 Rickinder Sethi1
Rickinder Sethi1 Nieves Gómez-Coronado2
Nieves Gómez-Coronado2 Adam J. Walker3
Adam J. Walker3 Oliver D’Arcy Robertson3,4
Oliver D’Arcy Robertson3,4 Bruno Agustini3
Bruno Agustini3 Michael Berk3,4,5,6,7*
Michael Berk3,4,5,6,7* Seetal Dodd3,4,5,6
Seetal Dodd3,4,5,6- 1Department of Psychiatry, Western University, London, ON, Canada
- 2Unidad de Gestión Clinica Salud Mental, Hospital Universitario Virgen del Rocio, Sevilla, Spain
- 3IMPACT Strategic Research Centre, Deakin University, Geelong, VIC, Australia
- 4University Hospital Geelong, Barwon Health, Geelong, VIC, Australia
- 5Department of Psychiatry, The University of Melbourne, Parkville, VIC, Australia
- 6Orygen, The National Centre of Excellence in Youth Mental Health, Parkville, VIC, Australia
- 7Department of Psychiatry, Florey Institute of Neuroscience and Mental Health, Parkville, VIC, Australia
Neuropsychiatric disorders, such as depression, bipolar disorder, schizophrenia, obsessive-compulsive disorder, and neurodevelopmental disorders such as autism spectrum disorder, are associated with significant illness burden. Accumulating evidence supports an association between these disorders and inflammation. Consequently, anti-inflammatory agents, such as the cyclooxygenase-2 inhibitors, represent a novel avenue to prevent and treat neuropsychiatric illness. In this paper, we first review the role of inflammation in psychiatric pathophysiology including inflammatory cytokines’ influence on neurotransmitters, the hypothalamic–pituitary–adrenal axis, and microglial mechanisms. We then discuss how cyclooxygenase-2-inhibitors influence these pathways with potential therapeutic benefit, with a focus on celecoxib, due to its superior safety profile. A search was conducted in PubMed, Embase, and PsychINFO databases, in addition to Clinicaltrials.gov and the Stanley Medical Research Institute trial registries. The results were presented as a narrative review. Currently available outcomes for randomized controlled trials up to November 2017 are also discussed. The evidence reviewed here suggests cyclooxygenase-2 inhibitors, and in particular celecoxib, may indeed assist in treating the symptoms of neuropsychiatric disorders; however, further studies are required to assess appropriate illness stage-related indication.
Introduction
The immune system involves a complex array of cells, tissues, and organs working in concert to protect the body from foreign molecules at both the intracellular and extracellular level (1). Pro-inflammatory and anti-inflammatory cytokines, along with other mechanisms, balance the inflammatory response (1). External causes of inflammation include microbial or viral infections, cigarette smoking, poor dietary composition, air pollution, and trauma (both physical and psychological), among others (2). Internal causes may include ischemic events or malignancy (1). In instances where inflammatory mediators are unable to inhibit the pro-inflammatory immune reaction, a chronic inflammatory state may ensue. Chronic activation of this system can lower the allostatic load threshold, contributing to the development of neuropsychiatric disorders (2).
While monoaminergic dysregulation remains a prevailing hypothesis regarding neuropsychiatric disorders, refractory illnesses remain a significant challenge in addition to a relative paucity of novel treatment options (3). In recent years, the inflammatory model has been revisited due to the fragmented efficacy of the current management approaches. As a result, more attention is being paid to pharmacotherapies that lay outside the traditional vault of psychotropic agents such as antidepressants and antipsychotics. Cyclooxygenase-2 inhibitors, best known for their role in acute pain management, are a potent example of this pharmacological appropriation. Celecoxib and rofecoxib—selective cyclooxygenase-2 inhibitors—have been investigated for their efficacy as both stand-alone therapies and augmentation agents in psychiatry.
The purpose of this paper is twofold: first, to review the relationship between inflammation and neuropsychiatric illnesses, and second, to provide a review of randomized control trials (RCTs) that investigate the use of cyclooxygenase-2 inhibitors for the treatment of select neuropsychiatric illnesses.
Methods
A Boolean search was conducted for literature published up to November 19, 2017. We searched PubMed, Embase, and PsychINFO databases, and the Clinicaltrials.gov and The Stanley Medical Research Institute trial registries. Search terms included are attached in Appendix A. Articles were selected for human, randomized clinical trials and treatment efficacy. The search was augmented by manually searching the references of key papers and related literature. We adhered to PRISMA guidelines and flowsheet attached in Appendix A. Table 1 contains a summary chart of the results. The results were presented as a narrative review format.
