 Huilin Hu1†
Huilin Hu1† Yunqing Chen
Yunqing Chen Liang Shen
Liang Shen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 22 July 2021
Sec. Cardiovascular Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.710963
This article is part of the Research Topic Highlights in Cardiovascular Therapeutics: 2021 View all 8 articles
Ferroptosis is an iron-dependent cell death, which is characterized by iron overload and lipid peroxidation. Ferroptosis is distinct from apoptosis, necroptosis, autophagy, and other types of cell death in morphology and function. Ferroptosis is regulated by a variety of factors and controlled by several mechanisms, including mitochondrial activity and metabolism of iron, lipid, and amino acids. Accumulating evidence shows that ferroptosis is closely related to a majority of cardiovascular diseases (CVDs), including cardiomyopathy, myocardial infarction, ischemia/reperfusion injury, heart failure, and atherosclerosis. This review summarizes the current status of ferroptosis and discusses ferroptosis as a potential therapeutic target for CVDs.
Cell death plays a vital role throughout the whole life stage. There are two main types of cell death, accidental cell death (ACD) and regulated cell death (RCD) (1). Apoptosis was the first discovered manner of RCD (2). Since then, a growing number of RCD forms were unveiled, including mitochondrial permeability transition (MPT)-derived necrosis, necroptosis, pyroptosis, and ferroptosis (3–7). Ferroptosis is a new type of RCD proposed in 2012 (6), which is induced by the accumulation of iron-triggered lipid peroxidation. Ferroptosis is apparently distinct from apoptosis, necroptosis, and autophagy in several aspects (Table 1). Over the past few years, the research field of ferroptosis has been extraordinary expanded; various mechanisms and regulators were identified in the process of ferroptosis. Ferroptosis is also implicated in an amount of diseases, such as cancer, neurodegenerative diseases, and ischemia–reperfusion injury (8–12).
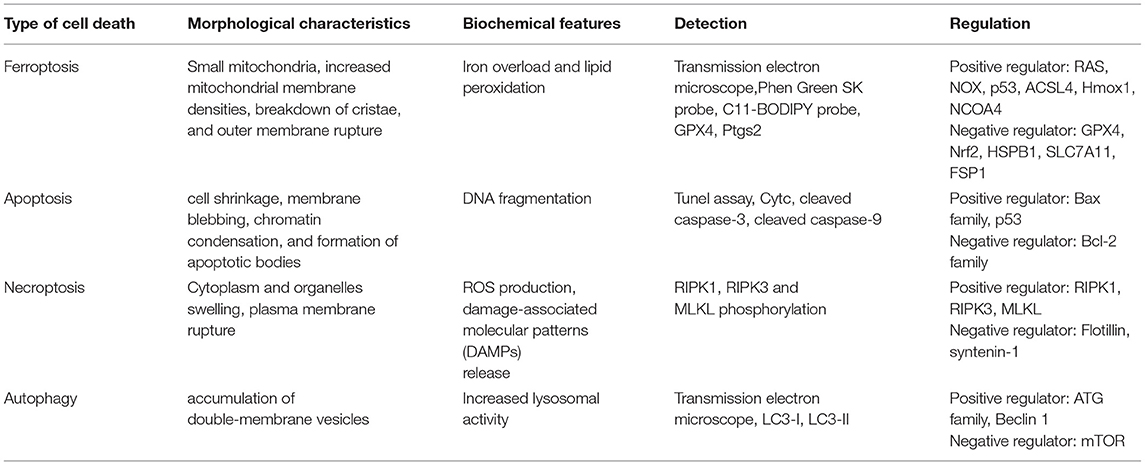
Table 1. Characteristics of different types of cell death.
Recently, ferroptosis has been discovered to contribute to the occurrence and progression of a variety of cardiovascular diseases (CVDs), such as cardiomyopathy, myocardial infarction (MI), ischemia/reperfusion injury (IRI), heart failure (HF), and atherosclerosis (AS). In this review, we summarize the regulatory mechanisms of ferroptosis and discuss the impact of ferroptosis on CVDs.
Ferroptosis is an distinctive type of non-apoptotic cell death defined by Dixon in 2012 (6), it is an iron-dependent form of RCD, which is characterized by imbalance of iron homeostasis and accumulation of lipid reactive oxygen species (ROS). Actually, ferroptotic cell death can be traced back to the 1950s. Eagle found that deficiency of cystine inhibits cell growth and causes cell death with a distinct morphology; endogenous synthesis of cysteine prevents cells from such cell death (13–15). Following studies further confirmed the importance of cystine deprivation in the process of cell death (16, 17). In 2003, Dolma et al. (18) discovered that a new compound named erastin applied a selectively lethal effect on human foreskin fibroblasts (BJeLR cells) with the expression of the engineered mutant RAS oncogene, but the type of cell death was different from what had been noticed before, with no nuclear morphological changes and DNA breakup. Subsequently, studies further demonstrated that the cell death manner mentioned above could be suppressed by iron chelators and initiated by Ras-selective lethal small molecule (RSL) (19). Then, in 2012, Dixon et al. finally proposed that the non-apoptotic form of cell death was named ferroptosis.
Unlike cell shrinkage, chromatin condensation and apoptotic body formation presented in apoptosis, cytoplasm, and organelle swelling and cell membrane rupture seen in necrosis, classical double-membrane enclosed vesicles observed in autophagy, pyroptosome assembly, and membrane pore formation detected in pyroptosis and ferroptosis are morphologically, biochemically, and genetically different from these types of RCD. The morphological characteristics of ferroptosis include the shrunken mitochondria with increased mitochondrial membrane density as well as degeneration and breakdown of mitochondrial crista, while the morphology of the nucleus remains unchanged. Ferroptosis can be induced by many chemical compounds and drugs and regulated by a variety of proteins and genes (Table 2). The ferroptosis process also involve multiple metabolic pathways, including iron metabolism, and metabolism of amino acids and lipids (Figure 1).
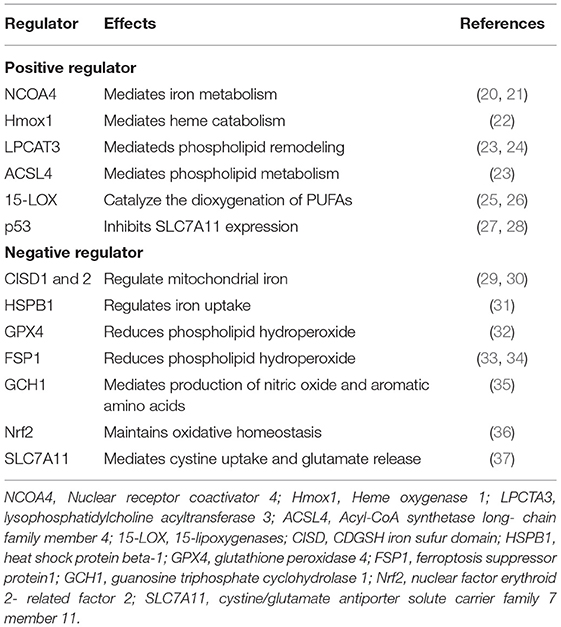
Table 2. Factors involved in ferroptosis regulation.
