
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Vet. Sci., 07 October 2020
Sec. Veterinary Pharmacology and Toxicology
Volume 7 - 2020 | https://doi.org/10.3389/fvets.2020.00589
 Renge Hu1Xue Wang1Inam Muhamamd1Yiming Wang1Wenlong Dong1Haipeng Zhang1Yu Wang1Shuming Liu1Yunhang Gao1,2Lingcong Kong1,2*
Renge Hu1Xue Wang1Inam Muhamamd1Yiming Wang1Wenlong Dong1Haipeng Zhang1Yu Wang1Shuming Liu1Yunhang Gao1,2Lingcong Kong1,2* Hongxia Ma1,2*
Hongxia Ma1,2*To evaluate the antimicrobial resistance and virulence gene characteristics of highly pathogenic Proteus mirabilis. In this study, we isolated P. mirabilis CC15031 from diarrhea dogs in China, tested the median lethal dose (LD50), and measured the minimum inhibitory concentration (MIC) of 10 different antibiotics commonly used in veterinary clinic. Meanwhile, we presented the complete genome sequence annotations to analyze the virulence and resistance formation mechanism. The results showed that the CC15031 presented relatively potent pathogenicity in mice (LD50 = 0.57 × 106 CFU) and exhibited a high degree of resistance to all the tested antimicrobial agents. The CC15031 genome of 4,031,742 bp with 3,745 predicted genes had an average gene length of 917 bp and 38.99% guanine-cytosine content. A new variant of an integrative and conjugative element with a type IV secretion system (217,446 bp) conferring multidrug resistance was identified and characterized by structural analysis in CC15031. These data provide a foundation for understanding the genomic features and antimicrobial resistance mechanisms of this pathogen.
Proteus mirabilis (P. mirabilis) has been recognized as a significant zoonotic pathogen, causing a variety of diseases, including diarrhea, urinary tract infections, and keratitis and considered the second most common zoonotic bacterium after enteropathogenic Escherichia coli in human medicine (1). In recent years, it has been considered to be a repository for virulence and resistance genes and has become a potential public health concern. Previous studies indicated that severe drug resistance had been developed in P. mirabilis against commonly used antibiotics. Thus, the emergence of drug-resistant strains has posed clinical difficulties and become a potential threat to public health (2).
Generally, P. mirabilis accounts for about 90% of Proteus infections and is considered a community-acquired infection (3). Virulence determinants acquired by P. mirabilis induce infections successfully (4). Drug resistance genes and drug resistance gene islands, such as the extended-spectrum β-lactamase gene (blaVEB−6), have been found in P. mirabilis in many countries (5, 6). Also, the acquired quinolone resistance gene (qnrA1) was isolated from humans and animals in France (6). Recently, two novel Salmonella genomic island 1 (SGI1) variants, SGI1-PmBC1123, and SGI1-PmSC1111, which carried multiple drug resistance genes, were described in P. mirabilis strains isolated from Food Animals in China (7). Also, some studies used PCR sequencing and demonstrated that P. mirabilis carries variant virulence islands (8, 9).
Since 2015–2017, we have found that the diarrhea disease of dogs caused by P. mirabilis in the Changchun area of China is relatively severe, but the underlying antimicrobial resistance and pathogenicity are yet unknown. Although the genomes of many human-sourced P. mirabilis (for example, NCTC1178, 81–176) have been sequenced, to date, P. mirabilis with multidrug resistance and virulence in dogs have not been sequenced and reported. In this study, one strain of P. mirabilis with high pathogenicity and drug resistance, which was isolated, was selected and analyzed by whole-genome sequence, and a new SXT/R391 ICE variant was found.
The samples were collected from diarrheal specimens of 62 dogs suffering from bacterial enteritis during 2015–2017. The samples were inoculated on lysogeny broth (LB) agar medium and brain heart infusion (BHI) agar medium and cultured at 37 °C for 12 h. The bacterial growth was observed, and the colonies with the same morphology and migration ability were selected for streak culture to obtain the pure culture strain. Then, the genomes of the isolates were extracted, amplified, and sequenced by PCR using 16S ribosomal RNA (rRNA) primers. The sequencing results were subjected to BLAST analysis at the National Center for Biotechnology Information website. Strains with 99% sequence homology to the P. mirabilis 16S rRNA gene were selected for further analysis.
