounour, S. (2016). The influence of skin microorganisms on cutaneous immunity. Nat. Rev. Immunol. 16, 353–366. doi: 10.1038/NRI.2016.48
Biagi, E., D’Amico, F., Soverini, M., Angelini, V., Barone, M., Turroni, S., et al. (2019). Faecal bacterial communities from Mediterranean loggerhead sea turtles (Caretta caretta). Environ. Microbiol. Rep. 11, 361–371. doi: 10.1111/1758-2229.12683
Bierlich, K. C., Miller, C., DeForce, E., Friedlaender, A. S., Johnston, D. W., and Apprill, A. (2018). Temporal and regional variability in the skin microbiome of humpback whales along the Western Antarctic Peninsula. Appl. Environ. Microbiol. 84:e02574-17. doi: 10.1128/AEM.02574-17
Blasi, M. F., Rotini, A., Bacci, T., Targusi, M., Ferraro, G. B., Vecchioni, L., et al. (2022). On Caretta caretta’s shell: first spatial analysis of micro- and macro-epibionts on the Mediterranean loggerhead sea turtle carapace. Mar. Biol. Res. 17, 762–774. doi: 10.1080/17451000.2021.2016840
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 1–17. doi: 10.1186/S40168-018-0470-Z
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Boone, D. R., Castenholz, R. W., and Garrity, G. M. (2001). Bergey’s Manual Of Systematic Bacteriology. New York, NY: Springer.
Broderick, A. C., Glen, F., Godley, B. J., and Hays, G. C. (2002). Estimating the number of green and loggerhead turtles nesting annually in the Mediterranean. Oryx 36, 227–235. doi: 10.1017/S0030605302000431
Brooks, A. W., Kohl, K. D., Brucker, R. M., van Opstal, E. J., and Bordenstein, S. R. (2016). Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 14:e2000225. doi: 10.1371/journal.pbio.2000225
Byrd, A. L., Belkaid, Y., and Segre, J. A. (2018). The human skin microbiome. Nat. Rev. Microbiol. 16, 143–155. doi: 10.1038/nrmicro.2017.157
Cafarchia, C., Paradies, R., Figueredo, L. A., Iatta, R., Desantis, S., Di Bello, A. V. F., et al. (2020). Fusarium spp. in Loggerhead sea turtles (Caretta caretta): from colonization to infection. Vet. Pathol. 57, 139–146. doi: 10.1177/0300985819880347
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/NMETH.3869
Casale, P., Broderick, A. C., Camiñas, J. A., Cardona, L., Carreras, C., Demetropoulos, A., et al. (2018). Mediterranean sea turtles: current knowledge and priorities for conservation and research. Endanger. Species Res. 36, 229–267. doi: 10.3354/ESR00901
Cognetti, G., Lardicci, C., Abbiati, M., and Castelli, A. (2000). The Adriatic Sea and the Tyrrhenian Sea. Seas Millenn. Environ. Eval. 1, 267–284.
Cordier, T., Lanzén, A., Apothéloz-Perret-Gentil, L., Stoeck, T., and Pawlowski, J. (2019). Embracing environmental genomics and machine learning for routine biomonitoring. Trends Microbiol. 27, 387–397. doi: 10.1016/J.TIM.2018.10.012
del Campo, J., Kolisko, M., Boscaro, V., Santoferrara, L. F., Nenarokov, S., Massana, R., et al. (2018). EukRef: phylogenetic curation of ribosomal RNA to enhance understanding of eukaryotic diversity and distribution. PLoS Biol. 16:e2005849. doi: 10.1371/JOURNAL.PBIO.2005849
Duperron, S., Halary, S., Habiballah, M., Gallet, A., Huet, H., Duval, C., et al. (2019). Response of fish gut microbiota to toxin-containing cyanobacterial extracts: a microcosm study on the medaka (Oryzias latipes). Environ. Sci. Technol. Lett. 6, 341–347. doi: 10.1021/ACS.ESTLETT.9B00297
Faith, D. P. (1992). Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10. doi: 10.1016/0006-3207(92)91201-3
Ferentinos, G., Papatheodorou, G., Geraga, M., Iatrou, M., Fakiris, E., Christodoulou, D., et al. (2010). Fjord water circulation patterns and dysoxic/anoxic conditions in a Mediterranean semi-enclosed embayment in the Amvrakikos Gulf, Greece. Estuar. Coast. Shelf Sci. 88, 473–481. doi: 10.1016/j.ecss.2010.05.006
Filek, K., Trotta, A., Gračan, R., Di Bello, A., Corrente, M., and Bosak, S. (2021). Characterization of oral and cloacal microbial communities of wild and rehabilitated loggerhead sea turtles (Caretta caretta). Anim. Microbiome 3:59. doi: 10.1186/S42523-021-00120-5
Frazão, B., Martins, R., and Vasconcelos, V. (2010). Are known cyanotoxins involved in the toxicity of picoplanktonic and filamentous north atlantic marine cyanobacteria? Mar. Drugs 8:1908. doi: 10.3390/MD8061908
Frick, M., and Pfaller, J. B. (2013). “Sea turtle epibiosis,” in The Biology of Sea Turtles, Vol. III, eds J. Wyneken, K. J. Lohmann, and J. A. Musick (Boca Raton, FL: CRC Press), 304–347.
