Explore article hub
 Takashi Hisatomi1,2,3
Takashi Hisatomi1,2,3 Qian Wang4,5Fuxiang Zhang6Shane Ardo7,8,9Erwin Reisner10Hiroshi Nishiyama11Akihiko Kudo12,13
Qian Wang4,5Fuxiang Zhang6Shane Ardo7,8,9Erwin Reisner10Hiroshi Nishiyama11Akihiko Kudo12,13 Taro Yamada11
Taro Yamada11 Kazunari Domen1,3,11*
Kazunari Domen1,3,11*- 1Research Initiative for Supra-Materials, Shinshu University, Nagano, Japan
- 2Precursory Research for Embryonic Science and Technology - Japan Science and Technology Agency (PRESTO-JST), Nagano, Japan
- 3Institute for Aqua Regeneration, Shinshu University, Nagano, Japan
- 4Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Japan
- 5Institute for Advanced Research, Nagoya University, Nagoya, Japan
- 6State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences (CAS), Dalian National Laboratory for Clean Energy, Dalian, China
- 7Department of Chemistry, University of California, Irvine, Irvine, CA, United States
- 8Department of Chemical and Biomolecular Engineering, University of California, Irvine, Irvine, CA, United States
- 9Department of Materials Science and Engineering, University of California, Irvine, Irvine, CA, United States
- 10Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge, United Kingdom
- 11Office of University Professors, The University of Tokyo, Tokyo, Japan
- 12Department of Applied Chemistry, Faculty of Science, Tokyo University of Science, Shinjuku, Japan
- 13Carbon Value Research Center, Research Institute for Science and Technology, Tokyo University of Science, Noda, Japan
Abstract
Sunlight-driven water splitting allows renewable hydrogen to be produced from abundant and environmentally benign water. Large-scale societal implementation of this green fuel production technology within energy generation systems is essential for the establishment of sustainable future societies. Among various technologies, photocatalytic water splitting using particulate semiconductors has attracted increasing attention as a method to produce large amounts of green fuels at low cost. The key to making this technology practical is the development of photocatalysts capable of splitting water with high solar-to-fuel energy conversion efficiency. Furthermore, advances that enable the deployment of water-splitting photocatalysts over large areas are necessary, as is the ability to recover hydrogen safely and efficiently from the produced oxyhydrogen gas. This lead article describes the key discoveries and recent research trends in photosynthesis using particulate semiconductors and photocatalyst sheets for overall water splitting, via one-step excitation and two-step excitation (Z-scheme reactions), as well as for direct conversion of carbon dioxide into renewable fuels using water as an electron donor. We describe the latest advances in solar water-splitting and carbon dioxide reduction systems and pathways to improve their future performance, together with challenges and solutions in their practical application and scalability, including the fixation of particulate photocatalysts, hydrogen recovery, safety design of reactor systems, and approaches to separately generate hydrogen and oxygen from water.
Key points
- The urgent need for humanity to reduce its dependence on fossil fuels and shift to renewable fuels has driven interest in the large-scale implementation of photocatalytic water splitting using solar energy to produce green hydrogen.
- Near-perfect conversion yield for photocatalytic water splitting was achieved under irradiation using ultraviolet light and the feasibility of scaling up photocatalytic solar hydrogen production by photocatalyst sheet was demonstrated using a 100 m2 outdoor prototype panel reactor system. These breakthroughs provide key steps toward large-scale implementation.
- Proactive development of visible light-responsive photocatalysts with high solar-to-hydrogen energy conversion efficiencies, and improvement and further scale-up of photocatalytic water-splitting systems, are priorities for their large-scale deployment to be competitive with existing fuel production technologies. In this regard, photocatalyst sheets are a game-changer.
- Safety is of utmost importance when handling oxyhydrogen gas that can result from photocatalytic water splitting; risk can be reduced to an acceptable level by ensuring the safety of the entire process and complying with relevant laws and regulations. An alternative to the production of oxyhydrogen gas is the separate generation of hydrogen and oxygen, which is inherently safe.
- An accrediting organization composed of experts with a deep understanding of photocatalysis would be instrumental in streamlining the hydrogen production process and certifying reported solar-to-hydrogen energy conversion efficiencies, thereby supporting further scale-up and implementation of photocatalytic water-splitting systems.
Introduction
There is a continued growing concern that human activity is affecting Earth’s climate, which is amplified by the increasing global energy demand (1). As the industrial sector is one of the largest consumers of primary energy, derived mainly from fossil resources, there is societal pressure to reduce the heavy dependence on such resources to address the climate crisis. Hydrogen (H2) has garnered increasing interest as a versatile energy carrier and as a fundamental chemical used, for example, for the hydrogenation of carbon dioxide (CO2) to hydrocarbons and nitrogen (N2) to ammonia (NH3). While current technologies largely derive hydrogen from fossil resources, hydrogen can also be produced renewably from water, including through sunlight-driven water splitting. Large-scale societal implementation of this new green fuel production technology within energy generation systems is essential for the establishment of sustainable future societies.
A key practical consideration of any new approach is the land area required for the technology to meet our energy demand. Estimated global primary energy consumption in 2021 equates to an average power usage of 18.9 TW (2). Assuming a solar-to-hydrogen energy conversion efficiency (STH efficiency) of 10% and a relatively strong annual solar irradiance of 2000 kWh m−2, the ground surface area needed to meet this demand would be 8 × 105 km2. This is much larger than the world’s largest solar power plant in Sweihan, United Arab Emirates (7.8 km2), and exceeds even the area of Japan (3.78 × 105 km2) or the United Kingdom (2.44 × 105 km²). Aside from the challenges of STH efficiency and scalability, solar-driven hydrogen production processes must meet stringent requirements for total system cost and service life if they are to be economically feasible. Assuming a total system cost of US$100 m−2, a service life of approximately 10 years, and an annual depreciation rate of 4%, the estimated cost of solar hydrogen is US$3.5 kg−1 (3). As this cost is higher than that for hydrogen derived from natural gas (US$0.5–1.7 kg−1) (4), further reductions in costs, improvements in STH efficiency, and extension of the system service life are needed.
Photovoltaic-assisted electrochemical systems, photoelectrochemical systems, and photocatalytic systems have each been used to generate hydrogen via sunlight-driven water splitting (5, 6). An elaborate laboratory-scale photovoltaic-powered electrolyzer can exhibit an STH efficiency of 30% (7) and pilot-scale plants based on this technology have been constructed worldwide. Even so, large-scale production of solar hydrogen is likely still more expensive than generating hydrogen from fossil resources (6, 8). Photoelectrochemical designs probably involve similar challenges in cost and scalability because current approaches incorporate photovoltaic-grade materials and/or solar cells to achieve high STH efficiencies. In addition, suitable reactors and operating conditions must be discovered to minimize the concentration overpotential between hydrogen-evolving photocathodes and oxygen (O2)-evolving photoanodes during scale-up while allowing them to receive sunlight effectively (9). Systems based on particulate photocatalysts are potentially much simpler and less expensive and can also be readily scaled up, although they exhibit lower STH efficiencies at present. Recently, many researchers have focused on improving the performance of photocatalysts in sunlight-driven water splitting (3, 10–13).
This article describes the key discoveries and recent research trends in photocatalysis using particulate semiconductors and photocatalyst sheets for overall water splitting (OWS), via one-step excitation and two-step excitation, or Z-scheme reactions, as well as for direct conversion of CO2 into renewable fuels through artificial photosynthesis. We describe the latest advances in water-splitting systems and pathways to improve their future performance, together with challenges and solutions in their practical application and scalability, including fixation of particulate photocatalysts, efficient recovery of hydrogen from the raw oxyhydrogen gas product, safety design of reactor systems, and approaches to separately generate hydrogen and oxygen from water.
Photocatalytic materials and systems
The process of photocatalytic water splitting is considered a scalable and cost-effective method for producing renewable solar hydrogen. However, STH efficiencies are currently insufficient for practical implementation. Therefore, extensive research has aimed to enhance the performance of photocatalysts in sunlight-driven water splitting. This section describes two representative methods of photocatalytic water splitting: one-step and two-step excitation systems.
In one-step excitation systems, electrons and holes generated in the conduction and valence bands of a photocatalyst upon photoexcitation reduce protons to hydrogen and oxidize water to oxygen, respectively (14). In this system, in the steady state under sunlight exposure, electrons and holes must have sufficient electrochemical potentials to drive the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), respectively (15), where this OWS reaction is often kinetically rapid when the band gap of the photocatalyst straddles the hydrogen evolution potential [0 V versus reversible hydrogen electrode (RHE)] and the oxygen evolution potential (+1.23 V versus RHE). The one-step excitation system is simple in components and easily spread over large areas. However, it is challenging to prepare high-quality particulate photocatalysts that have a band gap narrow enough to absorb visible light sufficiently while meeting the aforementioned thermodynamic requirement and to construct active sites for hydrogen and oxygen evolution on single photocatalyst particles simultaneously. Consequently, the current STH efficiency is markedly low, especially when considering visible light-responsive photocatalysts.
In two-step excitation, or Z-scheme, systems, a hydrogen evolution photocatalyst (HEP) reduces water to hydrogen and an oxygen evolution photocatalyst (OEP) oxidizes water to oxygen, and the holes and electrons remaining in the HEP and OEP, respectively, recombine via aqueous redox-based or solid-state electron mediators (14). Therefore, a narrow band gap photocatalyst can be used as a HEP if conduction band electrons can drive the HER and as an OEP if valence band holes can drive the OER. Separating the thermodynamic requirement of OWS between two semiconductors significantly expands the range of photocatalysts applicable for water splitting to narrow band gap materials. However, this does not always result in larger STH efficiencies because use of two semiconductors also doubles the number of photon needed for OWS. Attention must also be paid to the need for electron transfer from the OEP to the HEP and the inclusion of more redox reactions involving electron mediators during the water-splitting reaction. To achieve highly efficient water-splitting reactions, it is necessary to find an efficient way to transfer charge between the HEP and OEP and to control the selectivity of the reaction.
Photocatalytic water splitting involves many processes that span a wide range of timescales. Typically, photophysical processes such as photoexcitation, charge separation, and charge recombination occur in the nanosecond timespan or shorter, whereas redox reactions at the surface and chemical reactions occur in the timescale of a microsecond or longer (16). Therefore, electrons and holes must be separated spatially by loading photocatalysts with cocatalysts. Furthermore, charge separation and redox reactions must be promoted, otherwise charge recombination will be dominant. As the water-splitting reaction is greatly endergonic, and backward reactions are thus thermodynamically favored, the reaction selectivity of photoexcited electrons and holes must be controlled. Moreover, safe and efficient hydrogen recovery methods are essential because the raw product of photocatalytic water splitting is usually an explosive oxyhydrogen gas containing water vapor.
One-step excitation systems
Oxide photocatalysts
Photocatalyst materials for splitting water into hydrogen and oxygen in stoichiometric amounts have been extensively studied since the Honda-Fujishima effect was reported in the late 1960s (17). Materials such as titanium dioxide (TiO2) and cadmium sulfide (CdS) were examined in the 1970s–80s, but many studies showed a lack of oxygen evolution and extremely low activity, and most attempts to make TiO2 photocatalysts responsive to visible light through transition metal doping failed. In the late 1980s, photocatalysts consisting of d0 metal cations—including zirconium dioxide (ZrO2), tantalum pentoxide (Ta2O5), K4Nb6O17, and K2La2Ti3O10—and various mixed oxide photocatalysts consisting of d10 metal cations, such as gallium (Ga3+), indium (In3+), germanium (Ge4+), tin (Sn4+), and antimony (Sb5+), were reported. (18). In particular, layered oxide materials such as K4Nb6O17 (19) and K2La2Ti3O10 (20) arose as a new photocatalyst family. In the late 1990s, NiO/NaTaO3:La (21) and RhCrOx/Ga2O3:Zn (22) photocatalysts that gave high quantum yields were found. Moreover, studies in the 2020s showed that aluminum (Al)-doped RhCrOx/SrTiO3 (RhCrOx/SrTiO3:Al) exhibited nearly a 100% quantum yield (23), and silver tantalate (AgTaO3) (24) and Na0.5Bi0.5TiO3 (25) photocatalysts gave high quantum yields for OWS. Although these oxide materials only respond to ultraviolet (UV) light owing to their large band gaps, these studies proved that powdered photocatalysts can give a high quantum yield for endergonic OWS.
Strontium titanate (SrTiO3) photocatalysts have long been studied using various approaches to improve their performance, and this led to important advances toward efficient OWS. Treating SrTiO3 powder in a molten strontium chloride (SrCl2) flux with an aluminum oxide (Al2O3) powder resulted in morphology-controlled, single-crystalline SrTiO3:Al particles with a size of approximately 200–500 nm. The doping of aluminum into SrTiO3 resulted in high photocatalytic activity. The photocatalytic activity of SrTiO3:Al was further improved by optimizing the cocatalyst components and loading methods (23). The stepwise photodeposition of rhodium (Rh) and subsequent chromium trioxide (Cr2O3) onto SrTiO3:Al resulted in the formation of a Rh/Cr2O3-core/shell cocatalyst that enabled higher water-splitting activity (26). The rhodium promotes both the HER and the oxygen reduction reaction (ORR) as an undesired back electron transfer process. Cr2O3, however, only inhibits the ORR without suppressing the HER on the rhodium core, thus allowing the OWS reaction to successfully proceed. Finally, the photodeposition of cobalt oxyhydroxide (CoOOH) as an oxygen-evolving cocatalyst onto the Cr2O3/Rh-loaded SrTiO3:Al led to an approximately 96% apparent quantum yield (AQY) in the wavelength region of 350–365 nm and a 0.65% STH efficiency. Although this efficiency was still lower than that required for commercialization, this allowed the fabrication and operation of a prototype of a solar hydrogen station with a 100 m2 total light-accepting area (27).
For a practical application of solar hydrogen production using a powdered photocatalyst, developing visible light-responsive photocatalysts that split water efficiently under solar irradiation is indispensable. Although an oxide material is attractive due to its stability and ease of production, the valence band is usually formed by O 2p orbitals located at a largely more positive level than the oxidation potential of water. Therefore, the band gap of the metal oxide needs to widen to satisfy the reduction potential of water. The formation of a new sub-valence band operating at an electron donor level is an effective way to solve the issue. A doping strategy can be utilized for the design of a visible light-responsive photocatalyst, as shown in Figure 1 (19). Doping refers to the introduction of a foreign element at the lattice point of a host material (28). The element forms an impurity level in a forbidden band of the semiconductor, enabling a response to visible light. Although a dopant contributes to the sensitization of a photocatalyst to visible light, it also introduces a recombination center and thereby decreases photocatalytic activity. Thus, a suitable combination of a dopant and a host photocatalyst, and optimization of the doping concentration, are important.
