
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 May 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.870535
This article is part of the Research Topic The Role of Monocytes/Macrophages in Autoimmunity and Autoinflammation View all 19 articles
 Takayuki Tanaka1,2*
Takayuki Tanaka1,2* Takeshi Shiba3
Takeshi Shiba3 Yoshitaka Honda4,5
Yoshitaka Honda4,5 Kazushi Izawa1
Kazushi Izawa1 Takahiro Yasumi1
Takahiro Yasumi1 Megumu K. Saito6
Megumu K. Saito6 Ryuta Nishikomori7
Ryuta Nishikomori7The concept of autoinflammation, first proposed in 1999, refers to a seemingly unprovoked episode of sterile inflammation manifesting as unexplained fever, skin rashes, and arthralgia. Autoinflammatory diseases are caused mainly by hereditary abnormalities of innate immunity, without the production of autoantibodies or autoreactive T cells. The revolutionary discovery of induced pluripotent stem cells (iPSCs), whereby a patient’s somatic cells can be reprogrammed into an embryonic pluripotent state by forced expression of a defined set of transcription factors, has the transformative potential to enable in vitro disease modeling and drug candidate screening, as well as to provide a resource for cell replacement therapy. Recent reports demonstrate that recapitulating a disease phenotype in vitro is feasible for numerous monogenic diseases, including autoinflammatory diseases. In this review, we provide a comprehensive overview of current advances in research into autoinflammatory diseases involving iPSC-derived monocytes/macrophages. This review may aid in the planning of new studies of autoinflammatory diseases.
The concept of autoinflammation, which was first proposed in 1999, refers to seemingly unprovoked and episodic sterile inflammation manifesting as unexplained fever, skin rashes, and arthralgia (1). Autoinflammatory diseases are caused mainly by hereditary abnormalities of innate immunity, without the production of autoantibodies or autoreactive T cells. Analysis of blood cells from patients with autoinflammatory diseases has expanded our understanding of these conditions; however, there are several limitations to this approach: i) collecting enough patient blood samples for analysis is difficult because autoinflammatory diseases are rare and, to make matters worse, many patients are infants, and ii) the in vitro phenotype of hematopoietic cells in these patients is affected by existing inflammation or by the prescribed drugs.
The revolutionary discovery of induced pluripotent stem cells (iPSCs), whereby a patient’s somatic cells can be reprogrammed into an embryonic pluripotent state by forced expression of a defined set of transcription factors (2, 3), has the transformative potential to enable in vitro disease modeling and drug candidate screening, as well as to provide a resource for cell replacement therapy. iPSC-derived monocytes/macrophages provide an opportunity to analyze the effect of genetic variants in the absence of the limitations described above. Recent reports demonstrate that recapitulating a disease phenotype in vitro is feasible for numerous monogenic diseases, including autoinflammatory diseases (4–16).
One of the main obstacles to disease studies based on iPSCs is that directed differentiation of iPSCs is time- and labor-intensive, and the results of functional analysis usually show high variation (even among iPSC clones). To overcome these issues and to obtain a stable and scalable number of mature monocytic cells from iPSC clones, immortalized proliferating myeloid cell lines have been utilized (6, 10). Recent advances in genome editing technology, such as the CRISPR system (17), facilitate functional comparisons between isogenic pairs of mutant and control iPSC clones.
In this review, we provide a comprehensive overview of current advances in research into the role of iPSC-derived monocytes/macrophages in autoinflammatory diseases. We will outline how iPSC-derived blood cells contribute to i) elucidation of disease pathogenesis, ii) functional analyses to facilitate correct diagnosis, iii) evaluation of the disease relevancies of newly identified mutations, and iv) discovery of new drug candidates.
The availability of tissue-resident macrophages isolated directly from human tissues is limited due to ethical issues; therefore, monocyte-derived macrophages (MDMs) are used widely for research into human macrophages. This approach involves isolating CD14+ monocytes from peripheral blood mononuclear cells (PBMCs) and exposing them to macrophage colony-stimulating factor (M-CSF) to induce differentiation into macrophages (Figure 1) (18). An advantage of the MDM model is that human peripheral blood samples can be obtained without an invasive procedure; indeed, several million monocytes can be obtained from a single venipuncture. Because the in vitro culture period is only 1 week, MDMs are most likely free from artifacts that appear after long-term culture; therefore, they should be more representative of the patients’ macrophages. However, MDMs have drawbacks, including limited proliferative capacity and a short culture period, which limit the options when it comes to genetic modification. In an era when pluripotent stem cell (PSC)-derived macrophages (PSC-MPs) are available, MDMs remain an important research tool for autoinflammatory diseases because they are likely more “physiological” than PSC-MPs. For example, MDMs have been used to examine the clinical relevance of findings obtained using PSC-MPs (6) and to examine functional differences between primary monocytes and macrophages with respect to cytokine secretion (12) (see below).
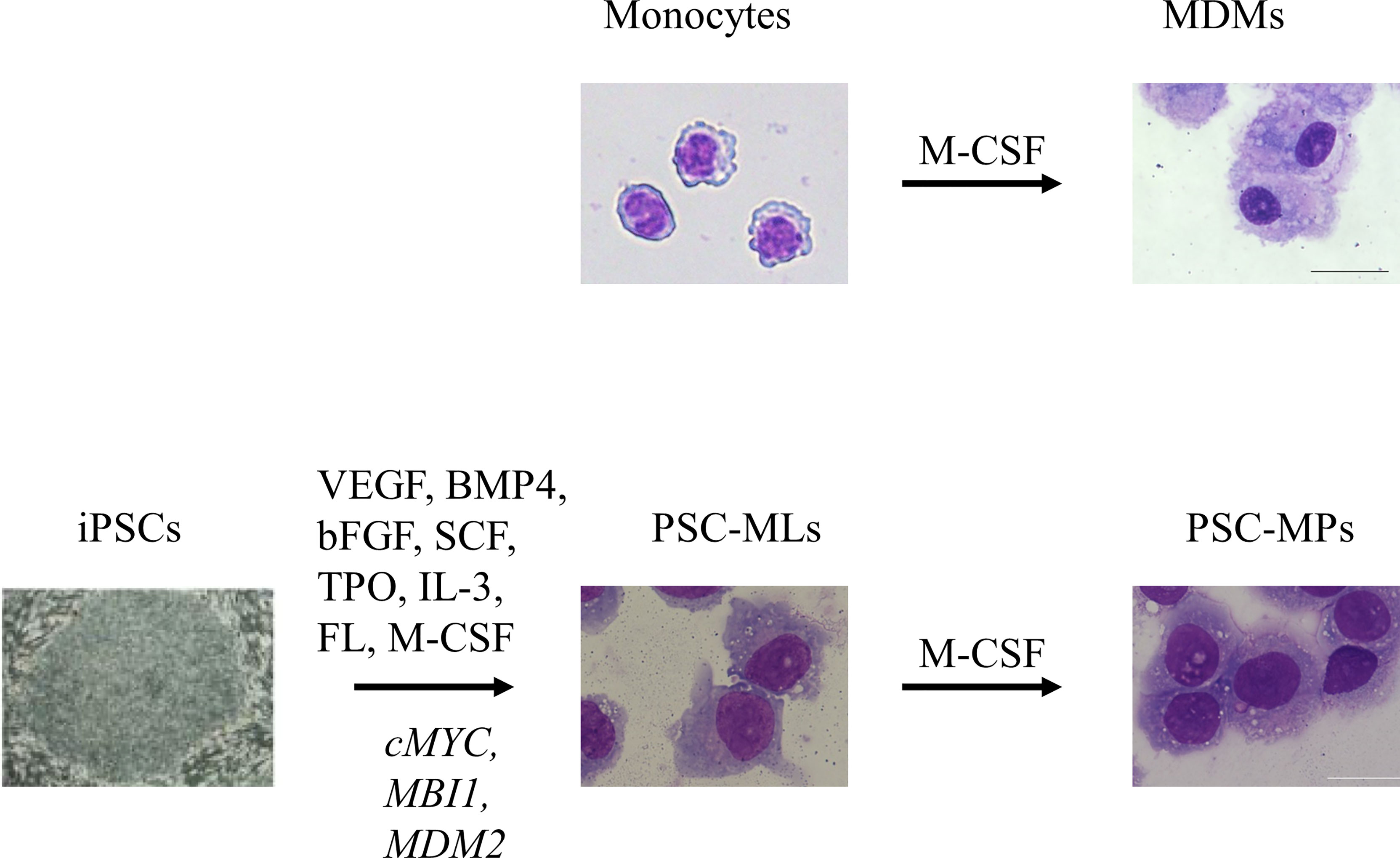
Figure 1 In vitro culture of primary monocytes for 7 days in the presence of macrophage colony-stimulating factor (M-CSF) gives rise to adherent monocyte-derived macrophages (MDMs). Sequential stimulation with VEGF, BMP4, bFGF, SCF, TPO, IL-3, FL, and M-CSF differentiates induced pluripotent stem cells (iPSCs) into floating monocyte-like cells. After introduction of three transgenes, namely, cMYC, MBI1, and MDM2, into the floating cells, PSC-derived immortalized myeloid cell lines (PSC-MLs) begin to proliferate. In vitro culture of PSC-MLs for 7 days in the presence of M-CSF gives rise to adherent PSC-derived macrophages (PSC-MPs). Scale bars, 20 µm. VEGF, vascular endothelial growth factor; BMP4, bone morphogenic protein type 4; bFGF, basic fibroblast growth factor; SCF, stem cell factor; TPO, thrombopoietin; IL-3, interleukin-3; FL, FLT3 ligand.
The immortalized cell lines THP-1 and U937 are used as alternative sources of macrophages because they expand spontaneously and are amenable to genetic manipulation. These cell lines originate from the peripheral blood of patients with acute monocytic leukemia and contain highly proliferative floating CD14+ “monocyte-like” cells that can differentiate into “macrophage-like” cells upon culture in the presence of phorbol myristate acetate or M-CSF (19). In the field of autoinflammatory disease research, THP-1 cells are used to analyze cell death caused by the expression of mutant NLRP3 (20, 21). However, as these cells are derived from malignant tumor cells, their biological relevance to non-malignant monocytes/macrophages is limited (22). For example, THP-1 cells secrete only small amounts of cytokines in response to lipopolysaccharide (LPS) stimulation (22), and there are no reports describing increased inflammasome activation in these immortalized cell lines.
