 Isabel N. Schellinger
Isabel N. Schellinger
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 15 April 2021
Sec. Cardiovascular Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.571076
This article is part of the Research Topic Highlights in Cardiovascular Therapeutics: 2021 View all 8 articles
New technologies have greatly shaped the scientific and medical landscape within the last years. The unprecedented expansion of data and information on RNA biology has led to the discovery of new RNA classes with unique functions and unexpected modifications. Today, the biggest challenge is to transfer the large number of findings in basic RNA biology into corresponding clinical RNA-based therapeutics. Lately, this research begins to yield positive outcomes. RNA drugs advance to the final phases of clinical trials or even receive FDA approval. Furthermore, the introduction of the RNA-guided gene-editing technology CRISPR and advances in the delivery of messenger RNAs have triggered a major progression in the field of RNA-therapeutics. Especially short interfering RNAs and antisense oligonucleotides are promising examples for novel categories of therapeutics. However, several issues need to be addressed including intracellular delivery, toxicity, and immune responses before utilizing RNAs in a clinical setting. In this review, we provide an overview on opportunities and challenges for clinical translation of RNA-based therapeutics, with an emphasis on advances in novel delivery technologies and abdominal aortic aneurysm disease where non-coding RNAs have been shown to play a crucial regulatory role.
We believe that through the hereby provided review, we can give the interested readership a thoughtfully cured overview regarding the following topics:
1. A broad review on the ever-growing spectrum of RNAs with a focus on non-coding RNAs (including small and long non-coding RNAs).
2. An introduction to a widespread range of translational methods to apply RNAs as therapeutics (including nanocarriers and extracellular vesicles).
3. A comprehensive insight into challenges of translation in the field of RNAs.
4. Instructive examples where the mentioned challenges have been accepted and first results have been reached (including atherosclerosis and aneurysm disease).
Abdominal aortic aneurysm (AAA) is a common disease and defined as an abnormal focal dilation of the infrarenal aorta over 50% of the normal diameter (1). The normal diameter dependents on various factors such as age, gender, and body composition (2). Typically, an infrarenal aortic diameter ≥3.0 cm is considered aneurysmal (3). AAAs are subjected to treatment when the diameter reaches >5.5 cm, if rapid progression occurs (>0.5 cm/year) or if the AAA becomes symptomatic (4). Yet, AAAs are considered ticking time bombs as most patients remain asymptomatic until rupture. In case of rupture, bleeding is often fatal (5).
Factors increasing the risk for AAA development include smoking, male sex, and age >65 years (5). Interestingly the risk of AAA rupture is higher in females, although a smaller percentage of women generally suffer from AAA (6).
When a large pulsatile abdominal mass is found during physical examination, imaging is required to confirm that an aneurysm is present. Usually, abdominal ultrasound is performed to diagnose AAA (7) and lately ultrasound screening programs have been implemented in many countries of the European Union and in the USA to promote early disease diagnosis (8, 9).
Up to date, no pharmacological agent is available to prevent or treat AAA development and progression. Current therapies are limited to open surgical or interventional stent-graft based techniques. Unfortunately, these treatment options hold a high risk for procedural complications and are therefore reserved for larger aneurysms with a diameter >5.5 cm (4). Mortality regarding surgical treatment may be up to 5%, while endovascular repair displays endoleaks, stent graft migration and continuing AAA growth that may lead to secondary interventions or prostheses explanation (10).
On a molecular level, AAA pathology is characterized by smooth muscle cell (SMC) apoptosis, vascular inflammation, and extra cellular matrix (ECM) remodeling (11). Over the last couple of years, research has demonstrated that non-coding RNAs are significant modulators of these AAA-driving processes (12, 13). Thus, innovative RNA-targeting approaches hold great promise for revolutionizing clinical treatment of AAA disease.
When discussing the clinical potential of novel RNA-based approaches, there are several therapeutic categories that need to be distinguished. Most obviously, RNAs can serve as direct therapeutic targets to modify disease (14). Additionally, RNAs can act as mediators to indirectly achieve differential modulation of downstream regulatory networks. Moreover, RNAs can be used as tools for therapeutic intervention. In particular, short interfering RNA (siRNA) can be used to alter mRNA levels of particular target genes (14).
As of 2020, 10 nucleic acid based drugs have gained FDA-approval, of which two are implicated in cardiovascular conditions. Mipomersen, a gapmeR targeting ApoB, has been approved for homozygous familial hypercholesterolemia. The agent aids to reduce cardiovascular disease risk by reducing low-density lipoprotein (LDL) cholesterol levels. Defibrotide is a polydisperse oligo-nucleotide purified from porcine intestinal mucosa that has been approved as a drug against severe hepatic veno-occlusive disease, where it acts in a complex sequence-independent mechanism on fibrosis, angiogenesis, apoptosis, and aggregation of thrombocytes (15, 16). Other nucleotide-based therapeutic approaches are currently being tested in clinical trials (17, 18). So far, no nucleic acic based drug has been approved for treatment of aneurysmal disease.
During the last years of the RNA revolution, small non-coding RNA molecules that are only ~20–30 nucleotides long, have emerged as important regulators of the eukaryotic genome (for overview see Figure 1). Two major classes are short interfering RNAs (siRNAs) and microRNAs (miRs). Traditionally, it was believed that siRNAs only originate from exogenous sources (e.g., viruses) while miRs are of endogenous origin (19). However, extensive research showed that this distinction was rather artificial (20).
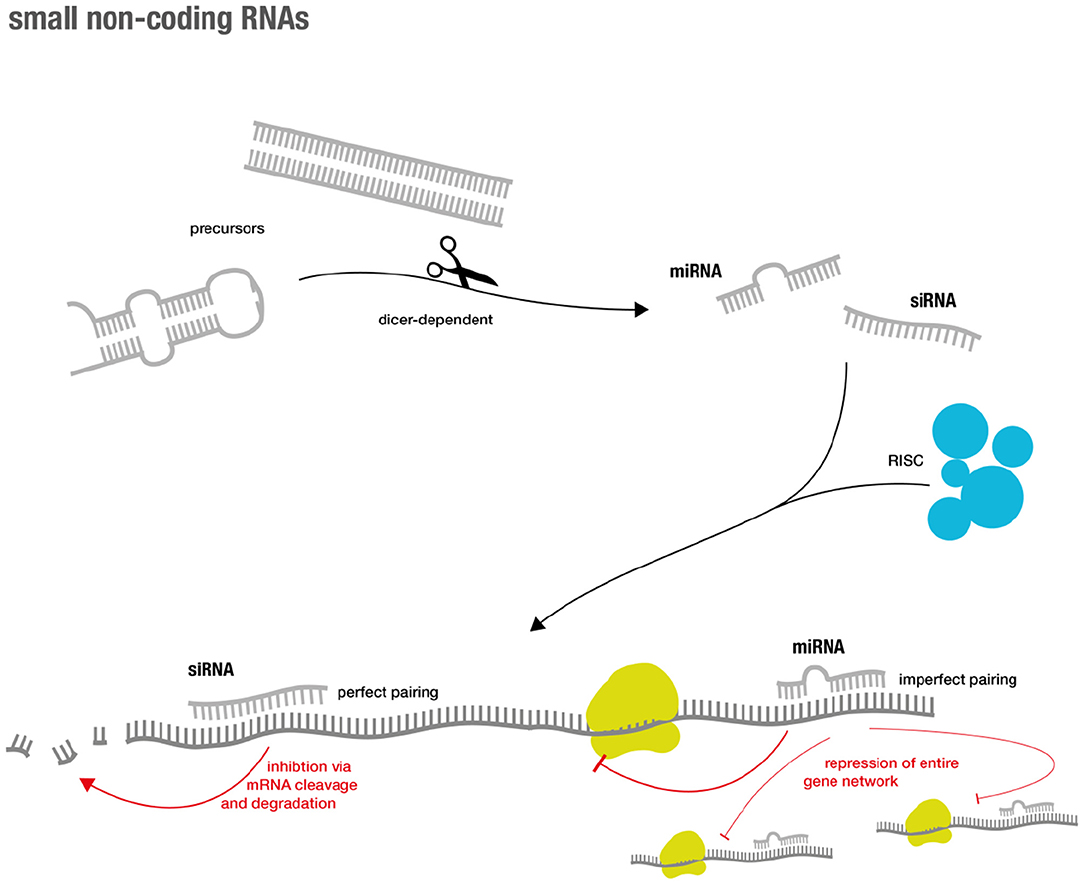
Figure 1. Overview over small RNA genesis and mode of action.
Both classes generally inhibit gene expression and their effector functions are commonly summarized as RNA silencing. The latter is achieved via a complex succession of molecular sequences. In short, precursors (such as double-stranded RNA (dsRNA) and hairpin precursors) of both classes are processed via a Dicer-dependent mechanism that then enter the silencing machinery of the RNA-induced silencing complex (RISC) in their single-stranded form. The mRNAs of interest are targeted via base pairing of the small RNA component. This eventually results in sequence specific gene silencing (19). Perfect base-pair matching between the siRNA strand and the target mRNA is required for cleavage and subsequent degradation, while imperfect base-pair matching can result in miR-like repression (19, 21). The latter accounts for advantages and challenges in miR-based therapeutics. As miRNAs not only target one specific gene, but instead regulate entire functional gene networks (22), they might be able to address endogenous redundancy or compensatory mechanisms that cause difficulties for classical drugs (23). However, the broad regulation via miRs might also lead to unexpected and unwanted off-target effects (24).
Fire and Mello were the first to proof that experimental introduction of dsRNAs into a cell can be used to synthetically interfere with the function of an endogenous gene (25). Today, the RNA interference (RNAi) technology holds great promise to silence “non-druggable” targets that cannot be addressed using small-molecules, monoclonal antibodies, or other conventional therapeutic approaches (26).
On the opposite site of the non-coding RNA spectrum, we find so called long non-coding RNAs (lncRNAs) (27). These comprise a diverse class of RNA molecules that do not code for proteins and that consist of >200 nucleotides and that are frequently polyadenylated (28).
Some lncRNAs are pseudogenic, copies of coding genes harboring mutations rendering them non-coding (29). Many lncRNAs overlap coding genes or are described as intergenic [known as long intergenic ncRNAs (lincRNAs) (30)]. But they also share many features with coding transcripts such as the occurrence of introns, the existence of splice variants and the presence of epigenetic marks indicating differential expression (29, 31). Additionally—without the need for translation—ncRNA expression is highly dynamic and can be rapidly up- or down-regulated to modulate gene expression. Their ability to fold into complex secondary structures also provides them with flexibility to interact with various substrates (such as proteins, RNAs, and DNAs) in a highly-specific manner (28, 32).