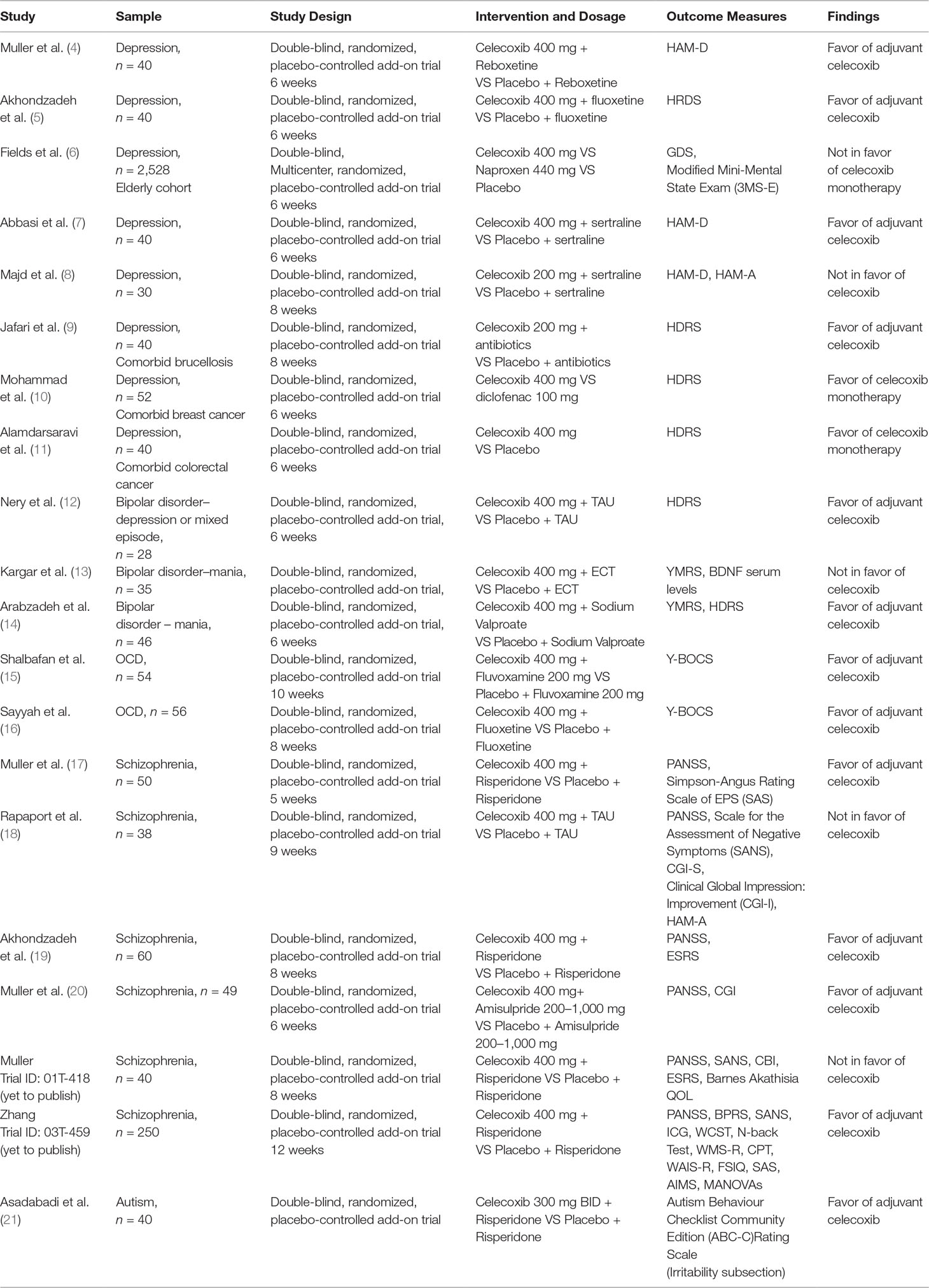
Table 1 Clinical trials investigating celecoxib in neuropsychiatric disorders.
The Link Between Inflammatory System, the Brain, and Mental Illness
In 1927, Julius Wagner-Jauregg became the first and the only psychiatrist thus far to win a Nobel Prize in Medicine. His impactful discovery involved the association with inflammation via malaria inoculation to cure neuropsychiatric symptoms of syphilis (22). Unfortunately, this inflammatory etiological theory was set aside during the advent of the psychotropic revolution (23). While support for the monoamine hypothesis in neuropsychiatric disorders continued to gain traction in subsequent decades, a residual group of patients exhibited persistent treatment-refractory illnesses and chronic debilitating symptoms suggestive of alternate hypotheses for neuropsychiatric conditions (3).