The median lethal dose (LD50) of P. mirabilis was determined by the modified Kirschner method, and the colony count of each strain was carried out (10). Then, three dose groups of 1 × 107-1 × 109 CFU/ml were set through the pre-experiment. Eight mice, an equal number of males and females, were randomly selected from each group. Each dose group was injected intraperitoneally with a 0.2 ml bacterial solution at different gradient concentration. The mice in each group were kept in isolation, the death rate was observed for 3 days, and the LD50 was calculated (11). The data were statistically analyzed using SPSS (19.0) software. All the methods were carried out by the National Institutes of Health guidelines and protocols for laboratory animal use and proper care, approved by the Animal Care and Use Committee of Jilin Agricultural University.
The antimicrobial sensitivity phenotypes of P. mirabilis were determined by the broth dilution method using Muller–Hinton (MH) agar plates following Clinical and Laboratory Standards Institute guidelines M100-26. The resistance breakpoint was as follows: enrofloxacin > 2 μg/ml, clindamycin > 4 μg/ml, ceftriaxone > 4 μg/ml, sulfamethoxazole > 512 μg/ml, gentamicin > 16 μg/ml, chloramphenicol > 32 μg/ml, florfenicol > 16 μg/ml, doxycycline > 16 μg/ml, ciprofloxacin > 4 μg/ml, and erythromycin > 32 μg/ml. E. coli ATCC25922 strain was used for quality control.
The total DNA of P. mirabilis CC15031 was extracted using a genomic DNA kit (A&A Biotechnology, Gdansk, Poland). Then, the SMRT Bell library was constructed. The purified fragments were solubilized in the buffer, and the specific size fragments were screened with Blue Pipin and purified with AMpure PB magnetic beads. The library was constructed quantitatively based on Qubit estimation, and the insert size was detected with Agilent 2100. Then, the fragments were sequenced on the PacBio platform. The sequencing data were deposited in GenBank, with a CP031532 accession number.
The gene function was subsequently annotated with GO (Gene Ontology), KEGG (Kyoto Encyclopedia of Genes and Genomes), COG (Cluster of Orthologous Groups of proteins), and NR (Non-Redundant Protein Database). ARG (antibiotic resistance gene) analysis was performed using the Resistance Gene Identifier (v4.1.0) tool of the CARD (The Comprehensive Antibiotic Resistance Database), CGE ResFinder 3.1, and IslandPath-DIOMB software (version 0.2) was used to predict the gene islands by detecting the dinucleotide biases and mobility of genes to identify the gene islands and the potential horizontal gene transfers.
One P. mirabilis strain was selected from diarrheal specimens of dogs and identified by the 16S rRNA sequencing. Antibiotic resistance and pathogens of the isolates were evaluated by the median lethal dose (LD50) and the minimum inhibitory concentration (MIC) test (Tables S1, S2). The LD50 of the bacterial strain P. mirabilis CC15031 isolated in this study was determined by the modified Kirschner method and showed relatively strong virulence to mice (LD50 = 0.57 × 106CFU). Following standardized international definitions, the MICs of different antimicrobial agents against the P. mirabilis CC15031 showed a high-level antibiotic resistance, representing a multidrug-resistant strain (Table 1).

Table 1. MICs (microgram per milliliter) of different antibiotics agains P. mirabilis CC15031.
Based on the above LD50 and MICs studies, CC15031 was selected for whole-genome sequencing to elucidate potential mechanisms underlying the multidrug resistance and high virulence, a Circos circular representation of the CC15031 genome with annotated genes was constructed (Figure S2). The CC15031 genome is comprised of one chromosome and a plasmid. It is 4,031,742 bp long, containing 3,745 predicted genes. The average length of each gene was 917 bp. Guanine-cytosine content of this genome was 38.99%, which was similar to other reported P. mirabilis genomes. The complete nucleotide sequence of CC15031 characterized in this study was submitted to GenBank and assigned accession number CP048787.