Regarding target sites, it is possible to distinguish between cis- and trans-acting lncRNAs. Cis-acting lncRNAs control the expression of genes in close neighborhood, whereas trans-acting lncRNAs function from a distance (28).
Regarding lncRNA location, one can differentiate between cytoplasmatic lncRNAs that modulate translational control and nuclear lncRNAs that guide chromatin modifiers to specific genomic loci (33–35). The latter usually results in DNA and histone modifications that yield inhibitory heterochromatin and transcriptional repression (34).
However, transcriptional activation has also been reported especially via activation of specific enhancer regions or by “sponging” miRs. LncRNAs that act as negative regulators of miRs and thereby achieve mRNA upregulation function as competing endogenous RNAs (ceRNAs) (36, 37). Linear ceRNAs have a short half-life that allow for quick control over sponge activity, while circular sponges—such as circRNAs—have much greater stability due to their resistance against endonuclease degradation (38).
Moreover, some of the best-characterized lncRNAs are involved in processes like X chromosomal inactivation (XIST) (39) and genomic imprinting (H19) (40). Yet, much more research will be necessary to characterize the many diverse functions and modes of action related to lncRNAs [for more detailed review see (28) and for overview see Figure 2].
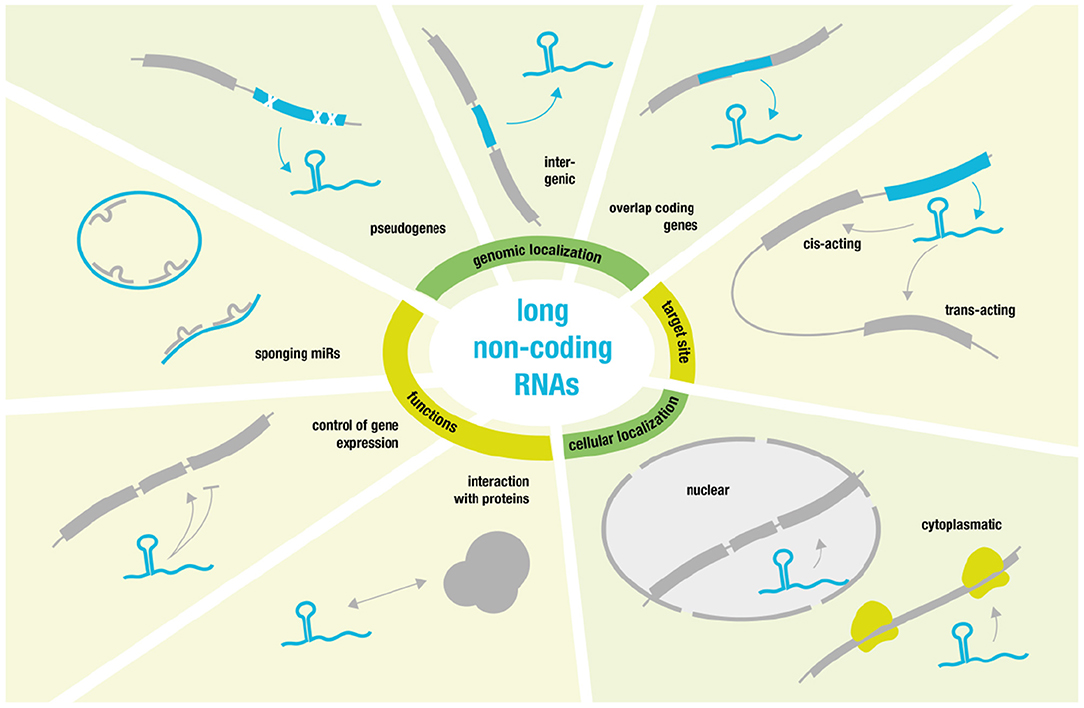
Figure 2. Overview over long non-coding RNA function and behavior.
In the context of aneurysm disease H19 has been identified to boost aneurysm progression (41).
Using RNAs for therapeutic approaches has many advantages due to their unique biology. Targeting and editing the undruggable human genome, modulating gene expression and altering epigenetic processes are just some of the many opportunities (26).
However, current RNA-related methods are restraint by a couple of significant factors. RNAs are predisposed to endonuclease degradation (26), can cause adverse immunological reactions (42), and their large size and negative charge prevent them from passively crossing the cell membrane's lipid bilayer (43). Generally, RNA molecules are taken up by endocytosis and remain trapped inside the endosome, but outside the cytoplasm and nucleus (44). During the last decade, significant research has therefore focused on addressing the many challenges associated with RNA-based delivery.
Today, single-stranded antisense oligonucleotides (ASOs) usually contain a phophorythioate backbone replacing the early phophodiester bonds. This results in increased stability and hydrophobicity, thereby facilitating cellular uptake, enhancing bioavailability, and improving pharmacokinetics by binding serum proteins like albumin (45, 46).
ASOs usually function via cleavage of a formed mRNA:ASO duplex by a recognition enzyme termed RNase H (47). One setback in this regard is the reduced binding affinity of phosphorothiorate ASOs toward the mRNA target (48). Binding affinity can be improved by incorporating 2′ modifications [such as 2′-O-methyl (2′-OMe), 2′-O-methoxyethyl (2′-MOE), and 2′-Fluoro (2′-F)] (49). These modifications however reduce nuclease degradation (50).
Naked (unconjugated) ASOs—just like other RNAs—enter the cell via endocytosis (51). Yet, they have the ability to slowly cross the lipid bilayer and escape into the cytoplasm and nucleus (gymnosis) due to their hydrophobic characteristics (45, 46, 52). The latter render ASOs independent of additional delivery agents (53).
The specific delivery to many cell and tissue types remains difficult via current ASO methods. In this context, conjugating cell-type-specific targeting domains to ASOs might help to overcome this huge challenge. Successful examples for the mentioned approach include the N-acetylgalactosamine (GalNAc) ligand that is used for targeted ASO delivery to hepatocytes via the hepatocyte-specific asialoglycoprotein receptor (54, 55).
ASOs that include a 2′,4′-O-methylene link between two joint rings (rendering the molecule bicyclic) are known as locked nucleic acids (LNAs). LNAs have an overall enhanced resistance toward degradation by 3′-exonucleases and show a great affinity to their mRNA target. The latter decreasing molecule length and improving delivery (56, 57).
LNAs can be further divided into two main groups: mixmers and gapmers. Mixmers contain LNAs and DNA nucleosides that are spread throughout the oligonucleotide sequence. Mixmers block protein translation but are not able to support mRNA cleavage (48). Conversely, gapmers comprise a central segment (gap) that is flanked by two LNA segments at both ends of the oligonucleotide and inhibit mRNA expression (by recruiting RNase H) and protein translation alike (58). One prime example for a novel FDA-approved RNA drug is developed as a gapmer. Mipomersen is directed against ApoB to counteract homozygous familial hypercholesterolemia (59).
Another subclass of ASOs are so called “antagomirs” or “anti-miRs.” These terms are used to describe single-stranded oligonucleotides that prevent miRs from exerting their inhibitory effect on their respective target mRNAs (60). Antagomirs are able to reach many different tissues without crossing the blood-brain barrier and their effects last at least 3 weeks after intravenous injection (60). However, their way of functioning has not been completely understood, yet. Chemically, antagomirs show a close relation to their double-stranded siRNA counterpart (61).
ASOs and siRNAs show prominent differences regarding their mode of action. As described above, ASOs bind their mRNA target in a direct and unassisted manner, therefore permitting for various chemical modifications. SiRNAs rely on supporting molecules such as the dicer enzyme and the RNA-induced silencing complex (RISC) (62). This dependency restricts the extent of possible chemical modifications greatly (63, 64).
Introduction of phosphorothioates has improved siRNA stability and shortening of the siRNA double strand has reduced overall charge for enhanced local delivery (65–67). Other modifications include a “Trojan horse approach” via short-interfering ribonucleic neutral (siRNN) molecules that contain a phosphotriester backbone. Initially, siRNNs mimic a neutrally charged protein surface. After entering the cell, they are cleaved into negatively charged phosphodiester siRNAs that induce a RNAi response (68). Despite all modifications, siRNAs remain macromolecules that cannot cross the cell's lipid bilayer. Thus, siRNAs depend on aiding delivery agents (43).
Lipid nanoparticles (LNPs) and synthetic nanoparticles were early attempts to master the siRNA delivery dilemma. Both systems were originally used for DNA-mediated gene therapy and required optimization for a siRNA approach (69, 70). But supplementing additional components to LNPs increases toxicity and decreases solubility, thus leading the RNAi field away from large LNPs to smaller, more tailored methods (44, 71).
When it comes to hepatic targeting, GalNac conjugation—similar to the ASOs approach described in the section above—has been successfully applied (72). For extra-hepatic delivery, antibodies hold great promise for a more specific delivery approach (73). However, difficulties regarding the antibody-RNAi conjugate's expression, aggregation, and activity need to be addressed, before entering the clinical trial stage (74).
A novel and utmost elegant way of using RNAs for therapeutic modulation is genome engineering via Clustered Regulatory Interspaced Short Palindromic Repeats (CRISPR) and the CRISPR-associated Proteins (Cas) proteins. Originally, CRISPR/Cas is a bacterial defense mechanism that prokaryotes use against viral and plasmid intruders. Prokaryotes integrate DNA sequences that match past intruders into their genome as part of their adaptive immune response. This intruder library or memory helps the organism to recognize and defend itself during future attacks (75).
Today, the CRISPR/Cas9 type II system is commonly used in the research set-up. There have been other nuclease-based systems that contain genome editing capabilities but CRISPR is highly user friendly due to its simplicity and flexibility (76). In short, the Cas9 nuclease is guided to the target sequence adjacent to a protospacer-adjacent motif (PAM) by a single guide RNA (sgRNA). There, a double-strand break is produced that can be repaired by non-homologous end joining (77). The latter usually leads to insertions or deletions resulting in loss of function (knock-out). However, gain of function (knock-in) can also been achieved via exogenous donor DNAs.
Just as with the above described techniques there are several delivery hurdles to take when transferring CRISPR/Cas9 into a clinical context. Both Cas9 and the sgRNA are charged macromolecules that are difficult to introduce into the cell's nucleus (78).
Other concerns include off-target effects that could be reduced by adapting the sgRNA design (79, 80). Design algorithms have already been implemented focusing on the 5' sequence of the sgRNA to maximize on-target action (81). Another reason for off-target effects are unintended high levels of Cas9 nuclease associated with unspecific cleavage (82). There are different strategies to control the activity of Cas9 such as generation of Tet-controlled promoters (83), fusion of Cas9 with an estrogen receptor domain (84, 85) or usage of light-responsive elements (86) to control Cas9 activity.