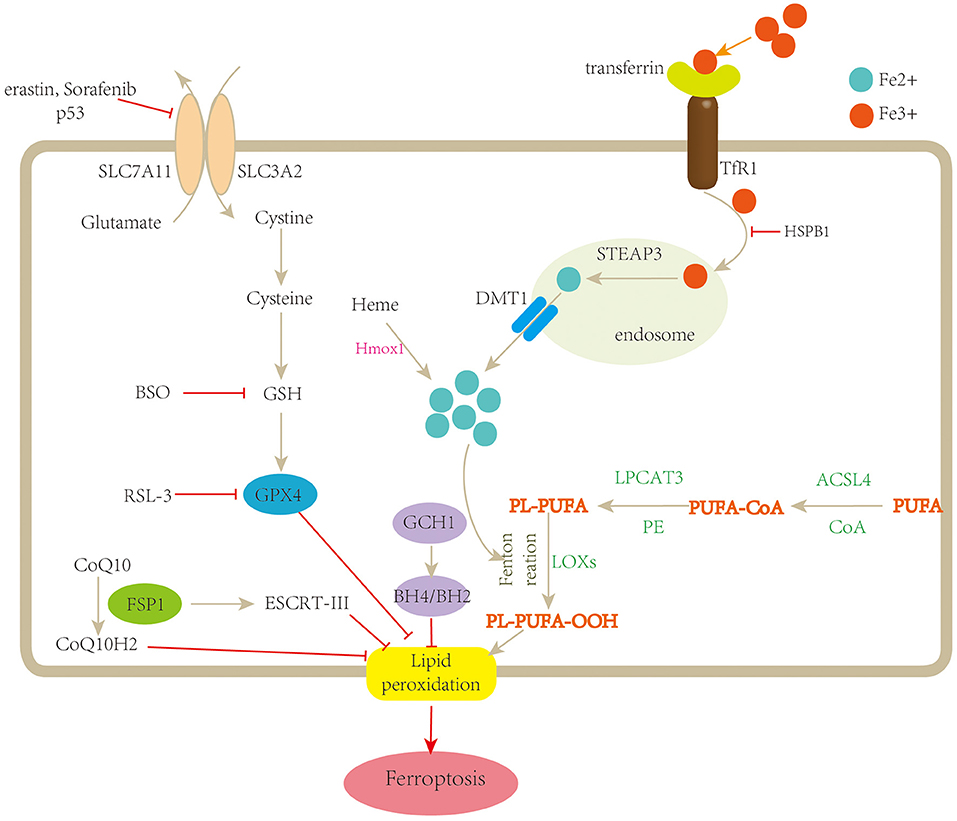
Figure 1. Schematic description of the mechanisms of ferroptosis. Ferroptosis is a type of regulated cell death characterized by iron overload and lipid peroxidation. Fe3+ is transferred into the cell by TfR1, then converted to Fe2+ in the endosome, and released from the endosome by divalent metal transporter 1 (DMT1). Fenton reaction promotes lipid peroxidation by activating lipoxygenases (LOXs). Cystine is uptaken by system xc− for the synthesis of GSH, which further enhances the anti-lipid peroxidation activity of GPX4. In addition, FSP1-CoQ10 and GCH1-BH4/BH2 are two parallel GPX4-independent pathways in the suppression of ferroptosis. TfR1, transferrin receptor 1; STEAP3, STEAP family member 3; Hmox1, heme oxygenase-1; BSO, buthionine sulfoximine; PUFA, polyunsaturated fatty acid; CoA, coenzyme A; PL, lysophosphatide; ACSL4, acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; LOOH, polyunsaturated fatty acid hydroperoxides; GSH, glutathione; SLC7A11, solute carrier family 7 member 11; SLC3A2, solute carrier family 3 member 2; FSP1, ferroptosis suppressor protein1; ESCRT-III, endosomal sorting complex required for transport-III; GCH-1, guanosine triphosphate cyclohydrolase 1; BH4/BH2, tetrahydrobiopterin/dihydrobiopterin.
Iron homeostasis is necessary during various biological processes and essential for cell viability. Both iron overload and deficiency cause pathological conditions. Ferroptosis is a type of RCD mediated by excessive iron; iron excess causes lipid peroxidation and triggers cell death.
Although the exact mechanisms between iron metabolism and ferroptosis remain obscure, there is no doubt that iron metabolism plays a key role during ferroptosis. Erastin-induced death is dependent on intracellular iron, but not on other metal ions; co-treatment with the iron chelator deferoxamine suppresses ROS accumulation and ferroptosis (6).
Fe3+ is transferred into the cell by transferrin receptor 1 (TfR1), then converted to Fe2+ in the endosome by six-transmembrane epithelial antigens of the prostate 3 (STEAP3) and released from the endosome by divalent metal transporter 1 (DMT1). Fe2+ is stored in the unstable iron pool (LIP) and ferritin and exported by ferroportin-1 (FPN1) (38). Once the balance between iron absorption, utilization, and recycling was disrupted, free iron ions might accumulate and catalyze the Fenton reaction in the presence of hydrogen peroxide (H2O2), leading to the formation of lipid peroxides and eventually ferroptosis (39). For instance, silencing the gene encoding TfR 1 can prevent ferroptosis induced by erastin, while depletion of FPN1 increases the sensitivity of cells to ferroptosis (40).
Recent studies have found multiple regulators of iron metabolism involved in the process of ferroptosis. CDGSH iron–sulfur domains 1 and 2 (CISD1 and 2) are both mitochondrial iron regulators; inhibition of CISD1 increases iron-mediated mitochondrial lipid peroxidation, promoting erastin-induced ferroptosis (29), deficiency of CISD2 increases levels of mitochondrial ferrous iron and lipid ROS, facilitating sulfasalazine-induced ferroptosis as well (30). Iron is a cofactor for enzymes involved in many metabolic processes; excess iron could prompt the overproduction of mitochondrial ROS and ferroptosis. Ferritinophagy is a critical mechanism to maintain iron homeostasis in cells. It could induce ferroptosis by promoting labile iron overload and lipid peroxidation (41). Impaired ferritinophagy is related to the resistance of ferroptosis in senescent cells (42); during renal IRI, cold-inducible RNA-binding protein (CIRIB) promotes ferroptosis via activating ferritinophagy (43). Moreover, in cancer cells, ferritinophagy is required for the initiation of ferroptosis through bromodomain protein BRD4 inhibitor (44), and it is also involved in sepsis-induced cardiac injury and zinc oxide nanoparticle (ZnONP)-induced vascular endothelial cytotoxicity (45, 46). Nuclear receptor coactivator 4 (NCOA4) is a cargo receptor for the degradation of ferritin. It maintains intracellular iron homeostasis by mediating ferritinophagy as well (20, 21). Silencing of NCOA4 abolishes the accumulation of reactive iron and ROS, and eventual ferroptotic cell death as well, while overexpression of NCOA4 promotes ferroptosis and increases ferritin degradation (47, 48). Heat shock protein beta-1 (HSPB1), a member of small heat shock proteins (HSPs), has the ability to inhibit iron uptake (31). Upregulation of HSPB1 suppresses erastin-induced ferroptosis. HSPB1 phosphorylation protects cells against ferroptosis by reducing iron-dependent production of lipid ROS (49). Hmox1 is an essential enzyme in heme catabolism. Hmox1 knockout mice present iron accumulation in the liver and kidneys, indicating that Hmox1 is involved in iron metabolism. The impact of Hmox1 on ferroptosis is controversial. Inhibition of Hmox1 by ZnPP represses doxorubicin-induced ferroptosis in the myocardium (22). However, in renal proximal tubule cells, knockout of Hmox1 enhances erastin- or RSL3-induced ferroptosis (50), suggesting that the role of Hmox1 in ferroptosis might be context dependent. Ferritin, a spherical heteropolymer formed by 24 subunits of heavy (FTH) and light (FTL) chain, plays a crucial role in iron metabolism by storing extracellular iron (51). In Hippo mutant cells, increased expression of ferritin heavy chain protects against ROS accumulation and inhibits ferroptosis, and loss of cardiac ferritin heavy chain facilitates cardiomyocyte death and heart failure via ferroptosis (52). Most recently, ceruloplasmin (CP), lactotranferrin (LTF), and prominin-2 (PROM2), which mediates oxidation of Fe2+ to Fe3+, iron transportation and ferritin exportation, respectively, are all demonstrated to be involved in the process of ferroptosis (53–55). All these studies suggest that iron metabolism plays a crucial role during ferroptosis.
