 Jun Sun1*
Jun Sun1* Masanori Aikawa2
Masanori Aikawa2 Hassan Ashktorab3
Hassan Ashktorab3 Noam D. Beckmann4,5
Noam D. Beckmann4,5 Michael L. Enger6
Michael L. Enger6 Joaquin M. Espinosa7
Joaquin M. Espinosa7 Xiaowu Gai8,9
Xiaowu Gai8,9 Benjamin D. Horne10,11
Benjamin D. Horne10,11 Paul Keim12,13,14Jessica Lasky-Su15
Paul Keim12,13,14Jessica Lasky-Su15 Rebecca Letts16Cheryl L. Maier17
Rebecca Letts16Cheryl L. Maier17 Meisha Mandal6Lauren Nichols16
Meisha Mandal6Lauren Nichols16 Nadia R. Roan18,19
Nadia R. Roan18,19 Mark W. Russell20
Mark W. Russell20 Jacqueline Rutter16George R. Saade21,22Kumar Sharma23,24
Jacqueline Rutter16George R. Saade21,22Kumar Sharma23,24 Stephanie Shiau25
Stephanie Shiau25 Stephen N. Thibodeau26
Stephen N. Thibodeau26 Samuel Yang27
Samuel Yang27 Lucio Miele28* NIH Researching COVID to Enhance Recovery (RECOVER) Consortium
Lucio Miele28* NIH Researching COVID to Enhance Recovery (RECOVER) Consortium- 1Department of Medicine, Division of Gastroenterology and Hepatology, University of Illinois Chicago, Chicago, IL, United States
- 2Cardiovascular Division and Channing Division of Network Medicine, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, United States
- 3Department of Medicine, Howard University, Washington, DC, United States
- 4Department of Medicine, Division of Data Driven and Digital Medicine (D3M), New York, NY, United States
- 5Charles Bronfman Institute for Personalized Medicine, Mount Sinai Clinical Intelligence Center, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 6RTI International, Durham, NC, United States
- 7Linda Crnic Institute for Down Syndrome, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 8Department of Pathology and Laboratory Medicine, Children’s Hospital Los Angeles, Los Angeles, CA, United States
- 9Department of Pathology, Keck School of Medicine, University of Southern California, Los Angeles, CA, United States
- 10Intermountain Medical Center Heart Institute, Murray, UT, United States
- 11Department of Medicine, Division of Cardiovascular Medicine, Stanford University, Stanford, CA, United States
- 12Department of Biology, Northern Arizona University, Flagstaff, AZ, United States
- 13Pathogens Genomics Program, Translational Genomics Institute (TGen), Phoenix, AZ, United States
- 14Department of Biology, University of Oxford, Oxford, United Kingdom
- 15Channing Department of Network Medicine, Brigham and Women’s Hospital, Harvard University, Boston, MA, United States
- 16RECOVER patient representative, Durham, NC, United States
- 17Department of Pathology and Laboratory Medicine, Emory University School of Medicine, Atlanta, GA, United States
- 18Gladstone Institute of Virology, Gladstone Institutes, San Francisco, CA, United States
- 19Department of Urology, University of California San Francisco, San Francisco, CA, United States
- 20Department of Pediatrics, Division of Pediatric Cardiology, University of Michigan, Ann Arbor, MI, United States
- 21Department of Obstetrics and Gynecology, University of Texas Medical Branch, Galveston, TX, United States
- 22Department of Obstetrics and Gynecology, Eastern Virginia Medical School, Norfolk, VA, United States
- 23Center for Precision Medicine, University of Texas San Antonio Health Sciences Center, San Antonio, TX, United States
- 24Department of Medicine, Division of Nephrology, University of Texas San Antonio Health Sciences Center, San Antonio, TX, United States
- 25Department of Biostatistics and Epidemiology, Rutgers School of Public Health, Piscataway, NJ, United States
- 26Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, United States
- 27Department of Emergency Medicine, Stanford University, Stanford, CA, United States
- 28Department of Genetics, School of Medicine, Louisiana State University Health Sciences, Center New Orleans, New Orleans, LA, United States
Post-Acute Sequelae of SARS-CoV-2 infection (PASC or “Long COVID”), includes numerous chronic conditions associated with widespread morbidity and rising healthcare costs. PASC has highly variable clinical presentations, and likely includes multiple molecular subtypes, but it remains poorly understood from a molecular and mechanistic standpoint. This hampers the development of rationally targeted therapeutic strategies. The NIH-sponsored “Researching COVID to Enhance Recovery” (RECOVER) initiative includes several retrospective/prospective observational cohort studies enrolling adult, pregnant adult and pediatric patients respectively. RECOVER formed an “OMICS” multidisciplinary task force, including clinicians, pathologists, laboratory scientists and data scientists, charged with developing recommendations to apply cutting-edge system biology technologies to achieve the goals of RECOVER. The task force met biweekly over 14 months, to evaluate published evidence, examine the possible contribution of each “omics” technique to the study of PASC and develop study design recommendations. The OMICS task force recommended an integrated, longitudinal, simultaneous systems biology study of participant biospecimens on the entire RECOVER cohorts through centralized laboratories, as opposed to multiple smaller studies using one or few analytical techniques. The resulting multi-dimensional molecular dataset should be correlated with the deep clinical phenotyping performed through RECOVER, as well as with information on demographics, comorbidities, social determinants of health, the exposome and lifestyle factors that may contribute to the clinical presentations of PASC. This approach will minimize lab-to-lab technical variability, maximize sample size for class discovery, and enable the incorporation of as many relevant variables as possible into statistical models. Many of our recommendations have already been considered by the NIH through the peer-review process, resulting in the creation of a systems biology panel that is currently designing the studies we proposed. This system biology strategy, coupled with modern data science approaches, will dramatically improve our prospects for accurate disease subtype identification, biomarker discovery and therapeutic target identification for precision treatment. The resulting dataset should be made available to the scientific community for secondary analyses. Analogous system biology approaches should be built into the study designs of large observational studies whenever possible.
1 Introduction
The term Post-Acute Sequelae of SARS-CoV-2 infection (PASC), also known as “Long COVID”, refers to numerous conditions associated with widespread morbidity and rising healthcare costs. PASC has highly variable clinical presentations, and likely includes multiple molecular subtypes (Thompson et al., 2023; Sherif et al., 2023). The NIH-sponsored “Researching COVID to Enhance Recovery” (RECOVER) initiative includes retrospective/prospective cohort studies including an adult cohort (Horwitz et al., 2023), a cohort of pregnant adults (Metz et al., 2023; Reel et al., 2021) and a pediatric cohort (Gross et al., 2024; Reel et al., 2021). These studies aim to enroll a total of 12,580 adult non-pregnant patients, 2,300 adult pregnant patients and 19,300 pediatric patients to rapidly improve our understanding of and ability to predict, treat, and prevent Post-Acute Sequelae of SARS-CoV-2 infection (PASC, or “Long COVID”) through deep clinical phenotyping and laboratory studies. THE RECOVER “OMICS” Task Force was charged with developing recommendations based on published evidence and the experiences of its members, to incorporate multi-omics into the analysis of RECOVER results.
2 Methods
2.1 Objectives
The “OMICS” task force of the RECOVER study, a multi-disciplinary committee including clinicians, pathologists, laboratory scientists and data scientists, was charged with developing recommendations to apply cutting-edge system biology technologies to achieve the goals of RECOVER. The task force met biweekly over 14 months, to evaluate published evidence, examine the possible contribution of each “omics” technique to the study of PASC, as well as the potential limitations of each technique, and develop a consensus recommendation. The work was divided into two stages. During the first stage, sub-committees with specific expertise on an “omics” technique examined evidence supporting the use of that technique to study PASC, the type of data it could generate and the mechanistic questions it could answer, based on published evidence and the experiences of its members, to incorporate multi-omics into the analysis of RECOVER results. Each sub-committee presented to the entire task force. During the second stage, the task force combined the findings of each sub-committee into a comprehensive study design recommendation.
3 Results and discussion
The OMICS task force recommended that integrated, longitudinal, simultaneous multi-omics studies of participant biospecimens be performed on the entire RECOVER cohort through centralized laboratories, as opposed to multiple smaller studies using one or few analytical techniques.
The RECOVER adult protocol (Horwitz et al., 2023) includes multiple biospecimen collections: nasopharyngeal or nasal swab, 2 8.5 mL aliquots of blood in serum separation tubes, 4 × 8 ml aliquots of blood in cell preparation tubes, 2 × 2.7 mL aliquots of blood in sodium citrate tubes for plasma proteomics, 1 × 10 ml aliquot of blood in EDTA tube, 1 2.5 mL aliquot of blood in PAXgene RNA tube, 1 × 10 ml urine (no additives), 1 × 2mL aliquot of saliva in Oragene OGR 600 and 1 25 mL aliquot of stool. Of these, stool is sent by participants while the other samples are processed locally as per protocol specifications and shipped in batches to the central tissue bank. Participants who consent to biospecimen donation for future research are asked to provide blood and nasopharyngeal/nasal swab biospecimens at enrollment, 90 and 180 days after the index date (date of first infection or negative COVID test), and then annually (Horwitz et al., 2023). Saliva is collected upon enrollment for genetic analysis. Urine and stool are collected biannually. Additionally, a battery of clinical laboratory tests is performed in CLIA-certified laboratories at enrollment, 90 and 189 days after the index date, and thereafter, abnormal tests are repeated annually. Specific symptoms or abnormal study results trigger “Tier 2” or “Tier 3” assessments (see (Horwitz et al., 2023) for details). A SARS-CoV-2 PCR test is performed at enrollment for all “uninfected” participants, who are also tested for SARS-CoV-2 nucleocapsid antibodies spike protein antibodies for unvaccinated participants.
In addition to study visits, imaging and laboratory tests, participants complete multiple surveys, using validated survey instruments whenever possible, at 90-day intervals throughout the study. At enrollment, data are collected on demographics, social determinants of health (SDOH), disability, characteristics of the initial SARS-CoV-2 infection (if applicable), pregnancy (if applicable), vaccination status, comorbidities, medications, and PASC symptoms. Subsequently, at 90-day intervals, data are collected on interim infections, time-varying social determinants, vaccinations, comorbidities, medications and symptoms (Horwitz et al., 2023). The PASC symptom survey was developed for RECOVER and includes an overall quality of life instrument (PROMIS-10) and screening for core symptoms (43 for biological males and 46 for biological females) drawn from existing literature plus input from patient representatives and investigators. Questions about depression, anxiety, post-traumatic stress disorder (PTSD), and grief are also included. Report of a symptom may trigger additional questions about that symptom. Details of survey instruments are in the original reference (Horwitz et al., 2023).