Guillou, L., Bachar, D., Audic, S., Bass, D., Berney, C., Bittner, L., et al. (2013). The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41, D597–D604. doi: 10.1093/NAR/GKS1160
Hollenberg, G. J. (1971). Phycological notes. V. New species of marine algae from California. Phycologia 10, 11–16.
Hooper, R., Brealey, J., van der Valk, T., Alberdi, A., Durban, J., Fearnbach, H., et al. (2019). Host-derived population genomics data provides insights into bacterial and diatom composition of the killer whale skin. Mol. Ecol. 28, 484–502. doi: 10.1111/MEC.14860
Kanjer, L., Majewska, R., Van de Vijver, B., Gračan, R., Lazar, B., and Bosak, S. (2020). Diatom diversity on the skin of frozen historic loggerhead sea turtle specimens. Diversity 12:383. doi: 10.3390/d12100383
Kapsimalis, V., Pavlakis, P., Poulos, S. E., Alexandri, S., Tziavos, C., Sioulas, A., et al. (2005). Internal structure and evolution of the Late Quaternary sequence in a shallow embayment: the Amvrakikos Gulf, NW Greece. Mar. Geol. 222-223, 399–418.
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/NAR/GKF436
Keeley, N., Wood, S. A., and Pochon, X. (2018). Development and preliminary validation of a multi-trophic metabarcoding biotic index for monitoring benthic organic enrichment. Ecol. Indic. 85, 1044–1057. doi: 10.1016/J.ECOLIND.2017.11.014
Komárek, J., Kaštovský, J., Mareš, J., and Johansen, J. R. (2014). Taxonomic classification of cyanoprokaryotes (cyanobacterial genera) 2014, using a polyphasic approach. Preslia 86, 295–335.
Lau, K. E. M., Washington, V. J., Fan, V., Neale, M. W., Lear, G., Curran, J., et al. (2015). A novel bacterial community index to assess stream ecological health. Freshw. Biol. 60, 1988–2002. doi: 10.1111/FWB.12625
Li, J., Chen, C., Zhang, T., Liu, W., Wang, L., Chen, Y., et al. (2019). μEvaluation of microcystin-LR absorption using an in vivo intestine model and its effect on zebrafish intestine. Aquat. Toxicol. 206, 186–194. doi: 10.1016/J.AQUATOX.2018.11.014
Lind, A. L., and Pollard, K. S. (2021). Accurate and sensitive detection of microbial eukaryotes from whole metagenome shotgun sequencing. Microbiome 9:58. doi: 10.1186/s40168-021-01015-y
Lozupone, C. A., Hamady, M., Kelley, S. T., and Knight, R. (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. doi: 10.1128/AEM.01996-06
Majewska, R., Santoro, M., Bolaños, F., Chaves, G., and De Stefano, M. (2015). Diatoms and other epibionts associated with Olive Ridley (Lepidochelys olivacea) sea turtles from the Pacific Coast of Costa Rica. PLoS One 10:e0130351. doi: 10.1371/journal.pone.0130351
Margaritoulis, D., and Rees, A. F. (2011). Loggerhead turtles nesting at Rethymno, Greece, prefer the Aegean Sea as their main foraging area. Mar. Turt. Newsl. 131, 12–14.