Some of the main questions linked to genome engineering via CRISPR/Cas are of ethical nature as editing a patient's genome would cause a life-long alteration of the human “code” (87). At the moment, the CRISPR/Cas method is still at a very early step of its development and the next years and decades will show how to apply this exciting, new system to achieve the best possible outcome for every single patient.
The following section will give an overview on different delivery systems used for RNA therapy including viral and non-viral vectors (for overview see Figure 3).
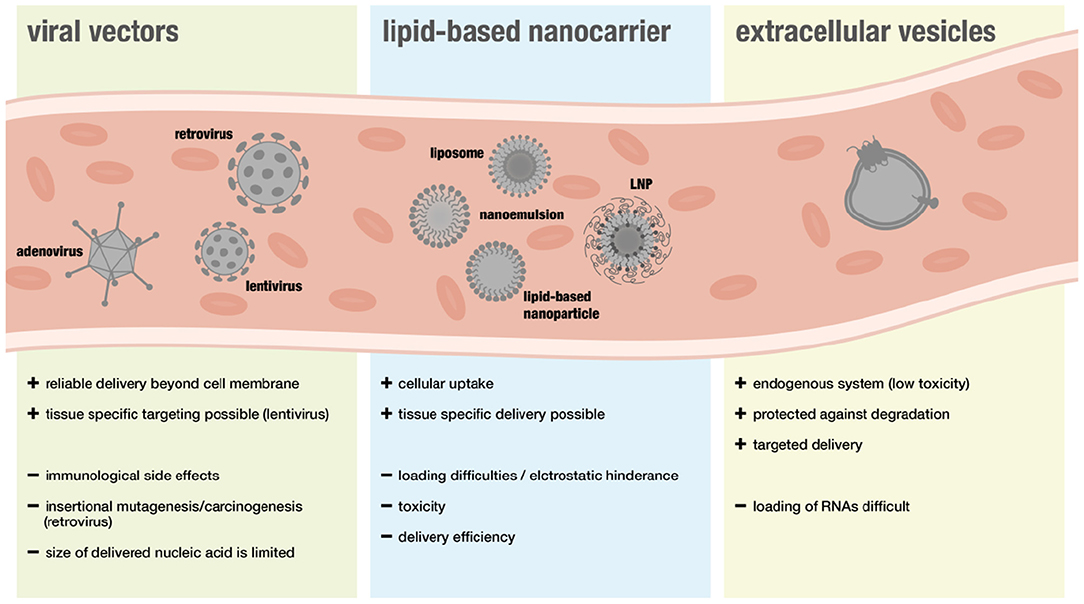
Figure 3. Overview over different delivery system in the context of RNA therapeutics.
Viral vectors are considered a suitable approach for gene therapy because they can deliver nucleic acids easily beyond the cell membrane. However, safety concerns have been limiting translational efforts. In particular immunological side effects and insertional mutagenesis leading to cancer development raised concerns (88). Furthermore, some people show immunity toward specific viral vectors and the size of nucleic acids that can be transported via viral systems is more restricted than with non-viral systems (89).
Retroviruses were among the first viral vectors used for cell transfection with RNA-expressing plasmids. These viruses provided great results in vitro, while they were linked to safety concerns and limited efficiency in vivo—especially in the human setting (90). Retroviruses integrate their viral genome into the host genome for viral replication. This mode of action can cause insertional mutagenesis and even carcinogenesis (91, 92). Furthermore, retroviruses need replicating cells for RNA therapy. However, most mammalian cells are in resting state and not actively replicating. This causes difficulties for retroviruses in reaching the cell type of interest (93).
Thus, lentiviruses, a subclass of retroviruses, have emerged as appealing vectors for in vivo application. While replication competent lentiviruses—virus particles capable of infecting cells and replicating to produce additional infectious particles—remain a safety concern, compared to other retroviruses, lentiviruses have reduced risk of insertional mutagenesis (94) and are able to transfect non-dividing cells (95). Additionally, they are less immunogenic than adenoviral vectors (96) and can be used for tissue-specific targeting (97). The latter can be achieved via integration of envelope material from other viruses. Experiments with murine embryos showed that, e.g., Mokola or Ebola virus glycoprotein can confer certain tissue tropism toward heart and skeletal muscle cells (MVG and EVG) (98).
Adenoviral vectors are a popular type of viral vector for gene therapy trials that have been widely studied (99). Different to other viral vectors the genetic information does not integrate into the host cell's genome but persists in episomal form in the nucleus (100). On one side this reduces the risk for viral DNA integration into the host cell's genome, on the other side this leads to more transient effects (100). To achieve long-term transfection in vivo, all viral coding sequences were removed and immune responses against transduced cells limited (101). Nevertheless, the high (also dose dependent) immunogenicity and hepatotoxcity of certain adenoviral proteins remains a concern (102, 103). Traditionally, human adenovirus are classified in unevenly prevalent serotypes based on immune cross-reactivity. Some of the immunity is broadly spread thereby diminishing the potency of adenoviral vectors. To bypass this challenge, adenoviral vectors are mostly derived from rare serotypes. Other classifications are based on sequence homology for the identification of new adenoviruses (104). Targeting specific cell populations, apart from the natural tropism of vectors, was achieved by specific re-targeting approaches leading to an expansion in transduction of specific cell populations (105).
Through upgraded vector design the safety of the delivery systems could be improved [as reviewed in (88, 99)] and today more than two thirds of all gene therapeutically clinical trials registered until the end of 2018 (a total amount of 2,600) have been conducted using viral vectors (17).
Some viral vector based therapies have been cleared by the FDA including adenovirus associated vectors and many clinical trials are active.
Although many advances have been made regarding safety, viral vectors are still associated with a higher risk of triggering adverse immunogenic responses. Thus, non-viral nanocarriers have been widely favored when it comes to RNA-delivery. Lipid-based nanocarriers can enable cellular uptake of RNAs, as their natural structure is ideal when it comes to interactions with the cellular phospholipid membrane (106).
Nanoemulsions consist of two immiscible liquids that are mixed by using specific surfactants to form a single phase (107). Although it is relatively easy to mix several compounds into a single nanoemulison formulation, this method shows some challenges when applied to the RNA field. One advantage of nanoemulsions is the relative ease to mix multiple ingredients in a single formulation. By convention, nano-emulsions are made of negatively charged ingredients. This will make it problematic to trap RNA molecules within a nanoemulsion (108). Furthermore, keeping the nanoemulsion's fine lipid droplets small and uniform while stored after inserting the quite large and hydrophilic RNA molecules (108).
Another form of nanocarriers are lipid-based nanoparticles. They encounter difficulties regarding RNA delivery due to their lipophilic core that does not match the hydrophilic, poly-anionic RNA molecules. However, this structure allows to encapsulate lipophilic drugs into the core and coat the RNA on the nanoparticle surface thereby creating unique complexes of high translational potential (109).
Liposomes are easily prepared and consist of an aqueous core surrounded by a phospholipid bilayer (110). Tissue-specific delivery and extended circulation time can be achieved via functionalization (e.g., PEG-lipids) (111, 112). The aqueous core renders liposomes suitable for loading hydrophilic and ionic drug molecules, distinguishing them from other lipid-based systems that are usually loaded with lipophilic compounds (110). Subsequently, positively charged (cationic) liposomes can readily encapsulate RNAs and are therefore considered most suitable when it comes to RNA delivery. Unfortunately, cationic lipids are also the main driver of toxicity regarding lipid-based RNA nanocarriers (113). Alteration strategies [detailed review in (114, 115)] have been implemented to improve both transfection performance and cytotoxicity. Phospholipid molecules typically consist of three main components: a cationic head, a hydrophobic hydrocarbon backbone and a linker region (116). Each component can be modified to achieve certain molecule properties. DOTAP that is considered today's gold standard for RNA transfection of cationic liposomes has a linker consisting of biodegradable ester bonds thereby limiting toxic effects (117, 118).
In terms of clinical translation, LNPs (lipid nanoparticles) represent the most successful type of RNA nanocarrier (119). LNPs are ~120 nm in diameter or less, composed of neutral lipids, cationic lipids, and PEG-conjugated lipids and form by spontaneous vesicle formation (120). LNPs have integrated many of the mentioned design strategies to overcome multiple challenges associated with RNA delivery, including limited in vivo circulation, inefficient transfection, and unacceptable toxicity (120).
A new type of RNA-delivery for therapeutic purposes are extracellular vesicles (EVs) that have recently been discovered to be endogenous RNA carriers. EVs are released by cells and serve as means of intracellular communication. Since cells use miR-loaeded EVs to regulate gene expression in other cells and different sorts of RNAs, including lncRNAs, were found to be presented in these vesicles, they might represent a safer and more effective method of RNA-delivery (121–126).
Interestingly, there seems to be a clear pattern when it comes to RNA vesicle packaging. Certain sequence motifs are favorably integrated into EVs (127). Moreover, there is evidence, that proteins of the RISC complex are involved in the packaging process since they were detected inside of the EVs as well (128).
One major advantage of RNAs that are loaded into EVs is that they are protected from degradation by extracellular RNases (129). EVs serving as delivery systems are also considered superior with regards to targeted delivery and cytotoxic side effects. Yet, a major problem remains in loading the RNA therapeutics into the vesicles. Due to the negative charge of the RNAs, crossing the vesicle membrane represents a major challenge. Therefore, most studies investigating EVs as delivery tools are focused on siRNAs and miRs rather than lncRNAs due to their restricted size that facilitates vesicle packaging (126).
RNAs have successfully been loaded into EVs prior to isolation which raised concerns about the purity of the generated RNA-loaded vesicles. Consequently, several different mechanisms have been established to incorporate RNAs into EVs after isolation, including electroporation, sonication, and incubation with varying degree of success, as extensively reviewed in Jiang et al. (126).
There are two major types of off-target effects that can be distinguished (for overview see Figure 4). On the one hand, off-target effects result from ubiquitous expression of targeted coding and non-coding RNAs in different tissues and cell types, resulting in unintended systemic treatment effects (130). On the other hand, off target effects may be due to the promiscuous mode of action of miRNAs that usually do not target one but several mRNAs (22, 131).