Innate and Adaptive Immunity
Immune system responses are typically classified as either innate or adaptive. The innate immune system features elements that are both genetically heritable and evolutionarily ancient, found in all multicellular organisms (24,). The innate system’s principal phagocytes include neutrophils, monocytes, and macrophages, which work in synergy to establish the first-line barrier of immunity (26). This line of defense is supplemented by the adaptive immune system, which includes specialized cells, B-lymphocytes and T-lymphocytes. Both response sectors produce a composite operation of moderating immunotransmitters, defined by cytokines. These immunomodulatory cytokines are typically categorized as pro-inflammatory or anti-inflammatory on the basis of their general effects. Pro-inflammatory cytokines such as tumor necrosis factor alpha (TNFα), interferon gamma (IFNγ), and interleukin (IL)-1 and IL-6 are primarily secreted by monocytes and macrophages, promoting additional complex inflammatory response systems, discussed in detail elsewhere [e.g., Ref. (27)]. Anti-inflammatory cytokines include IL-4, IL-10, IL-11, and IL-13 (26,). In simplistic terms, imbalanced pro-inflammatory over anti-inflammatory cytokine load will preferentially increase the throughput of pathological cellular pathways.
Central Nervous System Immunity
The blood–brain barrier is the brain’s primary defense against chemical insult. During peripheral inflammatory activation, there is increased permeability of the blood–brain barrier (29,). Such increases in blood–brain permeability may exacerbate or possibly even initiate neuropsychiatric and neurological disorders [see Ref. (31) for a review]. Furthermore, recent identification of lymphatic vessels within the central nervous system (CNS) reveals an alternate route of communication with the immune system to the brain (32). Once activated, a host of cellular and chemical pathways within the brain can result in significant structural change.
Microglia are specialized macrophages localized to the CNS that also play an important regulatory role in inflammatory response. They secrete neurotrophic factors important for cellular repair and signal recruitment for immune cells (26,,). The role microglia play in inflammation-driven neuronal damage and degeneration is also well established (35). Microglia have been shown to express different phenotypes or polarizations, classified as M1 and M2. M1 polarization is influenced by the pro-inflammatory state and acts in neuronal apoptosis, while the M2 polarization, in contrast, promotes neurogenesis (32,–39). Interferon alpha (IFNα) has been shown to induce a pro-inflammatory shift in microglial phenotype, from M2 to M1, resulting in depressive symptoms in mice (40,).
Inflammation and Neuropsychiatric Symptoms
When anti-inflammatory regulators are unable to balance pro-inflammatory reactions, inflammation can persist, in conjunction with sub-threshold neuropsychiatric symptoms (42). The following mechanisms describe the cytokine communication with neuropsychiatric symptoms and thus support the inflammatory hypothesis.
Pro-inflammatory markers have been associated with the development of neuropsychiatric symptoms (43). IL-6 activates the type 2 immune response, prompting the B-cell maturation pathway, consequently producing antibodies directed against extracellular pathogens. In addition, IL-6 activates the release of C-reactive protein (CRP) from the liver. Elevated levels of CRP and IL-6 in childhood were associated with an increased risk of developing depressive and psychotic symptoms in the future (44). Consistent with this, a significant association has been reported between CRP and several neuropsychiatric disorders, including depression, anxiety, and schizophrenia (45,). CRP is a well-established biomarker for an active inflammatory process and is a significant independent predictor of coronary heart disease risk (47–53). Furthermore, elevated CRP and IL-6 levels have also been associated with cognitive dysfunction (54). Studies have shown that biomarkers such as CRP and IL-6 may shed light on subtyping depression (55,). Several meta-analyses have demonstrated significant evidence of elevated pro-inflammatory cytokines in patients with depressive symptoms (43,,), bipolar disorder (58–60), schizophrenia (58,), obsessive compulsive disorder (OCD) (62), and autism spectrum disorders (ASDs) (63). These pro-inflammatory markers include CRP, IL-6, TNFα, and the IL-1 receptor antagonist (IL-1Ra) (43, 57–63). In addition, pro-inflammatory cytokines have been shown to trend toward normalization with symptom improvement indicating treatment response (64). Participation from microglia and peripheral macrophages are identified in activated inflammatory networks (65). An exaggerated immune response can be responsible for neuronal damage and decreased brain derived neurotrophic factor (BDNF), a protein integral to neuronal growth, plasticity, and survival (66–70). Excessive activation of the immune system may exacerbate mental illness in a subgroup of vulnerable individuals (71, 72). The inflammatory hypothesis suggests that hyperactivation of the immune system may produce nitro-oxidative stress and alterations of the kynurenine pathway, subsequently dysregulating monoamine levels and activating the glutamatergic system (2, 15, 73).