To overcome the limitations described above, several methods have been developed to generate macrophages from PSCs. In this approach, the culture conditions drive embryonic stem cells or iPSCs to differentiate through a pathway that recapitulates embryonic hematopoiesis (23–26). The advantages of PSC-MPs include easy availability, scalability, standardizability, and easy genetic manipulation (27, 28).
While a few reports have described protocols for differentiating PSCs into monocytes (29), more research has been performed with PSC-MPs than with PSC-derived monocytes, particularly within the field of autoinflammatory diseases (Table 1). Many studies report that PSC-MPs and MDMs have similar phenotypes, functions, and transcriptomes (26, 30–33). However, stable differences between PSC-MPs and MDMs were also identified; indeed, it is suggested that PSC-MPs recapitulate embryonic-origin macrophages rather than MDMs, which are derived from definitive hematopoiesis (23–26). Therefore, even though PSC-MPs have been applied successfully to the functional analysis of macrophages in the context of many diseases (Table 1), we need to keep in mind that the phenotype of PSC-MPs sometimes differs from that of MDMs or tissue-resident macrophages; where necessary, the validity of findings based on PSC-MPs should be confirmed using MDMs.

Table 1 Disease modeling and application of iPSCs to autoinflammatory diseases.
In early studies, whenever we needed PSC-MPs, we repeated the whole process of differentiating PSCs into macrophages; this process took almost 1 month to generate enough cells for our experiments (4). The differentiation efficiency was not consistent, and the protocol was laborious. To improve the efficiency of differentiation and to standardize macrophage products, we developed a method of cryopreserving PSC-MP (34). For this purpose, we established PSC-derived immortalized myeloid cell lines (PSC-MLs) by introducing MYC, BMI1, and MDM2 into iPSC-derived floating monocytic cells (Figure 1) (6, 10). The resulting PSC-MLs proliferated vigorously and continuously and were amenable to freeze-and-thaw cycles. After a 1-week culture in the presence of M-CSF, PSC-MLs differentiated into adherent macrophages. Both PSC-MLs and PSC-MPs expressed monocyte/macrophage markers CD45, CD11b, and CD14 and secreted cytokines in response to various stimuli. While immature PSC-MLs were more proliferative than PSC-MPs, terminally differentiated PSC-MPs secreted higher levels of cytokines than PSC-MLs. Therefore, we used both PSC-MLs and PSC-MPs for our research, depending on the goal of each experiment (4, 6–8, 10–12, 15, 16) (Table 1). Next, we will outline how PSC-derived blood cells contribute to i) elucidation of disease pathogenesis, ii) functional analysis to facilitate correct diagnosis, iii) evaluation of the disease relevancies of newly identified mutations, and iv) discovery of new drug candidates.
Chronic infantile neurologic cutaneous and articular (CINCA; MIM 607115) syndrome, also known as a neonatal-onset multisystem inflammatory disease (NOMID), is an autoinflammatory syndrome characterized by systemic inflammation accompanied by an urticarial rash, neurologic manifestations, and arthropathy that begins during the neonatal period (35, 36); it is the most severe form of the autoinflammatory spectrum called cryopyrin-associated periodic fever syndrome. Patients carry a heterozygous gain-of-function mutation in NLRP3 gene and present with systemic inflammation due to excessive IL-1β production caused by hyperactivation of the NLRP3 inflammasome (37, 38). While approximately half of CINCA patients carry heterozygous gain-of-function mutations in NLRP3 gene (39), 30%–40% harbor NLRP3 mutations in only a small number of somatic cells (4.2%–35.8% of blood cells) (40, 41). Despite the small percentage of mutant cells, the clinical phenotype of mosaic patients is comparable with that of patients with germline mutations. Therefore, it remains controversial whether these low-frequency NLRP3 mutant-positive cells alone are responsible for the disease phenotype or whether cells other than those harboring NLRP3 mutations also contribute to pathogenesis.
Since each iPSC clone originates from a single cell (42), iPSC lines can be used as a discovery tool to evaluate the impact of low-frequency somatic mosaicism mutations. Taking advantage of this feature, NLRP3-mutant and wild-type (WT) iPSCs were established from patients with CINCA syndrome harboring a somatic NLRP3 mutation (4). When these iPSCs were differentiated into macrophages and their phenotypes were compared, only NLRP3-mutant macrophages produced a large amount of IL-1β. Interestingly, when mutant macrophages were co-cultured with WT macrophages to create a pseudo-mosaic state, the production of IL-1β was significantly higher than that of mutant macrophages alone. In other words, in cases of somatic mosaicism, NLRP3-mutant cells are the main producers of IL-1β, although WT cells also contribute to inflammation in some way. Later, Baroja-Mazo et al. used patient-derived MDMs to show that upon activation of caspase-1, oligomeric NLRP3 inflammasome particles are released from activated macrophages and phagocytosed by surrounding macrophages, leading to further activation of caspase-1 (43). Thus, iPSCs can be used for a detailed analysis of the unique pathology associated with somatic mosaicism.
Regarding the pathogenesis of cartilage overgrowth in CINCA syndrome patients, different methods have been used to assess the contribution of chondrocytes and hematopoietic cells. After differentiating WT or mutant iPSCs into chondrocytes, we compared the size of the chondrocyte tissues produced; we found that mutant iPSCs produced larger chondrocyte masses than WT iPSCs owing to the overproduction of glycosaminoglycans, which correlated with increased expression of the chondrocyte master regulator SOX9; this was independent of caspase 1 and IL-1 and, thus, the NLRP3 inflammasome (5). By contrast, Wang et al. used a model mouse exhibiting global NLRP3 activation and several characteristics of the human disease (i.e., systemic inflammation and cartilage dysplasia) to show that activation of NLRP3 in myeloid cells, but not in mesenchymal cells, triggers chronic inflammation, which ultimately causes growth plate and epiphyseal dysplasia (44). Mechanistically, inflammation causes severe anemia and hypoxia in the bone environment but downregulates the HIF-1α pathway in chondrocytes, thereby promoting the demise of these cells. It is theoretically possible to obtain both PSC-derived chondrocytes and macrophages and evaluate their interaction in vitro co-cultures; however, mouse models may provide the opportunity to observe physiological interactions over a longer term than PSC-derived somatic cell models.
Blau syndrome (MIM 186580) is a disease caused by a heterozygous gain-of-function mutation in NOD2 gene, which leads to granulomatous lesions in the skin, joints, and eyes during childhood and can cause severe complications such as blindness and joint contractures later in life (45, 46). The NOD2 protein is an intracellular pathogen recognition receptor, which upon recognition of the ligand MDP activates the nuclear factor-κB (NF-κB) pathway, thereby upregulating the production of proinflammatory cytokines and chemokines. Although pathological studies reveal that granulomas in Blau syndrome patients are accompanied by a prominent expression of IFN-γ (47), the details regarding the molecular mechanism(s) by which NOD2 mutations drive the pathogenesis of Blau syndrome are unclear. The treatment for Blau syndrome has long been non-specific immunosuppressive therapies such as corticosteroids and/or methotrexate; however, recent studies report the effectiveness of biologics targeting TNF, IL-6, and IL-1 (48–53). Among these, anti-TNFα agents are used most widely, although the pharmacologic mechanism is unknown. Therefore, investigation of the cellular phenotypes of patients is necessary to evaluate the mechanism(s) underlying anti-TNFα therapy.
Studies based on mouse models have not reproduced the disease-related phenotype sufficiently (54). Therefore, we investigated the phenotypes of human macrophages carrying mutant NOD2 by establishing iPSCs from patients with a NOD2 mutation and obtaining isogenic iPSC clones in which the mutation was repaired by CRISPR/Cas9; we then differentiated them into macrophages (6). We found that IFN-γ-primed PSC-MPs harboring mutant NOD2 demonstrated ligand-independent activation of NF-kB translocation to the nucleus, followed by the production of proinflammatory cytokines such as IL-6 and IL-8. We also confirmed this phenotype in more physiological cells; indeed, MDMs derived from Blau syndrome patients showed IFN-γ-induced ligand-independent activation of NF-kB translocation to the nucleus and subsequent cytokine production.
Next, we tried to clarify how anti-TNF treatment helps to control inflammation by comparing characteristics such as transcriptome profiling and cytokine secretion by MDMs from untreated and anti-TNF-treated Blau syndrome patients (7). We found that TNF-dependent NF-kB signaling reduces the threshold for IFN-γ-mediated inflammatory responses in Blau syndrome and that resetting of this primed state by anti-TNF treatment contributes to the prevention of the autoinflammatory loop, even in the presence of a NOD2 mutation and IFN-γ stimulation. Thus, the iPSC-based macrophage study enabled us to elucidate disease pathogenesis and to identify the mechanism underlying the efficacy of anti-TNF treatment at the cellular level. To ascertain whether blocking IFN-γ signaling is a potential treatment for chronic inflammation in Blau syndrome patients, we still need to determine whether the IFN-γ pathway is actually activated in these patients and whether IFN-γ signaling is the principal priming pathway among the stimulants known to upregulate NOD2 expression (i.e., TNF-α, LPS, and other Toll-like receptor ligands) (55–57).
Nakajo–Nishimura syndrome (NNS)/chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome/joint contractures, muscular atrophy, microcytic anemia, and panniculitis-induced lipodystrophy (JMP) syndrome is a form of proteasome-associated autoinflammatory syndrome (PRAAS1/MIM 256040) characterized by chronic inflammation and lipomuscular atrophy caused by homozygous loss-of-function mutations in PSMB8 gene encoding β5i, a component of the immunoproteasome (58, 59). Based on the finding of a strong type I interferon (IFN) response gene signature in patient peripheral blood cells (60, 61), Janus kinase (JAK) inhibitors are an effective treatment for PRAAS (62) because they inhibit the JAK/STAT pathway, the principal signaling pathway downstream of cytokines and growth factor receptors (including the IFN-α/β receptor) (63, 64). However, the precise mechanism underlying autoinflammation remains unclear. To elucidate the impact of PSMB8 mutations on monocyte/macrophage function, we generated iPSCs from an NNS patient and repaired the PSMB8 mutation using the CRISPR/Cas9 system (8). We then generated iPSC lines that share the same genetic background but without the PSMB8 mutation. When immunoproteasome assembly in PSC-MLs was induced by IFN-γ and TNF-α, immunoproteasome activity in PSMB8-mutant PSC-MLs was impaired significantly compared with that in the WT counterparts. As a consequence, secretion of the proinflammatory cytokine IL-6, and chemokines MCP-1 and IP-10, by mutant PSC-MLs increased. Furthermore, we revealed that the production of intracellular reactive oxygen species also increased, that mutant cells had higher levels of p38 MAPK and phosphorylated STAT1, and that addition of antioxidants, a p38 MAPK inhibitor, or JAK inhibitors suppressed the production of proinflammatory cytokines and chemokines. This demonstrates that PSC-MLs is a useful tool for modeling proteasome-associated autoinflammatory diseases.