The results of comparing the identity values ≥ 80% were statistically analyzed based on the Virulence Factor Database. A large number of virulence-associated genes were predicted in the genome of CC15031 (Table S3). Among the selected genes involved in the capsular polysaccharide, elongation factor Tu (EF-Tu) and adhesion were identified. In the biofilm formation system, the genome of CC15031 contained 26 virulence factors, which involved 44 genes (Table 2). As for motility characteristics, 9 different diverse flagellum virulence factors related to 51 genes and 12 fimbriae virulence factors related to 47 genes were identified in the CC15031 genome (Tables 3, 4). Among all the obtained virulence factors, two exogenous adhesive molecules, AI396 (M. catarrhalis) and CVF228 (Listeria), and three exogenous pilus virulence factors, AI102, CVF825 (Hemorrhagic coli pili), and CVF6259 (E. coli common pilus) were identified. Genes associated with flagella formation flaA, flgD were observed in CC15031 genome and no flaB found. Compared with the genes of other P. mirabilis, CC15031 contained several typical secretion systems (TNSSs), including one T1SS, six T3SS, and nine T6SS. A total of 15 gene islands with a total length of 248,432 bp and an average length of 16,562 bp of each gene were predicted using IslandPath-DIOMB software (version 0.2) (Figure S3).
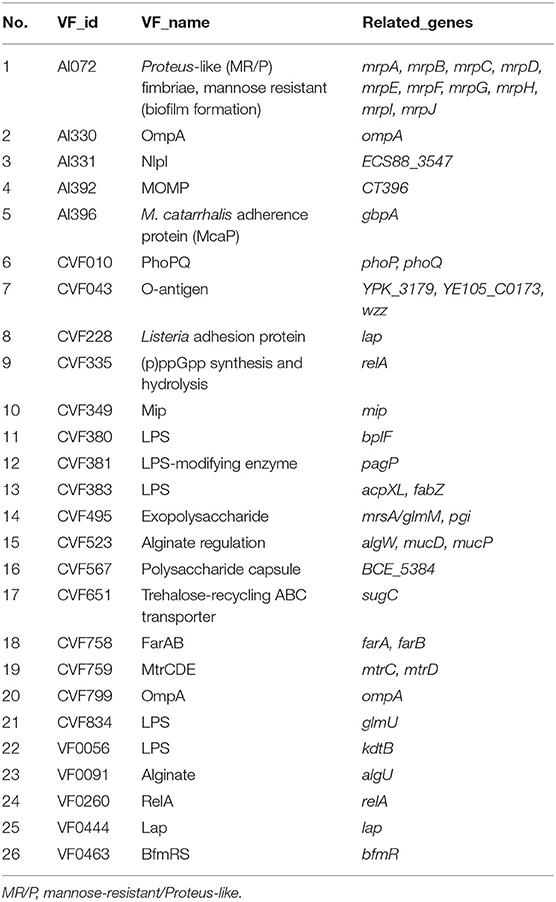
Table 2. Biofilm formation-related gene annotations of CC15031 based on the Virulence Factor Database.
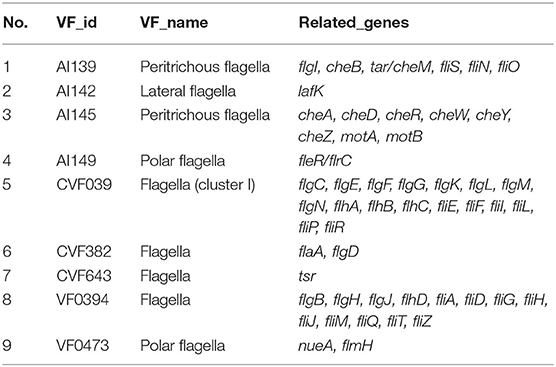
Table 3. Flagellum-related gene annotation of CC15031.