The pregnancy study (Metz et al., 2023) follows a similar design to the non-pregnant adult study, enrolling participants with suspected, probable or confirmed SARS-CoV-2 infection during pregnancy, or documented lack of exposure to SARS-CoV-2 during pregnancy. Study procedures and biospecimen collections are analogous to those in the non-pregnant adult study (Metz et al., 2023), with modifications for breastfeeding or postpartum participants, and additional health and developmental assessments for babies exposed in utero to SARS-CoV-2.
The RECOVER pediatric study (Gross et al., 2024) has a similar design, with limitations due to the age range of participants. All pediatric participants complete a single Tier 1 visit including PROMIS global health measures and symptom screening. This visit includes a donation of saliva and capillary blood. Depending on infection status, clinical history, symptoms and probability of PASC, pediatric participants are promoted to Tier 2 or Tier 3, which include additional biospecimen donations during the acute and post-acute phase of PASC, as well as additional clinical assessments and surveys. The types, aliquot numbers, and cadence of biospecimen collections are described in detail in (Gross et al., 2024).
In summary, each RECOVER study will generate vast longitudinal datasets including clinical, demographic, medication, SDOH and lifestyle data for each participant, as well as sufficient types and numbers of biospecimen aliquots to permit a comprehensive, longitudinal multi-omics investigation. Potential environmental exposures can furthermore be estimated from census tract or ZIP code data.
The multi-dimensional molecular dataset generated by the multi-omics investigation should be correlated with the deep clinical phenotyping performed through RECOVER, as well as with information on demographics, comorbidities, social determinants of health, the exposome and lifestyle factors collected through RECOVER surveys, that may contribute to the clinical presentations of PASC. Data generation and analytical strategies should leverage integrative bioinformatics and machine learning.
A major advantage, and a potential challenge, of multi-omics approaches is that datasets derived from different analytical techniques and measured using different scales must be integrated. Approaches including multi-omics integration paired with ML have been gaining popularity in clinical and biomedical research (see (Reel et al., 2021; Niranjan et al., 2023)), though this field is rapidly evolving. An important advantage inherent in multi-dimensional measurements is that the extent to which different measurements agree with each other or not is potentially informative. For instance, transcriptomic data may or may not be reflected in the relative abundance of protein products, or quantitative differences in non-coding RNA expression may or may not translate into relative abundance of potential target mRNAs, the proteins they encode or the metabolites that these proteins may process. System biology approaches based on multi-omics have been used successfully in the study of cardiovascular disease (Joshi et al., 2021).
With respect to PASC, strategies similar to what we propose have been used on a smaller scale. ML has been used in the context of a multi-step analytical strategy to combine proteomic and metabolomic data to generate a multi-omics biomarker predictive of the risk of PASC (Wang et al., 2023) and give insights on the metabolic pathways altered during PASC. Dimensionality reduction was achieved through unsupervised cluster analysis followed by autoencoder (AE), using a three-layer neural network. Supervised ML was then used to identify the minimal number of molecules predictive of adverse clinical outcomes. This study, though very promising, was limited by small sample size (117, of whom 105 were used as a training cohort for model development and 10% as a validation cohort), the severity of acute COVIDs in the patients enrolled and the absence of a vaccinated group. Despite these limitations, these results indicate that similar analytical strategies can be used successfully on a much larger sample with broader phenotyping, to discover predictive biomarkers, therapeutic targets and risk factors and to generate mechanistic hypotheses.
Within the RECOVER study, an unsupervised ML approach has been used to identify clinical subtypes of PASC, after symptoms differentiating infected from uninfected patients were identified using LASSO (least absolute shrinkage and selection operator) (Thaweethai et al., 2023).
Highly multiplexed “omics” approaches measure common clinical analytes and many more parameters (Table 1) at a fraction of the cost of traditional clinical tests, oftentimes using similar quantities of specimens (Table 2). In a multi-omics approach, analytes within each category (e.g., proteins, lipids, nucleic acids, metabolites, and microbes) are all measured simultaneously, generating high-content data that is more than the sum of its parts. This approach allows the discovery of new molecular signatures to enhance our understanding of complex disease pathophysiology. These signatures may occur within a single analyte category, but more likely cover more complex patterns that span multiple molecular layers, e.g., genomics, epigenomics, transcriptomics, proteomics, lipidomics, metabolomics, and microbiomics (Figure 1). Deep multi-omics profiling will allow us to explore a broad spectrum of pathophysiological mechanisms (Table 3), define gene-environment interactions involved in the pathogenesis of PASC, identify molecular subtypes and candidate biomarkers and propose mechanism-based therapeutic strategies. The relative contributions of each “omics” we evaluated and considerations on data generation and analysis are described below.
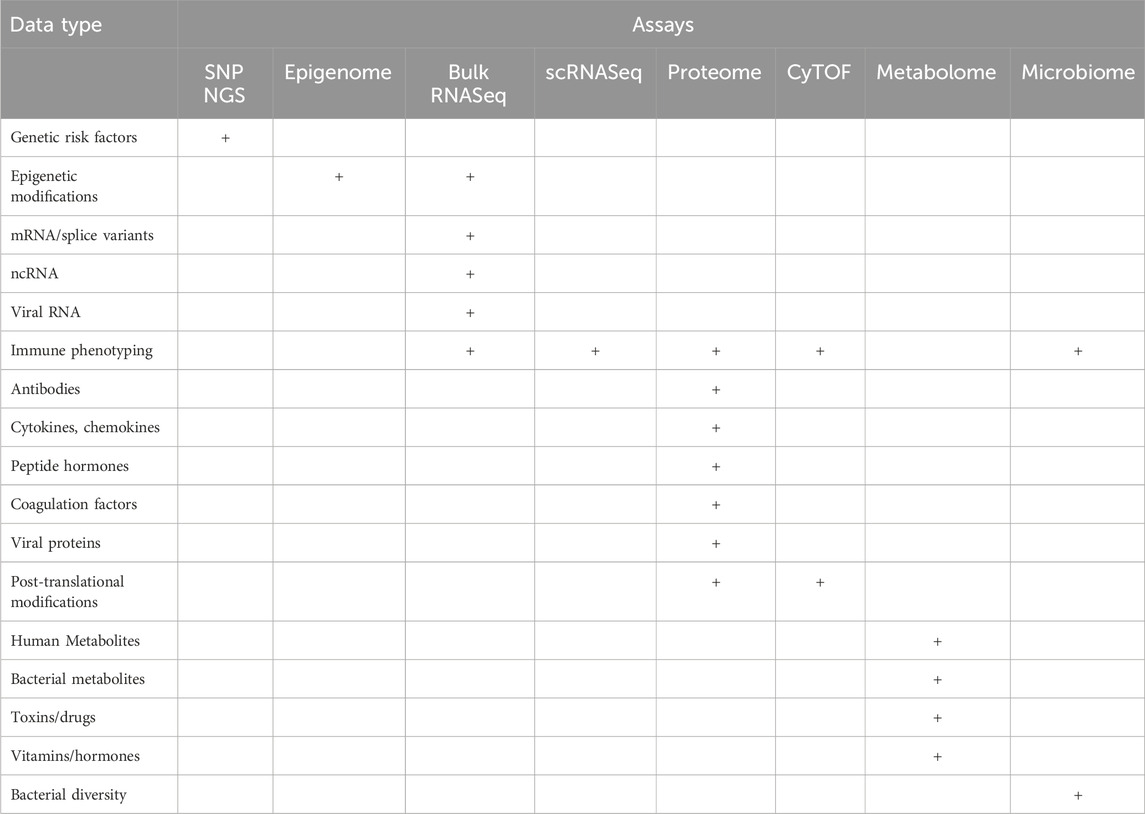
Table 1. Complementary data types captured by multi-omics assays.
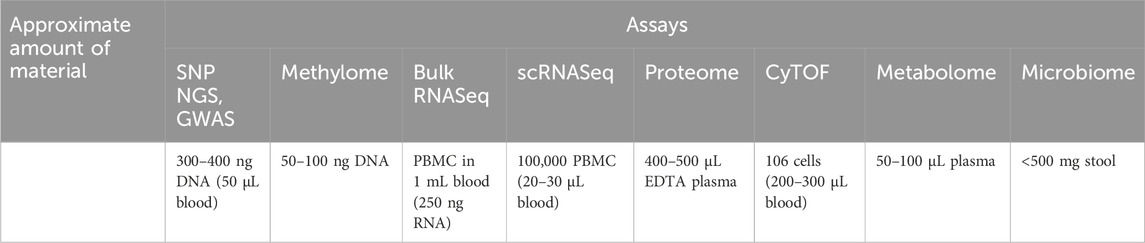
Table 2. Approximate sample requirements for multi-omics assays.
