 Ryan R. Kelly
Ryan R. Kelly Sara J. Sidles
Sara J. Sidles Amanda C. LaRue
Amanda C. LaRue
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Psychol. , 30 November 2020
Sec. Psychopathology
Volume 11 - 2020 | https://doi.org/10.3389/fpsyg.2020.612366
This article is part of the Research Topic Psychological Factors as Determinants of Medical Conditions, Volume II View all 18 articles
Neurological diseases, particularly in the context of aging, have serious impacts on quality of life and can negatively affect bone health. The brain-bone axis is critically important for skeletal metabolism, sensory innervation, and endocrine cross-talk between these organs. This review discusses current evidence for the cellular and molecular mechanisms by which various neurological disease categories, including autoimmune, developmental, dementia-related, movement, neuromuscular, stroke, trauma, and psychological, impart changes in bone homeostasis and mass, as well as fracture risk. Likewise, how bone may affect neurological function is discussed. Gaining a better understanding of brain-bone interactions, particularly in patients with underlying neurological disorders, may lead to development of novel therapies and discovery of shared risk factors, as well as highlight the need for broad, whole-health clinical approaches toward treatment.
The skeleton is necessary for facilitating movement, providing a framework for muscles and soft tissues, protecting vital organs, storage of minerals and fat, harboring the bone marrow, and regulating blood cell formation. Maintaining bone health is, therefore, important for overall health and well-being. In recent years, we have begun to better dissect the relationship between the brain and skeleton and how they regulate one another. One way to better appreciate this relationship is to determine how it becomes altered with disease. Osteoporosis is the most common form of metabolic bone disease and is characterized by low bone mass (≤ 2.5 standard deviations below peak bone mass) and micro-architectural bone deterioration that can lead to debilitating fractures. In the United States, osteoporosis accounts for over 1.5 million fractures annually. By 2050, the costs associated with osteoporotic fractures are expected to exceed $130 billion (Bartl and Bartl, 2019). In regard to brain disease, more than one million U.S. adults are diagnosed annually with a chronic brain disease or disorder at a healthcare cost of > $800 billion (Gooch et al., 2017). In addition, many of these disorders are associated with increased age, a shared risk factor with osteoporosis. Osteoporosis is more prevalent in people with neurological conditions, such as multiple sclerosis (MS) and Parkinson’s disease (PD), while sudden neurological events, such as stroke and spinal cord injury (SCI), can cause rapid loss in bone mineral density (BMD). Further, lack of physical activity in degenerative neurological conditions can lead to mineral loss and osteoporosis, particularly in aging populations and/or those with comorbidities (e.g., obesity, diabetes, smoking). A better understanding of the mechanisms that cause bone loss and how neurological conditions uniquely impact bone health is of clinical importance and will guide treatment options, encourage lifestyle change, and aid in development of novel osteoporosis therapies. Likewise, gaining a better understanding of how bones regulate the brain may provide novel insights into how neurological disorders develop. This review will provide an overview of literature related to neurological disorders and their effects on bone health with a goal to promote recognition of the mechanisms by which changes in the brain can impart changes in the bones. We also discuss how certain treatments for neurological disorders can negatively affect bone health. This will encourage a broader view of disease management toward improved patient health and outcomes.
As an adaptation to stress and to maintain calcium homeostasis, the skeleton undergoes constant remodeling. In adult humans, the skeleton replaces itself almost entirely every 10 years [Office of the Surgeon General (US), 2004]. During this remodeling process, the new bone laid down by osteoblasts must replace the bone resorbed by osteoclasts in a fine-tuned manner. However, with age, net bone loss occurs as a result of increased osteoclastic activity and/or decreased osteoblastic activity at a rate of approximately 1% per year after 30 years of age. The bone remodeling process is regulated by endocrine, paracrine, mechanical, and, as the subject of more recent focus, neuronal factors.
Investigation into the contribution of neuronal signaling in regulating bone remodeling and homeostasis began with a focus on leptin, an adipose-derived hormone involved in energy regulation and metabolism. Mice deficient for leptin (ob/ob mice) were found to have a high vertebral trabecular bone mass phenotype (Ducy et al., 2000). This phenotype was, surprisingly, rescued by infusion of small amounts of leptin in the third hypothalamic ventricle (Thomas et al., 1999; Cornish et al., 2002; Takeda et al., 2002). It became evident that the pathways connecting brain and bone play critical roles in energy regulation and bone metabolism. Since this landmark finding, a rapid expansion in studies attempting to understanding this brain-bone interface occurred, with both the sympathetic (SNS) and parasympathetic nervous systems (PNS) having now been shown to regulate bone through various pathways, including leptin, serotonin, adiponectin, circadian genes, neuropeptide Y, muscarinic receptors, nicotinic receptors, beta-adrenergic receptors, and sensory nerve innervation of bone (Marenzana and Chenu, 2008; Dimitri and Rosen, 2017; Elefteriou, 2018). The importance of autonomic tone in regulating bone mass is supported by the observed benefit of beta-blockers on BMD and fracture risk (Schlienger et al., 2004; Bonnet et al., 2007; Graham et al., 2008; Reid, 2008).
Psychological stress can also impart changes in bone. We have previously discussed the relationship between osteoporosis and psychological stress, which is partially regulated through activation of the hypothalamic-pituitary-adrenal (HPA) axis, glucocorticoid signaling, and blunted response of growth factors that contribute to bone mass, such as insulin-like growth factor-1 (IGF-1) (Kelly et al., 2019). Conversely, bone has been found to regulate the brain, with osteocalcin (OCN), an osteogenic hormone, being essential for activation of the acute stress response via inhibition of the PNS (Berger et al., 2019). Thus, the brain-bone axis is critically important for skeletal metabolism, sensory innervation, and endocrine cross-talk between these organs.
Low BMD and cognitive impairment/reduced executive function often occur together, with osteoporosis and related fracture being associated with progression of cognitive impairment, likely, in part, due to increased fall risk (Liu-Ambrose et al., 2007; Cauley et al., 2016). In community-dwelling adults aged > 50 years without history of stroke or dementia, low BMD correlated with cognitive impairment, even after adjusting for confounding factors (Kang et al., 2018). Cognitive impulsivity is significantly related to BMD in elderly women, while verbal working memory has been associated with BMD and may alter fracture risk (Martino et al., 2019; Catalano et al., 2020b). The incidence of physical frailty and dementia follow similar patterns in the aging population, providing further evidence that frailty and cognitive function may be mechanistically and pathologically linked (Halil et al., 2015). Possible mechanisms for the association between BMD and cognitive impairment have been proposed. Mineral concentrations in osteoporosis may facilitate formation of senile plaques and neurofibrillary tangles (NFT) in Alzheimer’s disease (AD) (Lopez et al., 2008; Loskutova et al., 2009; Zhou et al., 2011). Estrogen, involved in bone homeostasis, may also affect cognitive function through inhibition of neuronal apoptosis, promotion of synaptic plasticity, reduction of oxidative stress, and improvement in cerebral blood flow by increasing high-density lipoprotein cholesterol levels (Funk et al., 1991; Brownbill and Ilich, 2004; Zhou et al., 2011; Kang et al., 2018). Further, estrogen replacement may lead to benefits in cognitive function (Luine, 2014). Age-related alterations in inflammatory processes, including increased levels of circulating IL-6 and diminished T regulatory cell activity, may dually contribute to bone loss (via local activation of osteoclasts) and cognitive decline (via increasing levels of neuronal α2-macroglobulin, a protease inhibitor implicated in AD plaques) (Ershler, 1993; Licastro et al., 2000; Lui et al., 2003). Lastly, parathyroid hormone (PTH) may represent a clinically important link between cognitive dysfunction and bone health, as a regulator of calcium and phosphate metabolism that can cross the blood-brain barrier (Lourida et al., 2015; Murthy et al., 2018). Hyperparathyroidism has been associated with physical frailty, bone mass loss via osteoclast activation, as well as poor cognitive function, potentially through calcium overload, disruption of neuronal signaling, and hippocampal atrophy (Numann et al., 1984; Roman et al., 2005; Marcocci et al., 2012; Murthy et al., 2018). It is clear that the bone and brain play key roles in regulating each other. Understanding how bone phenotypes are altered with neurological disorders may provide further mechanistic insights.
To date, there are more than 600 classified neurological disorders with distinct etiology, neuro-pathophysiology, and symptomology. Herein, we discuss major categories of neurological disorders, including autoimmune, dementia-related, infection-related, movement-related, neural developmental, neuromuscular, psychological, trauma-related, vascular, and other neuronal diseases. Although the etiology of many neurological disorders is highly complex and not fully understood, genetic, epigenetic, and external (e.g., trauma, infection, environment) factors have been implicated in neurological disease initiation and progression. Considerable clinical evidence across many neurological disease categories indicates that changes in neurophysiology lead to changes in bone physiology, resulting in decreased BMD and bone mineral content (BMC), altered bone microarchitecture, and decreased bone strength, ultimately leading to the development of osteopenia/osteoporosis and increased fracture risk. Recent clinical and preclinical studies have begun to shed light on some of the shared hormonal, immune, and molecular/cellular pathways that are impacted in neurological disorders and may mediate secondary effects in bone (Table 1). These include sustained activation of peripheral and central nervous systems (CNS), inflammatory pathways, changes in glutamate signaling, dysregulation of the SNS and PNS, and HPA axis dysregulation. In addition to shared pathophysiology, lifestyle changes secondary to neurological disorders, including modifications in diet and overall physical activity, can contribute to deleterious bone effects. Further, drug treatments for many neurological disorders (e.g., selective serotonin reuptake inhibitors (SSRIs), antipsychotics, anti-epilepsy drugs) have independent and overlapping roles in mediating bone loss. Since osteopenia/osteoporosis and resulting fracture have debilitating effects on patient quality-of-life, it is imperative that we understand how neurological diseases lead to secondary effects in bone to move toward more informed, comprehensive clinical treatment.
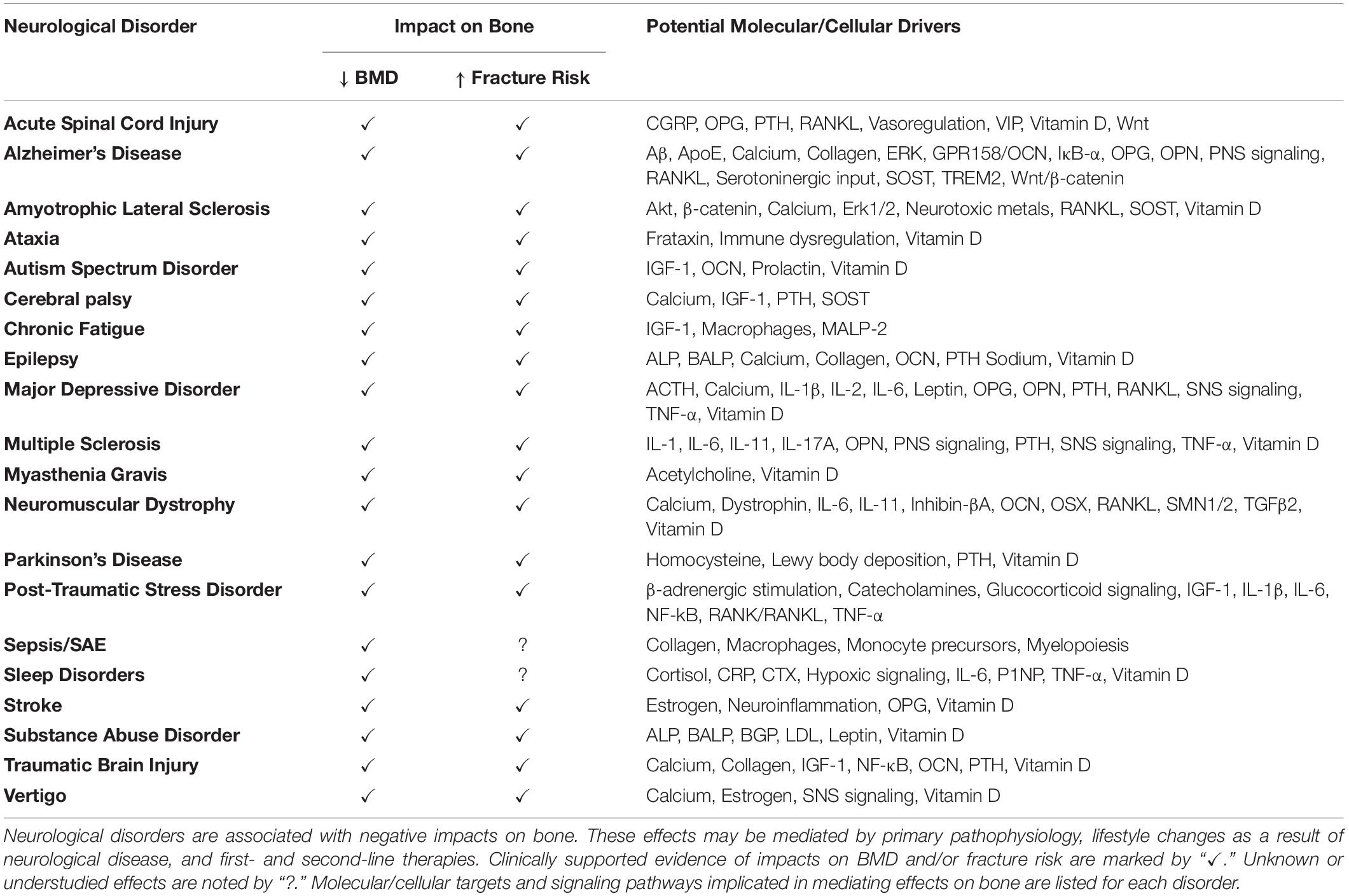
Table 1. Neurological disorders, impacts on bone, and potential mechanisms.
Multiple sclerosis is the most common, non-traumatic disease impacting young adults and can be divided into two stages: an inflammatory phase leading to relapsing-remitting disease and a secondary phase leading to prolonged neurodegeneration and non-relapsing, progressive disease (Dobson and Giovannoni, 2019). Early disease is characterized by neural inflammation, leading to the production of demyelinating plaques in the CNS that results in episodes of vision loss, fatigue, and tingling/numbness. As MS progresses, irreversible axon damage occurs and patients experience pain, muscle spasms, vision loss, and trouble walking. While the etiology of MS has not been fully elucidated, it likely involves multiple factors in addition to genetic predeterminants, including smoking, vitamin D, obesity, gut microbiota, and Epstein-Barr virus (EBV) infection (Dobson et al., 2012; Michel, 2018).
Clinical evidence suggests that MS is associated with detrimental bone effects, including osteopenia/osteoporosis and increased fracture risk (Moen et al., 2011; Sioka et al., 2011; Coskun Benlidayi et al., 2015; Huang et al., 2015a; Bisson et al., 2019). Meta-analyses show that MS patients have reduced BMD in lumbar spine, femur neck, and hip regions compared to healthy controls (Huang et al., 2015a). In a large clinical study, the prevalence of osteoporosis was found to be significantly higher in MS patients (16–26%) compared to healthy controls (6–15%) after adjusting for potential confounding factors, including age, sex, previous fracture history, and comorbidities (Bisson et al., 2019). Assessment of bone microarchitecture in MS patients, reported as a trabecular bone score (TBS), revealed no significant changes compared to healthy controls, suggesting that bone density, but not microarchitecture, is impacted in MS (Olsson et al., 2018).
Potential risk factors for reduced BMD in MS patients include vitamin D insufficiency, disease duration (> 7 years), total steroid dose (> 15 g), disease severity, decreased ambulation, altered parasympathetic signaling, and inflammation (Coskun Benlidayi et al., 2015; Huang et al., 2015b; Murphy et al., 2016). In female MS patients, BMD of lumbar spine and femoral neck was found to be 1-2 SDs lower than in age-matched, healthy controls, with lowest BMD values observed in patients with most severe disease (Nieves et al., 1994). BMD was directly correlated to levels of circulating vitamin D and inversely correlated with levels of parathyroid hormone (PTH), pointing to vitamin D deficiency and hyperparathyroidism as potential contributors to bone loss. In another study of both female (pre- and post-menopausal) and male MS patients, significant bone loss was observed in the femoral neck region, with female patients also presenting with bone loss in the spine. Compared to 2% of healthy controls, 22% of these patients had experienced a non-trauma-related fracture since the age of 35, highlighting a significant clinical outcome (Cosman et al., 1998). Both ambulatory status and steroid treatment > 5 months were shown to be predictors of bone loss, and patients with low levels of vitamin D exhibited greater bone loss overall. Importantly, bone loss in the spine was only observed in patients with low vitamin D levels and was insignificant in patients with normal vitamin D levels. In relatively young, ambulatory patients experiencing acute MS relapses, 51% were found to have low BMD and 62% were found to be vitamin D-deficient, providing further support for a potential link between bone health and vitamin D status (Murphy et al., 2016).
Disease severity and duration, as well as decreased ambulation, have been associated with bone loss and increased risk of fracture in MS patients (Tyblova et al., 2015; Olsson et al., 2018). MS patients with moderate disease, defined by a score of < 4.5–6.5 > on the Expanded Disability Status Scale (EDSS), had lower BMC and BMD in total and regional scans compared to patients with mild disease (score < 1.0–4.0 >) (Pilutti and Motl, 2019). In studies of pre-menopausal women with MS, a significant inverse relationship was observed between EDSS score, disease duration, and BMD, also pointing to disease severity as a primary predictor of bone loss (Zorzon et al., 2005; Terzi et al., 2010). Importantly, lifetime dosage of glucocorticoids was not associated with bone loss in this population, but only with EDSS score (Zorzon et al., 2005). In male patients, decreased mobility and lower EDSS scores were associated with decreased BMD in the femur and muscle wasting in the lower extremities (Zikán et al., 2012). Chronic use of glucocorticoids was not associated with bone loss in this population.
Inflammation has been implicated as a driver in MS-related bone loss. Several inflammatory factors implicated in the pathogenesis of MS, including IL-1, IL-17A, TNF-α, IL-6, and IL-11, have been shown to play a role in osteoporosis (Rifas, 1999; Kasper and Shoemaker, 2010). Levels of proinflammatory osteopontin (OPN) are increased in MS patients compared to healthy controls, with levels directly correlating with femur neck bone loss (Altıntaş et al., 2009). In relapsing-remitting MS patients, increased levels of circulating OPN were shown to correlate with levels of circulating IgG and markers of bone turnover, pointing to OPN as a potential driver of MS-related bone loss (Vogt et al., 2010). Increased levels of OPN were also observed in the cerebrospinal fluid (CSF) of MS patients and correlate with cognitive impairment, suggesting OPN may be a key player in the dysregulated bone-brain axis in MS and may serve as a biomarker of disease progression (Wen et al., 2012). In addition to pathological inflammatory processes, alterations in SNS and PNS signaling have been observed in MS patients and, in some cases, correlate with disease severity and progression (Flachenecker et al., 1999, 2001; Elefteriou, 2008). In a cross-sectional study, patients with active relapsing-remitting MS exhibited impaired sympathetic function and decreased levels of circulating catecholamines compared to healthy control and clinically stable patients (Bartl and Bartl, 2019). Longitudinal follow-up of a subset of these patients revealed a progressive decline in parasympathetic, but not sympathetic, function, suggesting that autonomic tone is differentially impacted as the disease progresses. Future studies in MS patients will be required to delineate the direct and indirect impacts of autonomic dysfunction on bone, as well as how bone innervation and/or signaling may change with disease progression. Together, these studies indicate that pathological mechanisms mediating primary MS symptoms and impaired mobility play major roles in mediating bone loss in MS patients, while the effects of glucocorticoids on bone in MS may be minimal and potentially offset by the positive impact of restored mobility.
Alzheimer’s disease is a progressive neurodegenerative disorder and the primary cause of dementia. AD currently affects over 5.8 million Americans age 65 and older, and this is anticipated to grow to 13.8 million by mid-century (Alzheimer’s Association, 2020). AD is characterized by progressive loss of cognitive function and pathologically by extracellular senile plaques enriched in amyloid-β peptide (Aβ) and intracellular NFTs formed by hyperphosphorylated tau protein (Mandelkow and Mandelkow, 2012; Selkoe et al., 2012). For over 20 years, skeletal fragility has been recognized as a comorbidity in AD (Birge et al., 1994; Melton et al., 1994; Johansson and Skoog, 1996; Weller, 2004; Looker et al., 2012). Low BMD and osteoporosis occur at twice the rate in AD patients as neurotypical adults, and this is independent of age, sex, body mass index, physical activity, and disease stage (Melton et al., 1994; Weller, 2004; Loskutova et al., 2009; Zhou et al., 2011, 2014; Zhao et al., 2012). Bone loss has been shown to occur in pre-clinical AD, often preceding diagnosis, thus it may have predictive value in estimating AD risk and likelihood of progression to full AD in patients with mild cognitive impairment (MCI) (Tan et al., 2005; Zhou et al., 2011, 2014; Chang et al., 2014; Sohrabi et al., 2015). Low BMD values have been suggested to predict a faster and more severe rate of cognitive decline (Zhou et al., 2014). Thus, while bone loss cannot be used as an independent risk factor for AD, as it frequently occurs in the non-dementia population, it may add predictive value to models used to assess dementia risk.
Regarding molecular pathways that intersect brain and bone in AD, the major genetic risk factor for AD is the apolipoprotein E (ApoE) 4 allele (Raber et al., 2004). ApoE also plays a critical role in maintaining bone mass by promoting osteogenesis and inhibiting osteoclastogenesis (Noguchi et al., 2018). Transgenic mouse models of AD, including the htau, amyloid precursor protein (APP)/presenilin1 mutant, and Swedish mutation APP strains exhibit low BMD (Cui et al., 2011; Yang et al., 2011; Xia et al., 2013; Peng et al., 2014; Dengler-Crish et al., 2016). Studies using the htau mouse model showed evidence of low BMD that preceded the onset of widespread tauopathy and memory deficits (Dengler-Crish et al., 2016, 2018). Tauopathy in the dorsal raphe nucleus (DRN) localized within serotonergic neurons and was associated with a 70% reduction in the overall number of serotonergic neurons in htau DNR, suggesting a link between serotoninergic input, bone loss, and AD. In AD mouse models that overexpress Aβ (e.g., APPswe and APP/PS1ΔE9), low BMD and osteoporosis were reported (Yang et al., 2011; Zhou et al., 2014). APP and its cleavage fragment Aβ are expressed in both neural and non-neural tissues, including osteoblasts and osteoclasts, and studies show that Aβ can directly impair osteoblast proliferation and promote osteoclast activity (Cui et al., 2011; Xia et al., 2013). Mechanistically, Aβ was shown to enhance RANKL-induced osteoclast activation through IκB-α degradation, ERK phosphorylation, and calcium oscillation signaling pathways (Li et al., 2014).
Clinical evidence suggests that dysfunctional autonomic signaling contributes to AD-related bone loss. AD patients commonly exhibit increased sympathetic tone and reduced parasympathetic flow, marked by reduced cholinergic innervation in the aging population (Aharon-Peretz et al., 1992; de Vilhena Toledo and Junqueira, 2010; Schliebs and Arendt, 2011). Further, AD patients treated with AChE inhibitors exhibit reduced risk of hip fracture and improved bone healing, suggesting that impaired parasympathetic signaling impacts bone homeostasis in AD patients and may be targeted to improve bone health (Weller, 2004; Tamimi et al., 2012; Eimar et al., 2013).
A potential common contributor to both AD and bone loss is the wingless-type murine-mammary-tumor virus integration site (Wnt) signaling pathway. In brain, Wnt signaling plays a role in neuronal survival and formation of synaptic connections and has been reported to play a neuroprotective role in AD (Oliva et al., 2013). In bone, Wnt signaling through the canonical (i.e., Wnt/β-catenin) pathway promotes osteoblast differentiation and increased bone mass (Krishnan, 2006). Mechanistically, this pathway influences renewal of stem cells, stimulation of pre-osteoblast replication, induction of osteoblastogenesis, and inhibition of osteoblast and osteocyte apoptosis (Krishnan, 2006). Loss-of-function mutations in the Wnt signaling pathway results in skeletal fragility and decreased bone mass (Shah et al., 2015). Preclinical studies in the htau mouse demonstrated Wnt signaling deficiencies in both the brain and bones of mice with low BMD (Dengler-Crish et al., 2018). Triggering receptor expressed on myeloid cells-2 (TREM2) is one potential activator of the canonical Wnt/β-catenin pathway that may tie together bone and brain effects. TREM2 is expressed on microglia, where it is neuroprotective, and on osteoclasts, where it controls the rate of osteoclastogenesis (Otero et al., 2012; Jay et al., 2015; Bemiller et al., 2017). Homozygous loss-of-function mutations in TREM2 are associated with an autosomal recessive form of early-onset dementia presenting with bone cysts and consequent fractures called Nasu–Hakola disease (Paloneva et al., 2000). Meta-analysis has shown heterozygous rare variants in TREM2 are associated with a significant increase in the risk of AD (Guerreiro et al., 2013). Together, these studies suggest a common potential target for addressing bone loss in AD.
While the studies above highlight the potential mechanisms regulating brain’s influence on bone, an understanding of bone’s effects on brain is emerging. Bone is considered an endocrine organ that influences other organs through the secretion of proteins, such as OCN, sclerostin (SOST), and OPN. Blood biomarkers associated with osteoporosis, including C-terminal collagen fragments, OPG, and OCN, are increased in AD (Emanuele et al., 2004; Luckhaus et al., 2009). OCN is a bone-derived hormone that can regulate brain development and function (Greenhill, 2013). Circulating OCN inversely correlates with age, and cognitive function of aged mice can be improved with injection of plasma from young mice (Villeda et al., 2014). Most recently, Karsenty’s group has identified the G protein-coupled receptor 158 (GPR 158) as a receptor for OCN in the brain (Khrimian et al., 2017; Obri et al., 2018).