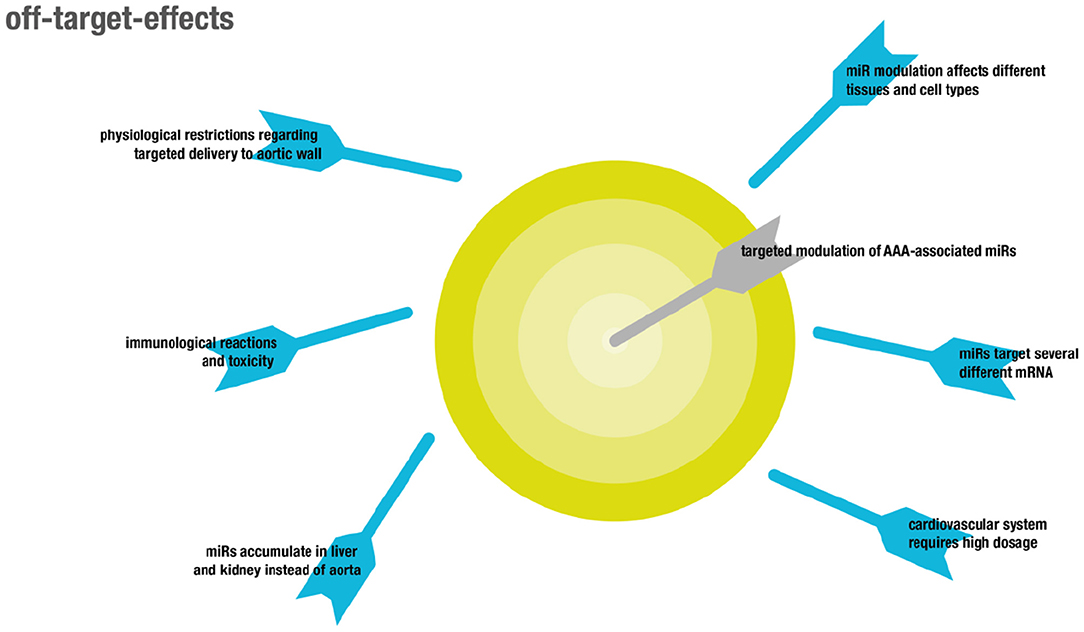
Figure 4. Overview showing off-target effects in the context of RNA therapeutics.
Although miRNAs were shown to target functional gene networks (23), unwanted off-target effects are likely to occur, due to inherently imperfect binding to mRNA sequences (24). In particular, some microRNAs relevant in the cardiovascular system might play differential roles in different pathologies, like cancer. For example, overexpression of miR-21 was shown to be protective in two models of AAA by influencing apoptosis and proliferation of vascular smooth muscle cells in the aortic wall via PTEN and AKT signaling, resulting in attenuated expansion of AAA (132). However, miR-21 is also well-established as an onco-miR with upregulated levels in several different kinds of solid and leukemic malignancies, likewise attributed to its regulatory function on PTEN (133).
Another example is miR-29b. While LNA-mediated inhibition of miR-29b increased fibrosis as indicated by collagen expression and thus resulted in attenuated growth of AAA, induction of fibrosis is pathological in other organs, which makes systemic delivery of miR29b inhibitors non-feasible (134). Targeted delivery to the aorta is complicated by the inherent feature of the aorta that any compound locally delivered to the blood will immediately be distributed throughout the entire circulation, which is in contrast to targeting other vascular structures like the coronary arteries (135). This process results in accumulation of the delivered compounds in highly perfused capillary beds in non-target organs like the liver. Adverse effects in general primarily affect both liver and kidneys, since exogenously administered RNAs preferentially accumulate in these organs (136). Moreover, both liver and kidney are responsible for clearance of naked RNA from the bloodstream, on the one hand through accumulation in the liver and on the other hand through glomerular filtration in the kidney (137). As a consequence, the first therapeutic approaches involving RNAs these days target hepatic diseases since therapeutic concentrations of RNA can be reached most easily in this organ. In particular, nanoparticles and cationic lipid-dependent systems strongly accumulate in the liver (138). Accumulation in other organs might be achieved by use of specific ligands binding to receptors on the target cells which was already shown for vascular targets (139). However, as already mentioned this might be difficult in the case of aortic aneurysms because receptors on relevant cell types like SMCs are not in direct contact with the blood stream. The dose commonly administered in animal models to target the cardiovascular system is about 3–10-fold higher than that used in animal models of liver pathologies. This increases toxicity as well as potential unspecific binding and thus off-target effects, consequentially narrowing the therapeutic window (140).
AAA pathology is characterized by three distinct mechanisms on a molecular level (for overview see Figure 5): ECM remodeling, vascular inflammation and SMC apoptosis (11). Lately research has demonstrated that non-coding RNAs are significant modulators of these processes (12, 13).
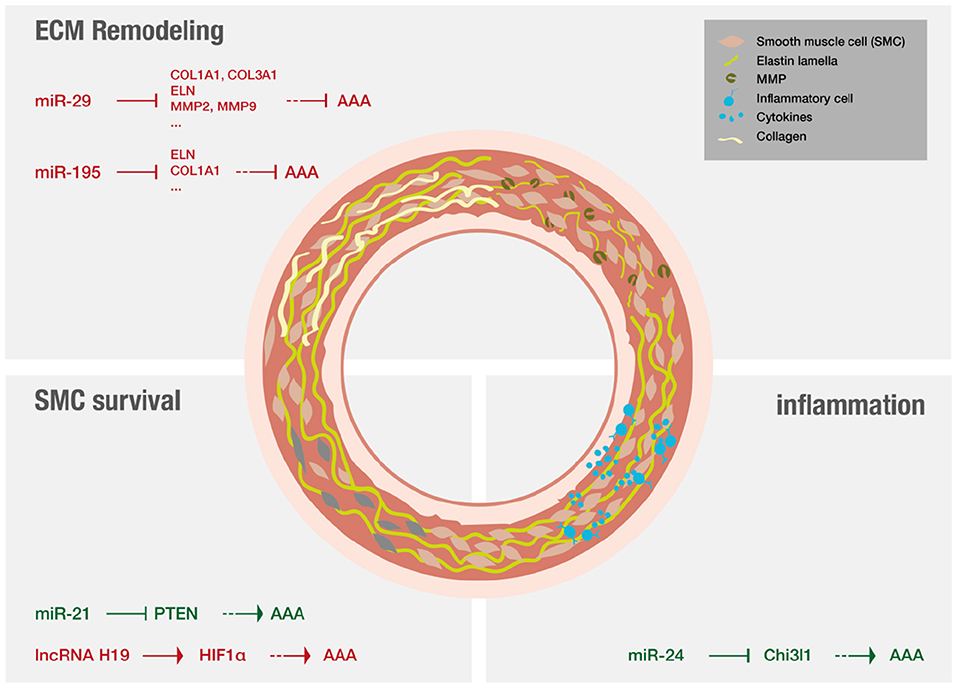
Figure 5. Non-coding RNAs that have been shown to play a major role in AAA disease.
MiRs are of special interest in a translational context as they are able to modify whole gene networks (23). Concerning fibrosis miR-29 has evolved as an important player (134, 141) and the effect of LNA-anti-miR directed against miR-29 on the formation of aneurysms was studied extensively. Mägdefessel et al. (134) found miR-29b to be significantly downregulated during murine AAA development as well as in human AAA tissue (134). The study found increased expression of several different collagen isoforms as wells as elastin upon treatment with LNA-anti-miR-29b in two different murine models of AAA, using either Angiotensin II infusion in apoE−/− mice or porcine-pancreatic-elastase infusion in C57Bl/6 mice. In both models, LNA-anti-miR-29b treatment resulted in reduced formation of AAA (134). The opposite results were found when the mice were treated with miR-29b mimics. However, this study also demonstrated increased collagen mRNA levels in the heart, kidney, and liver upon systemic administration of anti-miR-29b, indicating off-target effects that demand for local delivery of the anti-miR-29b (134). In line with these results, another study evaluated the therapeutic potential of miR-29b in a mouse model of Marfan syndrome (142). LNA-anti-miR-29b was administered both prenatal and postnatally by retro-orbital injection. The investigators found that anti-miR-29b could reduce formation of AAA in the long-term if administered prior to formation of AAA but did not ameliorate the outcome of already formed AAA. Anti-miR29b treatment, if administered prenatally resulted in increased expression and decreased breakdown of elastin in the aorta, potentially attributed to decreased matrix-metalloproteinase-2 (MMP-2) activity and expression (142). Furthermore, collagen levels were found to be increased upon treatment in both studies (134, 142). The manipulation (either inhibiton or overexpression) of other miRs can also ameliorate AAA disease by increasing collagen content and reducing elastin fragmentation. Two examples are miR-181b that is effective by regulating macrophage timp3 expression and miR-126-5p that signals via ADAMTS-4, an enzyme that cleaves various ECM proteins (143, 144).
Regarding inflammation in the context of AAA, miR-24 was identified as key regulator in both established murine models as well as in human aortic tissue and plasma analysis (145). Inhibition of miR-24 showed enhanced AAA development, while upregulation of miR-24 ameliorated disease. It was further revealed that chitinase 3-like 1 (Chi3l1) is a major target of miR-24. In vivo and in vitro experiments additionally showed that Chi3l1 not only regulates cytokine synthesis and survival of macrophages but also promotes aortic smooth muscle cell migration and cytokine production, and stimulates adhesion molecule expression in vascular endothelial cells (145). A recent review focused on myeloic cells and aneurysm disease (146).
Other studies investigated RNA effects on SMC survival. MiR-21 levels were found to increase during AAA development in two different murine models, miR-21 overexpression was shown to increase SMC proliferation via reduction of phosphatase and tensin homolog (PTEN) protein, leading to increased phosphorylation and activation of AKT thus having protective effect on AAA development (132). The results could be confirmed using LNA-anti-miR-21 constructs that were applied systemically and resulted in increased apoptosis and decreased proliferation of SMCs, thus strongly aggravating AAA (132). Interestingly, miR-21 levels were shown to be increased in a nicotine-dependent manner. Upregulation of miR-21 in AAA as well as increased upregulation of miR-21 in AAA of smokers could be confirmed in human samples (132). Additionally, a recent study reported that inhibition of long non-coding RNA H19, utilizing LNA-GapmeRs in vivo, significantly limited aneurysm growth in established murine models by influencing SMC survival. This study proofed especially translational as it investigated similar patterns in human AAA tissue samples, and in a novel preclinical LDLR−/− Yucatan mini-pig aneurysm model (41).
Zampetaki et al. (147) evaluated the effect of local as well as systemic delivery of antagomiR-195 on the development of aortic aneurysm in ApoE−/− mice treated with Angiotensin II. AntagomiR-195 was found to de-repress aortic expression of elastin and other extracellular matrix genes after systemic application while antagomiR-29b mainly affected gene expression in the liver, thus already indicating potential off-target effects. The antagomiR-29b effect on the development of AAA was more pronounced than the effect of antagomiR-195 (147). The antagomiRs used were cholesterol-conjugated and delivered by peritoneal injection. For evaluation of local effects, an aortic isograft model using aorta incubated with cholesterol-bound LNA-antimiR-195 was used. Local delivery showed additional de-repressive effects on MMP expression. This study also evaluated the effect of antagomiR-195 on closely related miRs of the miR-15 family and did not find any off-target effects in that regard (147).