Lipid peroxide accumulation is the main feature of ferroptosis. Accumulation of lipid peroxides degrades into reactive toxic aldehydes or hydroxy fatty acids, such as malondialdehyde (MDA), which induces cell toxicity and initiates cell death cascades. Polyunsaturated fatty acids (PUFAs) are components of the cell membrane. ROS react with PUFAs in lipid membranes and induce lipid peroxidation. PUFAs are the most susceptible lipids to peroxidation during ferroptosis; lipoxygenases and phosphorylase kinase G2 are two key regulators of lipid peroxidation during ferroptosis (56). In contrast to PUFAs, monounsaturated fatty acids (MUFAs) exert anti-ferroptotic effects by inhibiting lipid peroxidation. In ovarian cancer cells, overexpression of stearoyl-CoA desaturase 1 (SCD1), an essential enzyme in biosynthesis of MUFAs, shows resistance to ferroptosis (57); besides, SCD1 expressed by cancer cells leads to fatty acid (FA) desaturation and contributes to the protection of oxidative stress-induced ferroptosis, eventually promoting tumor recurrence (58).
Acyl-CoA synthetase long-chain family member 4 (ACSL4) is an enzyme involved in phospholipid metabolism, which plays a key role in the synthesis of long-chain PUFA-CoA. Compared with wild-type cells, ACSL4-deficient cells are accompanied by a significant decrease in AA- and AdA-containing phosphatidylethanolamine (PE), while supplementation with exogenous AA/AdA protects the sensitivity to ferroptosis in ACSL4 knockout cells, indicating that ACSL4 is required for the generation of ferroptosis by accumulating oxidized membrane phospholipids (23). In addition to PE, phosphatidylserine (PS) and phosphatidylinositol (PI) are also involved in ferroptosois (59). Lysophosphatidylcholine acyltransferase 3 (LPCAT3) is closed related to phospholipid remodeling. Downregulation of LPCTA3 also contributes to suppression of ferroptosis, but the function of LPCTA3 in ferroptosis might be less efficient than ACSL4 (23, 24). Lipoxygenases (LOXs) are a family of iron-containing enzymes that catalyze the dioxygenation of PUFAs. However, the role of LOXs in lipid peroxidation and ferroptosis is controversial. The specific inhibitor of 12/15-LOX, but not the inhibitors of 5-LOX or COX, protects GPX4-deficient cells against ferroptosis, suggesting that 12/15-LOX is responsible for ferroptosis (25). Moreover, overexpression of 15-LOX promotes lipid peroxidation, while 15-LOX suppressors, but not COX or CYP450 inhibitors, prevent ferroptosis (26). However, in tert-butyl hydroperoxide (TBH)-induced ferroptosis, p53 upregulates 15-LOX, but not 5-LOX or 12-LOX (60), while in another study, depletion of 12-LOX blocks TBH-induced ferroptosis (27). Therefore, the role of LOXs in ferroptosis needs to be further investigated. In addition to LOXs, other oxygenases, such as NOXs and cytochrome P450 oxidoreductase (POR), are also suggested to be involved in ferroptosis (61–63). Additionally, the HMG-CoA reductase pathway might also participate in the process of ferroptosis. Inhibition of HMG-CoA reductase could induce ferroptosis by inactivating GPX4 (64). Most recently, Chen et al. found that the calcium-independent phospholipase iPLA2β suppresses p53-induced ferroptosis via detoxification of peroxidized lipids. Taken together, the modulation of lipid peroxidation might provide novel therapeutic target for ferroptosis-associated disease.
Mitochondria play a vital role in the regulation of cellular metabolism and signal transduction, and they are the main organelles of iron, lipid, and amino acid metabolism. The contribution of mitochondria to ferroptosis is controversial. In melanoma cell lines, inhibition of complex I (CI) of the mitochondrial respiratory chain promotes the opening of the mitochondrial permeability transition pore (mPTP) and cellular ROS levels, as well as accumulation of lipid peroxidation and ferroptosis (65). In cysteine-deprivation-induced ferroptosis, cysteine deficiency leads to hyperpolarization of mitochondrial membrane potential and accumulation of lipid ROS. Inhibition of mitochondrial tricarboxylic acid (TCA) cycle or electron transfer chain (ETC) alleviates lipid ROS accumulation and ferroptosis (66). Moreover, in DOX-induced cardiomyopathy, ferroptosis is the major form of RCD and is mainly triggered in the mitochondria (67). Furthermore, Jelinek et al. (68) and Neitemeier et al. (69) both found that knockout or inhibition of BID preserved mitochondrial integrity and function, therefore, protecting cells against ferroptosis, suggesting the mitochondria as a promising therapeutic target for preventing ferroptosis. However, Gaschler et al. found that cell deficiency of the mitochondria remains sensitive to ferroptotic inducers. Additionally, both ferrostatin-1 (Fer-1) and iron chelators are able to prevent erastin-induced ferroptosis in cells depleted of mitochondria, suggesting that the mitochondria are not necessary for ferroptosis (70). More recently, Song et al. demonstrated that mitochondrial protein PDK4 suppressed ferroptosis by blocking pyruvate oxidation and fatty acid production in pancreatic ductal carcinoma cells, indicating the importance of mitochondria-dependent lipid metabolism in ferroptotic cell death (71). Therefore, further studies are needed to determine the relationship between mitochondrial activity and ferroptosis.
System xc−/glutathione (GSH)/glutathione peroxidase 4 (GPX4) axis is considered as the main pathway involved in ferroptosis. System xc− is a cystine/glutamate antiporter comprised of two subunits, SLC7A11 and SLC3A2, which enable the exchange of glutamate and cystine across the cell membrane (37). Once uptaken by system xc− and transported into the cell, cystine is reduced to cysteine, which is essential for GSH biosynthesis. GSH is a potent reductant and a cofactor for GPX4. GSH depletion could induce ferroptosis by increasing lipid ROS (72). Inhibiting the activity of system xc− disturbs the combination of GSH and ultimately decreases the activity of GPX4. Moreover, p53 can inhibit system xc− by suppressing the expression of SLC7A11, thus, impacting the activity of GPX4, leading to an accumulation of lipid ROS and ferroptotic cell death (28).
GPX4 is a member of the glutathione peroxidases (GPXs). It has been shown that GPX4 plays an important role in the process of ferroptosis and is regarded as the key regulator (32). Erastin inhibits system xc− and leads to the depletion of GSH, which inhibits the activity of GPX4 and then promotes the formation of ROS and causes ferroptosis, while RSL3 directly inhibits GPX4 without the depletion of GSH (6). Similar to erastin, sorafenib, a multikinase inhibitor approved for the therapy of hepatic carcinoma (73), also induces ferroptosis through the blockage of GSH synthesis (74). In GPX4 knockout mice, Friedmann Angeli et al. demonstrated an important role of the GSH/GPX4 axis in protecting acute renal failure and related ferroptosis (12). Furthermore, they discovered that liproxstatin-1, a potent spiroquinoxalinamine derivative, suppresses ferroptosis in GPX4 deficiency kidney. In lymphocyte T cells, GPX4 depletion also results in membrane lipid peroxide accumulation and ferroptosis (75).