Several different mechanisms have been postulated to explain the lipodystrophy in PRAAS. On the one hand, lipophagia can result from the proinflammatory state of adipose tissue macrophages (65, 66). Alternatively, high IFN levels may be toxic to adipocytes (67). PSC-derived blood cells alone cannot reproduce the complex interactions within the human body. Verhoeven et al. reported that hematopoietic stem cell transplantation halted the progression of lipodystrophy in a PRAAS patient during a 7-year follow-up, demonstrating that hematopoietic cells play a role in the lipodystrophy (68). It is theoretically possible to obtain both macrophages and adipocytes from PSCs and to evaluate their interaction in in vitro co-culture; however, to examine long-term effects, clinical observation of the patients may be more appropriate.
IL-10, one of the most important cytokines for the maintenance of intestinal homeostasis, regulates inflammation by inhibiting macrophage activation (69, 70). While the protective role of IL-10 is relatively well established in the context of inflammatory bowel disease (IBD) and other inflammatory diseases, its role in susceptibility to infections is less well understood. To study the impact of IL-10 on the inflammatory and microbicidal activities of macrophages, Mukhopadhyay et al. established iPSCs from a patient with homozygous loss-of-function mutations in the IL-10 receptor β (IL-10RB) and differentiated them into macrophages (9). They showed that IL-10RB−/− patient PSC-MPs were deficient in the IL-10 signaling pathway and that suppression of proinflammatory cytokine secretion was not observed upon simultaneous stimulation with IL-10 and LPS. IL-10RB−/− macrophages also exhibited a defect in bactericidal activity. Genes involved in synthesis and receptor pathways for PGE2 were more highly induced in IL-10RB−/− PSC-MPs, and these macrophages produced more PGE2 than controls after LPS stimulation. Furthermore, combined inhibition of PGE2 synthesis and receptor binding increased bactericidal activity. These results indicate the presence of crosstalk between the IL-10 and PGE2 pathways, dysregulation of which may drive aberrant macrophage activation and impaired host defense, thereby contributing to IBD pathogenesis.
As mentioned above, while about 90% of CINCA syndrome patients have constitutive or somatic mosaic mutations in NLRP3, the remaining 10% do not (41). Since most CINCA patients lacking NLRP3 mutations respond to anti-IL-1 therapy, activation of some kind of inflammasome is suspected. Therefore, we established iPSCs from a CINCA syndrome patient in whom an NLRP3 mutation was not identified by conventional Sanger sequencing, differentiated them into PSC-MLs, and measured the production of IL-1β in response to NLRP3 inflammasome activation (10). PSC-ML clones were categorized as “normal” clones that secreted IL-1β after LPS and ATP stimulation and “pathological” clones that secreted IL-1β after LPS stimulation alone. To elucidate the phenotypic heterogeneity of IL-1β secretion among the clones, we performed whole-exome sequencing of representative iPSC clones and identified a novel mutation in NLRC4 gene in the diseased clones. The mutant allele was observed in the patient’s fibroblasts (34.1%) and PBMCs (30.3%). When we knocked out the mutant NLRC4 gene in PSC-MLs, the production of IL-1β normalized. These results show that somatic mosaicism of the NLRC4 gene mutation caused the clinical phenotype of CINCA syndrome in this patient. To date, no other case of somatic mosaicism of NLRC4 has been reported. Collectively, these data show that iPSC technology can be used to diagnose a novel somatic mosaic mutation.
NF-κB essential modulator (NEMO), also known as an inhibitor of NF-κB kinase subunit gamma (IKK-γ), encoded by IKBKG gene (71, 72), is the third regulatory subunit of the IκB kinase (IKK) complex (73, 74). Amorphic mutations of IKBKG, which abolish canonical NF-κB activation, are lethal in men, whereas in women, they cause X-linked dominant incontinentia pigmenti (IP) (MIM 308300), a multisystem disorder affecting the skin and its appendages (75, 76). Hypomorphic IKBKG mutations that impair IκBα phosphorylation and sequential NF-κB activation cause X-linked recessive (XR) anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) (MIM 300291) (77, 78). Affected men display typical signs of EDA, including sparse hair, eyebrows, and eyelashes; hypohidrosis; hypodontia; and conical incisors, together with immunodeficiency or inflammatory colitis (79). The main immune phenotype of EDA-ID is immunodeficiency rather than inflammation, but both EDA-ID and autoinflammatory diseases are categorized within the broad spectrum of primary immunodeficiency (80). Since iPSC-derived macrophages and ectodermal cells contributed substantially to establishing a correct diagnosis, we would like to describe the following study. While most variants underlying XR-EDA-ID are missense mutations or in-frame indels, approximately 10% of sporadic and familial cases of EDA-ID remain genetically unexplained. Therefore, we investigated three male patients from two families whose ID phenotype was much more severe than the manifestations of EDA (11), leading to an early death before the age of 1 year. Whole-genome sequencing identified the same deep intronic mutation in IKBKG. Next, we found that this deep intronic IKBKG mutation created a novel splicing donor site for a pseudoexon inclusion, which led to a severe decrease in NEMO protein expression and inflammatory cytokine secretion by patient PBMCs. Using patient-derived iPSCs, we revealed a cell type-dependent effect of the mutation on aberrant IKBKG splicing, which explains the reason for the discrepancy between the severe ID phenotype and the more subtle EDA symptoms. When we measured the levels of WT and alternative IKBKG transcripts in undifferentiated iPSCs, PSC-MLs, and iPSC-derived neuronal precursor cells (iPSC-NPs), we found that iPSCs produced 17% WT transcripts, PSC-MLs produced only 3% WT transcripts, and iPSC-NPs produced 35%. Patient-derived PSC-MLs showed much lower WT NEMO protein levels, along with impaired NF-κB activation upon LPS stimulation. Complementation of patient-iPSCs with WT NEMO restored NF-κB pathway activation in PSC-MLs. Thus, iPSCs contribute to the correct diagnosis of the deep intronic IKBKG mutation and to the identification of the cell type-dependent quantitative NEMO deficiency, thereby expanding our understanding of this disease.
Monocytes and macrophages play similar roles in the pathogenesis of most inflammatory diseases. For example, both NLRP3-mutant monocytes (37) and macrophages (4) exhibit spontaneous NLRP3 inflammasome activation without secondary signals and secrete IL-1β after priming with LPS alone. However, cytokine secretion by monocytes and macrophages from familial Mediterranean fever (FMF) patients is clearly different (12).
FMF, the most common hereditary autoinflammatory disorder, is characterized by recurrent episodes of fever, polyserositis, and abdominal pain (MIM 249100). FMF is associated with mutations in MEFV gene, which encodes the inflammasome adaptor pyrin (81). Pyrin is an inflammasome sensor that detects imbalances in Rho GTPase activity, which can be caused by bacterial effectors or bacterial toxins (82). Almost 400 MEFV variants have been recorded in Infevers, an online database of autoinflammatory disease mutations. Among MEFV variants, a systematic review revealed that M694V and M694I in exon 10 are related to a severe phenotype of FMF (83). Other MEFV variants are associated with variable clinical phenotypes, including pyrin-associated autoinflammation with neutrophilic dermatosis (84, 85) and autosomal-dominant FMF-like diseases (86–88). Consequently, the novel umbrella term, pyrin-associated autoinflammatory diseases, has been proposed to define all autoinflammatory diseases caused by MEFV mutations (89). Although a consensus-driven pathogenicity classification was proposed recently to support the uniform diagnosis of FMF worldwide (90), the complexity of the clinical phenotype and its association with MEFV variants led to difficulty in assessing the pathogenicity of variants identified in clinical settings. Despite the successful use of colchicine and IL-1β-blocking therapies as treatments for FMF, in vitro pyrin inflammasome activation (and its inhibition by colchicine) in patients’ hematopoietic cells remains controversial (91–94).
To clarify this issue, we evaluated cytokine secretion by primary monocytes and MDMs obtained from FMF patients carrying the heterozygous M694I mutation. In response to TcdA stimulation, levels of IL-1β secreted by FMF monocytes were similar to those of control monocytes, and colchicine failed to inhibit IL-1β secretion by FMF monocytes. By contrast, IL-1β secretion by FMF MDMs was significantly higher than that by control MDMs in response to LPS and TcdA, and IL-1β secretion by FMF MDMs was inhibited by colchicine. These results suggest that MDMs, rather than monocytes, reflect the clinical features of FMF patients (e.g., hyperactivation of the pyrin inflammasome and subsequent inhibition by colchicine). After confirming that macrophages derived from patients’ iPSCs (PSC-MPs) recapitulate the phenotype of FMF MDMs, we evaluated two rare MEFV variants, T577N and N679H, identified in two families in which autoinflammatory disease with dominant inheritance was suspected (95, 96). No T577N patients met the Tel-Hashomer criteria, whereas two N679H patients fulfilled the criteria (97). Whereas the amount of IL-1β secreted by T577N iPS-MPs was comparable with that secreted by WT cells, N679H iPS-MPs secreted significantly more IL-1β (like the M694I variant). Thus, MEFV variants causing FMF, namely, N679H and M694I, induced IL-1β secretion after pyrin inflammasome activation. Thus, we established a method for evaluating MEFV variants by obtaining mutant PSC-MPs and measuring cytokine secretion in response to pyrin inflammasome stimulation (12).
In addition, we characterized cytokine secretion by primary monocytes and macrophages isolated from typical FMF patients. Gene expression differs considerably between monocytes and macrophages, including expression of the tubulin-related genes (98). Given the vital role of microtubule polymerization in pyrin inflammasome activation (99), we speculate that greater expression of tubulin-related genes in macrophages might be related to the differential response to colchicine inhibition between monocytes and macrophages. Moreover, monocytes and macrophages are somewhat different in terms of inflammasome activation pathways. For example, although both cell types use the canonical NLRP3 inflammasome activation pathway, the alternative (100) or non-canonical (101) NLRP3 inflammasome pathway is functional only in monocytes. It is possible that an undiscovered pyrin inflammasome activation pathway is functioning in either monocytes or macrophages, but not in both. The precise reason underlying the distinct mechanisms of pyrin inflammasome activation in these cells remains to be elucidated.
