 Taseer Ahmad
Taseer Ahmad- 1Department of Pharmacology, College of Pharmacy, University of Sargodha, Sargodha, Pakistan
- 2Division of Clinical Pharmacology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
- 3Division of Nephrology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, United States
- 4Department of Physiological Sciences, School of Medicine and Health Sciences, Mulungushi University, Livingstone, Zambia
- 5Department of Medicine, University of Pittsburgh, Pittsburgh, PA, United States
- 6Department of Cell Biology, University of Pittsburgh, Pittsburgh, PA, United States
- 7Department of Pharmacology and Chemical Biology, University of Pittsburgh, Pittsburgh, PA, United States
A major regulator of blood pressure and volume homeostasis in the kidney is the epithelial sodium channel (ENaC). ENaC is composed of alpha(α)/beta(β)/gamma(γ) or delta(δ)/beta(β)/gamma(γ) subunits. The δ subunit is functional in the guinea pig, but not in routinely used experimental rodent models including rat or mouse, and thus remains the least understood of the four subunits. While the δ subunit is poorly expressed in the human kidney, we recently found that its gene variants are associated with blood pressure and kidney function. The δ subunit is expressed in the human vasculature where it may influence vascular function. Moreover, we recently found that the δ subunit is also expressed human antigen presenting cells (APCs). Our studies indicate that extracellular Na+ enters APCs via ENaC leading to inflammation and salt-induced hypertension. In this review, we highlight recent findings on the role of extra-renal ENaC in inflammation, vascular dysfunction, and blood pressure modulation. Targeting extra-renal ENaC may provide new drug therapies for salt-induced hypertension.
Introduction
Hypertension affects more than 1 billion people around the globe. While our understanding of hypertension and the available treatments has improved vastly over the decades, blood pressure control remains a challenge in many hypertensive patients (1, 2). The etiology of hypertension is multifactorial, including environmental, genetic, and demographic factors (3). High Na+ intake is one of the key environmental risk factors for elevated blood pressure (4). However, salt sensitivity of blood pressure, which is a phenotype characterized by changes in blood pressure that correspond to dietary salt intake, is not uniform in humans (5). Many mechanisms have been described to explain the variability of salt sensitivity. These include genetic variations related to the renin-angiotensin-aldosterone system (RAAS), renal Na+ transporters, sympathetic nervous system and vascular dysfunction (6, 7). Recently, inflammation has been found to be a key modulator of salt sensitive blood pressure response (8). Indeed, inflammatory cytokines and reactive oxygen species (ROS) induce vascular endothelial dysfunction and impair renal Na+ excretion, resulting in blood pressure elevation (9, 10).
ENaC dependent reabsorption of Na+ in the aldosterone-sensitive distal nephron (ASDN) has a role in regulating extracellular fluid volume and blood pressure (11). The channel also facilitates K+ secretion in the ASDN (12). ENaC is thought to be composed of αβγ or δβγ subunits, encoded by genes SCNN1A (α subunit), SCNN1B (β subunit), SCNN1G (γ subunit), and SCNN1D (δ subunit) respectively, that are members of the ENaC/Degenerin superfamily (13). Other subunit stoichiometries have been reported in specific tissues. For example, the channel in mouse dendritic cells (DCs) has only α and γ subunits (14–16).
In the aldosterone-sensitive distal nephron of human kidney, ENaC is composed of the α, β, and γ, as the δ subunit is poorly expressed in this nephron segment (5). Besides its role in renal Na+ and K+ handling in the kidneys, ENaC affects blood pressure through its actions in various extrarenal tissues. ENaC in the lingual epithelium mediates salt taste and influences Na+ ingestion, while ENaC in the distal colon serves as the final site for absorption of ingested Na+. Neurons in the rostral ventral medulla of the brain sense increases in [Na+] in an ENaC dependent manner, leading to increase in sympathetic nerve activity and high blood pressure (17, 18). In animal models, such as the mouse and rat, ENaC has been proposed as a key protein for salt taste, but its contribution to human salt taste is less clear (19, 20). Recently, we found that extracellular Na+ enters APCs via ENaC leading to formation of lipid peroxidation products known as Isolevuglandins (IsoLGs), release of proinflammatory cytokines and salt-induced hypertension (14, 16, 21). Moreover, we found that the δ subunit is the most expressed subunit in human APCs (15). In a recent analysis of phenotypic and whole genome sequence data within the Trans-Omics in Precision Medicine project (TOPMED), we found that low frequency and rare variants of α, β and δ subunits of ENaC are associated with blood pressure, and β, δ subunits were associated with estimated glomerular filtration rate (22). Although the δ subunit is poorly expressed in the human kidney, it was the subunit where variants were associated with all blood pressure parameters analyzed, including pulse pressure, systolic, diastolic, and mean arterial pressure as well as kidney function (22). These studies indicate that extrarenal the δ subunit plays an important role in blood pressure regulation. In this review, we provide an overview of the latest findings relating to the roles of extrarenal ENaC, and its δ subunit in inflammation, vascular dysfunction, and blood pressure modulation (21, 23).
Basic structure of ENaC
Ion channels are found in all cells of the body serve to selectively transport ions such as Na+, K+ and Ca2+ across cell membranes (24). ENaC is an amiloride-sensitive, voltage-independent, trimeric constitutively-active ion channel formed by structurally related subunits with two transmembrane-spanning regions, intracellular COOH and NH2 termini, connected by a large extracellular domain/loop as shown in Figure 1 (24). The extracellular loop has a “hand -like” structure, consisting of a “palm”, “ball”, “finger”, “thumb”, “β-ball” and “knuckle” domain (25). These extracellular regions interact with various stimuli that modulate channel activity (26–28). The ENaC structure reveals that it assembles with a 1:1:1 stoichiometry of α, β, γ subunits arranged in a counter-clockwise manner (29).
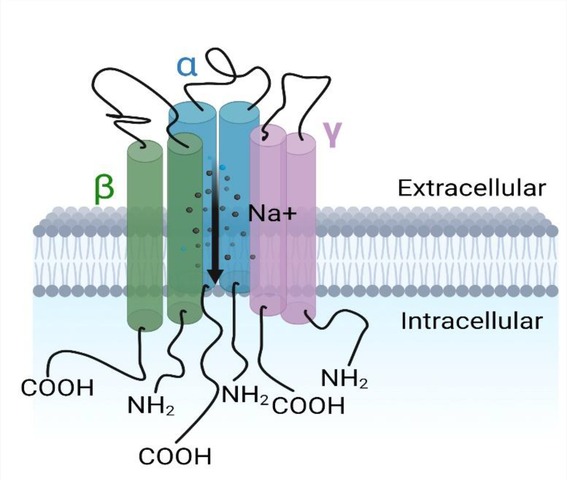
Figure 1. The epithelial Na+ channel. Each subunit (α, β, γ) having the carboxyl and amino termini groups intracellular with two (2) transmembrane segments each and a large extracellular domain between them.