Figure 1
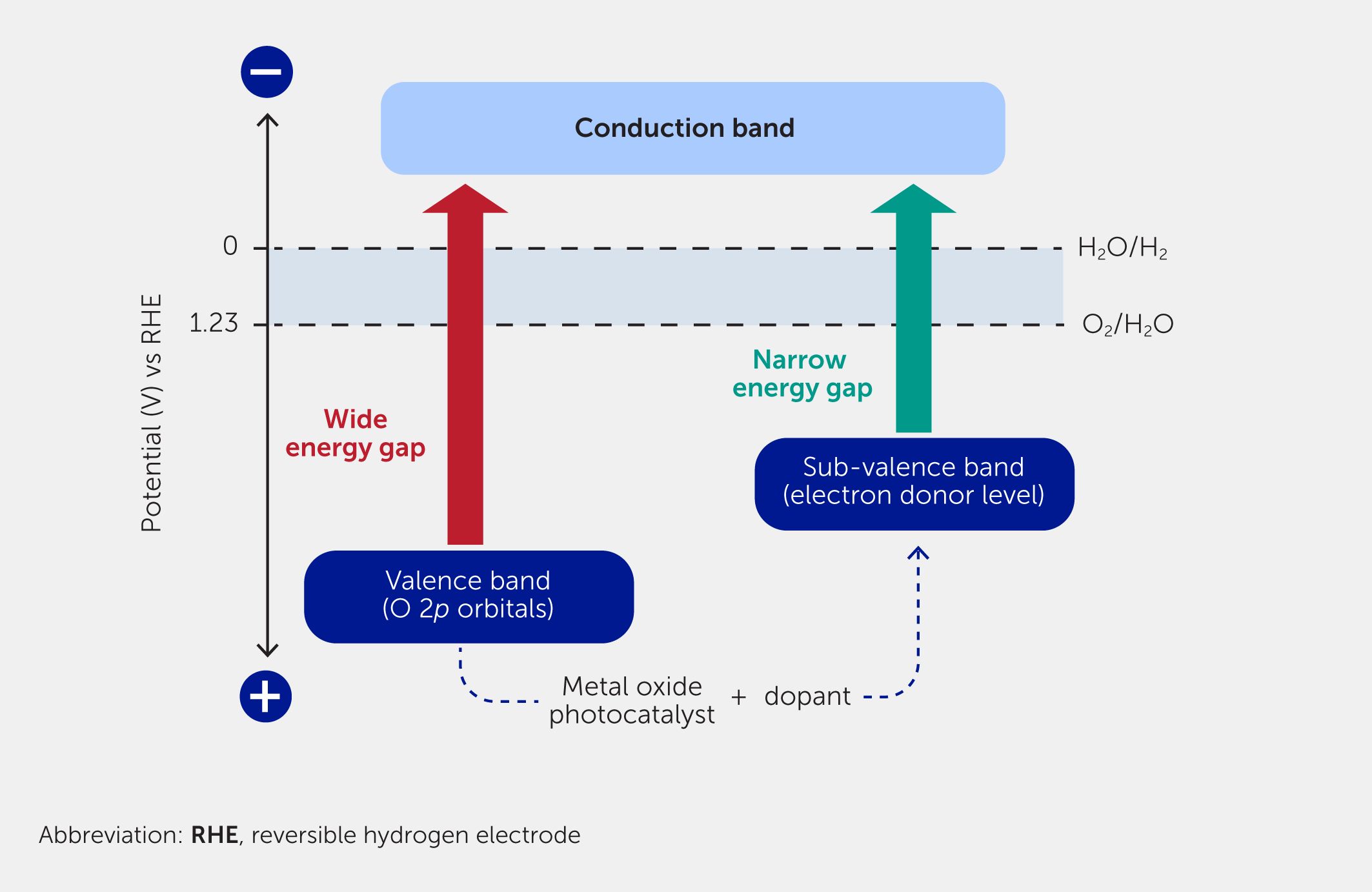
Figure 1. Sensitization of a wide band gap photocatalyst to visible light by doping with transition metal(s). Doped elements form impurity levels in the forbidden band of a wide band gap semiconductor, narrowing the energy gap required for photoexcitation and making it responsive to visible light.
Various visible light-responsive metal oxide photocatalysts have been developed by doping transition metal ions such as nickel (Ni), chromium (Cr), ruthenium (Ru), rhodium, and iridium (Ir) into wide band gap photocatalysts (29). The impurity levels formed with Cr3+, Ru3+, Rh3+, and Ir3+ work as a sub-valence band to form a narrow energy gap for visible light response. SrTiO3 doped with these metal cations and an antimony codopant shows sacrificial hydrogen and oxygen evolutions under visible light irradiation. The Sb5+ codopant provides charge compensation to control the oxidation number of the dopants and suppress recombination (30). When the chromium-, ruthenium-, rhodium-, iridium-, and/or antimony-codoped SrTiO3 powders are treated with a molten SrCl2 flux containing an Al2O3 powder and loaded with RhCrOx and CoOOH cocatalysts (as done for SrTiO3:Al), they become active for water splitting into hydrogen and oxygen—responding to visible light with wavelengths of 540–660 nm as a single particulate photocatalyst requiring only one-step photoexcitation (31–33). The RhCrOx/SrTiO3:Ir,Sb,Al/CoOOH shows the highest activity among them, giving an AQY of 0.73% at 420 nm and an STH efficiency of 0.33%.
Non-oxide photocatalysts
Metal (oxy)nitrides and oxysulfides have been studied as photocatalytic materials because they have a band gap suitable for one-step excitation OWS under visible light (14). The valence band maximum of these materials is more negative than that of the corresponding oxides owing to the higher electronegativity of nitride and sulfide ions, respectively, while the conduction band minimum (originating mainly from orbitals of metal cations) is hardly influenced by these anions. Polymeric semiconductors free from metals, such as carbon nitride (C3N4) and related materials, have also been shown to function as photocatalysts for OWS. Metal (oxy)nitrides and oxysulfides are synthesized in non-oxidizing reaction environments in the presence of nitride and sulfide ion sources, respectively. However, the products tend to be inhomogeneous and contain considerable concentrations of defects and impurities because nitride and sulfide ions are susceptible to oxidation. In addition, non-oxide photocatalysts inevitably have lower reaction-driving forces due to their narrow band gap and tend to exhibit lower quantum efficiencies than wide band gap oxides. Therefore, recent research on non-oxide photocatalysts focuses on devising and improving synthesis methods for highly crystalline and morphology-controlled semiconductor particles and on effectively coloading hydrogen and oxygen evolution cocatalysts, as in the case of the highly-active SrTiO3:Al photocatalyst (23).
Some wurtzite-type oxynitride solid solutions of typical metals, such as Ga1−xZnxN1−xOx and Zn1+xGeN2Ox, have been known to be active photocatalysts for OWS via one-step excitation since 2005 (34, 35). Ga1−xZnxN1−xOx absorbs visible light although gallium nitride (GaN) and zinc oxide (ZnO) are both responsive only to UV light. Experimentally, the absorption edge wavelength of Ga1−xZnxN1−xOx lengthens with increasing ZnO content. However, the ZnO content is generally low when Ga1−xZnxN1−xOx is synthesized by thermal nitridation under ammonia flow because of the volatilization of ZnO, and the absorption edge wavelength of active Ga1−xZnxN1−xOx had been at most approximately 500 nm (36). Recently, it was reported that well crystallized Ga1−xZnxN1−xOx with a high ZnO content and a long absorption edge wavelength (up to ~600 nm) can be synthesized by heating a mixture of gallium oxide (Ga2O3), zinc, and ammonium chloride (NH4Cl) in a sealed evacuated tube (37). The resultant Ga1−xZnxN1−xOx exhibited OWS activity under visible light via one-step excitation when loaded with appropriate cocatalysts.
Transition metal oxynitride, mainly tantalum (Ta)-based, photocatalysts are also active in visible-light-driven OWS. ZrO2−modified tantalum oxynitride (TaON) and LaMg1/3Ta2/3O2N absorb visible light with wavelengths of up to approximately 500 nm and 600 nm, respectively (38, 39). Some C3N4-based materials also exhibit water-splitting activity under visible light. However, their visible light absorption is weak (40), and materials showing decent AQYs are currently only responsive to UV light (41). A feature of the LaMg1/3Ta2/3O2N photocatalyst is that the surface is coated with an amorphous TiO2 layer after cocatalyst loading. This effectively suppresses the oxidative decomposition of the oxynitride and the reverse reaction of water splitting. Most recently, tantalum nitride (Ta3N5) (42), zirconium-doped TaON (43), magnesium--doped BaTaO2N (44), and SrTaO2N (45) have also been reported to be active in OWS under visible light. A reexamination of starting materials and the coloading of composite cocatalysts played important roles in these achievements.
Among oxysulfide photocatalysts, to date, only Y2Ti2O5S2 has been reported to split water into hydrogen and oxygen under visible light stably (46). Y2Ti2O5S2 absorbs visible light up to approximately 640 nm and exhibits outstanding thermal and chemical stability due to the hybridization of the S 3p and O 2p orbitals. To realize photocatalytic OWS using this oxysulfide, it was essential to coload a hydrogen evolution cocatalyst [chromium oxide (Cr2O3)-coated Rh] and an oxygen evolution cocatalyst (iridium dioxide; IrO2) and to tune the pH of the reaction solution to between 8 and 9. The absence of IrO2 accelerates the self-oxidation of the oxysulfide. Moreover, the driving force for oxygen evolution on this oxysulfide photocatalyst is too small at lower pH values, whereas the Cr2O3 component required to suppress the reverse reactions dissolves at higher pH values. The Y2Ti2O5S2 photocatalyst active in OWS was synthesized by a solid-state reaction in a sealed evacuated tube. The resultant Y2Ti2O5S2 photocatalyst exhibited extremely long photoexcited charge carrier lifetimes, which should be favorable for the realization of OWS (47). However, certain defects still exist in the Y2Ti2O5S2 material (48). In addition, Y2Ti2O5S2 grows into excessively large particles during the approximately 1 week of heating. To reduce the particle size, short-time synthesis of Y2Ti2O5S2 was investigated using a flux method (49) and a sulfurization method using hydrogen sulfide (H2S), (50), but the resultant Y2Ti2O5S2 products were unable to split water due to various kinds of defects.
Currently, most narrow band gap non-oxide photocatalysts rely on cocatalyst components containing precious metals and hazardous materials. For example, rhodium and ruthenium are used as the main components of hydrogen evolution cocatalysts, which are further modified with a Cr2O3 shell using a hazardous Cr6+ species as a precursor, while iridium and ruthenium are commonly used as the main components of oxygen evolution cocatalysts to compensate for the low driving force for water oxidation (37, 43–46). To mitigate the economic and environmental impact of using these precious or hazardous elements, abundant and environmentally friendly alternatives have been investigated, although the performance of these alternative materials is mostly inferior to that of conventional noble or hazardous materials. For example, copper species have been shown to have reasonable performance as hydrogen evolution cocatalysts (51), while oxides or oxyhydroxides of manganese (Mn) and cobalt (Co) have been shown to act as oxygen evolution cocatalysts in OWS similarly to iridium species (52) or often better in the presence of sacrificial electron acceptors such as silver (Ag+) cations (53). In addition, a thin layer of some metal oxides such as TiO2, Ta2O5, and silicon dioxide (SiO2) has been shown to exhibit permselectivity, suppressing undesirable backward reactions while retaining the ability for forward reactions (54, 55).
Future directions for improving photocatalyst performance
Improvements in the preparation of cocatalyst/photocatalyst composites have enabled OWS under visible light using some oxide, (oxy)nitride, and oxysulfide photocatalysts. Metal oxides are an attractive materials class for photocatalysts due to their stability under operation and ease of mass production. An important issue is how to form a new valence band above the inherent valence band of an oxide consisting of O 2p orbitals and confer visible-light activity. The component forming the new valence band should allow for rapid tranport of photogenerated holes in the band and not serve as a recombination center. Metal (oxy)nitrides and oxysulfides are endowed with adequate visible light absorption capacity. However, their AQY has been below 1% at 420 nm, and STH efficiency has only been 0.01% at most (6) (Figure 2). Dramatic improvements in STH efficiency, regardless of whether oxide or non-oxide, are required to make this technology useful in the real world. Photocatalytic OWS is a non-equilibrium phenomenon that occurs under photoexcitation. Therefore, it is important to deepen our understanding of, and ultimately control over, charge transfer and recombination processes to upgrade the reaction efficiency. This can be achieved by precisely controlling the defect formation and morphology of particulate photocatalysts, as well as the interface with cocatalysts. Materials development may be accelerated by advances in technology capable of tracking the photophysical and electrochemical processes occurring on particulate photocatalysts under photoexcitation with high spatial and temporal resolution (3, 56).
Figure 2
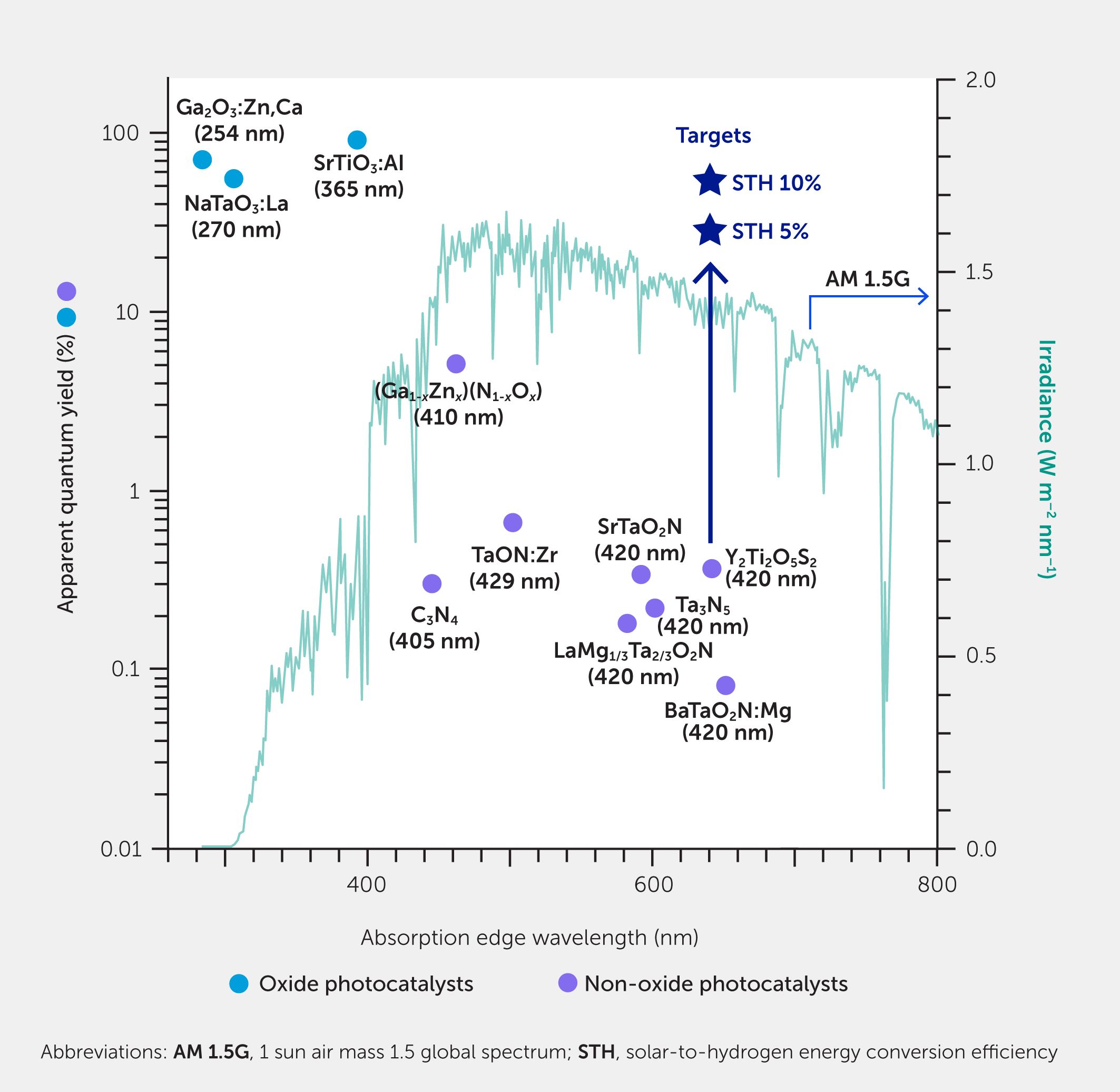
Figure 2. Performance of some photocatalysts for overall water splitting via one-step excitation. The apparent quantum yield (AQY) as a function of the absorption edge wavelength of oxide and non-oxide photocatalysts is shown. The wavelengths at which the AQY values were measured are indicated in parentheses. The irradiance spectrum of the reference sunlight (AM 1.5G) is also plotted on the right axis for comparison. To achieve solar-to-hydrogen efficiencies of 5% and 10%, it is necessary to achieve AQYs of 25.3% and 50.6% up to 640 nm, referred to as targets for guidance.