Sclerostin is an osteocyte-specific secreted glycoprotein, encoded by the SOST gene that binds to low-density lipoprotein-receptor-related protein-5 or -6 (LRP5/6) to regulate Wnt signaling. Through this binding, SOST prevents Wnt ligand binding to LRP5/6 and its co-receptor, Frizzled, leading to decreased bone formation and increased bone resorption (Shah et al., 2015). While these data suggest SOST may impact Wnt signaling, which, in turn, affects the brain and AD pathophysiology (Inestrosa et al., 2002; Inestrosa and Varela-Nallar, 2014), further research needs to be conducted, as it is unclear if circulating SOST can cross the blood-brain barrier (BBB). One potential mechanism by which SOST could influences brain physiology may be through vascular regulation. SOST has recently been shown to influence vascular pathophysiology, a known risk factor for dementia, with high levels of SOST having been linked to cardiovascular mortality (Justin et al., 2013; Novo-Rodríguez et al., 2013; Catalano et al., 2020a). However, whether or not SOST plays an associative or causative role in vascular pathophysiology, and how this may influence development of dementia, remains unknown.
Osteopontin is thought to enhance bone resorption by anchoring osteoclasts to bone matrix, and high serum levels of OPN correlate with low BMD in post-menopausal women (Reinholt et al., 1990; Cho et al., 2013; Fodor et al., 2013). OPN also acts as a cytokine with upregulated production in response to inflammation and injury, including neuronal damage (Denhardt et al., 2001; Wang and Denhardt, 2008). OPN levels increase in patients with MCI progressing to AD, suggesting OPN could be a marker of neuroinflammation and early clinical stages of AD (Simonsen et al., 2007; Sun et al., 2013). Thus, it is possible that, early on, increased OPN expression may be neuroprotective in AD. This is supported by studies demonstrating a more marked increase in OPN levels in AD subjects in early stages of disease (Comi et al., 2010).
Shared risk factors for AD and bone loss include aging, systemic inflammation, depression, genetics, sex, and physical inactivity. However, the relationship between bone loss and AD is complex and cannot be solely attributed to aging, osteoporosis, or dementia. Rather, data suggest there are common pathophysiological mechanisms contributing to both diseases. Further adding to this complexity is the reciprocal crosstalk that occurs between brain and bone. Thus, bidirectional signaling between brain and bone tissue should be considered in the context of AD and its treatments.
Septicemia, the entry of bacteria into the bloodstream, leads to rapid immune activation and can result in a systemic reaction (sepsis), which can lead to death (Taeb et al., 2017). While the pathophysiology of sepsis is complex, it involves an impaired immune response in which an initial, rapid increase in inflammation is followed by sustained dysregulation of immune activation/suppression, impacting multiple organ systems and resulting in long-term morbidity (Uhle et al., 2016; Taeb et al., 2017; Cecconi et al., 2018). Increased activation of the peripheral immune system can also lead to CNS inflammation, owing to a disrupted BBB, which can result in sepsis-associated encephalopathy (SAE) (Meneses et al., 2019). As SAE impacts roughly 70% of septic patients and is a leading cause of brain dysfunction, it is critical to understand the mechanisms by which sepsis-induced dysregulation of the neuroimmune-endocrine response may impact organ systems like bone (Lamar, 2011).
Clinical evidence suggests sepsis negatively impacts bone health. In a retrospective study of patients initially treated for sepsis with absolute increase > 2 in Sequential Organ Failure Assessment score, significant bone loss was observed in the thoracic, lumbar, and sacral spine regions (Hongo et al., 2019). A second retrospective study showed that sepsis patients had an increased risk for developing osteoporosis compared to non-sepsis patients (Lee et al., 2020). In addition to bone loss, sepsis has been linked to heterotopic ossification, the abnormal formation of lamellar bone in connective tissue. In a retrospective study of patients hospitalized for burn injuries, sepsis following burn injury was associated with the development of heterotopic ossification ∼37 days after admittance, suggesting sepsis temporally modulates bone physiology (Orchard et al., 2015). Together, these studies indicate that the mechanisms by which sepsis impacts bone are complex and likely dependent on many factors, including type, duration, and location of initial infection.
Studies in preclinical models have shown sepsis rapidly reduces bone strength, impacts cellular differentiation in bone marrow (BM), and induces prolonged changes in peripheral macrophage populations. In a rat model of cecal ligation-puncture, trabecular bone strength was significantly reduced beginning 24 h following sepsis induction and was associated with decreased collagen and mineral elastic modulus at 24- and 96-h post-sepsis induction, respectively. These results suggest sepsis rapidly impacts biomechanical properties of bone and may lead to lasting changes in bone microarchitecture (Puthucheary et al., 2017). In addition to altered biomechanics, data indicate sepsis impacts differentiation of BM myeloid cells. In a combined rodent model of burn injury and sepsis, a shift in myeloid differentiation toward monocytopoiesis 72 h following thermal injury was reported, indicating sepsis can lead to rapid changes in differentiation of HSCs in BM (Santangelo et al., 2001). Alterations in the BM compartment were also observed in a rodent model of cecal ligation-puncture, in which epigenetic modifications in BM-derived monocyte precursors were found to impact the function and wound-healing capabilities of circulating macrophages (Davis et al., 2019). These studies point to modulation of monocyte differentiation and macrophage function as additional mechanisms by which sepsis may induce long-lasting effects in bone, possibly through dysregulated osteoclast differentiation and/or alterations in osteal macrophage function.
Parkinson’s disease (PD) is a progressive neurodegenerative basal ganglia syndrome characterized by bradykinesia and rigidity, resulting in limited daily activity and increased fall risk (Latt et al., 2009; Tassorelli et al., 2017). A number of studies have examined impacts of PD on bone, with PD being associated with decreased BMD and increased fracture risk (Vaserman, 2005; Wood and Walker, 2005; Fink et al., 2008; Gnädinger et al., 2011; Raglione et al., 2011; van den Bos et al., 2013; Gao et al., 2015; Sleeman et al., 2016). Meta-analysis indicates PD patients are at a higher risk for osteoporosis and have lower hip, lumbar spine, and femoral neck BMD compared to healthy controls (Zhao et al., 2013). Women with PD have 7.3% lower total hip BMD and an increased risk of hip fracture (Schneider et al., 2008). Vitamin D concentrations and weight loss are reduced in early PD patients and associated with bone loss (van den Bos et al., 2013; Ozturk et al., 2020). No difference was found in BMD between male PD subjects with short disease duration (0 to 5 years) compared to those with longer disease duration (5 to 10 years), suggesting PD progression may not correlate directly to decreasing BMD and that early detection is key to addressing PD-induced bone loss (Daniel et al., 2012). Thus, PD patients should be closely monitored for vitamin D levels and weight, as well as receiving routine dual-energy X-ray absorptiometry (DEXA) scans and fracture risk assessment (FRAX) (Henderson et al., 2019).
Evidence for molecular mechanisms of PD related to bone loss and increased fracture risk is scant. Lewy body deposition in areas of the brain that regulate bone growth and strength may play a role (Litvan et al., 2007; Daniel et al., 2012). Lifestyle changes associated with PD may result in vitamin D deficiency, which can impact bone loss via compensatory hyperparathyroidism (Invernizzi et al., 2009; van den Bos et al., 2013; De Pablo-Fernández et al., 2017). PD also alters levels of bone metabolism markers (Sato et al., 2004; Bezza et al., 2008). Elevated homocysteine levels from levodopa treatment, the central drug treatment for PD, may also impact bone, as homocysteine can induce osteoclast differentiation and osteoblast apoptosis (Koh et al., 2006; Lee et al., 2010). A significant proportion of PD patients suffer from depression, and concomitant use of antidepressants with levodopa results in a 3- to 5-fold increase in risk of hip and femur fracture (Lieberman, 2006; Arbouw et al., 2011). In general, several factors may be involved in development of bone loss associated with PD, including limited mobility/activity, malnutrition, low body mass index, decreased muscle strength, medication use, and vitamin D deficiency (Invernizzi et al., 2009; Malochet-Guinamand et al., 2015). Larger and more powerful studies are needed to determine effects of PD on osteoporosis risk and to stratify this risk by various confounding factors.
Ataxia, a degenerative CNS disease, results in impaired balance and coordination. It is usually caused by damage to the cerebellum but can be caused by damage to the spinal cord or other nerves. There have been few studies on subtypes of ataxia and their effects on bone. In Friedreich ataxia (FDRA), the most common inherited ataxic disorder in the Caucasian population caused by a GAA triplet expansion in the first intron of the frataxin gene on chromosome 9q13, scoliosis and foot deformities are frequent (Labelle et al., 1986; Delatycki et al., 2005; Milbrandt et al., 2008). A strong negative correlation between ataxia severity, GAA repeat length, and BMD was reported in the femoral neck of FDRA patients (Eigentler et al., 2014). The low observed BMD may be due to disease-related falls, mobility restrictions, and/or wheelchair-dependency. Additionally, vitamin D levels were low in the patient cohort, but it is unclear as to why. Suboptimal bone growth and mineralization in FRDA patients during childhood and adolescence may also impact adult BMD and growth. Farias et al. assessed BMD in patients with spinocerebellar ataxia type 3, also known as Machado-Joseph disease (MJD), which is a progressive ataxia resulting in movement restriction caused by an abnormal cytosine-adenine-guanine (CAG) expansion on chromosome 14q32.1. Ten patients out of thirty showed low BMD in at least one of the sites studied, while five patients had at least one lumbar fracture and seven patients reported more than ten falls per month (Farias et al., 2019). This study also found a correlation between CAG expansion and low femoral neck score, providing further evidence that gene alterations may be related to lower BMD. Simonsen et al. found that 75.3% of patients with hereditary ataxia had osteopenia or osteoporosis (Simonsen et al., 2016). These studies suggest the need for routine BMD measurements in ataxia patients to initiate prophylactic osteoporosis treatments. Further studies are needed to determine molecular and genetic mechanisms as opposed to lifestyle changes (e.g., reduced exercise/mobility, increased falls) that may be causing bone loss. In addition, patients with ataxia telangiectasia show immune dysregulation and premature aging, both of which can affect bone loss (Ambrose and Gatti, 2013). Examining immune markers and comparing bones from young ataxia patients to bones from the elderly may provide new pathways for study.
Autism spectrum disorder (ASD) is a neurodevelopmental disorder with heterogeneous origin and symptomology, including atypical autism, autism, and Asperger Syndrome, that disproportionately impacts males (3:1) (Kim et al., 2011; Sinha et al., 2015; Bhandari et al., 2020). Main symptoms include lack of social interaction, abnormal emotional/sensory processing, and repetitive, restricted behaviors, while secondary symptoms can include irritability, anxiety, aggression, and comorbid disorders. The etiology of ASD is complex and likely involves variations in genes regulating synaptogenesis and signaling pathways, as well as epigenetic and environmental factors, that ultimately lead to neural plasticity dysfunction and the precipitation of social, emotional, and sensory processing symptoms.
Poor nutrition, decreased physical activity, vitamin D deficiency, and use of antipsychotic therapies (APTs) have been associated with poor bone health in ASD. In a cross-sectional study of adolescent boys aged 8–17 years, ASD patients exhibited lower BMD in the lumbar spine, femoral neck, total hip, and whole-body regions compared to age-matched controls (Neumeyer et al., 2013). In addition to lower BMD scores, adolescent boys with ASD had lower consumption levels of protein, calcium, and phosphorus, and were less physically active than typically developing adolescents. In another study of prepubertal boys, patients with ASD were shown to have lower BMD at both the hip and femoral neck regions compared to healthy control patients, as well as lower levels of serum vitamin D and decreased physical activity, pointing to vitamin D deficiency and decreased overall activity as potential risk factors for ASD-related bone loss in the adolescent ASD population (Neumeyer et al., 2013).
Autism spectrum disorder also impacts bone microarchitecture and fracture risk. In a cross-sectional study of adolescent boys, ASD patients exhibited lower trabecular thickness, compressive stiffness, and failure load at the ultradistal radius, as well as a 61% reduction in cortical area compared to typically developing controls, with similar effects observed in the distal tibia (Neumeyer et al., 2017). These ASD patients also exhibited increased body fat, increased serum IGF-1, lower lean mass, and decreased whole body and femoral neck BMD, suggesting physical activity, nutrition, and changes in IGF-1 responsiveness may contribute to ASD-related changes in bone density and microarchitecture. Increased fracture risk has also been observed in the ASD patient population. In a national study of emergency room visits, a higher rate of hip fractures was observed in both children/adolescents (3–22 years) and adults (23–50 years) with ASD compared to patients without ASD, with a higher rate of forearm and spine fractures also observed in adult women (Neumeyer et al., 2015).
While not all studies in the ASD population control for use of APTs, such as risperidone, there is evidence that patients taking APTs have decreased BMD. In a study of adolescent boys with ASD, ∼49% of patients taking an APT had hyperprolactinemia with decreased lumbar spine BMD, as well as decreased levels of the bone turnover marker, carboxyterminal cross-linking telopeptide of bone collagen, compared to patients taking APT without hyperprolactinemia (Roke et al., 2012). Another study of ASD boys aged 5–17 years taking risperidone showed decreased trabecular BMD and decreased radius bone strength compared to healthy controls (Calarge and Schlechte, 2017). Preclinical studies have shed light on the biological mechanisms by which ASD, as well as APT treatment, may impact bone health. In a genetic mouse model for human 15q11-13 duplication, decreased bone mass was observed and associated with osteoblast reduction and decreased bone formation (Lewis et al., 2017). Osteoblasts from ASD mice exhibited decreased proliferation, differentiation, and mineralization, whereas osteoclasts were minimally impacted. In a rat model of ASD, based on maternal exposure to LPS, ASD rats showed decreased bone stiffness and strength, in addition to a reduced number of OCN-positive cells compared to control rats, also indicating that ASD leads to impaired osteoblast proliferation and/or differentiation (Amini et al., 2020). Treatment of ASD rats with risperidone led to more extreme impacts on bone strength, providing evidence that APT treatment exacerbates ASD-related bone effects. Additional studies will help delineate the influence of APTs on bone health in the context of ASD.
Cerebral palsy (CP) is the most common motor disorder in children and causes a wide range of symptoms impacting neurological (e.g., ataxia, impaired gross motor coordination), orthopedic (e.g., hip dysplasia/dislocation), cognitive (e.g., autism, epilepsy), and visual/hearing systems (Brandenburg et al., 2019; Vitrikas, 2020). CP etiology is complex and involves pathophysiology in the brain and spinal cord. Spastic CP, which accounts for > 80% of cases, is thought to result from dysfunction in the spinal cord, leading to disinhibition of motor neurons and causing symptoms of spasticity, impaired coordination/movement, hyperreflexia, muscle contracture, and weakness (Sheean and McGuire, 2009; Brandenburg et al., 2019). While the pathological development of spastic CP is complex, it has been linked to asphyxia, prenatal/neonatal hemorrhagic or ischemic stroke, infection, brain malformation, trauma, and genetic factors (Brandenburg et al., 2019).
In addition to neuromuscular dysfunction, children and adolescents with CP often present with impaired bone health. Children with quadriplegic CP had reduced BMD of the lumbar spine compared to age-matched healthy children, with the most severe bone impacts observed in patients at level V of the Gross Motor Function Classification System (GMFCS) and in malnourished patients, pointing to disease severity, mobility, and malnutrition as potential risk factors for bone loss (Alvarez Zaragoza et al., 2018). In addition to disease severity, changes in the IGF-1 axis have been implicated in CP-induced bone loss. BMD was significantly decreased in children with spastic CP compared to healthy children and was associated with low circulating IGF-1 levels, severe GMFCS level, and use of anticonvulsive drugs. Children with CP also exhibited increased fracture risk (Nazif et al., 2017).
There is conflicting evidence for the role of anticonvulsive drugs as a risk factor for low BMD. In a study of non-ambulatory children with CP, no significant differences in BMD were found between children taking and not taking anticonvulsant therapies (Cheng et al., 2016). However, nutritional status was implicated as a risk factor. Studies comparing ambulatory and non-ambulatory children with CP have shown that the main predictor of low BMD in the distal femur is impaired mobility (Finbråten et al., 2015). Adolescents and young adults with CP have shown similar deficits in areal BMD, which were associated with reduced mobility by the GMFCS (Trinh et al., 2019).
Although less well-studied, there is clinical evidence that adult CP patients also exhibit bone loss. In a study of premenopausal women and men under 50 years old, BMD was significantly decreased at the lumbar spine, total hip, and femoral neck regions compared to healthy controls. For the lumbar spine and hip, BMD was associated with impaired motor ability by the GMFCS (Fowler et al., 2015). In a demographically similar population, BMD at the second metacarpal bone (mBMD) was found to be decreased and correlated with use of anticonvulsant drugs (Nakano et al., 2003). In men, abnormal calcium metabolism was also associated with lower mBMD, whereas, impaired mobility was associated with mBMD in women, suggesting the mechanisms by which CP impacts bone health may be sex-dependent and include both physical and biomolecular factors. In a study of ambulatory versus non-ambulatory adult CP patients, non-ambulatory patients had decreased BMD, lower PTH levels, and higher SOST levels compared to ambulatory patients, implicating systemic changes in hormones and bone remodeling factors in CP-related bone loss in adult patients (Shin et al., 2017). Taken together, these studies implicate disease severity, mobility status, and alterations in hormones and bone remodeling factors as critical risk factors for CP-associated bone loss.
Epilepsy is a complex neurological disorder characterized by repeated, unprovoked seizures. Diagnosis is made upon the occurrence of two or more unprovoked seizures more than 24 h apart or one unprovoked seizure with high probability of recurrence (> 60%) over the next 10 years (Beghi et al., 2015). While the etiology of epilepsy is unknown, genetic predisposition, brain injury, and infection have been implicated as causal factors (Thijs et al., 2019). Epilepsy is a highly heterogeneous disorder, with four major types (focal, generalized, combined focal/generalized, and unknown) that can be divided based on nature of seizure onset, as well as level of awareness, motor symptoms, and non-motor symptoms (Thijs et al., 2019).
Patients with epilepsy have increased risk of developing osteoporosis and increased fracture risk that is 2–6 times higher than the general population, independent of seizure-related fractures (Diemar et al., 2019a). Decreased exercise, a more sedentary indoor lifestyle, and use of anti-epileptic drugs (AEDs), which can lead to impaired coordination and disrupted calcium/vitamin D metabolism, may contribute to poor bone health (Kobau et al., 2004; Shellhaas and Joshi, 2010; Diemar et al., 2019b). A meta-analysis of epileptic children showed a significant BMD decrease at lumbar spine, trochanter, femoral neck, and total body regions (Zhang et al., 2015). Decreased serum vitamin D and increased serum alkaline phosphatase (ALP) were also observed, suggesting abnormal vitamin D and/or calcium metabolism may contribute to decreased BMD in children with epilepsy.
Use of AEDs, especially cytochrome P450 enzyme-inducing AEDs (EIAEDs), have been associated with poor bone health in children and adult patients with epilepsy. In ambulant children with epilepsy, use of > 2 EIAEDs was shown to be a significant risk factor for low lumbar BMD (Fong et al., 2018). In a case-control study of matched-pair adolescents, epileptic patients taking AEDs had a significantly increased fracture risk and a 14% reduction in trabecular volumetric BMD compared to matched controls, demonstrating a link between AED use and poor bone health (Simm et al., 2017). A similarly designed study of same-sex twin/age-matched sibling pairs showed EIAED users exhibited a greater reduction in hip and total body BMD, which was not observed in non-enzyme-inducing AED (NEIAED) users, pointing to EIAEDs as the primary driver of bone loss in this patient population (Shiek Ahmad et al., 2017). Similarly, patients on carbamazepine (CBZ), a widely used EIAED, have increased hip and femoral neck BMD loss during initial years of therapy compared to nonusers and have increased hip BMD loss compared to users on NEIAEDs, including levetiracetam and valproate. While the mechanisms by which EIAEDs impact bone have not been fully elucidated, increased catabolism of vitamin D to inactive metabolites, decreased calcium, increased PTH, and increased bone turnover have been implicated (Pack, 2008). In a study of epilepsy patients taking CBZ for > 12 months, decreased BMD and decreased serum vitamin D were observed. A concomitant increase in OCN was observed in CBZ users, suggesting that, in addition to modulation of vitamin D, CBZ may impact bone turnover (Suljic et al., 2018). In an epilepsy rat model, CBZ was associated with decreased serum vitamin D and elevated PTH, as well as decreased BMC, impaired collagen crosslinks, and decreased microhardness, indicating CBZ therapy may affect bone strength and microarchitecture (Garip Ustaoglu et al., 2018). Although less well documented, there is evidence to suggest that some NEIAEDs, including valproate, also impact bone health in patients with epilepsy. In a meta-analysis of valproate users, BMD was found to be decreased in spine and femoral neck regions compared to healthy controls and was associated with increased serum bone-specific alkaline phosphate (BALP) (Fan et al., 2016). In addition to vitamin D deficiency, use of AEDs has been associated with hyponatremia in epilepsy patients. In a cross-sectional study of patients with epilepsy, hyponatremia was observed in ∼10% of the population and was independently associated with decreased BMD and increased risk of osteoporosis, providing evidence that altered sodium metabolism may contribute to AED-associated bone loss in epilepsy (Diemar et al., 2019a). While additional mechanistic studies are needed, current evidence points to altered vitamin D and sodium metabolism, elevated PTH, and dysregulated bone turnover in epilepsy patients taking AEDs.
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder characterized by loss of cortical, brainstem, and spinal motor neurons that results in progressive muscle atrophy. There is currently no cure for ALS. Most patients eventually become dependent on mechanical ventilation and usually die due to respiratory failure (Portaro et al., 2018). How ALS may affect the skeleton and modify osteoporosis risk is under-studied. Altered calcium metabolism, hypovitaminosis D, reduced cortical bone mass, and vertebral defects have been noted in ALS patients (Mallette et al., 1977; Yanagihara et al., 1984; Joyce et al., 2012). In clinical studies, ALS patients had 14% more fractures than controls, and, in a Swedish population, fracture was associated with higher incidence of ALS (Parfitt, 1994; Peters et al., 2017). ALS has also been associated with increased bone turnover markers in the blood (Fang et al., 2010). In a case report of an 81-year-old man with ALS, multiple hidden vertebral fractures were found, with a low Z score but normal TBS, suggesting normal bone structure (Portaro et al., 2018). It is possible that reduced muscle strength from ALS resulted in an unsupported spinal column, leading to these fractures. In a mouse model of ALS, SOD1G93A mice demonstrated decreased bone mass with notable whole bone biomechanical deficits (Ko et al., 2018). Osteoblasts isolated from SOD1G93A mice with muscle atrophy had impaired differentiation capacity, while osteoclast activity was increased compared to wildtype mice (Zhu et al., 2015). Aberrant Akt, Erk1/2, SOST, RANKL, and β-catenin signaling pathways were noted, which could be further links between ALS-induced muscle atrophy and bone loss. Muscle and bone are known to cross-talk extensively, with skeletal muscle providing an important source of osteogenic growth factors (e.g., IGF-1, FGF-2), as well as driving bone morphogenesis through mechanical load (Hamrick et al., 2010; Sharir et al., 2011; Zhou et al., 2015). Thus, it is likely that muscle atrophy is the key component linking ALS to bone loss and increased fracture risk.
Another interesting mechanism that may link ALS to bone is accumulation of neurotoxic metals. Neurotoxic metals, such as lead, have been found in the brain and CSF of ALS patients and have been shown to affect bone mineralization, whereby they accumulate in the bone and act as substitutes for calcium in hydroxyapatite (Roos et al., 2013; Chen X. et al., 2014; Roos, 2014). It is also possible that osteoporosis can worsen neurodegenerative disease outcomes, as the bones and CSF share circulation. Bone may act as a sink for neurotoxic metals, releasing them during osteoporosis, thereby facilitating neurodegeneration (Roos, 2014). Patients exposed to high concentrations of neurotoxic metals should be more closely monitored for osteoporosis and frequently tested for neurotoxic metals. It is still unclear how any specific molecular mechanism tied to ALS may influence bone homeostasis or if increased risk is due primarily to lifestyle factors, as age, reduced mobility, increased falls, and weight loss are all prevalent in ALS and are common osteoporosis risk factors. Thus, more studies are needed examining molecular mechanisms linking ALS to bone, particularly any associated with muscle atrophy or neurotoxic metals.
Myasthenia gravis (MG) is a neuromuscular disorder that results in weakening of the skeletal muscles. It is frequently a product of autoimmune disease, resulting in attack on nicotinic acetylcholine receptors or on muscle-specific tyrosine kinase (MuSK). The exact cause of this autoimmune reaction is still under investigation. Symptoms include difficulty breathing or swallowing, fatigue, drooping of eyelids, problems walking or lifting objects, trouble talking, and double vision. Regarding impacts on bone, a study found that MG resulted in a 1.96-fold increased risk of developing osteoporosis, likely due to lack of outdoor activity leading to decreased sunlight exposure/vitamin D and physical inactivity (Yeh et al., 2014). However, altered acetylcholine signaling may also play a role, as osteoblasts express acetylcholine receptors and elevated acetylcholine levels induce osteoblast proliferation (En-Nosse et al., 2009; Sato et al., 2010). In addition, MG patients prescribed antidepressants, anxiolytics, or anticonvulsants had increased fracture risk (Pouwels et al., 2013). The mechanism underlying this finding is unknown but may be due to altered neuronal signaling, as use of SSRIs reduces BMD in humans (Ducy and Karsenty, 2010; Haney et al., 2010; Brinton et al., 2019). The use of anticonvulsants increases vitamin D catabolism, leading to increased bone resorption (Kinjo et al., 2005). However, how these mechanisms coincide with MG to increase fracture risk remains unknown.