Since its cloning in 1993 and 1994 by Canessa and colleagues (30), key features regarding ENaC's structure and regulation have been described in detail (31, 32). Expressed in the apical plasma membrane of epithelia, ENaC mediates the first step of transepithelial Na+ reabsorption in the ASDN of the kidney, airway and alveolar cells, distal colon and sweat ducts. It is involved in the regulation of blood pressure and extracellular [K+], and gene variants within genes encoding ENaC subunits (SCNN1A, SCNN1B, SCNN1G and SCNN1D) influence blood pressure (22, 33). ENaC also has crucial roles in the homeostasis of lung alveolar fluid (34, 35).
The delta subunit of the epithelial sodium channel
ENaC was initially thought to consist of three subunits: α, β, γ (36). The fourth subunit, δ, was later identified (37, 38). While ENaC is formed by the combination of three homologous αβγ subunits in kidney (36, 39), the δ-subunit may replace α to form δβγ in other tissues (40). It is generally accepted that for proper channel function, ENaC must be composed of an α or δ subunit. Co-expression of β and γ subunits augment channel activity in heterologous expression systems, contribute to the channel pore and influence channel properties (38, 41). The α, β and γ subunits are primarily expressed in epithelial cells of the kidney and colon. While the δ subunit is expressed in lung, it is also expressed in non-epithelial tissues including, heart, brain, vasculature, and immune cells (38). All four subunits (αβγδ) of ENaC are also expressed in the normal human eye (42). Interestingly, in human lung epithelial cells the δ subunit may form multimeric channels with the β and γ subunits that could account for functional heterogeneity of the channel in this tissue (43). The biophysical and pharmacological properties of human αβγ and δβγ differ (44). Human αβγ channels are activated by proteases and inhibited by extracellular Na+, effects that are largely absent in δβγ channels. Furthermore, human δβγ channels have a higher single channel Na+ ion conductance and a higher amiloride IC50, compared to human αβγ channels (2, 45).
The δ subunit is expressed at sites, including specific locations in the brain, APCs and vasculature, where it likely has a role in Na+ sensing rather than transepithelial Na+ transport (38, 46). While αβγ ENaC has been extensively studied in mice and rats, studies of δ ENaC have lagged, in large part due to its expression as a pseudogene in these rodents albeit guinea pigs express functional δβγ channels (47). Furthermore, studies using humanized rodent models may provide a solution to understand the role of the δ subunit in blood pressure regulation.
ENaC in the vasculature
Recent work suggests that δ subunit variants are associated with vascular function and blood pressure (22, 40, 47–49). ENaC in endothelial cells influences vascular tone by increasing intracellular Na+, stabilizing f-actin, and inhibiting endothelial nitric oxide synthase (eNOS), leading to endothelial stiffening, and reduced nitric oxide production (12, 50–52). This work has largely been performed in cultured cells. This regulatory pathway may be relevant in vivo, as mice with an endothelial γ subunit knockout have increased eNOS levels and eNOS activation (52). In addition, elevation of intracellular Na+ concentration hindered the transportation of l-arginine, resulting in impaired generation of nitric oxide (53, 54). However, the exact role of endothelial ENaC in the regulation of blood pressure is still unclear. Elevated expression and increased activity of ENaC can result in vascular dysfunction in some rodent animal models (40). While there is limited knowledge on the expression and function of ENaC channels in human vasculature, the δ-subunit has been reported to be expressed in human endothelial cells, where functional αβγ and δβγ channels are been observed at the single channel level (40, 55). Vascular endothelium is a target for aldosterone, where it regulates ENaC expression in a mineralocorticoid-dependent manner (50). ENaC expression affects endothelial cell stiffness and nitric oxide synthesis in cultured human endothelial cells (56).
In vitro studies in human endothelial cell lines suggested that elevated levels of extracellular Na+ results in enhanced Na+ entry into endothelial cells via ENaC, increasing endothelial stiffness and reducing nitric oxide generation, potentially altering vascular tone (Figure 2) (50). However, to date there are no publications demonstrating that a knockout of ENaC subunits in endothelial cells affects blood pressure. Recently it was reported that functional ENaC subunits are expressed in the human aorta and internal mammary artery and their expression levels are associated with hypertension (48, 50). Reduced expression of the γ subunit was observed in the aorta of hypertensives with controlled blood pressure compared to aorta from normotensive individuals, while reduced expression of δ subunit was observed in internal mammary arteries from controlled hypertensives compared to normotensive individuals (40, 57). While interesting, the observations are correlative, based on small numbers and need to be confirmed.
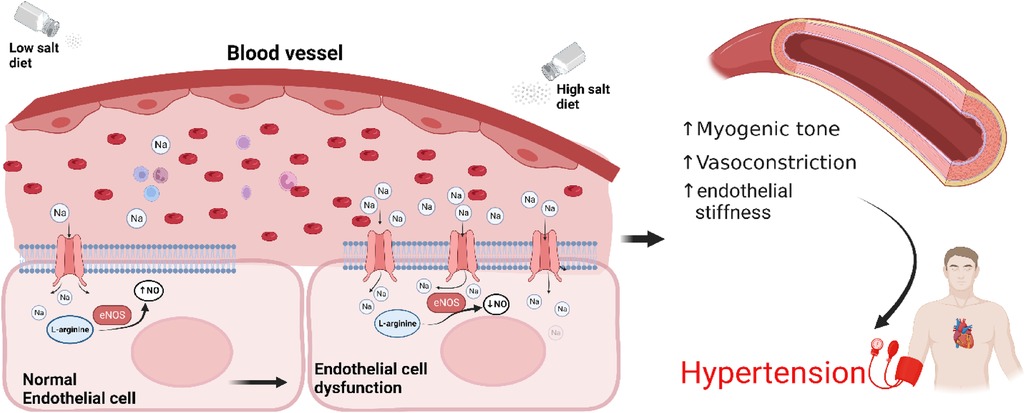
Figure 2. Sodium induces an ENaC-dependent transition from endothelial function to dysfunction. High salt diet leads to overexpression of ENaC and reduced nitric oxide production, increased myogenic tone, vasoconstriction, arterial stiffness resulting in hypertension. Endothelial cells at the bottom have been enlarged for illustration purposes. NO, nitric oxide; eNOS, endothelial nitric oxide synthase.