Two-step excitation systems
Suspension Z-scheme systems
Inspired by natural photosynthesis, the concept of two-step artificial photosynthesis (Z-scheme) was proposed by Bard (57) and first realized experimentally in 2001 (58). In this system, the OWS reaction is composed of two stages on two photocatalysts: one for hydrogen evolution and the other for oxygen evolution, both of which need to complete an entire photocatalytic process involving light absorption, charge separation, and surface reactions (Figure 3). A diffusional aqueous redox couple or solid-state electron mediator has been used to accomplish the charge transfer between the HEP and OEP. Compared with the one-step photoexcitation system, the Z-scheme system not only lowers the thermodynamic requirement for each semiconductor to drive the photocatalytic process, extending the available wavelength range, but can also potentially allow in situ separation of hydrogen and oxygen through a separator. The relaxation of the thermodynamic requirement enables more types of semiconductors to be candidates for photocatalytic OWS. Thus far, oxides—especially metal-doped and dye-sensitized oxides—(oxy)nitrides, and (oxy)sulfides have been employed as HEPs, and oxides including metal-doped oxides, oxychlorides, and (oxy)nitrides have been used as OEPs (59). Besides inorganic semiconductor materials, polymers, metal-organic frameworks, and some natural photosynthetic enzymes such as Photosystem (PS) I and PSII membrane fragments have also been explored for the construction of Z-scheme OWS systems (60–62). The selection of electron mediators is important to the success of Z-scheme systems, thus we discuss design guidelines and challenges for two types of electron mediators: redox-based and solid-state-based electron mediators.
Figure 3
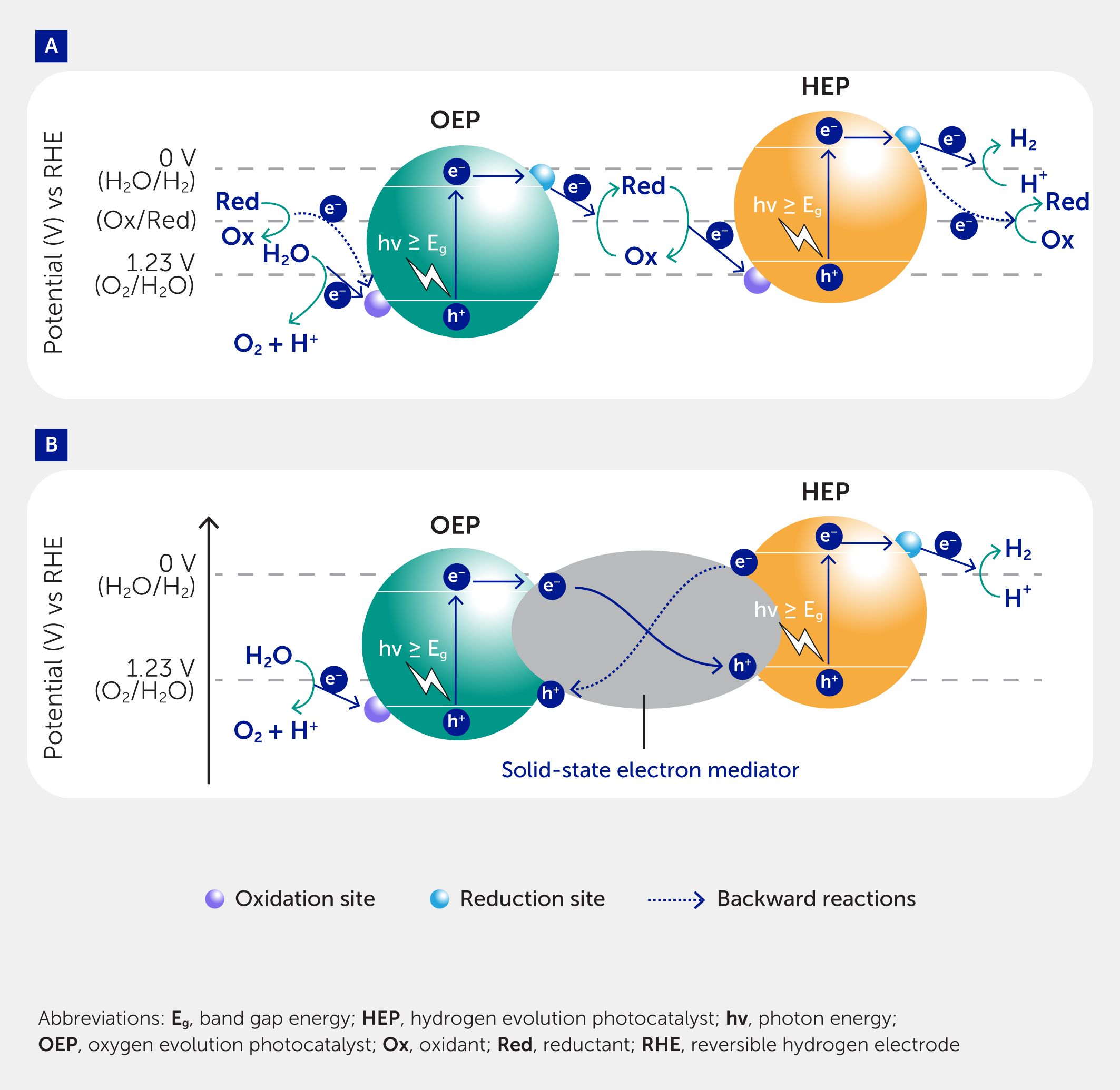
Figure 3. A typical model of suspended Z-scheme overall water splitting systems. Systems based on (A) redox mediators and (B) solid electron mediators. Charge transfer occurs via reversible valence changes of the redox mediator in the former and via charge injection through contacts and junctions between solids in the latter.
Redox-based electron mediators
In the redox-based Z-scheme OWS system (Figure 3A), charge transfer relies on reversible valence changes of the redox mediator. Up to now, several aqueous redox couples, such as triiodide/iodide/ (I3−/I−), iodate/iodide (IO3−/I−), iron-based (Fe3+/Fe2+), [Fe(CN)6]3−/4− and [Co(bpy/phen)3]3+/2+ (bpy = 2,2’-bipyridine; phen = 1,10-phenanthroline) have been exploited. Different redox couples often require different semiconductors to drive them. As far as iodine is concerned, rutile-type TiO2 and WO3 are chosen as effective OEPs because of their selective surface adsorption of IO3– ions and negligible adsorption of I– ions. In addition, the Z-scheme OWS system is mainly driven by the HEP when the initial reaction solution only contains I– ions. For iron-based redox, SrTiO3:Rh is a typical HEP and the OWS system is mostly constructed in aqueous solutions containing Fe3+ because the reduction of Fe3+ can be efficiently inhibited on SrTiO3:Rh. For redox couples based on metal complexes, such as [Fe(CN)6]3−/4− or [Co(bpy/phen)3]3+/2+, bismuth vanadate (BiVO4) has been predominantly used as the OEP due to its high oxygen evolving activity in in-situ formed low concentrations of [Fe(CN)6]3− and [Co(bpy/phen)3]3+.
Charge transfer between the redox mediator and OEP is the key to improving efficiency, and the loading of cocatalysts is important to accelerate reduction of the redox mediator. For example, platinum oxides (PtOx), iridium, and RuOx cocatalysts can activate the reduction of IO3– to I–, and gold (Au) and iridium efficiently promote the reduction of [Fe(CN)6]3−. An efficient Z-scheme OWS system with an AQY of 12.3% at 420 nm was recently achieved based on site-selective deposition of novel dual-cocatalysts for synergetic promotion of charge separation in BiVO4 (63). Another important consideration for the redox-based Z-scheme OWS is how to inhibit the undesired backward reaction (the dotted lines in Figure 3A). This is because the reduction and oxidation of the redox mediator are thermodynamically more favorable than HER and OER, and so are likely to proceed preferentially. To solve this problem, the regulation of the adsorption properties of the semiconductor or cocatalyst surface is particularly significant. For instance, the modification of rutile-type TiO2 or magnesium oxide (MgO) on Ta3N5 hinders the access of I– to inhibit its oxidation. Furthermore, the core/shell-structure of the Cr2O3/Pt cocatalyst structure prevents IO3– from reaching the surface of platinum (Pt) to suppress its reduction as a result of the steric hindrance effect.
Solid-state electron mediators
In the solid-state Z-Scheme OWS system (Figure 3B), physical contact or a solid electron mediator is applied to complete the interparticle charge transfer between the HEP and OEP. In early research, the physical mixture of Ru/SrTiO3:Rh and BiVO4 was proven active for Z-scheme OWS but the performance was low. This demonstrates that the contact interface has a significant influence on the charge transfer efficiency. Thus, some electrical conductors (e.g., Ir, Rh, and reduced graphene oxide) have been used. However, in most cases, the contact between the HEP and OEP mainly depends on the electrostatic attractive force resulting from different isoelectric points through the pH adjustment. High interfacial resistance is a problem owing to loose contact. Moreover, the backward charge transfer process (whereby photo-generated electrons from the HEP are injected into the solid conductor and sequentially recombine with photo-generated holes from the OEP) must be prevented to avoid a short circuit effect (the dotted line in Figure 3B). Band bending in the space-charge layer may help to promote forward charge transfer. In light of this, the combination of n-type and p-type semiconductors is an ideal model due to the matched surface band bending direction. As a special case, a BiVO4–Ru/SrTiO3:Rh (n-type/p-type) composite was successfully constructed through in situ growth, which provided suitable contact and consequently demonstrated higher activity with respect to the physical mixture under neutral pH conditions (AQY: 1.6% at 420 nm) (64).
Immobilized Z-scheme systems
Photocatalyst sheets producing hydrogen from water
Although Z-scheme systems using redox couples (e.g., Fe3+/Fe2+ and IO3−/I−) to mediate electron transfer between HEPs and OEPs have been extensively explored, they have limitations, such as competitive side reactions and shading of photocatalysts from incident light (59). Photocatalyst sheet systems were developed to overcome these hurdles. These comprise two redox-complementary semiconductor particles embedded in a conductive layer such as gold, carbon, or indium tin oxide (ITO) (Figure 4A) (65–67). The conductive layer serves to connect the photocatalytic particles, facilitating effective interparticle electron transfer and photocatalytic performance in the Z-scheme OWS reaction, resulting in an STH efficiency >1%. The standalone and wireless photocatalyst sheet systems stand out from conventional Z-scheme and photoelectrochemical systems as they operate in pure water without the need for pH adjustment, redox mediators, electrolytes, or buffering agents. Additionally, the photosynthetic activity of the photocatalyst sheet, achieved by fixing the photocatalyst powder on a substrate, is scalable and overcomes the constraints of photoelectrochemical or artificial leaf systems (such as difficulties in large-scale fabrication and pH gradients under neutral pH conditions) without solution convection and resistive losses caused by the electrolyte (68). This is due to the reduction and oxidation reactions occurring in close proximity on the photocatalyst sheet, resulting in the suppression of the pH gradient and ohmic potential drop. Moreover, large photocatalyst sheets can be easily prepared via a cost-effective and accessible screen-printing technique using the ink containing photocatalyst powder and gold or ITO nanoparticles as the conductive mediator (65, 67).
Figure 4
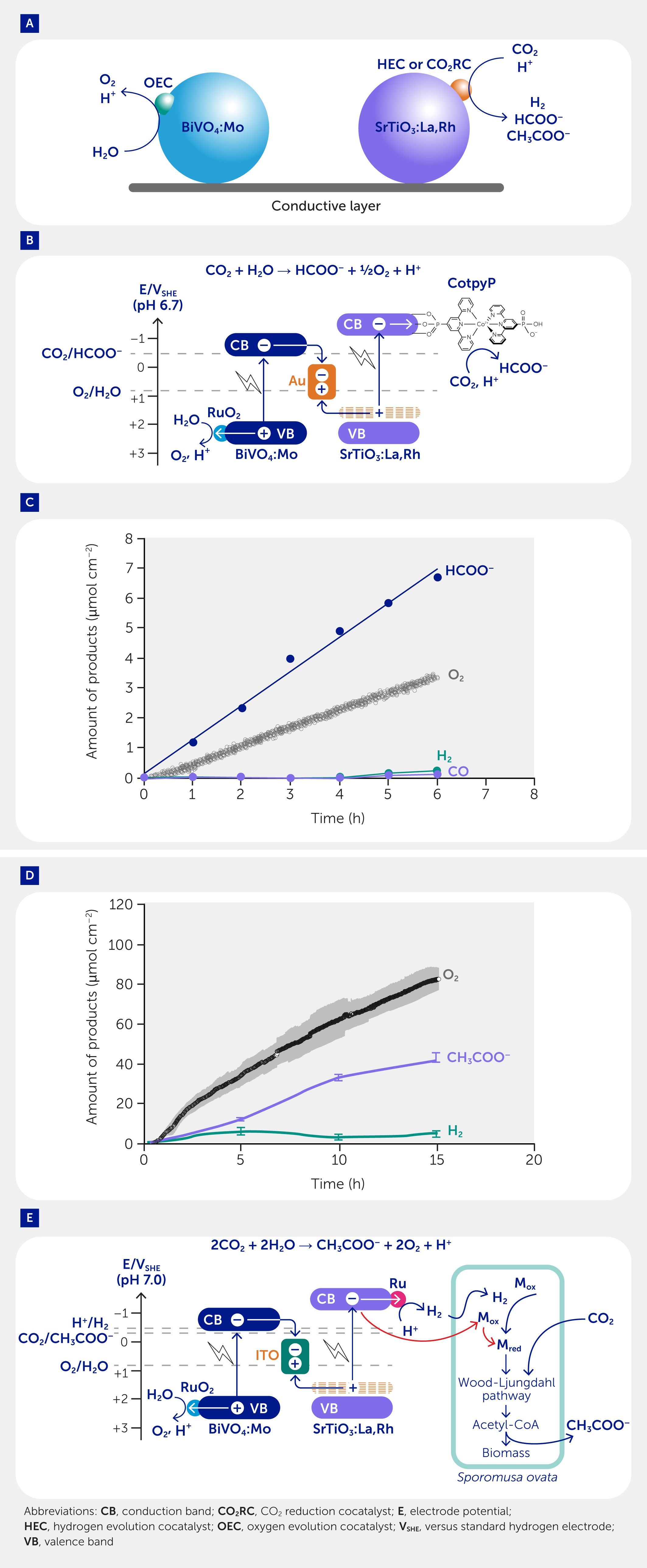
Figure 4. Photocatalyst sheets for solar-to-fuel conversion. (A) Photocatalyst sheets for artificial photosynthesis to produce fuels from sunlight, water, and carbon dioxide (CO2). (B) An energy diagram depicting the photosynthetic CO2 reduction coupled with water oxidation over a CotypP-SrTiO3:La,Rh|Au|RuO2-BiVO4:Mo photocatalyst sheet. (C) Formate (HCOO−), oxygen (O2), hydrogen (H2), and carbon monoxide (CO) production over a CotypP-SrTiO3:La,Rh|Au|RuO2-BiVO4:Mo photocatalyst sheet under simulated sunlight (AM 1.5 G, 100 mW cm−2). (D) CH3COO–, O2, and H2 production over S. ovata|Cr2O3/Ru-SrTiO3:La,Rh|ITO|RuO2-BiVO4:Mo hybrids from water and CO2 under simulated sunlight (AM 1.5 G, 100 mW cm−2). (E) Sporomusa ovata|Cr2O3/Ru-SrTiO3:La,Rh|ITO|RuO2-BiVO4:Mo for photosynthetic CO2-to-acetate (CH3COO–) conversion coupled with water oxidation. Mox and Mred represent intracellularly oxidized and reduced redox mediators, respectively.
Photocatalyst sheets for selective CO2 reduction
In addition to hydrogen production through the OWS reaction, artificial photosynthetic systems that use sunlight and water to directly convert CO2 into renewable fuels are of growing interest and are undergoing rapid development (69). This is because the utilization of traditional fossil energy sources leads to excessive anthropogenic CO2 emissions, which accumulate in the atmosphere and cause severe environmental issues. Photocatalytic CO2 reduction results in the production of various liquid and gaseous products, including carbon monoxide (CO), methane (CH4), formate (HCOO−), acetate (CH3COO−), and methanol (CH3OH). The conversion of CO2 into liquid fuels, such as formate and methanol, using intermittent solar energy presents an alluring opportunity owing to their potential for fuels with high-energy densities, ease of storage and transportation, and the potential to support the sustainable production of commodity chemicals in the post-fossil fuel era (70). It can also prevent the formation of explosive oxyhydrogen gas from water and allow the spontaneous separation of CO2 reduction products and oxygen in the liquid and gas phases, respectively.