More evidence on vascular targeting of antagomiRs derives from studies on atherosclerosis and restenosis. Systemic antagormiR-92a delivery was already shown to be effective in downregulating vascular miR-92a and thereby leading to decreased atherosclerosis and endothelial dysfunction in LDLR−/− mice that were fed a high-fat diet (148). This provides evidence, that systemic antagomiR delivery can successfully target the aorta. Notably, the measured effects in this study were higher at the aortic arch than at the thoracic aorta. Furthermore, effect of antagomiR-92a was not assessed in other organs (148). In another study, antagomiR-92a was shown to potentially reduce restenosis in a rat model of carotid artery balloon injury. While also this study implies successful delivery of the antagomiR to the vasculature after systemic administration, no off-target effect were evaluated either (149).
Despite limitations, different studies examining the effect of miR inhibition or stimulation by systemic mechanisms successfully detected positive effects on AAA development. Unfortunately some studies also found systemic side-effects (134, 142) that need to be even more carefully evaluated in the future when clinical translation wants to be achieved.
While targeted delivery of anti-miR to treat AAA using nanoparticles to our knowledge has not been reported yet, this approach has been investigated for the treatment of atherosclerosis.
One study exploited the fact that along with miR-712, disturbed flow in blood vessels also leads to the upregulation of vascular cell adhesion molecule 1 (VCAM-1) on endothelial cells. Coated cationic lipoparticles were modified using a ligand for the VCAM-1 internalizing sequence (139). This approach tackles different challenges in miR delivery. It increases transfection efficiency due to the presence of cationic lipids in the lipoparticles, while simultaneously reducing toxicity and aggregation due to a coat of neutral lipids. Furthermore, it enables targeted delivery to inflamed and early-stages of atherosclerotic vessels by providing a ligand that first binds VCAM-1 and then initiates its internalization. The study examined the effect of their nanoparticles on the development of atherosclerosis in a partial carotid ligation model in apoE−/− mice. While leading to specific downregulation of miR-712 in endothelial cells of affected areas and prevention of atherosclerosis development, no impact on miR-712 expression was observed in other vascular regions or any other organ investigated. During in vivo experiments, no differences in blood proteins or blood cell counts were observed between treated and control mice. However, in vitro results showed increased C3a levels after treatment with targeted nanoparticles (139). Therefore, investigations of the immune response and consequences of chronic treatment remain issues that need to be addressed in this promising approach. As demonstrated in the study above, anti-miR-712 was undetectable in the aortic media (139).
With regards to treatment of AAA, targeted delivery will be more difficult, since a potential receptor on SMCs or fibroblasts will not be in direct contact with the systemic blood stream. However, recent studies show progress in side-specific administration (150).
Another study used microspheres to deliver antagomiR-92 into the coronary arteries in a pig model of perfused acute myocardial infarction to limit post-infarct remodeling, dysfunction of vessel wall motion, and promote vessel growth (135). This was successfully accomplished by a single intracoronary injection of antagomiR-92 loaded microsphereses shortly after reperfusion. The antagomiR was designed as a LNA to increase stability in the blood, and did not have any ligands to mediate cell-specific delivery. After injection, the microspheres were shown to be present in the capillaries of the damaged area and were not detectable in other organs, thus demonstrating successful local delivery without major spill-out into the circulation (135). However, this also leads to the conclusion that this approach is not feasible for treatment of aortic aneurysms since the microspheres are likely to accumulate in the capillary beds of downstream organs, thus increasing off-site effects.
To our knowledge, there is no study using extracellular vesicles to target RNA therapeutics to AAA. However, it was shown that when administering mesenchymal stem cell derived extracellular vesicles to a murine model of intracranial aneurysm the rate of aneurysm rupture was decreased (151). While the extracellular vesicles were detected in brain tissue, they were also present in spleen, liver, and lung of the investigated mice. Of interest, administration of the extracellular vesicles resulted in decreased mRNA levels of target genes like tumor necrosis factor α (TNFα), increased cyclooxygenase 2 (COX2), and E-prostanoid-4 mRNA levels, thus contributing to decreased aneurysmal rupture. Changed mRNA expression patterns might be indicative of miRNA as one of the effective agents within the extracellular vesicles in this study (151).
Several studies investigating the role of different miRNAs employed viral vectors to investigate the effects on gene expression in vivo. For example, one study found miR-24 to be protective against AAA development using lentiviral vectors to deliver pre-miR-24 in two different murine models of AAA. In both models decreased aneurysm development was demonstrated. The results could be confirmed using lentiviral vectors expressing anti-miR-24 which led to aggravated aneurysm development (145). A more recent study demonstrated decreased AAA formation after infection with miR-145 containing lentivirus in an aneurysm model of Ang II infused ApoE−/− mice. This therapeutic effect was mediated by miR-145-dependent downregulation of MMP-2 expression (152). In a different approach, bone-marrow derived leukocytes were infected with lentiviral vectors containing siRNA against C-C chemokine receptor type 2 (CCR-2) which is known to be involved in the initial pathogenesis of AAA as a part of the immunological degradation of ECM components (153). The transduced cells were delivered by bone marrow transplantation prior to Ang II infusion in an aneurysm model of Ang II infused ApoE−/− mice. This siRNA approach significantly reduced the aneurysmal damage to the aortae in these mice (153).
Another possibility of local delivery that is particularly feasible for AAA (and clinical translation) are drug-eluting stents. This stent approach has been tested in a humanized myointimal hyperplasia model for in-stent restenosis using balloon-injured human internal mammary arteries that were transplanted into Nowett-nude rats (154). The miR under investigation for its therapeutic relevance regarding in-stent restenosis was miR-21. Due to substantial off-target effects in liver, lung, heart, and particularly kidney after systemic delivery of LNA-anti-miR-21, anti-miR-21 coated stents were tested. While the anti-miR eluting stents showed significantly reduced neointima formation compared to conventional stents, no off-target effects were observable, not even in the kidney that showed marked LNA-anti-miR accumulation after systemic application (154). With regard to AAA, these study demonstrates that local delivery of miR inhibitors to the vasculature is possible and is potentially advantageous over systemic delivery not only with regards to off-target effects, but also regarding treatment efficiency. Drug-eluting stents are a promising route of delivery to the thoracic and abdominal aorta.
In a pig model of acute ischemia and reperfusion injury, delivery of LNA-anti-miR-92a was tested in a catheter-based approach. The study compared systemic, retrograde, and anterograde delivery to the heart, all resulting in significant downregulation of miR-92a (155). MiR-92a appears to target mRNAs corresponding to several proteins in the context of angiogenesis, thereby leading to improved capillary density after ischemic challenge. The miR-92a family, including miR-25, miR-92a-1, miR-92a-2, and miR-363, originate from paralog clusters (miR-17-92, miR-106a-363, and miR-106b-25) that are highly conserved. The latter partly explains side effects that were observed during delivery of LNA-anti-miR-92a: miR-25 was downregulated just as miR-92a. Yet, other miRs of the miR-17-92a cluster were unaffected.
Despite local delivery, miR-92a levels were found to be affected in liver, kidney, and spleen, while the impact in lung tissue was lower compared to systemic delivery. However, significant decrease in infarct size and cell death as well as significantly improved global heart function were only observed after catheter-mediated anti-miR-92a delivery and not after systemic application (155). Furthermore, this study indicates that local delivery potentially enables reduction of anti-miR dose, thus resulting in reduced but not abrogated off-site effects (155). Again, this approach is in general feasible for targeting the vasculature but is less feasible for treatment of AAA since the compounds will be distributed with the circulation and are likely to accumulate in downstream capillary beds of non-target organs.
While many revolutionary developments regarding RNA knowledge and technology have been mentioned in the sections above, this review will close with some short notes on a method that has just started to evolve in the DNA/RNA field and that might be promising in the context of AAA disease as well.
The DNA origami method is named after a famous Japanese paper folding art. Origami artists aim to sculpture a flat sheet of paper into a desired object by applying complex folding sequences. DNA origami aims to create a long single-stranded DNA molecule (scaffold) out of hundreds of short synthetic DNA nucleotides (staples), thereby folding specifically shaped objects at a nanoscale (156).
At first it was only possible to generate planar, two-dimensional objects (156), but since 2009 complicated three-dimensional structures can be effortlessly produced due to the introduction of novel design strategies (157). These powerful computational tools for designing and analyzing DNA nanostructures enable custom-made shapes (158) and lately even scaffold-free approaches (159, 160).
DNA-origami objects can load drugs and thereby function as miniature transporters. These so called “nanorobots” have been experimentally tested in the oncology field (161). But origami objects could (even more excitingly) also serve as miniature devices and, e.g., assemble critical molecules in cells, such as proteins and nucleic acids (162).
Although RNA molecules display many similarities to DNA molecules, it was not until 2013 that RNA origami had been reported by the Sugiyama and Mao groups (163, 164). In 2014, Sugiyama et al. (165) generated pure RNA origami objects by using RNA staples to fold the RNA scaffold. It is obvious that still a lot of research effort will be necessary to translate the RNA origami method into the cardiovascular field but it will be an exciting journey nevertheless.
The RNA revolution has taken the modern research world by storm. It presented some major successes when it comes to discovering new biological paradigms and clinical-translatable technology. One disease that benefited from this extensive research effort—by reaching a better understanding of disease mechanisms—was AAA disease. Discovering underlying pathological mechanisms helped to outline solution strategies. Still, years of further research will be needed to take several technological hurdles and achieve the ultimate goal of clinical translation.
IS and AD drafted the manuscript. IS, UR, and PT contributed the initial idea, funding, and drafting. KM contributed ideas to the manuscript and created the figures. All authors contributed to the article and approved the submitted version.
This work was supported by research grants from the Deutsche Forschungsgemeinschaft (Sche 2125/2-1 to IS), the University of Leipzig Medical Faculty (934300-022 to IS) and the German Center for Cardiovascular Research (DZHK) e.V. (81X3300104 to UR).