Renal ENaC and hypertension
ENaC gain- and loss-of-function mutations have profound effects on blood pressure in individuals with Liddle syndrome and pseudohypoaldosteronism type I, respectively (23, 58–63). Mutations that result in activation of αβγ channels cause Liddle syndrome, a hereditary form of hypertension (64). In contrast, ENaC loss of function mutations are observed in pseudohypoaldosteronism type 1 and cause salt-wasting and hypotension. Based on these observations, factors that increase ENaC activity in the kidney have been suggested to contribute to the genesis of salt-sensitive hypertension (63, 65–67). Aldosterone is a major regulator of ENaC activity through the mineralocorticoid receptor, which tightly controls renal Na+ absorption and K+ secretion in the ASDN, thus regulation both the extracellular fluid volume (ECFV) and serum [K+]. Primary hyperaldosteronism, which is due to poorly regulated aldosterone secretion in the adrenal gland, is a well-known cause of secondary hypertension. It is estimated that about 10% of hypertensives have primary hyperaldosteronism (68, 69) and this number increases to 20% in drug-resistant hypertensives (64, 70). As approximately 90% of the US adult population consumes significantly more salt than the daily recommendation of <2,300 mg (71), the vast majority of patients with primary hyperaldosteronism are estimated to have high salt intake (72).
Aldosterone also has been associated with proinflammatory immune effects. Dietary salt and aldosterone also have synergistic effects on the expression of proinflammatory cytokines (73, 74). In addition to mineralocorticoid antagonists, pharmacological blockade of ENaC with amiloride is an important therapeutic option for patients with refractory hypertension and primary hyperaldosteronism (75, 76). Interestingly, ENaC inhibition by amiloride is effective in reducing blood pressure in patients with hypertension resistant to mineralocorticoid antagonism (77). This phenomenon may be explained by ENaC's expression in the vascular smooth muscle of renal, mesenteric and cerebral arteries (8, 78, 79) where its inhibition may result into the loss of myogenic response and pressure-induced vasoconstriction (49). Emerging evidence also suggest that systemic ENaC inhibition may modulate salt-induced immune activation and inflammation-mediated end-organ damage. However, its unclear whether amiloride achieves sufficient levels in the systemic circulation to affect non-renal ENaCs in humans.
Extra-renal ENaC in inflammation and salt-sensitive hypertension
While the role of ENaC in the pathogenesis of salt-sensitive hypertension through renal volume and Na+ homeostasis has been extensively studied, emerging evidence points to a role of extra-renal ENaC in blood pressure regulation through inflammatory pathways. Immune activation and inflammation play a well-established role in hypertension. Both innate and adaptive immunity are fundamental in the development of hypertensive responses to salt and related vascular and renal dysfunction, as previously reviewed elsewhere (80, 81). Pro-hypertensive stimuli first activate APCs, including DCs and macrophages, which, in turn, activate T-cells through antigen-MHC receptor interaction (82). Inhibition of this interaction abolishes deoxycorticosterone acetate (DOCA)-salt induced hypertension (83). High dietary salt intake results in infiltration of APCs and T-lymphocytes into the kidneys that cause vascular remodeling, renal Na+ retention and subsequent hypertension (8, 84–86).
Importantly, recent evidence suggests that ENaC plays a critical role in the association between inflammation and salt-sensitive hypertension. Antigen-presenting DCs and monocytes in humans express all ENaC subunits, with the δ subunit exhibiting highest expression levels (21). In contrast, the mouse splenic DCs express the α and γ subunits of ENaC, but β subunit is absent (12, 16).
We previously found a pathway by which ENaC-mediated Na+ entry into APCs leads to T-cell activation and subsequent elevation in blood pressure (87). Elevated extracellular Na+ enters APCs via ENaC (88). Once inside the cell, Na+ is exchanged with Ca2+, leading to an increase in intracellular Ca2+ and activation of protein kinase C (PKC). PKC phosphorylates and activates NADPH oxidase and leads to the formation of isolevuglandins (IsoLGs; also called Isoketals or γ-ketoaldehydes). IsoLGs are highly reactive oxidative products of arachidonic acid metabolism and adduct to proteins through the lysine residues. The resulting IsoLG-protein adducts are highly immunogenic and are presented on the MHC-II cell surface receptors that activates T cells. This salt-induced, ENaC-mediated immune cell activation leads to the secretion of pro-inflammatory cytokines including IL-1β and IL-6 from the APCs and IFN-γ and IL-17A from the T cells. Tissue infiltration by these cells and the release of inflammatory cytokines results in vascular and kidney dysfunction leading to hypertension (Figure 3) (85). Recent evidence indicates that ENaC-mediated Na+ entry also triggers the NOD [nucleotide-binding and oligomerization domain]-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation in APCs, another important instigator of hypertensive response (15).