The integration of a cocatalyst is commonly required for photocatalytic CO2 conversion to control the desired catalytic pathway, induce selectivity into the system, and enhance the conversion efficiency (71, 72). For example, inorganic metallic nanoparticles, such as silver and gold, have been widely employed as cocatalysts on photocatalysts for photocatalytic CO2 reduction to produce CO and formate (73–75). However, the selectivity and efficiency of these systems remain constrained by the limited overpotential that semiconductor light absorbers can provide and many CO2-reducing solid-state materials rely on precious metals. In recent years, immobilized molecular catalysts—often containing inexpensive 3d transition metals—and biocatalysts—including microorganisms and isolated enzymes—have emerged as promising cocatalysts in (bio)molecular-semiconductor hybrid systems owing to their high product selectivity and ability to form multi-carbon products (72, 76).
Small-molecule catalysts display discrete active sites that can be readily tuned to improve activity and can thus reduce the overpotential by stabilizing intermediate transition states between linear CO2 molecules and the desired product, overcoming kinetic barriers for the reaction (77). Transition metal complexes are frequently used as homogeneous catalysts for CO2 reduction as they can facilitate multi-proton/electron-transfer processes and exhibit multiple accessible redox states (72). Enzymes are evolutionarily optimized catalysts that often function with “ideal” performance, thus delivering high rates of catalysis at a minimal overpotential (78–80). Immobilization of (bio)molecular catalysts on semiconductor light absorbers not only provides active sites for selective CO2 reduction but also enables efficient operation in aqueous solutions. Additionally, immobilization enhances photocatalyst recycling from the reaction medium (81–85). In recent years, several studies have demonstrated the effectiveness of molecular catalysts in selective photocatalytic CO2 reduction in aqueous solutions containing sacrificial electron donors such as triethanolamine (81, 83, 86–89). These studies demonstrate the potential of coupling molecular catalysts with photocatalysts. However, to achieve carbon neutrality and solar fuel production on a global scale via artificial photosynthesis, the process must be coupled with the OER, resulting in the release of oxygen that can be used as an oxidant for fuel consumption and to regenerate the initial species (90).
To achieve photocatalytic CO2 reduction without the need for sacrificial electron donors, a monolithic device was constructed by immobilizing a CO2 reduction molecular catalyst onto the surface of a photocatalyst sheet. Specifically, a sheet comprising of SrTiO3:La,Rh|Au|BiVO4:Mo was utilized to immobilize a phosphonated cobalt bis(terpyridine) molecular catalyst (CotpyP) instead of using a deposited hydrogen evolution cocatalyst, leading to conversion of CO2 to formate with a selectivity of 97 ± 3% and a solar-to-formate energy conversion efficiency of 0.08 ± 0.01% (Figures 4B, C) (91). Irradiation with simulated sunlight generated electron-hole pairs in both SrTiO3:La,Rh and BiVO4:Mo. The gold layer facilitated electron transfer from BiVO4:Mo to SrTiO3:La,Rh. The immobilized molecular complex on SrTiO3:La,Rh functioned as a CO2 reduction cocatalyst, reducing CO2 to HCOO− with electrons, while the holes in BiVO4:Mo oxidized water to oxygen, with RuO2 serving as an oxygen evolution cocatalyst. Stoichiometric production of oxygen gas during the oxidation reaction confirms that the conversion takes place cleanly without the requirement for sacrificial reagents. The scalability of the hybrid system was demonstrated as the solar-to-formate energy conversion efficiency was largely maintained (0.06%) even when the sheet area was increased 20-fold.
Some microorganisms possess the remarkable ability to synthesize complex compounds from water and CO2 via carbon assimilation pathways, exemplified by the Wood-Ljungdahl pathway, a feat that remains challenging to achieve with abiotic cocatalysts (76, 92). Furthermore, whole-cell microorganisms have the advantage of self-replication and self-repair, making them robust for long-term applications. Photocatalyst sheets demonstrate a suppressed pH gradient during redox reactions and maintain pH-independent activity within a neutral pH range, thus enabling them to achieve artificial photosynthesis without pH adjustment or buffer solution addition (93). This property makes the sheets particularly suitable for constructing hybrid bio-abiotic systems and performing semi-artificial photosynthesis as they can achieve high solar-to-fuel energy conversion efficiency at a biocompatible neutral pH (94).
To create a biohybrid system to produce fuels utilizing sunlight, CO2, and water, CO2-fixing acetogenic bacteria (Sporomusa ovata) were combined with an OWS photocatalyst sheet (Cr2O3/Ru-SrTiO3:La,Rh|ITO|RuO2-BiVO4:Mo) (Figure 4D) (92). Prepared using a relatively inexpensive and readily accessible drop-casting method, this synthetic sheet achieved photocatalytic OWS to generate hydrogen. Combining the photocatalyst sheet with S. ovata resulted in a hybrid prototype that produced CH3COO– and oxygen in a 1:2 stoichiometric ratio from CO2 and water using sunlight as the sole energy input, achieving a solar-to-acetate energy conversion efficiency of 0.70 ± 0.04% (Figure 4D). The hydrogen produced by the photocatalyst sheet was used by S. ovata to reduce CO2 and produce CH3COO– via the acetyl-CoA Wood-Ljungdahl pathway. Additionally, S. ovata can directly use photoexcited electrons from SrTiO3:La,Rh to conduct photosynthesis for acetate formation, albeit at a significantly reduced efficiency (Figure 4E). As CH3COO– production is accompanied by oxygen evolution, it is important to carefully control the concentration of accumulated oxygen in the reaction system to avoid harm to the anaerobic bacterium.
Furthermore, the CH3COO– produced by this semi-artificial photosynthesis system can be fed into a microbial fuel cell—for example, comprising an inverse opal-ITO electrode inoculated with Geobacter sulfurreducens—to generate renewable electricity, thereby producing power in the dark or upon demand and turning the CH3COO– back into CO2 and water (92). These bio-abiotic hybrid systems thus offer a promising strategy for sustainably and cleanly fixing CO2 and closing the carbon cycle.
Pathways for advancing the performance of Z-scheme systems
Various photocatalytic materials—especially those with a long-wavelength photoresponse—can be efficiently applied to solar-to-chemical energy conversion reactions using the Z-scheme system. The development of water-compatible, precious-metal-free molecular catalysts and their immobilization on semiconductors enables their operation in water, an environmentally benign solvent, without the need for organic solvents (72). Furthermore, microbes can be readily grown and self-repair and self-replicate, while fuel-producing enzymes employ abundant metals and are biodegradable. The biocatalysts operate under environmentally benign conditions, such as moderate temperatures and around neutral aqueous pH, although special buffer and anaerobic conditions may be required (95).
The STH efficiency and the solar-to-fuel energy conversion efficiency of CO2 reduction are still approximately 1% at most, which is below the practical target. In addition, the long-term durability of the system using molecular catalysts and biocatalysts has not been extensively examined. Therefore, research and development are still needed to improve the performance of the Z-scheme system. Regardless of whether redox mediators or solid-state electron mediators are used for electron transfer, high performance in suspension Z-scheme OWS systems depends on transferring electrons and holes to the surface and driving the forward reactions (namely, HER, OER, and CO2 reduction) in a highly selective manner, in addition to improving the efficiency of light absorption and charge separation. Most current research focuses on the exploration of new redox mediators and semiconductors, and little attention has been paid to the suppression of undesired backward reactions. New strategies for achieving efficient charge separation and promoting forward reactions are highly desirable. Photocatalyst sheets have proven to be efficient and scalable in their ability to drive OWS and convert water and CO2 into valuable chemicals and fuels under simple reaction conditions. Moreover, molecular catalysts and biocatalysts can be easily assembled on the sheets, making it possible to construct organic-inorganic and bio-abiotic hybrid systems. This versatility allows for the production of a wide range of solar fuels and chemicals in the future by combining various molecular catalysts, biocatalysts, and semiconductors. Such hybrids inherit both the high light-harvesting efficiency of semiconductors and the superior catalytic performance of molecular catalysts and biocatalysts. One of the key challenges with this approach is to understand and manipulate the hybrid interface for effective electron transfer, which represents the next frontier of research.
Reaction systems and processes
The previous section described laboratory-scale studies of particulate photocatalysis, where photocatalysts are exposed to an artificial excitation light source under well-controlled conditions in a dedicated reactor, and typically trace amounts of products are separated and quantified using dedicated analyzers. However, even if particulate photocatalysts are developed that efficiently split water into hydrogen and oxygen in such an environment, this does not mean that the hydrogen produced is immediately available for use: it must be recovered and purified to a usable level. In particular, the crude product of the OWS reaction is explosive oxyhydrogen gas, which requires careful handling for safety reasons. Therefore, in addition to the development of photocatalytic materials and their composite systems, it is necessary to develop systems and processes that enable the large-scale application of photocatalysts and the safe and efficient recovery of the products. This section presents a panel reactor system as an example of a large-scale application of water-splitting photocatalysts and the latest technology for the separation and recovery of hydrogen from the oxyhydrogen gas produced. A concept for producing hydrogen and oxygen separately from water, from the outset, is also discussed.
Photocatalyst sheets and panel reactors
Scaling up photocatalytic water splitting
Supplying low-cost hydrogen globally by photocatalytic OWS would require a photocatalyst reactor system on a scale of at least 1 ha to 1 km2 per plant (3). Thus far, photocatalysts have primarily been evaluated under specific experimental conditions, e.g., for light irradiation intensity, reaction temperature, and pressure. Obviously, it is exceedingly difficult to implement photocatalysts under ideal conditions in plant operations in the natural environment. For example, the irradiation intensity varies significantly between dawn and dusk, ranging from nearly 0 to 1 kW m−2, and the reaction temperature varies considerably between winter and summer. Therefore, to scale up the ideal small-scale laboratory experiments to a practical scale it is critical to conduct outdoor experiments on a specific scale and gain knowledge from them.
Multiple options may exist for photocatalyst reactor structures. Domen et al. constructed an array of panel reactors, using a particulate SrTiO3:Al photocatalyst, measuring 100 m2—the largest scale reported among photocatalytic solar hydrogen production systems—and operated the system for several months (27, 96). The study specifically focused on developing a reactor structure that was simple. One crucial factor to consider is how to introduce photocatalysts into the reactor system. While it is feasible to use the photocatalyst slurry utilized in small-scale laboratory experiments, maintaining a uniform slurry in large-scale systems is challenging (97). First, it is difficult to minimize the cost of the reactor being able to sustain large amounts of water. For example, even at a depth of only 1 cm, the mass of water in a reactor can reach 10 kg m−2. As a result, reactors tend to be bulky and unfeasibly costly on a large scale—the maximum allowable cost of an entire hydrogen production system is estimated at ~$US100 m−2 (3). Second, the particulate photocatalyst settles to the bottom of the reactor and cannot effectively receive incident light unless the reactor is completely level or stirred. Third, collecting the suspended photocatalyst powder by filtration or centrifugation is both energy- and time-consuming. Given the large footprint of photocatalytic water-splitting systems, the processes associated with replacing spent photocatalysts should be simpler. The use of fixed particulate photocatalysts can solve all of these problems. Therefore, in the aforementioned system, the photocatalyst was immobilized on a suitable substrate and introduced into a large-scale reactor. The lifetime of photocatalysts is also critical in the development of photocatalytic reactors and systems, as it takes a long time to build and test large-scale photocatalytic plants. The SrTiO3:Al photocatalyst is ideal in this regard because it was able to maintain STH efficiency for over 1600 h under continuous exposure to simulated sunlight (27).
The SrTiO3:Al photocatalyst was produced in large quantities using the SrCl2 flux method (27). Cocatalysts were applied by impregnating the photocatalyst in an aqueous solution of Na3RhCl6, Cr(NO3)3, and Co(NO3)2, followed by calcination in the air (27, 97, 98). Photocatalyst sheets were fabricated by spraying a slurry of modified SrTiO3:Al mixed with chained colloidal silica and calcium chloride onto glass sheets and heated at 90°C with a relative humidity of 50% for 4 h. The loading of modified SrTiO3:Al was controlled to 0.89 mg cm−2 (27). The silica matrix was embedded with SrTiO3:Al particles with a diameter of a few hundred nanometers. The photocatalyst layer was ~10 μm thick. Note that at the end of the photocatalyst life cycle, the whole photocatalyst can be processed for recycling rhodium, the precious cocatalyst material, in standard industrial noble-metal recovery methods.
The photocatalyst sheet was arranged into a panel reactor (625 cm2), which was tested under UV light (λ = 365 nm) produced by a light-emitting diode array. It was found that the oxyhydrogen gas generated by OWS was smoothly discharged from the reactor even when the gap between the photocatalyst sheet and the panel window was merely 0.1 mm. The 48-panel reactor units were then combined into a module with a light-receiving area of 3 m2 supported by a single scaffold (27). The racks for the reactors were tilted towards the equator (southward in Japan) at an angle of 30°. An operational photocatalytic panel reactor system with a 100 m2 light-receiving area was completed by arraying these 3 m2 modules (Figure 5).
Figure 5

Figure 5. Aerial view of an operational 100 m2 photocatalyst array system for solar hydrogen production. The system uses 1,600 panel reactor units (each 625 cm2), where 48 panel reactor units are integrated to fabricate a 3 m2 module, and 33 and one-third (33 + 1/3) modules are linked to form one system. The panel reactor is connected to a gas separation facility.
The oxyhydrogen product gas was transferred from each reactor unit via a Teflon tube (inner diameter 1.6 mm) and subsequently passed through polyurethane tubes (8.6 mm) using daisy-chained plastic connectors across the entire panel reactor. The outgoing gas flow was measured using a soap film gas flowmeter and a mass flow meter combined with a membrane gas dryer. The STH efficiency measured over 30 min reached 0.76% at the end of summer in Japan, a value somewhat higher than that measured using simulated sunlight irradiation in the laboratory. This is because the SrTiO3 photocatalyst could use a larger fraction of UV light from whole solar radiation in the summer, as compared with standard simulated sunlight. The voluminous size of this system enabled it to achieve an outstanding solar hydrogen output. The application of plastic materials for the major parts of the reactor panel arrays, namely, photoreactor vessels and fluid tubing, is essential for the rational lifecycle operation of the plant in the future. A certain part of the product hydrogen can be dispensed for the synthesis of olefins from CO2, and the olefins can be converted to polymeric materials to embody the plant. The CO2 fixation ability of the whole plant will be rationally higher than the CO2 exhaust of the waste plastic from the plant components after combustion. This mechanism of self-regeneration will be a characteristic feature of photo-reaction devices of this type. As for the operational safety of the 100 m2 plant generating oxyhydrogen, no instances of autonomous product gas combustion were observed during its operation for approximately 3 years, confirming the safety and capacity of an appropriately designed large-scale photocatalyst system for further practical deployment (27). The results of this experiment demonstrate that it is theoretically possible to scale up solar hydrogen production using photocatalysts safely without reducing their efficiency. Nonetheless, when designing an oxyhydrogen-generating system, it is necessary to incorporate devices and design tactics to prevent system destruction caused by accidental ignition.
Handling of oxyhydrogen gas
Since the photocatalytic water-splitting panel reactor can safely evolve and transfer the stoichiometric mixture of oxyhydrogen by photocatalytic OWS onsite and is suitable for scaling up, the next key question is whether hydrogen can be recovered safely and efficiently from oxyhydrogen. Oxyhydrogen is explosive, thus supporting why there are few studies on its use and handling (99). Oxyhydrogen has a low ignition threshold energy (100), ignites by itself in a region of high temperature and high pressure, and ignition leads to detonation (propagation of a combustion shock wave), depending on the size and shape of the containing vessel. The safe operation of OWS photocatalysts therefore requires the establishment of design criteria for gas-handling equipment to avoid ignition/explosion, and to limit the damage to equipment caused by occasional explosions.