IS and UR are cofounders of Angiolutions. Angiolutions is a company developing vascular devices for aneurysm diseases.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Johnston KW, Rutherford RB, Tilson MD, Shah DM, Hollier L, Stanley JC. Suggested standards for reporting on arterial aneurysms. J Vasc Surg. (1991) 13:452–8. doi: 10.1067/mva.1991.26737
2. Lederle FA, Johnson GR, Wilson SE, Gordon IL, Chute EP, Littooy FN, et al. Relationship of age, gender, race, and body size to infrarenal aortic diameter. J Vasc Surg. (1997) 26:595–601. doi: 10.1016/S0741-5214(97)70057-0
4. Golledge J, Norman PE. Current status of medical management for abdominal aortic aneurysm. Atherosclerosis. (2011) 217:57–63. doi: 10.1016/j.atherosclerosis.2011.03.006
5. Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. (2011) 8:92–102. doi: 10.1038/nrcardio.2010.180
6. Norman PE, Powell JT. Abdominal aortic aneurysm: the prognosis in women is worse than in men. Circulation. (2007) 115:2865–9. doi: 10.1161/CIRCULATIONAHA.106.671859
7. Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation. (2005) 111:816–28. doi: 10.1161/01.CIR.0000154569.08857.7A
8. Pande RL, Beckman JA. Abdominal aortic aneurysm: populations at risk and how to screen. J Vasc Interv Radiol. (2008) 19(6 Suppl):S2–8. doi: 10.1016/j.jvir.2008.03.010
9. Mussa FF. Screening for abdominal aortic aneurysm. J Vasc Surg. (2015) 62:774–8. doi: 10.1016/j.jvs.2015.05.035
10. Greenhalgh RM, Brown LC, Powell JT, Thompson SG, Epstein D, Sculpher MJ, et al. Endovascular versus open repair of abdominal aortic aneurysm. N Engl J Med. (2010) 362:1863–71. doi: 10.1056/NEJMoa0909305
11. Norman PE, Powell JT. Site specificity of aneurysmal disease. Circulation. (2010) 121:560–8. doi: 10.1161/CIRCULATIONAHA.109.880724
12. Milewicz DM. MicroRNAs, fibrotic remodeling, and aortic aneurysms. J Clin Invest. (2012) 122:490–3. doi: 10.1172/JCI62204
13. Lu H, Rateri DL, Bruemmer D, Cassis LA, Daugherty A. Novel mechanisms of abdominal aortic aneurysms. Curr Atherosc Rep. (2012) 14:402–12. doi: 10.1007/s11883-012-0271-y
14. Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. (2016) 16:167. doi: 10.1038/nrd.2016.117
16. Stein CA, Castanotto D. FDA-approved oligonucleotide therapies in 2017. Mol Ther. (2017) 25:1069–75. doi: 10.1016/j.ymthe.2017.03.023
17. Ginn SL, Amaya AK, Alexander IE, Edelstein M, Abedi MR. Gene therapy clinical trials worldwide to 2017: an update. J Gene Med. (2018) 20:e3015. doi: 10.1002/jgm.3015
18. Dammes N, Peer D. Paving the road for RNA therapeutics. Trends Pharmacol Sci. (2020) 41:755–75. doi: 10.1016/j.tips.2020.08.004
19. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. (2009) 136:642–55. doi: 10.1016/j.cell.2009.01.035
20. Golden DE, Gerbasi VR, Sontheimer EJ. An inside job for siRNAs. Mol Cell. (2008) 31:309–12. doi: 10.1016/j.molcel.2008.07.008
21. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. (2008) 9:102–14. doi: 10.1038/nrg2290
22. Small EM, Frost RJA, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation. (2010) 121:1022. doi: 10.1161/CIRCULATIONAHA.109.889048
23. Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell. (2010) 18:510–25. doi: 10.1016/j.devcel.2010.03.010
24. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 136:215–33. doi: 10.1016/j.cell.2009.01.002
25. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. (1998) 391:806–11. doi: 10.1038/35888
26. Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. (2004) 432:173. doi: 10.1038/nature03121
27. Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. (2012) 489:101–8. doi: 10.1038/nature11233
28. Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. (2018) 172:393–407. doi: 10.1016/j.cell.2018.01.011
29. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. (2012) 22:1775–89. doi: 10.1101/gr.132159.111
30. Ulitsky I, Bartel David P. lincRNAs: genomics, evolution, and mechanisms. Cell. (2013) 154:26–46. doi: 10.1016/j.cell.2013.06.020
31. Kapranov P, Drenkow J, Cheng J, Long J, Helt G, Dike S, et al. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. (2005) 15:987–97. doi: 10.1101/gr.3455305
32. Novikova IV, Dharap A, Hennelly SP, Sanbonmatsu KY. 3S: shotgun secondary structure determination of long non-coding RNAs. Methods (San Diego, Calif). (2013) 63:170–7. doi: 10.1016/j.ymeth.2013.07.030
33. Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci U S A. (2008) 105:716–21. doi: 10.1073/pnas.0706729105
34. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. (2007) 129:1311–23. doi: 10.1016/j.cell.2007.05.022
35. Zhang K, Shi ZM, Chang YN, Hu ZM, Qi HX, Hong W. The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene. (2014) 547:1–9. doi: 10.1016/j.gene.2014.06.043
36. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. (2011) 146:353–8. doi: 10.1016/j.cell.2011.07.014
37. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. (2014) 505:344–52. doi: 10.1038/nature12986
38. Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. (2013) 495:384–8. doi: 10.1038/nature11993
39. Brockdorff N, Ashworth A, Kay GF, McCabe VM, Norris DP, Cooper PJ, et al. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell. (1992) 71:515–26. doi: 10.1016/0092-8674(92)90519-I
40. Bartolomei MS, Zemel S, Tilghman SM. Parental imprinting of the mouse H19 gene. Nature. (1991) 351:153. doi: 10.1038/351153a0
41. Li DY, Busch A, Jin H, Chernogubova E, Pelisek J, Karlsson J, et al. H19 induces abdominal aortic aneurysm development and progression. Circulation. (2018) 138:1551–68. doi: 10.1161/CIRCULATIONAHA.117.032184
42. Gantier MP, Williams BR. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. (2007) 18:363–71. doi: 10.1016/j.cytogfr.2007.06.016
43. Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. (2017) 35:222–9. doi: 10.1038/nbt.3802
44. Juliano RL, Ming X, Carver K, Laing B. Cellular uptake and intracellular trafficking of oligonucleotides: implications for oligonucleotide pharmacology. Nucleic Acid Ther. (2014) 24:101–13. doi: 10.1089/nat.2013.0463
45. Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. (2014) 24:374–87. doi: 10.1089/nat.2014.0506
46. Stec WJ, Zon G, Egan W. Automated solid-phase synthesis, separation, and stereochemistry of phosphorothioate analogs of oligodeoxyribonucleotides. J Am Chem Soc. (1984) 106:6077–9. doi: 10.1021/ja00332a054
47. Larrouy B, Blonski C, Boiziau C, Stuer M, Moreau S, Shire D, et al. RNase H-mediated inhibition of translation by antisense oligodeoxyribonucleotides: use of backbone modification to improve specificity. Gene. (1992) 121:189–94. doi: 10.1016/0378-1119(92)90121-5
48. Kurreck J, Wyszko E, Gillen C, Erdmann VA. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. (2002) 30:1911–8. doi: 10.1093/nar/30.9.1911
49. Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. (2020) 19:673–94. doi: 10.1038/s41573-020-0075-7
50. Shen LX, Kandimalla ER, Agrawal S. Impact of mixed-backbone oligonucleotides on target binding affinity and target cleaving specificity and selectivity by Escherichia coli RNase H. Bioorg Med Chem. (1998) 6:1695–705. doi: 10.1016/S0968-0896(98)00131-X
51. Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. (2009) 78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540
52. Crooke ST, Wang S, Vickers TA, Shen W, Liang XH. Cellular uptake and trafficking of antisense oligonucleotides. Nat Biotechnol. (2017) 35:230–7. doi: 10.1038/nbt.3779
53. Liang XH, Shen W, Sun H, Kinberger GA, Prakash TP, Nichols JG, et al. Hsp90 protein interacts with phosphorothioate oligonucleotides containing hydrophobic 2'-modifications and enhances antisense activity. Nucleic Acids Res. (2016) 44:3892–907. doi: 10.1093/nar/gkw144
54. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, . editors. Essentials of Glycobiology. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press (2009).