Besides the classical GPX4-dependent ferroptotic pathway, recent studies have uncovered a new regulatory pathway independent of GPX4, including the ferroptosis suppressor protein (FSP1)–CoQ10 pathway, endosomal sorting complex required for transport-III (ESCRT-III)-mediated membrane repair pathway and guanosine triphosphate cyclohydrolase 1-tetrahydrobiopterin (GCH1-BH4) pathway.
FSP1 was first defined as a p53-responsive gene, once named apoptosis-inducing factor mitochondria-associated 2 (AIFM2). Recently, two studies simultaneously reported that FSP1 is an effective ferroptosis suppressor and protects cells from ferroptotic cell death. Meanwhile, the inhibition of ferroptosis by FSP1 is mediated by CoQ10 (33, 34). ESCRT-III belong to the family of ESCRT complex, which is composed of five subcomplexes and plays a context-dependent role in membrane remodeling. Dai et al. demonstrated that classical ferroptosis activators such as erastin and RSL3 increase the accumulation of ESCRT-III subunits, while knockdown of components of ESCRT-III machinery enhances ferroptosis (76). BH4 is a cofactor involved in the production of aromatic amino acids and nitric oxide. GCH1 is the rate-limiting enzyme for BH4 synthesis. Through using CRISPR-mediated whole-genome activation screens, Kraft et al. found that GCH1 is the most prominent gene involved in the suppression of ferroptosis. Upregulation or silence of GCH1 causes cancer cells correspondingly resistant or sensitive to ferroptosis by controlling endogenous production of the antioxidant BH4, suggesting a distinctive mechanism of ferroptosis protection that is independent of GPX4 (35).
Cardiomyopathy is a progressive heart disease with multifactorial pathogenesis and high mortality. Cardiomyopathy is always accompanied by cardiomyocyte death, which contributes to pathological ventricular remodeling and heart failure.
Doxorubicin (DOX) is a second-generation anthracycline chemotherapeutic drug that causes cardiotoxicity, leading to cardiomyopathy named DOX-induced cardiomyopathy (77). Fang et al. (22) found that ferroptosis plays a crucial role in the DOX-induced cardiomyopathy. DOX treatment induced a robust increase in prostaglandin endoperoxide synthase 2 (Ptgs2) mRNA in the heart. Mice pretreated with Fer-1 or dexrazoxane (DXZ, the prodrug for iron chelation) significantly improved cardiac function and reduced DOX-induced cardiac injury mortality. RNA sequencing (RNA-seq) analysis showed that Hmox1 was significantly upregulated, accompanied by activation of the Nrf2, and the Nrf2/Hmox1 pathway causes heme degradation and free iron release in the myocardium. Besides, the authors also determined the effects of MitoTEMPO, a mitochondrially targeted antioxidant, and found that MitoTEMPO suppressed lipid peroxidation and cardiac ferroptosis, which eventually attenuate DOX-induced cardiomyopathy.
Tadokoro et al. (67) also investigated the role of ferroptosis in DOX-induced cardiomyopathy and demonstrated that mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. In this study, the authors found that in DOX-induced cardiomyopathy mice and DOX-cultured cardiomyocytes, GPX4 was downregulated, and lipid peroxides were accumulated especially in the mitochondria. Overexpression of GPX4 or iron chelation targeting Fe2+ in mitochondria prevented ferroptosis in cultured cardiomyocytes. They concluded that ferroptosis is the major form of RCD in DOX-induced cardiomyopathy, and DOX-induced ferroptosis is triggered in the mitochondria.
In another study, Liu et al. also detected ferroptosis in DOX-induced cardiomyopathy. Suppression of ferroptosis by Fer-1 effectively prevented cardiac injury. Moreover, the authors found that acyl-CoA thioesterase 1 (Acot1), one of the leading-edge core genes, protects the deterioration of DOX-induced cardiomyopathy by suppressing ferroptosis (78).
Diabetic cardiomyopathy (DCM) is described as a specific abnormality of the myocardial structure and function in diabetic patients and increases the risk of HF independent of coronary artery disease and hypertension (79, 80). Cell death is considered as the terminal pathway of cardiomyocytes in the process of DCM (81). As a new RCD form, whether ferroptosis participates in the pathology of DCM is unclear.
ROS are excessively produced in diabetes mellitus and regarded as the predominant pathogenesis for the progression of DCM (82). Since ROS production promotes ferroptosis, it is reasonable to believe that ferroptosis is involved in DCM. Therapeutic inhibition of mitochondrial ROS by MitoTEMPO abrogated high glucose-induced cell death and mitigated myocardial dysfunction in diabetic mice, suggesting that MitoTEMPO might have been a therapeutic strategy for DCM (83).
Nrf2 is a crucial factor in maintaining oxidative homeostasis and has been proposed as a potential therapeutic target for DCM (36). There is a growing number of evidences that indicate that Nrf2 reduces high glucose-induced oxidative damage and prevents the development of DCM. In type 1 diabetic mice, Zang et al. found that Nrf2 knockout inhibits cardiac pathological remodeling and oxidative stress. Meanwhile, chronic type 1 diabetes leads to autophagy deficiency, which promotes Nrf2-mediated ferroptosis, thereby exaggerating the progression of DCM (84). Considering that Nrf2 plays a vital role in regulating genes for glutathione regulation and lipid peroxidation, which are implicated in ferroptosis (85, 86), altered ferroptosis by Nrf2 activators may play an important role in the development of DCM.
Vascular endothelial dysfunction is thought to be one of the major causes of DCM (87). Recently, Luo et al. (88) found that high glucose induces ferroptosis in human umbilical vein endothelial cells (HUVECs) and concluded that ferroptosis is involved in endothelial dysfunction via p53-xCT (the substrate-specific subunit of system xc−)–GSH axis, suggesting that ferroptosis might participate in the process of DCM.
MI defines cardiomyocyte necrosis in a clinical situation consistent with acute myocardial ischemia, which causes ventricular remodeling and HF (89, 90). Myocardial iron is a risk factor for adverse left ventricular (LV) remodeling after MI.
Baba et al. (91) examined the role of cardiac mTOR in iron-mediated cell death and found that mTOR overexpression inhibits ROS production and ferroptosis, while mTOR deficiency promotes iron-induced ferroptosis. Cardiac mTOR might be an effective therapeutic target in acute MI through specifically managing iron homeostasis. However, in diabetic rats, Wang et al. demonstrated that diabetes aggravates myocardial IRI through programmed cell death such as apoptosis and ferroptosis, while AMPK attenuates post-ischemic myocardial injury and ferroptosis (92). Besides, AMPK activation is also involved in energy stress-induced ferroptosis (93). As we know, AMPK might involve mTOR inhibition; hence, the role of mTOR in cardiac ferroptosis needs further investigation.
In addition to mTOR, exosome-mediated ferroptosis was also investigated in myocardial injury. Song et al. (94) demonstrated that ferroptosis was generated in the infarcted myocardium and in cardiomyocyte following hypoxia-induced injury. Human umbilical cord blood-derived MSC (HUCB-MSCs) exosomes inhibit ferroptosis and attenuate myocardial injury by suppressing DMT1 expression. DMT1 is responsible for iron import. Upregulation of DMT1 might increase iron uptake, resulting in lipid peroxidation and ferroptosis (95). In Parkinson's disease mice model, the expression of DMT1 was significantly increased, leading to iron deposition and ferroptosis (96). Besides, overexpression of NF-κB-regulated 1b isoform (1B)/DMT1 contributes to cell death during brain ischemia (97), suggesting that DMT1-mediated ferroptosis might be a therapeutic target for myocardial ischemia.