Domen et al. have assessed the damage caused by intentional ignition/explosion of oxyhydrogen confined in the parts of photocatalytic reactors, gas transfer tubes, gas reservoirs, and gas filter modules—component by component (27). The safety design criteria gained from these experiments for oxyhydrogen at 1 atm are: (a) ignition in a small compartment does not destroy the vessel, either completely sealed or open to the air; (b) ignition in a small-diameter, long tube causes no damage, whereas if the vessel is three-dimensionally extended (e.g., cubes, boxes, and cylindrical vessels) a destructive explosion may occur, even at a volume of a few hundred cm3; and (c) ignition in soft containers, such as those made of soft polyvinyl chloride (PVC) or polyurethane, will not cause a destructive explosion. These criteria were reflected in the design of the aforementioned 100 m2 photocatalytic reactor arrays and the hydrogen filtering apparatus (27) to avoid the hazards of incidental explosions. Notably, (b) is related to the one-dimensional propagation of the supersonic detonation of oxyhydrogen without a destructive explosion. An ignition experiment in a 100 m soft PVC tube (inner diameter 10 mm) exhibited propagation of a flame at 2,800 m s−1, recorded by a high-speed video camera. However, the tube was undamaged, and the flame was arrested at the end of the tube which was immersed in water. In the 100 m2 photoreactor experiment, hydrogen gas was produced safely for approximately 3 years without any accident by spontaneous ignition.
Extracting hydrogen from oxyhydrogen
The extraction of hydrogen from oxyhydrogen is an essential engineering aspect. Hydrogen can be separated from oxyhydrogen by gas filtration driven by input gas pressure at a relatively low energetic cost compared with liquefaction/distillation or pressure-swing adsorption processes. The material used for the gas filter element should be highly permeable to hydrogen gas and minimally permeable to oxygen. Commercial filter modules containing polyimide hollow tubes (101) can be used. Crucially, these tubes can withstand the moisture contained in oxyhydrogen—the product gas of OWS has 100% relative humidity. Their permeability ratio for hydrogen/oxygen is ~10 or more, and the filtrate hydrogen is anticipated to be concentrated by more than 90% by applying a transmembrane pressure to the filter element. According to the principle of entropy, gas separation costs a certain amount of energy to keep the operational pressure and the speed of gas flow. Moreover, the filtering element requires optimum pressure and flow-speed conditions for good efficiency. The gas filter module exhibits a trade-off in the filtrate hydrogen purity and recovery ratio of hydrogen in the filtrate. If the feed gas flow rate is high, the filtrate hydrogen purity will be high but the hydrogen recovery will be low. If the feed gas flow rate is low, hydrogen purity decreases but the recovery ratio increases. The feed gas flow rate must therefore be fixed at a value providing the desired filtrate hydrogen purity and recovery.
In a test operation of hydrogen reparation and recovery from moist oxyhydrogen gas from the 100 m2 scale photocatalytic water-splitting reactor, a vacuum pump maintained the filtrate pressure at nearly zero and released the hydrogen-enriched gas at 1 atm (27). This allowed for a steady-state enrichment of hydrogen in the filtrate gas flow and oxygen in the residue gas flow. Under these real-world conditions, the constantly changing weather resulted in a variable rate at which oxyhydrogen was produced. Therefore, in order to control and optimize the filtration feed rate, the oxyhydrogen was stored in an intermediary vessel: once this was full, the gas was discharged into the filter at an optimal, high flow rate, controlled by a throttle valve. The oxyhydrogen vessel should be carefully designed following the safety design criteria to avoid destructive explosions. This recovery system has undergone whole-daytime tests in the photoreactor array under variable weather conditions. The filtrate hydrogen purity was >92%, and the hydrogen recovery was ~73% during early development (27). The hydrogen purity can be further improved by catalytically quenching the remaining oxygen with hydrogen to exceed the explosion limit (102). Notably, atmospheric hydrogen recovered from the photocatalytic panel reactor can be used directly for CO2 methanation when connected to a methanation catalyst (Figure 6) (102). Such a reaction system received the highest evaluation in the Horizon Prize competition on “Fuel from the sun: artificial photosynthesis” organized by the European Innovation Council (103). The competition entry involved automated operation of integrated photocatalytic OWS using a panel reactor, hydrogen separation and recovery using a separation membrane module, and catalytic CO2 methanation, all performed outdoors under natural sunlight for 3 days. It was possible to power a Stirling engine with the crude methane produced.
Figure 6
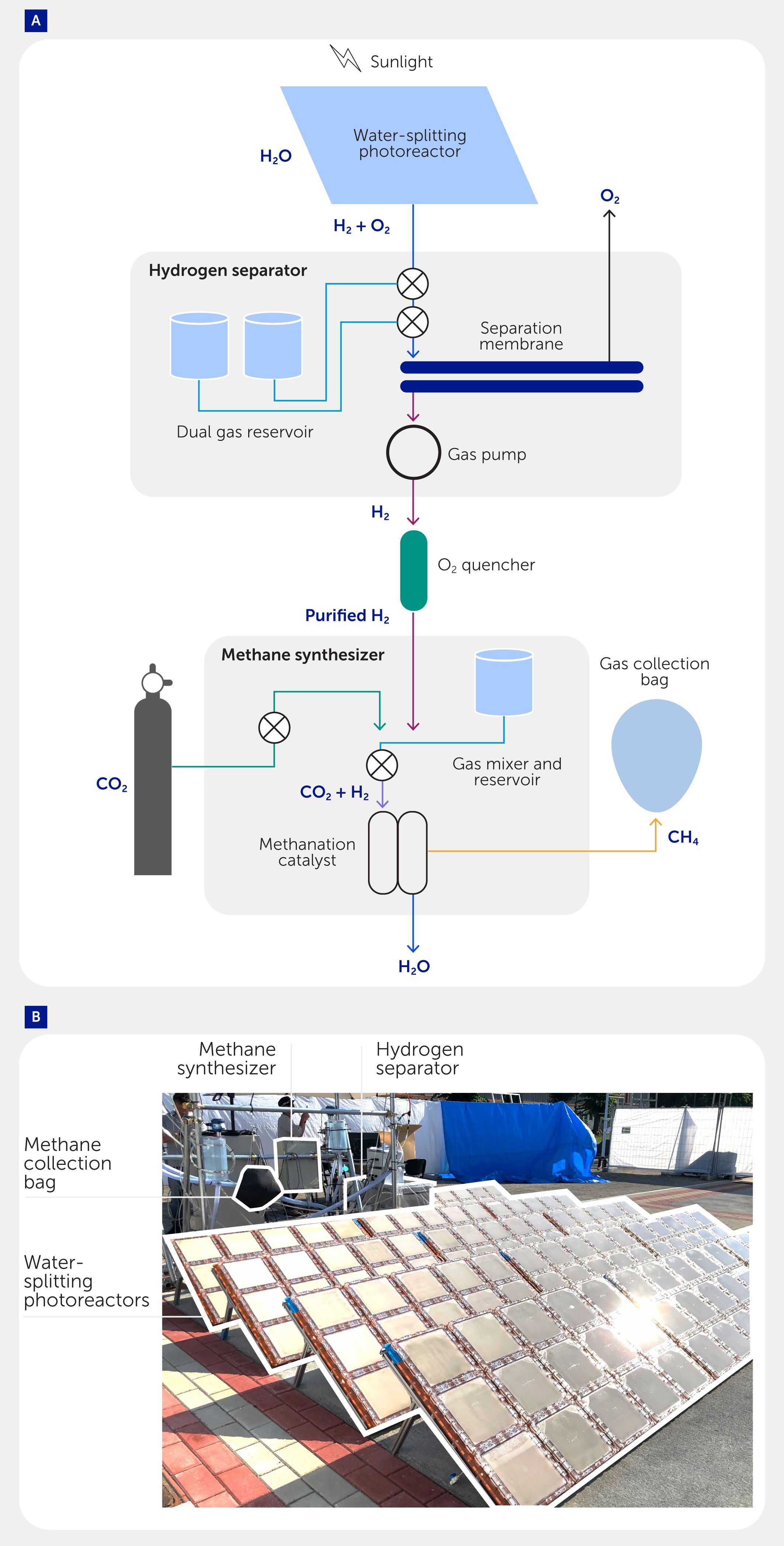
Figure 6. A mini-pilot system for green methane (CH4) production. (A) Schematic and (B) photograph of the system. The system consists of (i) a water-splitting photoreactor to produce oxyhydrogen gas from water, (ii) a hydrogen (H2) separator to recover purified H2 from oxyhydrogen, an oxygen (O2) quencher to increase the H2 purity, and (iii) a CH4 synthesizer to convert carbon dioxide (CO2) to methane. The methane produced is recovered in (iv) a balloon. Byproduct H2O of the CO2 methanation is discharged. CO2 is supplied externally.
Prospects for optimizing reactor systems and safety measures
The societal-level implementation of large-scale solar-powered hydrogen production plants will require low-cost, large-scale reactor systems equipped with highly active photocatalysts. The panel reactors described above were manufactured for academic research purposes and are robust enough to withstand long-term outdoor operation without considering manufacturing or operating costs. However, a practical solar fuel production system must be cost-competitive with existing fuel production technologies. Simpler reactors, made of lighter and less expensive materials while ensuring safety and durability, need to be developed.
Technical advances necessary for the safe handling of oxyhydrogen and efficient separation/purification of hydrogen are (i) the development of high-performance gas-filter materials for better purity and recovery ratio of hydrogen; (ii) measures allowing energy-saving operation of the ancillary devices in the plant, such as pumps, valves, and temperature control; and (iii) application of soft, low-cost plastic materials that can tolerate detonation within the gas tubes and vessels. The risk of hydrogen explosions in photocatalytic panel reactors for solar-driven OWS and associated hydrogen separation and recovery systems can likely be reduced to acceptable levels. However, because safety is critical, it is recommended that in the research of oxyhydrogen handling, operators stay away from areas where oxyhydrogen is collected and that all operations be remotely commanded and monitored for safety. In addition, newly developed units should be inspected and approved by chemical engineering safety specialists to ensure compliance with safety and liability requirements.
Once hydrogen is separated and recovered, it can be combined with various catalytic reactions to synthesize useful and easy-to-handle chemicals and fuels (e.g., CO2 to methane, nitrogen to ammonia, and toluene to methylcyclohexane.). Such product materials are more realistic in terms of safe handling and ease of transportation compared to hydrogen as the final product. The key to this process is a catalyst that can operate at low temperatures. Hydrogen is produced intermittently and in smaller quantities in sunlight-driven photocatalytic OWS reactions than in conventional chemical industrial processes. In particular, no hydrogen is produced at night. Solar energy is inherently limited, and ancillary energy for the chemical processes, such as pumping and heating, must be as small as possible. Therefore, the development of highly active and durable catalysts that can withstand daily start-up and shut-down operations will become increasingly important in establishing solar-to-chemical energy conversion technologies.
Separate production of hydrogen and oxygen
Most uses of hydrogen require that it be of ultra-high purity and great care must be taken to ensure any produced oxyhydrogen gas mixtures do not contain >5% oxygen in hydrogen, which is explosive (104). These purity demands support rational design and control over product separation during solar water splitting. For example, in photoelectrochemical designs, an ion-permeable (ion-exchange) polymer electrolyte membrane is often positioned between sites of light-driven oxidation and reduction, such that initial redox events inherently generate separated chemical products (6) (Figure 7A). More generally, ion-permeable membranes are used commercially in electrochemical devices to carry ionic current as charge equivalents to complete the electrochemical circuit, while attenuating the mixing of chemical products. However, photoelectrochemical designs containing an ion-exchange membrane are unlikely to meet the technoeconomic targets necessary for the implementation of large-scale renewable energy storage systems (8, 105). Alternatively, downstream product separation and purification is possible, as explained in the previous section.
Figure 7
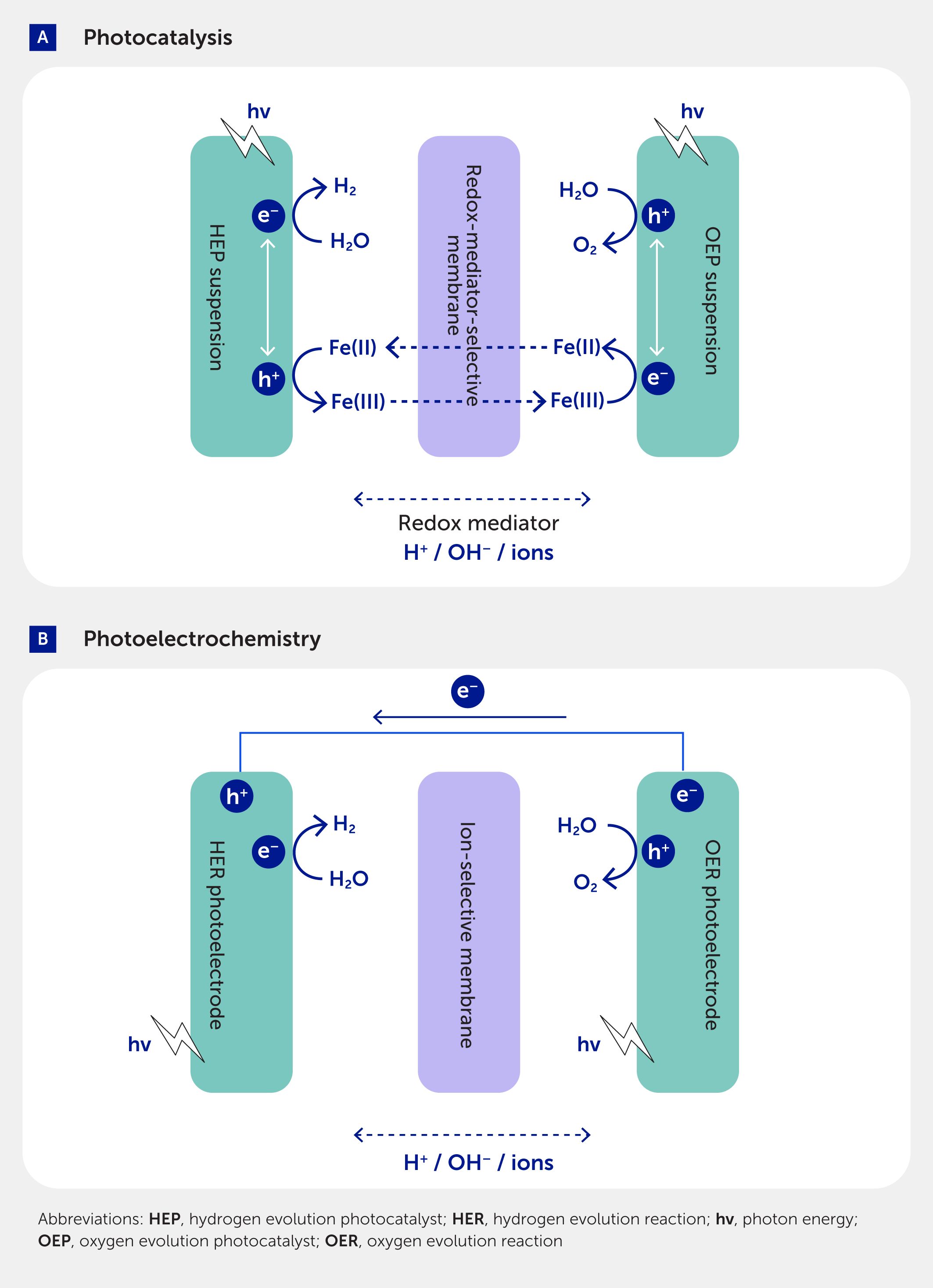
Figure 7. Separation processes at the device level during tandem solar water splitting. (A) Dual-compartment Z-scheme photocatalysis, with a membrane for selective transport of redox mediator species, is shown here for a soluble iron-based redox couple (Fe(III)/Fe(II)). (B) Dual-photoelectrode photoelectrochemistry, with a membrane for selective transport of ions, such as protons (H+) or hydroxides (OH–).