In addition to the effects of MG itself, corticosteroids, a common treatment for MG, are known to cause bone loss, with long-term corticosteroid use the most common mediator of secondary osteoporosis (Buehring et al., 2013). Corticosteroids have been shown to increase osteoporosis risk in MG (Pascuzzi et al., 1984; Konno et al., 2015; Braz et al., 2017). A case study reported an MG patient with eight spinal compression fractures due to intensive and prolonged prednisone treatment, but DEXA scanning and/or bisphosphonate treatment were not mentioned as being used prophylactically (Raibagkar et al., 2017). This situation may, unfortunately, not be uncommon among neurologists, as there may be a lack of awareness and non-implementation of iatrogenic osteoporosis treatment guidelines, with limited requested DEXA scanning and a lack of understanding in how to interpret T scores leading to over- or under-treatment with bisphosphonates (Lewis and Smith, 2001; Lozsadi et al., 2006; Gallagher and Sturrock, 2007). To demonstrate the benefits of prophylactic osteoporosis treatment, BMD in 36 MG patients who had undergone long-term prednisolone administration with concurrent treatment with elcatonin was measured, and a decrease in BMD was found in 31% of female patients and osteoporosis in 11.5% compared to a presumptive rate of 22.6% in the general population. No osteoporosis was detected in male patients (Wakata et al., 2004). This suggests that prednisolone-treated MG patients have an acceptable bone loss risk when monitored and provided prophylactic osteoporosis treatment. Likewise, MG patients with history of glucocorticoid treatment who were treated with alendronate combined with alfacalcidol showed increased BMD and decreased bone turnover biomarker levels (Lv et al., 2018). Further studies are needed to dissect the mechanistic roles by which MG impacts bone health independent of corticosteroid use.
Neuromuscular dystrophy (NMD) is a group of degenerative muscle diseases in which genetic mutations result in loss of muscle mass and progressive weakness. Muscle-bone interactions have been extensively studied, and it is thought that alterations in muscle-derived myokines, bone deformation as a result of muscle weakness, and direct effects of genetic defects on bone cells may negatively impact bone in NMD, as well as low vitamin D, nutritional deficits, immobility, and drug treatments (Kurek et al., 1996; Febbraio and Pedersen, 2002; Veilleux and Rauch, 2017).
Duchenne muscular dystrophy (DMD) is the most common form of NMD and is an X-linked recessive disorder linked to a mutation in the dystrophin gene that is characterized by progressive muscle weakness due to reduction of dystrophin and destabilizing effects on the sarcolemmal membrane, ultimately leading to premature death (Hoffman et al., 1987). There is currently no cure. DMD is the most studied form of NMD in regards to bone health, with reports dating back to 1941 (Maybarduk and Levine, 1941; Joyce et al., 2012). Boys with DMD have abnormalities in bone geometry, presenting with slender long-bone shafts, a likely risk factor for long bone fracture (Veilleux and Rauch, 2017). Up to 90% of patients with DMD have scoliosis, providing strong evidence for the role of dystrophin in regulating bone health and development (Pecak et al., 1980). Decreased BMD, increased fracture rate, and vitamin D deficiency have been reported in DMD (Siegel, 1977; Larson and Henderson, 2000; Vestergaard et al., 2001; McDonald et al., 2002; Bianchi et al., 2003; Hawker et al., 2005; Perera et al., 2016; Joseph et al., 2019). Aparicio et al. found that eight out of ten boys aged 6–11 years with DMD years had osteoporosis in the proximal femur, while the remaining two boys had osteopenia (Aparicio et al., 2002). Bianchi et al. showed that DMD patients had reductions in spine BMD, hypocalciuria, increased bone turnover markers, and low vitamin D levels (Bianchi et al., 2003). Fall risk is also increased with DMD, thereby further increasing fracture risk (McDonald et al., 2002).
Using the dystrophin-null mdx mouse, Rufo et al. found mdx mice displayed changes in BMD in a manner similar to that observed in humans. Osteoclasts and IL-6 levels were increased, while RANKL:OPG ratio was altered in favor of increased bone resorption. Human primary osteoblasts incubated with sera from DMD patients showed decreased nodule mineralization, downregulation of OSX and OCN, and upregulation of IL6, IL11, inhibin-βA, and TGFβ2, suggesting DMD can directly impact bone at a cellular and molecular level (Rufo et al., 2011). This study also proposed anti-IL-6 therapy as a possible treatment of bone disease in DMD. In regard to current DMD treatment, increasing routine use of corticosteroids to treat DMD is concerning, due to the known effects of chronic glucocorticoid treatment on bone health (Bell et al., 2017). In DMD, corticosteroid use may be increasing prevalence of vertebral fracture (King et al., 2007; Houde et al., 2008; Annexstad et al., 2019). However, robust evidence that corticosteroid use increases fracture risk in DMD, specifically, is lacking. Teriparatide has also been used with success in DMD, showing improvements in BMD and quality of life with few, if any, side effects (Catalano et al., 2016; Nasomyont et al., 2020). It is clear that routine imaging, particularly of the spine, clinical monitoring, consideration of skeletal delay, possible vitamin D supplementation and/or teriparatide treatment are needed when evaluating bone health and BMD Z-scores in patients with DMD (Ko et al., 2020).
Another form of NMD, spinal muscular atrophy (SMA), an autosomal recessive disorder due to mutations in survival motor neuron 1 and (SMN1/2) that involves selective destruction of anterior horn cells, is associated with fractures at birth and increased rates of fracture throughout life. There are multiple subtypes that present with different degrees of severity. Both long bone and vertebral fractures are relatively common, as well as scoliosis (Vai et al., 2015). Animal studies have shown that the exon 7 splice variant of SMN upregulates osteoclast-stimulating factor, promotes osteoclast formation, and enhances bone resorption (Kurihara et al., 2001; Shanmugarajan et al., 2009). However, more in depth mechanistic studies are needed in humans to determine direct effects of SMA mutations on bone.
Major Depressive Disorder (MDD) is a highly prevalent, heterogeneous mood disorder characterized by biases in attending to and recalling negative emotional stimuli that align with a negative mood state (Belmaker, 2008). The etiology of MDD is complex and likely involves dysregulation of serotonin and norepinephrine neurotransmission, increased neuroinflammation, and environmental factors (Woelfer et al., 2019). Significant clinical evidence shows MDD is associated with poor bone health, including decreased BMD and increased risk for osteoporosis. Patients with MDD were found to be 1.3 times more likely to develop osteoporosis compared to patients without MDD (Lee et al., 2015). In a population-based cohort study, patients diagnosed with a depressive disorder were found to have higher risk of vertebral fracture compared to healthy patients (Lee et al., 2017). In a meta-analysis, MDD was associated with lower BMD in the lumbar spine, femur, and total hip regions in all age groups compared to healthy controls (Schweiger et al., 2016). Differences in lumbar spine and femur BMD were more prevalent in women, whereas differences in hip BMD were more prevalent in men, suggesting sex-dependent effects of MDD on bone. Clinical studies also point to age-dependent differential effects of MDD on bone. Adolescent boys with MDD had lower hip and femoral neck BMD compared to age-matched controls, whereas no differences in BMD were found in girls with MDD compared to healthy controls (Fazeli et al., 2013).
Although many clinical studies examining the impact of MDD on bone do not stratify based on MDD subtype, different subtypes of MDD may be associated with greater risk for low BMD. In a study of premenopausal woman with melancholic, atypical, or undifferentiated MDD, women with undifferentiated MDD were more likely to exhibit low BMD at the femoral neck compared to healthy control patients. Elevated levels of adrenocorticotropic hormone (ACTH) were observed in women with atypical MDD, while higher levels of leptin were observed in women with melancholic MDD, suggesting differences in the pathophysiology between MDD subtypes may also account for observed differences in bone health (Cizza et al., 2010; Woelfer et al., 2019). Psychological conditions commonly co-occurring with MDD, including anxiety, may also contribute to bone loss. In a study of postmenopausal women, patients with higher anxiety levels exhibited decreased lumbar and femoral neck BMD and increased fracture risk compared to patients with lower anxiety levels (Catalano et al., 2018). A separate study in postmenopausal women showed that anxiety level, as determined by the Hamilton Anxiety Rating Scale (HAMA), was inversely correlated with levels of vitamin D and this association was independent of patient level of depression, suggesting that anxiety independently impacts vitamin D metabolism and this may be one mechanism by which anxiety contributes to bone loss and decreased patient quality of life in the aging population (Martino et al., 2018a,b). As SSRIs are used as a first-line therapy for MDD patients and have negative bone effects, it is difficult to parse out how MDD pathophysiology, versus SSRI treatment, impacts bone health. Recurrent MDD in adult men was associated with decreased forearm and total body BMD compared to men with no history of MDD (Rauma et al., 2015). Further, antidepressant use in this population was associated with lower BMD in lower-weight men only. Interestingly, acute MDD episodes were found to be associated with higher BMD at total hip, pointing to differential effects of acute versus recurrent MDD. In a study of adolescents and young adults who were either unmedicated or within 1 month of starting SSRI treatment, SSRI use was associated with increased lumbar spine areal BMD in females and decreased lumbar spine areal BMD in males, suggesting SSRIs independently impact bone health in a sex-dependent manner in MDD (Calarge et al., 2014). In young to middle-aged patients with an acute episode of depression, SSRI use had no impact on BMD (Malik et al., 2013). However, increased levels of OPG and increased levels of physical activity were observed in MDD patients, suggesting there may be protective/compensatory mechanisms in acute MDD that temper SSRI-mediated bone effects. In a study examining levels of bone turnover markers in medication-free inpatients with recurrent MDD, patients with MDD exhibited decreased levels of baseline OPG/RANKL and plasma OPN, indicating MDD may modulate bone physiology independent of SSRI use (Kadriu et al., 2018).
Bone loss in MDD patients may also be associated with increased activation of the ACTH and PTH axis, dysregulation of the OPG-RANK-RANKL axis, increased inflammation, and autonomic dysfunction (Rosenblat et al., 2016; Elefteriou, 2018). In postmenopausal women, patients with MDD exhibited decreased BMD and increased levels of PTH and RANKL compared to healthy controls (Atteritano et al., 2013). In premenopausal women, patients with MDD exhibited lower BMD at baseline, as well as increased PTH and ACTH at baseline and 6-month follow-up compared to healthy controls (Cizza et al., 2012). Levels of PTH remained higher in MDD patients at 24 months, suggesting sustained activation of the PTH axis may perpetuate negative bone effects. Calcium and vitamin D levels were also significantly decreased in patients with MDD compared to controls, indicating vitamin deficiency is a contributing factor. Increased prevalence of low BMD at the femoral neck and hip was observed in premenopausal women with MDD compared to controls, with a concomitant increase in circulating proinflammatory cytokines (e.g., IL-1β, IL-2, IL-6, TNF-α) (Eskandari, 2007). Significant increases in plasma IL-6 and changes in its diurnal release were also observed in patients with active MDD and found to correlate with mood ratings, providing additional evidence that inflammation may dually mediate neurocognitive effects and bone effects in MDD (Alesci et al., 2005). Patients with MDD also commonly present with autonomic dysfunction (Kemp et al., 2010; Yang et al., 2011; Brunoni et al., 2013). Studies in preclinical models show that MDD-associated trabecular bone loss is associated with increased levels of bone norepinephrine and can be attenuated by treatment with propranolol, a beta-adrenergic antagonist (Yirmiya et al., 2006). Together, these studies implicate increased sympathetic output as a driver of MDD-associated bone loss, although additional clinical studies will be necessary to determine the mechanisms by which altered sympathetic signaling impacts bone in MDD patients. Further, additional clinical studies that carefully control for concomitant SSRI use, MDD subtype and nature (acute versus recurrent), and comorbid conditions such as anxiety, will better our understanding of MDD-driven bone pathophysiology.
Post-traumatic stress disorder (PTSD) is characterized by an abnormal, persistent response to a traumatic event (Seal, 2007; Heron-Delaney et al., 2013). Patients with PTSD exhibit re-experiencing symptoms related to the initial traumatic event, such as flashbacks and nightmares, as well as avoidance, arousal/reactivity, cognitive, and mood symptoms, which can manifest as being easily startled, difficulty sleeping, negative thoughts, and angry outbursts (Qi et al., 2016). While the pathophysiology of PTSD is complex and not fully understood, dysregulations in the HPA axis and glucocorticoid signaling, as well as increased neural and systemic inflammation, have been implicated in disease initiation and development (Girgenti et al., 2017; Dunlop and Wong, 2019).
In addition to primary psychological symptoms, studies in adult, elderly adult, Veteran, and Prisoner of War populations provide substantial evidence that PTSD negatively impacts bone health (Glaesmer et al., 2011; Hain et al., 2011; Huang et al., 2018). A large nationwide longitudinal study showed adults with PTSD were more likely to develop osteoporosis at an earlier age, compared to age-matched, healthy controls (Huang et al., 2018). Similarly, the National Health and Resilience in Veterans study showed that U.S. Veterans diagnosed with PTSD had significantly increased risk for developing osteoporosis or osteopenia (El-Gabalawy et al., 2018). Evidence also supports that PTSD patients have an increased fracture risk (Jiang et al., 2018).
While the mechanisms by which PTSD impacts bone health have not been fully elucidated, several molecular, hormonal, and immune-related pathways implicated in PTSD pathophysiology negatively impact bone and have been extensively reviewed elsewhere (Kelly et al., 2019). In response to a stressor, stimulation of the glucocorticoid axis can lead to activation of inflammatory and RANK pathways via NF-kB, decreased osteoblast function, and prolonged osteoclast viability (Vega et al., 2007; Briot and Roux, 2015). Stress-induced increases in catecholamines, the “fight-or-flight” hormones, can also lead to activation of β-adrenergic receptors, stimulating RANKL expression and promoting osteoclast differentiation (Rodrigues et al., 2012). In a mouse model of PTSD, PTSD was associated with significantly decreased BMC and BMD in the femur, lumbar vertebra, and tibia 3 weeks following initial trauma, providing evidence that activation of the stress response in PTSD mice leads to bone loss (Yu et al., 2012). In addition to dysregulated glucocorticoid and stress hormone signaling, changes in IGF signaling and immune activation may contribute to PTSD-related bone loss. IGFs have been implicated in stress signaling and are known to be important regulators of the osteoblast-osteoclast balance (Canalis, 2009; Zegarra-Valdivia, 2017). IGF-1 supports osteoblast differentiation, with decreasing levels of IGF-1 in the aging population being associated with osteoporosis (Perrini et al., 2010; Crane et al., 2013). Increased levels of inflammatory factors, including IL-1β, IL-6, and TNF-α, are upregulated in patients with PTSD and have been implicated in mediating neurocognitive effects of the disorder (Passos et al., 2015; Lindqvist et al., 2017; Imai et al., 2018). Proinflammatory cytokines also promote osteoclast formation and amplify bone resorption (Sims, 2016; Weitzmann, 2017). These studies suggest PTSD-induced inflammation may negatively impact bone health, although additional studies are needed to delineate these effects.
In addition to shared mechanisms of pathophysiology, therapies used to treat PTSD may also negatively impact bone health. First- and second-line treatment options for PTSD patients, including SSRIs and antipsychotic drugs, are associated with increased risk of developing osteoporosis and fracture (Vestergaard et al., 2006, 2008; Haney et al., 2010; Alexander, 2012; Rabenda et al., 2013; Seifert and Wiltrout, 2013; Rauma et al., 2015). Therefore, it is reasonable to speculate that SSRI use may exacerbate PTSD-associated bone loss. However, clinical evidence of the combined effects of PTSD and SSRI use on bone health is currently lacking.
Substance use disorders (SUDs), including addictions to alcohol, heroin, cannabis, ecstasy, cocaine, and amphetamines, are a major global health concern. While addiction to different substances can produce different neurobiological effects, there are shared layers of symptomology between different substance groups. These include a reduction in response to normal biological activities like social cooperation and a loss of ability to control drug-seeking behavior. Progressive changes in dopamine transmission in the corticolimbic brain regions, alterations in glutamatergic synapses, and environmental stress have been implicated in the initiation and development of SUDs (Reid et al., 2012).
In addition to changes in neural circuitry and plasticity, SUDs have been associated with impaired bone health. The impact of alcohol addiction on bone has been well studied and will be the focus of this section, although opioid, cannabis, and amphetamine addictions have also been associated with decreased BMD and increased fracture risk (Gozashti et al., 2011; Mosti et al., 2016; Gotthardt et al., 2017; Heydari et al., 2017; Sophocleous et al., 2017). Chronic-excessive use of alcohol has been shown to negatively impact bone health, resulting in decreased BMD, decreased BMC, and increased fracture risk. In a study of males classified as heavy drinkers, osteopenia was observed in 23% of the population, with a significant inverse correlation observed between total alcohol intake and BMD (González-Calvin et al., 2009). In another study of chronic alcoholic males, 34% of patients had osteoporosis, with low BMD in the femoral neck and lumbar spine compared to age-matched, healthy controls (Peris et al., 1995). In addition to increased risk of vertebral and non-vertebral fractures, chronic male alcoholics exhibited increased ALP and BALP, as well as significantly reduced vitamin D and BGP levels (Santori et al., 2008). Together, these studies point to impaired bone formation as the major mechanism by which alcohol addiction impacts bone. Although less well studied, chronic female alcohol users also exhibit impaired bone health. In a cross-sectional study of women aged 18–70 years, chronic alcohol users exhibited decreased femoral neck and lumbar spine BMD and had a higher prevalence of fractures compared to non-alcohol-abusing women, although fracture risk could not be attributed to alcohol use alone (Clark et al., 2003). In a study of both male and female chronic alcohol users, BMD directly correlated with total cholesterol and LDL-cholesterol, independent of liver function, providing evidence that alcohol addiction may mediate changes in lipid profiles that have separate effects on bone health. Together, these studies provide evidence that alcohol abuse negatively impacts bone health and is closely intertwined with sex, overall nutrition, and vitamin D deficiency.
Due to the nature of SUDs, it is difficult to parse out the effects of substance versus substance-induced neurological changes on bone. In vitro studies provide evidence that alcohol can directly impact the osteoblast:osteoclast balance. Treatment of BM cultures with alcohol or acetaldehyde directly impaired osteoblast differentiation, decreased osteogenesis, and promoted adipogenesis (Giuliani et al., 1999; Cui et al., 2011). Further, ethanol treatment increased bone resorption by osteoclasts in vitro (Cheung et al., 1995). In a rabbit model of alcoholism, increased triglyceride-bearing osteocytes and increased empty lacunae were observed, suggesting alcohol leads to impaired osteocyte function and may promote osteocyte apoptosis (Wang et al., 2003). Preclinical studies also point to a role for alcohol-induced changes in leptin signaling, resulting in impaired osteogenesis and increased adipogenesis (Wang et al., 2003; Otaka et al., 2007; Maurel et al., 2011). In alcoholism and other SUDs, it is likely that both direct effects of the substance itself and indirect effects (e.g., neurobiological impacts on bone turnover) are responsible for impaired bone health and will require further study to fully dissect.
The global incidence of spinal cord injury (SCI) is 10.4–83 cases/million/year. Bone loss below the level of the lesion is rapid and as high as 4% per month in trabecular bone and 2% per month in cortical bone (Wilmet et al., 1995; Szollar et al., 1997; Dauty et al., 2000). This bone loss persists for ∼2 years post-SCI, with peak loss at 3–5 months, resulting in increased risk of fracture and osteoporosis (Roberts et al., 1998; Maïmoun et al., 2006; Smith and Carroll, 2011). Post-SCI, bone resorption markers increase with a lack of concomitant increase in bone formation markers (Smith and Carroll, 2011; Thakkar et al., 2020). Numerous mechanisms have been implicated in SCI-related bone loss, including mechanical unloading from loss of motor function, as well as metabolic, endocrine, neural denervation, and vascular changes (Jiang et al., 2006b). Each of these can result in osteoblast:osteoclast imbalance, leading to bone loss, osteoporosis, and fragility fractures. In the absence of mechanical strain, osteocytes signal to reduce osteoblast activity, resulting in reduced bone formation (Jiang et al., 2006a,b). SCI also impacts the OPG-RANKL system and Wnt signaling, shifting the balance to bone resorption over bone formation (Maïmoun et al., 2006; Bonewald and Johnson, 2008). Altered vasoregulation due to injury impacts viability of oxygen and nutrients to bone, promoting osteoclast formation and bone resorption (Jiang et al., 2006a,b). Decreased innervation due to SCI may also affect availability of neuropeptides, such as vasoactive intestinal peptide (VIP) and calcitonin gene-related peptide (CGRP), which suppress bone resorptive activities through RANKL/OPG pathway (Yoon et al., 2004). Decreased PTH has also been reported 4–12 months post-SCI, which can lead to decreased vitamin D and subsequent impaired absorption of dietary calcium (Giangregorio and Blimkie, 2002; Jiang et al., 2006a,b). Thus, while disuse is considered to be the most impactful factor in post-SCI osteoporosis and associated fractures, it is clearly a more complex, multi-factorial process, and this should be considered when developing therapeutic strategies.
Traumatic brain injury (TBI) results in temporary to permanent neurological damage and dysfunction and is associated with increased mortality and morbidity. TBI is often referred to as the “silent epidemic,” and, while the incidence has been difficult to determine, reports suggest 69 million individuals suffer TBI from all causes annually (Dewan et al., 2019). Patients with TBI exhibit an elevated risk for fracture and reduced BMD. A recent study demonstrated that patients recovering from TBI had suboptimal BMD measurements that were low for their age and gender, with 18% of the participants meeting criteria for osteopenia measured at the radius and 51% meeting the criteria for osteopenia/osteoporosis measured at the tibia (Banham-Hall et al., 2013). Markers of bone turnover, including OCN, type I collagen, and PTH, were dysregulated during the early post-traumatic period, suggesting an imbalance between bone formation and resorption that occurred rapidly post-injury (Trentz et al., 2005). Recent preclinical studies suggest inflammatory stress on bone and BM following TBI leads to NFκB activation, which, in turn, induces osteoclastogenesis and bone resorption (Singleton et al., 2019). Pre-clinical studies from Mohan’s group using a repetitive mouse TBI model have shown reduced BMC, bone area, bone strength, and BMD in TBI mice accompanied by negative impacts on cortical structure and trabecular architecture (Yu et al., 2014a,b). Their work also demonstrated that mild TBI and bony effects were associated with decreased circulating IGF-1 levels (Yu et al., 2014a). Pituitary dysfunctions post-TBI are common, present in 25–70% of patients (Rosario et al., 2013). Deficiencies in pituitary hormones, including those that impact bone formation and contribute to peak bone mass (e.g., IGF-1), can result in negative effects on skeletal maintenance (Mohan et al., 2003; Mohan and Baylink, 2005; Xing et al., 2012). An additional contributor to low BMD risk in TBI patients is the use of anti-epileptic drugs, which induce the cytochrome P450 system and likely increase the conversion of vitamin D to its inactive forms, resulting in less biologically active vitamin D, decreased calcium absorption, hypocalcemia, increased PTH levels, and compensatory mobilization of calcium stores from bone (Smith et al., 2016). Polytrauma with TBI and concomitant fracture has been shown to result in higher functional deficits and mortality rates (Albrecht et al., 2019). Studies using two mouse models of TBI showed that neurological inflammation and brain damage was increased in animals with fracture and that this damage could be alleviated by blocking the inflammatory effects of fracture (Shultz et al., 2015; Yang et al., 2016). While mechanisms driving the effects of fracture on TBI outcomes are still being elucidated, these studies suggest that exacerbated neuroinflammation may be an important contributing factor.
Stroke is one of the most common neurologic problems, with hemiplegia being a common outcome that results in loss of voluntary movement, immobilization, and sensory disturbances. There is a robust and long literature examining the effects of stroke on bone health and fracture, notably in the hip (Peszczynski, 1957). The relative risk of fracture after hospitalization for stroke is greater than 7 times the rate of fracture in age- and sex-matched control populations (Kanis et al., 2001). BMD is lower following stroke, while low BMD may also be a prospective risk factor for stroke (Ramnemark et al., 1999; Jørgensen et al., 2000; Myint et al., 2007, 2014; Lee et al., 2013; Lam et al., 2016). Low BMD may increase stroke risk through altered estrogen/OPG signaling that leads to increased risk of intracerebral hemorrhage (Strand et al., 2007). Cibelli et al. showed that aseptic long bone fracture caused neuroinflammation and cognitive decline (Cibelli et al., 2010). Thus, determining the cause-and-effect relationship between stroke and fracture can be complex. A meta-analysis showed that, in subacute and chronic stroke, skeletal sites in the affected/paretic limbs had greater decline in bone quality and deleterious changes in bone geometry compared to unaffected/non-paretic limbs. This rate of change slowed as post-stroke duration increased, with the greatest changes occurring in the first few months post-stroke. A strong relationship between bone density/strength index and muscle strength/mass was also noted, demonstrating the importance of muscle-bone interactions and how they may act as a functional unit, as proposed by Schoenau (2005). These findings suggest muscle strength training and early intervention are key to minimizing negative bone effects of stroke (Borschmann et al., 2018; Yang et al., 2020). To this end, a mix of resistance, aerobic, and dynamic loading exercises resulted in better bone outcomes in the hip and tibia on the affected side in chronic stroke patients (Pang et al., 2005, 2006).