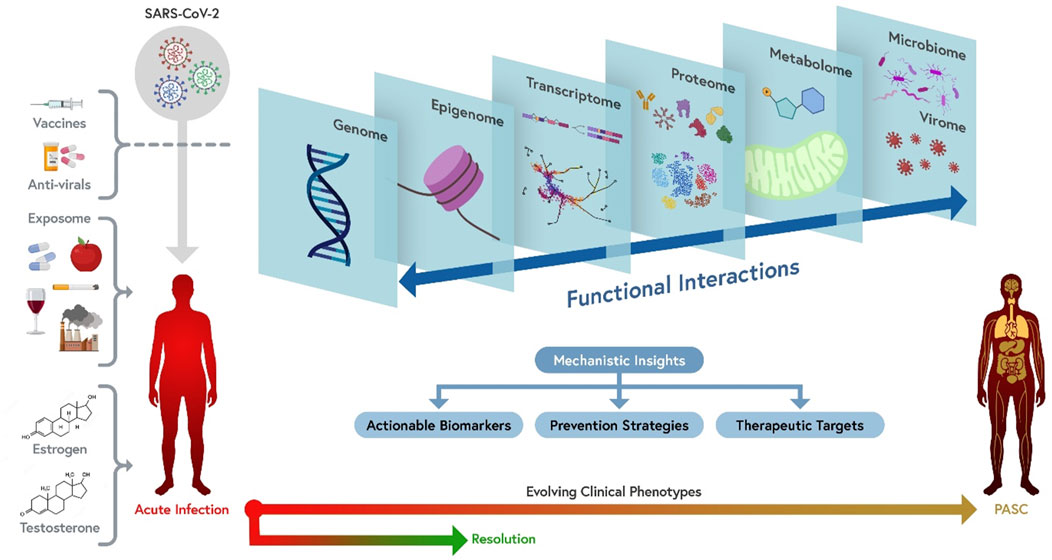
Figure 1. A comprehensive multi-omics approach to the mechanism(s) of PASC. From left to right: PASC is a consequence of infection with SARS-CoV-2. Different viral variants or sub-variants (represented in different colors) may have different probability of causing PASC or be associated with different presentations (e.g., due to different ability to cause persistent infection, to trigger pathogenic antibody responses, or to damage vascular endothelium). Vaccines and anti-viral agents can decrease the risk of PASC by interfering with viral persistence and replication. Multiple exposures, including diet, medications, tobacco, alcohol, environmental pollutants and co-morbidities, socioeconomic and psychosocial exposures, as well as sex hormones, can potentially affect the risk and clinical presentations of PASC. The combined effect of these factors results into evolving clinical phenotypes ranging from acute COVID-19 resolution to PASC through a number of mechanisms that can be best understood by simultaneously interrogating the multi-omics landscape of patients, including individual genomics, epigenomics, bulk and single-cell transcriptomics, plasma and cellular proteomics, metabolomics, and microbiome/virome. These different dimensions functionally interact with one another to determine pathogenetic mechanisms (e.g., persistent viral infection, modulated by individual genetics, triggers immune, inflammatory and metabolic changes that are in turn modulated by the intestinal and respiratory microbiomes and potentially by reactivation of other viruses). Insights generated by an integrated multi-omics investigation of patients with well-characterized clinical phenotypes are likely to identify actionable biomarkers (which may discriminate between PASC molecular subtypes), as well as therapeutic targets and prevention strategies. Orthogonal multi-omics tests repeated over time are the most informative approach to capture the pathogenesis of the different clinical presentations of PASC and their evolution over time.
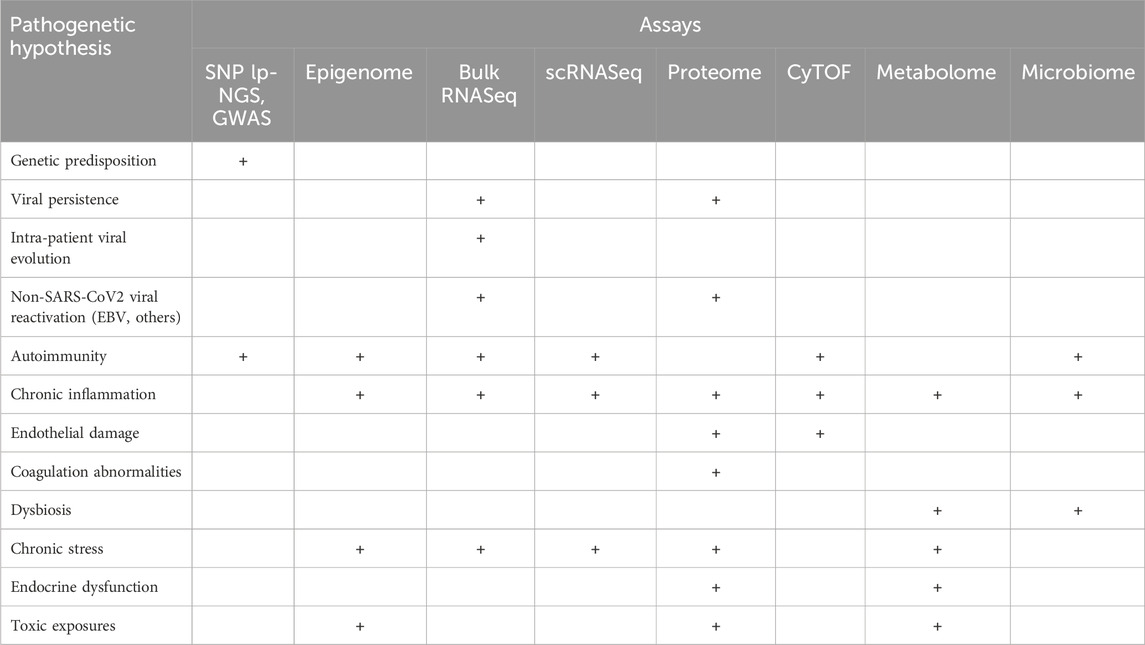
Table 3. Multi-omics assays generate information relevant to testing multiple mechanistic hypotheses for PASC.
3.1 Evidence supporting multi-omics technologies used in COVID-19 and PASC studies
3.1.1 Genomics
Genomics is an invaluable asset to understand disease risk, mechanism and etiology, and to serve as a backbone to allow for better modeling of multi-omics profiles in patient populations. Several genome-wide association studies (GWAS) have identified reproducible associations between specific loci and risk and outcomes of acute COVID-19 (Ferreira et al., 2022) with the most reproducible being with LZTFL1 and contiguous regions on 3q21.31 and ABO on 9q34.2. A recent GWAS study, currently in pre-print, detected an association between a locus near the FOXP4 gene and risk of developing PASC (Lammi et al., 2023). That study analyzed data from 24 studies conducted in 16 countries, totaling 6,450 PASC cases and 1,093,995 controls. However, most of the patients were of European ancestry, and this study should be replicated in a more diverse cohort. In GWAS studies, sample size and composition of study population (e.g., case/control ratio, ancestry, genetic admixture, etc.) are critical. FOXP4 is a broadly expressed transcription factor. Lammi et al. (Lammi et al., 2023) analyzed single-cell RNASeq data to confirm the expression of FOXP4 in surfactant-producing Type II alveolar cells and granulocytes. This correlation supports a possible mechanistic link, and demonstrates the importance of integrative multi-omics approaches. A recent computational study analyzed the evolution of predicted CD8 T-cell epitopes in SARS-CoV-2 variants and its correlation with clinical outcomes of acute COVID-19 in patients with different HLA genotypes, illustrating the importance of integrated analysis of viral and patient genomic data with clinical data (Kim et al., 2024). A similar approach could be used with PASC, and/or PASC clinical subtype, as an outcome. Beyond GWAS or other genetic analyses, genotyping data can be used in conjunction with other multi-omics profiles to increase the likelihood of discovery. Identification of molecular quantitative trait loci (QTL) can be used to identify possible pathogenetic pathways (Debnath et al., 2020). Different technologies can be used to obtain genotyping information in PASC cases: high-density SNP-chips or low-pass sequencing are established platforms. Emerging technologies, such as nanopore long-read sequencing (Cuppen et al., 2022; Pervez et al., 2022), may also reduce the cost of whole-genome and whole-transcriptome sequencing.
3.1.2 Epigenomics
Epigenomics measure molecular events that regulate chromatin accessibility and expression, which can reflect long-term physiological states. Using methods such as chromatin immunoprecipitation sequencing (ChIP-seq), CUT&RUN, or assay for transposase-accessible chromatin using sequencing (ATAC-seq) (Yan et al., 2020; Sun et al., 2019), which can also be used in single-cell applications (Kashima et al., 2020), many epigenetic processes have been identified and associated with complex traits. One of the best characterized is DNA methylation (5 methyl-cytosine), which is altered in numerous human diseases. There is compelling evidence that changes in DNA methylation profiles are detectable in viral infections such as HIV (Bednarik et al., 1990; Zhang et al., 2016) and MERS (Menachery et al., 2018). In an epigenome-wide association study (EWAS) (Bhat and Jones, 2022), DNA methylation differences associated with a phenotype can be assessed at hundreds of thousands of cytosine-phosphate-guanine (CpG) sites across the epigenome. Several EWAS of COVID-19 in the literature found distinct patterns of DNA methylation associated with disease severity early in the disease course (Castro de Moura et al., 2021; Corley et al., 2021; Balnis et al., 2021; Zhou et al., 2021). EWAS in the ongoing Norwegian Corona Cohort Study also assessed whether there were differentially methylated CpGs between those with PASC (N = 41) compared to a remission group (N = 63), but did not find significant differences. However, the study was not longitudinal, and the authors point out that their sample size for PASC was small (Lee et al., 2022). The same study identified 3 differentially methylated sites associated with acute COVID-19 severity, including hypomethylation of IFI44L, an interferon response gene also associated with COVID-19 severity (Castro de Moura et al., 2021).
3.1.3 Transcriptomics
a) Bulk Transcriptomics: RNA transcripts act as intermediary components between genetic information and protein synthesis, and carry specific functions themselves. Transcripts are a regulation hub that responds to both environmental and genetic control, thus playing a major role in the molecular characterization of diseases. Non-coding RNAs fine-tune the expression levels of coding RNAs and their protein products, providing an additional level of regulation. Given its role as an ‘integration hub’ between genetic variation and environmental exposures, the transcriptome dataset is a key layer in multi-omics approaches. Bulk RNA sequencing (RNASeq) can measure the relative abundance of individual transcripts, and determine differences in mRNA splicing isoforms and RNA editing. Whole blood transcriptome analysis can accurately measure the expression levels of >16,000-20,000 RNA species, both protein-coding and non-coding, thus providing one of the most high-quality and high-content multi-omics datasets. Bulk transcriptomics integrates the effects of multiple key variables that can dynamically affect gene expression in blood cells (e.g., metabolic state, epigenetic variation, exposure to medications, stress etc.). Transcriptional signatures in the blood or cells of COVID-19 patients can help identify causal factors for acute or chronic complications as well as potential therapeutic targets (Jha et al., 2020; Asano et al., 2022). Recently, a long non-coding RNA-based ML model has been used to identify an RNA (LEF1-AS1) predictive of acute COVID-19 mortality in a ML-driven study of 1,286 patients in 15 institutions (Devaux et al., 2024). Additionally, a candidate signature of acute COVID-19 including 3 long non-coding RNA, 2 cytokines and 2 proteins in peripheral blood mononuclear cells (PMBCs) has been identified using a ML approach (Heydari et al., 2024). This study had a fairly small sample size (28 COVID-19 patients and 17 controls), but it illustrates the promise of multi-analyte biomarkers including RNAs in COVID-19. Current bioinformatics deconvolution approaches enable effective estimation of cell-type fraction and cell type specific gene expression in the peripheral immune system from bulk transcriptome data, offering a powerful tool for immune-phenotyping (Chen et al., 2018) that is complementary to plasma and cellular proteomics. Bulk samples employed for RNAseq can also be used for in-depth immune repertoire analyses (Galson et al., 2020). These data also allow prediction of physiological states, such as PANoptosis (Yang et al., 2024; Dai et al., 2023) or innate immunity activation (Karki and Kanneganti, 2022), and upstream regulators of these states (e.g., transcription factors, protein kinases, hormones), thus enabling the identification of potential therapeutic targets. Critically, transcriptomic data can also allow for the identification of circulating SARS-CoV-2 viral load from whole blood (including viral variant calling and, given sufficient sequence coverage, detection of intra-patient viral evolution), thus constituting an important tool to assess persistent viremia from sources such as vascular beds. Further virome/microbiome analyses of these data can capture other viruses/bacteria that may contribute to PASC pathophysiology (e.g., EBV). Several such transcriptome analyses have been completed for acute COVID-19 and PASC (Thompson et al., 2023; Hadjadj et al., 2020; Lucas et al., 2020; Sposito et al., 2021; Sullivan et al., 2021; Ziegler et al., 2021; Galbraith et al., 2022), but without integration with other omics. This supports the need for further transcriptome analyses in the RECOVER cohort in the context of a multi-omics approach.