Any separation step to enrich product purity requires additional energy (106), and thus designs with inherent product separation are particularly attractive from a cost perspective (107, 108). On the scale of the single sub-ten-micron-sized particles used in photocatalytic water splitting, it is cost-prohibitive to position an ion-permeable membrane between each site of oxidation and reduction. Instead, in dual-compartment Z-scheme designs, redox mediators store oxidizing equivalents from HEPs and shuttle them to OEPs for their reduction. In this case, a membrane permeable to redox mediator species replaces the membrane permeable to ions in (photo)electrochemical designs (Figure 7B). Using cationic redox mediators, the commercial Nafion™ cation-exchange membrane has been demonstrated for use in dual-compartment Z-scheme photocatalytic water splitting (109) and CO2 reduction (85, 92, 110).
Redox mediator properties
Redox mediator species are large compared to hydrogen and oxygen. Thus, it is difficult to engineer fast rates of permeation for redox mediator species, yet slow rates of permeation for product hydrogen and oxygen in an aqueous solution. An additional concern is the pressure differential that forms due to the 2:1 molar stoichiometry of gaseous hydrogen and oxygen produced from water splitting, which, if poorly controlled, can result in hydrogen- or oxygen-saturated water transport across the membrane, and ultimately an explosive mixture of gases (111). Thus, control over the transport of species in aqueous solutions is a critical consideration, challenging the feasibility of large-scale implementation of porous separators (111). Notwithstanding, the solubility of hydrogen and oxygen in water is poor, which limits concentrations to <1 mM and slows entropically-driven diffusive crossover. In comparison, redox mediators can exist at concentrations exceeding 1 M, meaning that if partitioning and diffusion—and thus permeability—are the same for hydrogen, oxygen, and both redox states of the redox mediator, the use of 100 mM redox mediator will result in only 2% Faradaic crossover of undesired species, thus circumventing an explosive mixture of gases. Dual-compartment Z-scheme photocatalytic water splitting has been demonstrated using a microporous separator (112) and using a two-stage process where the solution of aqueous redox mediators was cycled (113–115).
Choices for redox mediators are often historical. Both the dye-sensitized solar cell community and, more recently, the redox flow battery community have developed highly soluble molecules to store and transport redox equivalents. Yet requirements for photocatalysis differ (e.g., significant sunlight transparency) therefore requiring disparate redox mediator needs. For example, dual-compartment Z-scheme photocatalytic water splitting often requires transport distances of ~1–10 cm. This is approximately three orders of magnitude larger than transport distances in dye-sensitized solar cells, where redox mediators can be used that exhibit significant sunlight absorption, rather slow diffusion, and/or short-lived stability in one oxidation state, e.g., I3–/I– or those based on large coordination compounds of cobalt or copper (116). Insight from energy storage molecules used in redox flow batteries is also limited, because some common molecules (e.g., quinones) are generally unstable in the presence of water and/or oxygen (117), two species necessarily present in photocatalytic reactors for solar water splitting. Notwithstanding, vanadium-based redox mediators common in redox flow batteries have been demonstrated for use in dual-compartment Z-scheme photocatalytic water splitting (113, 115).
Tradeoffs in the aforementioned properties suggest that IO3–/I– can result in large STH efficiencies in dual-compartment Z-scheme designs (118), where IO3−/I− has near-ideal energetics (119) and near-zero parasitic absorption of sunlight (120). Notably, demonstrated aqueous iodine redox in photocatalysis (IO3–/I–) differs from that used in nonaqueous dye-sensitized solar cells (I3–/I–), due to the use of near-neutral or weakly alkaline aqueous electrolytes, which results in the predominance of the 6-electron, 6-proton redox couple (121). Large diffusivity, optical transparency, and stability aside, redox mediators for use in dual-compartment Z-scheme photocatalytic water splitting must undergo highly selective and stable redox reactions, or else converted solar energy is undesirably dissipated as heat. There are two dominant means by which this occurs, both in the light and in the dark: (i) Faradaic interfacial charge-transfer reactions at solid|liquid interfaces and (ii) bulk redox reactions.
Designing hydrogen and oxygen redox selectivity and stability
Process (i) can be slowed using surface chemical modifications or membrane coatings (122–124) that impart species-selective permeabilities. Photodeposition of coatings from soluble molecular precursors allows their spatial positioning at sites where they are needed the most (26, 39, 54, 55). These interfacial surface modifications serve the purpose of an ion-exchange membrane, akin to those used in photoelectrochemical designs. However, these surface modifications must exhibit exceptional redox selectivity—defined as the ratio of the desired to total photocurrent—because of the small operating photocurrent densities at the single-particle level, even for highly efficient photocatalysts (125). The impact of operation at low current densities is a research area that is underexplored and will likely present new challenges and unanswered questions that do not apply to photoelectrochemical designs, such as the impact of low concentrations of impurities and stochastic photon absorption events (126, 127). Process (ii) can be slowed using mediators that exhibit sluggish redox kinetics, e.g., those with multiple proton-coupled-electron-transfer reaction steps such as IO3–/I–. Sluggish redox kinetics will slow reactivity in bulk solutions, but may then also necessitate the use of cocatalysts to catalyze desired redox mediator reactivity at particle surfaces.
Prospects for reactor-level demonstrations of the separate production of hydrogen and oxygen
The separate generation of hydrogen and oxygen is inherently safe when scaling up because no oxyhydrogen is made and the risk of an explosion is small. However, irrespective of the process, membranes and cocatalysts are important for solar water-splitting designs. Membranes should prevent hydrogen-oxygen crossover while allowing rapid transport of redox mediator species and cocatalysts must exhibit high selectivity for redox reactions. The next key step to validate the feasibility of a dual-compartment Z-scheme design that inherently produces separated hydrogen and oxygen is to build and test a reactor that is beyond the benchtop scale. While technoeconomic analyses and lifecycle assessments support the future cost-effectiveness of such an approach (107, 108), unexpected challenges in overall system and reactor designs should be identified early. Demonstration systems are possible today using off-the-shelf components and model photocatalyst particles (e.g., SrTiO3, TiO2, and BiVO4) under multi-sun irradiance to mimic high-efficiency solar water-splitting operation.
Conclusion
Photocatalytic OWS is one of the simplest reactions that can produce hydrogen from water in an environmentally friendly manner. This reaction can be further combined directly or indirectly using thermal/biocatalytic reactions at a later stage with the reduction of CO2. Societal-level implementation of this new green fuel production technology will be indispensable for the establishment of a sustainable and fossil-fuel-free society in the future. We have described the status of photocatalytic OWS for large-scale solar-to-chemical energy conversion from the perspective of materials, reaction systems, and processes. Photocatalysts have been developed that split water into hydrogen and oxygen without recombination losses in the near-UV region, revealing favorable structural and functional properties of cocatalyst/photocatalyst composites for OWS. In addition, several photocatalytic materials have been shown to split water into hydrogen and oxygen under long-wavelength visible light irradiation up to approximately 600 nm, although their AQY is still low owing to the difficulty in controlling material properties. Methods have also been developed to effectively utilize long-wavelength visible light for Z-scheme OWS by combining multiple different photocatalysts. Moreover, reactors suitable for large-scale deployment and processes to recover pure hydrogen from hydrogen/oxygen mixtures saturated with water vapor are under development. Most of these important and symbolic advances have been made in the past decade. Such rapid developments hold promise for further progress toward large-scale solar and chemical energy conversion through photocatalytic OWS reactions. However, many challenges remain regarding the practical application of this technology.
Technoeconomic evaluations of photocatalytic water splitting suggest that an STH efficiency of at least 5% is required for this technology to be impactful in the real world. In addition, to produce solar fuels in sufficient quantities, it is necessary to develop low-cost mass-production technologies for photocatalyst preparation and reactor construction, as well as energy-efficient processes for hydrogen separation and recovery (128). Regrettably, photocatalytic materials exhibiting such high STH efficiency have not yet been developed, and it appears that only a few researchers are pursuing scaling up in a serious manner. This may be due in part to the lack of standardization in the implementation of photocatalytic hydrogen production and the evaluation conditions for STH efficiency. Particulate photocatalysts are inherently heterogeneous due to their powdery nature, making standardized evaluation difficult. As a result, researchers have developed materials under overly customized (often impractical) and incomparable conditions on a very small scale. This situation underscores the need to establish an efficiency accreditation organization comprising well-regarded researchers with a thorough understanding of the mechanisms of photocatalysis (129). In developing and introducing new technologies, ensuring the safety of the entire process and compliance with laws and regulations must also be considered. Governments should take the lead in establishing clear and reliable international frameworks for green fuel-related technologies, including laws, regulations, and licensing procedures (130), which will help ensure compatibility among technologies, avoid market fragmentation, and allow smooth technology development and societal deployment. However, photocatalytic green fuel production is still technologically underdeveloped, with low STH efficiencies, high product costs, and many uncertainties in its development. Given that most efforts are consistent with a low technology readiness level, it is first necessary to standardize STH efficiency measurements under the initiative of leading photocatalyst researchers. For example, we propose that the STH efficiency of photocatalytic water splitting should be determined by quantifying the product gases of the reaction by the upward water displacement method using a panel reactor that is inherently suitable for large-area application (131).
Innovative and unconventional approaches will be needed to overcome these various problems. The authors hope that this lead article, in conjunction with the global movement toward carbon-neutral energy policies and sustainable development, will encourage experts from various fields to participate in photocatalysis research and jointly push the development of photocatalysts for large-scale solar-to-chemical energy conversion via OWS reactions.
Statements
Author contributions
TH: Visualization, Writing – original draft, Writing – review & editing. QW: Writing – original draft, Writing – review & editing. FZ: Writing – original draft, Writing – review & editing. SA: Writing – original draft, Writing – review & editing. ER: Writing – original draft, Writing – review & editing. HN: Writing – original draft, Writing – review & editing. AK: Writing – original draft, Writing – review & editing. TY: Writing – original draft, Writing – review & editing. KD: Writing – original draft, Writing – review & editing, Conceptualization, Visualization.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. QW, HN, AK, TY, and KD thank the New Energy and Industrial Technology Development Organization (NEDO, project no. P21021) for financial support. TH thanks the Japan Science and Technology Agency (JST, grant no. JPMJPR20T9) for financial support. FZ thanks the National Natural Science Foundation of China (21925206) for financial support. ER acknowledges the United Kingdom Research & Innovation (UKRI) for a European Research Council (ERC) Advanced Grant (EP/X030563/1) and the United Kingdom’s Department of Science, Innovation and Technology and the Royal Academy of Engineering Chair in Emerging Technologies programme (CIET-2324-83). SA acknowledges support as part of Ensembles of Photosynthetic Nanoreactors (EPN), an Energy Frontier Research Center funded by the United States Department of Energy, Office of Science under Award Number DE-SC0023431, and the HydroGEN Advanced Water Splitting Materials Consortium, established as part of the Energy Materials Network under the United States Department of Energy, Office of Energy Efficiency and Renewable Energy, Hydrogen and Fuel Cell Technologies Office under Award Number DE-EE0008838. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of interest
SA is a co-inventor on the following pending patent applications that are relevant to this article: US11912589 (125), WO2020093059 (125), and US20230390724 (132).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MA declared a past collaboration with the authors ER and his co-reviewer CP declared a past collaboration with the authors ER, TY, TH, KD to the handling editor.
Generative AI statement
The authors declared that no generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Eyring V, Gillett NP, Achuta Rao KM, Barimalala R, Barreiro Parrillo M, Bellouin N, et al. Human influence on the climate system. In: Masson-Delmotte V, Zhai P, Pirani A, Connors SL, Péan C, Berger S, et al., editors. Climate change 2021: the physical science basis. Contribution of Working Group I to the sixth assessment report of the Intergovernmental Panel on Climate Change. Cambridge: Cambridge University Press (2021). 423–552. doi: 10.1017/9781009157896.005
2. British Petroleum. bp statistical review of world energy (71st ed). British Petroleum (2022). Available at: https://www.bp.com/content/dam/bp/business-sites/en/global/corporate/pdfs/energy-economics/statistical-review/bp-stats-review-2022-full-report.pdf
3. Hisatomi T, Domen K. Reaction systems for solar hydrogen production via water splitting with particulate semiconductor photocatalysts. Nat Catal (2019) 2(5):387–99. doi: 10.1038/s41929-019-0242-6
4. International Energy Agency. Global hydrogen review 2021. Paris: IEA (2021). Available at: https://www.iea.org/reports/global-hydrogen-review-2021
5. Hisatomi T, Domen K. Introductory lecture: sunlight-driven water splitting and carbon dioxide reduction by heterogeneous semiconductor systems as key processes in artificial photosynthesis. Faraday Discuss (2017) 198:11–35. doi: 10.1039/c6fd00221h
6. Segev G, Kibsgaard J, Hahn C, Xu ZJ, Cheng W-H, Deutsch TG, et al. The 2022 solar fuels roadmap. J Phys D: Appl Phys (2022) 55(32):323003. doi: 10.1088/1361-6463/ac6f97
7. Jia J, Seitz LC, Benck JD, Huo Y, Chen Y, Ng JWD, et al. Solar water splitting by photovoltaic-electrolysis with a solar-to-hydrogen efficiency over 30%. Nat Commun (2016) 7:13237. doi: 10.1038/ncomms13237
8. Shaner MR, Atwater HA, Lewis NS, McFarland EW. A comparative technoeconomic analysis of renewable hydrogen production using solar energy. Energy Environ Sci (2016) 9(7):2354–71. doi: 10.1039/C5EE02573G
9. Modestino MA, Hashemi SMH, Haussener S. Mass transport aspects of electrochemical solar-hydrogen generation. Energy Environ Sci (2016) 9(5):1533–51. doi: 10.1039/C5EE03698D
10. Schneidewind J. How much technological progress is needed to make solar hydrogen cost-competitive? Adv Energy Mater (2022) 12(18):2200342. doi: 10.1002/aenm.202200342
11. Song H, Luo S, Huang H, Deng B, Ye J. Solar-driven hydrogen production: recent advances, challenges, and future perspectives. ACS Energy Lett (2022) 7(3):1043–65. doi: 10.1021/acsenergylett.1c02591
12. Wang Y, Shang X, Shen J, Zhang Z, Wang D, Lin J, et al. Direct and indirect Z-scheme heterostructure-coupled photosystem enabling cooperation of CO2 reduction and H2O oxidation. Nat Commun (2020) 11(1):3043. doi: 10.1038/s41467-020-16742-3
13. Tao X, Zhao Y, Wang S, Li C, Li R. Recent advances and perspectives for solar-driven water splitting using particulate photocatalysts. Chem Soc Rev (2022) 51(9):3561–608. doi: 10.1039/d1cs01182k
14. Hisatomi T, Kubota J, Domen K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem Soc Rev (2014) 43(22):7520–35. doi: 10.1039/c3cs60378d
15. Kaufman AJ, Nielander AC, Meyer GJ, Maldonado S, Ardo S, Boettcher SW. Absolute band-edge energies are over-emphasized in the design of photoelectrochemical materials. Nat Catal (2024) 7(6):615–23. doi: 10.1038/s41929-024-01161-0
16. Hisatomi T, Takanabe K, Domen K. Photocatalytic water-splitting reaction from catalytic and kinetic perspectives. Catal Lett (2015) 145(1):95–108. doi: 10.1007/s10562-014-1397-z
17. Fujishima A, Honda K. Electrochemical photolysis of water at a semiconductor electrode. Nature (1972) 238(5358):37. doi: 10.1038/238037a0
18. Kudo A, Miseki Y. Heterogeneous photocatalyst materials for water splitting. Chem Soc Rev (2009) 38(1):253–78. doi: 10.1039/b800489g
19. Kudo A, Tanaka A, Domen K, Maruya K, Aika K, Onishi T. Photocatalytic decomposition of water over NiO-K4Nb6O17 catalyst. J Catal (1988) 111(1):67–76. doi: 10.1016/0021-9517(88)90066-8
20. Takata T, Furumi Y, Shinohara K, Tanaka A, Hara M, Kondo JN, et al. Photocatalytic decomposition of water on spontaneously hydrated layered perovskites. Chem Mater (1997) 9(5):1063–4. doi: 10.1021/cm960612b
21. Kato H, Asakura K, Kudo A. Highly efficient water splitting into H2 and O2 over lanthanum-doped NaTaO3 photocatalysts with high crystallinity and surface nano-structure. J Am Chem Soc (2003) 125(10):3082–9. doi: 10.1021/ja027751g
22. Sakata Y, Matsuda Y, Nakagawa T, Yasunaga R, Imamura H, Teramura K. Remarkable improvement of the photocatalytic activity of Ga2O3 towards the overall splitting of H2O. ChemSusChem (2011) 4(2):181–4. doi: 10.1002/cssc.201000258
23. Takata T, Jiang J, Sakata Y, Nakabayashi M, Shibata N, Nandal V, et al. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature (2020) 581(7809):411–4. doi: 10.1038/s41586-020-2278-9
24. Watanabe K, Iwase A, Kudo A. Solar water splitting over Rh0.5Cr1.5O3-loaded AgTaO3 of a valence-band-controlled metal oxide photocatalyst. Chem Sci (2020) 11(9):2330–4. doi: 10.1039/c9sc05909a
25. Watanabe K, Iikubo Y, Yamaguchi Y, Kudo A. Highly crystalline Na0.5Bi0.5TiO3 of a photocatalyst valence-bandcontrolled with Bi(III) for solar water splitting. Chem Commun (Camb) (2021) 57(3):323–6. doi: 10.1039/d0cc07371g
26. Maeda K, Teramura K, Lu D, Saito N, Inoue Y, Domen K. Noble-metal/Cr2O3 core/shell nanoparticles as a cocatalyst for photocatalytic overall water splitting. Angew Chem Int Ed (2006) 45(46):7806–9. doi: 10.1002/anie.200602473
27. Nishiyama H, Yamada T, Nakabayashi M, Maehara Y, Yamaguchi M, Kuromiya Y, et al. Photocatalytic solar hydrogen production from water on a 100-m2 scale. Nature (2021) 598(7880):304–7. doi: 10.1038/s41586-021-03907-3
28. Kudo A, Niishiro R, Iwase A, Kato H. Effects of doping of metal cations on morphology, activity, and visible light response of photocatalysts. Chem Phys (2007) 339(1–3):104–10. doi: 10.1016/j.chemphys.2007.07.024
29. Yamaguchi Y, Kudo A. Visible light responsive photocatalysts developed by substitution with metal cations aiming at artificial photosynthesis. Front Energy (2021) 15(3):568–76. doi: 10.1007/s11708-021-0774-8
30. Furuhashi K, Jia Q, Kudo A, Onishi H. Time-resolved infrared absorption study of SrTiO3 photocatalysts codoped with rhodium and antimony. J Phys Chem C (2013) 117(37):19101–6. doi: 10.1021/jp407040p
31. Kawamoto H. Development of visible light driven photocatalysts for water splitting by Cr, Mn, and Ru doping into metal oxides with perovskite structure [thesis; Japanese]. Tokyo: Tokyo University of Science (2022).