Evaluation of pyrin inflammasome activation using PSC-MPs led to the discovery of a role for enhanced pyrin inflammasome activation in the pathogenesis of CDC42-associated autoinflammation (102). Nishitani-Isa et al. used PSC-MPs to show that aberrant palmitoylation of CDC42 protein carrying the mutation caused its retention in the Golgi apparatus and triggered overactivation of the pyrin inflammasome. By contrast, subsequent ex vivo or in vitro studies established methods for functional evaluation of FMF-related MEFV variants. THP-1 cells transfected with FMF-related MEFV variants showed higher levels of UCN-01/TcdA-induced cell death than THP-1 cells expressing other MEFV variants (103). Similar studies focusing on the evaluation of primary cells have been reported; indeed, Magnotti et al. showed that UCN-01-induced cell death was much faster in FMF monocytes than in monocytes from healthy donors or patients suffering from other inflammatory disorders. They also established an assay that can be used for rapid diagnosis of FMF with high sensitivity and specificity (104). By focusing on unresponsiveness to colchicine inhibition, van Gorp et al. reported an assay that robustly segregated FMF patients from healthy donors and patients with other inflammatory disorders (105). One advantage of this assay is that the test may be performed on PBMCs and even whole blood. While cell line-based approaches are free from the influence of existing inflammation or prescribed drugs, as well as being more suitable for the evaluation of a specific mutation, primary cell assays have an advantage in that they take less time and the results reflect the influence of the genetic background, or the combined effects of multiple MEFV variants, in a single patient. Therefore, we need to select the most appropriate approach for each situation.
Type I IFN-inducible oligoadenylate synthetase 1 (OAS1) initiates an antiviral immune response upon recognition of cytoplasmic viral double-stranded RNA (dsRNA) (106, 107). OAS1 is a template-independent nucleotidyltransferase that produces the second messengers 2′-5′-oligoadenylate (2-5A) (108, 109). In turn, 2-5A activates ribonuclease L (RNase L), which degrades viral and cellular RNA, thereby interfering with viral propagation (110).
Okano et al. described a polymorphic autoinflammatory immunodeficiency with recurrent fever, dermatitis, IBD, PAP, and hypogammaglobulinemia caused by de novo heterozygous OAS1 gain-of-function mutations; they named the disease OAS1-associated polymorphic autoinflammatory immunodeficiency (OPAID) (13). The expression of mutant OAS1 in response to common infectious agents resulted in an inappropriate synthesis of 2-5A independent of dsRNA binding; this induced RNase L-mediated cleavage of cellular RNA, leading to transcriptomic alteration, translational arrest, and dysfunction and apoptosis of primary monocytes, PSC-MPs, and B cells. To overcome the scarcity of primary monocytes in that study, the authors differentiated patient-derived iPSCs into macrophages (i.e., PSC-MPs). IFNα-stimulated PSC-MPs carrying mutant OAS1 displayed impaired cell adhesion and clustering, scavenger receptor expression, and phagocytosis in an RNase L-dependent manner. While mutant OAS1-knock-in mice failed to reproduce the disease phenotype (111), the characteristics of PSC-MPs were consistent with those of primary monocytes. Given the reported differences in immune responses between species (112), human iPSC-derived hematopoietic cells may be a more relevant source of primary patient-derived cells than animal models for research into certain diseases.
The NF-κB protein complex is integral to the initiation of inflammation, and NF-κB activation is controlled by inhibitors of κB (IκBα, IκBβ, and IκBϵ) and by the IκB kinase (IKK) complex, which comprises NEMO, IKK1, and IKK2 (described above). Patients with genetic defects (e.g., in NEMO and NFKBIA) in the NF-κB signaling pathway usually display severe immunodeficiency, with impaired cellular responses to immune stimuli such as LPS or TNF-α (113, 114).
Tan et al. reported an infant with a clinical pathology comprising neutrophil-mediated autoinflammation and recurrent bacterial infections caused by a de novo heterozygous missense mutation in NFKBIA (14). The resulting L34P IκBα variant caused a severe reduction in NF-κB nuclear translocation and, consequently, downstream production of IL-6 or IL-8 by the patient’s fibroblasts. Paradoxically, IL-1β concentrations in the patient’s blood were elevated. To determine whether myeloid cells were the major source of elevated IL-1β levels, they generated iPSC-derived macrophages from the patient’s fibroblasts. Despite the patient’s PSC-MPs showing defective nuclear translocation of NF-κB in response to LPS stimulation, they produced significantly more IL-1β than control PSC-MPs. The patient’s hypersecretion of IL-1β correlated with activated neutrophilia and liver fibrosis with neutrophil accumulation. Hematopoietic stem cell transplantation reversed the neutrophilia, restored neutrophils to a resting state, and normalized IL-1β release from stimulated leukocytes. These data suggest that NF-κB in humans plays an unexpected role as an anti-inflammatory agent by regulating IL-1β secretion, thereby preventing myeloid inflammation.
Why are iPSC-derived monocytes/macrophages a useful platform for high-throughput screening of drug candidates? Because of their pluripotency and proliferative potential, iPSCs can serve as an unlimited source of patient-derived somatic cells. Two studies provide proof of the concept that iPSC-derived monocytes/macrophages are a useful tool for drug screening (15, 16).
In one study, we searched for compounds that inhibit NLRP3 inflammasome activation in PSC-MPs (15). The NLPR3 inflammasome is an attractive drug target because NLRP3 inflammasome activation is associated not only with rare autoinflammatory disorders such as CINCA syndrome but also with the pathogenesis of various chronic inflammatory conditions (115). NLRP3-mutant macrophages were used for this assay because LPS-mediated stimulation in the absence of a second signal was sufficient to activate the NLRP3 inflammasome (4). High-throughput screening of 4,825 compounds, including Food and Drug Administration (FDA)-approved drugs and compounds with known bioactivity, identified seven that selectively inhibited IL-1β secretion without affecting IL-6 production. All seven compounds inhibit the NLRP3 inflammasome (116–119). Before selecting the cell types for high-throughput screening, we compared three types of PSC-derived blood cells in terms of the coefficient of variation (CV) for IL-1β secretion; this is because blood cells with a low CV value enabled us to accurately assess the potency of candidate compounds. The first type of PSC-derived floating cells was obtained after a 2-week culture of iPSCs in a differentiation medium. The second cell type of PSC-MLs was established by the lentiviral-based introduction of three genes into PSC-derived floating cells (6, 10). After a 1-week culture of PSC-MLs in a fetal calf serum (FCS)-containing medium in the presence of M-CSF, we obtained a third type of terminally differentiated PSC-MP (Figure 1). The levels of released cytokines were more consistent, and the CV value became lower as differentiation progressed; the CV value was lowest for PSC-MPs. Therefore, we used PSC-MPs to screen NLRP3 inhibitors (15).
In another study, we screened potential therapeutic candidates using PSC-MLs derived from NNS patients (16); screening was based on consistent overproduction of MCP-1 and IL-10 from PSC-MLs derived from NNS patients in the preceding study (8). We screened 5,821 compounds, including FDA-approved drugs, kinase inhibitors, and bioactive chemicals, and we identified CUDC-907 as an effective inhibitor of MCP-1 and IP-10 release (16). While hit compound CUDC-907 seemed a promising drug candidate in terms of efficacy, there were concerns regarding adverse effects because CUDC-907 inhibited NNS fibroblast proliferation during a 2-week culture. Therefore, CUDC-907 was not directly applicable to the clinical study.
In both studies, we started screening compound libraries with known bioactivity and provided proof of concept that PSC-derived monocytes/macrophages can serve as an effective tool for screening drug candidates, although the hit compounds could not be applied directly to clinical studies. Combining high-throughput screening using PSC-monocytes/macrophages with pharmaceutical techniques (to generate a more potent compound from known substances by modifying the chemical structure) may pave the way to novel drug discovery.
The PSC–macrophage system would not necessarily be suitable for modeling all autoinflammatory diseases. Therefore, we would like to mention two points that should be considered before starting a study using PSC-MPs.
PSC-derived macrophages are a homogeneous population, and modeling the whole human body using only macrophages is impossible. For example, RELA (120) or RIPK 1 (121) mutations were identified among patients with early-onset inflammatory diseases. Both proteins are involved in the NF-κB activation pathway in response to TNF stimulation (122). Since they noted enhanced cytotoxicity caused by TNF stimulation in fibroblasts derived from the RELA-haploinsufficiency patient (but not in hematopoietic cells) (120), PSC-MPs alone would be insufficient for modeling such diseases; rather, studies of the interaction among epithelial cells, stromal cells, and hematopoietic cells would be necessary. As described above, clinical observation provided novel findings of lipodystrophy in a PRAAS patient (68), and a knock-in mouse model revealed the contribution of NLRP3-mutant hematopoietic cells to cartilage overgrowth (44). Thus, elucidation of the pathologic interactions between hematopoietic and non-hematopoietic cells is usually difficult when using PSC-derived somatic cells alone.
Many researchers have identified similarities between MDMs and PSC-MPs with regard to global gene expression (23, 24, 26, 30, 32, 33), cytokine secretion (26), and phagocytosis of infectious organisms (32). Polarization of macrophages from M0 to M1 or M2 is accompanied by changes in gene expression similar to those observed in blood-derived counterparts (26, 33). However, certain differences in the expression of genes involved in chemokine production, antigen presentation, and tissue remodeling were identified (30). We need to be cautious when applying PSC-MPs for disease modeling because these differences may affect their responses.
The list of autoinflammatory diseases continues to expand and now includes over 40 genetically defined disorders categorized according to defects affecting the inflammasome, type 1 interferonopathies, and non-inflammasome-related conditions (123). Although the genetic basis of many autoinflammatory diseases is now known, the molecular etiology frequently remains unclear. Given the rapid progress in the application of PSC-MPs to research into autoinflammatory diseases, further discoveries are expected. Takata et al. reported differentiation of PSCs into tissue-resident macrophage-like cells upon receipt of organ-specific cues (24). Co-culturing human PSC-MPs with iPSC-derived neurons in vitro promoted differentiation into microglia-like cells. Furthermore, murine PSC-MPs differentiated in vivo into functional alveolar macrophages after engraftment in the lung. Novel methods of driving differentiation into tissue-resident macrophages will enable modeling and elucidation of organ-specific inflammation. In addition to monocytes/macrophages, neutrophils are important innate immune cells that are involved in the pathogenesis of autoinflammatory disorders. While it is now possible to cryopreserve PSC-MPs at the progenitor level (6, 10), or as the final product (34), cryopreservation of PSC-derived neutrophils has yet to be reported. Improvements in the differentiation protocol may enable the utilization of PSC-derived neutrophils for autoinflammatory disease research. We would like to describe two other promising examples of PSC-MP applications.