Martino, C., Morton, J. T., Marotz, C. A., Thompson, L. R., Tripathi, A., Knight, R., et al. (2019). A novel sparse compositional technique reveals microbial perturbations. mSystems 4:e00016-19. doi: 10.1128/mSystems.00016-19
McAllister, T. G., Wood, S. A., and Hawes, I. (2016). The rise of toxic benthic Phormidium proliferations: a review of their taxonomy, distribution, toxin content and factors regulating prevalence and increased severity. Harmful Algae 55, 282–294. doi: 10.1016/j.hal.2016.04.002
McKnight, D. T., Zenger, K. R., Alford, R. A., and Huerlimann, R. (2020). Microbiome diversity and composition varies across body areas in a freshwater turtle. Microbiology 166, 440–452. doi: 10.1099/mic.0.000904
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/JOURNAL.PONE.0061217
Nowlan, J. P., Lumsden, J. S., and Russell, S. (2020). Advancements in characterizing tenacibaculum infections in Canada. Pathogens 9:1029. doi: 10.3390/pathogens9121029
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Lagendre, P., McGlinn, D., et al. (2020). vegan community ecology package version 2.5-7. Available online at: https://www.researchgate.net/publication/346579465_vegan_community_ecology_package_version_25-7_November_2020 (accessed November 16, 2021).
Pace, A., Dipineto, L., Fioretti, A., and Hochscheid, S. (2019). Loggerhead sea turtles as sentinels in the western Mediterranean: antibiotic resistance and environment-related modifications of Gram-negative bacteria. Mar. Pollut. Bull. 149:110575. doi: 10.1016/J.MARPOLBUL.2019.110575
Pang, W., Bosak, S., and de Vijver, B. V. (2021). First report of the marine chrysophycean stomatocysts from the carapace biofilm of a Mediterranean loggerhead sea turtle. Nova Hedwig 113, 33–44. doi: 10.1127/NOVA_HEDWIGIA/2021/0643
Parada, A. E., Needham, D. M., and Fuhrman, J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Parks, M., Kedy, C., and Skalla, C. (2020). Consistent patterns in 16S and 18S microbial diversity from the shells of the common and widespread red-eared slider turtle (Trachemys scripta). PLoS One 15:e0244489. doi: 10.1371/journal.pone.0244489
Pinou, T., Domènech, F., Lazo-Wasem, E., Majewska, R., Pfaller, J., Zardus, J., et al. (2019). Standardizing sea turtle epibiont sampling: outcomes of the Epibiont Workshop at the 37 th International Sea Turtle Symposium. Mar. Turtle Newslett. 157, 22–32. doi: 10.13140/RG.2.2.31843.81440
Piredda, R., Tomasino, M. P., D’Erchia, A. M., Manzari, C., Pesole, G., Montresor, M., et al. (2017). Diversity and temporal patterns of planktonic protist assemblages at a Mediterranean Long Term Ecological Research site. FEMS Microbiol. Ecol. 93:200. doi: 10.1093/FEMSEC/FIW200
Pohlner, M., Dlugosch, L., Wemheuer, B., Mills, H., Engelen, B., and Reese, B. K. (2019). The majority of active rhodobacteraceae in marine sediments belong to uncultured genera: a molecular approach to link their distribution to environmental conditions. Front. Microbiol. 10:659. doi: 10.3389/FMICB.2019.00659
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/JOURNAL.PONE.0009490
Pujalte, M. J., Lucena, T., Ruvira, M. A., Arahal, D. R., and Macián, M. C. (2014). “The Family Rhodobacteraceae,” in The Prokaryotes: Alphaproteobacteria and Betaproteobacteria, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), 439–512.
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/NAR/GKS1219
Rees, A. F., Margaritoulis, D., Newman, R., Riggall, T. E., Tsaros, P., Zbinden, J. A., et al. (2013). Ecology of loggerhead marine turtles Caretta caretta in a neritic foraging habitat: movements, sex ratios and growth rates. Mar. Biol. 160, 519–529. doi: 10.1007/s00227-012-2107-2
Rivera, S. F., Vasselon, V., Ballorain, K., Carpentier, A., Wetzel, C. E., Ector, L., et al. (2018). DNA metabarcoding and microscopic analyses of sea turtles biofilms: complementary to understand turtle behavior. PLoS One 13:e0195770. doi: 10.1371/journal.pone.0195770
Robinson, N. J., Majewska, R., Lazo-Wasem, E. A., Nel, R., Paladino, V., Rojas, L., et al. (2016). Epibiotic diatoms are universally present on all sea turtle species. PLoS One 11:e0157011. doi: 10.1371/journal.pone.0157011
Ross, A. A., Müller, K. M., Weese, J. S., and Neufeld, J. D. (2018). Comprehensive skin microbiome analysis reveals the uniqueness of human skin and evidence for phylosymbiosis within the class Mammalia. Proc. Natl. Acad. Sci. U.S.A. 115, E5786–E5795. doi: 10.1073/PNAS.1801302115
Ross, A. A., Rodrigues Hoffmann, A., and Neufeld, J. D. (2019). The skin microbiome of vertebrates. Microbiome 7, 1–14. doi: 10.1186/S40168-019-0694-6
Round, F. E., Crawford, R. M., and Mann, D. G. (1990). The Diatoms: Biology and Morphology of the Genera. Cambridge, MA: Cambridge University Press.