32. Ueki Y, Kawamoto H, Kaiya K, Yoshino S, Yamaguchi Y, Kudo A. A. Water splitting under visible light irradiation over Rh and Ru-doped SrTiO3 photocatalysts treated with a flux [abstract P4-004; Japanese]. 11th Chemical Society of Japan Chemical Festa (2021).
33. Kaiya K, Watanabe K, Yoshino S, Yamaguchi Y, Kudo A. Photocatalytic water splitting under visible light irradiation over SrTiO3:Ir,Sb with response to wide range of visible light improved by flux treatment [abstract P1-007; Japanese]. 126th Catalysis Society of Japan meeting (2020).
34. Maeda K, Teramura K, Lu D, Takata T, Saito N, Inoue Y, et al. Photocatalyst. Nature (2006) 440(7082):295. doi: 10.1038/440295a
35. Lee Y, Terashima H, Shimodaira Y, Teramura K, Hara M, Kobayashi H, et al. Zinc germanium oxynitride as a photocatalyst for overall water splitting under visible light. J Phys Chem C (2007) 111(2):1042–8. doi: 10.1021/jp0656532
36. Maeda K, Domen K. Solid solution of GaN and ZnO as a stable photocatalyst for overall water splitting under visible light. Chem Mater (2010) 22(3):612–23. doi: 10.1021/cm901917a
37. Liu K, Zhang B, Zhang J, Lin W, Wang J, Xu Y, et al. Synthesis of narrow-band-gap GaN: ZnO solid solution for photocatalytic overall water splitting. ACS Catal (2022) 12(23):14637–46. doi: 10.1021/acscatal.2c04361
38. Maeda K, Lu D, Domen K. Direct water splitting into hydrogen and oxygen under visible light by using modified TaON photocatalysts with d(0) electronic configuration. Chem Eur J (2013) 19:4986–91. doi: 10.1002/chem.201300158
39. Pan C, Takata T, Nakabayashi M, Matsumoto T, Shibata N, Ikuhara Y, et al. A complex perovskite-type oxynitride: the first photocatalyst for water splitting operable at up to 600 nm. Angew Chem Int Ed Engl (2015) 54(10):2955–9. doi: 10.1002/anie.201410961
40. Zhang G, Lan Z-A, Lin L, Lin S, Wang X. Overall water splitting by Pt/g-C3N4 photocatalysts without using sacrificial agents. Chem Sci (2016) 7(5):3062–6. doi: 10.1039/c5sc04572j
41. Wang Q, Zhang G, Xing W, Pan Z, Zheng D, Wang S, et al. Bottom-up synthesis of single-crystalline poly (triazine imide) nanosheets for photocatalytic overall water splitting. Angew Chem Int Ed Engl (2023) 62(37):e202307930. doi: 10.1002/anie.202307930
42. Wang Z, Inoue Y, Hisatomi T, Ishikawa R, Wang Q, Takata T, et al. Overall water splitting by Ta3N5 nanorod single crystals grown on the edges of KTaO3 particles. Nat Catal (2018) 1(10):756–63. doi: 10.1038/s41929-018-0134-1
43. Xiao J, Nishimae S, Vequizo JJM, Nakabayashi M, Hisatomi T, Li H, et al. Enhanced overall water splitting by a zirconium-doped TaON-based photocatalyst. Angew Chem Int Ed Engl (2022) 61(17):e202116573. doi: 10.1002/anie.202116573
44. Li H, Xiao J, Vequizo JJM, Hisatomi T, Nakabayashi M, Pan Z, et al. One-step excitation overall water splitting over a modified Mg-doped BaTaO2N photocatalyst. ACS Catal (2022) 12(16):10179–85. doi: 10.1021/acscatal.2c02394
45. Chen K, Xiao J, Vequizo JJM, Hisatomi T, Ma Y, Nakabayashi M, et al. Overall water splitting by a SrTaO2N-based photocatalyst decorated with an Ir-promoted Ru-based cocatalyst. J Am Chem Soc (2023) 145(7):3839–43. doi: 10.1021/jacs.2c11025
46. Wang Q, Nakabayashi M, Hisatomi T, Sun S, Akiyama S, Wang Z, et al. Oxysulfide photocatalyst for visible-light-driven overall water splitting. Nat Mater (2019) 18(8):827–32. doi: 10.1038/s41563-019-0399-z
47. Nandal V, Shoji R, Matsuzaki H, Furube A, Lin L, Hisatomi T, et al. Unveiling charge dynamics of visible light absorbing oxysulfide for efficient overall water splitting. Nat Commun (2021) 12(1):7055. doi: 10.1038/s41467-021-27199-3
48. Li R, Zha Z, Zhang Y, Yang M, Lin L, Wang Q, et al. Band-tail states meditated visible-light-driven overall water splitting in Y2Ti2O5S2 photocatalyst. J Mater Chem A (2023) 10(45):24247–57. doi: 10.1039/D2TA06315H
49. Nakabayashi M, Nishiguchi K, Liang X, Hisatomi T, Takata T, Tsuchimochi T, et al. Characterization of planar defect in layered perovskite photocatalyst Y2Ti2O5S2 by electron microscopy and first-principles calculations. J Phys Chem C (2023) 127:7887–93. doi: 10.1021/acs.jpcc.3c00820
50. Pan Z, Yoshida H, Lin L, Xiao Q, Nakabayashi M, Shibata N, et al. Synthesis of Y2Ti2O5S2 by thermal sulfidation for photocatalytic water oxidation and reduction under visible light irradiation. Res Chem Intermed (2021) 47(1):225–34. doi: 10.1007/s11164-020-04329-y
51. Maeda K, Ohno T, Domen K. A copper and chromium based nanoparticulate oxide as a noble-metal-free cocatalyst for photocatalytic water splitting. Chem Sci (2011) 2(7):1362–8. doi: 10.1039/c1sc00177a
52. Xiong A, Yoshinaga T, Ikeda T, Takashima M, Hisatomi T, Maeda K, et al. Effect of hydrogen and oxygen evolution cocatalysts on photocatalytic activity of GaN:ZnO. Eur J Inorg Chem (2014) 2014(4):767–72. doi: 10.1002/ejic.201300439
53. Zhang F, Yamakata A, Maeda K, Moriya Y, Takata T, Kubota J, et al. Cobalt-modified porous single-crystalline LaTiO2N for highly efficient water oxidation under visible light. J Am Chem Soc (2012) 134(20):8348–51. doi: 10.1021/ja301726c
54. Takata T, Pan C, Nakabayashi M, Shibata N, Domen K. Fabrication of a core–shell-type photocatalyst via photodeposition of Group IV and V transition metal oxyhydroxides: an effective surface modification method for overall water splitting. J Am Chem Soc (2015) 137(30):9627–34. doi: 10.1021/jacs.5b04107
55. Bau JA, Takanabe K. Ultrathin microporous SiO2 membranes photodeposited on hydrogen evolving catalysts enabling overall water splitting. ACS Catal (2017) 7(11):7931–40. doi: 10.1021/acscatal.7b03017
56. Kranz C, Wächtler M. Characterizing photocatalysts for water splitting: from atoms to bulk and from slow to ultrafast processes. Chem Soc Rev (2021) 50(2):1407–37. doi: 10.1039/d0cs00526f
57. Bard AJ. Photoelectrochemistry and heterogeneous photo-catalysis at semiconductors. J Photochem (1979) 10(1):59–75. doi: 10.1016/0047-2670(79)80037-4
58. Abe R, Sayama K, Domen K, Arakawa H. A new type of water splitting system composed of two different TiO2 photocatalysts (anatase, rutile) and a IO3-/I- shuttle redox mediator. Chem Phys Lett (2001) 344(3–4):339–44. doi: 10.1016/S0009-2614(01)00790-4
59. Wang Y, Suzuki H, Xie J, Tomita O, Martin DJ, Higashi M, et al. Mimicking natural photosynthesis: solar to renewable H2 fuel synthesis by Z-scheme water splitting systems. Chem Rev (2018) 118(10):5201–41. doi: 10.1021/acs.chemrev.7b00286
60. Wang W, Chen J, Li C, Tian W. Achieving solar overall water splitting with hybrid photosystems of photosystem II and artificial photocatalysts. Nat Commun (2014) 5:4647. doi: 10.1038/ncomms5647
61. Hu H, Wang Z, Cao L, Zeng L, Zhang C, Lin W, et al. Metal–organic frameworks embedded in a liposome facilitate overall photocatalytic water splitting. Nat Chem (2021) 13(4):358–66. doi: 10.1038/s41557-020-00635-5
62. Zhang JZ, Reisner E. Advancing photosystem II photoelectrochemistry for semi-artificial photosynthesis. Nat Rev Chem (2020) 4(1):6–21. doi: 10.1038/s41570-019-0149-4
63. Qi Y, Zhang J, Kong Y, Zhao Y, Chen S, Li D, et al. Unraveling of cocatalysts photodeposited selectively on facets of BiVO4 to boost solar water splitting. Nat Commun (2022) 13(1):484. doi: 10.1038/s41467-022-28146-6
64. Jia Q, Iwase A, Kudo A. BiVO4–Ru/SrTiO3:Rh composite Z-scheme photocatalyst for solar water splitting. Chem Sci (2014) 5(4):1513–9. doi: 10.1039/c3sc52810c
65. Wang Q, Hisatomi T, Jia Q, Tokudome H, Zhong M, Wang C, et al. Scalable water splitting on particulate photocatalyst sheets with a solar-to-hydrogen energy conversion efficiency exceeding 1%. Nat Mater (2016) 15(6):611–5. doi: 10.1038/nmat4589
66. Wang Q, Hisatomi T, Suzuki Y, Pan Z, Seo J, Katayama M, et al. Particulate photocatalyst sheets based on carbon conductor layer for efficient Z-scheme pure-water splitting at ambient pressure. J Am Chem Soc (2017) 139(4):1675–83. doi: 10.1021/jacs.6b12164
67. Wang Q, Okunaka S, Tokudome H, Hisatomi T, Nakabayashi M, Shibata N, et al. Printable photocatalyst sheets incorporating a transparent conductive mediator for Z-scheme water splitting. Joule (2018) 2(12):2667–80. doi: 10.1016/j.joule.2018.08.003
68. Andrei V, Wang Q, Uekert T, Bhattacharjee S, Reisner E. Solar panel technologies for light-to-chemical conversion. Acc Chem Res (2022) 55(23):3376–86. doi: 10.1021/acs.accounts.2c00477
69. Fang S, Rahaman M, Bharti J, Reisner E, Robert M, Ozin GA, et al. Photocatalytic CO2 reduction. Nat Rev Methods Primers (2023) 3(1):61. doi: 10.1038/s43586-023-00243-w
70. Wang L, Nitopi S, Wong AB, Snider JL, Nielander AC, Morales-Guio CG, et al. Electrochemically converting carbon monoxide to liquid fuels by directing selectivity with electrode surface area. Nat Catal (2019) 2(8):702–8. doi: 10.1038/s41929-019-0301-z
71. Wagner A, Sahm CD, Reisner E. Towards molecular understanding of local chemical environment effects in electro- and photocatalytic CO2 reduction. Nat Catal (2020) 3(10):775–86. doi: 10.1038/s41929-020-00512-x
72. Dalle KE, Warnan J, Leung JJ, Reuillard B, Karmel IS, Reisner E. Electro- and solar-driven fuel synthesis with first row transition metal complexes. Chem Rev (2019) 119(4):2752–875. doi: 10.1021/acs.chemrev.8b00392
73. Li X, Yu J, Jaroniec M, Chen X. Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem Rev (2019) 119(6):3962–4179. doi: 10.1021/acs.chemrev.8b00400
74. Wang Q, Pan Z. Advances and challenges in developing cocatalysts for photocatalytic conversion of carbon dioxide to fuels. Nano Res (2022) 15(12):10090–109. doi: 10.1007/s12274-022-4705-8
75. Iwase A, Yoshino S, Takayama T, Ng YH, Amal R, Kudo A. Water splitting and CO2 reduction under visible light irradiation using Z-scheme systems consisting of metal sulfides, CoOx-loaded BiVO4, and a reduced graphene oxide electron mediator. J Am Chem Soc (2016) 138(32):10260–4. doi: 10.1021/jacs.6b05304
76. Fang X, Kalathil S, Reisner E. Semi-biological approaches to solar-to-chemical conversion. Chem Soc Rev (2020) 49(14):4926–52. doi: 10.1039/c9cs00496c
77. Kumar B, Llorente M, Froehlich J, Dang T, Sathrum A, Kubiak CP. Photochemical and photoelectrochemical reduction of CO2. Annu Rev Phys Chem (2012) 63:541–69. doi: 10.1146/annurev-physchem-032511-143759
78. Armstrong FA, Belsey NA, Cracknell JA, Goldet G, Parkin A, Reisner E, et al. Dynamic electrochemical investigations of hydrogen oxidation and production by enzymes and implications for future technology. Chem Soc Rev (2008) 38(1):36–51. doi: 10.1039/b801144n
79. Armstrong FA, Hirst J. Reversibility and efficiency in electrocatalytic energy conversion and lessons from enzymes. Proc Natl Acad Sci U.S.A. (2011) 108(34):14049–54. doi: 10.1073/pnas.1103697108
80. Miller M, Robinson WE, Oliveira AR, Heidary N, Kornienko N, Warnan J, et al. Interfacing formate dehydrogenase with metal oxides for the reversible electrocatalysis and solar-driven reduction of carbon dioxide. Angew Chem Int Ed Engl (2019) 58(14):4601–5. doi: 10.1002/anie.201814419
81. Leung JJ, Warnan J, Ly KH, Heidary N, Nam DH, Kuehnel MF, et al. Solar-driven reduction of aqueous CO2 with a cobalt bis(terpyridine)-based photocathode. Nat Catal (2019) 2(4):354–65. doi: 10.1038/s41929-019-0254-2
82. Suzuki TM, Yoshino S, Takayama T, Iwase A, Kudo A, Morikawa T. Z-schematic and visible-light-driven CO2 reduction using H2O as an electron donor by a particulate mixture of a Ru-complex/(CuGa)1–xZn2xS2 hybrid catalyst, BiVO4 and an electron mediator. Chem Commun (Camb) (2018) 54(72):10199–202. doi: 10.1039/c8cc05505j
83. Kuehnel MF, Orchard KL, Dalle KE, Reisner E. Selective photocatalytic CO2 reduction in water through anchoring of a molecular Ni catalyst on CdS nanocrystals. J Am Chem Soc (2017) 139(21):7217–23. doi: 10.1021/jacs.7b00369
84. Kumagai H, Sahara G, Maeda K, Higashi M, Abe R, Ishitani O. Hybrid photocathode consisting of a CuGaO2 p-type semiconductor and a Ru(ii)–Re(i). Chem Sci (2017) 8(6):4242–9. doi: 10.1039/c7sc00940b