Oxidative stress is known to contribute the progression of MI. During MI, both glutathione metabolic pathway and ROS pathway were significantly inhibited (98). GPX4 expression was repressed in the early and middle stages of MI, accompanied by Nrf2, and epithelial–mesenchymal transition (EMT) pathways were significantly altered. In particular, the Nrf2–Hmox1 pathway was enhanced in the early and middle stages of MI, and EMT- Zeb1/2 was increased in the early stage of MI. However, not all studies come to the same conclusion. In a study conducted by Tang et al. (99), ferroptosis was found mainly occurring in the phase of myocardial reperfusion. Therefore, further research is needed to explore the association between ferroptosis and MI, and provides a basis for precise therapy of MI.
Myocardial IRI is a pathological event characterized by an initial restriction of blood supply to the myocardium, usually as a result of severe stenosis or thrombosis, followed by restoration of blood flow and concomitant reoxygenation to the ischemic region. The mechanisms of IRI include oxidative stress, calcium overload, mitochondrial dysfunction, inflammation, microvascular dysfunction, and activation of cell death pathways (100). Cell death is frequently associated with IRI; however, IRI-induced cardiac myocyte death has not been fully explicated. Recent studies indicate that ferroptosis might participate in IRI. Compared with sham-operated mice, mice suffered to myocardial I/R had significantly higher levels of cardiac Ptgs2 mRNA expression (22). Pretreatment with Fer-1 and DXZ reduced myocardial infarct size. Besides, liproxstatin-1 decreased mitochondrial ROS production and increased GPX4 protein level in the I/R model (101). Overexpression of USP22 could inhibit ferroptotic cardiomyocyte death to protect against IRI (102). Most recently, Stamenkovic et al. demonstrated that oxidized phosphatidylcholines (OxPCs) increased in the process of myocardial IRI, leading to decreased GPX4 activity and increased ferroptosis. Notably, cell death induced by OxPCs could be suppressed by Fer-1 (103). In another study, the expression of embryonic lethal-abnormal vision-like protein 1 (ELAVL1) was upregulated during myocardial I/R. Knockdown of ELAVL1 decreased ferroptosis and ameliorated IRI. The authors also showed that ELAVL1 was transcriptionally activated by forkhead box C1 (FOXC1), and the process of ferroptosis was modulated by autophagy (104).
Ferroptosis is involved in diabetes myocardial IRI as well. Li et al. (105) investigated the effect of ferroptosis in the process of diabetes myocardial IRI and revealed that inhibition of ferroptosis could reduce endoplasmic reticulum stress and myocardial injury in the rat I/R model and reduce cell injury in H9c2 cells. Additionally, as mentioned above, diabetes exaggerates myocardial IRI through activating Nox2-related oxidative stress and ferroptosis, while suppression of Nox2 protects diabetic rats from myocardial IRI (92), providing a therapeutic regent for myocardial IRI in diabetic patients.
The association between ferroptosis and heart transplantation-related IRI has been investigated as well (106). The results showed that ferroptosis recruited neutrophil to the injured myocardium by releasing damage-associated molecular patterns (DAMPs). Fer-1 reduces cardiomyocyte death and blocks neutrophil recruitment following heart transplantation. Targeting ferroptosis might provide therapeutic strategies for patients who are vulnerable to IRI following restoration of coronary blood flow after heart transplantation.
HF is a clinical syndrome caused by a structure and/or function cardiac abnormality, leading to a reduced cardiac output and/or elevated intracardiac pressures at rest or during stress (107). Iron homeostasis is important for maintaining the function of cardiomyocytes. Both iron deficiency and overload are associated with HF. Fang et al. (52) discovered that mice lacking ferritin H (Fth) have increased ROS production and developed HF at 6 months of age. High-iron diet caused ferroptosis and myocardial hypertrophy in cardiac Fth loss mice, which was suppressed by Fer-1. They also found that overexpressing Slc7a11 in cardiomyocytes suppressed high-iron diet-induced cardiac ferroptosis and protected against HF.
In aortic banding-induced heart failure, ferroptosis was also observed. Puerarin, an antioxidant reagent, exhibited cardioprotective function by significantly blocking iron overload and lipid peroxidation, indicating that puerarin can alleviate HF partly through ferroptosis mitigation (108). In addition, Chen et al. (109) also investigated the role of ferroptosis in pressure overload-induced HF. The results showed that ferroptosis was detected in HF rats caused by aortic banding. Either TLR4 and NADPH oxidase 4 (NOX4) knock-down through lentiviral delivery of siRNA remarkably inhibited the activation of ferroptosis and relieved the symptoms of HF, suggesting TLR4-NOX4 as a potential therapeutic target for HF by inhibiting ferroptosis-mediated cell death. Most recently, the relationship between circRNA, iron metabolism, and heart failure was also investigated, and the results showed that ferroptosis was increased in transverse aortic constriction (TAC)-induced HF mice. Furthermore, the interrelationships of miR-224-5p with circSnx12 and Fth1 was established. During HF, the expression of Fth1 was downregulated, which, in turn, releases a large amount of ferrous ions and eventually contributes to ROS accumulation and ferroptotic cell death (110).
Atherosclerosis (AS) is a chronic inflammatory disorder characterized by disorders of lipid metabolism. It is well-known that lipid peroxidation plays a significant role in AS. Guo et al. (111) found that overexpressing GPX4 decreased lipid peroxidation and alleviated atherosclerotic lesions in the aorta of ApoE-deficient mice. As excessive lipid peroxidation is the main feature of ferroptosis, it is reasonable to believe that ferroptosis might participate in the initiation and progression of AS. Recently, a study by Bai et al. (112) explored the potential effects of ferroptosis on AS. The results showed that inhibition of ferroptosis could inhibit lipid peroxidation and alleviate high fat food-induced AS lesion in ApoE–/– mice. Additionally, ox-LDL causes mitochondrial damage and downregulates the expressions of SLC7A11 and GPX4 in mouse aortic endothelial cells (MAECs), while Fer-1 could repress ox-LDL-induced lipid peroxidation and endothelial dysfunction in MAECs. They concluded that ferroptosis might be involved in the pathological process of AS and potentially to be a therapeutic target for AS. In addition, ferroptosis is detected in human coronary artery specimens. In the advanced stages of AS, the expression of Ptgs2 and ACSL4 were upregulated, while GPX4 was downregulated. The severity of AS was associated with Ptgs2 and ACSL4 positively and GPX4 negatively. They concluded that ferroptosis and pyroptosis may regulate the occurrence and progress of AS, providing potential target for the prevention and therapy of coronary artery atherosclerosis (113).
In sepsis-induced cardiac injury, lipopolysaccharide (LPS) increased the levels of Ptgs2 and lipid ROS, which were attenuated by Fer-1 and DXZ (45). LPS upregulated the expression of NCOA4 and the level of intracellular Fe2+; cytoplasmic Fe2+ further increased the level of siderofexin (SFXN1) on mitochondrial membrane, which in turn carried cytoplasmic Fe2+ into the mitochondria, leading to the generation of mitochondrial ROS and ferroptosis. Ferroptosis might be a therapeutic target for preventing sepsis in the future.
Arrhythmias are particularly common in patients with heart disease. Studies have shown that excessive ROS can cause arrhythmia (114). To the best of our knowledge, ferroptosis has not yet been investigated in arrhythmia. A recent study revealed that iron overload resulted in mitochondrial ROS generation and mitochondrial membrane potential (ΔΨm) depolarization, thus, opening the mPTP, thereby promoting cardiac arrhythmias (115). As we know, mitochondrial ROS generation and ΔΨm depolarization are features of ferroptosis as well. Targeting ferroptosis might provide a new idea for the treatment of arrhythmia.