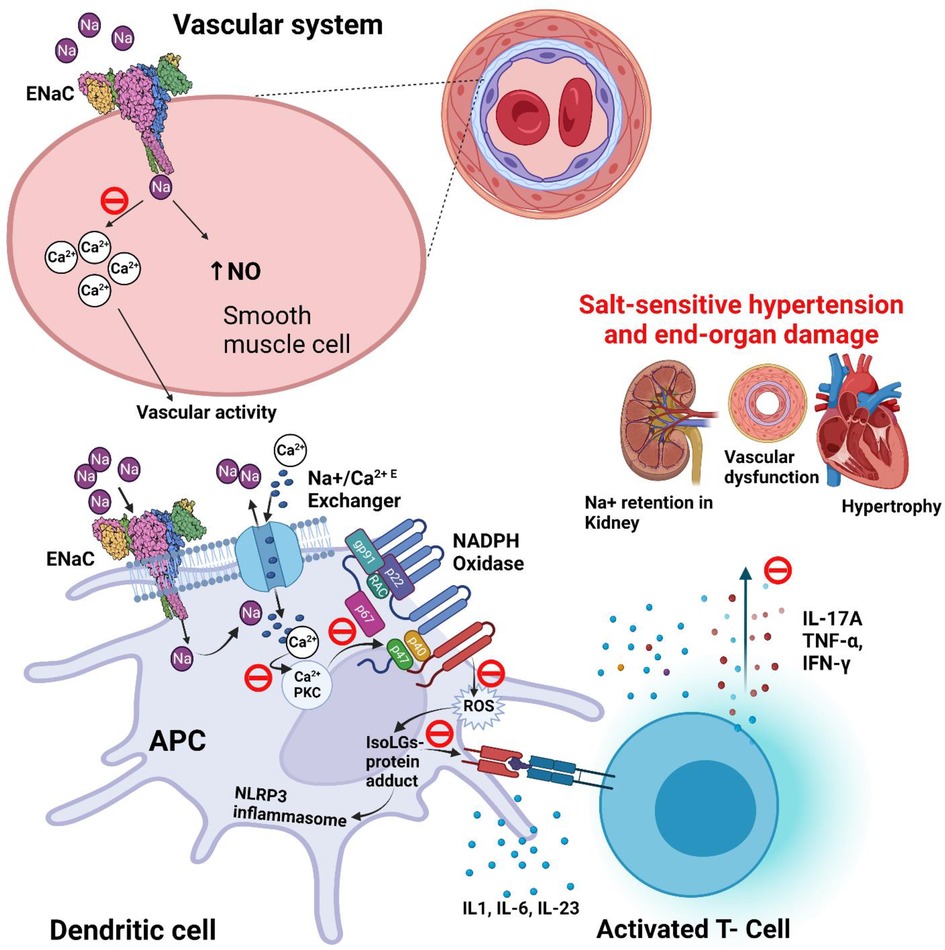
Figure 3. Potential role and proposed blockade mechanisms of ENaC in salt-sensitive hypertension. ENaC (epithelial sodium channel) and IsoLG (isolevuglandins) dependent activation of NLRP3 [NOD (nucleotide-binding and oligomerization domain)-like receptor family pyrin domain containing 3] inflammasome in salt-sensitive hypertension. Also shows the proposed blockade of signaling pathway of ENaC (θ; blockade icon), and epithelial sodium channel (ENaC) role in the regulation of blood pressure in smooth muscle of blood vessel. APC, antigen presenting cell; IsoLG, Isolevuglandins; NO, nitric oxide; NADPH, nicotinamide adenine dinucleotide phosphate; P, phosphorylation; PKC, protein kinase C; ROS, reactive oxygen species.
Inflammasomes are intracellular sensors of pathogen-associated molecular patterns (PAMPs) and endogenous host-derived damage-associated molecular patterns (DAMPs). The NLRP3 inflammasome can be activated through the canonical and non-canonical pathways and results in the release of pro-inflammatory cytokines IL-1β and IL-18, as well as gasdermin D induced pyroptotic cell death (89). The canonical pathway includes a priming and an activation signal. The priming signal, which is provided by Toll-like receptors (TLRs), the nucleotide-binding oligomerization domain (NOD) 1 and 2 or cytokine receptors, activates the nuclear factor kappa B (NF-κB), which results in the expression of NLRP3 and pro-IL-1β (90). After priming, various signals including pathogen components, microbial toxins, and cellular signals such as ion influx, reactive oxygen species (ROS), mitochondrial and lysosomal damage, can serve as the second signal for activation of NLRP3 (91). Patients with hypertension are characterized by high plasma levels of IL-1β and IL-18, the main end-products of NLRP3 inflammasome activation (92–94), which also associates with end-organ damage (95).
Further evidence suggesting a link between NLRP3 inflammasome and hypertension has been provided by genetic studies showing an association between high blood pressure and single nucleotide polymorphism in NLRP3 gene, rs7512998 (96) as well as tandem repeat polymorphism in NLRP3 gene CIAS1 (97, 98).
In animal models of salt sensitive hypertension, absence, or inhibition of NLRP3 inflammasome has been found to prevent the development of hypertension and associated renal damage (99, 100). Similarly, NLRP3 inflammasome, IL-1 receptor and inflammasome-mediated immune cell activation were shown to be essential in the development of aldosterone-induced vascular damage (101). Inhibition of NF-κB, an essential component of canonical pathway for inflammasome activation, induces vasodilation (102), decreases blood pressure and protects against hypertensive end-organ damage (103, 104).
Recent studies in our laboratory have also shown a crucial role of NLRP3 inflammasome in ENaC- and IsoLG-dependent APCs activation and subsequent inflammation in salt-sensitive hypertension (105). Using cell hashing, and cellular indexing of transcriptomes and epitopes of peripheral blood mononuclear cells, we found that NLRP3 inflammasome expression and IL-1β echo changes in blood pressure induced by salt depletion. In vitro exposure of human monocytes to high Na+ increases the expression of caspase-1, IL-1β and IL-18. In further studies using mouse models of salt-sensitive hypertension, we found that APCs from salt-sensitive mice have higher salt-induced intracellular IL-1β production. Following 4-weeks of high salt diet, DCs and monocytes exhibited increased accumulation of NLRP3, IL-1β along with IsoLG-protein adducts. Pharmacological inhibition of NLRP3 inflammasome attenuates salt-induced increase in APC NLRP3, IL-1β and IsoLG-adducts as well as salt-sensitive blood pressure elevation. These findings show that ENaC mediated Na+ entry in the APCs induce immune cell activation and inflammation through IsoLG-formation, NLRP3 inflammasome activation and release of pro-inflammatory cytokines that orchestrate vascular and renal dysfunction and ensuing salt-sensitive hypertension (21, 85). These new insights imply that ENaC may play a pivotal role in the regulation of blood pressure through its actions in immune cells in addition to its well-recognized function in the nephron (106). So, the recent pivotal discoveries related to the presence and functioning of extra-renal ENaC in immune cells may illuminate additional therapeutic targets for ENaC in salt-induced blood pressure (8).
Conclusion
Recent evidence from human and animal studies has expanded our understanding of ENaC and its subunit δ in relation to its functional roles in non-epithelial tissues and blood pressure regulation. It is now well-recognized that the immune system plays an active role in the development and progression of hypertension, and high salt intake not only drives hemodynamic changes but is also associated with inflammation. Thus, inhibition of extra-renal ENaC, including the δ subunit, or an intermediate step in this signaling pathway (as shown in Figure 3), may improve blood pressure control in patients with salt-sensitive hypertension (107). A major side effect of ENaC inhibitors is hyperkalemia (108). The development of drugs that selectively target δβγ channels, while not blocking αβγ channels in the human kidney should prevent drug-induced hyperkalemia. Furthermore, future studies should investigate potential novel drugs specifically targeting APCs, ENaC, NLRP3, PKC or oxidative stress for safety and efficacy (as shown in Figure 3). Lastly, as high salt intake is consistently associated with a rise in blood pressure (109) and has a more pronounced influence on salt-sensitive populations, reductions in dietary salt could provide a cost effective approach to decrease the risk of hypertension and related cardiovascular disease (110).