85. Andrei V, Ucoski GM, Pornrungroj C, Uswachoke C, Wang Q, Achilleos DS, et al. Floating perovskite-BiVO4 devices for scalable solar fuel production. Nature (2022) 608(7923):518–22. doi: 10.1038/s41586-022-04978-6
86. Sato S, Arai T, Morikawa T, Uemura K, Suzuki TM, Tanaka H, et al. Selective CO2 conversion to formate conjugated with H2O oxidation utilizing semiconductor/complex hybrid photocatalysts. J Am Chem Soc (2011) 133(39):15240–3. doi: 10.1021/ja204881d
87. Kuriki R, Yamamoto M, Higuchi K, Yamamoto Y, Akatsuka M, Lu D, et al. Robust binding between carbon nitride nanosheets and a binuclear ruthenium(II) complex enabling durable, selective CO2 reduction under visible light in aqueous solution. Angew Chem Int Ed Engl (2017) 56(17):4867–71. doi: 10.1002/anie.201701627
88. Kuriki R, Matsunaga H, Nakashima T, Wada K, Yamakata A, Ishitani O, et al. Nature-inspired, highly durable CO2 reduction system consisting of a binuclear ruthenium(II) complex and an organic semiconductor using visible light. J Am Chem Soc (2016) 138(15):5159–70. doi: 10.1021/jacs.6b01997
89. Rosser TE, Hisatomi T, Sun S, Antón-García D, Minegishi T, Reisner E, et al. La5Ti2Cu0.9Ag0.1S5O7 modified with a molecular Ni catalyst for photoelectrochemical H2 generation. Chemistry (2018) 24(69):18393–7. doi: 10.1002/chem.201801169
90. Lewis NS, Nocera DG. Powering the planet: chemical challenges in solar energy utilization. Proc Natl Acad Sci U.S.A. (2006) 103(43):15729–35. doi: 10.1073/pnas.0603395103
91. Wang Q, Warnan J, Rodríguez-Jiménez S, Leung JJ, Kalathil S, Andrei V, et al. Molecularly engineered photocatalyst sheet for scalable solar formate production from carbon dioxide and water. Nat Energy (2020) 5(9):703–10. doi: 10.1038/s41560-020-0678-6
92. Wang Q, Kalathil S, Pornrungroj C, Sahm CD, Reisner E. Bacteria–photocatalyst sheet for sustainable carbon dioxide utilization. Nat Catal (2022) 5(7):633–41. doi: 10.1038/s41929-022-00817-z
93. Wang Q, Li Y, Hisatomi T, Nakabayashi M, Shibata N, Kubota J, et al. Z-scheme water splitting using particulate semiconductors immobilized onto metal layers for efficient electron relay. J Catal (2015) 328:308–15. doi: 10.1016/j.jcat.2014.12.006
94. Kornienko N, Zhang JZ, Sakimoto KK, Yang P, Reisner E. Interfacing nature’s catalytic machinery with synthetic materials for semi-artificial photosynthesis. Nat Nanotechnol (2018) 13(10):890–9. doi: 10.1038/s41565-018-0251-7
95. Lozano P, García-Verdugo E. From green to circular chemistry paved by biocatalysis. Green Chem (2023) 25(18):7041–57. doi: 10.1039/D3GC01878D
96. Lyu H, Hisatomi T, Goto Y, Yoshida M, Higashi T, Katayama M, et al. An Al-doped SrTiO3 photocatalyst maintaining sunlight-driven overall water splitting activity over 1000 h of constant illumination. Chem Sci (2019) 10(11):3196–201. doi: 10.1039/c8sc05757e
97. Goto Y, Hisatomi T, Wang Q, Higashi T, Ishikiriyama K, Maeda T, et al. A particulate photocatalyst water-splitting panel for large-scale solar hydrogen generation. Joule (2018) 2(3):509–20. doi: 10.1016/j.joule.2017.12.009
98. Ham Y, Hisatomi T, Goto Y, Moriya Y, Sakata Y, Yamakata A, et al. Flux-mediated doping of SrTiO3 photocatalysts for efficient overall water splitting. J Mater Chem A (2016) 4(8):3027–33. doi: 10.1039/C5TA04843E
99. Schroeder V, Holtappels K. Explosion characteristics of hydrogen-air and hydrogen-oxygen mixtures at elevated pressures [abstract 120001]. International Conference on Hydrogen Safety (2005). Available at: https://conference.ing.unipi.it/ichs2005/Papers/120001.pdf.
100. Kumamoto A, Iseki H, Ono R, Oda T. Measurement of minimum ignition energy in hydrogenoxygen-nitrogen premixed gas by spark discharge. J Phys Conf S (2011) 301:012039. doi: 10.1088/1742-6596/301/1/012039
101. Tanihara N, Nakanishi S, Yoshinaga T. Gas and vapor separation through polyimide membranes. J Jpn Petrol Inst (2016) 59(6):276–82. doi: 10.1627/jpi.59.276
102. Yamada T, Nishiyama H, Akatsuka H, Nishimae S, Ishii Y, Hisatomi T, et al. Production of methane by sunlight-driven photocatalytic water splitting and carbon dioxide methanation as a means of artificial photosynthesis. ACS Eng Au (2023) 3(5):352–63. doi: 10.1021/acsengineeringau.3c00034
103. European Commission Directorate-General for Research and Innovation. EIC horizon prize on artificial photosynthesis: prototype of an artificial photosynthetic fuel production system wins €5 million [online] (2020). Available at: https://research-and-innovation.ec.europa.eu/news/all-research-and-innovation-news/eic-horizon-prize-artificial-photosynthesis-prototype-artificial-photosynthetic-fuel-production-2022-12-05_en
104. Grigoriev SA, Porembskiy VI, Korobtsev SV, Fateev VN, Auprêtre F, Millet P. High-pressure PEM water electrolysis and corresponding safety issues. Int J Hydrog Energy (2011) 36(3):2721–8. doi: 10.1016/j.ijhydene.2010.03.058
105. Pinaud BA, Benck JD, Seitz LC, Forman AJ, Chen Z, Deutsch TG, et al. Technical and economic feasibility of centralized facilities for solar hydrogen production via photocatalysis and photoelectrochemistry. Energy Environ Sci (2013) 6(7):1983–2002. doi: 10.1039/c3ee40831k
106. Fabian DM, Hu S, Singh N, Houle FA, Hisatomi T, Domen K, et al. Particle suspension reactors and materials for solar-driven water splitting. Energy Environ Sci (2015) 8(10):2825–50. doi: 10.1039/C5EE01434D
107. James BD, DeSantis DA, Huya-Kouadio JM, Houchins C, Acevedo Y, Saur G. Hydrogen production and delivery analysis [online] (2021). Available at: https://www.hydrogen.energy.gov/pdfs/review21/p102_james_2021_p.pdf
108. James BD, Huya-Kouadio JM, Houchins C, Acevedo Y, McNamara K, Saur G. Hydrogen production cost and performance analysis. DOE Hydrogen Program. 2022 annual merit review and peer evaluation meeting [online]. Available at: https://www.hydrogen.energy.gov/pdfs/review22/p204_james_2022_p.pdf
109. Lo C-C, Huang C-W, Liao C-H, Wu JCS. Novel twin reactor for separate evolution of hydrogen and oxygen in photocatalytic water splitting. Int J Hydrog Energy (2010) 35(4):1523–9. doi: 10.1016/j.ijhydene.2009.12.032
110. Sokol KP, Robinson WE, Oliveira AR, Warnan J, Nowaczyk MM, Ruff A, et al. Photoreduction of CO2 with a formate dehydrogenase driven by photosystem II using a semi-artificial Z-scheme architecture. J Am Chem Soc (2018) 140(48):16418–22. doi: 10.1021/jacs.8b10247
111. Haussener S, Xiang C, Spurgeon JM, Ardo S, Lewis NS, Weber AZ. Modeling, simulation, and design criteria for photoelectrochemical water-splitting systems. Energy Environ Sci (2012) 5:9922–35. doi: 10.1039/c2ee23187e
112. Sasaki Y, Kato H, Kudo A. [Co(bpy)3]3+/2+ and [Co(phen)3]3+/2+ electron mediators for overall water splitting under sunlight irradiation using Z-scheme photocatalyst system. J Am Chem Soc (2013) 135(14):5441–9. doi: 10.1021/ja400238r
113. Linkous CA, McKaige GT, Slattery DK, Ouellette AJA, Austin BCN. Solar photocatalytic hydrogen production from water using a dual bed photosystem. Task 2 report; Annual report. United States Department of Energy Office of Scientific and Technical Information (1995). doi: 10.2172/564090
114. Bae SW, Ji SM, Hong SJ, Jang JW, Lee JS. Photocatalytic overall water splitting with dual-bed system under visible light irradiation. Int J Hydrog Energy (2009) 34(8):3243–9. doi: 10.1016/j.ijhydene.2009.02.022
115. Miseki Y, Fujiyoshi S, Gunji T, Sayama K. Photocatalytic Z-scheme water splitting for independent H2/O2 production via a stepwise operation employing a vanadate redox mediator under visible light. J Phys Chem C (2017) 121(18):9691–7. doi: 10.1021/acs.jpcc.7b00905
116. Ardo S, Meyer GJ. Photodriven heterogeneous charge transfer with transition-metal compounds anchored to TiO2 semiconductor surfaces. Chem Soc Rev (2009) 38(1):115–64. doi: 10.1039/b804321n
117. Ji Y, Goulet M-A, Pollack DA, Kwabi DG, Jin S, De Porcellinis D, et al. A phosphonate-functionalized quinone redox flow battery at near-neutral pH with record capacity retention rate. Adv Ener Mater (2019) 9(12):1900039. doi: 10.1002/aenm.201900039
118. Bala Chandran RB, Breen S, Shao Y, Ardo S, Weber AZ. Evaluating particle-suspension reactor designs for Z-scheme solar water splitting via transport and kinetic modeling. Energy Environ Sci (2018) 11(1):115–35. doi: 10.1039/C7EE01360D
119. Keene S, Bala Chandran RB, Ardo S. Calculations of theoretical efficiencies for electrochemically-mediated tandem solar water splitting as a function of bandgap energies and redox shuttle potential. Energy Environ Sci (2019) 12(1):261–72. doi: 10.1039/C8EE01828F
120. Linkous CA, Slattery DK. Solar photocatalytic hydrogen production from water using a dual bed photosystem - Phase I final report and Phase II proposal. United States Department of Energy Office of Scientific and Technical Information (2000). doi: 10.2172/771058
121. Pourbaix M. Atlas of electrochemical equilibria in aqueous solutions. (2nd edition). Houston, TX: National Association of Corrosion Engineers (1974).
122. Yoshida M, Maeda K, Lu D, Kubota J, Domen K. Lanthanoid oxide layers on rhodium-loaded (Ga1–xZnx)(N1–xOx) photocatalyst as a modifier for overall water splitting under visible-light irradiation. J Phys Chem C (2013) 117(27):14000–6. doi: 10.1021/jp402240d
123. Stinson WDH, Brayton KM, Ardo S, Talin AA, Esposito DV. Quantifying the influence of defects on selectivity of electrodes encapsulated by nanoscopic silicon oxide overlayers. ACS Appl Mater Interfaces (2022) 14(50):55480–90. doi: 10.1021/acsami.2c13646
124. Li Z, Li R, Jing H, Xiao J, Xie H, Hong F, et al. Blocking the reverse reactions of overall water splitting on a Rh/GaN–ZnO photocatalyst modified with Al2O3. Nat Catal (2023) 6(1):80–8. doi: 10.1038/s41929-022-00907-y
125. Ardo S, Keene ST, Phun GS. Optically thin light-absorbers for increasing photochemical energy-conversion efficiencies [U.S. Patent Publication No. US11912589 (awarded) and WO2020093059 (published)]. United States Patent and Trademark Office (2020).
126. Utterback JK, Wilker MB, Brown KA, King PW, Eaves JD, Dukovic G. Competition between electron transfer, trapping, and recombination in CdS nanorod–hydrogenase complexes. Phys Chem Chem Phys (2015) 17(8):5538–42. doi: 10.1039/c4cp05993j
127. Tkaczibson K, Ardo S. Numerical Monte Carlo simulations of charge transport across the surface of dye and cocatalyst modified spherical nanoparticles under conditions of pulsed or continuous illumination. Sustain Energy Fuels (2019) 3(6):1573–87. doi: 10.1039/C9SE00009G
128. Hisatomi T, Domen K. Overall water splitting: what’s next? Next Energy (2023) 1(1):100006. doi: 10.1016/j.nxener.2023.100006
129. Wang Z, Hisatomi T, Li R, Sayama K, Liu G, Domen K, et al. Efficiency accreditation and testing protocols for particulate photocatalysts toward solar fuel production. Joule (2021) 5(2):344–59. doi: 10.1016/j.joule.2021.01.001
130. International Energy Agency. Global hydrogen review 2023. Paris: IEA. Available at: https://www.iea.org/reports/global-hydrogen-review-2023
131. Hisatomi T, Domen K. Best practices for photocatalytic water splitting. Nat Sustain (2024) 7:1082–4. doi: 10.1038/s41893-024-01382-y
Keywords: photocatalyst, water splitting, green hydrogen production, carbon dioxide conversion, scale up, panel reactor
Citation: Hisatomi T, Wang Q, Zhang F, Ardo S, Reisner E, Nishiyama H, Kudo A, Yamada T and Domen K. Photocatalytic water splitting for large-scale solar-to-chemical energy conversion and storage. Front Sci (2024) 2:1411644. doi: 10.3389/fsci.2024.1411644
Received: 03 April 2024; Accepted: 12 September 2024;
Published: 03 December 2024.
Edited by:
Yufei Zhao, Beijing University of Chemical Technology, ChinaReviewed by:
Detlef W. Bahnemann, Leibniz University Hannover, GermanyMarkus Antonietti, Max Planck Institute of Colloids and Interfaces, Germany
Christian Mark Pelicano, Max Planck Institute of Colloids and Interfaces, Germany, in collaboration with reviewer MA
Copyright © 2024 Hisatomi, Wang, Zhang, Ardo, Reisner, Nishiyama, Kudo, Yamada and Domen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazunari Domen, ZG9tZW5AY2hlbXN5cy50LnUtdG9reW8uYWMuanA=; ZG9tZW5Ac2hpbnNodS11LmFjLmpw