The scalability and standardizability of PSC-MPs make them particularly suitable for screening drug candidates. In addition to the diseases described above (15, 16), PSC-MPs may be useful for identifying pyrin inflammasome inhibitors other than the traditional colchicine. Colchicine has been the standard treatment for FMF for more than 100 years. However, in 5%–10% of patients, it is either ineffective or associated with unacceptable side effects (124). The efficacy and safety of canakinumab, an anti-IL-1β monoclonal antibody, have been shown in patients with colchicine-resistant FMF (125). However, given the drawbacks of canakinumab, such as high cost and limited safety information about administration to pregnant women, novel alternative treatments are needed. Recent reports about the association between microtubule polymerization and pyrin inflammasome activation (99, 126) have focused attention on microtubule inhibitors. Microtubules are the target of anticancer drugs such as paclitaxel or vinblastine. Potent and safe colchicine binding-site inhibitors such as CA4P and ABT-751 have entered clinical trials as anticancer agents (127). Although many other candidates have not entered clinical trials due to toxicity at the high concentrations needed for anticancer treatment, they may be effective and safe pyrin inflammasome inhibitors when used at lower concentrations. Among the tubulin inhibitors that were previously developed as anticancer medicines, we may be able to identify a more potent and less toxic pyrin inflammasome inhibitor than conventional colchicine.
Originally, “autoinflammatory disorders” covered inborn errors of innate immunity (1). Classification of autoinflammatory diseases has been updated periodically and now covers not only abnormalities in innate immunity but also those in adaptive immunity, including STING-associated vasculopathy with onset in infancy (SAVI) or COPA syndrome. A severe inflammatory phenotype of SAVI is induced by gain-of-function mutations in STING1, which encodes STING (stimulator of IFN genes); this phenotype is characterized clinically by skin vasculopathy, systemic inflammation, and lung involvement (e.g., interstitial lung disease or diffuse alveolar hemorrhage), which is associated with high morbidity and mortality (128, 129). In addition to constitutive activation of the type I IFN pathway, an autoimmune component (e.g., high titers of autoantibodies) is frequently detected in SAVI (130). Self-DNA sensing through cGAS-STING is involved in many processes, including autoimmunity (131); however, the precise mechanism linking STING gain of function to the production of autoantibodies has not yet been defined.
While various groups have established methods for obtaining innate immune macrophages from PSCs (as described above), differentiating PSCs into T cells is difficult. However, a recent report describes a protocol for differentiating PSCs to T-cell receptor (TCR)-expressing innate lymphoid-like helper cells (132). These innate lymphoid helper-like cells induce bcr-abl-specific TCR signaling, which mediates effective anti-leukemic cytotoxic T lymphocyte responses via dendritic cell (DC) maturation. Further deciphering of STING-mediated autoimmunity is awaited, and further investigations into PSC-derived macrophages and T cells may provide an opportunity to study aberrant interactions between innate and adaptive immune cells.
In this review, we provide a comprehensive overview of current advances in the use of iPSC-derived monocytes/macrophages for research into autoinflammatory diseases. We describe the advantages and characteristics of PSC-MPs, current applications to research into autoinflammatory diseases, and future directions. We hope that this review will provide clues that facilitate further research into autoinflammatory diseases and contribute to the development of new treatments for patients.
This article is mainly written by TT. YH wrote part of the manuscript. TS, KI, TY, and MS proofread the manuscript. RN revised the manuscript. The corresponding author had final responsibility for the decision to submit for publication. All authors contributed to the article and approved the submitted version.
This research was supported by the following grant: Grants-in-Aids for Scientific Research (C) (grant JP19K08320 to T.T.).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline Mutations in the Extracellular Domains of the 55 kDa TNF Receptor, TNFR1, Define a Family of Dominantly Inherited Autoinflammatory Syndromes. Cell (1999) 97(1):133–44. doi: 10.1016/S0092-8674(00)80721-7
2. Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells From Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell (2006) 126(4):663–76. doi: 10.1016/j.cell.2006.07.024
3. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of Pluripotent Stem Cells From Adult Human Fibroblasts by Defined Factors. Cell (2007) 131(5):861–72. doi: 10.1016/j.cell.2007.11.019
4. Tanaka T, Takahashi K, Yamane M, Tomida S, Nakamura S, Oshima K, et al. Induced Pluripotent Stem Cells From CINCA Syndrome Patients as a Model for Dissecting Somatic Mosaicism and Drug Discovery. Blood (2012) 120(6):1299–308. doi: 10.1182/blood-2012-03-417881
5. Yokoyama K, Ikeya M, Umeda K, Oda H, Nodomi S, Nasu A, et al. Enhanced Chondrogenesis of Induced Pluripotent Stem Cells From Patients With Neonatal-Onset Multisystem Inflammatory Disease Occurs via the Caspase 1-Independent cAMP/Protein Kinase A/CREB Pathway. Arthritis Rheumatol (2015) 67(1):302–14. doi: 10.1002/art.38912
6. Takada S, Kambe N, Kawasaki Y, Niwa A, Honda-Ozaki F, Kobayashi K, et al. Pluripotent Stem Cell Models of Blau Syndrome Reveal an IFN-Gamma-Dependent Inflammatory Response in Macrophages. J Allergy Clin Immunol (2018) 141(1):339–49.e11. doi: 10.1016/j.jaci.2017.04.013
7. Kitagawa Y, Kawasaki Y, Yamasaki Y, Kambe N, Takei S, Saito MK. Anti-TNF Treatment Corrects IFN-Gamma-Dependent Proinflammatory Signatures in Blau Syndrome Patient-Derived Macrophages. J Allergy Clin Immunol (2021) 149(1):176–88.e7. doi: 10.1016/j.jaci.2021.05.030
8. Honda-Ozaki F, Terashima M, Niwa A, Saiki N, Kawasaki Y, Ito H, et al. Pluripotent Stem Cell Model of Nakajo-Nishimura Syndrome Untangles Proinflammatory Pathways Mediated by Oxidative Stress. Stem Cell Rep (2018) 10(6):1835–50. doi: 10.1016/j.stemcr.2018.04.004
9. Mukhopadhyay S, Heinz E, Porreca I, Alasoo K, Yeung A, Yang HT, et al. Loss of IL-10 Signaling in Macrophages Limits Bacterial Killing Driven by Prostaglandin E2. J Exp Med (2020) 217(2):e20180649. doi: 10.1084/jem.20180649
10. Kawasaki Y, Oda H, Ito J, Niwa A, Tanaka T, Hijikata A, et al. Identification of a High-Frequency Somatic NLRC4 Mutation as a Cause of Autoinflammation by Pluripotent Cell-Based Phenotype Dissection. Arthritis Rheumatol (2017) 69(2):447–59. doi: 10.1002/art.39960
11. Boisson B, Honda Y, Ajiro M, Bustamante J, Bendavid M, Gennery AR, et al. Rescue of Recurrent Deep Intronic Mutation Underlying Cell Type-Dependent Quantitative NEMO Deficiency. J Clin Invest (2019) 129(2):583–97. doi: 10.1172/JCI124011
12. Shiba T, Tanaka T, Ida H, Watanabe M, Nakaseko H, Osawa M, et al. Functional Evaluation of the Pathological Significance of MEFV Variants Using Induced Pluripotent Stem Cell-Derived Macrophages. J Allergy Clin Immunol (2019) 144(5):1438–41.e12. doi: 10.1016/j.jaci.2019.07.039
13. Magg T, Okano T, Koenig LM, Boehmer DFR, Schwartz SL, Inoue K, et al. Heterozygous OAS1 Gain-of-Function Variants Cause an Autoinflammatory Immunodeficiency. Sci Immunol (2021) 6(60):eabf9564. doi: 10.1126/sciimmunol.abf9564
14. Tan EE, Hopkins RA, Lim CK, Jamuar SS, Ong C, Thoon KC, et al. Dominant-Negative NFKBIA Mutation Promotes IL-1beta Production Causing Hepatic Disease With Severe Immunodeficiency. J Clin Invest (2020) 130(11):5817–32. doi: 10.1172/JCI98882
15. Seki R, Ohta A, Niwa A, Sugimine Y, Naito H, Nakahata T, et al. Induced Pluripotent Stem Cell-Derived Monocytic Cell Lines From a NOMID Patient Serve as a Screening Platform for Modulating NLRP3 Inflammasome Activity. PloS One (2020) 15(8):e0237030. doi: 10.1371/journal.pone.0237030
16. Kase N, Terashima M, Ohta A, Niwa A, Honda-Ozaki F, Kawasaki Y, et al. Pluripotent Stem Cell-Based Screening Identifies CUDC-907 as an Effective Compound for Restoring the In Vitro Phenotype of Nakajo-Nishimura Syndrome. Stem Cells Transl Med (2021) 10(3):455–64. doi: 10.1002/sctm.20-0198
17. Alagoz M, Kherad N. Advance Genome Editing Technologies in the Treatment of Human Diseases: CRISPR Therapy (Review). Int J Mol Med (2020) 46(2):521–34. doi: 10.3892/ijmm.2020.4609
18. Brugger W, Kreutz M, Andreesen R. Macrophage Colony-Stimulating Factor is Required for Human Monocyte Survival and Acts as a Cofactor for Their Terminal Differentiation to Macrophages In Vitro. J Leukoc Biol (1991) 49(5):483–8. doi: 10.1002/jlb.49.5.483