b) Single-cell transcriptomics: Bulk transcriptomics measures RNA expression as an average of all cell types present in a sample. This can potentially mask the contribution of rare cell types or cellular states to the transcriptome. Single cell RNA sequencing (scRNAseq) can add further detail to immune phenotyping by measuring the transcriptomes of up to 20,000 individual cells simultaneously. This can provide highly detailed information, albeit at higher cost than bulk transcriptomics. scRNAseq protocols relevant to PASC can include Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq) and single cell VDJ sequencing (scVDJ) analyses, which can provide advanced immune phenotyping and T cell receptor/B cell receptor (TCR/BCR) repertoire data, respectively, on the same cell (Cadot et al., 2020; Kim et al., 2020; Lian et al., 2020; Saigusa and Ley, 2020; Frangieh et al., 2021; Mercatelli et al., 2021; Rodahl et al., 2021; Shi et al., 2021; Fan et al., 2022; Xu et al., 2022; He et al., 2022).
3.1.4 Proteomics
a) Soluble proteins: Protein-based biomarkers are commonly used for the diagnosis and management of myriad medical conditions and are likely to be useful for the prediction, diagnosis, prognosis and clinical management of PASC. Cytokines, chemokines, antibodies, coagulation factors, growth factors, complement cascade components, peptide hormones, and viral proteins can all be measured by high-content proteomic methods in plasma. Multiple technologies are now available to identify hundreds to thousands of individual proteins from very small volumes of serum, plasma, tissues, or cells, including peripheral blood mononuclear cells (PBMCs). These include mass spectrometry, SOMAscan® assays, Olink® proteomics, and PhIP-seq (phage immunoprecipitation sequencing), to name a few. Furthermore, some of these technologies (e.g., mass spectrometry conjugated with the newest search algorithms such as MSFragger (Kong et al., 2017)) enable the identification of protein isoforms and post-translational modifications, including novel ones. For example, while still under development, the latest SOMAscan® platform measures >7,000 proteins from a mere 125 μL of plasma or serum (Gold et al., 2012). Of critical importance for the study of autoimmunity in PASC, the PhIP-seq technology enables the identification of virtually all auto-antibodies produced by an individual (Mohan et al., 2018). A recent PhIP-seq study in a relatively small cohort identified a common autoreactive pattern in PASC patients and patients who had recovered from acute COVID-19 (Bodansky et al., 2023), raising important questions about the possible role of autoantibodies in PASC.
Plasma proteomic biosignatures can inform on multiple pathophysiological processes at once, including but not restricted to various forms of inflammation (e.g., systemic, organ-specific, vascular), organ injury, vascular disorders, neurodegeneration, dysregulation of coagulation and fibrinolysis, and remote organ crosstalk via the blood. A wealth of proteomics data are already available for acute COVID-19 (Sullivan et al., 2021; Galbraith et al., 2022; Galbraith et al., 2021; D’Alessandro et al., 2020), as well as myriad auto-inflammatory conditions. In the context of an integrated multi-omics strategy, proteomics data could maximize the opportunities to discover mechanisms underlying PASC pathophysiology as well as molecular subtypes, clinically actionable biomarkers and treatment targets.
b) Cellular proteomics-based immunophenotyping: Immunophenotyping, which allows for the precise detection of membrane and intracellular proteins using antibodies, has identified signatures that are predictive of subsequent PASC (Peluso et al., 2021). Cellular proteomics-based immunophenotyping technologies can simultaneously quantify, at the single-cell level, expression levels of 40–50 surface and intracellular proteins of immune cells. These include high-parameter flow cytometry (e.g., BD X50), spectral flow cytometry (e.g., Cytek Aurora), and CyTOF (cytometry by time-of-flight, a.k.a. mass cytometry). CyTOF, the most common of these approaches, is a powerful high-dimensional immunophenotyping method that can, in a single specimen, quantify all major subsets of cells using its ∼40 available channels. Alternatively, it can be used to deeply characterize one immune subset of interest (e.g., to interrogate phenotypes, homing properties, effector functions, and self-renewal capacities of T cells) (Neidleman et al., 2021a; Ma et al., 2021; Neidleman et al., 2021b; Neidleman et al., 2020). Phospho-CyTOF also enable analyses of signaling states of individual cells (Bendall et al., 2011). It can also be used to characterize the glycan features of immune cells at the single-cell level, informing on immune functions which are very much modulated by cell-surface glycosylation (Ma et al., 2022). As PASC is multifactorial and heterogeneous, approaches such as CyTOF which allow for broad and specific studies of immune subsets, will be key. Studies can, for example, examine how the global immune landscape is altered during PASC, as well as whether specific subsets implicated in COVID-19 disease progression or PASC (e.g., T cells, myeloid cells, neutrophils) exhibit subset-specific changes that can inform on mechanism of action. Studies that have begun to use CyTOF to explore immunological differences between fully recovered vs individuals with PASC have revealed a dysregulated adaptive immune response in the latter, e.g., global differences in T-cell subsets, sex-specific differences in cytolytic T-cells, increased frequency of T-cells migrating to inflamed tissues but also exhausted T-cells, as well as increased frequency of exhausted T-cells (Yin et al., 2023; Yin et al., 2024). Further studies using larger cohorts are warranted. From a practical standpoint, for the amount of data generated CyTOF is cost-effective and requires relatively few cells relative to if samples were to be analyzed using multiple low-parameter panels implemented in conventional flow cytometry.
3.1.5 Metabolomics
Metabolites are the end products of multiple pathways and often indicate the major phenotype(s) of metabolic and genetic disorders. From diabetes to inborn errors of metabolism, metabolites can often define the key pathways underlying complex diseases and serve as potential biomarkers. Metabolites may also mediate the downstream effects of genomic, epigenomic and transcriptomic processes, and in turn influence these processes to modify PASC phenotypes. As a measure of the status of hundreds of metabolic pathways, the overall metabolome and the lipidome represent biologically and mechanistically informative data streams. The endogenous metabolome captures a broad range of inflammatory processes, energy production, microbial metabolites, organ-specific biomarkers, lipids, carbohydrates, steroids, and amino acids, among other relevant information on physiologic processes. Furthermore, exogenous metabolites capture environmental exposures, including but not restricted to food and supplement intake, toxins (e.g., per-and polyfluoroalkylic substances, also known as PFAS, tobacco byproducts, illicit drugs), and medications (e.g., statins, ibuprofen, selective serotonin reuptake inhibitors), all of which may be important in the development or modification of PASC phenotypes, and cannot be easily predicted by other omics but can potentially impact the results of other omics tests. Importantly, these exogenous metabolites are not measurable by any other mechanisms. Microbial metabolites, also measured by metabolomics assays, may serve as important connectors to microbiome data. The interconnections between the metabolome and other multi-omics profiling illustrates an important aspect of multi-omics strategies: while metabolomics can provide crucial information as a single platform, it acts synergistically with other omics data in elucidating important functional relationships to PASC. Several small studies have demonstrated strong dysregulation of endogenous metabolites associated with particular PASC phenotypes (Valdés et al., 2022). For example, tryptophan metabolism was found to be dysregulated by several groups using metabolomic analyses in blood and urine studies (Bustamante et al., 2022; Dewulf et al., 2022), but the pathogenesis of this phenomenon is unclear. We believe that a comprehensive, longitudinal metabolomics investigation of PASC in the context of a multi-omics strategy in a sufficiently large cohort of patients with deep clinical phenotypes will help define and prioritize functional pathways.
3.1.6 Microbiome
The microbiome has multiple physiological roles in human health, including: i) extracting indigestible ingredients from food and synthesizing nutritional factors; ii) affecting host metabolism; iii) developing systemic and intestinal immunity; vi) providing signals for epithelial renewal and maintaining gut integrity; and iv) secreting anti-microbial products. Alterations of the microbiome may often be an initial disturbance with far-reaching ramifications on disease progression. The gut microbiomes of hospitalized COVID-19 patients were enriched with opportunistic pathogens such as Clostridium hathewayi, Bacteroides nordii, and Actinomyces viscosus (Zuo et al., 2020). In acute COVID-19, the gut microbiome is associated with immune responses and disease severity (Zhang et al., 2021; Maeda et al., 2022; Zuo et al., 2021) and also interacts with the lung microbiome (Zhu et al., 2022). Changes in the gut microbiome could influence respiratory tract infections through the common mucosal immune system. Conversely, respiratory tract dysbiosis and functional disorders due to COVID-19 also affect the digestive tract (Zhu et al., 2022). Studies have demonstrated SARS-CoV-2 interactions with host microbiome/virome communities, clotting/coagulation issues, dysfunctional brainstem/vagus nerve signaling, and immune cells (reviewed in (Proal and VanElzakker, 2021)). There is observational evidence of gut microbiome compositional alterations in patients with long-term complications of COVID-19 (Liu et al., 2022). However, the current studies have sample sizes varying from 8 to 130 patients and few studies followed patients beyond 6 months post-infection (reviewed in (Zhang et al., 2023)). A recent study (Xiong et al., 2023) using multi-omics of microbiome-host interactions identified phenotypic, intestinal microbial, and metabolic biomarkers for short-and long-term myalgic encephalomyelitis/chronic fatigue syndrome. Large amounts of microbiome data can be easily generated at low-cost in the RECOVER adult and pediatric cohorts. These data, when integrated with other multi-omics data, will allow for a better understanding of the virus-microbiome-host interactions and identifying microbial and metabolic biomarkers for PASC. Further studies are also needed to investigate whether microbiota modulation can prevent or facilitate the recovery from PASC.