Muscle imbalance may not be the only mechanism by which stroke affects bone health. Lower bone turnover markers have been noted in serum from stroke patients, suggesting dysregulated remodeling at the bone multicellular unit (Sato et al., 2000). Stroke patients also have higher energy expenditure, with this interruption of energy homeostasis potentially negatively impacting the skeleton during bone remodeling (Detrembleur et al., 2003; Lee et al., 2007; Driessler and Baldock, 2010). Additionally, reduced vitamin D levels, degree of recovery, increased fall risk, and use of anticoagulants may increase bone loss post-stroke (Jørgensen et al., 2000; Smith and Carroll, 2011; Batchelor et al., 2012; Signorelli et al., 2019). Thus, increased bone screening measures are needed in stroke patients, as screening may currently be infrequent (Kapoor et al., 2019). Osteoporosis treatments, such as bisphosphonates, may be beneficial for preserving BMD post-stroke, but there is little evidence to date (Hsieh et al., 2020). More studies are needed to dissect the molecular mechanisms at the intersection of bone and stroke to guide treatment and screening recommendations.
Chronic fatigue syndrome (CFS) is a complex neurological disorder associated with persistent, overwhelming fatigue that affects > 3% of the population in Western countries and is more prevalent in women (Griffith and Zarrouf, 2008). Diagnostic criteria include severe, persistent fatigue for at least 6 months, exclusion of other medical disorders, and observation of at least four minor symptoms, including impaired memory, nausea, extreme post-exertion fatigue, headaches, muscle pain, sore throat, and poor sleep (Committee on the Diagnostic Criteria for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome et al., 2015). There remains a lack of treatment and diagnostics tools for CFS, although glucocorticoids have been used (McKenzie et al., 2000). Bone loss and increased fracture risk have been reported in individuals with CFS, independent of glucocorticoid use. Hoskin et al. found that hip BMD was approximately 7% lower in women with CFS (Hoskin et al., 2006). A prospective study reported a 1.16-fold increased risk of fracture in the CFS cohort without osteoporosis compared to the non-CFS cohort (Chen C.-S. et al., 2014). However, no mechanistic insights were provided in these studies. Other studies have reported that IGF-1 levels are altered in CFS patients (Buchwald et al., 1996; Berwaerts et al., 1998; Cleare et al., 2000; Nijs et al., 2003). IGF-1 is essential for osteoblast proliferation, thus impaired secretion could lead to bone loss. Studies are needed to further characterize those CFS patients with low serum IGF-1 to determine if these subgroups have increased fracture risk compared to CFS patients with normal or high levels of IGF-1. High prevalence of mycoplasma infections has also been reported in CFS patients (Choppa et al., 1998; Nasralla et al., 1999), which can stimulate macrophage activation and release of pro-inflammatory cytokines that enhance osteoclast activity. M. fermentans has been shown to produce 2-kDa macrophage-activating lipopeptide (MALP-2), which stimulates macrophages and bone resorption in a dose-dependent manner and is increased with CFS (Piec et al., 1999). Thus, chronic fatigue may induce bone loss or increase fracture risk through increased inflammation and/or dysregulation of growth factors.
Chronic sleep deprivation is becoming a widespread problem, with at least one-third of adults reporting less than 6.5 h of sleep per night compared to about 9 h of average sleep in the early 1900s (Bonnet and Arand, 1995; Spiegel et al., 1999; Specker et al., 2007). Sleep deprivation can negatively impact health by decreasing cardiovascular health and increasing risk for development of diabetes and obesity. Since circadian rhythm has been extensively shown to regulate bone, sleep disorders may also affect bone homeostasis (Swanson et al., 2015, 2018; Song et al., 2018). Spiegel et al. found that cortisol concentrations were higher in the evening with sleep deprivation, a known risk factor for bone loss (Spiegel et al., 1999). Three weeks of sleep restriction has been shown to cause a decline in N-terminal propeptide of type 1 procollagen (P1NP), a bone formation marker, with stable resorption markers (e.g., CTX), suggesting an uncoupling of bone remodeling. Importantly, this observed decrease in P1NP was not rescued with ongoing exposure, suggesting BMD could be lowered over time when paired with poor sleeping habits (Swanson et al., 2017, 2019). Sleep-deprived women had lower cortical BMD compared to women with normal sleeping habits, and sleep quality, sleep latency, and sleep timing, but not sleep duration, were associated with osteopenia and sarcopenia in middle-aged individuals (Specker et al., 2007; Lucassen et al., 2017). Shiftwork may also lower BMD, providing evidence that circadian rhythm, and not just amount of sleep, can significantly impact bone (Quevedo and Zuniga, 2010; Kim et al., 2013).
Obstructive sleep apnea (OSAS) has also been shown to lower BMD and vitamin D levels, possibly due to increased hypoxia, which can cause oxidative stress, SNS activity, endothelial dysfunction, and stimulation of osteoclasts (Shahar et al., 2001; Arnett, 2010; Uzkeser et al., 2013; Terzi and Yılmaz, 2016; Eimar et al., 2017). Further, OSAS increases systemic inflammation, with resultant increased IL-6, TNF-α, and C-reactive protein (CRP) production, known risk factors for bone loss (Tomiyama et al., 2008). Fracture can also affect sleep disturbance, likely due to effects on emotional well-being (Shulman et al., 2015). Likewise, vertebral fractures have been associated with poor sleep, and those with osteoporosis were 67% more likely to report decreased sleep. These studies suggest a negative feedback loop may be occurring between reduced sleep and poor bone health (Silverman, 1992; Foley et al., 2004). Interestingly, Cikrikcioglu et al. found that women with restless legs syndrome had increased lumbar BMD, despite lower vitamin D levels, possibly due to unconsciously performing exercise (Cikrikcioglu et al., 2016).
Thus, there are many mechanisms that may link disrupted sleep to bone loss, including increased systemic inflammation, hypoxia, insulin resistance, and oxidative stress, as well as altered circadian rhythm, decreased growth hormone secretion, and physical inactivity. Large-scale, prospective studies are needed to elucidate if sleep loss and/or OSAS are independent risk factors for osteoporosis (Schiza et al., 2013). Further, other sleep disorders, such as narcolepsy or somnambulism, need to be studied in more detail for their potential effects on bone.
Vertigo is a symptom in which someone feels like they are moving or surrounded by moving objects when they are not. This can be associated with nausea, sweating, vomiting, hearing loss, and/or difficulties in walking and balance. As bone mediates hearing and movement and vestibular changes alter SNS output, vertigo may be linked to alterations in bone (Radaei and Gharibzadeh, 2013; Mendy et al., 2014; Yates et al., 2014). Although there are many types of vertigo, one of the most common types, benign paroxysmal positional vertigo (BPPV), has been studied in the context of BMD and vitamin D changes. BPPV is a vestibular dysfunction that is typically unilateral and characterized by short, intense episodes of vertigo. BPPV represents 20–30% of dizziness diagnoses, with no current consensus on its etiology and pathogenesis (Grill et al., 2014; Bazoni et al., 2020). However, the incidence of BPPV increases with age and is believed to involve abnormal stimulation of the cupula by otoliths in any of the three semicircular canals upon changes in head position (Furman and Cass, 1999).
Some studies have noted that reductions in bone mass correlate with both occurrence and recurrence of BPPV (Vibert et al., 2003; Jang and Kang, 2009; Jeong et al., 2009; Kim et al., 2017; Wu et al., 2018; Wang et al., 2020). A meta-analysis found significantly higher incidence of osteoporosis and osteopenia in BPPV patients (He et al., 2019). In addition, BPPV has been associated with vitamin D deficiency, which can affect both bone and the inner ear (Jeong et al., 2013). BPPV frequently occurs in females over 50 years old, suggesting estrogen loss may be involved in onset (Vibert et al., 2003). Incidence of BPPV recurrence was significantly higher in post-menopausal women with osteoporosis (56.3%) than those with normal BMD (16.1%), and frequency of recurrence increased with decreasing BMD (Yamanaka et al., 2013). Estrogen deficiency can cause low bone mass by altering calcium metabolism, inducing a calcium insufficiency (Riggs et al., 1998). Calcium is important in the synthesis and absorption of otoconia and otoliths, which mature by absorbing calcium. Thus, if there is a shortage of calcium with reduced bone mass, incomplete maturation of otoliths could occur, leading to BPPV. The otolith also acts as a calcium reservoir to maintain calcium homeostasis when necessary, such as in postmenopausal women with osteoporosis (Campos et al., 1999). Thus, altered calcium metabolism caused by decreased estrogen secretion may be a pathophysiological mechanism shared by both BPPV and osteoporosis. Additionally, as electrical signals from the inner ear are relayed to the CNS to maintain body balance and vestibular dysfunction alters SNS output, vertigo may affect bone remodeling and bone mass, as well as fall risk, leading to increased fracture risk. In support of this, fracture risk has been shown to be increased with BPPV (Nakada et al., 2018). Likewise, the reduction in bone mass caused by vertigo may further alter calcium metabolism, increasing vertigo incidence and creating a negative feedback loop. Osteoporosis is, therefore, a risk factor for BPPV recurrence and prognosis may be clinically predicted by BMD reduction, while BPPV itself may increase osteoporosis-related fracture incidence.
It is becomingly increasingly clear that bone is a dynamic organ with complex signaling responses throughout the body. Recent studies have led to a better understanding of the brain-bone axis, which regulates skeletal metabolism, hormonal response, and sensory innervation. In this review, we discussed how different neurological disorders impact bone health and how bone itself can affect cognitive function and development. Across many subcategories of neurological disease, there is direct pre-clinical and clinical evidence that deficits in the brain can cause deficits in the bone, including osteopenia/osteoporosis and increased fracture risk. This is due to a complex mixture of neuronal (e.g., SNS/PNS dysregulation), psychological (e.g., HPA/stress), mechanical (e.g., muscle-bone interactions), cellular (e.g., macrophage, neuron, osteoblast, osteoclast), molecular (e.g., IGF-1, IL-6, PTH, Wnt), lifestyle (e.g., falls, malnutrition, physical inactivity, vitamin D deficiency), and treatment (e.g., AED, glucocorticoid, SSRI) factors. Current treatments for osteoporosis, including bisphosphonates, estrogen replacement therapy, and anabolic therapies (e.g., teriparatide, romosozumab) may be beneficial for certain subsets of patients with neurological diseases (Figure 1). A better understanding of the mechanisms that lead to bone loss in neurological disorders is of clinical importance and may better inform treatment approaches, encourage lifestyle change, and aid in development of novel osteoporosis therapies. Similarly, a better understanding of how bone regulates the brain will provide new insights into the etiology and development of neurological disorders. Clinicians should consider taking a whole-body approach when treating neurological patients and ensure that treatments directed at the brain (e.g., glucocorticoids) are not causing deleterious effects elsewhere in the body, such as in the skeleton, which may concurrently signal through negative feedback loops to impact disease severity.
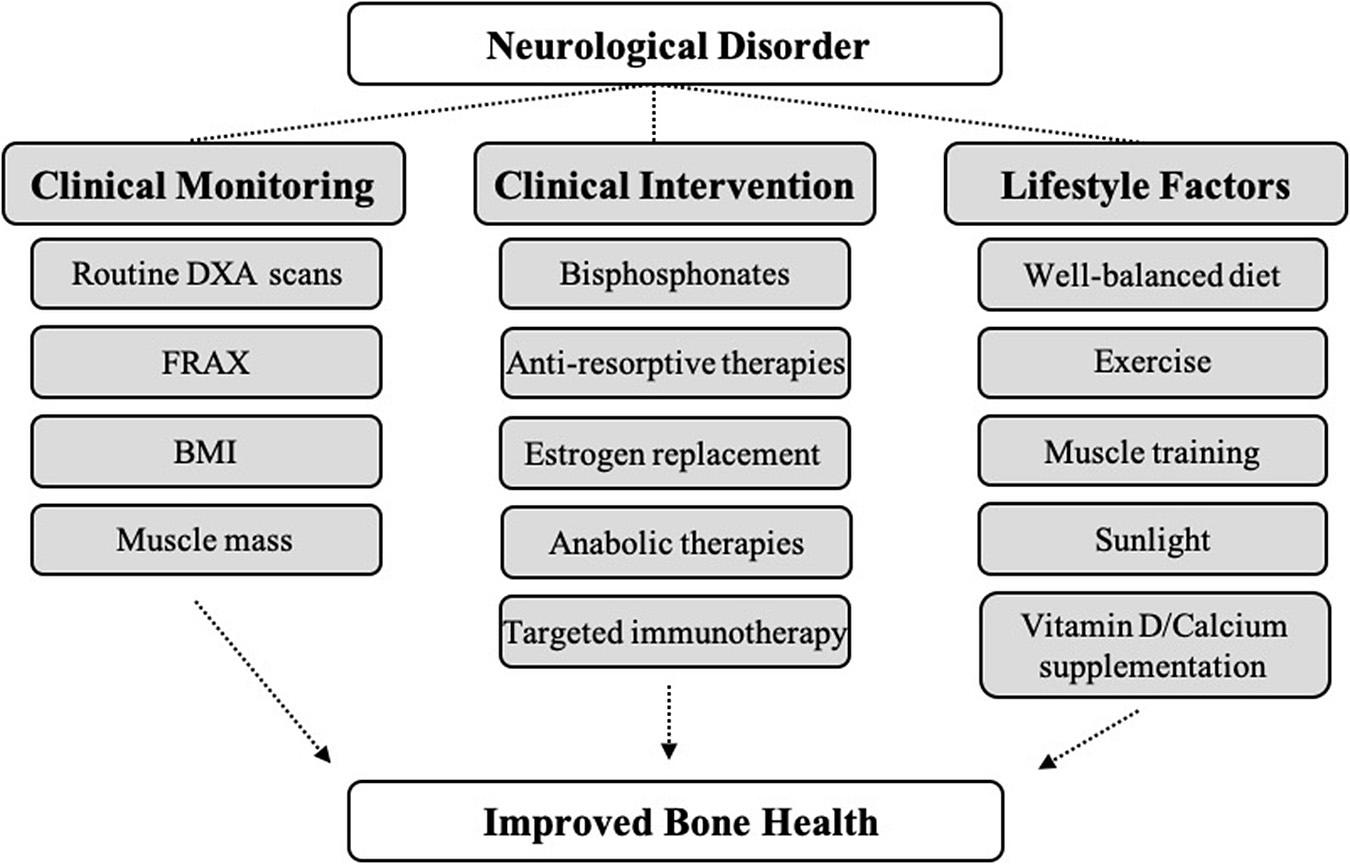
Figure 1. Proposed methods to improve bone health in patients with neurological disorders. There is considerable clinical evidence demonstrating negative impacts of neurological diseases on bone across many disease categories. Patients with neurological diseases exhibit decreased BMD, as well as increased risk for osteoporosis and fracture. Careful clinical monitoring, clinical intervention, and positive lifestyle changes may lead to better bone outcomes in certain subsets of neurological disease patients.
RK and SS: conception and design, drafting, and revising of the manuscript. AL: drafting and revising of the manuscript. All authors read and approved the final manuscript.
Development of this manuscript was supported by the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs (VA Merit Award to AL, BX000333).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aβ, Amyloid-β; ACTH, Adrenocorticotropic Hormone; AD, Alzheimer’s Disease; AED, Anti-Epileptic Drug; AKT, Protein Kinase B; ALP, Alkaline Phosphatase; ALS, Amyotrophic Lateral Sclerosis; ApoE, Apolipoprotein E; App, Amyloid Precursor Protein; APT, Antipsychotic Therapy; ASD, Autism Spectrum Disorder; BALP, Bone-Specific Alkaline Phosphatase; BBB, Blood-Brain Barrier; BGP, Bone GLA Protein; BM, Bone Marrow; BMC, Bone Mineral Content; BMD, Bone Mineral Density; CBZ, Carbamazepine; CFS, Chronic Fatigue Syndrome; CGRP, Calcitonin Gene-Related Peptide; CNS, Central Nervous System; CP, Cerebral Palsy; CRP, C-Reactive Protein; CSF, Cerebrospinal Fluid; CTX, C-Terminal Telopeptide Of Type I Collagen; DEXA, Dual-Energy X-Ray Absorptiometry; DMD, Duchenne Muscular Dystrophy; DRN, Dorsal Raphe Nucleus; EBV, Epstein-Barr Virus; EDSS, Expanded Disability Status Scale; EIAED, Enzyme-Inducing Anti-Epileptic Drug; ERK, Extracellular Signal-Regulated Kinase; FDRA, Friedreich Ataxia; FRAX, Fracture Risk Assessment; GMFCS, Gross Motor Function Classification System; GPR158, G Protein-Coupled Receptor 158; HAMA, Hamilton Anxiety Rating Scale; HPA, Hypothalamic-Pituitary-Adrenal; IκB-α, Nuclear Factor Of Kappa Light Polypeptide Gene Enhancer In B-Cells Inhibitor, Alpha; IGF-1, Insulin-Like Growth Factor-1; IL, Interleukin; LDL, Low-Density Lipoprotein; LRP5/6, Lipoprotein-Receptor-Related Protein-5/6; MALP-2, 2-kDa Macrophage-Activating Protein; mBMD, Metacarpal Bone Mineral Density; MCI, Mild Cognitive Impairment; MDD, Major Depressive Disorder; MG, Myasthenia Gravis; MJD, Machado-Joseph Disease; MS, Multiple Sclerosis; MuSK, Muscle-Specific Tyrosine Kinase; NEIAED, Non-Enzyme-Inducing Antiepileptic Drug; NF-κB, Nuclear Factor Kappa B; NFT, Neurofibrillary Tangle; NMD, Neuromuscular Dystrophy; OCN, Osteocalcin; OPG, Osteoprotegerin; OPN, Osteopontin; OSAS, Obstructive Sleep Apnea; OSX, Osterix; P1NP, N-Terminal Propeptide Of Type I Collagen; PD, Parkinson’s Disease; PNS, Parasympathetic Nervous System; PTH, Parathyroid Hormone; PTSD, Post-Traumatic Stress Disorder; RANK, Receptor Activator of Nuclear Factor Kappa B; RANKL, Receptor Activator of Nuclear Factor Kappa B Ligand; SAD, Substance Use Disorder; SAE, Sepsis-Associated Encephalopathy; SCI, Spinal Cord Injury; SMA, Spinal Muscular Atrophy; SMN, Survival Motor Neuron; SNS, Sympathetic Nervous System; SOST, Sclerostin; SSRI, Selective Serotonin Reuptake Inhibitor; SUD, Substance Use Disorder; TBI, Traumatic Brain Injury; TBS, Trabecular Bone Score; TGF-β2, Transforming Growth Factor Beta 2; TNF-α, Tumor Necrosis Factor Alpha; TREM2, Triggering Receptor Expressed On Myeloid Cells-2; VIP, Vasoactive Intestinal Peptide; Wnt, Wingless-Type Murine-Mammary-Tumor Virus Integration Site.
Aharon-Peretz, J., Harel, T., Revach, M., and Ben-Haim, S. A. (1992). Increased sympathetic and decreased parasympathetic cardiac innervation in patients with Alzheimer’s disease. Arch. Neurol. 49, 919–922. doi: 10.1001/archneur.1992.00530330041013
Albrecht, J. S., Al Kibria, G., Gruber-Baldini, A., and Magaziner, J. (2019). Risk of mortality in individuals with hip fracture and traumatic brain injury: TBI and mortality in individuals with hip fracture. J. Am. Geriatr. Soc. 67, 124–127. doi: 10.1111/jgs.15661
Alesci, S., Martinez, P. E., Kelkar, S., Ilias, I., Ronsaville, D. S., Listwak, S. J., et al. (2005). Major depression is associated with significant diurnal elevations in plasma interleukin-6 levels, a shift of its circadian rhythm, and loss of physiological complexity in its secretion: clinical implications. J. Clin. Endocrinol. Metab. 90, 2522–2530. doi: 10.1210/jc.2004-1667
Alexander, W. (2012). Pharmacotherapy for post-traumatic stress disorder in combat veterans. P T 37, 32–38.
Altıntaş, A., Saruhan-Direskeneli, G., Benbir, G., Demir, M., and Purisa, S. (2009). The role of osteopontin: a shared pathway in the pathogenesis of multiple sclerosis and osteoporosis? J. Neurol. Sci. 276, 41–44. doi: 10.1016/j.jns.2008.08.031
Alvarez Zaragoza, C., Vasquez Garibay, E. M., García Contreras, A. A., Larrosa Haro, A., Romero Velarde, E., Rea Rosas, A., et al. (2018). Bone mineral density and nutritional status in children with quadriplegic cerebral palsy. Arch. Osteoporos. 13:17.
Alzheimer’s Association (2020). 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 16, 391–460. doi: 10.1002/alz.12068
Ambrose, M., and Gatti, R. A. (2013). Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood 121, 4036–4045. doi: 10.1182/blood-2012-09-456897
Amini, A., Namvarpour, Z., Namvarpour, M., and Raoofi, A. (2020). Risperidone accelerates bone loss in rats with autistic-like deficits induced by maternal lipopolysaccharides exposure. Life Sci. 258:118197. doi: 10.1016/j.lfs.2020.118197
Annexstad, E. J., Bollerslev, J., Westvik, J., Myhre, A. G., Godang, K., Holm, I., et al. (2019). The role of delayed bone age in the evaluation of stature and bone health in glucocorticoid treated patients with Duchenne muscular dystrophy. Int. J. Pediatr. Endocrinol. 2019:4.
Aparicio, L. F., Jurkovic, M., and DeLullo, J. (2002). Decreased bone density in ambulatory patients with duchenne muscular dystrophy. J. Pediatr. Orthop. 22, 179–181. doi: 10.1097/01241398-200203000-00008
Arbouw, M. E. L., Movig, K. L. L., van Staa, T. P., Egberts, A. C. G., Souverein, P. C., and de Vries, F. (2011). Dopaminergic drugs and the risk of hip or femur fracture: a population-based case-control study. Osteoporos. Int. 22, 2197–2204. doi: 10.1007/s00198-010-1455-3
Arnett, T. R. (2010). Acidosis, hypoxia and bone. Arch. Biochem. Biophys. 503, 103–109. doi: 10.1016/j.abb.2010.07.021
Atteritano, M., Lasco, A., Mazzaferro, S., Macrì, I., Catalano, A., Santangelo, A., et al. (2013). Bone mineral density, quantitative ultrasound parameters and bone metabolism in postmenopausal women with depression. Intern. Emerg. Med. 8, 485–491. doi: 10.1007/s11739-011-0628-1
Banham-Hall, N., Kothwal, K., Pipkin, J., Bentley, J., and Dickens, G. L. (2013). Prevalence of low bone mineral density in inpatients with traumatic brain injury receiving neurobehavioural rehabilitation: a postoperative, observational study. Physiotherapy 99, 328–334. doi: 10.1016/j.physio.2012.12.009
Bartl, R., and Bartl, C. (2019). “Epidemiology of osteoporotic fractures,” in The Osteoporosis Manual: Prevention, Diagnosis and Management [Internet], eds R. Bartl and C. Bartl (Cham: Springer International Publishing).
Batchelor, F. A., Mackintosh, S. F., Said, C. M., and Hill, K. D. (2012). Falls after stroke. Int. J. Stroke 7, 482–490. doi: 10.1111/j.1747-4949.2012.00796.x
Bazoni, J. A., Ciquinato, D. S. A., Marquez, A. D. S., Costa, V. D. S. P., Marchiori, G. D. M., and Marchiori, L. L. D. M. (2020). Hypovitaminosis D, low bone mineral density, and diabetes mellitus as probable risk factors for benign paroxysmal positional vertigo in the elderly. Int. Arch. Otorhinolaryngol. 24, e272–e277.
Beghi, E., Giussani, G., and Sander, J. W. (2015). The natural history and prognosis of epilepsy. Epileptic Disord. 17, 243–253. doi: 10.1684/epd.2015.0751
Bell, J. M., Shields, M. D., Watters, J., Hamilton, A., Beringer, T., Elliott, M., et al. (2017). Interventions to prevent and treat corticosteroid-induced osteoporosis and prevent osteoporotic fractures in Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 24:CD010899.
Bemiller, S. M., McCray, T. J., Allan, K., Formica, S. V., Xu, G., Wilson, G., et al. (2017). TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 12:74.
Berger, J. M., Singh, P., Khrimian, L., Morgan, D. A., Chowdhury, S., Arteaga-Solis, E., et al. (2019). Mediation of the acute stress response by the skeleton. Cell Metab. 30, 890–902.e8.
Berwaerts, J., Moorkens, G., and Abs, R. (1998). Secretion of growth hormone in patients with chronic fatigue syndrome. Growth Horm. IGF Res. 8, 127–129. doi: 10.1016/s1096-6374(98)80036-1
Bezza, A., Ouzzif, Z., Naji, H., Achemlal, L., Mounach, A., Nouijai, M., et al. (2008). Prevalence and risk factors of osteoporosis in patients with Parkinson’s disease. Rheumatol. Int. 28, 1205–1209.