Coronavirus disease 2019 (COVID-19), caused by the SARS-CoV-2 virus, has caused a major health problem around the world. Multiple organs affected in COVID-19 include the lungs, kidneys, liver, and heart. As a novel RCD, ferroptosis might be an important cause of cardiac injury in COVID-19 and serves as a new therapeutic target. In one COVID-19 patient, Jacobs et al. discovered lipid peroxidation in the myocardium and concluded that ferroptosis is a detrimental factor in COVID-19 cardiac damage (116). Iron overload, which is associated with inflammation, hypercoagulation, and immune dysfunction, has been considered as a vital player in the pathogenesis of COVID-19, leading to acute respiratory distress syndrome (ARDS), thromboembolic complications, and cardiovascular damage (117, 118). Iron chelators or ferroptosis inhibitors might be potential treatment candidates for COVID-19-related multiple organ failure.
Death of cardiomyocyte results in dramatic impairments to cardiac function and eventually in HF (119); therefore, preventing and reducing cardiomyocyte death is crucial to the improvement of cardiac function. Recently, ferroptosis, an iron-mediated RCD, has received growing attention and is found to be associated with cardiac pathology. Targeting ferroptosis may provide potential treatment strategy for CVDs (Table 3).
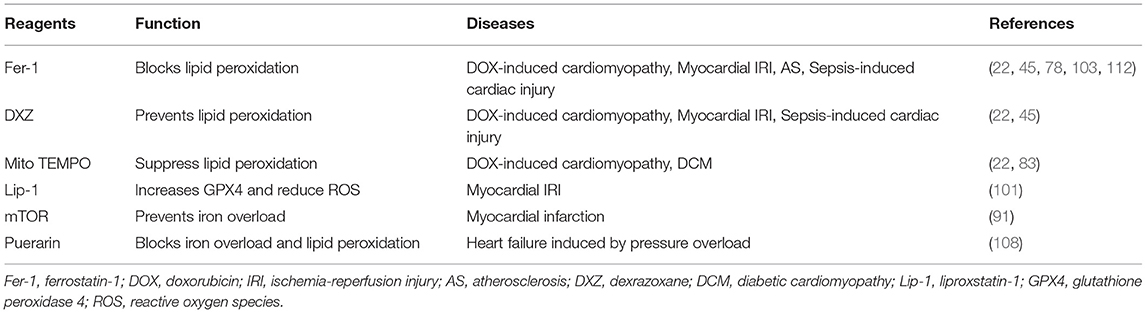
Table 3. Potential therapeutic strategy for CVDs.
As discussed above, ferroptosis is involved in the process of DOX-induced cardiomyopathy, diabetic cardiomyopathy, MI, myocardial IRI, HF, AS, sepsis-induced cardiac injury, cardiac arrhythmia, and COVID-19-associated cardiac injury, while the relationship between ferroptosis and other cardiovascular disorders, such as dilated cardiomyopathy, valvular heart disease, myocarditis, and aortic disease (including aortic dissection and aortic aneurysm), has not ever been investigated. Further studies are needed to establish the web between ferroptosis and various CVDs.
Although ferroptosis occurs in various diseases, whether it acts as an automatic response to the stimuli that disrupt the metabolic balance or it is the stimuli that directly interrupt the balance is controversial. In other words, we still have no idea whether ferroptosis is achieved “actively” or “passively.” Further studies are needed to explore the exact association between ferroptosis and the different pathophysiological conditions.
Increasing evidence has shown the crosstalk between ferroptosis and other types of cell death, such as autophagy and apoptosis (21, 28). Whether these various modes of cell death can be integrated into a complete regulatory network remains to be explored. Further elucidating the interrelation between ferroptosis and other types of cell death is important to clarify the mechanisms of ferroptosis and developing treatments.
At present, there are no specific biomarkers for ferroptosis. Since ferroptosis occurs in the early stage of MI and cardiomyopathy, discovery of specific markers of ferroptosis may provide great assistance in early diagnosis of CVDs. Moreover, the cardioprotection of ferroptosis suppressors is just limited in animal studies. How basic research results should be applied in a clinical situation also needs further exploration. Nevertheless, inhibition of ferroptosis is thought to be a potential therapeutic option for CVDs (22, 120).
In conclusion, ferroptosis plays a critical role in the pathogenesis of various CVDs, but its mechanism needs to be further explored. Suppression of ferroptosis is expected to become an effective therapeutic strategy for CVDs.
LS designed the study and revised the manuscript. LJ generated the figure and tables. CZ edited the manuscript. HH and YC drafted the manuscript. All authors have read and approved the content of the manuscript.
This research was funded by the Key Medicine Disciplines Co-construction Project of Jiaxing Municipal (Grant No. 2019-ss-xxgbx).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
2. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. (1972) 26:239–57. doi: 10.1038/bjc.1972.33
3. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. (2014) 15:135–47. doi: 10.1038/nrm3737
4. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. (2013) 38:209–23. doi: 10.1016/j.immuni.2013.02.003
5. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. (2001) 9:113–4. doi: 10.1016/S0966-842X(00)01936-3
6. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
7. Vercammen D, Vandenabeele P, Beyaert R, Declercq W, Fiers W. Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine. (1997) 9:801–8. doi: 10.1006/cyto.1997.0252
8. Hao S, Yu J, He W, Huang Q, Zhao Y, Liang B, et al. Cysteine dioxygenase 1 mediates erastin-induced ferroptosis in human gastric cancer cells. Neoplasia. (2017) 19:1022–32. doi: 10.1016/j.neo.2017.10.005
9. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. (2017) 551:639–43. doi: 10.1038/nature24637
10. Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, et al. Ferroptosis, a newly characterized form of cell death in Parkinson's disease that is regulated by PKC. Neurobiol Dis. (2016) 94:169–78. doi: 10.1016/j.nbd.2016.05.011
11. Lane DJR, Ayton S, Bush AI. Iron and Alzheimer's disease: an update on emerging mechanisms. J Alzheimers Dis. (2018) 64:S379–95. doi: 10.3233/JAD-179944
12. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. (2014) 16:1180–91. doi: 10.1038/ncb3064
13. Eagle H. Nutrition needs of mammalian cells in tissue culture. Science. (1955) 122:501–14. doi: 10.1126/science.122.3168.501
14. Eagle H. Amino acid metabolism in mammalian cell cultures. Science. (1959) 130:432–7. doi: 10.1126/science.130.3373.432
15. Eagle H, Piez KA, Oyama VI. The biosynthesis of cystine in human cell cultures. J Biol Chem. (1961) 236:1425–8. doi: 10.1016/S0021-9258(18)64190-0
16. Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ. Cystine deprivation induces oligodendroglial death: rescue by free radical scavengers and by a diffusible glial factor. J Neurochem. (1996) 67:566–73. doi: 10.1046/j.1471-4159.1996.67020566.x
17. Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, et al. 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur J Neurosci. (2004) 20:2049–58. doi: 10.1111/j.1460-9568.2004.03650.x
18. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. (2003) 3:285–96. doi: 10.1016/S1535-6108(03)00050-3
19. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
20. Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. (2014) 16:1069–79. doi: 10.1038/ncb3053
21. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. (2014) 509:105–9. doi: 10.1038/nature13148
22. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. (2019) 116:2672–80. doi: 10.1073/pnas.1821022116
23. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
24. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. (2015) 10:1604–9. doi: 10.1021/acschembio.5b00245
25. Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. (2008) 8:237–48. doi: 10.1016/j.cmet.2008.07.005
26. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. (2017) 13:81–90. doi: 10.1038/nchembio.2238
27. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. (2019) 21:579–91. doi: 10.1038/s41556-019-0305-6
28. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
29. Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. (2016) 478:838–44. doi: 10.1016/j.bbrc.2016.08.034
30. Kim EH, Shin D, Lee J, Jung AR, Roh JL. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. (2018) 432:180–90. doi: 10.1016/j.canlet.2018.06.018
31. Chen H, Zheng C, Zhang Y, Chang YZ, Qian ZM, Shen X. Heat shock protein 27 downregulates the transferrin receptor 1-mediated iron uptake. Int J Biochem Cell Biol. (2006) 38:1402–16. doi: 10.1016/j.biocel.2006.02.006
32. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
33. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
34. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
35. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
36. Ge ZD, Lian Q, Mao X, Xia Z. Current status and challenges of NRF2 as a potential therapeutic target for diabetic cardiomyopathy. Int Heart J. (2019) 60:512–20. doi: 10.1536/ihj.18-476
37. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. (1999) 274:11455–8. doi: 10.1074/jbc.274.17.11455
38. Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. (2020) 8:590226. doi: 10.3389/fcell.2020.590226
39. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
40. Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci. (2018) 22:3826–36. doi: 10.26355/eurrev_201806_15267
41. Tang M, Chen Z, Wu D, Chen L. Ferritinophagy/ferroptosis: iron-related newcomers in human diseases. J Cell Physiol. (2018) 233:9179–90. doi: 10.1002/jcp.26954
42. Masaldan S, Clatworthy SAS, Gamell C, Meggyesy PM, Rigopoulos AT, Haupt S, et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. (2018) 14:100–15. doi: 10.1016/j.redox.2017.08.015
43. Sui M, Xu D, Zhao W, Lu H, Chen R, Duan Y, et al. CIRBP promotes ferroptosis by interacting with ELAVL1 and activating ferritinophagy during renal ischaemia-reperfusion injury. J Cell Mol Med. (2021) 25:6203–216. doi: 10.1111/jcmm.16567
44. Sui S, Zhang J, Xu S, Wang Q, Wang P, Pang D. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. (2019) 10:331. doi: 10.1038/s41419-019-1564-7
45. Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. (2020) 160:303–18. doi: 10.1016/j.freeradbiomed.2020.08.009
46. Qin X, Zhang J, Wang B, Xu G, Yang X, Zou Z, et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy. 2021:1–20. doi: 10.1080/15548627.2021.1911016
47. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
48. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. (2016) 26:1021–32. doi: 10.1038/cr.2016.95
49. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. (2015) 34:5617–25. doi: 10.1038/onc.2015.32
50. Adedoyin O, Boddu R, Traylor A, Lever JM, Bolisetty S, George JF, et al. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Renal Physiol. (2018) 314:F702–14. doi: 10.1152/ajprenal.00044.2017
51. Ganz T. Systemic iron homeostasis. Physiol Rev. (2013) 93:1721–41. doi: 10.1152/physrev.00008.2013
52. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res. (2020) 127:486–501. doi: 10.1161/CIRCRESAHA.120.316509
53. Shang Y, Luo M, Yao F, Wang S, Yuan Z, Yang Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal. (2020) 72:109633. doi: 10.1016/j.cellsig.2020.109633
54. Wang Y, Liu Y, Liu J, Kang R, Tang D. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun. (2020) 531:581–7. doi: 10.1016/j.bbrc.2020.07.032
55. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. (2019) 51:575–86. e4. doi: 10.1016/j.devcel.2019.10.007
56. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. (2016) 113:E4966–75. doi: 10.1073/pnas.1603244113
57. Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. (2019) 79:5355–66. doi: 10.1158/0008-5472.CAN-19-0369
58. Luis G, Godfroid A, Nishiumi S, Cimino J, Blacher S, Maquoi E, et al. Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. (2021) 43:102006. doi: 10.1016/j.redox.2021.102006
59. Wiernicki B, Dubois H, Tyurina YY, Hassannia B, Bayir H, Kagan VE, et al. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. (2020) 11:922. doi: 10.1038/s41419-020-03118-0
60. Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. (2016) 113:E6806–12. doi: 10.1073/pnas.1607152113
61. Yang WH, Huang Z, Wu J, Ding CC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol Cancer Res. (2020) 18:79–90. doi: 10.1158/1541-7786.MCR-19-0691
62. Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. (2019) 28:2501–8. e4. doi: 10.1016/j.celrep.2019.07.107
63. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. (2020) 16:302–9. doi: 10.1038/s41589-020-0472-6
64. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. (2017) 547:453–57. doi: 10.1038/nature23007
65. Basit F, van Oppen LM, Schockel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. (2017) 8:e2716. doi: 10.1038/cddis.2017.133
66. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354–63. e3. doi: 10.1016/j.molcel.2018.10.042
67. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.132747
68. Jelinek A, Heyder L, Daude M, Plessner M, Krippner S, Grosse R, et al. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic Biol Med. (2018) 117:45–57. doi: 10.1016/j.freeradbiomed.2018.01.019
69. Neitemeier S, Jelinek A, Laino V, Hoffmann L, Eisenbach I, Eying R, et al. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. (2017) 12:558–70. doi: 10.1016/j.redox.2017.03.007
70. Gaschler MM, Hu F, Feng H, Linkermann A, Min W, Stockwell BR. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem Biol. (2018) 13:1013–20. doi: 10.1021/acschembio.8b00199
71. Song X, Liu J, Kuang F, Chen X, Zeh HJ 3rd, Kang R, et al. PDK4 dictates metabolic resistance to ferroptosis by suppressing pyruvate oxidation and fatty acid synthesis. Cell Rep. (2021) 34:108767. doi: 10.1016/j.celrep.2021.108767
72. Sun Y, Zheng Y, Wang C, Liu Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. (2018) 9:753. doi: 10.1038/s41419-018-0794-4
73. Zhang L, Gong F, Zhang F, Ma J, Zhang P, Shen J. Targeted therapy for human hepatic carcinoma cells using folate-functionalized polymeric micelles loaded with superparamagnetic iron oxide and sorafenib in vitro. Int J Nanomedicine. (2013) 8:1517–24. doi: 10.2147/IJN.S43263
74. Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, et al. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. (2014) 34:6417–22.
75. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. (2015) 212:555–68. doi: 10.1084/jem.20140857
76. Dai E, Meng L, Kang R, Wang X, Tang D. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun. (2020) 522:415–21. doi: 10.1016/j.bbrc.2019.11.110
77. Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. (1998) 339:900–5. doi: 10.1056/NEJM199809243391307
78. Liu Y, Zeng L, Yang Y, Chen C, Wang D, Wang H. Acyl-CoA thioesterase 1 prevents cardiomyocytes from doxorubicin-induced ferroptosis via shaping the lipid composition. Cell Death Dis. (2020) 11:756. doi: 10.1038/s41419-020-02948-2
79. Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D, et al. Diabetic cardiomyopathy—a comprehensive updated review. Prog Cardiovasc Dis. (2019) 62:315–26. doi: 10.1016/j.pcad.2019.03.003
80. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
81. Chen Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G. Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol. (2020) 11:42. doi: 10.3389/fphar.2020.00042
82. Wilson AJ, Gill EK, Abudalo RA, Edgar KS, Watson CJ, Grieve DJ. Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart. (2018) 104:293–9. doi: 10.1136/heartjnl-2017-311448
83. Ni R, Cao T, Xiong S, Ma J, Fan GC, Lacefield JC, et al. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic Biol Med. (2016) 90:12–23. doi: 10.1016/j.freeradbiomed.2015.11.013
84. Zang H, Wu W, Qi L, Tan W, Nagarkatti P, Nagarkatti M, et al. Autophagy inhibition enables Nrf2 to exaggerate the progression of diabetic cardiomyopathy in mice. Diabetes. (2020) 69:2720–34. doi: 10.2337/db19-1176
85. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. (2017) 6:e371. doi: 10.1038/oncsis.2017.65
86. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
87. Knapp M, Tu X, Wu R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol Sin. (2019) 40:1–8. doi: 10.1038/s41401-018-0042-6
88. Luo EF, Li HX, Qin YH, Qiao Y, Yan GL, Yao YY, et al. Role of ferroptosis in the process of diabetes-induced endothelial dysfunction. World J Diabetes. (2021) 12:124–37. doi: 10.4239/wjd.v12.i2.124
89. Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Fourth universal definition of myocardial infarction 2018. J Am Coll Cardiol. (2018) 72:2231–64. doi: 10.1161/CIR.0000000000000617
90. Jenca D, Melenovsky V, Stehlik J, Stanek V, Kettner J, Kautzner J, et al. Heart failure after myocardial infarction: incidence and predictors. ESC Heart Fail. (2020) 8:222–37. doi: 10.1002/ehf2.13144
91. Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. (2018) 314:H659–68. doi: 10.1152/ajpheart.00452.2017
92. Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang Z, et al. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J Cell Mol Med. (2020) 24:6670–9. doi: 10.1111/jcmm.15318
93. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. (2020) 22:225–34. doi: 10.1038/s41556-020-0461-8
94. Song Y, Wang B, Zhu X, Hu J, Sun J, Xuan J, et al. Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol Toxicol. (2021) 37:51–64. doi: 10.1007/s10565-020-09530-8
95. Li LB, Chai R, Zhang S, Xu SF, Zhang YH, Li HL, et al. Iron exposure and the cellular mechanisms linked to neuron degeneration in adult mice. Cells. (2019) 8:198. doi: 10.3390/cells8020198
96. Song LM, Xiao ZX, Zhang N, Yu XQ, Cui W, Xie JX, et al. Apoferritin improves motor deficits in MPTP-treated mice by regulating brain iron metabolism and ferroptosis. iScience. (2021) 24:102431. doi: 10.1016/j.isci.2021.102431
97. Ingrassia R, Lanzillotta A, Sarnico I, Benarese M, Blasi F, Borgese L, et al. 1B/(-)IRE DMT1 expression during brain ischemia contributes to cell death mediated by NF-kappaB/RelA acetylation at Lys310. PLoS ONE. (2012) 7:e38019. doi: 10.1371/journal.pone.0038019
98. Park TJ, Park JH, Lee GS, Lee JY, Shin JH, Kim MW, et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. (2019) 10:835. doi: 10.1038/s41419-019-2061-8
99. Tang LJ, Luo XJ, Tu H, Chen H, Xiong XM, Li NS, et al. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn Schmiedebergs Arch Pharmacol. (2021) 394:401–10. doi: 10.1007/s00210-020-01932-z
100. Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. (2011) 17:1391–401. doi: 10.1038/nm.2507
101. Feng Y, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun. (2019) 520:606–11. doi: 10.1016/j.bbrc.2019.10.006
102. Ma S, Sun L, Wu W, Wu J, Sun Z, Ren J. USP22 Protects against myocardial ischemia-reperfusion injury via the SIRT1-p53/SLC7A11-dependent inhibition of ferroptosis-induced cardiomyocyte death. Front Physiol. (2020) 11:551318. doi: 10.3389/fphys.2020.551318
103. Stamenkovic A, O'Hara KA, Nelson DC, Maddaford TG, Edel AL, Maddaford G, et al. Oxidized phosphatidylcholines trigger ferroptosis in cardiomyocytes during ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. (2021) 320:H1170–84. doi: 10.1152/ajpheart.00237.2020
104. Chen HY, Xiao ZZ, Ling X, Xu RN, Zhu P, Zheng SY. ELAVL1 is transcriptionally activated by FOXC1 and promotes ferroptosis in myocardial ischemia/reperfusion injury by regulating autophagy. Mol Med. (2021) 27:14. doi: 10.1186/s10020-021-00271-w
105. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. (2020) 39:210–25. doi: 10.1089/dna.2019.5097
106. Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. (2019) 129:2293–304. doi: 10.1172/JCI126428
107. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. (2016) 37:2129–200. doi: 10.1093/eurheartj/ehw128
108. Liu B, Zhao C, Li H, Chen X, Ding Y, Xu S. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem Biophys Res Commun. (2018) 497:233–40. doi: 10.1016/j.bbrc.2018.02.061
109. Chen X, Xu S, Zhao C, Liu B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem Biophys Res Commun. (2019) 516:37–43. doi: 10.1016/j.bbrc.2019.06.015
110. Zheng H, Shi L, Tong C, Liu Y, Hou M. circSnx12 is involved in ferroptosis during heart failure by targeting miR-224-5p. Front Cardiovasc Med. (2021) 8:656093. doi: 10.3389/fcvm.2021.656093
111. Guo Z, Ran Q, Roberts LJ 2nd, Zhou L, Richardson A, Sharan C, et al. Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein E-deficient mice. Free Radic Biol Med. (2008) 44:343–52. doi: 10.1016/j.freeradbiomed.2007.09.009
112. Bai T, Li M, Liu Y, Qiao Z, Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med. (2020) 160:92–102. doi: 10.1016/j.freeradbiomed.2020.07.026
113. Zhou Y, Zhou H, Hua L, Hou C, Jia Q, Chen J, et al. Verification of ferroptosis and pyroptosis and identification of PTGS2 as the hub gene in human coronary artery atherosclerosis. Free Radic Biol Med. (2021) 171:55–68. doi: 10.1016/j.freeradbiomed.2021.05.009
114. Munzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. J Am Coll Cardiol. (2017) 70:212–29. doi: 10.1016/j.jacc.2017.05.035
115. Gordan R, Fefelova N, Gwathmey JK, Xie LH. Iron overload, oxidative stress and calcium mishandling in cardiomyocytes: role of the mitochondrial permeability transition pore. Antioxidants. (2020) 9:758. doi: 10.3390/antiox9080758
116. Jacobs W, Lammens M, Kerckhofs A, Voets E, Van San E, Van Coillie S, et al. Fatal lymphocytic cardiac damage in coronavirus disease 2019 (COVID-19): autopsy reveals a ferroptosis signature. ESC Heart Fail. (2020) 7:3772–81. doi: 10.1002/ehf2.12958
117. Yang M, Lai CL. SARS-CoV-2 infection: can ferroptosis be a potential treatment target for multiple organ involvement? Cell Death Discov. (2020) 6:130. doi: 10.1038/s41420-020-00369-w
118. Habib HM, Ibrahim S, Zaim A, Ibrahim WH. The role of iron in the pathogenesis of COVID-19 and possible treatment with lactoferrin and other iron chelators. Biomed Pharmacother. (2021) 136:111228. doi: 10.1016/j.biopha.2021.111228
119. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99:1765–817. doi: 10.1152/physrev.00022.2018
Keywords: cardiovascular diseases, ferroptosis, iron, therapeutic target, lipid peroxidation
Citation: Hu H, Chen Y, Jing L, Zhai C and Shen L (2021) The Link Between Ferroptosis and Cardiovascular Diseases: A Novel Target for Treatment. Front. Cardiovasc. Med. 8:710963. doi: 10.3389/fcvm.2021.710963
Received: 17 May 2021; Accepted: 21 June 2021;
Published: 22 July 2021.
Edited by:
Qian Yang, First Affiliated Hospital of Chinese PLA General Hospital, ChinaReviewed by:
Carsten Culmsee, University of Marburg, GermanyCopyright © 2021 Hu, Chen, Jing, Zhai and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Shen, c2hlbmxpYW5nODAxMUAxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.