19. Rodell CB, Koch PD, Weissleder R. Screening for New Macrophage Therapeutics. Theranostics (2019) 9(25):7714–29. doi: 10.7150/thno.34421
20. Fujisawa A, Kambe N, Saito M, Nishikomori R, Tanizaki H, Kanazawa N, et al. Disease-Associated Mutations in CIAS1 Induce Cathepsin B-Dependent Rapid Cell Death of Human THP-1 Monocytic Cells. Blood (2007) 109(7):2903–11. doi: 10.1182/blood-2006-07-033597
21. Nakagawa K, Gonzalez-Roca E, Souto A, Kawai T, Umebayashi H, Campistol JM, et al. Somatic NLRP3 Mosaicism in Muckle-Wells Syndrome. A Genetic Mechanism Shared by Different Phenotypes of Cryopyrin-Associated Periodic Syndromes. Ann Rheum Dis (2015) 74(3):603–10. doi: 10.1136/annrheumdis-2013-204361
22. Bosshart H, Heinzelmann M. THP-1 Cells as a Model for Human Monocytes. Ann Transl Med (2016) 4(21):438. doi: 10.21037/atm.2016.08.53
23. Buchrieser J, James W, Moore MD. Human Induced Pluripotent Stem Cell-Derived Macrophages Share Ontogeny With MYB-Independent Tissue-Resident Macrophages. Stem Cell Rep (2017) 8(2):334–45. doi: 10.1016/j.stemcr.2016.12.020
24. Takata K, Kozaki T, Lee CZW, Thion MS, Otsuka M, Lim S, et al. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity (2017) 47(1):183–98.e6. doi: 10.1016/j.immuni.2017.06.017
25. Tasnim F, Xing J, Huang X, Mo S, Wei X, Tan MH, et al. Generation of Mature Kupffer Cells From Human Induced Pluripotent Stem Cells. Biomaterials (2019) 192:377–91. doi: 10.1016/j.biomaterials.2018.11.016
26. Cui D, Franz A, Fillon SA, Jannetti L, Isambert T, Fundel-Clemens K, et al. High-Yield Human Induced Pluripotent Stem Cell-Derived Monocytes and Macrophages Are Functionally Comparable With Primary Cells. Front Cell Dev Biol (2021) 9:656867. doi: 10.3389/fcell.2021.656867
27. Lee CZW, Kozaki T, Ginhoux F. Studying Tissue Macrophages In Vitro: Are iPSC-Derived Cells the Answer? Nat Rev Immunol (2018) 18(11):716–25. doi: 10.1038/s41577-018-0054-y
28. Lyadova I, Gerasimova T, Nenasheva T. Macrophages Derived From Human Induced Pluripotent Stem Cells: The Diversity of Protocols, Future Prospects, and Outstanding Questions. Front Cell Dev Biol (2021) 9:640703. doi: 10.3389/fcell.2021.640703
29. Karlsson KR, Cowley S, Martinez FO, Shaw M, Minger SL, James W. Homogeneous Monocytes and Macrophages From Human Embryonic Stem Cells Following Coculture-Free Differentiation in M-CSF and IL-3. Exp Hematol (2008) 36(9):1167–75. doi: 10.1016/j.exphem.2008.04.009
30. Alasoo K, Martinez FO, Hale C, Gordon S, Powrie F, Dougan G, et al. Transcriptional Profiling of Macrophages Derived From Monocytes and iPS Cells Identifies a Conserved Response to LPS and Novel Alternative Transcription. Sci Rep (2015) 5:12524. doi: 10.1038/srep12524
31. Mukherjee C, Hale C, Mukhopadhyay S. A Simple Multistep Protocol for Differentiating Human Induced Pluripotent Stem Cells Into Functional Macrophages. Methods Mol Biol (2018) 1784:13–28. doi: 10.1007/978-1-4939-7837-3_2
32. Yeung ATY, Hale C, Lee AH, Gill EE, Bushell W, Parry-Smith D, et al. Exploiting Induced Pluripotent Stem Cell-Derived Macrophages to Unravel Host Factors Influencing Chlamydia Trachomatis Pathogenesis. Nat Commun (2017) 8:15013. doi: 10.1038/ncomms15013
33. Zhang H, Xue C, Shah R, Bermingham K, Hinkle CC, Li W, et al. Functional Analysis and Transcriptomic Profiling of iPSC-Derived Macrophages and Their Application in Modeling Mendelian Disease. Circ Res (2015) 117(1):17–28. doi: 10.1161/CIRCRESAHA.117.305860
34. Munn C, Burton S, Dickerson S, Bakshy K, Strouse A, Rajesh D. Generation of Cryopreserved Macrophages From Normal and Genetically Engineered Human Pluripotent Stem Cells for Disease Modelling. PloS One (2021) 16(4):e0250107. doi: 10.1371/journal.pone.0250107
35. Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De Novo CIAS1 Mutations, Cytokine Activation, and Evidence for Genetic Heterogeneity in Patients With Neonatal-Onset Multisystem Inflammatory Disease (NOMID): A New Member of the Expanding Family of Pyrin-Associated Autoinflammatory Diseases. Arthritis Rheumatol (2002) 46(12):3340–8. doi: 10.1002/art.10688
36. Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic Infantile Neurological Cutaneous and Articular Syndrome is Caused by Mutations in CIAS1, a Gene Highly Expressed in Polymorphonuclear Cells and Chondrocytes. Am J Hum Genet (2002) 71(1):198–203. doi: 10.1086/341357
37. Gattorno M, Tassi S, Carta S, Delfino L, Ferlito F, Pelagatti MA, et al. Pattern of Interleukin-1beta Secretion in Response to Lipopolysaccharide and ATP Before and After Interleukin-1 Blockade in Patients With CIAS1 Mutations. Arthritis Rheumatol (2007) 56(9):3138–48. doi: 10.1002/art.22842
38. Saito M, Fujisawa A, Nishikomori R, Kambe N, Nakata-Hizume M, Yoshimoto M, et al. Somatic Mosaicism of CIAS1 in a Patient With Chronic Infantile Neurologic, Cutaneous, Articular Syndrome. Arthritis Rheumatol (2005) 52(11):3579–85. doi: 10.1002/art.21404
39. Aksentijevich I, Putnam CD, Remmers EF, Mueller JL, Le J, Kolodner RD, et al. The Clinical Continuum of Cryopyrinopathies: Novel CIAS1 Mutations in North American Patients and a New Cryopyrin Model. Arthritis Rheumatol (2007) 56(4):1273–85. doi: 10.1002/art.22491
40. Saito M, Nishikomori R, Kambe N, Fujisawa A, Tanizaki H, Takeichi K, et al. Disease-Associated CIAS1 Mutations Induce Monocyte Death, Revealing Low-Level Mosaicism in Mutation-Negative Cryopyrin-Associated Periodic Syndrome Patients. Blood (2008) 111(4):2132–41. doi: 10.1182/blood-2007-06-094201
41. Tanaka N, Izawa K, Saito MK, Sakuma M, Oshima K, Ohara O, et al. High Incidence of NLRP3 Somatic Mosaicism in Patients With Chronic Infantile Neurologic, Cutaneous, Articular Syndrome: Results of an International Multicenter Collaborative Study. Arthritis Rheumatol (2011) 63(11):3625–32. doi: 10.1002/art.30512
42. Abyzov A, Mariani J, Palejev D, Zhang Y, Haney MS, Tomasini L, et al. Somatic Copy Number Mosaicism in Human Skin Revealed by Induced Pluripotent Stem Cells. Nature (2012) 492(7429):438–42. doi: 10.1038/nature11629
43. Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 Inflammasome is Released as a Particulate Danger Signal That Amplifies the Inflammatory Response. Nat Immunol (2014) 15(8):738–48. doi: 10.1038/ni.2919
44. Wang C, Xu CX, Alippe Y, Qu C, Xiao J, Schipani E, et al. Chronic Inflammation Triggered by the NLRP3 Inflammasome in Myeloid Cells Promotes Growth Plate Dysplasia by Mesenchymal Cells. Sci Rep (2017) 7(1):4880. doi: 10.1038/s41598-017-05033-5
45. Blau EB. Familial Granulomatous Arthritis, Iritis, and Rash. J Pediatr (1985) 107(5):689–93. doi: 10.1016/S0022-3476(85)80394-2
46. Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-Onset Sarcoidosis and CARD15 Mutations With Constitutive Nuclear factor-kappaB Activation: Common Genetic Etiology With Blau Syndrome. Blood (2005) 105(3):1195–7. doi: 10.1182/blood-2004-07-2972
47. Janssen CE, Rose CD, De Hertogh G, Martin TM, Bader Meunier B, Cimaz R, et al. Morphologic and Immunohistochemical Characterization of Granulomas in the Nucleotide Oligomerization Domain 2-Related Disorders Blau Syndrome and Crohn Disease. J Allergy Clin Immunol (2012) 129(4):1076–84. doi: 10.1016/j.jaci.2012.02.004
48. Chen J, Luo Y, Zhao M, Wu D, Yang Y, Zhang W, et al. Effective Treatment of TNFalpha Inhibitors in Chinese Patients With Blau Syndrome. Arthritis Res Ther (2019) 21(1):236. doi: 10.1186/s13075-019-2017-5
49. Matsuda T, Kambe N, Ueki Y, Kanazawa N, Izawa K, Honda Y, et al. Clinical Characteristics and Treatment of 50 Cases of Blau Syndrome in Japan Confirmed by Genetic Analysis of the NOD2 Mutation. Ann Rheum Dis (2020) 79(11):1492–9. doi: 10.1136/annrheumdis-2020-217320
50. Nagakura T, Wakiguchi H, Kubota T, Yamatou T, Yamasaki Y, Nonaka Y, et al. Tumor Necrosis Factor Inhibitors Provide Longterm Clinical Benefits in Pediatric and Young Adult Patients With Blau Syndrome. J Rheumatol (2017) 44(4):536–8. doi: 10.3899/jrheum.160672
51. Otsubo Y, Okafuji I, Shimizu T, Nonaka F, Ikeda K, Eguchi K. A Long-Term Follow-Up of Japanese Mother and Her Daughter With Blau Syndrome: Effective Treatment of Anti-TNF Inhibitors and Useful Diagnostic Tool of Joint Ultrasound Examination. Mod Rheumatol (2017) 27(1):169–73. doi: 10.3109/14397595.2014.964388
52. Simonini G, Xu Z, Caputo R, De Libero C, Pagnini I, Pascual V, et al. Clinical and Transcriptional Response to the Long-Acting Interleukin-1 Blocker Canakinumab in Blau Syndrome-Related Uveitis. Arthritis Rheumatol (2013) 65(2):513–8. doi: 10.1002/art.37776
53. Lu L, Shen M, Jiang D, Li Y, Zheng X, Li Y, et al. Blau Syndrome With Good Reponses to Tocilizumab: A Case Report and Focused Literature Review. Semin Arthritis Rheumatol (2018) 47(5):727–31. doi: 10.1016/j.semarthrit.2017.09.010