Bhandari, R., Paliwal, J. K., and Kuhad, A. (2020). Neuropsychopathology of autism spectrum disorder: complex interplay of genetic, epigenetic, and environmental factors. Adv. Neurobiol. 24, 97–141. doi: 10.1007/978-3-030-30402-7_4
Bianchi, M. L., Mazzanti, A., Galbiati, E., Saraifoger, S., Dubini, A., Cornelio, F., et al. (2003). Bone mineral density and bone metabolism in Duchenne muscular dystrophy. Osteoporos. Int. 14, 761–767. doi: 10.1007/s00198-003-1443-y
Birge, S. J., Morrow-Howell, N., and Proctor, E. K. (1994). Hip fracture. Clin. Geriatr. Med. 10, 589–609.
Bisson, E. J., Finlayson, M. L., Ekuma, O., Leslie, W. D., and Marrie, R. A. (2019). Multiple sclerosis is associated with low bone mineral density and osteoporosis. Neurol. Clin. Pract. 9, 391–399.
Bonewald, L. F., and Johnson, M. L. (2008). Osteocytes, mechanosensing and Wnt signaling. Bone 42, 606–615. doi: 10.1016/j.bone.2007.12.224
Bonnet, M. H., and Arand, D. L. (1995). We are chronically sleep deprived. Sleep 18, 908–911. doi: 10.1093/sleep/18.10.908
Bonnet, N., Gadois, C., McCloskey, E., Lemineur, G., Lespessailles, E., Courteix, D., et al. (2007). Protective effect of beta blockers in postmenopausal women: influence on fractures, bone density, micro and macroarchitecture. Bone 40, 1209–1216. doi: 10.1016/j.bone.2007.01.006
Borschmann, K., Iuliano, S., Ghasem-Zadeh, A., Churilov, L., Pang, M. Y. C., and Bernhardt, J. (2018). Upright activity and higher motor function may preserve bone mineral density within 6 months of stroke: a longitudinal study. Arch. Osteoporos. 13:5.
Brandenburg, J. E., Fogarty, M. J., and Sieck, G. C. A. (2019). Critical evaluation of current concepts in cerebral palsy. Physiology 34, 216–229. doi: 10.1152/physiol.00054.2018
Braz, N. F. T., Rocha, N. P., Vieira, ÉL. M., Gomez, R. S., Barbosa, I. G., Malheiro, O. B., et al. (2017). Negative impact of high cumulative glucocorticoid dose on bone metabolism of patients with myasthenia gravis. Neurol. Sci. 38, 1405–1413. doi: 10.1007/s10072-017-2964-z
Brinton, D. L., Simpson, A. N., Fominaya, C. E., and LaRue, A. C. (2019). Impact of selective serotonin reuptake inhibitors in the veteran population: 10-year risk outcomes. J. Comp. Eff. Res. 8, 431–440. doi: 10.2217/cer-2018-0085
Brownbill, R. A., and Ilich, J. Z. (2004). Cognitive function in relation with bone mass and nutrition: cross-sectional association in postmenopausal women. BMC Womens Health. 4:2. doi: 10.1186/1472-6874-4-2
Brunoni, A. R., Kemp, A. H., Dantas, E. M., Goulart, A. C., Nunes, M. A., Boggio, P. S., et al. (2013). Heart rate variability is a trait marker of major depressive disorder: evidence from the sertraline vs. electric current therapy to treat depression clinical study. Int. J. Neuropsychopharmacol. 16, 1937–1949. doi: 10.1017/s1461145713000497
Buchwald, D., Umali, J., and Stene, M. (1996). Insulin-like growth factor-I (somatomedin C) levels in chronic fatigue syndrome and fibromyalgia. J. Rheumatol. 23, 739–742.
Buehring, B., Viswanathan, R., Binkley, N., and Busse, W. (2013). Glucocorticoid-induced osteoporosis: an update on effects and management. J. Allergy Clin. Immunol. 132, 1019–1030. doi: 10.1016/j.jaci.2013.08.040
Calarge, C. A., and Schlechte, J. A. (2017). Bone mass in boys with autism spectrum disorder. J. Autism Dev. Disord. 47, 1749–1755. doi: 10.1007/s10803-017-3097-1
Calarge, C. A., Butcher, B. D., Burns, T. L., Coryell, W. H., Schlechte, J. A., and Zemel, B. S. (2014). Major depressive disorder and bone mass in adolescents and young adults. J. Bone Miner Res. 29, 2230–2237. doi: 10.1002/jbmr.2249
Campos, A., Crespo, P. V., García, J. M., Sánchez-Quevedo, M. C., and Ciges, M. (1999). The crystalline pattern of calcium in different topographical regions of the otoconial membrane. An electron probe and spectroscopic diffraction study. Acta Otolaryngol. 119, 203–206. doi: 10.1080/00016489950181675
Canalis, E. (2009). Growth factor control of bone mass. J. Cell Biochem. 108, 769–777. doi: 10.1002/jcb.22322
Catalano, A., Bellone, F., Morabito, N., and Corica, F. (2020a). Sclerostin and vascular pathophysiology. Int. J. Mol. Sci. 21:4779. doi: 10.3390/ijms21134779
Catalano, A., Martino, G., Bellone, F., Gaudio, A., Lasco, C., Langher, V., et al. (2018). Anxiety levels predict fracture risk in postmenopausal women assessed for osteoporosis. Menopause 25, 1110–1115. doi: 10.1097/gme.0000000000001123
Catalano, A., Sardella, A., Bellone, F., Lasco, C. G., Martino, G., and Morabito, N. (2020b). Executive functions predict fracture risk in postmenopausal women assessed for osteoporosis. Aging Clin. Exp. Res. 32, 2251–2257. doi: 10.1007/s40520-019-01426-w
Catalano, A., Vita, G. L., Russo, M., Vita, G., Lasco, A., Morabito, N., et al. (2016). Effects of teriparatide on bone mineral density and quality of life in Duchenne muscular dystrophy related osteoporosis: a case report. Osteoporos. Int. 27, 3655–3659. doi: 10.1007/s00198-016-3761-x
Cauley, J. A., Cawthon, P. M., Peters, K. E., Cummings, S. R., Ensrud, K. E., Bauer, D. C., et al. (2016). Risk factors for hip fracture in older men: the osteoporotic fractures in men study (MrOS). J. Bone Miner Res. 31, 1810–1819. doi: 10.1002/jbmr.2836
Cecconi, M., Evans, L., Levy, M., and Rhodes, A. (2018). Sepsis and septic shock. Lancet 392, 75–87.
Chang, K.-H., Chung, C.-J., Lin, C.-L., Sung, F.-C., Wu, T.-N., and Kao, C.-H. (2014). Increased risk of dementia in patients with osteoporosis: a population-based retrospective cohort analysis. AGE 36, 967–975. doi: 10.1007/s11357-013-9608-x
Chen, C.-S., Lin, W.-M., Yang, T.-Y., Chen, H.-J., Kuo, C.-N., and Kao, C.-H. (2014). Chronic fatigue syndrome is associated with the risk of fracture: a nationwide cohort study. QJM 107, 635–641. doi: 10.1093/qjmed/hcu037
Chen, X., Wang, K., Wang, Z., Gan, C., He, P., Liang, Y., et al. (2014). Effects of lead and cadmium co-exposure on bone mineral density in a Chinese population. Bone. 63, 76–80. doi: 10.1016/j.bone.2014.02.017
Cheng, S. W., Ko, C. H., and Lee, C. Y. (2016). The effect of anticonvulsant use on bone mineral density in non-ambulatory children with cerebral palsy. Hong Kong Med. J. Xianggang Yi Xue Za Zhi. 22, 242–248.
Cheung, R. C., Gray, C., Boyde, A., and Jones, S. J. (1995). Effects of ethanol on bone cells in vitro resulting in increased resorption. Bone 16, 143–147. doi: 10.1016/8756-3282(95)80025-l
Cho, E.-H., Cho, K.-H., Lee, H. A., and Kim, S.-W. (2013). High serum osteopontin levels are associated with low bone mineral density in postmenopausal women. J. Korean Med. Sci. 28:1496. doi: 10.3346/jkms.2013.28.10.1496
Choppa, P. C., Vojdani, A., Tagle, C., Andrin, R., and Magtoto, L. (1998). Multiplex PCR for the detection of Mycoplasma fermentans, M. hominis and M. penetrans in cell cultures and blood samples of patients with chronic fatigue syndrome. Mol. Cell Probes. 12, 301–308. doi: 10.1006/mcpr.1998.0186
Cibelli, M., Fidalgo, A. R., Terrando, N., Ma, D., Monaco, C., Feldmann, M., et al. (2010). Role of interleukin-1beta in postoperative cognitive dysfunction. Ann. Neurol. 68, 360–368.
Cikrikcioglu, M. A., Sekin, Y., Halac, G., Kilic, E., Kesgin, S., Aydin, S., et al. (2016). Reduced bone resorption and increased bone mineral density in women with restless legs syndrome. Neurology 86, 1235–1241. doi: 10.1212/wnl.0000000000002521
Cizza, G., Primma, S., Coyle, M., Gourgiotis, L., and Csako, G. (2010). Depression and osteoporosis: a research synthesis with meta-analysis. Horm. Metab. Res. 42, 467–482. doi: 10.1055/s-0030-1252020
Cizza, G., Ronsaville, D. S., Kleitz, H., Eskandari, F., Mistry, S., Torvik, S., et al. (2012). Clinical subtypes of depression are associated with specific metabolic parameters and circadian endocrine profiles in women: the power study. PLoS One 7:e28912. doi: 10.1371/journal.pone.0028912
Clark, M. K., Sowers, M. R., Dekordi, F., and Nichols, S. (2003). Bone mineral density and fractures among alcohol-dependent women in treatment and in recovery. Osteoporos. Int. 14, 396–403. doi: 10.1007/s00198-003-1387-2
Cleare, A. J., Sookdeo, S. S., Jones, J., O’Keane, V., and Miell, J. P. (2000). Integrity of the growth hormone/insulin-like growth factor system is maintained in patients with chronic fatigue syndrome. J. Clin. Endocrinol. Metab. 85, 1433–1439. doi: 10.1210/jc.85.4.1433
Comi, C., Carecchio, M., Chiocchetti, A., Nicola, S., Galimberti, D., Fenoglio, C., et al. (2010). Osteopontin is increased in the cerebrospinal fluid of patients with Alzheimer’s disease and its levels correlate with cognitive decline. J. Alzheimers Dis. 19, 1143–1148. doi: 10.3233/jad-2010-1309
Committee on the Diagnostic Criteria for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome, Board on the Health of Select Populations, and Institute of Medicine (2015). Current Case Definitions and Diagnostic Criteria, Terminology, and Symptom Constructs and Clusters. Washington, DC: National Academies Press (US).
Cornish, J., Callon, K. E., Bava, U., Lin, C., Naot, D., Hill, B. L., et al. (2002). Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J. Endocrinol. 175, 405–415. doi: 10.1677/joe.0.1750405
Coskun Benlidayi, I., Basaran, S., Evlice, A., Erdem, M., and Demirkiran, M. (2015). Prevalence and risk factors of low bone mineral density in patients with multiple sclerosis. Acta Clin. Belg. 70, 188–192. doi: 10.1179/2295333715y.0000000002
Cosman, F., Nieves, J., Komar, L., Ferrer, G., Herbert, J., Formica, C., et al. (1998). Fracture history and bone loss in patients with MS. Neurology 51, 1161–1165. doi: 10.1212/wnl.51.4.1161
Crane, J. L., Zhao, L., Frye, J. S., Xian, L., Qiu, T., and Cao, X. I. G. F. - (2013). 1 Signaling is essential for differentiation of mesenchymal stem cells for peak bone mass. Bone Res. 1, 186–194. doi: 10.4248/br201302007
Cui, S., Xiong, F., Hong, Y., Jung, J.-U., Li, X.-S., Liu, J.-Z., et al. (2011). APPswe/Aβ regulation of osteoclast activation and RAGE expression in an age-dependent manner. J. Bone Miner Res. 26, 1084–1098. doi: 10.1002/jbmr.299
Daniel, S. K., Lansang, M. C., and Okun, M. S. (2012). Bone mineral density (BMD) in male patients with Parkinson’s disease. Int. J. Neurosci. 122, 523–527. doi: 10.3109/00207454.2012.685530
Dauty, M., Perrouin Verbe, B., Maugars, Y., Dubois, C., and Mathe, J. F. (2000). Supralesional and sublesional bone mineral density in spinal cord-injured patients. Bone 27, 305–309. doi: 10.1016/s8756-3282(00)00326-4
Davis, F. M., Schaller, M. A., Dendekker, A., Joshi, A. D., Kimball, A. S., Evanoff, H., et al. (2019). Sepsis induces prolonged epigenetic modifications in bone marrow and peripheral macrophages impairing inflammation and wound healing. Arterioscler Thromb. Vasc. Biol. 39, 2353–2366. doi: 10.1161/atvbaha.119.312754
De Pablo-Fernández, E., Breen, D. P., Bouloux, P. M., Barker, R. A., Foltynie, T., and Warner, T. T. (2017). Neuroendocrine abnormalities in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 88, 176–185.
de Vilhena Toledo, M. A., and Junqueira, L. F. (2010). Cardiac autonomic modulation and cognitive status in Alzheimer’s disease. Clin. Auton. Res. 20, 11–17. doi: 10.1007/s10286-009-0035-0
Delatycki, M. B., Holian, A., Corben, L., Rawicki, H. B., Blackburn, C., Hoare, B., et al. (2005). Surgery for equinovarus deformity in Friedreich’s ataxia improves mobility and independence. Clin. Orthop. 430, 138–141. doi: 10.1097/01.blo.0000150339.74041.0e
Dengler-Crish, C. M., Ball, H. C., Lin, L., Novak, K. M., and Cooper, L. N. (2018). Evidence of Wnt/β-catenin alterations in brain and bone of a tauopathy mouse model of Alzheimer’s disease. Neurobiol. Aging 67, 148–158. doi: 10.1016/j.neurobiolaging.2018.03.021
Dengler-Crish, C. M., Smith, M. A., and Wilson, G. N. (2016). Early evidence of low bone density and decreased serotonergic synthesis in the dorsal raphe of a tauopathy model of Alzheimer’s disease. Grinberg L, editor. J. Alzheimers Dis. 55, 1605–1619. doi: 10.3233/jad-160658
Denhardt, D. T., Noda, M., O’Regan, A. W., Pavlin, D., and Berman, J. S. (2001). Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J. Clin. Invest. 107, 1055–1061. doi: 10.1172/jci12980
Detrembleur, C., Dierick, F., Stoquart, G., Chantraine, F., and Lejeune, T. (2003). Energy cost, mechanical work, and efficiency of hemiparetic walking. Gait Posture 18, 47–55. doi: 10.1016/s0966-6362(02)00193-5
Dewan, M. C., Rattani, A., Gupta, S., Baticulon, R. E., Hung, Y.-C., Punchak, M., et al. (2019). Estimating the global incidence of traumatic brain injury. J. Neurosurg. 130, 1080–1097.
Diemar, S. S., Sejling, A.-S., Eiken, P., Andersen, N. B., and Jørgensen, N. R. (2019a). An explorative literature review of the multifactorial causes of osteoporosis in epilepsy. Epilepsy Behav. 100:106511. doi: 10.1016/j.yebeh.2019.106511
Diemar, S. S., Sejling, A.-S., Eiken, P., Suetta, C., Jørgensen, N. R., and Andersen, N. B. (2019b). Hyponatremia and metabolic bone disease in patients with epilepsy. Bone 123, 67–75. doi: 10.1016/j.bone.2019.03.017
Dimitri, P., and Rosen, C. (2017). The central nervous system and bone metabolism: an evolving story. Calcif. Tissue Int. 100, 476–485. doi: 10.1007/s00223-016-0179-6
Dobson, R., Ramagopalan, S., and Giovannoni, G. (2012). Bone health and multiple sclerosis. Mult. Scler J. 18, 1522–1528. doi: 10.1177/1352458512453362
Driessler, F., and Baldock, P. A. (2010). Hypothalamic regulation of bone. J. Mol. Endocrinol. 45, 175–181. doi: 10.1677/jme-10-0015
Ducy, P., Amling, M., Takeda, S., Priemel, M., Schilling, A. F., Beil, F. T., et al. (2000). Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell 100, 197–207. doi: 10.1016/s0092-8674(00)81558-5
Ducy, P., and Karsenty, G. (2010). The two faces of serotonin in bone biology. J. Cell Biol. 191, 7–13. doi: 10.1083/jcb.201006123
Dunlop, B. W., and Wong, A. (2019). The hypothalamic-pituitary-adrenal axis in PTSD: pathophysiology and treatment interventions. Prog. Neuropsychopharmacol. Biol. Psychiatry 89, 361–379. doi: 10.1016/j.pnpbp.2018.10.010
Eigentler, A., Nachbauer, W., Donnemiller, E., Poewe, W., Gasser, R. W., and Boesch, S. (2014). Low bone mineral density in Friedreich Ataxia. Cerebellum 13, 549–557. doi: 10.1007/s12311-014-0568-1
Eimar, H., Lara, A. P., Tamimi, I., Sánchez, P. M., Talavera, I. G., and Tomba, F. R. (2013). Acetylcholinesterase inhibitors and healing of hip fracture in Alzheimer’s disease patients: a retrospective cohort study. J. Musculoskelet Neuronal Interact. 13, 454–463.
Eimar, H., Saltaji, H., Ghorashi, S., Isfeld, D., MacLean, J. E., Gozal, D., et al. (2017). Association between sleep apnea and low bone mass in adults: a systematic review and meta-analysis. Osteoporos. Int. 28, 1835–1852. doi: 10.1007/s00198-017-3912-8
Elefteriou, F. (2008). Regulation of bone remodeling by the central and peripheral nervous system. Arch. Biochem. Biophys. 473, 231–236. doi: 10.1016/j.abb.2008.03.016
Elefteriou, F. (2018). Impact of the autonomic nervous system on the skeleton. Physiol. Rev. 98, 1083–1112. doi: 10.1152/physrev.00014.2017
El-Gabalawy, R., Blaney, C., Tsai, J., Sumner, J. A., and Pietrzak, R. H. (2018). Physical health conditions associated with full and subthreshold PTSD in U.S. military veterans: results from the National Health and resilience in veterans study. J. Affect. Disord. 227, 849–853. doi: 10.1016/j.jad.2017.11.058
Emanuele, E., Peros, E., Scioli, G., D’Angelo, A., Olivieri, C., Montagna, L., et al. (2004). Plasma osteoprotegerin as a biochemical marker for vascular dementia and Alzheimer’s disease. Int. J. Mol. Med. 13, 849–853.
En-Nosse, M., Hartmann, S., Trinkaus, K., Alt, V., Stigler, B., Heiss, C., et al. (2009). Expression of non-neuronal cholinergic system in osteoblast-like cells and its involvement in osteogenesis. Cell Tissue Res. 338, 203–215. doi: 10.1007/s00441-009-0871-1
Ershler, W. B. (1993). Interleukin-6: a cytokine for gerontologists. J. Am. Geriatr. Soc. 41, 176–181.
Eskandari, F. (2007). Low bone mass in premenopausal women with depression. Arch. Intern. Med. 167:2329. doi: 10.1001/archinte.167.21.2329
Fan, H.-C., Lee, H.-S., Chang, K.-P., Lee, Y.-Y., Lai, H.-C., Hung, P.-L., et al. (2016). The impact of anti-epileptic drugs on growth and bone metabolism. Int. J. Mol. Sci. 17:1242. doi: 10.3390/ijms17081242
Fang, F., Kwee, L. C., Allen, K. D., Umbach, D. M., Ye, W., Watson, M., et al. (2010). Association between blood lead and the risk of amyotrophic lateral sclerosis. Am. J. Epidemiol. 171, 1126–1133.
Farias, A. M., Appenzeller, S., França, M. C., Martinez, A. R., Etchebehere, E. E., Souza, T. F., et al. (2019). Assessment of bone mineral density of patients with spinocerebellar ataxia type 3. J. Mov. Disord. 12, 43–46. doi: 10.14802/jmd.18041
Fazeli, P. K., Mendes, N., Russell, M., Herzog, D. B., Klibanski, A., and Misra, M. (2013). Bone density characteristics and major depressive disorder in adolescents. Psychosom. Med. 75, 117–123. doi: 10.1097/psy.0b013e3182821e91
Febbraio, M. A., and Pedersen, B. K. (2002). Muscle-derived interleukin-6: mechanisms for activation and possible biological roles. FASEB J. 16, 1335–1347. doi: 10.1096/fj.01-0876rev
Finbråten, A.-K., Syversen, U., Skranes, J., Andersen, G. L., Stevenson, R. D., and Vik, T. (2015). Bone mineral density and vitamin D status in ambulatory and non-ambulatory children with cerebral palsy. Osteoporos. Int. 26, 141–150. doi: 10.1007/s00198-014-2840-0
Fink, H. A., Kuskowski, M. A., Taylor, B. C., Schousboe, J. T., Orwoll, E. S., Ensrud, K. E., et al. (2008). Association of Parkinson’s disease with accelerated bone loss, fractures and mortality in older men: the Osteoporotic Fractures in Men (MrOS) study. Osteoporos. Int. 19, 1277–1282. doi: 10.1007/s00198-008-0584-4
Flachenecker, P., Reiners, K., Krauser, M., Wolf, A., and Toyka, K. V. (2001). Autonomic dysfunction in multiple sclerosis is related to disease activity and progression of disability. Mult. Scler. 7, 327–334. doi: 10.1191/135245801681138031
Flachenecker, P., Wolf, A., Krauser, M., Hartung, H.-P., and Reiners, K. (1999). Cardiovascular autonomic dysfunction in multiple sclerosis: correlation with orthostatic intolerance. J. Neurol. 246, 578–586. doi: 10.1007/s004150050407
Fodor, D., Bondor, C., Albu, A., Simon, S., Craciun, A., and Muntean, L. (2013). The value of osteopontin in the assessment of bone mineral density status in postmenopausal women. J. Investig. Med. 61, 15–21. doi: 10.2310/jim.0b013e3182761264
Foley, D., Ancoli-Israel, S., Britz, P., and Walsh, J. (2004). Sleep disturbances and chronic disease in older adults: results of the 2003 National Sleep Foundation Sleep in America Survey. J. Psychosom. Res. 56, 497–502.
Fong, C. Y., Kong, A. N., Noordin, M., Poh, B. K., Ong, L. C., and Ng, C. C. (2018). Determinants of low bone mineral density in children with epilepsy. Eur. J. Paediatr. Neurol. 22, 155–163. doi: 10.1016/0169-6009(92)91958-l
Fowler, E. G., Rao, S., Nattiv, A., Heberer, K., and Oppenheim, W. L. (2015). Bone density in premenopausal women and men under 50 years of age with cerebral palsy. Arch. Phys. Med. Rehabil. 96, 1304–1309. doi: 10.1016/j.apmr.2015.03.012
Funk, J. L., Mortel, K. F., and Meyer, J. S. (1991). Effects of estrogen replacement therapy on cerebral perfusion and cognition among postmenopausal women. Dement. Geriatr. Cogn. Disord. 2, 268–272. doi: 10.1159/000107215
Furman, J. M., and Cass, S. P. (1999). Benign paroxysmal positional vertigo. N. Engl. J. Med. 341, 1590–1596.
Gallagher, D. A., and Sturrock, A. (2007). Corticosteroid-induced osteoporosis in neurology patients. Clin. Neurol. Neurosurg. 109, 218–219. doi: 10.1016/j.clineuro.2006.07.005
Gao, H., Wei, X., Liao, J., Wang, R., Xu, J., Liu, X., et al. (2015). Lower bone mineral density in patients with Parkinson’s disease: a cross-sectional study from chinese mainland. Front. Aging Neurosci. 7:203. doi: 10.3389/fnagi.2015.00203
Garip Ustaoglu, S., Evis, Z., Ilbay, G., Boskey, A. L., and Severcan, F. (2018). Side-effects of convulsive seizures and anti-seizure therapy on bone in a rat model of epilepsy. Appl. Spectrosc. 72, 689–705. doi: 10.1177/0003702817734617
Giangregorio, L., and Blimkie, C. J. (2002). Skeletal adaptations to alterations in weight-bearing activity: a comparison of models of disuse osteoporosis. Sports Med. 32, 459–476. doi: 10.2165/00007256-200232070-00005
Girgenti, M. J., Hare, B. D., Ghosal, S., and Duman, R. S. (2017). Molecular and cellular effects of traumatic stress: implications for PTSD. Curr. Psychiatry Rep. 19:85.
Giuliani, N., Girasole, G., Vescovi, P. P., Passeri, G., and Pedrazzoni, M. (1999). Ethanol and acetaldehyde inhibit the formation of early osteoblast progenitors in murine and human bone marrow cultures. Alcohol. Clin. Exp. Res. 23, 381–385. doi: 10.1111/j.1530-0277.1999.tb04126.x
Glaesmer, H., Brähler, E., Gündel, H., and Riedel-Heller, S. G. (2011). The association of traumatic experiences and posttraumatic stress disorder with physical morbidity in old age: a german population-based study. Psychosom. Med. 73, 401–406. doi: 10.1097/psy.0b013e31821b47e8
Gnädinger, M., Mellinghoff, H.-U., and Kaelin-Lang, A. (2011). Parkinson’s disease and the bones. Swiss Med. Wkly. 141:w13154.