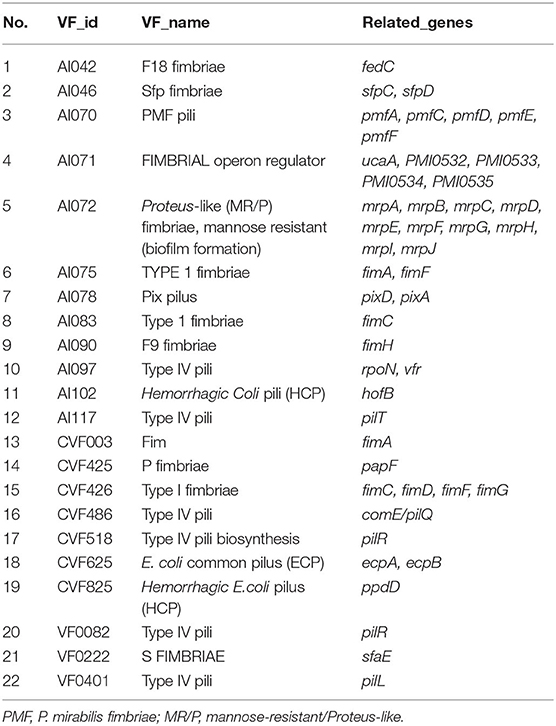
Table 4. Fimbriae-related gene annotation of CC15031.
The genome sequencing data showed that CC15031 contained 18 resistance genes (catA4, tetJ, ant(2″)-Ia, ereA, ble, sul1, catB, blaOXA-1, AAC(6′)-Ib, aac(3)IV, SJL2, cml_e3, blaCTX-M, aph(3′)-Ia, tetC, aph(3′)-Ia, ant(3″)-Ia, and dfrA1). These genes may confer resistance to penicillins, cephalosporins, macrolides, aminoglycosides, tetracyclines, and sulfonamides in CC15031 (Table 5).
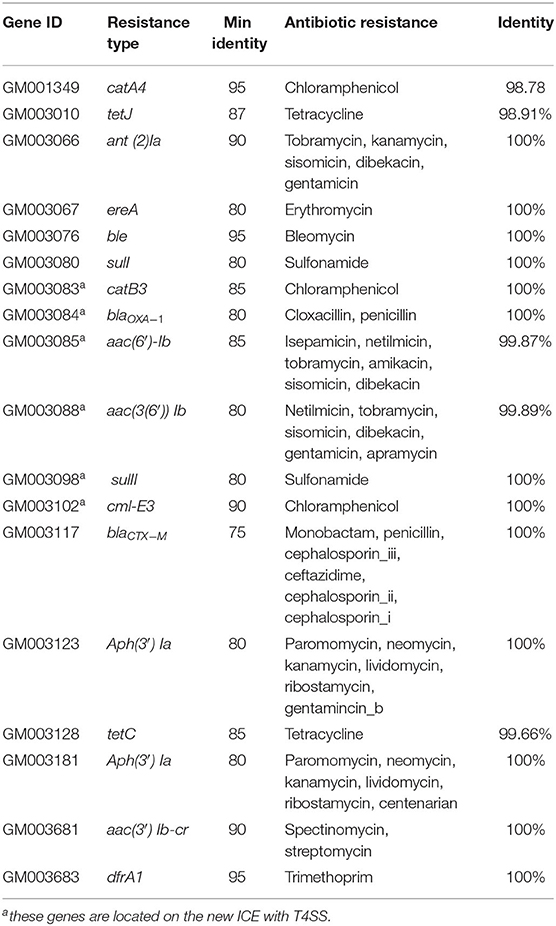
Table 5. Drug resistance gene statistics of chromosome.