Author contributions
TA, AK: investigation and writing—original draft. TA, AK, LE: conceptualization, and writing—review and editing. SM, TK: Figures, manuscript review and editing. AK, TK: validation, resources, and supervision. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Institutes of Health grants R01HL147818 (AK and TK), R03HL155041 (AK), R01HL144941 (AK), P30 DK079307 (TK).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mills KT, Bundy JD, Kelly TN, Reed JE, Kearney PM, Reynolds K, et al. Global disparities of hypertension prevalence and control: a systematic analysis of population-based studies from 90 countries. Circulation. (2016) 134(6):441–50. doi: 10.1161/CIRCULATIONAHA.115.018912
2. Kleyman TR, Kashlan OB, Hughey RP. Epithelial Na+ channel regulation by extracellular and intracellular factors. Annu Rev Physiol. (2018) 80:263–81. doi: 10.1146/annurev-physiol-021317-121143
3. Ruppert V, Maisch B. Genetics of human hypertension. Herz. (2003) 28(8):655–62. doi: 10.1007/s00059-003-2516-6
4. Blumenthal JA, Sherwood A, Smith PJ, Hinderliter A. The role of salt reduction in the management of hypertension. J Am Coll Cardiol. (2018) 71(14):1597–8. doi: 10.1016/j.jacc.2018.01.071
5. Mutchler SM, Kirabo A, Kleyman TR. Epithelial sodium channel and salt-sensitive hypertension. Hypertension. (2021) 77(3):759–67. doi: 10.1161/HYPERTENSIONAHA.120.14481
6. Ralph AF, Grenier C, Costello HM, Stewart K, Ivy JR, Dhaun N, et al. Activation of the sympathetic nervous system promotes blood pressure salt-sensitivity in C57BL6/J mice. Hypertension. (2021) 77(1):158–68. doi: 10.1161/HYPERTENSIONAHA.120.16186
7. Hall JE. Renal dysfunction, rather than nonrenal vascular dysfunction, mediates salt-induced hypertension. Circulation. (2016) 133(9):894–906. doi: 10.1161/CIRCULATIONAHA.115.018526
8. Pitzer AL, Van Beusecum JP, Kleyman TR, Kirabo A. ENaC in salt-sensitive hypertension: kidney and beyond. Curr Hypertens Rep. (2020) 22(9):69. doi: 10.1007/s11906-020-01067-9
9. McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. (2015) 116(6):1022–33. doi: 10.1161/CIRCRESAHA.116.303697
10. Rucker AJ, Rudemiller NP, Crowley SD. Salt, hypertension, and immunity. Annu Rev Physiol. (2018) 80:283–307. doi: 10.1146/annurev-physiol-021317-121134
11. Mamenko M, Zaika O, Doris PA, Pochynyuk O. Salt-dependent inhibition of epithelial Na+ channel–mediated sodium reabsorption in the aldosterone-sensitive distal nephron by bradykinin. Hypertension. (2012) 60(5):1234–41. doi: 10.1161/HYPERTENSIONAHA.112.200469
12. Mutchler SM, Kleyman TR. New insights regarding epithelial Na+ channel regulation and its role in the kidney, immune system and vasculature. Curr Opin Nephrol Hypertens. (2019) 28(2):113–9. doi: 10.1097/MNH.0000000000000479
13. Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: phylogeny, structure–function, tissue distribution, and associated inherited diseases. Gene. (2016) 579(2):95–132. doi: 10.1016/j.gene.2015.12.061
14. Van Beusecum JP, Barbaro NR, McDowell Z, Aden LA, Xiao L, Pandey AK, et al. High salt activates CD11c(+) antigen-presenting cells via SGK (serum glucocorticoid kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertension. (2019) 74(3):555–63. doi: 10.1161/HYPERTENSIONAHA.119.12761
15. Pitzer A, Elijovich F, Laffer CL, Ertuglu LA, Sahinoz M, Saleem M, et al. DC ENaC-dependent inflammasome activation contributes to salt-sensitive hypertension. Circ Res. (2022) 131(4):328–44. doi: 10.1161/CIRCRESAHA.122.320818
16. Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. (2017) 21(4):1009–20. doi: 10.1016/j.celrep.2017.10.002
17. Kinsman BJ, Browning KN, Stocker SD. Nacl and osmolarity produce different responses in organum vasculosum of the lamina terminalis neurons, sympathetic nerve activity and blood pressure. J Physiol. (2017) 595(18):6187–201. doi: 10.1113/JP274537
18. Kinsman BJ, Nation HN, Stocker SD. Hypothalamic signaling in body fluid homeostasis and hypertension. Curr Hypertens Rep. (2017) 19(6):50. doi: 10.1007/s11906-017-0749-7
19. Desor J, Finn J. Effects of amiloride on salt taste in humans. Chem Senses. (1989) 14(6):793–803. doi: 10.1093/chemse/14.6.793
20. Bigiani A. Does ENaC work as sodium taste receptor in humans? Nutrients. (2020) 12(4):1195. doi: 10.3390/nu12041195
21. Pitzer A, Elijovich F, Laffer CL, Ertuglu LA, Sahinoz M, Saleem M, et al. DC ENaC-dependent inflammasome activation contributes to salt-sensitive hypertension. Circ Res. (2022) 131(4):328–44. doi: 10.1161/CIRCRESAHA.122.320818
22. Blobner BM, Kirabo A, Kashlan OB, Sheng S, Arnett DK, Becker LC, et al. Rare variants in genes encoding subunits of the epithelial Na+ channel are associated with blood pressure and kidney function. Hypertension. (2022) 79(11):2573–82. doi: 10.1161/HYPERTENSIONAHA.121.18513
23. Rossier BC. Epithelial sodium channel (ENaC) and the control of blood pressure. Curr Opin Pharmacol. (2014) 15:33–46. doi: 10.1016/j.coph.2013.11.010
24. Tien J, Young DM, Jan YN, Jan LY. Molecular properties of ion channels. From Mol to Networks: Elsevier. (2014) 2:323–48. doi: 10.1016/B978-0-12-397179-1.00011-7
25. Stockand JD, Staruschenko A, Pochynyuk O, Booth RE, Silverthorn DU. Insight toward epithelial Na+ channel mechanism revealed by the acid-sensing ion channel 1 structure. IUBMB Life. (2008) 60(9):620–8. doi: 10.1002/iub.89
26. Alvarez de la Rosa D, Canessa CM, Fyfe GK, Zhang P. Structure and regulation of amiloride-sensitive sodium channels. Annu Rev Physiol. (2000) 62:573–94. doi: 10.1146/annurev.physiol.62.1.573
27. Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev. (2002) 82(3):735–67. doi: 10.1152/physrev.00007.2002
28. Ben-Shahar Y. Sensory functions for degenerin/epithelial sodium channels (DEG/ENaC). Adv Genet. (2011) 76:1–26. doi: 10.1016/B978-0-12-386481-9.00001-8
29. Noreng S, Bharadwaj A, Posert R, Yoshioka C, Baconguis I. Structure of the human epithelial sodium channel by cryo-electron microscopy. Elife. (2018) 7:e39340. doi: 10.7554/eLife.39340
30. Canessa CM, Horisberger J-D, Schild L, Rossier BC. Expression cloning of the epithelial sodium channel. Kidney Int. (1995) 48(4):950–5. doi: 10.1038/ki.1995.376
31. Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. (1997) 77(2):359–96. doi: 10.1152/physrev.1997.77.2.359
32. Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflugers Arch. (2009) 458(1):111–35. doi: 10.1007/s00424-009-0656-0
33. Liu F, Yang X, Mo X, Huang J, Chen J, Kelly TN, et al. Associations of epithelial sodium channel genes with blood pressure: the GenSalt study. J Hum Hypertens. (2015) 29(4):224–8. doi: 10.1038/jhh.2014.78
34. Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol. (2009) 71:403–23. doi: 10.1146/annurev.physiol.010908.163250
35. Althaus M, Clauss WG, Fronius M. Amiloride-sensitive sodium channels and pulmonary edema. Pulm Med. (2011) 2011:830320. doi: 10.1155/2011/830320
36. Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger J-D, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. (1994) 367(6462):463–7. doi: 10.1038/367463a0
37. Lingueglia E, Voilley N, Waldmann R, Lazdunski M, Barbry P. Expression cloning of an epithelial amiloride-sensitive Na+ channel. A new channel type with homologies to caenorhabditis elegans degenerins. FEBS Lett. (1993) 318(1):95–9. doi: 10.1016/0014-5793(93)81336-X
38. Ji HL, Zhao RZ, Chen ZX, Shetty S, Idell S, Matalon S. δ ENaC: a novel divergent amiloride-inhibitable sodium channel. Am J Physiol Lung Cell Mol Physiol. (2012) 303(12):L1013–26. doi: 10.1152/ajplung.00206.2012
39. Canessa CM, Horisberger J-D, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature. (1993) 361(6411):467–70. doi: 10.1038/361467a0
40. Paudel P, van Hout I, Bunton RW, Parry DJ, Coffey S, McDonald FJ, et al. Epithelial sodium channel δ subunit is expressed in human arteries and has potential association with hypertension. Hypertension. (2022) 79(7):1385–94. doi: 10.1161/HYPERTENSIONAHA.122.18924
41. Fyfe GK, Canessa CM. Subunit composition determines the single channel kinetics of the epithelial sodium channel. J Gen Physiol. (1998) 112(4):423–32. doi: 10.1085/jgp.112.4.423
42. Krueger B, Schlötzer-Schrehardt U, Haerteis S, Zenkel M, Chankiewitz VE, Amann KU, et al. Four subunits (αβγδ) of the epithelial sodium channel (ENaC) are expressed in the human eye in various locations. Invest Ophthalmol Vis Sci. (2012) 53(2):596–604. doi: 10.1167/iovs.11-8581
43. McNicholas CM, Canessa CM. Diversity of channels generated by different combinations of epithelial sodium channel subunits. J Gen Physiol. (1997) 109(6):681–92. doi: 10.1085/jgp.109.6.681
44. Ji HL, Su XF, Kedar S, Li J, Barbry P, Smith PR, et al. Delta-subunit confers novel biophysical features to alpha beta gamma-human epithelial sodium channel (ENaC) via a physical interaction. J Biol Chem. (2006) 281(12):8233–41. doi: 10.1074/jbc.M512293200
45. Kashlan OB, Adelman JL, Okumura S, Blobner BM, Zuzek Z, Hughey RP, et al. Constraint-based, homology model of the extracellular domain of the epithelial Na+ channel α subunit reveals a mechanism of channel activation by proteases. J Biol Chem. (2011) 286(1):649–60. doi: 10.1074/jbc.M110.167098
46. Waldmann R, Champigny G, Bassilana F, Voilley N, Lazdunski M. Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J Biol Chem. (1995) 270(46):27411–4. doi: 10.1074/jbc.270.46.27411
47. Gettings SM, Maxeiner S, Tzika M, Cobain MR, Ruf I, Benseler F, et al. Two functional epithelial sodium channel isoforms are present in rodents despite pronounced evolutionary pseudogenization and exon fusion. Mol Biol Evol. (2021) 38(12):5704–25. doi: 10.1093/molbev/msab271
48. Suckling RJ, He FJ, Markandu ND, MacGregor GA. Dietary salt influences postprandial plasma sodium concentration and systolic blood pressure. Kidney Int. (2012) 81(4):407–11. doi: 10.1038/ki.2011.369
49. Paudel P, McDonald FJ, Fronius M. The δ subunit of epithelial sodium channel in humans-a potential player in vascular physiology. Am J Physiol Heart Circ Physiol. (2021) 320(2):H487–93. doi: 10.1152/ajpheart.00800.2020
50. Kusche-Vihrog K, Jeggle P, Oberleithner H. The role of ENaC in vascular endothelium. Pflugers Arch. (2014) 466(5):851–9. doi: 10.1007/s00424-013-1356-3
51. Tarjus A, González-Rivas C, Amador-Martínez I, Bonnard B, López-Marure R, Jaisser F, et al. The absence of endothelial sodium channel α (αENaC) reduces renal ischemia/reperfusion injury. Int J Mol Sci. (2019) 20(13):3132. doi: 10.3390/ijms20133132
52. Mutchler SM, Hasan M, Kohan DE, Kleyman TR, Tan RJ. Deletion of the gamma subunit of ENaC in endothelial cells does not protect against renal ischemia reperfusion injury. Int J Mol Sci. (2021) 22(20):1–15. doi: 10.3390/ijms222010914
53. Guo D, Liang S, Wang S, Tang C, Yao B, Wan W, et al. Role of epithelial Na+ channels in endothelial function. J Cell Sci. (2016) 129(2):290–7. doi: 10.1242/jcs.168831
54. Ni Z, Vaziri ND. Effect of salt loading on nitric oxide synthase expression in normotensive rats. Am J Hypertens. (2001) 14(2):155–63. doi: 10.1016/S0895-7061(00)01234-6