González-Calvin, J. L., Mundi, J. L., Casado-Caballero, F. J., Abadia, A. C., and Martin-Ibañez, J. J. (2009). Bone mineral density and serum levels of soluble tumor necrosis factors, estradiol, and osteoprotegerin in postmenopausal women with cirrhosis after viral hepatitis. J. Clin. Endocrinol. Metab. 94, 4844–4850. doi: 10.1210/jc.2009-0835
Gooch, C. L., Pracht, E., and Borenstein, A. R. (2017). The burden of neurological disease in the United States: a summary report and call to action. Ann. Neurol. 81, 479–484. doi: 10.1002/ana.24897
Gotthardt, F., Huber, C., Thierfelder, C., Grize, L., Kraenzlin, M., Scheidegger, C., et al. (2017). Bone mineral density and its determinants in men with opioid dependence. J. Bone Miner. Metab. 35, 99–107. doi: 10.1007/s00774-015-0732-9
Gozashti, M., Shahesmaeili, A., and Zadeh, N. A. (2011). Is opium addiction a risk factor for bone loss? Iran. Red Crescent Med. J. 13, 464–468.
Graham, S., Hammond-Jones, D., Gamie, Z., Polyzois, I., Tsiridis, E., and Tsiridis, E. (2008). The effect of beta-blockers on bone metabolism as potential drugs under investigation for osteoporosis and fracture healing. Expert Opin. Investig. Drugs 17, 1281–1299. doi: 10.1517/13543784.17.9.1281
Greenhill, C. (2013). Osteocalcin influences fetal brain development and adult brain function. Nat. Rev. Endocrinol. 9, 689–689. doi: 10.1038/nrendo.2013.200
Griffith, J. P., and Zarrouf, F. A. (2008). A systematic review of chronic fatigue syndrome: don’t assume it’s depression. Prim. Care Companion J. Clin. Psychiatry 10, 120–128. doi: 10.4088/pcc.v10n0206
Grill, E., Strupp, M., Müller, M., and Jahn, K. (2014). Health services utilization of patients with vertigo in primary care: a retrospective cohort study. J. Neurol. 261, 1492–1498. doi: 10.1007/s00415-014-7367-y
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127.
Hain, R. E., Hoyt, R. E., Moore, J. L., Linnville, S., Segovia, F., and Ambrose, M. R. (2011). Potential association of posttraumatic stress disorder and decreased bone mineral density in repatriated prisoners of war. Mil. Med. 176, 270–275. doi: 10.7205/milmed-d-10-00365
Halil, M., Cemal Kizilarslanoglu, M., Emin Kuyumcu, M., Yesil, Y., and Cruz Jentoft, A. J. (2015). Cognitive aspects of frailty: Mechanisms behind the link between frailty and cognitive impairment. J. Nutr. Health Aging 19, 276–283. doi: 10.1007/s12603-014-0535-z
Hamrick, M. W., McNeil, P. L., and Patterson, S. L. (2010). Role of muscle-derived growth factors in bone formation. J. Musculoskelet Neuronal Interact. 10, 64–70.
Haney, E. M., Warden, S. J., and Bliziotes, M. M. (2010). Effects of selective serotonin reuptake inhibitors on bone health in adults: time for recommendations about screening, prevention and management? Bone 46, 13–17. doi: 10.1016/j.bone.2009.07.083
Hawker, G. A., Ridout, R., Harris, V. A., Chase, C. C., Fielding, L. J., and Biggar, W. D. (2005). Alendronate in the treatment of low bone mass in steroid-treated boys with Duchennes muscular dystrophy. Arch. Phys. Med. Rehabil. 86, 284–288. doi: 10.1016/j.apmr.2004.04.021
He, L.-L., Li, X.-Y., Hou, M.-M., and Li, X.-Q. (2019). Association between bone mineral density and benign paroxysmal positional vertigo: a meta-analysis. Eur. Arch. Otorhinolaryngol. 276, 1561–1571.
Henderson, E. J., Lyell, V., Bhimjiyani, A., Amin, J., Kobylecki, C., and Gregson, C. L. (2019). Management of fracture risk in Parkinson’s: a revised algorithm and focused review of treatments. Park. Relat. Disord. 64, 181–187. doi: 10.1016/j.parkreldis.2019.03.021
Heron-Delaney, M., Kenardy, J., Charlton, E., and Matsuoka, Y. A. (2013). systematic review of predictors of posttraumatic stress disorder (PTSD) for adult road traffic crash survivors. Injury 44, 1413–1422. doi: 10.1016/j.injury.2013.07.011
Heydari, Z., Shahesmaeili, A., Khajeh-Bahrami, M. R., Rezazadeh-Mehrizi, M., Gozashti, M. H., and Moazed, V. (2017). An investigation of the risk factors of osteoporosis and the correlation between opium consumption and osteoporosis in adults. Addict. Health 9, 214–221.
Hoffman, E. P., Brown, R. H., and Kunkel, L. M. (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928. doi: 10.1016/0092-8674(87)90579-4
Hongo, T., Kotake, K., Muramatsu, H., Omura, D., Yano, Y., Hasegawa, D., et al. (2019). Loss of bone mineral density following sepsis using Hounsfield units by computed tomography. Acute Med. Surg. 6, 173–179. doi: 10.1002/ams2.401
Hoskin, L., Clifton-Bligh, P., Hansen, R., Fulcher, G., and Gates, F. (2006). Bone density and body composition in young women with chronic fatigue syndrome. Ann. N. Y. Acad. Sci. 904, 625–627. doi: 10.1111/j.1749-6632.2000.tb06528.x
Houde, S., Filiatrault, M., Fournier, A., Dubé, J., D’Arcy, S., Bérubé, D., et al. (2008). Deflazacort use in Duchenne muscular dystrophy: an 8-year follow-up. Pediatr. Neurol. 38, 200–206. doi: 10.1016/j.pediatrneurol.2007.11.001
Hsieh, C.-Y., Sung, S.-F., and Huang, H.-K. (2020). Drug treatment strategies for osteoporosis in stroke patients. Expert Opin. Pharmacother. 21, 811–821. doi: 10.1080/14656566.2020.1736556
Huang, W.-S., Hsu, J.-W., Huang, K.-L., Bai, Y.-M., Su, T.-P., Li, C.-T., et al. (2018). Post-traumatic stress disorder and risk of osteoporosis: a nationwide longitudinal study. Stress Health J. Int. Soc. Investig. Stress 34, 440–445. doi: 10.1002/smi.2806
Huang, Z., Qi, Y., Du, S., Chen, G., and Yan, W. (2015a). BMI levels with MS Bone mineral density levels in adults with multiple sclerosis: a meta-analysis. Int. J. Neurosci. 125, 904–912.
Huang, Z., Qi, Y., Du, S., Chen, G., and Yan, W. (2015b). BMI levels with MS Bone mineral density levels in adults with multiple sclerosis: a meta-analysis. Int. J. Neurosci. 125, 904–912. doi: 10.3109/00207454.2014.988332
Imai, R., Hori, H., Itoh, M., Lin, M., Niwa, M., Ino, K., et al. (2018). Inflammatory markers and their possible effects on cognitive function in women with posttraumatic stress disorder. J. Psychiatr. Res. 102, 192–200. doi: 10.1016/j.jpsychires.2018.04.009
Inestrosa, N. C., and Varela-Nallar, L. (2014). Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 6, 64–74. doi: 10.1093/jmcb/mjt051
Inestrosa, N. C., Ferrari, G. V. D., Garrido, J. L., Alvarez, A., Olivares, G. H., Barrıa, M. I., et al. (2002). Wnt signaling involvement in NL-amyloid-dependent neurodegeneration. Neurochem. Int. 41, 341–344. doi: 10.1016/s0197-0186(02)00056-6
Invernizzi, M., Carda, S., Viscontini, G. S., and Cisari, C. (2009). Osteoporosis in Parkinson’s disease. Parkinsonism Relat. Disord. 15, 339–346.
Jang, Y. S., and Kang, M.-K. (2009). Relationship between bone mineral density and clinical features in women with idiopathic benign paroxysmal positional vertigo. Otol. Neurotol. 30, 95–100. doi: 10.1097/mao.0b013e31818f5777
Jay, T. R., Miller, C. M., Cheng, P. J., Graham, L. C., Bemiller, S., Broihier, M. L., et al. (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 212, 287–295. doi: 10.1084/jem.20142322
Jeong, S.-H., Choi, S. H., Kim, J.-Y., Koo, J.-W., Kim, H. J., and Kim, J. S. (2009). Osteopenia and osteoporosis in idiopathic benign positional vertigo. Neurology 72, 1069–1076. doi: 10.1212/01.wnl.0000345016.33983.e0
Jeong, S.-H., Kim, J.-S., Shin, J. W., Kim, S., Lee, H., Lee, A. Y., et al. (2013). Decreased serum vitamin D in idiopathic benign paroxysmal positional vertigo. J. Neurol. 260, 832–838.
Jiang, S.-D., Dai, L.-Y., and Jiang, L.-S. (2006a). Osteoporosis after spinal cord injury. Osteoporos. Int. 17, 180–192. doi: 10.1007/s00198-005-2028-8
Jiang, S.-D., Jiang, L.-S., and Dai, L.-Y. (2006b). Mechanisms of osteoporosis in spinal cord injury. Clin. Endocrinol. 65, 555–565.
Jiang, T., Veres, K., Körmendiné Farkas, D., Lash, T. L., Toft Sørensen, H., and Gradus, J. L. (2018). Post-traumatic stress disorder and incident fractures in the Danish population. Osteoporos. Int. 29, 2487–2493. doi: 10.1007/s00198-018-4644-0
Johansson, C., and Skoog, I. (1996). A population-based study on the association between dementia and hip fractures in 85-year olds. Aging Milan Italy 8, 189–196. doi: 10.1007/bf03339676
Jørgensen, L., Jacobsen, B. K., Wilsgaard, T., and Magnus, J. H. (2000). Walking after stroke: does it matter? Changes in bone mineral density within the first 12 months after stroke. A longitudinal study. Osteoporos. Int. 11, 381–387. doi: 10.1007/s001980070103
Joseph, S., Wang, C., Di Marco, M., Horrocks, I., Abu-Arafeh, I., Baxter, A., et al. (2019). Fractures and bone health monitoring in boys with Duchenne muscular dystrophy managed within the Scottish Muscle Network. Neuromuscul. Disord. 29, 59–66. doi: 10.1016/j.nmd.2018.09.005
Joyce, N. C., Hache, L. P., and Clemens, P. R. (2012). Bone health and associated metabolic complications in neuromuscular diseases. Phys. Med. Rehabil. Clin. N. Am. 23, 773–799. doi: 10.1016/j.pmr.2012.08.005
Justin, B. N., Turek, M., and Hakim, A. M. (2013). Heart disease as a risk factor for dementia. Clin. Epidemiol. 5, 135–145. doi: 10.2147/clep.s30621
Kadriu, B., Gold, P. W., Luckenbaugh, D. A., Lener, M. S., Ballard, E. D., Niciu, M. J., et al. (2018). Acute ketamine administration corrects abnormal inflammatory bone markers in major depressive disorder. Mol. Psychiatry 23, 1626–1631. doi: 10.1038/mp.2017.109
Kang, H. G., Park, H. Y., Ryu, H. U., and Suk, S.-H. (2018). Bone mineral loss and cognitive impairment: the PRESENT project. Medicine (Baltimore) 97:e12755. doi: 10.1097/md.0000000000012755
Kanis, J., Oden, A., and Johnell, O. (2001). Acute and long-term increase in fracture risk after hospitalization for stroke. Stroke 32, 702–706. doi: 10.1161/01.str.32.3.702
Kapoor, E., Austin, P. C., Alibhai, S. M. H., Cheung, A. M., Cram, P., Casaubon, L. K., et al. (2019). Screening and treatment for osteoporosis after stroke: results from the ontario stroke registry. Stroke 50, 1564–1566. doi: 10.1161/strokeaha.118.024685
Kasper, L. H., and Shoemaker, J. (2010). Multiple sclerosis immunology: the healthy immune system vs the MS immune system. Neurology 74, S2–S8.
Kelly, R. R., McDonald, L. T., Jensen, N. R., Sidles, S. J., and LaRue, A. C. (2019). Impacts of psychological stress on osteoporosis: clinical implications and treatment interactions. Front. Psychiatry. 10:200. doi: 10.3389/fpsyt.2019.00200
Kemp, A. H., Quintana, D. S., Gray, M. A., Felmingham, K. L., Brown, K., and Gatt, J. M. (2010). Impact of depression and antidepressant treatment on heart rate variability: a review and meta-analysis. Biol. Psychiatry 67, 1067–1074. doi: 10.1016/j.biopsych.2009.12.012
Khrimian, L., Obri, A., Ramos-Brossier, M., Rousseaud, A., Moriceau, S., Nicot, A.-S., et al. (2017). Gpr158 mediates osteocalcin’s regulation of cognition. J. Exp. Med. 214, 2859–2873. doi: 10.1084/jem.20171320
Kim, B. K., Choi, Y. J., and Chung, Y.-S. (2013). Other than daytime working is associated with lower bone mineral density: the Korea National Health and Nutrition Examination Survey 2009. Calcif. Tissue Int. 93, 495–501. doi: 10.1007/s00223-013-9779-6
Kim, S. Y., Han, S. H., Kim, Y. H., and Park, M.-H. (2017). Clinical features of recurrence and osteoporotic changes in benign paroxysmal positional vertigo. Auris Nasus Larynx 44, 156–161. doi: 10.1016/j.anl.2016.06.006
Kim, Y. S., Leventhal, B. L., Koh, Y.-J., Fombonne, E., Laska, E., Lim, E.-C., et al. (2011). Prevalence of autism spectrum disorders in a total population sample. Am. J. Psychiatry 168, 904–912.
King, W. M., Ruttencutter, R., Nagaraja, H. N., Matkovic, V., Landoll, J., Hoyle, C., et al. (2007). Orthopedic outcomes of long-term daily corticosteroid treatment in Duchenne muscular dystrophy. Neurology 68, 1607–1613. doi: 10.1212/01.wnl.0000260974.41514.83
Kinjo, M., Setoguchi, S., Schneeweiss, S., and Solomon, D. H. (2005). Bone mineral density in subjects using central nervous system-active medications. Am. J. Med. 118:1414.
Ko, A., Kong, J., Samadov, F., Mukhamedov, A., Kim, Y. M., Lee, Y.-J., et al. (2020). Bone health in pediatric patients with neurological disorders. Ann. Pediatr. Endocrinol. Metab. 25, 15–23. doi: 10.6065/apem.2020.25.1.15
Ko, F. C., Li, J., Brooks, D. J., Rutkove, S. B., and Bouxsein, M. L. (2018). Structural and functional properties of bone are compromised in amyotrophic lateral sclerosis mice. Amyotroph. Lateral Scler Front. Degener. 19, 457–462. doi: 10.1080/21678421.2018.1452946
Kobau, R., DiIorio, C. A., Price, P. H., Thurman, D. J., Martin, L. M., Ridings, D. L., et al. (2004). Prevalence of epilepsy and health status of adults with epilepsy in Georgia and Tennessee: behavioral risk factor surveillance system, 2002. Epilepsy Behav. 5, 358–366. doi: 10.1016/j.yebeh.2004.02.007
Koh, J.-M., Lee, Y.-S., Kim, Y. S., Kim, D. J., Kim, H.-H., Park, J.-Y., et al. (2006). Homocysteine enhances bone resorption by stimulation of osteoclast formation and activity through increased intracellular ROS generation. J. Bone Miner Res. 21, 1003–1011. doi: 10.1359/jbmr.060406
Konno, S., Suzuki, S., Masuda, M., Nagane, Y., Tsuda, E., Murai, H., et al. (2015). Association between glucocorticoid-induced osteoporosis and myasthenia gravis: a cross-sectional study. PLoS One 10:e0126579. doi: 10.1371/journal.pone.0126579
Krishnan, V. (2006). Regulation of bone mass by Wnt signaling. J. Clin. Invest. 116, 1202–1209. doi: 10.1172/jci28551
Kurek, J. B., Nouri, S., Kannourakis, G., Murphy, M., and Austin, L. (1996). Leukemia inhibitory factor and interleukin-6 are produced by diseased and regenerating skeletal muscle. Muscle Nerve 19, 1291–1301. doi: 10.1002/(sici)1097-4598(199610)19:10<1291::aid-mus6>3.0.co;2-9
Kurihara, N., Menaa, C., Maeda, H., Haile, D. J., and Reddy, S. V. (2001). Osteoclast-stimulating factor interacts with the spinal muscular atrophy gene product to stimulate osteoclast formation. J. Biol. Chem. 276, 41035–41039. doi: 10.1074/jbc.m100233200
Labelle, H., Tohmé, S., Duhaime, M., and Allard, P. (1986). Natural history of scoliosis in Friedreich’s ataxia. J. Bone Joint Surg. Am. 68, 564–572.
Lam, F. M. H., Bui, M., Yang, F. Z. H., and Pang, M. Y. C. (2016). Chronic effects of stroke on hip bone density and tibial morphology: a longitudinal study. Osteoporos. Int. 27, 591–603. doi: 10.1007/s00198-015-3307-7
Lamar, C. D. (2011). Sepsis-associated encephalopathy: review of the neuropsychiatric manifestations and cognitive outcome. J. Neuropsychiatry Clin. Neurosci. 23, 237–241. doi: 10.1176/jnp.23.3.jnp237
Larson, C. M., and Henderson, R. C. (2000). Bone mineral density and fractures in boys with Duchenne muscular dystrophy. J. Pediatr. Orthop. 20, 71–74. doi: 10.1097/01241398-200001000-00016
Latt, M. D., Lord, S. R., Morris, J. G. L., and Fung, V. S. C. (2009). Clinical and physiological assessments for elucidating falls risk in Parkinson’s disease. Mov. Disord. 24, 1280–1289. doi: 10.1002/mds.22561
Lee, C. W.-S., Liao, C.-H., Lin, C.-L., Liang, J.-A., Sung, F.-C., and Kao, C.-H. (2015). Increased risk of osteoporosis in patients with depression. Mayo Clin. Proc. 90, 63–70.
Lee, N. K., Sowa, H., Hinoi, E., Ferron, M., Ahn, J. D., Confavreux, C., et al. (2007). Endocrine regulation of energy metabolism by the skeleton. Cell 130, 456–469.
Lee, S. H., Kim, M. J., Kim, B.-J., Kim, S. R., Chun, S., Kim, H.-K., et al. (2010). Hyperhomocysteinemia due to levodopa treatment as a risk factor for osteoporosis in patients with Parkinson’s disease. Calcif. Tissue Int. 86, 132–141. doi: 10.1007/s00223-009-9327-6
Lee, S.-B., Cho, A.-H., Butcher, K. S., Kim, T.-W., Ryu, S.-Y., and Kim, Y.-I. (2013). Low bone mineral density is associated with poor clinical outcome in acute ischemic stroke. Int. J. Stroke 8, 68–72. doi: 10.1111/j.1747-4949.2011.00714.x
Lee, S.-C., Hu, L.-Y., Huang, M.-W., Shen, C.-C., Huang, W.-L., Lu, T., et al. (2017). Risk of vertebral fracture in patients diagnosed with a depressive disorder: a nationwide population-based cohort study. Clinics 72, 44–50. doi: 10.6061/clinics/2017(01)08
Lee, Y.-F., Tsou, H.-K., Leong, P.-Y., Wang, Y.-H., and Wei, J. C.-C. (2020). Association of sepsis with risk for osteoporosis: a population-based cohort study. Osteoporos. Int. doi: 10.1007/s00198-020-05599-3 [Epub ahead of print].
Lewis, K. E., Sharan, K., Takumi, T., and Yadav, V. K. (2017). Skeletal site-specific changes in bone mass in a genetic mouse model for human 15q11-13 duplication seen in autism. Sci. Rep. 7:9902.
Lewis, S. J. G., and Smith, P. E. M. (2001). Osteoporosis prevention in myasthenia gravis: a reminder: osteoporosis prevention in MG: a reminder. Acta Neurol. Scand. 103, 320–322. doi: 10.1034/j.1600-0404.2001.103005320.x
Li, S., Liu, B., Zhang, L., and Rong, L. (2014). Amyloid beta peptide is elevated in osteoporotic bone tissues and enhances osteoclast function. Bone 61, 164–175. doi: 10.1016/j.bone.2014.01.010
Licastro, F., Pedrini, S., Caputo, L., Annoni, G., Davis, L. J., Ferri, C., et al. (2000). Increased plasma levels of interleukin-1, interleukin-6 and a-1-antichymotrypsin in patients with Alzheimer’s disease: peripheral inflammation or signals from the brain? J. Neuroimmunol. 103, 97–102. doi: 10.1016/s0165-5728(99)00226-x
Lindqvist, D., Dhabhar, F. S., Mellon, S. H., Yehuda, R., Grenon, S. M., Flory, J. D., et al. (2017). Increased pro-inflammatory milieu in combat related PTSD – a new cohort replication study. Brain Behav. Immun. 59, 260–264. doi: 10.1016/j.bbi.2016.09.012
Litvan, I., Halliday, G., Hallett, M., Goetz, C. G., Rocca, W., Duyckaerts, C., et al. (2007). The etiopathogenesis of Parkinson disease and suggestions for future research. Part I. J. Neuropathol. Exp. Neurol. 66, 251–257. doi: 10.1097/nen.0b013e3180415e42
Liu-Ambrose, T., Pang, M. Y. C., and Eng, J. J. (2007). Executive function is independently associated with performances of balance and mobility in community-dwelling older adults after mild stroke: implications for falls prevention. Cerebrovasc. Dis. 23, 203–210. doi: 10.1159/000097642
Looker, A. C., Borrud, L. G., Dawson-Hughes, B., Shepherd, J. A., and Wright, N. C. (2012). Osteoporosis or low bone mass at the femur neck or lumbar spine in older adults: United States, 2005–2008. NCHS Data Brief 93, 1–8.
Lopez, J. R., Lyckman, A., Oddo, S., LaFerla, F. M., Querfurth, H. W., and Shtifman, A. (2008). Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J. Neurochem. 105, 262–271. doi: 10.1111/j.1471-4159.2007.05135.x
Loskutova, N., Honea, R. A., Vidoni, E. D., Brooks, W. M., and Burns, J. M. (2009). Bone density and brain atrophy in early Alzheimer’s disease. J. Alzheimers Dis. 18, 777–785. doi: 10.3233/jad-2009-1185
Lourida, I., Thompson-Coon, J., Dickens, C. M., Soni, M., Kuźma, E., Kos, K., et al. (2015). Parathyroid hormone, cognitive function and dementia: a systematic review. Quinn TJ, editor. PLoS One 10:e0127574. doi: 10.1371/journal.pone.0127574
Lozsadi, D. A., Peters, G., Sadik, H. Y., Kellett, M. W., Fox, S. H., and Smith, D. F. (2006). Prevention of osteoporosis in glucocorticoid-treated neurology patients. Clin. Neurol. Neurosurg. 108, 157–162. doi: 10.1016/j.clineuro.2005.05.008
Lucassen, E. A., de Mutsert, R., le Cessie, S., Appelman-Dijkstra, N. M., Rosendaal, F. R., van Heemst, D., et al. (2017). Poor sleep quality and later sleep timing are risk factors for osteopenia and sarcopenia in middle-aged men and women: The NEO study. PLoS One 12:e0176685. doi: 10.1371/journal.pone.0176685
Luckhaus, C., Mahabadi, B., Grass-Kapanke, B., Jänner, M., Willenberg, H., Jäger, M., et al. (2009). Blood biomarkers of osteoporosis in mild cognitive impairment and Alzheimer’s disease. J. Neural Transm. 116, 905–911.
Lui, L.-Y., Stone, K., Cauley, J. A., Hillier, T., and Yaffe, K. (2003). Bone loss predicts subsequent cognitive decline in older women: the study of osteoporotic fractures. J. Am. Geriatr. Soc. 51, 38–43. doi: 10.1034/j.1601-5215.2002.51007.x
Luine, V. N. (2014). Estradiol and cognitive function: Past, present and future. Horm. Behav. 66, 602–618. doi: 10.1016/j.yhbeh.2014.08.011
Lv, F., Guan, Y., Ma, D., Xu, X., Song, Y., Li, L., et al. (2018). Effects of alendronate and alfacalcidol on bone in patients with myasthenia gravis initiating glucocorticoids treatment. Clin. Endocrinol. 88, 380–387. doi: 10.1111/cen.13537
Maïmoun, L., Fattal, C., Micallef, J.-P., Peruchon, E., and Rabischong, P. (2006). Bone loss in spinal cord-injured patients: from physiopathology to therapy. Spinal Cord 44, 203–210. doi: 10.1038/sj.sc.3101832
Malik, P., Gasser, R. W., Moncayo, R. C., Kandler, C., Koudouovoh-Tripp, P., Giesinger, J., et al. (2013). Bone mineral density and bone metabolism in patients with major depressive disorder without somatic comorbidities. Prog. Neuropsychopharmacol. Biol. Psychiatry 44, 58–63. doi: 10.1016/j.pnpbp.2013.01.019
Mallette, L. E., Patten, B., Cook, J. D., and Engel, W. K. (1977). Calcium metabolism in amyotrophic lateral sclerosis. Dis. Nerv. Syst. 38, 457–461.
Malochet-Guinamand, S., Durif, F., and Thomas, T. (2015). Parkinson’s disease: A risk factor for osteoporosis. Joint Bone Spine 82, 406–410.
Mandelkow, E.-M., and Mandelkow, E. (2012). Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2:a006247. doi: 10.1101/cshperspect.a006247
Marcocci, C., Cianferotti, L., and Cetani, F. (2012). Bone disease in primary hyperparathyrodism. Ther. Adv. Musculoskelet. Dis. 4, 357–368. doi: 10.1177/1759720x12441869
Marenzana, M., and Chenu, C. (2008). Sympathetic nervous system and bone adaptive response to its mechanical environment. J. Musculoskelet Neuronal Interact. 8, 111–120.