3.2 Considerations on data generation and analysis
3.2.1 Data generation and randomization
Multi-omics data integration can generate valuable knowledge to understand disease pathogenesis. However, multi-omics data can often be burdened by large confounding signals that can prevent accurate modeling and successful discovery. It is therefore essential to appropriately design data collection and generation processes to ensure that such confounders are minimized, and that multi-omics data are amenable to address a large array of important biological questions aiming to characterize, understand and treat PASC. For example, it is usually better to reduce batch effects with a good study design that accurately accounts for them rather than attempting to correct for batch effects after the fact. One successful approach to minimize batch effects involves adequate randomization schemes that minimize risk of contamination of true signals by unwanted variation. Such an approach is powerful when biological questions are defined before data are collected, but often maximizes a distance metric between a measure of interest and the drivers of this unwanted variation at the cost of other potentially meaningful traits. In hypothesis-generating situations, other approaches, such as the inclusion of data generation controls (e.g., reference samples), profiled repeatedly within and across different multi-omics assays, have proven to be an important tool to control for unwanted variation, including confounding from technical variation. However, data generation controls can be complex to define, must contain enough material to be assayed repeatedly, need to capture the full range of biological variation in the multi-omics assessed, and depending on the number needed, can substantially increase data generation cost. These considerations are essential to design a successful multi-omics discovery effort, and it is therefore essential to include data generation experts as well as data scientists who know the biases of each multi-omics profiling technology in teams tasked with designing data generation strategies.
3.2.2 A multi-omics systems-biology approach to data analysis
Each of the omics assays generates vast datasets that require powerful analytical strategies (Gui et al., 2023; Martinez-Bartolome et al., 2018; Lee et al., 2019; Michelhaugh and Januzzi, 2023). Integrating data from multiple omics over time and with clinical, demographic and exposome data is the next level of analytical complexity. A multi-omics approach allows for the integration of multiple layers of information into systems biology models that capture the dynamic interplay between biological processes, allowing not only the study of the functional relationships between the molecular components of PASC, but the elucidation of their causal relationships (Beckmann et al., 2020; Kuijjer et al., 2019; Wang M. et al., 2021; Sonawane et al., 2022; Sonawane et al., 2019; Argelaguet et al., 2020). This approach is critical to understanding the pathogenesis of the clinical manifestations of PASC (Table 3). Integrating multi-omics data with the deep clinical and demographic phenotyping available via RECOVER will capture the most complete picture of disease pathophysiology, leading to more accurate identification and characterization of PASC subtypes as compared to individual omics studies of individual patient cohorts (Figure 1).
Multi-omics data are also important in substantiating and validating findings across individual omics platforms (e.g., a genetic polymorphism leading to transcriptomic, proteomic and metabolomics effects). Critical to this is the longitudinal capture of multi-omics data as the clinical presentations of PASC emerge and progress. Given the high-content nature of omics datasets, they support the development of machine learning (ML) class discovery approaches for identification of clinically relevant biosignatures. The rich datasets that will be produced as part of this effort will enable predictive and diagnostic algorithms to identify candidate biomarkers linked to disease outcomes. The thorough integration of these data into meaningful, queryable, and informative models is critical to understand the biological mechanisms, disease subtypes, progression and prognosis of PASC, to investigate the impact of modifiable risk factors and identify potential precision therapeutic approaches to PASC. Inherent in this, is the measure of these data at multiple time points throughout the disease process.
An example of the power of multi-omics approaches is the study of multisystem inflammatory syndrome in children (MIS-C), a serious complication of pediatric COVID-19. Longitudinal plasma bulk transcriptomics, combined with whole blood transcriptomics and plasma DNA epigenomics was recently used to develop multi-organ damage signatures indicative of MIS-C (Loy et al., 2022). This study complements previous genomic, proteomic and immunophenotyping investigations of MIS-C (Sacco et al., 2022; Gruber et al., 2020; Porritt et al., 2021; Carter et al., 2020) to delineate a clearer picture of its pathogenesis. We posit that a single comprehensive, integrated, longitudinal multi-omics approach would have reached similar conclusions as multiple consecutive studies focusing on 1-2 omics each. Such a comprehensive study, performed through centralized labs, would reduce lab-to-lab variability and pre-analytical variability, leveraging a large sample size with rich, highly standardized clinical phenotypes. Furthermore, it is difficult to predict ahead of time which omics would be the most consistent and/or most clinically informative, and which omics data are consistent with each other (e.g., a clinically informative RNA may predict the abundance of an enzyme that produces a metabolite, but if the protein abundance or the metabolite levels are not consistent with RNA abundance, perhaps because of short half-life of the protein or instability of the metabolite, that protein or its metabolite product would not be potential biomarkers or therapeutic targets).Another example of the importance of capturing multi-omics data across demographics and time points is the importance of sex and steroid hormones in the PASC population. Innate and adaptive, humoral and cell-mediated immune responses are impacted by hormones, and their dysregulation contributes to immune-mediated diseases including autoimmunity, a hallmark of PASC (Rojas et al., 2022; Moulton, 2018; Bereshchenko et al., 2018). Ovarian steroids recruit mast cells and T-regs to the uterus during pregnancy (Schumacher et al., 2014). Estradiol causes inflammasome activation in mast cells (Guo et al., 2021). Estradiol deficiency due to menopause and/or hypogonadism contributes to overactivity of the renin-angiotensin-aldosterone system (RAAS), while estrogen can contribute to mast cell activation syndrome (MCAS), which may contribute to the pathogenesis of PASC (O’Donnell et al., 2014; Szukiewicz et al., 2022; Arun et al., 2022). The SARS-CoV2 spike protein binds to and modulates both ACE2 and ERα receptors, and as sex hormones regulate the expression of ACE2 (Solis et al., 2022; Wang H. et al., 2021), the asymmetry in PASC development and clinical presentations between sexes - as well as across menstruation status and menstrual cycle time points - indicates that hormone measurements, which can be performed by metabolomics for non-peptide hormones and by proteomics for peptide hormones, are critical components of a multi-omics strategy.
3.2.3 Task force recommendations
We recommended the following strategy: germline whole genome sequencing (WGS) be performed on every RECOVER participant consented for genetic analysis to be used for GWAS studies. Epigenomics, bulk PBMC transcriptomics, plasma proteomics, plasma targeted metabolomics and stool proteomics should be performed on biospecimens from at least 2 time points per participant (baseline, 90 days and 180 days, or at a minimum baseline and 180 days) on biospecimens from as many participants as possible. Samples taken at later time points during the planned 4-year follow-up period may be analyzed as well in the future, particularly to investigate cases that persist long-term. However, the initial focus should be on the first 180 days post-enrollment, as the number of participants dropping out of the study or being lost to follow-up is likely to increase at later time points. This time window is likely to be long enough to compare COVID-19 cases that do result in PASC to cases that do not, which is one of the primary endpoints of the RECOVER studies, as well as PASC cases that resolve clinically within 180 days from cases that persist beyond that time, while maximizing sample size. Single-cell transcriptomics and/or single-cell immunophenotyping may be performed on subsets of participants from each arm of each cohort, to limit costs. Bioinformatics deconvolution of cellular populations based on bulk transcriptomics should be performed on all available PBMC biospecimens.
It must be pointed out that the adult and pregnant adult RECOVER cohorts include different arms: “acute infected” participants, who enroll within 30 days of a SARS-CoV-2 infection, “post-acute infected”, who enroll after 30 days post-infection, “acute uninfected” enrolled within 30 days of a negative COVID-19 test and “post-acute uninfected”, enrolled after 30 days post-negative test (Horwitz et al., 2023; Reel et al., 2021). This implies that baseline samples taken at enrollment are likely to reflect different pathobiological stages of disease in acute infected versus post-acute infected participants. It is also possible that a fraction of the “uninfected” participants will have experienced subclinical infections with SARS-CoV-2. Multi-omics analyses have the potential to identify these cases, particularly through proteomics-based identification of SARS-CoV-2 antibodies not detected by conventional tests. As there are significant differences in study design for the adult and pediatric cohorts (Horwitz et al., 2023; Reel et al., 2021), longitudinal biospecimens will only be available for Tier 2 pediatric participants, while baseline biospecimens will be available for all participants. Also, the amounts of blood/plasma available for the pediatric cohort will depend on the age of participants. With these considerations in mind, maximizing sample size should be the underlying principle. The main objective of this proposed multi-omics analysis is to generate a rich, multidimensional molecular profiling database to match the clinical, pathophysiological and socioeconomics data elements generated by the RECOVER studies. This data should be made available to the scientific community for secondary analyses.
4 Conclusion
Based on its analysis of the available evidence, the OMICS Task Force advocated an integrated “big data and systems biology” approach, using multi-omics to analyze biospecimens from the largest possible sample sizes in the RECOVER adult and pediatric cohorts, as opposed to single analyte assays or individual omics in multiple separate studies. This approach will maximize our ability to understand pathogenetic mechanisms in clinically defined patient subgroups, discover PASC molecular subtypes and guide precision therapeutic strategies. Centralized, streamlined omics analyses will limit potential inconsistencies associated with laboratory-to-laboratory and batch variations. In addition, multi-omics assays can capture most clinically assayed biomarkers at cheaper costs. Data generation on this scale can only be accomplished through highly multiplexed approaches, which will maximize opportunities to discover mechanisms underlying PASC pathophysiology.
PASC joins the number of poorly understood, chronic diseases that have been the bane of patients, healthcare providers and clinical researchers. While different clinical presentations of PASC have been described, traditional molecular approaches have thus far failed to produce a deep mechanistic understanding of the etiology, pathogenesis and molecular subtypes of PASC. Many of our recommendations have already been considered by the NIH through the peer-review process, resulting in the creation of a systems biology panel that is currently designing the studies we proposed. Currently, this panel is hammering down the details of the analytical strategies. The NIH RECOVER initiative offers an ideal opportunity to understand PASC in diverse populations, and can serve as a paradigm for the study of other complex, poorly-understood chronic diseases.