Sanford, J., and Gallo, R. (2013). Functions of the skin microbiota in health and disease. Semin. Immunol. 25, 370–377. doi: 10.1016/J.SMIM.2013.09.005
Scharschmidt, T., and Fischbach, M. (2013). What lives on our skin: ecology, genomics and therapeutic opportunities of the skin microbiome. Drug Discov. Today Dis. Mech. 10, e83–e89. doi: 10.1016/J.DDMEC.2012.12.003
Scheelings, T. F., Moore, R. J., Van, T. T. H., Klaassen, M., and Reina, R. D. (2020a). Microbial symbiosis and coevolution of an entire clade of ancient vertebrates: the gut microbiota of sea turtles and its relationship to their phylogenetic history. Anim. Microbiome 2, 1–12.
Scheelings, T. F., Moore, R. J., Van, T. T. H., Klaassen, M., and Reina, R. D. (2020b). The gut bacterial microbiota of sea turtles differs between geographically distinct populations. Endanger. Species Res. 42, 95–108. doi: 10.3354/ESR01042
Sehnal, L., Brammer-Robbins, E., Wormington, A. M., Blaha, L., Bisesi, J., Larkin, I., et al. (2021). Microbiome composition and function in aquatic vertebrates: small organisms making big impacts on aquatic animal health. Front. Microbiol. 12:567408. doi: 10.3389/fmicb.2021.567408
Stoeck, T., Bass, D., Nebel, M., Christen, R., Jones, M. D. M., Breiner, H.-W., et al. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19, 21–31. doi: 10.1111/j.1365-294X.2009.04480.x
Trotta, A., Marinaro, M., Sposato, A., Galgano, M., Ciccarelli, S., Paci, S., et al. (2021). Antimicrobial resistance in loggerhead sea turtles (Caretta caretta): a comparison between clinical and commensal bacterial isolates. Animals 11:2435. doi: 10.3390/ANI11082435
Tsui, C. K. M., Marshall, W., Yokoyama, R., Honda, D., Lippmeier, J. C., Craven, K. D., et al. (2009). Labyrinthulomycetes phylogeny and its implications for the evolutionary loss of chloroplasts and gain of ectoplasmic gliding. Mol. Phylogenet. Evol. 50, 129–140. doi: 10.1016/J.YMPEV.2008.09.027
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiome project. Nature 449, 804–810. doi: 10.1038/nature06244
Van de Vijver, B., Robert, K., Majewska, R., Frankovich, T. A., Panagopoulou, A., and Bosak, S. (2020). Geographical variation in the diatom communities associated with loggerhead sea turtles (Caretta caretta). PLoS One 15:e0236513. doi: 10.1371/journal.pone.0236513
Weitere, M., Erken, M., Majdi, N., Arndt, H., Norf, H., Reinshagen, M., et al. (2018). The food web perspective on aquatic biofilms. Ecol. Monogr. 88, 543–559. doi: 10.1002/ecm.1315
Keywords: epizoic, microbiome, reptile, Caretta caretta, skin, carapace, high throughput sequencing, metabarcoding
Citation: Kanjer L, Filek K, Mucko M, Majewska R, Gračan R, Trotta A, Panagopoulou A, Corrente M, Di Bello A and Bosak S (2022) Surface microbiota of Mediterranean loggerhead sea turtles unraveled by 16S and 18S amplicon sequencing. Front. Ecol. Evol. 10:907368. doi: 10.3389/fevo.2022.907368
Received: 29 March 2022; Accepted: 14 July 2022;
Published: 29 July 2022.
Edited by:
Jesse Robert Zaneveld, University of Washington Bothell, United StatesReviewed by:
Michael P. Doane, Flinders University, AustraliaSishuo Wang, The Chinese University of Hong Kong, Hong Kong SAR, China
Santo Caracappa, University of Camerino, Italy
Copyright © 2022 Kanjer, Filek, Mucko, Majewska, Gračan, Trotta, Panagopoulou, Corrente, Di Bello and Bosak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sunčica Bosak, suncica.bosak@biol.pmf.hr