Analysis of the chromosomal DNA of CC15031 revealed the presence of an integrative and conjugative element (ICE) region (217,446 bp in size with a guanine-cytosine content of 45%). The ICE flanked by a 17-bp target site duplication (TTAATAAAATAAAAACA). The ICE contained virulence factors, such as a T4SS, antibiotic resistance genes, such as the catB3 (encoding resistance to chloramphenicol), narrow-spectrum β-lactamase genes blaOXA−1 and blaCTX−M(encoding resistance to β-lactamase), catB3 (encoding resistance to chloramphenicol), aac(6′)-Ib, and aac(3′) Ib (encoding resistance to aminoglycoside), Cml-E3 (encoding resistance to chloramphenicol), and sulII (encoding resistance to sulfonamide) (Figure 1). The structural analysis showed that the ICE is a new ICE variant with T4SS. BLAST analysis showed that the new ICE had 70% nucleotide identity to P. mirabilis (MD20140904) and 56% nucleotide identity to Shewanella sp. (W3-18-1). Structures are drawn to scale from GenBank accession numbers. Shared regions with 99% identity are indicated by shading. Based on the comparative analysis, the ICE possessed many homologous genes in the gene cluster (link with dark line). Homologous regions include a series of DNA replication-related genes.
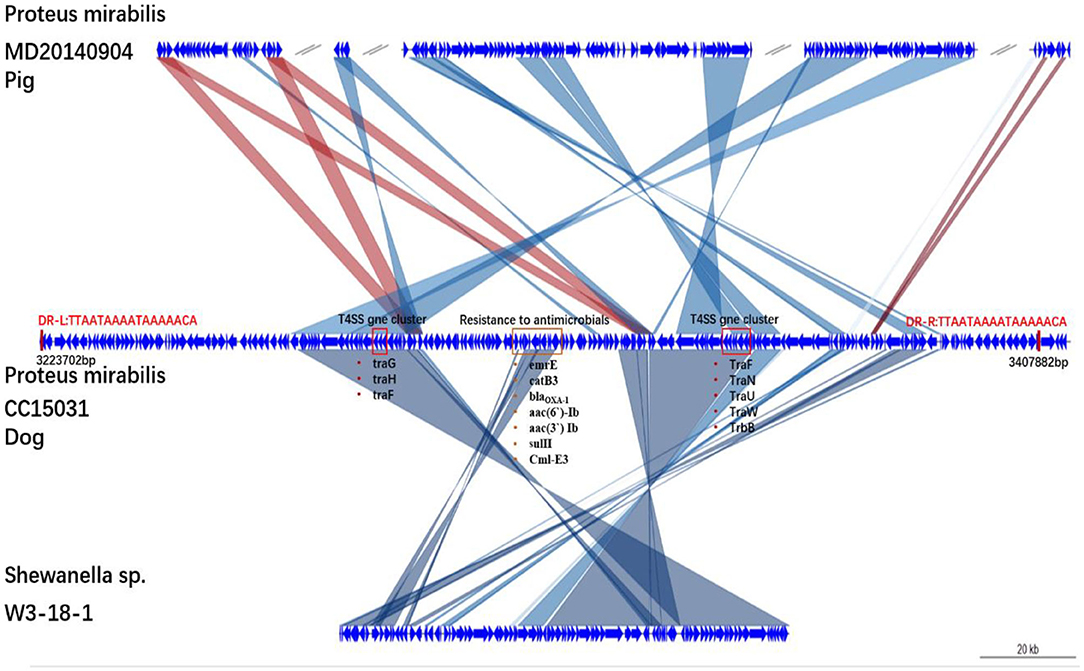
Figure 1. Genetic structures of SGI1-CC15031. Structures are drawn to scale from GenBank accession numbers MD20140904, W3-18-1. Genes and open reading frames are shown as arrows, and their orientations of transcription are indicated by the arrowheads. Shared regions with above 99% identity are indicated by shading. Resistance genes and integrase genes are in a red box. DR-L and DR-R represent the 18-bp direct repeats at the ends of SGI1.