Martino, G., Catalano, A., Bellone, F., Langher, V., Lasco, C., Penna, A., et al. (2018a). Quality of life in postmenopausal women: which role for vitamin D? Mediterr. J. Clin. Psychol. 6, 1–14.
Martino, G., Catalano, A., Bellone, F., Sardella, A., Lasco, C., Caprì, T., et al. (2018b). Vitamin D status is associated with anxiety levels in postmenopausal women evaluated for osteoporosis. Mediterr. J. Clin. Psychol. 6, 1–16.
Martino, G., Sardella, A., Bellone, F., Lasco, C., Langher, V., Cazzato, V., et al. (2019). Executive functions and bone health: a focus on cognitive impulsivity and bone mineral density. Mediterr. J. Clin. Psychol.7, 1–13. doi: 10.6092/2282-1619/2019.7.2167
Maurel, D. B., Jaffre, C., Rochefort, G. Y., Aveline, P. C., Boisseau, N., Uzbekov, R., et al. (2011). Low bone accrual is associated with osteocyte apoptosis in alcohol-induced osteopenia. Bone 49, 543–552. doi: 10.1016/j.bone.2011.06.001
Maybarduk, P. K., and Levine, M. (1941). Osseous atrophy associated with progressive muscular dystrophy. Am. J. Dis. Child 61, 565–576. doi: 10.1001/archpedi.1941.02000090141013
McDonald, D. G. M., Kinali, M., Gallagher, A. C., Mercuri, E., Muntoni, F., Roper, H., et al. (2002). Fracture prevalence in Duchenne muscular dystrophy. Dev. Med. Child Neurol. 44, 695–698.
McKenzie, R., Reynolds, J. C., O’Fallon, A., Dale, J., Deloria, M., Blackwelder, W., et al. (2000). Decreased bone mineral density during low dose glucocorticoid administration in a randomized, placebo controlled trial. J. Rheumatol. 27, 2222–2226.
Melton, L. J., Beard, C. M., Kokmen, E., Atkinson, E. J., and O’Fallon, W. M. (1994). Fracture risk in patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 42, 614–619.
Mendy, A., Vieira, E. R., Albatineh, A. N., Nnadi, A. K., Lowry, D., and Gasana, J. (2014). Low bone mineral density is associated with balance and hearing impairments. Ann. Epidemiol. 24, 58–62. doi: 10.1016/j.annepidem.2013.10.012
Meneses, G., Cárdenas, G., Espinosa, A., Rassy, D., Pérez-Osorio, I. N., Bárcena, B., et al. (2019). Sepsis: developing new alternatives to reduce neuroinflammation and attenuate brain injury: alternatives to modulate neuroinflammation. Ann. N. Y. Acad. Sci. 1437, 43–56. doi: 10.1111/nyas.13985
Michel, L. (2018). Environmental factors in the development of multiple sclerosis. Rev. Neurol. 174, 372–377. doi: 10.1016/j.neurol.2018.03.010
Milbrandt, T. A., Kunes, J. R., and Karol, L. A. (2008). Friedreich’s ataxia and scoliosis: the experience at two institutions. J. Pediatr. Orthop. 28, 234–238. doi: 10.1097/bpo.0b013e318164fa79
Moen, S. M., Celius, E. G., Sandvik, L., Nordsletten, L., Eriksen, E. F., and Holmoy, T. (2011). Low bone mass in newly diagnosed multiple sclerosis and clinically isolated syndrome. Neurology 77, 151–157. doi: 10.1212/wnl.0b013e3182242d34
Mohan, S., and Baylink, D. J. (2005). Impaired skeletal growth in mice with haploinsufficiency of IGF-I: genetic evidence that differences in IGF-I expression could contribute to peak bone mineral density differences. J. Endocrinol. 185, 415–420. doi: 10.1677/joe.1.06141
Mohan, S., Richman, C., Guo, R., Amaar, Y., Donahue, L. R., Wergedal, J., et al. (2003). Insulin-like growth factor regulates peak bone mineral density in mice by both growth hormone-dependent and -independent mechanisms. Endocrinology 144, 929–936. doi: 10.1210/en.2002-220948
Mosti, M. P., Flemmen, G., Hoff, J., Stunes, A. K., Syversen, U., and Wang, E. (2016). Impaired skeletal health and neuromuscular function among amphetamine users in clinical treatment. Osteoporos. Int. 27, 1003–1010. doi: 10.1007/s00198-015-3371-z
Murphy, O., Zandi, M. S., Lindenberg, N., Murphy, E., and Chataway, J. (2016). Bone health in patients with multiple sclerosis relapses. Mult. Scler Relat. Disord. 6, 75–80. doi: 10.1016/j.msard.2016.02.003
Murthy, L., Dreyer, P., Suriyaarachchi, P., Gomez, F., Curcio, C. L., Boersma, D., et al. (2018). Association between high levels of parathyroid hormone and frailty: the nepean osteoporosis and frailty (NOF) study. J. Frailty Aging 7, 253–257.
Myint, P. K., Clark, A. B., Kwok, C. S., Loke, Y. K., Yeong, J. K.-Y., Luben, R. N., et al. (2014). Bone mineral density and incidence of stroke: european prospective investigation into cancer-norfolk population–based study, systematic review, and meta-analysis. Stroke 45, 373–382. doi: 10.1161/strokeaha.113.002999
Myint, P. K., Poole, K. E. S., and Warburton, E. A. (2007). Hip fractures after stroke and their prevention. QJM 100, 539–545. doi: 10.1093/qjmed/hcm067
Nakada, T., Teranishi, M., Ueda, Y., and Sone, M. (2018). Fracture probability assessed using FRAX® in elderly women with benign paroxysmal positional vertigo. Auris Nasus Larynx. 45, 1173–1177. doi: 10.1016/j.anl.2018.05.002
Nakano, H., Aoyagi, K., Ohgi, S., and Akiyama, T. (2003). Factors influencing metacarpal bone mineral density in adults with cerebral palsy. J. Bone Miner Metab. 21, 409–414. doi: 10.1007/s00774-003-0436-4
Nasomyont, N., Keefe, C., Tian, C., Hornung, L., Khoury, J., Tilden, J. C., et al. (2020). Safety and efficacy of teriparatide treatment for severe osteoporosis in patients with Duchenne muscular dystrophy. Osteoporos. Int 31, 2449–2459. doi: 10.1007/s00198-020-05549-z
Nasralla, M., Haier, J., and Nicolson, G. L. (1999). Multiple mycoplasmal infections detected in blood of patients with chronic fatigue syndrome and/or fibromyalgia syndrome. Eur. J. Clin. Microbiol. Infect. Dis. 18, 859–865. doi: 10.1007/s100960050420
Nazif, H., Shatla, R., Elsayed, R., Tawfik, E., Osman, N., Korra, S., et al. (2017). Bone mineral density and insulin-like growth factor-1 in children with spastic cerebral palsy. Childs Nerv. Syst. 33, 625–630. doi: 10.1007/s00381-017-3346-9
Neumeyer, A. M., Cano Sokoloff, N., McDonnell, E., Macklin, E. A., McDougle, C. J., and Misra, M. (2017). Bone microarchitecture in adolescent boys with autism spectrum disorder. Bone 97, 139–146. doi: 10.1016/j.bone.2017.01.009
Neumeyer, A. M., Gates, A., Ferrone, C., Lee, H., and Misra, M. (2013). Bone density in peripubertal boys with autism spectrum disorders. J. Autism Dev. Disord. 43, 1623–1629. doi: 10.1007/s10803-012-1709-3
Neumeyer, A. M., O’Rourke, J. A., Massa, A., Lee, H., Lawson, E. A., McDougle, C. J., et al. (2015). Brief report: bone fractures in children and adults with autism spectrum disorders. J. Autism Dev. Disord. 45, 881–887. doi: 10.1007/s10803-014-2228-1
Nieves, J., Cosman, F., Herbert, J., Shen, V., and Lindsay, R. (1994). High prevalence of vitamin D deficiency and reduced bone mass in multiple sclerosis. Neurology 44, 1687–1692. doi: 10.1212/wnl.44.9.1687
Nijs, J., De Meirleir, K., Englebienne, P., and McGregor, N. (2003). Chronic fatigue syndrome: a risk factor for osteopenia? Med. Hypotheses. 60, 65–68. doi: 10.1016/s0306-9877(02)00332-8
Noguchi, T., Ebina, K., Hirao, M., Otsuru, S., Guess, A. J., Kawase, R., et al. (2018). Apolipoprotein E plays crucial roles in maintaining bone mass by promoting osteoblast differentiation via ERK1/2 pathway and by suppressing osteoclast differentiation via c-Fos, NFATc1, and NF-κB pathway. Biochem. Biophys. Res. Commun. 503, 644–650. doi: 10.1016/j.bbrc.2018.06.055
Novo-Rodríguez, C., García-Fontana, B., Luna-Del Castillo, J. D. D., Andújar-Vera, F., Ávila-Rubio, V., García-Fontana, C., et al. (2013). Circulating levels of sclerostin are associated with cardiovascular mortality. PLoS One 13:e0199504. doi: 10.1371/journal.pone.0199504
Numann, P. J., Torppa, A. J., and Blumetti, A. E. (1984). Neuropsychologic deficits associated with primary hyperparathyroidism. Surgery 96, 1119–1123.
Obri, A., Khrimian, L., Karsenty, G., and Oury, F. (2018). Osteocalcin in the brain: from embryonic development to age-related decline in cognition. Nat. Rev. Endocrinol. 14, 174–182. doi: 10.1038/nrendo.2017.181
Office of the Surgeon General (US) (2004). The Basics of Bone in Health and Disease. Roclville, MD: Office of the Surgeon General (US).
Oliva, C. A., Vargas, J. Y., and Inestrosa, N. C. (2013). Wnts in adult brain: from synaptic plasticity to cognitive deficiencies. Front. Cell Neurosci. 7:224. doi: 10.3389/fncel.2013.00224
Olsson, A., Oturai, A. B., Søndergaard, H. B., Sellebjerg, F., and Oturai, P. S. (2018). Bone microarchitecture and bone mineral density in multiple sclerosis. Acta Neurol. Scand. 137, 363–369. doi: 10.1111/ane.12884
Orchard, G. R., Paratz, J. D., Blot, S., and Roberts, J. A. (2015). Risk factors in hospitalized patients with burn injuries for developing heterotopic ossification–a retrospective analysis. J. Burn Care Res. 36, 465–470. doi: 10.1097/bcr.0000000000000123
Otaka, M., Konishi, N., Odashima, M., Jin, M., Wada, I., Matsuhashi, T., et al. (2007). Effect of alcohol consumption on leptin level in serum, adipose tissue, and gastric mucosa. Dig. Dis. Sci. 52, 3066–3069. doi: 10.1007/s10620-006-9635-x
Otero, K., Shinohara, M., Zhao, H., Cella, M., Gilfillan, S., Colucci, A., et al. (2012). TREM2 and β-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J. Immunol. 188, 2612–2621. doi: 10.4049/jimmunol.1102836
Ozturk, E. A., Gundogdu, I., Tonuk, B., Umay, E., Kocer, B. G., and Cakci, A. (2020). Bone mineral density and serum vitamin D status in Parkinson’s disease: are the stage and clinical features of the disease important? Neurol. India 68, 394–400. doi: 10.4103/0028-3886.283755
Pack, A. (2008). Bone health in people with epilepsy: is it impaired and what are the risk factors? Seizure 17, 181–186. doi: 10.1016/j.seizure.2007.11.020
Paloneva, J., Kestilä, M., Wu, J., Salminen, A., Böhling, T., Ruotsalainen, V., et al. (2000). Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 25, 357–361. doi: 10.1038/77153
Pang, M. Y. C., Ashe, M. C., Eng, J. J., McKay, H. A., and Dawson, A. S. A. (2006). 19-week exercise program for people with chronic stroke enhances bone geometry at the tibia: a peripheral quantitative computed tomography study. Osteoporos. Int. 17, 1615–1625. doi: 10.1007/s00198-006-0168-0
Pang, M. Y. C., Eng, J. J., Dawson, A. S., McKay, H. A., and Harris, J. E. A. (2005). community-based fitness and mobility exercise program for older adults with chronic stroke: a randomized, controlled trial. J. Am. Geriatr. Soc. 53, 1667–1674. doi: 10.1111/j.1532-5415.2005.53521.x
Parfitt, A. M. (1994). Osteonal and hemi-osteonal remodeling: the spatial and temporal framework for signal traffic in adult human bone. J. Cell Biochem. 55, 273–286. doi: 10.1002/jcb.240550303
Pascuzzi, R. M., Coslett, H. B., and Johns, T. R. (1984). Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann. Neurol. 15, 291–298. doi: 10.1002/ana.410150316
Passos, I. C., Vasconcelos-Moreno, M. P., Costa, L. G., Kunz, M., Brietzke, E., Quevedo, J., et al. (2015). Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry 2, 1002–1012. doi: 10.1016/s2215-0366(15)00309-0
Pecak, F., Trontelj, J. V., and Dimitrijevic, M. R. (1980). Scoliosis in neuromuscular disorders. Int. Orthop. 3, 323–328.
Peng, Y., Liu, J., Tang, Y., Liu, J., Han, T., Han, S., et al. (2014). High-fat-diet-induced weight gain ameliorates bone loss without exacerbating AÎ2PP processing and cognition in female APP/PS1 mice. Front. Cell Neurosci. 8:225. doi: 10.3389/fncel.2014.00225
Perera, N., Sampaio, H., Woodhead, H., and Farrar, M. (2016). Fracture in duchenne muscular dystrophy: natural history and vitamin D deficiency. J. Child Neurol. 31, 1181–1187. doi: 10.1177/0883073816650034
Peris, P., Guañabens, N., Monegal, A., Suris, X., Alvarez, L., Martinez de Osaba, M. J., et al. (1995). Aetiology and presenting symptoms in male osteoporosis. Br. J. Rheumatol. 34, 936–941. doi: 10.1093/rheumatology/34.10.936
Perrini, S., Laviola, L., Carreira, M. C., Cignarelli, A., Natalicchio, A., and Giorgino, F. (2010). The GH/IGF1 axis and signaling pathways in the muscle and bone: mechanisms underlying age-related skeletal muscle wasting and osteoporosis. J. Endocrinol. 205, 201–210. doi: 10.1677/joe-09-0431
Peters, T. L., Weibull, C. E., Fang, F., Sandler, D. P., Lambert, P. C., Ye, W., et al. (2017). Association of fractures with the incidence of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler Front. Degener. 18, 419–425. doi: 10.1080/21678421.2017.1300287
Piec, G., Mirkovitch, J., Palacio, S., Mühlradt, P. F., and Felix, R. (1999). Effect of MALP-2, a lipopeptide from mycoplasma fermentans, on bone resorption in vitro. Infect. Immun. 67, 6281–6285. doi: 10.1128/iai.67.12.6281-6285.1999
Pilutti, L. A., and Motl, R. W. (2019). Body composition and disability in people with multiple sclerosis: A dual-energy x-ray absorptiometry study. Mult. Scler Relat. Disord. 29, 41–47. doi: 10.1016/j.msard.2019.01.009
Portaro, S., Morini, E., Santoro, M. E., Accorinti, M., Marzullo, P., Naro, A., et al. (2018). Breathlessness in amyotrophic lateral sclerosis: a case report on the role of osteoporosis in the worsening of respiratory failure. Medicine (Baltimore) 97:e13026. doi: 10.1097/md.0000000000013026
Pouwels, S., de Boer, A., Javaid, M. K., Hilton-Jones, D., Verschuuren, J., Cooper, C., et al. (2013). Fracture rate in patients with myasthenia gravis: the general practice research database. Osteoporos. Int. 24, 467–476. doi: 10.1007/s00198-012-1970-5
Puthucheary, Z. A., Sun, Y., Zeng, K., Vu, L. H., Zhang, Z. W., Lim, R. Z. L., et al. (2017). Sepsis reduces bone strength before morphologic changes are identifiable. Crit. Care Med. 45, e1254–e1261.
Qi, W., Gevonden, M., and Shalev, A. (2016). Prevention of post-traumatic stress disorder after trauma: current evidence and future directions. Curr. Psychiatry Rep. 18:20.
Quevedo, I., and Zuniga, A. M. (2010). Low bone mineral density in rotating-shift workers. J. Clin. Densitom 13, 467–469. doi: 10.1016/j.jocd.2010.07.004
Rabenda, V., Nicolet, D., Beaudart, C., Bruyère, O., and Reginster, J.-Y. (2013). Relationship between use of antidepressants and risk of fractures: a meta-analysis. Osteoporos. Int. 24, 121–137. doi: 10.1007/s00198-012-2015-9
Raber, J., Huang, Y., and Ashford, J. W. (2004). ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 25, 641–650. doi: 10.1016/j.neurobiolaging.2003.12.023
Radaei, F., and Gharibzadeh, S. (2013). Relationship among bone mineral density reduction, hearing loss, and balance disorders in osteoporotic patients. Front. Bioeng. Biotechnol. 1:17. doi: 10.3389/fbioe.2013.00017
Raglione, L. M., Sorbi, S., and Nacmias, B. (2011). Osteoporosis and Parkinson’s disease. Clin. Cases Miner Bone Metab. 8, 16–18.
Raibagkar, P., Ferry, J. A., and Stone, J. H. (2017). Is MuSK myasthenia gravis linked to IgG4-related disease? J. Neuroimmunol. 305, 82–83. doi: 10.1016/j.jneuroim.2017.02.004
Ramnemark, A., Nyberg, L., Lorentzon, R., Olsson, T., and Gustafson, Y. (1999). Hemiosteoporosis after severe stroke, independent of changes in body composition and weight. Stroke 30, 755–760. doi: 10.1161/01.str.30.4.755
Rauma, P. H., Pasco, J. A., Berk, M., Stuart, A. L., Koivumaa-Honkanen, H., Honkanen, R. J., et al. (2015). The association between major depressive disorder, use of antidepressants and bone mineral density (BMD) in men. J. Musculoskelet Neuronal Interact. 15, 177–185.
Reid, A. G., Lingford-Hughes, A. R., Cancela, L. M., and Kalivas, P. W. (2012). Substance abuse disorders. Handb. Clin. Neurol. 106, 419–431.
Reid, I. R. (2008). Effects of beta-blockers on fracture risk. J. Musculoskelet Neuronal Interact. 8, 105–110.
Reinholt, F. P., Hultenby, K., Oldberg, A., and Heinegard, D. (1990). Osteopontin–a possible anchor of osteoclasts to bone. Proc. Natl. Acad. Sci. U.S.A. 87, 4473–4475. doi: 10.1073/pnas.87.12.4473
Rifas, L. (1999). Bone and cytokines: beyond IL-1, IL-6 and TNF-alpha. Calcif. Tissue Int. 64, 1–7. doi: 10.1007/s002239900570
Riggs, B. L., Khosla, S., and Melton, L. J. A. (1998). unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J. Bone Miner Res. 13, 763–773. doi: 10.1359/jbmr.1998.13.5.763
Roberts, D., Lee, W., Cuneo, R. C., Wittmann, J., Ward, G., Flatman, R., et al. (1998). Longitudinal study of bone turnover after acute spinal cord injury. J. Clin. Endocrinol. Metab. 83, 415–422. doi: 10.1210/jc.83.2.415
Rodrigues, W., Madeira, M., da Silva, T., Clemente-Napimoga, J., Miguel, C., Dias-da-Silva, V., et al. (2012). Low dose of propranolol down-modulates bone resorption by inhibiting inflammation and osteoclast differentiation. Br. J. Pharmacol. 165, 2140–2151. doi: 10.1111/j.1476-5381.2011.01686.x
Roke, Y., van Harten, P. N., Buitelaar, J. K., Tenback, D. E., Quekel, L. G. B. A., de Rijke, Y. B., et al. (2012). Bone mineral density in male adolescents with autism spectrum disorders and disruptive behavior disorder with or without antipsychotic treatment. Eur. J. Endocrinol. 167, 855–863. doi: 10.1530/eje-12-0521
Roman, S. A., Sosa, J. A., Mayes, L., Desmond, E., Boudourakis, L., Lin, R., et al. (2005). Parathyroidectomy improves neurocognitive deficits in patients with primary hyperparathyroidism. Surgery 138, 1121–1129. doi: 10.1016/j.surg.2005.08.033
Roos, P. M., Vesterberg, O., Syversen, T., Flaten, T. P., and Nordberg, M. (2013). Metal concentrations in cerebrospinal fluid and blood plasma from patients with amyotrophic lateral sclerosis. Biol. Trace Elem. Res. 151, 159–170. doi: 10.1007/s12011-012-9547-x
Rosario, E. R., Aqeel, R., Brown, M. A., Sanchez, G., Moore, C., and Patterson, D. (2013). Hypothalamic-pituitary dysfunction following traumatic brain injury affects functional improvement during acute inpatient rehabilitation. J. Head Trauma Rehabil. 28, 390–396. doi: 10.1097/htr.0b013e318250eac6
Rosenblat, J. D., Gregory, J. M., Carvalho, A. F., and McIntyre, R. S. (2016). Depression and disturbed bone metabolism: a narrative review of the epidemiological findings and postulated mechanisms. Curr. Mol. Med. 16, 165–178. doi: 10.2174/1566524016666160126144303
Rufo, A., Del Fattore, A., Capulli, M., Carvello, F., De Pasquale, L., Ferrari, S., et al. (2011). Mechanisms inducing low bone density in Duchenne muscular dystrophy in mice and humans. J. Bone Miner Res. 26, 1891–1903. doi: 10.1002/jbmr.410
Santangelo, S., Gamelli, R. L., and Shankar, R. (2001). Myeloid commitment shifts toward monocytopoiesis after thermal injury and sepsis. Ann. Surg. 233, 97–106. doi: 10.1097/00000658-200101000-00015
Santori, C., Ceccanti, M., Diacinti, D., Attilia, M. L., Toppo, L., D’Erasmo, E., et al. (2008). Skeletal turnover, bone mineral density, and fractures in male chronic abusers of alcohol. J. Endocrinol. Invest. 31, 321–326. doi: 10.1007/bf03346365
Sato, T., Abe, T., Chida, D., Nakamoto, N., Hori, N., Kokabu, S., et al. (2010). Functional role of acetylcholine and the expression of cholinergic receptors and components in osteoblasts. FEBS Lett. 584, 817–824. doi: 10.1016/j.febslet.2010.01.001
Sato, Y., Kanoko, T., Satoh, K., and Iwamoto, J. (2004). Risk factors for hip fracture among elderly patients with Alzheimer’s disease. J. Neurol. Sci. 223, 107–112. doi: 10.1016/j.jns.2004.03.033
Sato, Y., Kuno, H., Kaji, M., Etoh, K., and Oizumi, K. (2000). Influence of immobilization upon calcium metabolism in the week following hemiplegic stroke. J. Neurol. Sci. 175, 135–139. doi: 10.1016/s0022-510x(00)00298-7
Schiza, S. E., Bouloukaki, I., and Mermigkis, C. (2013). Bone mineral density in patients with obstructive sleep apnea syndrome. Sleep Breath 17, 17–18. doi: 10.1007/s11325-012-0699-x
Schliebs, R., and Arendt, T. (2011). The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 221, 555–563. doi: 10.1016/j.bbr.2010.11.058
Schlienger, R. G., Kraenzlin, M. E., Jick, S. S., and Meier, C. R. (2004). Use of beta-blockers and risk of fractures. JAMA 292, 1326–1332. doi: 10.1001/jama.292.11.1326
Schneider, J. L., Fink, H. A., Ewing, S. K., Ensrud, K. E., and Cummings, S. R. (2008). Study of osteoporotic fractures (SOF) research group. The association of Parkinson’s disease with bone mineral density and fracture in older women. Osteoporos. Int. 19, 1093–1097. doi: 10.1007/s00198-008-0583-5
Schoenau, E. (2005). From mechanostat theory to development of the “Functional Muscle-Bone-Unit.”. J. Musculoskelet Neuronal Interact. 5, 232–238.
Schweiger, J. U., Schweiger, U., Hüppe, M., Kahl, K. G., Greggersen, W., and Fassbinder, E. (2016). Bone density and depressive disorder: a meta-analysis. Brain Behav. 6:e00489.
Seal, K. H. (2007). Bringing the war back home: mental health disorders among 103 788 US veterans returning from Iraq and Afghanistan seen at department of veterans affairs facilities. Arch. Intern. Med. 167:476. doi: 10.1001/archinte.167.5.476
Seifert, C. F., and Wiltrout, T. R. (2013). Calcaneal bone mineral density in young adults prescribed selective serotonin reuptake inhibitors. Clin. Ther. 35, 1412–1417. doi: 10.1016/j.clinthera.2013.07.423
Selkoe, D., Mandelkow, E., and Holtzman, D. (2012). Deciphering Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2:a011460.