Author contributions
JS: Conceptualization, Writing–original draft, Writing–review and editing. MA: Conceptualization, Writing–original draft. HA: Conceptualization, Writing–original draft. NB: Conceptualization, Methodology, Visualization, Writing–original draft. ME: Project administration, Writing–original draft. JE: Conceptualization, Writing–original draft. XG: Conceptualization, Writing–original draft. BH: Conceptualization, Writing–original draft. PK: Conceptualization, Writing–original draft. JL-S: Conceptualization, Writing–original draft. RL: Conceptualization, Writing–original draft. CM: Conceptualization, Writing–original draft. MM: Conceptualization, Project administration, Writing–original draft. LN: Conceptualization, Writing–original draft. NR: Conceptualization, Writing–original draft. MR: Conceptualization, Writing–original draft. JR: Conceptualization, Writing–original draft. GS: Conceptualization, Writing–original draft. KS: Conceptualization, Writing–original draft. SS: Conceptualization, Methodology, Writing–original draft. ST: Conceptualization, Writing–original draft. SY: Conceptualization, Writing–original draft. LM: Conceptualization, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The RECOVER studies and the RECOVER OMICS task force are supported by the National Institutes of Health, through the parent grants OT2HL161847, OT2HL161841, OT2HL156812.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Argelaguet, R., Arnol, D., Bredikhin, D., Deloro, Y., Velten, B., Marioni, J. C., et al. (2020). MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biol. 21, 111. doi:10.1186/s13059-020-02015-1
Arun, S., Storan, A., and Myers, B. (2022). Mast cell activation syndrome and the link with long COVID. Br. J. Hosp. Med. (Lond) 83, 1–10. doi:10.12968/hmed.2022.0123
Asano, T., Chelvanambi, S., Decano, J. L., Whelan, M. C., Aikawa, E., and Aikawa, M. (2022). In silico drug screening approach using l1000-based connectivity map and its application to COVID-19. Front. Cardiovasc Med. 9, 842641. doi:10.3389/fcvm.2022.842641
Balnis, J., Madrid, A., Hogan, K. J., Drake, L. A., Chieng, H. C., Tiwari, A., et al. (2021). Blood DNA methylation and COVID-19 outcomes. Clin. Epigenetics 13, 118. doi:10.1186/s13148-021-01102-9
Beckmann, N. D., Lin, W. J., Wang, M., Cohain, A. T., Charney, A. W., Wang, P., et al. (2020). Multiscale causal networks identify VGF as a key regulator of Alzheimer's disease. Nat. Commun. 11, 3942. doi:10.1038/s41467-020-17405-z
Bednarik, D. P., Cook, J. A., and Pitha, P. M. (1990). Inactivation of the HIV LTR by DNA CpG methylation: evidence for a role in latency. EMBO J. 9, 1157–1164. doi:10.1002/j.1460-2075.1990.tb08222.x
Bendall, S. C., Simonds, E. F., Qiu, P., Amir el, A. D., Krutzik, P. O., Finck, R., et al. (2011). Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696. doi:10.1126/science.1198704
Bereshchenko, O., Bruscoli, S., and Riccardi, C. (2018). Glucocorticoids, sex hormones, and immunity. Front. Immunol. 9, 1332. doi:10.3389/fimmu.2018.01332
Bhat, B., and Jones, G. T. (2022). Data analysis of DNA methylation epigenome-wide association studies (EWAS): a guide to the principles of best practice. Methods Mol. Biol. 2458, 23–45. doi:10.1007/978-1-0716-2140-0_2
Bodansky, A., Wang, C. Y., Saxena, A., Mitchell, A., Takahashi, S., Anglin, K., et al. (2023). Autoantigen profiling reveals a shared post-COVID signature in fully recovered and Long COVID patients. medRxiv.
Bustamante, S., Yau, Y., Boys, V., Chang, J., Paramsothy, S., Pudipeddi, A., et al. (2022). Tryptophan metabolism 'hub' gene expression associates with increased inflammation and severe disease outcomes in COVID-19 infection and inflammatory bowel disease. Int. J. Mol. Sci. 23, 14776. doi:10.3390/ijms232314776
Cadot, S., Valle, C., Tosolini, M., Pont, F., Largeaud, L., Laurent, C., et al. (2020). Longitudinal CITE-Seq profiling of chronic lymphocytic leukemia during ibrutinib treatment: evolution of leukemic and immune cells at relapse. Biomark. Res. 8, 72. doi:10.1186/s40364-020-00253-w
Carter, M. J., Fish, M., Jennings, A., Doores, K. J., Wellman, P., Seow, J., et al. (2020). Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS-CoV-2 infection. Nat. Med. 26, 1701–1707. doi:10.1038/s41591-020-1054-6
Castro de Moura, M., Davalos, V., Planas-Serra, L., Alvarez-Errico, D., Arribas, C., Ruiz, M., et al. (2021). Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine 66, 103339. doi:10.1016/j.ebiom.2021.103339
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M., and Alizadeh, A. A. (2018). Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259. doi:10.1007/978-1-4939-7493-1_12
Corley, M. J., Pang, A. P. S., Dody, K., Mudd, P. A., Patterson, B. K., Seethamraju, H., et al. (2021). Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 110, 21–26. doi:10.1002/JLB.5HI0720-466R
Cuppen, E., Elemento, O., Rosenquist, R., Nikic, S., M, I. J., Zaleski, I. D., et al. (2022). Implementation of whole-genome and transcriptome sequencing into clinical cancer care. JCO Precis. Oncol. 6, e2200245. doi:10.1200/PO.22.00245
Dai, W., Zheng, P., Wu, J., Chen, S., Deng, M., Tong, X., et al. (2023). Integrated analysis of single-cell RNA-seq and chipset data unravels PANoptosis-related genes in sepsis. Front. Immunol. 14, 1247131. doi:10.3389/fimmu.2023.1247131
D’Alessandro, A., Thomas, T., Dzieciatkowska, M., Hill, R. C., Francis, R. O., Hudson, K. E., et al. (2020). Serum proteomics in COVID-19 patients: altered coagulation and complement status as a function of IL-6 level. J. Proteome Res. 19, 4417–4427. doi:10.1021/acs.jproteome.0c00365
Debnath, M., Banerjee, M., and Berk, M. (2020). Genetic gateways to COVID-19 infection: implications for risk, severity, and outcomes. FASEB J. 34, 8787–8795. doi:10.1096/fj.202001115R
Devaux, Y., Zhang, L., Lumley, A. I., Karaduzovic-Hadziabdic, K., Mooser, V., Rousseau, S., et al. (2024). Development of a long noncoding RNA-based machine learning model to predict COVID-19 in-hospital mortality. Nat. Commun. 15, 4259. doi:10.1038/s41467-024-47557-1
Dewulf, J. P., Martin, M., Marie, S., Oguz, F., Belkhir, L., De Greef, J., et al. (2022). Urine metabolomics links dysregulation of the tryptophan-kynurenine pathway to inflammation and severity of COVID-19. Sci. Rep. 12, 9959. doi:10.1038/s41598-022-14292-w
Fan, R., Liu, Y., DiStasio, M., Su, G., Asashima, H., Enninful, A., et al. (2022). Spatial-CITE-seq: spatially resolved high-plex protein and whole transcriptome co-mapping. Res. Sq. doi:10.21203/rs.3.rs-1499315/v1
Ferreira, L. C., Gomes, C. E. M., Rodrigues-Neto, J. F., and Jeronimo, S. M. B. (2022). Genome-wide association studies of COVID-19: connecting the dots. Infect. Genet. Evol. 106, 105379. doi:10.1016/j.meegid.2022.105379
Frangieh, C. J., Melms, J. C., Thakore, P. I., Geiger-Schuller, K. R., Ho, P., Luoma, A. M., et al. (2021). Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat. Genet. 53, 332–341. doi:10.1038/s41588-021-00779-1
Galbraith, M. D., Kinning, K. T., Sullivan, K. D., Araya, P., Smith, K. P., Granrath, R. E., et al. (2022). Specialized interferon action in COVID-19. Proc. Natl. Acad. Sci. U. S. A. 119. doi:10.1073/pnas.2116730119
Galbraith, M. D., Kinning, K. T., Sullivan, K. D., Baxter, R., Araya, P., Jordan, K. R., et al. (2021). Seroconversion stages COVID19 into distinct pathophysiological states. Elife 10, e65508. doi:10.7554/eLife.65508
Galson, J. D., Schaetzle, S., Bashford-Rogers, R. J. M., Raybould, M. I. J., Kovaltsuk, A., Kilpatrick, G. J., et al. (2020). Deep sequencing of B cell receptor repertoires from COVID-19 patients reveals strong convergent immune signatures. Front. Immunol. 11, 605170. doi:10.3389/fimmu.2020.605170
Gold, L., Walker, J. J., Wilcox, S. K., and Williams, S. (2012). Advances in human proteomics at high scale with the SOMAscan proteomics platform. N. Biotechnol. 29, 543–549. doi:10.1016/j.nbt.2011.11.016
Gross, R. S., Thaweethai, T., Rosenzweig, E. B., Chan, J., Chibnik, L. B., Cicek, M. S., et al. (2024). Researching COVID to enhance recovery (RECOVER) pediatric study protocol: rationale, objectives and design. PLoS One 19, e0285635. doi:10.1371/journal.pone.0285635
Gruber, C. N., Patel, R. S., Trachtman, R., Lepow, L., Amanat, F., Krammer, F., et al. (2020). Mapping systemic inflammation and antibody responses in multisystem inflammatory syndrome in children (MIS-C). Cell 183, 982–995. doi:10.1016/j.cell.2020.09.034
Gui, Y., He, X., Yu, J., and Jing, J. (2023). Artificial intelligence-assisted transcriptomic analysis to advance cancer immunotherapy. J. Clin. Med. 12, 1279. doi:10.3390/jcm12041279
Guo, X., Xu, X., Li, T., Yu, Q., Wang, J., Chen, Y., et al. (2021). NLRP3 inflammasome activation of mast cells by estrogen via the nuclear-initiated signaling pathway contributes to the development of endometriosis. Front. Immunol. 12, 749979. doi:10.3389/fimmu.2021.749979