P. mirabilis is a conditioned zoonotic pathogen. It has been isolated from a variety of animal species, such as chicken, ducks, dogs, foxes, goats, mink, and other animals, and considered to be the host of storing drug resistance and virulence genes. However, the characteristics of drug resistance and virulence genes of the typical strains of P. mirabilis isolated from dogs have not yet been reported. A total of 12 P. mirabilis strains were isolated from the diarrheal specimen of dogs in Changchun during 2015–2017. From which, the P. mirabilis CC15031 is closely related to RCAD120210 isolated from geese in Sichuan, China. The results of the virulence and drug sensitivity test showed CC15031 possessed strong pathogenicity and high multidrug resistance.
The result of the drug resistance spectrum of P. mirabilis CC15031 was slightly different from that of other animal and human sources. In another study from Africa, 73 strains of P. mirabilis isolated from a University hospital were found to be susceptible to ampicillin, chlorobenzicillin, amoxicillin, tetracycline, compound triazole, erythromycin, and chloramphenicol. Also, high resistance to ampicillin, chlorobenzicillin, amoxicillin, tetracycline, compound triazole, and erythromycin was observed. Furthermore, P. mirabilis has developed drug resistance in recent years, which might be related to the excessive use of antibiotics by clinicians. Also, the specific physiological characteristics of the bacterium promoted its drug resistance.
The improvement in the genomic data can provide a reference for the typing and diagnosis of pathogenic microorganisms (12). A highly resistant and pathogenic strain P. mirabilis CC15031 was isolated in this study; however, the gene–environment related to drug resistance and pathogenicity of CC15031 is not yet clarified. Therefore, we sequenced the whole genome of CC15031 and analyzed it by comparative genomics, obtained the genome map, analyzed its gene function, and screened the drug resistance and virulence genes. CC15031 had a 4,031,742-bp long genome, consisting of a total of 3,705 coding genes, including 363 virulence factors and 18 drug resistance genes.
The particular feature of P. mirabilis was the excellent bacterial motility; so far, it is the microorganism with the highest amount of potential fimbriae. The biofilm, pili, and flagella of P. mirabilis play an important role in bacterial movement, adhesion and colonization, host invasion capability, and so on. As for the virulence factors, 26 virulence factors related to biofilm formation, 9 virulence factors related to flagellum formation, and 12 virulence factors related to pili formation were identified by using the Virulence Factor Database.
The three most studied P. mirabilis fimbriae were identified in CC15031, P. mirabilis fimbriae (AI070), uroepithelial cell adhesion (AI071), and mannose-resistant/Proteus-like (AI072). The fimbriae with the most genes annotated were the mannose-resistant/Proteus-like fimbriae (AI072). It is the major contributor to urinary tract infection, and the most well-studied fimbriae encoded by P. mirabilis could agglutinate erythrocytes independent of D-mannose (13). Our strain presents two additional copies of ucaA and other fimbriae related-protein genes that are not present in HI4320.
Also, the detection of E. coli (CVF6259) and Hemorrhagic coli (AI102, CVF825) pilus indicated the integration of exogenous virulence factors; this might be part of the reason for the increased virulence of the CC15031.
More importantly, we have discovered a new ICE variant with T4SS. Based on the comparative analysis, the ICE possessed many homologous genes in the gene cluster (link with dark line). Homologous regions include a series of DNA replication-related genes. Previous studies have found that a large number of ICEs have been identified in several genera of Gammaproteobacteria (http://db-mml.sjtu.edu.cn/ICEberg/) (14); most of them carry a large number of drug-resistant genes. In recent years, many ICEs had been reported in P. mirabilis (7, 15–18); most of them carry SXT/R391 (19). Notably, two SXT/R391 ICEs, ICEPmiJpn1 and ICEPmiSpn1 (20, 21), carry the AmpC-lactamase gene blaCMY-2 mediating resistance to a broad spectrum of β-lactams and β-lactamase inhibitors, indicating that SXT/R391 ICEs can mediate the dissemination of clinically important resistance genes. Here, we characterized another novel ICE with T4SS that contained eight different antimicrobial resistance genes in P. mirabilis, including blaOXA1 and blaCTX-M, which could confer resistance to penicillin and ceftazidime, respectively. This new ICE has a longer sequence than previously reported. Whether the four-type secretory system it carries mediates the more serious pathogenicity of the bacterium needs further research.