Shah, A. D., Shoback, D., and Lewiecki, E. M. (2015). Sclerostin inhibition: a novel therapeutic approach in the treatment of osteoporosis. Int. J. Womens Health 7, 565–580. doi: 10.2147/ijwh.s73244
Shahar, E., Whitney, C. W., Redline, S., Lee, E. T., Newman, A. B., Nieto, F. J., et al. (2001). Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am. J. Respir. Crit. Care Med. 163, 19–25. doi: 10.1164/ajrccm.163.1.2001008
Shanmugarajan, S., Tsuruga, E., Swoboda, K. J., Maria, B. L., Ries, W. L., and Reddy, S. V. (2009). Bone loss in survival motor neuron (Smn(-/-) SMN2) genetic mouse model of spinal muscular atrophy. J. Pathol. 219, 52–60. doi: 10.1002/path.2566
Sharir, A., Stern, T., Rot, C., Shahar, R., and Zelzer, E. (2011). Muscle force regulates bone shaping for optimal load-bearing capacity during embryogenesis. Dev. Camb. Engl. 138, 3247–3259. doi: 10.1242/dev.063768
Sheean, G., and McGuire, J. R. (2009). Spastic hypertonia and movement disorders: pathophysiology, clinical presentation, and quantification. PM R 1, 827–833. doi: 10.1016/j.pmrj.2009.08.002
Shellhaas, R. A., and Joshi, S. M. (2010). Vitamin D and bone health among children with epilepsy. Pediatr. Neurol. 42, 385–393. doi: 10.1016/j.pediatrneurol.2009.12.005
Shiek Ahmad, B., Petty, S. J., Gorelik, A., O’Brien, T. J., Hill, K. D., Christie, J. J., et al. (2017). Bone loss with antiepileptic drug therapy: a twin and sibling study. Osteoporos. Int. 28, 2591–2600. doi: 10.1007/s00198-017-4098-9
Shin, Y.-K., Yoon, Y. K., Chung, K. B., Rhee, Y., and Cho, S.-R. (2017). Patients with non-ambulatory cerebral palsy have higher sclerostin levels and lower bone mineral density than patients with ambulatory cerebral palsy. Bone 103, 302–307. doi: 10.1016/j.bone.2017.07.015
Shulman, B. S., Liporace, F. A., Davidovitch, R. I., Karia, R., and Egol, K. A. (2015). Sleep disturbance after fracture is related to emotional well-being rather than functional result. J. Orthop. Trauma 29, e146–e150.
Shultz, S. R., Sun, M., Wright, D. K., Brady, R. D., Liu, S., Beynon, S., et al. (2015). Tibial fracture exacerbates traumatic brain injury outcomes and neuroinflammation in a novel mouse model of multitrauma. J. Cereb. Blood Flow Metab. 35, 1339–1347. doi: 10.1038/jcbfm.2015.56
Siegel, I. M. (1977). Fractures of long bones in Duchenne muscular dystrophy. J. Trauma 17, 219–222. doi: 10.1097/00005373-197703000-00006
Signorelli, S. S., Scuto, S., Marino, E., Giusti, M., Xourafa, A., and Gaudio, A. (2019). Anticoagulants and osteoporosis. Int. J. Mol. Sci. 20:5275. doi: 10.3390/ijms20215275
Silverman, S. L. (1992). The clinical consequences of vertebral compression fracture. Bone 13(Suppl. 2) S27–S31.
Simm, P. J., Seah, S., Gorelik, A., Gilbert, L., Nuguid, J., Werther, G. A., et al. (2017). Impaired bone and muscle development in young people treated with antiepileptic drugs. Epilepsia 58, 1931–1938. doi: 10.1111/epi.13893
Simonsen, A. H., McGuire, J., Hansson, O., Zetterberg, H., Podust, V. N., Davies, H. A., et al. (2007). Novel panel of cerebrospinal fluid biomarkers for the prediction of progression to Alzheimer dementia in patients with mild cognitive impairment. Arch. Neurol. 64:366. doi: 10.1001/archneur.64.3.366
Simonsen, C. S., Celius, E. G., Brunborg, C., Tallaksen, C., Eriksen, E. F., Holmøy, T., et al. (2016). Bone mineral density in patients with multiple sclerosis, hereditary ataxia or hereditary spastic paraplegia after at least 10 years of disease - a case control study. BMC Neurol. 16:252. doi: 10.1186/s12883-016-0771-4
Sims, N. A. (2016). Cell-specific paracrine actions of IL-6 family cytokines from bone, marrow and muscle that control bone formation and resorption. Int. J. Biochem. Cell Biol. 79, 14–23. doi: 10.1016/j.biocel.2016.08.003
Singleton, Q., Vaibhav, K., Braun, M., Patel, C., Khayrullin, A., Mendhe, B., et al. (2019). Bone marrow derived extracellular vesicles activate osteoclast differentiation in traumatic brain injury induced bone loss. Cells 8:63. doi: 10.3390/cells8010063
Sinha, S., McGovern, R. A., and Sheth, S. A. (2015). Deep brain stimulation for severe autism: from pathophysiology to procedure. Neurosurg. Focus 38:E3.
Sioka, C., Papakonstantinou, S., Fotopoulos, A., Alamanos, Y., Georgiou, A., Tsouli, S., et al. (2011). Bone mineral density in ambulatory patients with multiple sclerosis. Neurol. Sci. 32, 819–824.
Sleeman, I., Che, Z. C., and Counsell, C. (2016). Risk of fracture amongst patients with Parkinson’s disease and other forms of parkinsonism. Parkinsonism Relat. Disord. 29, 60–65. doi: 10.1016/j.parkreldis.2016.05.026
Smith, É, and Carroll, Á (2011). Bone mineral density in adults disabled through acquired neurological conditions: a review. J. Clin. Densitom. 14, 85–94. doi: 10.1016/j.jocd.2010.12.002
Smith, É, Comiskey, C., and Carroll, Á (2016). Prevalence of and risk factors for osteoporosis in adults with acquired brain injury. Iran. J. Med. Sci. 185, 473–481. doi: 10.1007/s11845-016-1399-5
Sohrabi, H. R., Bates, K. A., Weinborn, M., Bucks, R. S., Rainey-Smith, S. R., Rodrigues, M. A., et al. (2015). Bone mineral density, adiposity, and cognitive functions. Front. Aging Neurosci. 7:16. doi: 10.3389/fnagi.2015.00016
Song, C., Wang, J., Kim, B., Lu, C., Zhang, Z., Liu, H., et al. (2018). Insights into the role of circadian rhythms in bone metabolism: a promising intervention target? Biomed. Res. Int. 2018:9156478.
Sophocleous, A., Robertson, R., Ferreira, N. B., McKenzie, J., Fraser, W. D., and Ralston, S. H. (2017). Heavy cannabis use is associated with low bone mineral density and an increased risk of fractures. Am. J. Med. 130, 214–221. doi: 10.1016/j.amjmed.2016.07.034
Specker, B. L., Binkley, T., Vukovich, M., and Beare, T. (2007). Volumetric bone mineral density and bone size in sleep-deprived individuals. Osteoporos. Int. 18, 93–99. doi: 10.1007/s00198-006-0207-x
Spiegel, K., Leproult, R., and Van Cauter, E. (1999). Impact of sleep debt on metabolic and endocrine function. Lancet Lond. Engl. 354, 1435–1439. doi: 10.1016/s0140-6736(99)01376-8
Strand, M., Soderstrom, I., Wiklund, P.-G., Hallmans, G., Weinehall, L., Soderberg, S., et al. (2007). Polymorphisms at the osteoprotegerin and interleukin-6 genes in relation to first-ever stroke. Cerebrovasc. Dis. Basel Switz. 24, 418–425. doi: 10.1159/000108431
Suljic, E., Mehicevic, A., and Mahmutbegovic, N. (2018). Effect of long-term carbamazepine therapy on bone health. Med. Arch. 72:262. doi: 10.5455/medarh.2018.72.262-266
Sun, Y., Yin, X. S., Guo, H., Han, R. K., He, R. D., and Chi, L. J. (2013). Elevated osteopontin levels in mild cognitive impairment and Alzheimer’s disease. Mediat. Inflamm. 2013, 1–9. doi: 10.1097/00002093-200401000-00001
Swanson, C. M., Kohrt, W. M., Buxton, O. M., Everson, C. A., Wright, K. P., Orwoll, E. S., et al. (2018). The importance of the circadian system & sleep for bone health. Metabolism. 84, 28–43.
Swanson, C. M., Kohrt, W. M., Wolfe, P., Wright, K. P., Shea, S. A., Cain, S. W., et al. (2019). Rapid suppression of bone formation marker in response to sleep restriction and circadian disruption in men. Osteoporos. Int. 30, 2485–2493. doi: 10.1007/s00198-019-05135-y
Swanson, C. M., Shea, S. A., Stone, K. L., Cauley, J. A., Rosen, C. J., Redline, S., et al. (2015). Obstructive sleep apnea and metabolic bone disease: insights into the relationship between bone and sleep: obstructive sleep apnea and bone disease. J. Bone Miner Res. 30, 199–211. doi: 10.1002/jbmr.2446
Swanson, C. M., Shea, S. A., Wolfe, P., Cain, S. W., Munch, M., Vujoviæ, N., et al. (2017). Bone turnover markers after sleep restriction and circadian disruption: a mechanism for sleep-related bone loss in humans. J. Clin. Endocrinol. Metab. 102, 3722–3730. doi: 10.1210/jc.2017-01147
Szollar, S. M., Martin, E. M. E., Parthemore, J. G., Sartoris, D. J., and Deftos, L. J. (1997). Densitometric patterns of spinal cord injury associated bone loss. Spinal Cord 35, 374–382. doi: 10.1038/sj.sc.3100394
Taeb, A. M., Hooper, M. H., and Marik, P. E. (2017). Sepsis: current definition, pathophysiology, diagnosis, and management. Nutr. Clin. Pract. 32, 296–308. doi: 10.1177/0884533617695243
Takeda, S., Elefteriou, F., Levasseur, R., Liu, X., Zhao, L., Parker, K. L., et al. (2002). Leptin regulates bone formation via the sympathetic nervous system. Cell 111, 305–317. doi: 10.1016/s0092-8674(02)01049-8
Tamimi, I., Ojea, T., Sanchez-Siles, J. M., Rojas, F., Martin, I., Gormaz, I., et al. (2012). Acetylcholinesterase inhibitors and the risk of hip fracture in Alzheimer’s disease patients: a case-control study. J. Bone Miner Res. 27, 1518–1527. doi: 10.1002/jbmr.1616
Tan, Z. S., Seshadri, S., Beiser, A., Zhang, Y., Felson, D., Hannan, M. T., et al. (2005). Bone mineral density and the risk of Alzheimer disease. Arch. Neurol. 62:107.
Tassorelli, C., Berlangieri, M., Buscone, S., Bolla, M., De Icco, R., Baricich, A., et al. (2017). Falls, fractures and bone density in Parkinson’s disease – a cross-sectional study. Int. J. Neurosci. 127, 299–304. doi: 10.1080/00207454.2016.1206897
Terzi, R., and Yılmaz, Z. (2016). Bone mineral density and changes in bone metabolism in patients with obstructive sleep apnea syndrome. J. Bone Miner Metab. 34, 475–481. doi: 10.1007/s00774-015-0691-1
Terzi, T., Terzi, M., Tander, B., Cantürk, F., and Onar, M. (2010). Changes in bone mineral density and bone metabolism markers in premenopausal women with multiple sclerosis and the relationship to clinical variables. J. Clin. Neurosci. 17, 1260–1264. doi: 10.1016/j.jocn.2010.01.044
Thakkar, P., Prakash, N. B., Tharion, G., Shetty, S., Paul, T. V., Bondu, J., et al. (2020). Evaluating bone loss with bone turnover markers following acute spinal cord injury. Asian Spine J. 14, 97–105. doi: 10.31616/asj.2019.0004
Thijs, R. D., Surges, R., O’Brien, T. J., and Sander, J. W. (2019). Epilepsy in adults. Lancet 393, 689–701.
Thomas, T., Gori, F., Khosla, S., Jensen, M. D., Burguera, B., and Riggs, B. L. (1999). Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology 140, 1630–1638. doi: 10.1210/endo.140.4.6637
Tomiyama, H., Okazaki, R., Inoue, D., Ochiai, H., Shiina, K., Takata, Y., et al. (2008). Link between obstructive sleep apnea and increased bone resorption in men. Osteoporos. Int 19, 1185–1192. doi: 10.1007/s00198-007-0556-0
Trentz, O. A., Handschin, A. E., Bestmann, L., Hoerstrup, S. P., Trentz, O. L., and Platz, A. (2005). Influence of brain injury on early posttraumatic bone metabolism. Crit. Care Med. 33, 399–406. doi: 10.1097/01.ccm.0000152221.87477.21
Trinh, A., Wong, P., Fahey, M. C., Brown, J., Strauss, B. J., Ebeling, P. R., et al. (2019). Longitudinal changes in bone density in adolescents and young adults with cerebral palsy: a case for early intervention. Clin. Endocrinol. 91, 517–524. doi: 10.1111/cen.14052
Tyblova, M., Kalincik, T., Zikan, V., and Havrdova, E. (2015). Impaired ambulation and steroid therapy impact negatively on bone health in multiple sclerosis. Eur. J. Neurol. 22, 624–632. doi: 10.1111/ene.12479
Uhle, F., Chousterman, B. G., Grützmann, R., Brenner, T., and Weber, G. F. (2016). Pathogenic, immunologic, and clinical aspects of sepsis - update 2016. Expert Rev. Anti Infect. Ther. 14, 917–927. doi: 10.1080/14787210.2016.1224971
Uzkeser, H., Yildirim, K., Aktan, B., Karatay, S., Kaynar, H., Araz, O., et al. (2013). Bone mineral density in patients with obstructive sleep apnea syndrome. Sleep Breath 17, 339–342. doi: 10.1007/s11325-012-0698-y
Vai, S., Bianchi, M. L., Moroni, I., Mastella, C., Broggi, F., Morandi, L., et al. (2015). Bone and spinal muscular atrophy. Bone 79, 116–120.
van den Bos, F., Speelman, A. D., van Nimwegen, M., van der Schouw, Y. T., Backx, F. J. G., Bloem, B. R., et al. (2013). Bone mineral density and vitamin D status in Parkinson’s disease patients. J. Neurol. 260, 754–760.
Vega, D., Maalouf, N. M., and Sakhaee, K. (2007). The role of receptor activator of nuclear factor-κB (RANK)/RANK Ligand/Osteoprotegerin: clinical implications. J. Clin. Endocrinol. Metab. 92, 4514–4521. doi: 10.1210/jc.2007-0646
Veilleux, L.-N., and Rauch, F. (2017). Muscle-bone interactions in pediatric bone diseases. Curr. Osteoporos. Rep. 15, 425–432. doi: 10.1007/s11914-017-0396-6
Vestergaard, P., Glerup, H., Steffensen, B. F., Rejnmark, L., Rahbek, J., and Moseklide, L. (2001). Fracture risk in patients with muscular dystrophy and spinal muscular atrophy. J. Rehabil. Med. 33, 150–155. doi: 10.1080/16501970119066
Vestergaard, P., Rejnmark, L., and Mosekilde, L. (2006). Anxiolytics, sedatives, antidepressants, neuroleptics and the risk of fracture. Osteoporos. Int. 17, 807–816. doi: 10.1007/s00198-005-0065-y
Vestergaard, P., Rejnmark, L., and Mosekilde, L. (2008). Selective serotonin reuptake inhibitors and other antidepressants and risk of fracture. Calcif. Tissue Int. 82, 92–101. doi: 10.1007/s00223-007-9099-9
Vibert, D., Kompis, M., and Häusler, R. (2003). Benign paroxysmal positional vertigo in older women may be related to osteoporosis and osteopenia. Ann. Otol. Rhinol. Laryngol. 112, 885–889. doi: 10.1177/000348940311201010
Villeda, S. A., Plambeck, K. E., Middeldorp, J., Castellano, J. M., Mosher, K. I., Luo, J., et al. (2014). Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 20, 659–663. doi: 10.1038/nm.3569
Vogt, M. H., ten Kate, J., Drent, R. J., Polman, C. H., and Hupperts, R. (2010). Increased osteopontin plasma levels in multiple sclerosis patients correlate with bone-specific markers. Mult. Scler J. 16, 443–449. doi: 10.1177/1352458509359723
Wakata, N., Nemoto, H., Sugimoto, H., Nomoto, N., Konno, S., Hayashi, N., et al. (2004). Bone density in myasthenia gravis patients receiving long-term prednisolone therapy. Clin. Neurol. Neurosurg. 106, 139–141. doi: 10.1016/j.clineuro.2003.12.001
Wang, K. X., and Denhardt, D. T. (2008). Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev. 19, 333–345. doi: 10.1016/j.cytogfr.2008.08.001
Wang, Y., Li, Y., Mao, K., Li, J., Cui, Q., and Wang, G.-J. (2003). Alcohol-induced adipogenesis in bone and marrow: a possible mechanism for osteonecrosis. Clin. Orthop. 410, 213–224. doi: 10.1097/01.blo.0000063602.67412.83
Wang, Z., Yao, G., Tao, X., Zhang, J., Zhang, T., and Wu, Z. (2020). Evaluation of bone mineral density and 25-(OH) vitamin D levels in middle-aged and elderly women with recurrent benign paroxysmal positional vertigo. Acta Otolaryngol. 140, 89–93. doi: 10.1080/00016489.2019.1692146
Weller, I. (2004). Hip fractures and Alzheimer’s disease in elderly institutionalized Canadians. Ann. Epidemiol. 14, 319–324. doi: 10.1016/j.annepidem.2003.08.005
Wen, S.-R., Liu, G.-J., Feng, R.-N., Gong, F.-C., Zhong, H., Duan, S.-R., et al. (2012). Increased levels of IL-23 and osteopontin in serum and cerebrospinal fluid of multiple sclerosis patients. J. Neuroimmunol. 244, 94–96. doi: 10.1016/j.jneuroim.2011.12.004
Wilmet, E., Ismail, A. A., Heilporn, A., Welraeds, D., and Bergmann, P. (1995). Longitudinal study of the bone mineral content and of soft tissue composition after spinal cord section. Spinal Cord 33, 674–677. doi: 10.1038/sc.1995.141
Woelfer, M., Kasties, V., Kahlfuss, S., and Walter, M. (2019). The role of depressive subtypes within the neuroinflammation hypothesis of major depressive disorder. Neuroscience 403, 93–110. doi: 10.1016/j.neuroscience.2018.03.034
Wu, Y., Gu, C., Han, W., Lu, X., Chen, C., and Fan, Z. (2018). Reduction of bone mineral density in native Chinese female idiopathic benign paroxysmal positional vertigo patients. Am. J. Otolaryngol. 39, 31–33. doi: 10.1016/j.amjoto.2017.09.004
Xia, W.-F., Jung, J.-U., Shun, C., Xiong, S., Xiong, L., Shi, X.-M., et al. (2013). Swedish mutant APP suppresses osteoblast differentiation and causes osteoporotic deficit, which are ameliorated by N-acetyl-L-cysteine: app inhibits bone formation. J. Bone Miner Res. 28, 2122–2135. doi: 10.1002/jbmr.1954
Xing, W., Govoni, K. E., Donahue, L. R., Kesavan, C., Wergedal, J., Long, C., et al. (2012). Genetic evidence that thyroid hormone is indispensable for prepubertal insulin-like growth factor-I expression and bone acquisition in mice. J. Bone Miner Res. 27, 1067–1079. doi: 10.1002/jbmr.1551
Yamanaka, T., Shirota, S., Sawai, Y., Murai, T., Fujita, N., and Hosoi, H. (2013). Osteoporosis as a risk factor for the recurrence of benign paroxysmal positional vertigo: osteoporosis and BPPV. Laryngoscope 123, 2813–2816. doi: 10.1002/lary.24099
Yanagihara, R., Garruto, R. M., Gajdusek, D. C., Tomita, A., Uchikawa, T., Konagaya, Y., et al. (1984). Calcium and vitamin D metabolism in Guamanian Chamorros with amyotrophic lateral sclerosis and parkinsonism-dementia. Ann. Neurol. 15, 42–48. doi: 10.1002/ana.410150108
Yang, F. Z., Jehu, D. A. M., Ouyang, H., Lam, F. M. H., and Pang, M. Y. C. (2020). The impact of stroke on bone properties and muscle-bone relationship: a systematic review and meta-analysis. Osteoporos. Int. 31, 211–224. doi: 10.1007/s00198-019-05175-4
Yang, L., Guo, Y., Wen, D., Yang, L., Chen, Y., Zhang, G., et al. (2016). Bone fracture enhances trauma brain injury. Scand. J. Immunol. 83, 26–32. doi: 10.1111/sji.12393
Yang, M.-W., Wang, T.-H., Yan, P.-P., Chu, L.-W., Yu, J., Gao, Z.-D., et al. (2011). Curcumin improves bone microarchitecture and enhances mineral density in APP/PS1 transgenic mice. Phytomedicine 18, 205–213. doi: 10.1016/j.phymed.2010.05.011
Yates, B. J., Bolton, P. S., and Macefield, V. G. (2014). Vestibulo-sympathetic responses. Compr. Physiol. 4, 851–887. doi: 10.1002/cphy.c130041
Yeh, J.-H., Chen, H.-J., Chen, Y.-K., Chiu, H.-C., and Kao, C.-H. (2014). Increased risk of osteoporosis in patients with myasthenia gravis: a population-based cohort study. Neurology 83, 1075–1079. doi: 10.1212/wnl.0000000000000804
Yirmiya, R., Goshen, I., Bajayo, A., Kreisel, T., Feldman, S., Tam, J., et al. (2006). Depression induces bone loss through stimulation of the sympathetic nervous system. Proc. Natl. Acad. Sci. U.S.A. 103, 16876–16881. doi: 10.1073/pnas.0604234103
Yoon, Y.-S., Park, J.-S., Tkebuchava, T., Luedeman, C., and Losordo, D. W. (2004). Unexpected severe calcification after transplantation of bone marrow cells in acute myocardial infarction. Circulation 109, 3154–3157. doi: 10.1161/01.cir.0000134696.08436.65
Yu, H., Watt, H., and Mohan, S. (2014a). The negative impact of traumatic brain injury (TBI) on bone in a mouse model. Brain Inj. 28, 244–251. doi: 10.3109/02699052.2013.859735
Yu, H., Watt, H., Kesavan, C., Johnson, P. J., Wergedal, J. E., and Mohan, S. (2012). Lasting consequences of traumatic events on behavioral and skeletal parameters in a mouse model for post-traumatic stress disorder (PTSD). PLoS One 7:e42684. doi: 10.1371/journal.pone.0042684
Yu, H., Wergedal, J. E., Rundle, C. H., and Mohan, S. (2014b). Reduced bone mass accrual in mouse model of repetitive mild traumatic brain injury. J. Rehabil. Res. Dev. 51, 1427–1438. doi: 10.1682/jrrd.2014.04.0095
Zegarra-Valdivia, J. A. (2017). Insulin-like growth factor type 1 and its relation with neuropsychiatric disorders. Medwave 17:e7031. doi: 10.5867/medwave.2017.07.7031
Zhang, Y., Zheng, Y., Zhu, J., Zhang, J., and Zheng, Z. (2015). Effects of antiepileptic drugs on bone mineral density and bone metabolism in children: a meta-analysis. J. Zhejiang Univ. Sci. B. 16, 611–621. doi: 10.1631/jzus.b1500021
Zhao, Y., Shen, L., and Ji, H.-F. (2012). Alzheimer’s disease and risk of hip fracture: a meta-analysis study. Sci. World J. 2012, 1–5. doi: 10.1100/2012/872173
Zhao, Y., Shen, L., and Ji, H.-F. (2013). Osteoporosis risk and bone mineral density levels in patients with Parkinson’s disease: a meta-analysis. Bone 52, 498–505. doi: 10.1016/j.bone.2012.09.013
Zhou, J., Yi, J., and Bonewald, L. (2015). Muscle-bone crosstalk in amyotrophic lateral sclerosis. Curr. Osteoporos. Rep. 13, 274–279. doi: 10.1007/s11914-015-0281-0
Zhou, R., Deng, J., Zhang, M., Zhou, H.-D., and Wang, Y.-J. (2011). Association between bone mineral density and the risk of Alzheimer’s disease. J. Alzheimers Dis. 24, 101–108. doi: 10.3233/jad-2010-101467
Zhou, R., Zhou, H., Rui, L., and Xu, J. (2014). Bone loss and osteoporosis are associated with conversion from mild cognitive impairment to Alzheimer’s disease. Curr. Alzheimer Res. 11, 706–713. doi: 10.2174/1567205011666140812115818
Zhu, K., Yi, J., Xiao, Y., Lai, Y., Song, P., Zheng, W., et al. (2015). Impaired bone homeostasis in amyotrophic lateral sclerosis mice with muscle atrophy. J. Biol. Chem. 290, 8081–8094. doi: 10.1074/jbc.m114.603985
Zikán, V., Tıblová, M., Raška, I. Jr., Havrdová, E., Luchavová, M., Michalská, D., et al. (2012). Bone mineral density and body composition in men with multiple sclerosis chronically treated with low-dose glucocorticoids. Physiol. Res. 31, 405–417. doi: 10.33549/physiolres.932299
Keywords: Neurology, disease, osteoporosis, bone, mental health, depression, PTSD
Citation: Kelly RR, Sidles SJ and LaRue AC (2020) Effects of Neurological Disorders on Bone Health. Front. Psychol. 11:612366. doi: 10.3389/fpsyg.2020.612366
Received: 30 September 2020; Accepted: 11 November 2020;
Published: 30 November 2020.
Edited by:
Carmelo Mario Vicario, University of Messina, ItalyReviewed by:
Antonino Catalano, University of Messina, ItalyCopyright © 2020 Kelly, Sidles and LaRue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda C. LaRue, bGFydWVyY0BtdXNjLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.