55. Downs CA, Johnson NM, Coca C, Helms MN. Angiotensin II regulates δ-ENaC in human umbilical vein endothelial cells. Microvasc Res. (2018) 116:26–33. doi: 10.1016/j.mvr.2017.10.001
56. Oberleithner H, Riethmüller C, Schillers H, MacGregor GA, de Wardener HE, Hausberg M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci U S A. (2007) 104(41):16281–6. doi: 10.1073/pnas.0707791104
57. Li Q, Fung E. Multifaceted functions of epithelial Na+ channel in modulating blood pressure. Hypertension. (2019) 73(2):273–81. doi: 10.1161/HYPERTENSIONAHA.118.12330
58. Snyder PM, Price MP, McDonald FJ, Adams CM, Volk KA, Zeiher BG, et al. Mechanism by which liddle's syndrome mutations increase activity of a human epithelial Na+ channel. Cell. (1995) 83(6):969–78. doi: 10.1016/0092-8674(95)90212-0
59. Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of liddle syndrome. Nat Genet. (1995) 11(1):76–82. doi: 10.1038/ng0995-76
60. Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, et al. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci U S A. (1995) 92(25):11495–9. doi: 10.1073/pnas.92.25.11495
61. Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, Rossier BC. Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J Clin Invest. (1996) 97(7):1780–4. doi: 10.1172/JCI118606
62. Chang SS, Grunder S, Hanukoglu A, Rösler A, Mathew PM, Hanukoglu I, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. (1996) 12(3):248–53. doi: 10.1038/ng0396-248
63. Soundararajan R, Pearce D, Hughey RP, Kleyman TR. Role of epithelial sodium channels and their regulators in hypertension. J Biol Chem. (2010) 285(40):30363–9. doi: 10.1074/jbc.R110.155341
64. Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension. (2002) 40(6):892–6. doi: 10.1161/01.HYP.0000040261.30455.B6
65. Rossier BC, Pradervand S, Schild L, Hummler E. Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu Rev Physiol. (2002) 64:877–97. doi: 10.1146/annurev.physiol.64.082101.143243
66. Pratt JH. Central role for ENaC in development of hypertension. J Am Soc Nephrol. (2005) 16(11):3154–9. doi: 10.1681/ASN.2005050460
67. Bubien JK. Epithelial Na+ channel (ENaC), hormones, and hypertension. J Biol Chem. (2010) 285(31):23527–31. doi: 10.1074/jbc.R109.025049
68. Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol. (2006) 48(11):2293–300. doi: 10.1016/j.jacc.2006.07.059
69. Hundemer GL, Vaidya A. Primary aldosteronism diagnosis and management: a clinical approach. Endocrinol Metab Clin North Am. (2019) 48(4):681–700. doi: 10.1016/j.ecl.2019.08.002
70. Jaffe G, Gray Z, Krishnan G, Stedman M, Zheng Y, Han J, et al. Screening rates for primary aldosteronism in resistant hypertension: a cohort study. Hypertension. (2020) 75(3):650–9. doi: 10.1161/HYPERTENSIONAHA.119.14359
71. Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Circulation. (2018) 138(17):e426–83. doi: 10.1161/HYP.0000000000000066
72. Funder JW. Primary aldosteronism and salt. Pflüg Arch Eur J Physiol. (2015) 467(3):587–94. doi: 10.1007/s00424-014-1658-0
73. Afsar B, Kuwabara M, Ortiz A, Yerlikaya A, Siriopol D, Covic A, et al. Salt intake and immunity. Hypertension. (2018) 72(1):19–23. doi: 10.1161/HYPERTENSIONAHA.118.11128
74. Muñoz-Durango N, Vecchiola A, Gonzalez-Gomez LM, Simon F, Riedel CA, Fardella CE, et al. Modulation of immunity and inflammation by the mineralocorticoid receptor and aldosterone. Biomed Res Int. (2015) 2015:652738. doi: 10.1155/2015/652738
75. Carter AR, Zhou ZH, Calhoun DA, Bubien JK. Hyperactive ENaC identifies hypertensive individuals amenable to amiloride therapy. Am J Physiol Cell Physiol. (2001) 281(5):C1413–21. doi: 10.1152/ajpcell.2001.281.5.C1413
76. Sarafidis PA, Bakris GL. Resistant hypertension: an overview of evaluation and treatment. J Am Coll Cardiol. (2008) 52(22):1749–57. doi: 10.1016/j.jacc.2008.08.036
77. Laffer CL, Elijovich F, Eckert GJ, Tu W, Pratt JH, Brown NJ. Genetic variation in CYP4A11 and blood pressure response to mineralocorticoid receptor antagonism or ENaC inhibition: an exploratory pilot study in African Americans. J Am Soc Hypertens. (2014) 8(7):475–80. doi: 10.1016/j.jash.2014.04.011
78. Guan Z, Pollock JS, Cook AK, Hobbs JL, Inscho EW. Effect of epithelial sodium channel blockade on the myogenic response of rat juxtamedullary afferent arterioles. Hypertension. (2009) 54(5):1062–9. doi: 10.1161/HYPERTENSIONAHA.109.137992
79. Ge Y, Gannon K, Gousset M, Liu R, Murphey B, Drummond HA. Impaired myogenic constriction of the renal afferent arteriole in a mouse model of reduced βENaC expression. Am J Physiol Renal Physiol. (2012) 302(11):F1486–93. doi: 10.1152/ajprenal.00638.2011
80. Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med. (2018) 215(1):21–33. doi: 10.1084/jem.20171773
81. Harrison DG. The immune system in hypertension. Trans Am Clin Climatol Assoc. (2014) 125:130–38; discussion 8–40. doi: 10.1152/advan.00063.2013
82. Kambayashi T, Laufer TM. Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol. (2014) 14(11):719–30. doi: 10.1038/nri3754
83. Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. (2010) 122(24):2529–37. doi: 10.1161/CIRCULATIONAHA.109.930446
84. Caillon A, Mian MOR, Fraulob-Aquino JC, Huo KG, Barhoumi T, Ouerd S, et al. Γδ T cells mediate angiotensin II-induced hypertension and vascular injury. Circulation. (2017) 135(22):2155–62. doi: 10.1161/CIRCULATIONAHA.116.027058
85. Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. (2014) 124(10):4642–56. doi: 10.1172/JCI74084
86. Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, et al. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension. (2014) 64(5):1108–15. doi: 10.1161/HYPERTENSIONAHA.114.04147
87. Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. (2016) 126(1):50–67. doi: 10.1172/JCI80761
88. Fleysher L, Oesingmann N, Brown R, Sodickson DK, Wiggins GC, Inglese M. Noninvasive quantification of intracellular sodium in human brain using ultrahigh–field MRI. NMR Biomed. (2013) 26(1):9–19. doi: 10.1002/nbm.2813
89. Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
90. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20(13):1–24. doi: 10.3390/ijms20133328
91. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. (2013) 38(6):1142–53. doi: 10.1016/j.immuni.2013.05.016
92. Li Q-Z, Deng Q, Li J-Q, Yi G-H, Zhao S-P. Valsartan reduces interleukin-1β secretion by peripheral blood mononuclear cells in patients with essential hypertension. Clin Chim Acta. (2005) 355(1):131–6. doi: 10.1016/j.cccn.2004.12.006
93. Dorffel Y, Latsch C, Stuhlmuller B, Schreiber S, Scholze S, Burmester GR, et al. Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension. (1999) 34(1):113–7. doi: 10.1161/01.HYP.34.1.113
94. Rabkin SW. The role of interleukin 18 in the pathogenesis of hypertension-induced vascular disease. Nat Rev Cardiol. (2009) 6(3):192–9. doi: 10.1038/ncpcardio1453
95. De Miguel C, Pelegrín P, Baroja-Mazo A, Cuevas S. Emerging role of the inflammasome and pyroptosis in hypertension. Int J Mol Sci. (2021) 22(3):1064. doi: 10.3390/ijms22031064
96. Kunnas T, Määttä K, Nikkari ST. NLR Family pyrin domain containing 3 (NLRP3) inflammasome gene polymorphism rs7512998 (C > T) predicts aging-related increase of blood pressure, the TAMRISK study. Immun Ageing. (2015) 12(19):1–5. doi: 10.1186/s12979-015-0047-7
97. De Miguel C, Rudemiller NP, Abais JM, Mattson DL. Inflammation and hypertension: new understandings and potential therapeutic targets. Curr Hypertens Rep. (2015) 17(1):507. doi: 10.1007/s11906-014-0507-z
98. Omi T, Kumada M, Kamesaki T, Okuda H, Munkhtulga L, Yanagisawa Y, et al. An intronic variable number of tandem repeat polymorphisms of the cold-induced autoinflammatory syndrome 1 (CIAS1) gene modifies gene expression and is associated with essential hypertension. Eur J Hum Genet. (2006) 14(12):1295–305. doi: 10.1038/sj.ejhg.5201698
99. Krishnan SM, Dowling JK, Ling YH, Diep H, Chan CT, Ferens D, et al. Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice. Br J Pharmacol. (2016) 173(4):752–65. doi: 10.1111/bph.13230
100. Krishnan SM, Ling YH, Huuskes BM, Ferens DM, Saini N, Chan CT, et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc Res. (2019) 115(4):776–87. doi: 10.1093/cvr/cvy252
101. Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho F, Pequeno IO, Olivon VC, et al. NLRP3 Inflammasome mediates aldosterone-induced vascular damage. Circulation. (2016) 134(23):1866–80. doi: 10.1161/CIRCULATIONAHA.116.024369
102. Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduction and Targeted Therapy. (2017) 2(1):1–9. doi: 10.1038/sigtrans.2017.23
103. Rodriguez-Iturbe B, Ferrebuz A, Vanegas V, Quiroz Y, Mezzano S, Vaziri ND. Early and sustained inhibition of nuclear factor-kappaB prevents hypertension in spontaneously hypertensive rats. J Pharmacol Exp Ther. (2005) 315(1):51–7. doi: 10.1124/jpet.105.088062
104. Zambom FFF, Oliveira KC, Foresto-Neto O, Faustino VD, Ávila VF, Albino AH, et al. Pathogenic role of innate immunity in a model of chronic NO inhibition associated with salt overload. Am J Physiol Renal Physiol. (2019) 317(4):F1058–67. doi: 10.1152/ajprenal.00251.2019
105. Sahinoz M, Elijovich F, Ertuglu LA, Ishimwe J, Pitzer A, Saleem M, et al. Salt sensitivity of blood pressure in blacks and women: a role of inflammation, oxidative stress, and epithelial Na(+) channel. Antioxid Redox Signal. (2021) 35(18):1477–93. doi: 10.1089/ars.2021.0212
106. Ertuglu LA, Kirabo A. Dendritic cell ENaC in inflammation, salt-sensitive hypertension and kidney damage. Kidney. (2022) 360:1620–9. doi: 10.34067/KID.0001272022
107. Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C–dependent buffering mechanism. Nat Med. (2009) 15(5):545–52. doi: 10.1038/nm.1960
108. Vidt DG. Mechanism of action, pharmacokinetics, adverse effects, and therapeutic uses of amiloride hydrochloride, a new potassium-sparing diuretic. Pharmacotherapy. (1981) 1(3):179–87. doi: 10.1002/j.1875-9114.1981.tb02539.x
109. O'Donnell M, Mente A, Rangarajan S, McQueen MJ, Wang X, Liu L, et al. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N Engl J Med. (2014) 371(7):612–23. doi: 10.1056/NEJMoa1311889
Keywords: epithelial sodium channel (ENaC), δ-ENaC, antigen presenting cells, inflammation, endothelium dysfunction, blood pressure
Citation: Ahmad T, Ertuglu LA, Masenga SK, Kleyman TR and Kirabo A (2023) The epithelial sodium channel in inflammation and blood pressure modulation. Front. Cardiovasc. Med. 10:1130148. doi: 10.3389/fcvm.2023.1130148
Received: 22 December 2022; Accepted: 24 March 2023;
Published: 12 April 2023.
Edited by:
Rong Jin, Penn State Milton S. Hershey Medical Center, United StatesReviewed by:
Xuexin Zhang, The Pennsylvania State University (PSU), United StatesFiona McDonald, University of Otago, New Zealand
© 2023 Ahmad, Ertuglu, Masenga, Kleyman and Kirabo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annet Kirabo annet.kirabo@vumc.org
Specialty Section: This article was submitted to Hypertension, a section of the journal Frontiers in Cardiovascular Medicine