In conclusion, we found that P. mirabilis with explosive growth characteristics is the host of a large number of drug resistance and virulence genes, which might pose a threat to public health safety, requiring high attention. The biological and genetic evolutionary characteristics of P. mirabilis CC15031 will provide a reference for the prevention of human and animal diseases caused by P. mirabilis.
The datasets generated for this study can be found in the NCBI Database [accession: PRJNA596082].
The animal study was reviewed and approved by the Animal Care and Use Committee of the Jilin Agricultural University.
LK, RH, and XW performed the experiments. RH, IM, YiW, WD, HZ, YuW, and SL collected the samples. RH and LK wrote the manuscript. HM, YG, and LK approved the final manuscript.
This work was supported by the Jilin Province Science and Technology Department Theme Guide Project (20180101003JC), National Natural Science Foundation of China (31702293), Jilin Natural Science Foundation (20190201294JC), and Doctoral Fund of Jilin Agricultural University (201801).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Biomarker Technology Co., Ltd., Beijing, China, for high throughput sequencing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2020.00589/full#supplementary-material
1. Fonseca MRB, Sato JL, Lima-Noronha MA, Migliorini LB, Fernandez-Silva FS, Galhardo RS. Increased mutability to fosfomycin resistance in Proteus mirabilis clinical isolates. Infect Genet Evol. (2018) 58:27–33. doi: 10.1016/j.meegid.2017.12.012
2. Cernohorska L, Chvilova E. [Proteus mirabilis isolated from urine, resistance to antibiotics and biofilm formation]. Klin Mikrobiol Infekc Lek. (2011) 17:81–5.
3. Costantini E, Giannitsas K, Illiano E. The role of nonantibiotic treatment of community-acquired urinary tract infections. Curr Opin Urol. (2017) 27:120–6. doi: 10.1097/MOU.0000000000000366
4. Luthje P, Brauner A. Virulence factors of uropathogenic E. coli and their interaction with the host. Adv Microb Physiol. (2014) 65:337–72. doi: 10.1016/bs.ampbs.2014.08.006
5. de Curraize C, Siebor E, Varin V, Neuwirth C, Hall RM. Two new SGI1-LK variants found in Proteus mirabilis and evolution of the SGI1-HKL Group of Salmonella Genomic Islands. mSphere. (2020) 5:e00875-19. doi: 10.1128/mSphere.00875-19
6. Siebor E, Neuwirth C. The new variant of Salmonella genomic island 1 (SGI1-V) from a Proteus mirabilis French clinical isolate harbours blaVEB-6 and qnrA1 in the multiple antibiotic resistance region. J Antimicrob Chemother. (2011) 66:2513–20. doi: 10.1093/jac/dkr335
7. Wang XC, Lei CW, Kang ZZ, Zhang Y, Wang HN. IS26-mediated genetic rearrangements in Salmonella Genomic Island 1 of Proteus mirabilis. Front Microbiol. (2019) 10:2245. doi: 10.3389/fmicb.2019.02245
8. Lei CW, Zhang AY, Liu BH, Wang HN, Yang LQ, Guan ZB, et al. Two novel Salmonella genomic island 1 variants in Proteus mirabilis isolates from swine farms in China. Antimicrob Agents Chemother. (2015) 59:4336–8. doi: 10.1128/AAC.00120-15
9. Soliman AM, Ahmed AM, Shimamoto T, El-Domany RA, Nariya H, Shimamoto T. First report in Africa of two clinical isolates of Proteus mirabilis carrying Salmonella genomic island (SGI1) variants, SGI1-PmABB and SGI1-W. Infect Genet Evol. (2017) 51:132–7. doi: 10.1016/j.meegid.2017.03.029