54. Dugan J, Griffiths E, Snow P, Rosenzweig H, Lee E, Brown B, et al. Blau Syndrome-Associated Nod2 Mutation Alters Expression of Full-Length NOD2 and Limits Responses to Muramyl Dipeptide in Knock-in Mice. J Immunol (2015) 194(1):349–57. doi: 10.4049/jimmunol.1402330
55. Rosenstiel P, Fantini M, Brautigam K, Kuhbacher T, Waetzig GH, Seegert D, et al. TNF-Alpha and IFN-Gamma Regulate the Expression of the NOD2 (CARD15) Gene in Human Intestinal Epithelial Cells. Gastroenterology (2003) 124(4):1001–9. doi: 10.1053/gast.2003.50157
56. Totemeyer S, Sheppard M, Lloyd A, Roper D, Dowson C, Underhill D, et al. IFN-Gamma Enhances Production of Nitric Oxide From Macrophages via a Mechanism That Depends on Nucleotide Oligomerization Domain-2. J Immunol (2006) 176(8):4804–10. doi: 10.4049/jimmunol.176.8.4804
57. Lee KH, Biswas A, Liu YJ, Kobayashi KS. Proteasomal Degradation of Nod2 Protein Mediates Tolerance to Bacterial Cell Wall Components. J Biol Chem (2012) 287(47):39800–11. doi: 10.1074/jbc.M112.410027
58. Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, et al. Proteasome Assembly Defect Due to a Proteasome Subunit Beta Type 8 (PSMB8) Mutation Causes the Autoinflammatory Disorder, Nakajo-Nishimura Syndrome. Proc Natl Acad Sci USA (2011) 108(36):14914–9. doi: 10.1073/pnas.1106015108
59. Shi X, Xiang X, Wang Z, Ma L, Xu Z. Chinese Case of Nakajo-Nishimura Syndrome With a Novel Mutation of the PSMB8 Gene. J Dermatol (2019) 46(5):e160–e1. doi: 10.1111/1346-8138.14679
60. de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular Mechanisms in Genetically Defined Autoinflammatory Diseases: Disorders of Amplified Danger Signaling. Annu Rev Immunol (2015) 33:823–74. doi: 10.1146/annurev-immunol-032414-112227
61. Kim H, Sanchez GA, Goldbach-Mansky R. Insights From Mendelian Interferonopathies: Comparison of CANDLE, SAVI With AGS, Monogenic Lupus. J Mol Med (Berl) (2016) 94(10):1111–27. doi: 10.1007/s00109-016-1465-5
62. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 Inhibition With Baricitinib in the Treatment of Autoinflammatory Interferonopathies. J Clin Invest (2018) 128(7):3041–52. doi: 10.1172/JCI98814
63. Igaz P, Toth S, Falus A. Biological and Clinical Significance of the JAK-STAT Pathway; Lessons From Knockout Mice. Inflamm Res (2001) 50(9):435–41. doi: 10.1007/PL00000267
64. Leonard WJ, O’Shea JJ. Jaks and STATs: Biological Implications. Annu Rev Immunol (1998) 16:293–322. doi: 10.1146/annurev.immunol.16.1.293
65. Torrelo A, Noguera-Morel L, Hernandez-Martin A, Clemente D, Barja JM, Buzon L, et al. Recurrent Lipoatrophic Panniculitis of Children. J Eur Acad Dermatol Venereol (2017) 31(3):536–43. doi: 10.1111/jdv.13858
66. Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, et al. Distinct Macrophage Populations Direct Inflammatory Versus Physiological Changes in Adipose Tissue. Proc Natl Acad Sci USA (2018) 115(22):E5096–E105. doi: 10.1073/pnas.1802611115
67. Weise G, Hupp M, Kerstan A, Buttmann M. Lobular Panniculitis and Lipoatrophy of the Thighs With Interferon-Ss1a for Intramuscular Injection in a Patient With Multiple Sclerosis. J Clin Neurosci (2012) 19(9):1312–3. doi: 10.1016/j.jocn.2011.11.026
68. Verhoeven D, Schonenberg-Meinema D, Ebstein F, Papendorf JJ, Baars PA, van Leeuwen EMM, et al. Hematopoietic Stem Cell Transplantation in a Patient With Proteasome-Associated Autoinflammatory Syndrome (PRAAS). J Allergy Clin Immunol (2022) 149(3):1120–7 e8. doi: 10.1016/j.jaci.2021.07.039
69. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-Deficient Mice Develop Chronic Enterocolitis. Cell (1993) 75(2):263–74. doi: 10.1016/0092-8674(93)80068-P
70. Bogdan C, Vodovotz Y, Nathan C. Macrophage Deactivation by Interleukin 10. J Exp Med (1991) 174(6):1549–55. doi: 10.1084/jem.174.6.1549
71. Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-Gamma is an Essential Regulatory Subunit of the IkappaB Kinase Complex. Nature (1998) 395(6699):297–300. doi: 10.1038/26261
72. Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, et al. Complementation Cloning of NEMO, a Component of the IkappaB Kinase Complex Essential for NF-kappaB Activation. Cell (1998) 93(7):1231–40. doi: 10.1016/S0092-8674(00)81466-X
73. Ghosh S, Hayden MS. New Regulators of NF-kappaB in Inflammation. Nat Rev Immunol (2008) 8(11):837–48. doi: 10.1038/nri2423
74. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-Kappab: A Blossoming of Relevance to Human Pathobiology. Cell (2017) 168(1-2):37–57. doi: 10.1016/j.cell.2016.12.012
75. Fusco F, Pescatore A, Bal E, Ghoul A, Paciolla M, Lioi MB, et al. Alterations of the IKBKG Locus and Diseases: An Update and a Report of 13 Novel Mutations. Hum Mutat (2008) 29(5):595–604. doi: 10.1002/humu.20739
76. Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, et al. Genomic Rearrangement in NEMO Impairs NF-kappaB Activation and is a Cause of Incontinentia Pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature (2000) 405(6785):466–72. doi: 10.1038/35013114
77. Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, et al. X-Linked Anhidrotic Ectodermal Dysplasia With Immunodeficiency is Caused by Impaired NF-kappaB Signaling. Nat Genet (2001) 27(3):277–85. doi: 10.1038/85837
78. Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, et al. A Novel X-Linked Disorder of Immune Deficiency and Hypohidrotic Ectodermal Dysplasia is Allelic to Incontinentia Pigmenti and Due to Mutations in IKK-Gamma (NEMO). Am J Hum Genet (2000) 67(6):1555–62. doi: 10.1086/316914
79. Kawai T, Nishikomori R, Heike T. Diagnosis and Treatment in Anhidrotic Ectodermal Dysplasia With Immunodeficiency. Allergol Int (2012) 61(2):207–17. doi: 10.2332/allergolint.12-RAI-0446
80. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
81. French FMFC. A Candidate Gene for Familial Mediterranean Fever. Nat Genet (1997) 17(1):25–31. doi: 10.1038/ng0997-25
82. Heilig R, Broz P. Function and Mechanism of the Pyrin Inflammasome. Eur J Immunol (2018) 48(2):230–8. doi: 10.1002/eji.201746947
83. Gangemi S, Manti S, Procopio V, Casciaro M, Di Salvo E, Cutrupi M, et al. Lack of Clear and Univocal Genotype-Phenotype Correlation in Familial Mediterranean Fever Patients: A Systematic Review. Clin Genet (2018) 94(1):81–94. doi: 10.1111/cge.13223
84. Masters SL, Lagou V, Jeru I, Baker PJ, Van Eyck L, Parry DA, et al. Familial Autoinflammation With Neutrophilic Dermatosis Reveals a Regulatory Mechanism of Pyrin Activation. Sci Transl Med (2016) 8(332):332ra45. doi: 10.1126/scitranslmed.aaf1471
85. Moghaddas F, Llamas R, De Nardo D, Martinez-Banaclocha H, Martinez-Garcia JJ, Mesa-Del-Castillo P, et al. A Novel Pyrin-Associated Autoinflammation With Neutrophilic Dermatosis Mutation Further Defines 14-3-3 Binding of Pyrin and Distinction to Familial Mediterranean Fever. Ann Rheum Dis (2017) 76(12):2085–94. doi: 10.1136/annrheumdis-2017-211473
86. Aldea A, Campistol JM, Arostegui JI, Rius J, Maso M, Vives J, et al. A Severe Autosomal-Dominant Periodic Inflammatory Disorder With Renal AA Amyloidosis and Colchicine Resistance Associated to the MEFV H478Y Variant in a Spanish Kindred: An Unusual Familial Mediterranean Fever Phenotype or Another MEFV-Associated Periodic Inflammatory Disorder? Am J Med Genet A (2004) 124A(1):67–73. doi: 10.1002/ajmg.a.20296
87. Rowczenio DM, Youngstein T, Trojer H, Omoyinmi E, Baginska A, Brogan P, et al. British Kindred With Dominant FMF Associated With High Incidence of AA Amyloidosis Caused by Novel MEFV Variant, and a Review of the Literature. Rheumatol (Oxford) (2020) 59(3):554–8. doi: 10.1093/rheumatology/kez334
88. Stoffels M, Szperl A, Simon A, Netea MG, Plantinga TS, van Deuren M, et al. MEFV Mutations Affecting Pyrin Amino Acid 577 Cause Autosomal Dominant Autoinflammatory Disease. Ann Rheum Dis (2014) 73(2):455–61. doi: 10.1136/annrheumdis-2012-202580
89. Ben-Chetrit E, Gattorno M, Gul A, Kastner DL, Lachmann HJ, Touitou I, et al. Consensus Proposal for Taxonomy and Definition of the Autoinflammatory Diseases (AIDs): A Delphi Study. Ann Rheum Dis (2018) 77(11):1558–65. doi: 10.1136/annrheumdis-2017-212515
90. Van Gijn ME, Ceccherini I, Shinar Y, Carbo EC, Slofstra M, Arostegui JI, et al. New Workflow for Classification of Genetic Variants’ Pathogenicity Applied to Hereditary Recurrent Fevers by the International Study Group for Systemic Autoinflammatory Diseases (INSAID). J Med Genet (2018) 55(8):530–7. doi: 10.1136/jmedgenet-2017-105216
91. Chae JJ, Cho YH, Lee GS, Cheng J, Liu PP, Feigenbaum L, et al. Gain-Of-Function Pyrin Mutations Induce NLRP3 Protein-Independent Interleukin-1beta Activation and Severe Autoinflammation in Mice. Immunity (2011) 34(5):755–68. doi: 10.1016/j.immuni.2011.02.020