Hadjadj, J., Yatim, N., Barnabei, L., Corneau, A., Boussier, J., Smith, N., et al. (2020). Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724. doi:10.1126/science.abc6027
He, H., Li, Z., Lu, J., Qiang, W., Jiang, S., Xu, Y., et al. (2022). Single-cell RNA-seq reveals clonal diversity and prognostic genes of relapsed multiple myeloma. Clin. Transl. Med. 12, e757. doi:10.1002/ctm2.757
Heydari, R., Tavassolifar, M. J., Fayazzadeh, S., Sadatpour, O., and Meyfour, A. (2024). Long non-coding RNAs in biomarking COVID-19: a machine learning-based approach. Virol. J. 21, 134. doi:10.1186/s12985-024-02408-9
Horwitz, L. I., Thaweethai, T., Brosnahan, S. B., Cicek, M. S., Fitzgerald, M. L., Goldman, J. D., et al. (2023). Researching COVID to Enhance Recovery (RECOVER) adult study protocol: rationale, objectives, and design. PLoS One 18, e0286297. doi:10.1371/journal.pone.0286297
Jha, P. K., Vijay, A., Halu, A., Uchida, S., and Aikawa, M. (2020). Gene expression profiling reveals the shared and distinct transcriptional signatures in human lung epithelial cells infected with SARS-CoV-2, MERS-CoV, or SARS-CoV: potential implications in cardiovascular complications of COVID-19. Front. Cardiovasc Med. 7, 623012. doi:10.3389/fcvm.2020.623012
Joshi, A., Rienks, M., Theofilatos, K., and Mayr, M. (2021). Systems biology in cardiovascular disease: a multiomics approach. Nat. Rev. Cardiol. 18, 313–330. doi:10.1038/s41569-020-00477-1
Karki, R., and Kanneganti, T. D. (2022). Innate immunity, cytokine storm, and inflammatory cell death in COVID-19. J. Transl. Med. 20, 542. doi:10.1186/s12967-022-03767-z
Kashima, Y., Sakamoto, Y., Kaneko, K., Seki, M., Suzuki, Y., and Suzuki, A. (2020). Single-cell sequencing techniques from individual to multiomics analyses. Exp. Mol. Med. 52, 1419–1427. doi:10.1038/s12276-020-00499-2
Kim, G. J., Elnaggar, J. H., Varnado, M., Feehan, A. K., Tauzier, D., Rose, R., et al. (2024). A bioinformatic analysis of T-cell epitope diversity in SARS-CoV-2 variants: association with COVID-19 clinical severity in the United States population. Front. Sys. Biol. 15, 1357731. in press. doi:10.3389/fimmu.2024.1357731
Kim, H. J., Lin, Y., Geddes, T. A., Yang, J. Y. H., and Yang, P. (2020). CiteFuse enables multi-modal analysis of CITE-seq data. Bioinformatics 36, 4137–4143. doi:10.1093/bioinformatics/btaa282
Kong, A. T., Leprevost, F. V., Avtonomov, D. M., Mellacheruvu, D., and Nesvizhskii, A. I. (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520. doi:10.1038/nmeth.4256
Kuijjer, M. L., Tung, M. G., Yuan, G., Quackenbush, J., and Glass, K. (2019). Estimating sample-specific regulatory networks. iScience 14, 226–240. doi:10.1016/j.isci.2019.03.021
Lammi, V., Nakanishi, T., Jones, S. E., Andrews, S. J., Karjalainen, J., Cortés, B., et al. (2023). Genome-wide association study of long COVID medRxiv 2023.06.29.23292056. doi:10.1101/2023.06.29.23292056
Lee, L. H., Halu, A., Morgan, S., Iwata, H., Aikawa, M., and Singh, S. A. (2019). XINA: a workflow for the integration of multiplexed proteomics kinetics data with network analysis. J. Proteome Res. 18, 775–781. doi:10.1021/acs.jproteome.8b00615
Lee, Y., Riskedal, E., Kalleberg, K. T., Istre, M., Lind, A., Lund-Johansen, F., et al. (2022). EWAS of post-COVID-19 patients shows methylation differences in the immune-response associated gene, IFI44L, three months after COVID-19 infection. Sci. Rep. 12, 11478. doi:10.1038/s41598-022-15467-1
Lian, Q., Xin, H., Ma, J., Konnikova, L., Chen, W., Gu, J., et al. (2020). Artificial-cell-type aware cell-type classification in CITE-seq. Bioinformatics 36, i542–i550. doi:10.1093/bioinformatics/btaa467
Liu, Q., Mak, J. W. Y., Su, Q., Yeoh, Y. K., Lui, G. C., Ng, S. S. S., et al. (2022). Gut microbiota dynamics in a prospective cohort of patients with post-acute COVID-19 syndrome. Gut 71, 544–552. doi:10.1136/gutjnl-2021-325989
Loy, C. J., Sotomayor-Gonzalez, A., Servellita, V., Nguyen, J., Lenz, J., Bhattacharya, S., et al. (2022). Nucleic acid biomarkers of immune response and cell and tissue damage in children with COVID-19 and MIS-C. Cell Rep. Med. 4, 101034. doi:10.1016/j.xcrm.2023.101034
Lucas, C., Wong, P., Klein, J., Castro, T. B. R., Silva, J., Sundaram, M., et al. (2020). Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469. doi:10.1038/s41586-020-2588-y
Ma, T., McGregor, M., Giron, L., Xie, G., George, A. F., Abdel-Mohsen, M., et al. (2022). Single-cell glycomics analysis by CyTOF-Lec reveals glycan features defining cells differentially susceptible to HIV. Elife 11. doi:10.7554/elife.78870
Ma, T., Ryu, H., McGregor, M., Babcock, B., Neidleman, J., Xie, G., et al. (2021). Protracted yet coordinated differentiation of long-lived SARS-CoV-2-specific CD8(+) T cells during convalescence. J. Immunol. 207, 1344–1356. doi:10.4049/jimmunol.2100465
Maeda, Y., Motooka, D., Kawasaki, T., Oki, H., Noda, Y., Adachi, Y., et al. (2022). Longitudinal alterations of the gut mycobiota and microbiota on COVID-19 severity. BMC Infect. Dis. 22, 572. doi:10.1186/s12879-022-07358-7
Martinez-Bartolome, S., Medina-Aunon, J. A., Lopez-Garcia, M. A., Gonzalez-Tejedo, C., Prieto, G., Navajas, R., et al. (2018). PACOM: a versatile tool for integrating, filtering, visualizing, and comparing multiple large mass spectrometry proteomics data sets. J. Proteome Res. 17, 1547–1558. doi:10.1021/acs.jproteome.7b00858
Menachery, V. D., Schafer, A., Burnum-Johnson, K. E., Mitchell, H. D., Eisfeld, A. J., Walters, K. B., et al. (2018). MERS-CoV and H5N1 influenza virus antagonize antigen presentation by altering the epigenetic landscape. Proc. Natl. Acad. Sci. U. S. A. 115, E1012-E1021–E1021. doi:10.1073/pnas.1706928115
Mercatelli, D., Balboni, N., Giorgio, F., Aleo, E., Garone, C., and Giorgi, F. M. (2021). The transcriptome of SH-SY5Y at single-cell resolution: a CITE-seq data analysis workflow. Methods Protoc. 4, 28. doi:10.3390/mps4020028
Metz, T. D., Clifton, R. G., Gallagher, R., Gross, R. S., Horwitz, L. I., Jacoby, V. L., et al. (2023). Researching COVID to enhance recovery (RECOVER) pregnancy study: rationale, objectives and design. PLoS One 18, e0285351. doi:10.1371/journal.pone.0285351
Michelhaugh, S. A., and Januzzi, J. L. (2023). Using artificial intelligence to better predict and develop biomarkers. Clin. Lab. Med. 43, 99–114. doi:10.1016/j.cll.2022.09.021
Mohan, D., Wansley, D. L., Sie, B. M., Noon, M. S., Baer, A. N., Laserson, U., et al. (2018). PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat. Protoc. 13, 1958–1978. doi:10.1038/s41596-018-0025-6
Moulton, V. R. (2018). Sex hormones in acquired immunity and autoimmune disease. Front. Immunol. 9, 2279. doi:10.3389/fimmu.2018.02279
Neidleman, J., Luo, X., Frouard, J., Xie, G., Gill, G., Stein, E. S., et al. (2020). SARS-CoV-2-Specific T cells exhibit phenotypic features of helper function, lack of terminal differentiation, and high proliferation potential. Cell Rep. Med. 1, 100081. doi:10.1016/j.xcrm.2020.100081
Neidleman, J., Luo, X., George, A. F., McGregor, M., Yang, J., Yun, C., et al. (2021b). Distinctive features of SARS-CoV-2-specific T cells predict recovery from severe COVID-19. Cell Rep. 36, 109414. doi:10.1016/j.celrep.2021.109414
Neidleman, J., Luo, X., McGregor, M., Xie, G., Murray, V., Greene, W. C., et al. (2021a). mRNA vaccine-induced T cells respond identically to SARS-CoV-2 variants of concern but differ in longevity and homing properties depending on prior infection status. Elife 10, e72619. doi:10.7554/eLife.72619
Niranjan, V., Uttarkar, A., Kaul, A., and Varghese, M. (2023). A machine learning-based approach using multi-omics data to predict metabolic pathways. Methods Mol. Biol. 2553, 441–452. doi:10.1007/978-1-0716-2617-7_19
O’Donnell, E., Floras, J. S., and Harvey, P. J. (2014). Estrogen status and the renin angiotensin aldosterone system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 307, R498–R500. doi:10.1152/ajpregu.00182.2014
Peluso, M. J., Deitchman, A. N., Torres, L., Iyer, N. S., Munter, S. E., Nixon, C. C., et al. (2021). Long-term SARS-CoV-2-specific immune and inflammatory responses in individuals recovering from COVID-19 with and without post-acute symptoms. Cell Rep. 36, 109518. doi:10.1016/j.celrep.2021.109518
Pervez, M. T., Hasnain, M. J. U., Abbas, S. H., Moustafa, M. F., Aslam, N., and Shah, S. S. M. (2022). A comprehensive review of performance of next-generation sequencing platforms. Biomed. Res. Int. 2022, 3457806. doi:10.1155/2022/3457806
Porritt, R. A., Binek, A., Paschold, L., Rivas, M. N., McArdle, A., Yonker, L. M., et al. (2021). The autoimmune signature of hyperinflammatory multisystem inflammatory syndrome in children. J. Clin. Invest 131, e151520. doi:10.1172/JCI151520
Proal, A. D., and VanElzakker, M. B. (2021). Long COVID or post-acute sequelae of COVID-19 (PASC): an overview of biological factors that may contribute to persistent symptoms. Front. Microbiol. 12, 698169. doi:10.3389/fmicb.2021.698169