10. Zhang Y, Zeng J, Liu W, Zhao F, Hu Z, Zhao C, et al. Emergence of a hypervirulent carbapenem-resistant Klebsiella pneumoniae isolate from clinical infections in China. J Infect. (2015) 71:553–60. doi: 10.1016/j.jinf.2015.07.010
11. Almofti YA, Dai M, Sun Y, Haihong H, Yuan Z. Impact of erythromycin resistance on the virulence properties and fitness of Campylobacter jejuni. Microb Pathog. (2011) 50:336–42. doi: 10.1016/j.micpath.2011.02.009
12. Hasman H, Saputra D, Sicheritz-Ponten T, Lund O, Svendsen CA, Frimodt-Moller N, et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J Clin Microbiol. (2014) 52:139–46. doi: 10.1128/JCM.02452-13
13. Himpsl SD, Pearson MM, Mobley HL. Using hemagglutination, surface shearing, and acid treatment to study fimbriae in Proteus mirabilis. Methods Mol Biol. (2019) 2021:109–20. doi: 10.1007/978-1-4939-9601-8_11
14. Chen F, Zhang W, Schwarz S, Zhu Y, Li R, Hua X, et al. Genetic characterization of an MDR/virulence genomic element carrying two T6SS gene clusters in a clinical Klebsiella pneumoniae isolate of swine origin. J Antimicrob Chemother. (2019) 74:1539–44. doi: 10.1093/jac/dkz093
15. Bie L, Fang M, Li Z, Wang M, Xu H. Identification and characterization of new resistance-conferring SGI1s (Salmonella Genomic Island 1) in Proteus mirabilis. Front Microbiol. (2018) 9:3172. doi: 10.3389/fmicb.2018.03172
16. Siwinska M, Levina EA, Shashkov AS, Kalinchuk NA, Drzewiecka D, Knirel YA. Structural and serological characterization of the O82 antigen of a Proteus mirabilis strain isolated from a patient in Poland. Carbohydr Res. (2019) 486:107831. doi: 10.1016/j.carres.2019.107831
17. Sun L, Xu J, He F. Genomic characterisation of a Proteus mirabilis clinical isolate from China carrying blaNDM-5 on an IncX3 plasmid. J Glob Antimicrob Resist. (2019) 19:317–9. doi: 10.1016/j.jgar.2019.10.025
18. Tran J, Lessor L, O'Leary C, Gill J, Liu M. Complete genome sequence of Proteus mirabilis phage myduc. Microbiol Resour Announc. (2019) 8:e01313–19. doi: 10.1128/MRA.01313-19
19. Lei CW, Chen YP, Kang ZZ, Kong LH, Wang HN. Characterization of a novel SXT/R391 integrative and conjugative element carrying cfr, blaCTX-M-65, fosA3, and aac(6')-Ib-cr in Proteus mirabilis. Antimicrob Agents Chemother. (2018) 62:e00849–18. doi: 10.1128/AAC.00849-18
20. Abril C, Brodard I, Perreten V. Two novel antibiotic resistance genes, tet(44) and ant(6)-Ib, are located within a transferable pathogenicity island in Campylobacter fetus subsp. fetus. Antimicrob Agents Chemother. (2010) 54:3052–5. doi: 10.1128/AAC.00304-10
Keywords: Proteus mirabilis, antibiotic resistance, complete genome sequencing, resistance gene, virulence gene
Citation: Hu R, Wang X, Muhamamd I, Wang Y, Dong W, Zhang H, Wang Y, Liu S, Gao Y, Kong L and Ma H (2020) Biological Characteristics and Genetic Analysis of a Highly Pathogenic Proteus Mirabilis Strain Isolated From Dogs in China. Front. Vet. Sci. 7:589. doi: 10.3389/fvets.2020.00589
Received: 24 February 2020; Accepted: 22 July 2020;
Published: 07 October 2020.
Edited by:
Chi-Chung Chou, National Chung Hsing University, TaiwanReviewed by:
Paola Scavone, Instituto de Investigaciones Biológicas Clemente Estable (IIBCE), UruguayCopyright © 2020 Hu, Wang, Muhamamd, Wang, Dong, Zhang, Wang, Liu, Gao, Kong and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingcong Kong, Y29uZ3dic0AxMjYuY29t; Hongxia Ma, aG9uZ3hpYTA3MzEwMDFAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.