92. Jamilloux Y, Lefeuvre L, Magnotti F, Martin A, Benezech S, Allatif O, et al. Familial Mediterranean Fever Mutations are Hypermorphic Mutations That Specifically Decrease the Activation Threshold of the Pyrin Inflammasome. Rheumatol (Oxford) (2018) 57(1):100–11. doi: 10.1093/rheumatology/kex373
93. Park YH, Wood G, Kastner DL, Chae JJ. Pyrin Inflammasome Activation and RhoA Signaling in the Autoinflammatory Diseases FMF and HIDS. Nat Immunol (2016) 17(8):914–21. doi: 10.1038/ni.3457
94. Van Gorp H, Saavedra PH, de Vasconcelos NM, Van Opdenbosch N, Vande Walle L, Matusiak M, et al. Familial Mediterranean Fever Mutations Lift the Obligatory Requirement for Microtubules in Pyrin Inflammasome Activation. Proc Natl Acad Sci USA (2016) 113(50):14384–9. doi: 10.1073/pnas.1613156113
95. Fujimaki Y, Soutome T, Tanaka T, Shiba T, Watanabe M. A Familial Mediterranean Fever Girl Due to MEFV N679H Mutation With Gilbert’s Syndrome. Pediatr Int (2021) 63(8):982–4. doi: 10.1111/ped.14526
96. Nakaseko H, Iwata N, Izawa K, Shibata H, Yasuoka R, Kohagura T, et al. Expanding Clinical Spectrum of Autosomal Dominant Pyrin-Associated Autoinflammatory Disorder Caused by the Heterozygous MEFV P.Thr577Asn Variant. Rheumatol (Oxford) (2019) 58(1):182–4. doi: 10.1093/rheumatology/key283
97. Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, et al. Criteria for the Diagnosis of Familial Mediterranean Fever. Arthritis Rheumatol (1997) 40(10):1879–85. doi: 10.1002/art.1780401023
98. Dong C, Zhao G, Zhong M, Yue Y, Wu L, Xiong S. RNA Sequencing and Transcriptomal Analysis of Human Monocyte to Macrophage Differentiation. Gene (2013) 519(2):279–87. doi: 10.1016/j.gene.2013.02.015
99. Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, et al. HDAC6 Mediates an Aggresome-Like Mechanism for NLRP3 and Pyrin Inflammasome Activation. Science (2020) 369(6510):eaas8995. doi: 10.1126/science.aas8995
100. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity (2016) 44(4):833–46. doi: 10.1016/j.immuni.2016.01.012
101. Vigano E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human Caspase-4 and Caspase-5 Regulate the One-Step Non-Canonical Inflammasome Activation in Monocytes. Nat Commun (2015) 6:8761. doi: 10.1038/ncomms9761
102. Nishitani IM, Honda K, Nihira Y, Tanaka H, Shibata T, Kodama H, et al. Trapping of CDC42 C-Terminal Variants in the Golgi Drives Pyrin Inflammasome Hyperactivation. J Exp Med (2022) 219(6):e20211889. doi: 10.1084/jem.20211889
103. Honda Y, Maeda Y, Izawa K, Shiba T, Tanaka T, Nakaseko H, et al. Rapid Flow Cytometry-Based Assay for the Functional Classification of MEFV Variants. J Clin Immunol (2021) 41(6):1187–97. doi: 10.1007/s10875-021-01021-7
104. Magnotti F, Malsot T, Georgin-Lavialle S, Abbas F, Martin A, Belot A, et al. Fast Diagnostic Test for Familial Mediterranean Fever Based on a Kinase Inhibitor. Ann Rheum Dis (2021) 80(1):128–32. doi: 10.1136/annrheumdis-2020-218366
105. Van Gorp H, Huang L, Saavedra P, Vuylsteke M, Asaoka T, Prencipe G, et al. Blood-Based Test for Diagnosis and Functional Subtyping of Familial Mediterranean Fever. Ann Rheum Dis (2020) 79(7):960–8. doi: 10.1136/annrheumdis-2019-216701
106. Chebath J, Benech P, Revel M, Vigneron M. Constitutive Expression of (2’-5’) Oligo A Synthetase Confers Resistance to Picornavirus Infection. Nature (1987) 330(6148):587–8. doi: 10.1038/330587a0
107. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science (2013) 339(6121):786–91. doi: 10.1126/science.1232458
108. Clemens MJ, Williams BR. Inhibition of Cell-Free Protein Synthesis by Pppa2’p5’a2’p5’A: A Novel Oligonucleotide Synthesized by Interferon-Treated L Cell Extracts. Cell (1978) 13(3):565–72. doi: 10.1016/0092-8674(78)90329-X
109. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science (2013) 339(6121):826–30. doi: 10.1126/science.1229963
110. Han Y, Donovan J, Rath S, Whitney G, Chitrakar A, Korennykh A. Structure of Human RNase L Reveals the Basis for Regulated RNA Decay in the IFN Response. Science (2014) 343(6176):1244–8. doi: 10.1126/science.1249845
111. Okano T. [OAS1 Kinoukakutokugatahen’iniyoru Nyujikihasshohaihoutanpakushoto Jikoenshomen’ekifuzenshokogun]. Enshotomen’eki (2022) 30(2):159–63.
112. Bjornson-Hooper ZB, Fragiadakis GK, Spitzer MH, Chen H, Madhireddy D, Hu K, et al. A Comprehensive Atlas of Immunological Differences Between Humans, Mice, and Non-Human Primates. Front Immunol (2022) 13:867015. doi: 10.3389/fimmu.2022.867015
113. Picard C, Casanova JL, Puel A. Infectious Diseases in Patients With IRAK-4, MyD88, NEMO, or IkappaBalpha Deficiency. Clin Microbiol Rev (2011) 24(3):490–7. doi: 10.1128/CMR.00001-11
114. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic Bacterial Infections in Humans With MyD88 Deficiency. Science (2008) 321(5889):691–6. doi: 10.1126/science.1158298
115. Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 Inflammasome in Chronic Inflammatory Diseases: Current Perspectives. J Inflamm Res (2015) 8:15–27. doi: 10.2147/JIR.S51250
116. Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting Edge: Reactive Oxygen Species Inhibitors Block Priming, But Not Activation, of the NLRP3 Inflammasome. J Immunol (2011) 187(2):613–7. doi: 10.4049/jimmunol.1100613
117. Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandes-Alnemri T, Alnemri ES, et al. Inflammasome Priming by Lipopolysaccharide is Dependent Upon ERK Signaling and Proteasome Function. J Immunol (2014) 192(8):3881–8. doi: 10.4049/jimmunol.1301974
118. Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J. A Crucial Function of SGT1 and HSP90 in Inflammasome Activity Links Mammalian and Plant Innate Immune Responses. Nat Immunol (2007) 8(5):497–503. doi: 10.1038/ni1459
119. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical Role for Calcium Mobilization in Activation of the NLRP3 Inflammasome. Proc Natl Acad Sci USA (2012) 109(28):11282–7. doi: 10.1073/pnas.1117765109
120. Badran YR, Dedeoglu F, Leyva Castillo JM, Bainter W, Ohsumi TK, Bousvaros A, et al. Human RELA Haploinsufficiency Results in Autosomal-Dominant Chronic Mucocutaneous Ulceration. J Exp Med (2017) 214(7):1937–47. doi: 10.1084/jem.20160724
121. Cuchet-Lourenco D, Eletto D, Wu C, Plagnol V, Papapietro O, Curtis J, et al. Biallelic RIPK1 Mutations in Humans Cause Severe Immunodeficiency, Arthritis, and Intestinal Inflammation. Science (2018) 361(6404):810–3. doi: 10.1126/science.aar2641
122. Jarosz-Griffiths HH, Holbrook J, Lara-Reyna S, McDermott MF. TNF Receptor Signalling in Autoinflammatory Diseases. Int Immunol (2019) 31(10):639–48. doi: 10.1093/intimm/dxz024
123. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
124. Ozen S, Demirkaya E, Erer B, Livneh A, Ben-Chetrit E, Giancane G, et al. EULAR Recommendations for the Management of Familial Mediterranean Fever. Ann Rheum Dis (2016) 75(4):644–51. doi: 10.1136/annrheumdis-2015-208690
125. De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N Engl J Med (2018) 378(20):1908–19. doi: 10.1056/NEJMoa1706314
126. Magnotti F, Lefeuvre L, Benezech S, Malsot T, Waeckel L, Martin A, et al. Pyrin Dephosphorylation is Sufficient to Trigger Inflammasome Activation in Familial Mediterranean Fever Patients. EMBO Mol Med (2019) 11(11):e10547. doi: 10.15252/emmm.201910547
127. Sun K, Sun Z, Zhao F, Shan G, Meng Q. Recent Advances in Research of Colchicine Binding Site Inhibitors and Their Interaction Modes With Tubulin. Future Med Chem (2021) 13(9):839–58. doi: 10.4155/fmc-2020-0376
128. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING-Activating Mutation Underlies a Familial Inflammatory Syndrome With Lupus-Like Manifestations. J Clin Invest (2014) 124(12):5516–20. doi: 10.1172/JCI79100
129. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a Vascular and Pulmonary Syndrome. N Engl J Med (2014) 371(6):507–18. doi: 10.1056/NEJMoa1312625
130. Fremond ML, Crow YJ. STING-Mediated Lung Inflammation and Beyond. J Clin Immunol (2021) 41(3):501–14. doi: 10.1007/s10875-021-00974-z
131. Ablasser A, Chen ZJ. cGAS in Action: Expanding Roles in Immunity and Inflammation. Science (2019) 363(6431):eaat8657. doi: 10.1126/science.aat8657
Keywords: autoinflammatory diseases, induced pluripotent stem cells, disease modeling, drug screening, monocytes, macrophages
Citation: Tanaka T, Shiba T, Honda Y, Izawa K, Yasumi T, Saito MK and Nishikomori R (2022) Induced Pluripotent Stem Cell-Derived Monocytes/Macrophages in Autoinflammatory Diseases. Front. Immunol. 13:870535. doi: 10.3389/fimmu.2022.870535
Received: 07 February 2022; Accepted: 11 April 2022;
Published: 06 May 2022.
Edited by:
Naoki Iwamoto, Nagasaki University Hospital, JapanReviewed by:
Sinisa Savic, University of Leeds, United KingdomCopyright © 2022 Tanaka, Shiba, Honda, Izawa, Yasumi, Saito and Nishikomori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takayuki Tanaka, dGF0YW5ha2FAa3VocC5reW90by11LmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.