Reel, P. S., Reel, S., Pearson, E., Trucco, E., and Jefferson, E. (2021). Using machine learning approaches for multi-omics data analysis: a review. Biotechnol. Adv. 49, 107739. doi:10.1016/j.biotechadv.2021.107739
Rodahl, I., Gotley, J., Andersen, S. B., Yu, M., Mehdi, A. M., Christ, A. N., et al. (2021). Acquisition of murine splenic myeloid cells for protein and gene expression profiling by advanced flow cytometry and CITE-seq. Star. Protoc. 2, 100842. doi:10.1016/j.xpro.2021.100842
Rojas, M., Rodriguez, Y., Acosta-Ampudia, Y., Monsalve, D. M., Zhu, C., Li, Q. Z., et al. (2022). Autoimmunity is a hallmark of post-COVID syndrome. J. Transl. Med. 20, 129. doi:10.1186/s12967-022-03328-4
Sacco, K., Castagnoli, R., Vakkilainen, S., Liu, C., Delmonte, O. M., Oguz, C., et al. (2022). Immunopathological signatures in multisystem inflammatory syndrome in children and pediatric COVID-19. Nat. Med. 28, 1050–1062. doi:10.1038/s41591-022-01724-3
Saigusa, R., and Ley, K. (2020). CITE-seq hits vascular medicine. Clin. Chem. 66, 751–753. doi:10.1093/clinchem/hvaa016
Schumacher, A., Costa, S. D., and Zenclussen, A. C. (2014). Endocrine factors modulating immune responses in pregnancy. Front. Immunol. 5, 196. doi:10.3389/fimmu.2014.00196
Sherif, Z. A., Gomez, C. R., Connors, T. J., Henrich, T. J., and Reeves, W. B.RECOVER Mechanistic Pathway Task Force (2023). Pathogenic mechanisms of post-acute sequelae of SARS-CoV-2 infection (PASC). Elife 12, e86002. doi:10.7554/eLife.86002
Shi, X., Baracho, G. V., Lomas, W. E., Widmann, S. J., and Tyznik, A. J. (2021). Co-staining human PBMCs with fluorescent antibodies and antibody-oligonucleotide conjugates for cell sorting prior to single-cell CITE-Seq. Star. Protoc. 2, 100893. doi:10.1016/j.xpro.2021.100893
Solis, O., Beccari, A. R., Iaconis, D., Talarico, C., Ruiz-Bedoya, C. A., Nwachukwu, J. C., et al. (2022). The SARS-CoV-2 spike protein binds and modulates estrogen receptors. Sci. Adv. 8, eadd4150. doi:10.1126/sciadv.add4150
Sonawane, A. R., Aikawa, E., and Aikawa, M. (2022). Connections for matters of the heart: network medicine in cardiovascular diseases. Front. Cardiovasc Med. 9, 873582. doi:10.3389/fcvm.2022.873582
Sonawane, A. R., Tian, L., Chu, C. Y., Qiu, X., Wang, L., Holden-Wiltse, J., et al. (2019). Microbiome-transcriptome interactions related to severity of respiratory syncytial virus infection. Sci. Rep. 9, 13824. doi:10.1038/s41598-019-50217-w
Sposito, B., Broggi, A., Pandolfi, L., Crotta, S., Clementi, N., Ferrarese, R., et al. (2021). The interferon landscape along the respiratory tract impacts the severity of COVID-19. Cell 184, 4953–4968 e16. doi:10.1016/j.cell.2021.08.016
Sullivan, K. D., Galbraith, M. D., Kinning, K. T., Bartsch, K. W., Levinsky, N. C., Araya, P., et al. (2021). The COVIDome Explorer researcher portal. Cell Rep. 36, 109527. doi:10.1016/j.celrep.2021.109527
Sun, Y., Miao, N., and Sun, T. (2019). Detect accessible chromatin using ATAC-sequencing, from principle to applications. Hereditas 156, 29. doi:10.1186/s41065-019-0105-9
Szukiewicz, D., Wojdasiewicz, P., Watroba, M., and Szewczyk, G. (2022). Mast cell activation syndrome in COVID-19 and female reproductive function: theoretical background vs. Accumulating clinical evidence. J. Immunol. Res. 2022, 9534163. doi:10.1155/2022/9534163
Thaweethai, T., Jolley, S. E., Karlson, E. W., Levitan, E. B., Levy, B., McComsey, G. A., et al. (2023). Development of a definition of postacute sequelae of SARS-CoV-2 infection. JAMA 329, 1934–1946. doi:10.1001/jama.2023.8823
Thompson, R. C., Simons, N. W., Wilkins, L., Cheng, E., Del Valle, D. M., Hoffman, G. E., et al. (2023). Molecular states during acute COVID-19 reveal distinct etiologies of long-term sequelae. Nat. Med. 29, 236–246. doi:10.1038/s41591-022-02107-4
Valdés, A., Moreno, L. O., Rello, S. R., Orduña, A., Bernardo, D., and Cifuentes, A. (2022). Metabolomics study of COVID-19 patients in four different clinical stages. Sci. Rep. 12, 1650. doi:10.1038/s41598-022-05667-0
Wang, H., Sun, X., J, L. V., Kon, N. D., Ferrario, C. M., and Groban, L. (2021b). Estrogen receptors are linked to angiotensin-converting enzyme 2 (ACE2), ADAM metallopeptidase domain 17 (ADAM-17), and transmembrane protease serine 2 (TMPRSS2) expression in the human atrium: insights into COVID-19. Hypertens. Res. 44, 882–884. doi:10.1038/s41440-021-00626-0
Wang, K., Khoramjoo, M., Srinivasan, K., Gordon, P. M. K., Mandal, R., Jackson, D., et al. (2023). Sequential multi-omics analysis identifies clinical phenotypes and predictive biomarkers for long COVID. Cell Rep. Med. 4, 101254. doi:10.1016/j.xcrm.2023.101254
Wang, M., Li, A., Sekiya, M., Beckmann, N. D., Quan, X., Schrode, N., et al. (2021a). Transformative network modeling of multi-omics data reveals detailed circuits, key regulators, and potential therapeutics for alzheimer's disease. Neuron 109, 257–272 e14. doi:10.1016/j.neuron.2020.11.002
Xiong, R., Gunter, C., Fleming, E., Vernon, S. D., Bateman, L., Unutmaz, D., et al. (2023). Multi-‘omics of gut microbiome-host interactions in short-and long-term myalgic encephalomyelitis/chronic fatigue syndrome patients. Cell Host and Microbe 31, 273–287. e5. doi:10.1016/j.chom.2023.01.001
Xu, Z., Heidrich-O'Hare, E., Chen, W., and Duerr, R. H. (2022). Comprehensive benchmarking of CITE-seq versus DOGMA-seq single cell multimodal omics. Genome Biol. 23, 135. doi:10.1186/s13059-022-02698-8
Yan, F., Powell, D. R., Curtis, D. J., and Wong, N. C. (2020). From reads to insight: a hitchhiker's guide to ATAC-seq data analysis. Genome Biol. 21, 22. doi:10.1186/s13059-020-1929-3
Yang, Q., Song, W., Reheman, H., Wang, D., Qu, J., and Li, Y. (2024). PANoptosis, an indicator of COVID-19 severity and outcomes. Brief. Bioinform 25. doi:10.1093/bib/bbae124
Yin, K., Peluso, M. J., Luo, X., Thomas, R., Shin, M. G., Neidleman, J., et al. (2024). Long COVID manifests with T cell dysregulation, inflammation and an uncoordinated adaptive immune response to SARS-CoV-2. Nat. Immunol. 25 (2), 218–225. doi:10.1038/s41590-023-01724-6
Yin, K., Peluso, M. J., Thomas, R., Shin, M. G., Neidleman, J., Luo, X., et al. (2023). Long COVID manifests with T cell dysregulation, inflammation, and an uncoordinated adaptive immune response to SARS-CoV-2. bioRxiv.
Zhang, J., Garrett, S., and Sun, J. (2021). Gastrointestinal symptoms, pathophysiology, and treatment in COVID-19. Genes Dis. 8, 385–400. doi:10.1016/j.gendis.2020.08.013
Zhang, J., Zhang, Y., Xia, Y., and Sun, J. (2023). Microbiome and intestinal pathophysiology in post-acute sequelae of COVID-19. Genes Dis. 11, 100978. In press. doi:10.1016/j.gendis.2023.03.034
Zhang, X., Justice, A. C., Hu, Y., Wang, Z., Zhao, H., Wang, G., et al. (2016). Epigenome-wide differential DNA methylation between HIV-infected and uninfected individuals. Epigenetics 11, 750–760. doi:10.1080/15592294.2016.1221569
Zhou, S., Zhang, J., Xu, J., Zhang, F., Li, P., He, Y., et al. (2021). An epigenome-wide DNA methylation study of patients with COVID-19. Ann. Hum. Genet. 85, 221–234. doi:10.1111/ahg.12440
Zhu, T., Jin, J., Chen, M., and Chen, Y. (2022). The impact of infection with COVID-19 on the respiratory microbiome: a narrative review. Virulence 13, 1076–1087. doi:10.1080/21505594.2022.2090071
Ziegler, C. G. K., Miao, V. N., Owings, A. H., Navia, A. W., Tang, Y., Bromley, J. D., et al. (2021). Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell 184, 4713–4733 e22. doi:10.1016/j.cell.2021.07.023
Zuo, T., Wu, X., Wen, W., and Lan, P. (2021). Gut microbiome alterations in COVID-19. Genomics Proteomics Bioinforma. 19, 679–688. doi:10.1016/j.gpb.2021.09.004
Keywords: COVID-19, PASC, RECOVER, systems biology, multi-omics
Citation: Sun J, Aikawa M, Ashktorab H, Beckmann ND, Enger ML, Espinosa JM, Gai X, Horne BD, Keim P, Lasky-Su J, Letts R, Maier CL, Mandal M, Nichols L, Roan NR, Russell MW, Rutter J, Saade GR, Sharma K, Shiau S, Thibodeau SN, Yang S, Miele L and NIH Researching COVID to Enhance Recovery (RECOVER) Consortium (2025) A multi-omics strategy to understand PASC through the RECOVER cohorts: a paradigm for a systems biology approach to the study of chronic conditions. Front. Syst. Biol. 4:1422384. doi: 10.3389/fsysb.2024.1422384
Received: 23 April 2024; Accepted: 14 October 2024;
Published: 07 January 2025.
Edited by:
Yoram Vodovotz, University of Pittsburgh, United StatesReviewed by:
Elebeoba E. May, University of Wisconsin-Madison, United StatesSong Feng, Pacific Northwest National Laboratory (DOE), United States
Copyright © 2025 Sun, Aikawa, Ashktorab, Beckmann, Enger, Espinosa, Gai, Horne, Keim, Lasky-Su, Letts, Maier, Mandal, Nichols, Roan, Russell, Rutter, Saade, Sharma, Shiau, Thibodeau, Yang, Miele and NIH Researching COVID to Enhance Recovery (RECOVER) Consortium. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucio Miele, bG1pZWxlQGxzdWhzYy5lZHU=; Jun Sun, anVuc3VuN0B1aWMuZWR1