55. Prakash TP, Graham MJ, Yu J, Carty R, Low A, Chappell A, et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. (2014) 42:8796–807. doi: 10.1093/nar/gku531
56. Koshkin AA, Singh SK, Nielsen P, Rajwanshi VK, Kumar R, Meldgaard M, et al. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron. (1998) 54:3607–30. doi: 10.1016/S0040-4020(98)00094-5
57. Singh SK, Nielsen P, Koshkin AA, Wengel J. LNA (locked nucleic acids): synthesis and high-affinity nucleic acid recognition. Chem Commun. (1998) 1998:455–6. doi: 10.1039/a708608c
58. Monia BP, Lesnik EA, Gonzalez C, Lima WF, McGee D, Guinosso CJ, et al. Evaluation of 2'-modified oligonucleotides containing 2'-deoxy gaps as antisense inhibitors of gene expression. J Biol Chem. (1993) 268:14514–22. doi: 10.1016/S0021-9258(19)85268-7
59. Crooke RM, Graham MJ, Lemonidis KM, Whipple CP, Koo S, Perera RJ. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J Lipid Res. (2005) 46:872–84. doi: 10.1194/jlr.M400492-JLR200
60. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with 'antagomirs'. Nature. (2005) 438:685–9. doi: 10.1038/nature04303
61. Krutzfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, et al. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. (2007) 35:2885–92. doi: 10.1093/nar/gkm024
62. Ohrt T, Muetze J, Svoboda P, Schwille P. Intracellular localization and routing of miRNA and RNAi pathway components. Curr Top Med Chem. (2012) 12:79–88. doi: 10.2174/156802612798919132
63. Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. (1998) 17:7505–13. doi: 10.1093/emboj/17.24.7505
64. Rettig GR, Behlke MA. Progress toward in vivo use of siRNAs-II. Mol Ther J Am Soc Gene Ther. (2012) 20:483–512. doi: 10.1038/mt.2011.263
65. Jahns H, Roos M, Imig J, Baumann F, Wang Y, Gilmour R, et al. Stereochemical bias introduced during RNA synthesis modulates the activity of phosphorothioate siRNAs. Nat Commun. (2015) 6:6317. doi: 10.1038/ncomms7317
66. Sun X, Rogoff HA, Li CJ. Asymmetric RNA duplexes mediate RNA interference in mammalian cells. Nat Biotechnol. (2008) 26:1379–82. doi: 10.1038/nbt.1512
67. Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N Engl J Med. (2016) 376:41–51. doi: 10.1056/NEJMoa1609243
68. Meade BR, Gogoi K, Hamil AS, Palm-Apergi C, van den Berg A, Hagopian JC, et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat Biotechnol. (2014) 32:1256–61. doi: 10.1038/nbt.3078
69. Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, et al. RNAi-mediated gene silencing in non-human primates. Nature. (2006) 441:111–4. doi: 10.1038/nature04688
70. Semple SC, Akinc A, Chen J, Sandhu AP, Mui BL, Cho CK, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol. (2010) 28:172–6. doi: 10.1038/nbt.1602
71. Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. (2013) 12:967–77. doi: 10.1038/nmat3765
72. Nair JK, Willoughby JL, Chan A, Charisse K, Alam MR, Wang Q, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. (2014) 136:16958–61. doi: 10.1021/ja505986a
73. Song E, Zhu P, Lee S-K, Chowdhury D, Kussman S, Dykxhoorn DM, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. (2005) 23:709. doi: 10.1038/nbt1101
74. Cuellar TL, Barnes D, Nelson C, Tanguay J, Yu S-F, Wen X, et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. (2014) 43:1189–203. doi: 10.1093/nar/gku1362
75. Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. (2010) 327:167–70. doi: 10.1126/science.1179555
76. Xiao-Jie L, Hui-Ying X, Zun-Ping K, Jin-Lian C, Li-Juan J. CRISPR-Cas9: a new and promising player in gene therapy. J Med Genet. (2015) 52:289–96. doi: 10.1136/jmedgenet-2014-102968
77. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. (2012) 337:816–21. doi: 10.1126/science.1225829
78. Gori JL, Hsu PD, Maeder ML, Shen S, Welstead GG, Bumcrot D. Delivery and specificity of CRISPR-Cas9 genome editing technologies for human gene therapy. Hum Gene Ther. (2015) 26:443–51. doi: 10.1089/hum.2015.074
79. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. (2013) 31:822. doi: 10.1038/nbt.2623
80. Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol. (2014) 32:677. doi: 10.1038/nbt.2916
81. Gagnon JA, Valen E, Thyme SB, Huang P, Akhmetova L, Pauli A, et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One. (2014) 9:e98186. doi: 10.1371/journal.pone.0098186
82. Zhang X-H, Tee LY, Wang X-G, Huang Q-S, Yang S-H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids. (2015) 4:e264. doi: 10.1038/mtna.2015.37
83. Dow LE, Fisher J, O'Rourke KP, Muley A, Kastenhuber ER, Livshits G, et al. Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol. (2015) 33:390–4. doi: 10.1038/nbt.3155
84. Liu KI, Ramli MN, Woo CW, Wang Y, Zhao T, Zhang X, et al. A chemical-inducible CRISPR-Cas9 system for rapid control of genome editing. Nat Chem Biol. (2016) 12:980–7. doi: 10.1038/nchembio.2179
85. Oakes BL, Nadler DC, Flamholz A, Fellmann C, Staahl BT, Doudna JA, et al. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat Biotechnol. (2016) 34:646–51. doi: 10.1038/nbt.3528
86. Nihongaki Y, Yamamoto S, Kawano F, Suzuki H, Sato M. CRISPR-Cas9-based photoactivatable transcription system. Chem Biol. (2015) 22:169–74. doi: 10.1016/j.chembiol.2014.12.011
87. Caplan AL, Parent B, Shen M, Plunkett C. No time to waste–the ethical challenges created by CRISPR: CRISPR/Cas, being an efficient, simple, and cheap technology to edit the genome of any organism, raises many ethical and regulatory issues beyond the use to manipulate human germ line cells. EMBO Rep. (2015) 16:1421–6. doi: 10.15252/embr.201541337
88. Naldini L. Gene therapy returns to centre stage. Nature. (2015) 526:351. doi: 10.1038/nature15818
89. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. (2014) 15:541–55. doi: 10.1038/nrg3763
90. Escors D, Breckpot K. Lentiviral vectors in gene therapy: their current status and future potential. Arch Immunol Ther Exp. (2010) 58:107–19. doi: 10.1007/s00005-010-0063-4
91. Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. (2003) 348:255–6. doi: 10.1056/NEJM200301163480314
92. Stocking C, Bergholz U, Friel J, Klingler K, Wagener T, Starke C, et al. Distinct classes of factor-independent mutants can be isolated after retroviral mutagenesis of a human myeloid stem cell line. Growth Factors (Chur, Switzerland). (1993) 8:197–209. doi: 10.3109/08977199309011023
93. Miller DG, Adam MA, Miller AD. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol. (1990) 10:4239–42. doi: 10.1128/MCB.10.8.4239
94. Vigna E, Naldini L. Lentiviral vectors: excellent tools for experimental gene transfer and promising candidates for gene therapy. J Gene Med. (2000) 2:308–16. doi: 10.1002/1521-2254(200009/10)2:5<308::AID-JGM131>3.0.CO;2-3
95. Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science (New York, NY). (1996) 272:263–7. doi: 10.1126/science.272.5259.263
96. Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE, et al. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat Med. (2007) 13:419. doi: 10.1038/nm1549
97. Joglekar AV, Sandoval S. Pseudotyped lentiviral vectors: one vector, many guises. Hum Gene Ther Methods. (2017) 28:291–301. doi: 10.1089/hgtb.2017.084
98. MacKenzie TC, Kobinger GP, Kootstra NA, Radu A, Sena-Esteves M, Bouchard S, et al. Efficient transduction of liver and muscle after in utero injection of lentiviral vectors with different pseudotypes. Mol Ther J Am Soc Gene Ther. (2002) 6:349–58. doi: 10.1006/mthe.2002.0681
99. Lapteva L, Purohit-Sheth T, Serabian M, Puri RK. Clinical development of gene therapies: the first three decades and counting. Mol Ther Methods Clin Dev. (2020) 19:387–97. doi: 10.1016/j.omtm.2020.10.004
100. Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. (2001) 7:33. doi: 10.1038/83324
101. Ricobaraza A, Gonzalez-Aparicio M, Mora-Jimenez L, Lumbreras S, Hernandez-Alcoceba R. High-capacity adenoviral vectors: expanding the scope of gene therapy. Int J Mol Sci. (2020) 21:3643. doi: 10.3390/ijms21103643
102. Sun CP, Wu TH, Chen CC, Wu PY, Shih YM, Tsuneyama K, et al. Studies of efficacy and liver toxicity related to adeno-associated virus-mediated RNA interference. Hum Gene Ther. (2013) 24:739–50. doi: 10.1089/hum.2012.239
103. Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. (2003) 80:148–58. doi: 10.1016/j.ymgme.2003.08.016
104. Vannucci L, Lai M, Chiuppesi F, Ceccherini-Nelli L, Pistello M. Viral vectors: a look back and ahead on gene transfer technology. New Microbiol. (2013) 36:1–22.
105. Sun Y, Lv X, Ding P, Wang L, Sun Y, Li S, et al. Exploring the functions of polymers in adenovirus-mediated gene delivery: Evading immune response and redirecting tropism. Acta Biomater. (2019) 97:93–104. doi: 10.1016/j.actbio.2019.06.059
106. Stamatatos L, Leventis R, Zuckermann MJ, Silvius JR. Interactions of cationic lipid vesicles with negatively charged phospholipid vesicles and biological membranes. Biochemistry. (1988) 27:3917–25. doi: 10.1021/bi00411a005
107. Solans C, Izquierdo P, Nolla J, Azemar N, Garcia-Celma MJ. Nano-emulsions. Curr Opin Colloid Interf Sci. (2005) 10:102–10. doi: 10.1016/j.cocis.2005.06.004
108. Souto EB, Nayak AP, Murthy RS. Lipid nanoemulsions for anti-cancer drug therapy. Die Pharm. (2011) 66:473–8. doi: 10.1002/chin.201139266
109. Bae KH, Lee JY, Lee SH, Park TG, Nam YS. Optically traceable solid lipid nanoparticles loaded with siRNA and paclitaxel for synergistic chemotherapy with in situ imaging. Adv Healthcare Mater. (2013) 2:576–84. doi: 10.1002/adhm.201200338
110. Szoka F, Jr., Papahadjopoulos D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc Natl Acad Sci U S A. (1978) 75:4194–8. doi: 10.1073/pnas.75.9.4194
111. Allen TM, Hansen C, Martin F, Redemann C, Yau-Young A. Liposomes containing synthetic lipid derivatives of poly(ethylene glycol) show prolonged circulation half-lives in vivo. Biochim Biophys Acta Biomembr. (1991) 1066:29–36. doi: 10.1016/0005-2736(91)90246-5
112. Allen TM, Cullis PR. Liposomal drug delivery systems: From concept to clinical applications. Adv Drug Deliv Rev. (2013) 65:36–48. doi: 10.1016/j.addr.2012.09.037
113. Xue HY, Liu S, Wong HL. Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomedicine (London, England). (2014) 9:295–312. doi: 10.2217/nnm.13.204
114. Zhi D, Zhang S, Wang B, Zhao Y, Yang B, Yu S. Transfection efficiency of cationic lipids with different hydrophobic domains in gene delivery. Bioconjug Chem. (2010) 21:563–77. doi: 10.1021/bc900393r
115. Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. (2006) 114:100–9. doi: 10.1016/j.jconrel.2006.04.014
116. Mintzer MA, Simanek EE. Nonviral vectors for gene delivery. Chem Rev. (2009) 109:259–302. doi: 10.1021/cr800409e
117. Simberg D, Weisman S, Talmon Y, Barenholz Y. DOTAP (and other cationic lipids): chemistry, biophysics, and transfection. Crit Rev Ther Drug Carrier Syst. (2004) 21:257–317. doi: 10.1615/CritRevTherDrugCarrierSyst.v21.i4.10
118. Leventis R, Silvius JR. Interactions of mammalian cells with lipid dispersions containing novel metabolizable cationic amphiphiles. Biochim Biophys Acta Biomembranes. (1990) 1023:124–32. doi: 10.1016/0005-2736(90)90017-I
119. Xue HY, Guo P, Wen WC, Wong HL. Lipid-Based Nanocarriers for RNA delivery. Curr Pharm Des. (2015) 21:3140–7. doi: 10.2174/1381612821666150531164540
120. Li W, Szoka FC Jr. Lipid-based nanoparticles for nucleic acid delivery. Pharm Res. (2007) 24:438–49. doi: 10.1007/s11095-006-9180-5
121. Zhang Y, Liu D, Chen X, Li J, Li L, Bian Z, et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol Cell. (2010) 39:133–44. doi: 10.1016/j.molcel.2010.06.010
122. Lee JK, Park SR, Jung BK, Jeon YK, Lee YS, Kim MK, et al. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS One. (2013) 8:e84256. doi: 10.1371/journal.pone.0084256
123. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL, et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A. (2010) 107:6328–33. doi: 10.1073/pnas.0914843107
124. Iguchi H, Kosaka N, Ochiya T. Secretory microRNAs as a versatile communication tool. Commun Integr Biol. (2010) 3:478–81. doi: 10.4161/cib.3.5.12693
125. Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. (2010) 285:17442–52. doi: 10.1074/jbc.M110.107821
126. Jiang L, Vader P, Schiffelers RM. Extracellular vesicles for nucleic acid delivery: progress and prospects for safe RNA-based gene therapy. Gene Ther. (2017) 24:157–66. doi: 10.1038/gt.2017.8
127. Batagov AO, Kuznetsov VA, Kurochkin IV. Identification of nucleotide patterns enriched in secreted RNAs as putative cis-acting elements targeting them to exosome nano-vesicles. BMC Genom. (2011) 12(Suppl 3):S18. doi: 10.1186/1471-2164-12-S3-S18
128. Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol. (2009) 11:1143. doi: 10.1038/ncb1929
129. Tetta C, Ghigo E, Silengo L, Deregibus MC, Camussi G. Extracellular vesicles as an emerging mechanism of cell-to-cell communication. Endocrine. (2013) 44:11–9. doi: 10.1007/s12020-012-9839-0
130. Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. (2004) 5:11. doi: 10.1186/gb-2004-5-3-r13
131. Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. (2005) 433:769–73. doi: 10.1038/nature03315
132. Maegdefessel L, Azuma J, Toh R, Deng A, Merk DR, Raiesdana A, et al. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci Transl Med. (2012) 4:122ra22. doi: 10.1126/scitranslmed.3003441
133. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. (2009) 10:704. doi: 10.1038/nrg2634
134. Maegdefessel L, Azuma J, Toh R, Merk DR, Deng A, Chin JT, et al. Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J Clin Invest. (2012) 122:497–506. doi: 10.1172/JCI61598
135. Bellera N, Barba I, Rodriguez-Sinovas A, Ferret E, Asin MA, Gonzalez-Alujas MT, et al. Single intracoronary injection of encapsulated antagomir-92a promotes angiogenesis and prevents adverse infarct remodeling. J Am Heart Assoc. (2014) 3:e000946. doi: 10.1161/JAHA.114.000946
136. Braasch DA, Paroo Z, Constantinescu A, Ren G, Oz OK, Mason RP, et al. Biodistribution of phosphodiester and phosphorothioate siRNA. Bioorg Med Chem Lett. (2004) 14:1139–43. doi: 10.1016/j.bmcl.2003.12.074
137. Huang Y, Hong J, Zheng S, Ding Y, Guo S, Zhang H, et al. Elimination pathways of systemically delivered siRNA. Mol Ther. (2011) 19:381–5. doi: 10.1038/mt.2010.266
138. Lorenzer C, Dirin M, Winkler AM, Baumann V, Winkler J. Going beyond the liver: progress and challenges of targeted delivery of siRNA therapeutics. J Control Release. (2015) 203:1–15. doi: 10.1016/j.jconrel.2015.02.003
139. Kheirolomoom A, Kim CW, Seo JW, Kumar S, Son DJ, Gagnon MK, et al. Multifunctional nanoparticles facilitate molecular targeting and miRNA delivery to inhibit atherosclerosis in ApoE(-/-) mice. ACS nano. (2015) 9:8885–97. doi: 10.1021/acsnano.5b02611
140. Lucas T, Dimmeler S. RNA therapeutics for treatment of cardiovascular diseases: promises and challenges. Circ Res. (2016) 119:794–7. doi: 10.1161/CIRCRESAHA.116.308730
141. Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, Horrevoets AJ, et al. MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res. (2011) 109:1115–9. doi: 10.1161/CIRCRESAHA.111.255737
142. Okamura H, Emrich F, Trojan J, Chiu P, Dalal AR, Arakawa M, et al. Long-term miR-29b suppression reduces aneurysm formation in a Marfan mouse model. Physiol Rep. (2017) 5:e13257. doi: 10.14814/phy2.13257
143. Di Gregoli K, Mohamad Anuar NN, Bianco R, White SJ, Newby AC, George SJ, et al. MicroRNA-181b controls atherosclerosis and aneurysms through regulation of TIMP-3 and elastin. Circ Res. (2017) 120:49–65. doi: 10.1161/CIRCRESAHA.116.309321
144. Li L, Ma W, Pan S, Li Y, Wang H, Wang B, et al. MiR-126a-5p limits the formation of abdominal aortic aneurysm in mice and decreases ADAMTS-4 expression. J Cell Mol Med. (2020) 24:7896–906. doi: 10.1111/jcmm.15422
145. Maegdefessel L, Spin JM, Raaz U, Eken SM, Toh R, Azuma J, et al. miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat Commun. (2014) 5:5214. doi: 10.1038/ncomms6214
146. Knappich C, Spin JM, Eckstein HH, Tsao PS, Maegdefessel L. Involvement of myeloid cells and noncoding RNA in abdominal aortic aneurysm disease. Antioxid Redox Signal. (2020) 33:602–20. doi: 10.1089/ars.2020.8035
147. Zampetaki A, Attia R, Mayr U, Gomes RS, Phinikaridou A, Yin X, et al. Role of miR-195 in aortic aneurysmal disease. Circ Res. (2014) 115:857–66. doi: 10.1161/CIRCRESAHA.115.304361
148. Loyer X, Potteaux S, Vion AC, Guerin CL, Boulkroun S, Rautou PE, et al. Inhibition of microRNA-92a prevents endothelial dysfunction and atherosclerosis in mice. Circ Res. (2014) 114:434–43. doi: 10.1161/CIRCRESAHA.114.302213
149. Iaconetti C, Polimeni A, Sorrentino S, Sabatino J, Pironti G, Esposito G, et al. Inhibition of miR-92a increases endothelial proliferation and migration in vitro as well as reduces neointimal proliferation in vivo after vascular injury. Basic Res Cardiol. (2012) 107:296. doi: 10.1007/s00395-012-0296-y
150. Dhital S, Vyavahare NR. Nanoparticle-based targeted delivery of pentagalloyl glucose reverses elastase-induced abdominal aortic aneurysm and restores aorta to the healthy state in mice. PLoS One. (2020) 15:e0227165. doi: 10.1371/journal.pone.0227165
151. Liu J, Kuwabara A, Kamio Y, Hu S, Park J, Hashimoto T, et al. Human mesenchymal stem cell-derived microvesicles prevent the rupture of intracranial aneurysm in part by suppression of mast cell activation via a PGE2-dependent mechanism. Stem Cells (Dayton, Ohio). (2016) 34:2943–55. doi: 10.1002/stem.2448
152. Wu J, Wang J, Li X, Liu X, Yu X, Tian Y. MicroRNA-145 Mediates the formation of angiotensin II-induced murine abdominal aortic aneurysm. Heart Lung Circ. (2017) 26:619–26. doi: 10.1016/j.hlc.2016.10.009
153. de Waard V, Bot I, de Jager SC, Talib S, Egashira K, de Vries MR, et al. Systemic MCP1/CCR2 blockade and leukocyte specific MCP1/CCR2 inhibition affect aortic aneurysm formation differently. Atherosclerosis. (2010) 211:84–9. doi: 10.1016/j.atherosclerosis.2010.01.042
154. Wang D, Deuse T, Stubbendorff M, Chernogubova E, Erben RG, Eken SM, et al. Local microRNA modulation using a novel anti-miR-21-eluting stent effectively prevents experimental in-stent restenosis. Arteriosc Thromb Vasc Biol. (2015) 35:1945–53. doi: 10.1161/ATVBAHA.115.305597
155. Hinkel R, Penzkofer D, Zuhlke S, Fischer A, Husada W, Xu QF, et al. Inhibition of microRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation. (2013) 128:1066–75. doi: 10.1161/CIRCULATIONAHA.113.001904
156. Rothemund PW. Folding DNA to create nanoscale shapes and patterns. Nature. (2006) 440:297–302. doi: 10.1038/nature04586
157. Zheng J, Birktoft JJ, Chen Y, Wang T, Sha R, Constantinou PE, et al. From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nature. (2009) 461:74–7. doi: 10.1038/nature08274
158. Selnihhin D, Andersen ES. Computer-aided design of DNA origami structures. Methods Mol Biol (Clifton, NJ). (2015) 1244:23–44. doi: 10.1007/978-1-4939-1878-2_2
159. Ke Y, Ong LL, Shih WM, Yin P. Three-dimensional structures self-assembled from DNA bricks. Science (New York, NY). (2012) 338:1177. doi: 10.1126/science.1227268
160. Ong LL, Hanikel N, Yaghi OK, Grun C, Strauss MT, Bron P, et al. Programmable self-assembly of three-dimensional nanostructures from 10,000 unique components. Nature. (2017) 552:72. doi: 10.1038/nature24648
161. Li S, Jiang Q, Liu S, Zhang Y, Tian Y, Song C, et al. A DNA nanorobot functions as a cancer therapeutic in response to a molecular trigger in vivo. Nat Biotechnol. (2018) 36:258–64. doi: 10.1038/nbt.4071
162. Hong F, Zhang F, Liu Y, Yan H. DNA origami: scaffolds for creating higher order structures. Chem Rev. (2017) 117:12584–640. doi: 10.1021/acs.chemrev.6b00825
163. Wang P, Ko SH, Tian C, Hao C, Mao C. RNA-DNA hybrid origami: folding of a long RNA single strand into complex nanostructures using short DNA helper strands. Chem Commun. (2013) 49:5462–4. doi: 10.1039/c3cc41707g
164. Endo M, Yamamoto S, Tatsumi K, Emura T, Hidaka K, Sugiyama H. RNA-templated DNA origami structures. Chem Commun. (2013) 49:2879–81. doi: 10.1039/c3cc38804b
Keywords: innovation, RNA, therapeutics, cardiovascular medicine, epigenetics, non-coding RNAs, abdominal aortic aneurysm
Citation: Schellinger IN, Dannert AR, Mattern K, Raaz U and Tsao PS (2021) Unresolved Issues in RNA Therapeutics in Vascular Diseases With a Focus on Aneurysm Disease. Front. Cardiovasc. Med. 8:571076. doi: 10.3389/fcvm.2021.571076
Received: 09 June 2020; Accepted: 23 February 2021;
Published: 15 April 2021.
Edited by:
Claudio Santoro, University of Eastern Piedmont, ItalyReviewed by:
Maria Prat, University of Eastern Piedmont, ItalyCopyright © 2021 Schellinger, Dannert, Mattern, Raaz and Tsao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel N. Schellinger, Isabel.schellinger@medizin.uni-leipzig.de
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.