The evidence for cytokine-induced neuropsychiatric symptoms in healthy participants favors the inflammatory model. For example, healthy participants received an infusion of endotoxin to induce an inflammatory response, with resultant mood symptoms (74). Similarly, healthy individuals who received exogenous cytokines (IL-2, IFNα, and TNFα) also developed neuropsychiatric symptoms, including depression, mania, emotional dysregulation, cognitive impairment, and/or avolition (75). Elevated serum pro-inflammatory markers, TNFα, IL-6, and cortisol levels were observed by Salmonella abortus equi endotoxin injections (76). Subsequently, the subjects also exhibited neuropsychiatric symptoms of appetite changes, mood and anxiety symptoms, and cognitive decline without physical sickness symptoms (77). These findings were replicated with other vaccinations of healthy individuals (74, 78). Preclinical research has yielded similar findings, wherein lipopolysaccharide (LPS) and IL-1 injections in mice were found to result in sickness behavior, an analog to depressive symptoms (79).
Notably, pro-inflammatory agents such as recombinant IFNα and IL-2 have been used in the treatment of hepatitis C and carcinomas, respectively (80, 81). Interestingly, while effective for treating the targeted indication, IFNα was found to induce significant neuropsychiatric side effects with up to 80% of patients endorsing mild to moderate depressive symptoms (82–84). Grigoleit et al. (85) identified a positive dose-dependent association in IL-6, IL-10, TNFα, cortisol, and norepinephrine with neuropsychiatric symptoms. While some studies have reported that serum IL-6 and TNFα also appeared elevated in OCD patients compared to healthy controls [e.g., Ref. (86)], it should be noted that an earlier meta-analysis of OCD patients revealed no significant difference in TNFα or IL-6 (62). However, these authors did note a reduced level of pro-inflammatory IL-1β in OCD patients (62).
Autoimmune Conditions and Neuropsychiatric Disorders
Autoimmune and infectious conditions such as rheumatoid arthritis (87), type 1 diabetes (88), systemic lupus erythematosus (89), hepatitis, and sepsis (90) increase the risk of neuropsychiatric symptoms. An extensive Danish-based study found a 62% increased risk of mood disorders after infection-related hospitalizations (90). In patients who suffer from both Crohn’s disease and depression, exacerbations of both physical and mental illnesses tend to occur at the same time (91). Patients experiencing psoriasis and anxiety symptoms were shown to benefit from treatment with cytokine inhibitors (92–94). In addition, a greater prevalence of autoimmune conditions such as pemphigus in bipolar disorder is observed (95). In a large epidemiological study, multiple infections and autoimmune disorders were associated with the increased lifetime prevalence of schizophrenia spectrum disorders (96).
In summary, multiple studies have indicated a positive association between inflammation and neuropsychiatric symptoms. Elevated pro-inflammatory markers are consistently associated with neuropsychiatric symptoms and reveal a bidirectional relationship.
Cyclooxygenase-2 (COX-2) Inhibitors
The COX pathway involves the precursor substrate of arachidonic acid (AA) to produce thromboxane, prostacyclin, and prostaglandins (PG) D2, E2, F2, and I2. AA is extracted from cell membranes by phospholipases, predominately cytoplasmic phospholipase A2 (cPLA2), and metabolized by the COX enzymes. The two rate-limiting enzymes within the COX pathways are COX-1 and COX-2. COX-1 is constitutionally expressed, and thus responsible for baseline prostaglandin levels, whereas COX-2 is inducible and expressed exclusively in the CNS, kidney, thymus, GI tract, and possibly in the female reproductive system (97–100). However, during inflammatory processes, COX-2 expression is promoted by and regulated by inflammatory stimuli, including lipopolysaccharide (LPS), IL-1, IL-6, TNF, IFNγ, and AA (101–108). The variability of isotype expressions COX-1 and COX-2 might be explained by the target tissue and type of insult (109, 110). The COX-2 enzyme produces prostaglandins, thromboxanes, prostacyclins, and leukotrienes downstream, which are suspected to be culprits for inflammation and neoplastic growth, in particular PGE2 (111). Furthermore, COX-2 expression is supported by microglia (112) and is auto-regulated via its by-products, PGE2 and PGF2α, perpetuating the inflammation process (113, 114). Additionally, PGE2 promotes IL-6 production (115), subsequently reinforcing this positive inflammatory feedback loop (Figure 1). Notably, in COX-2 gene knockout mice, a decrease in PGE2 and nuclear factor (NF)-κB activity was observed (114, 116, 117).
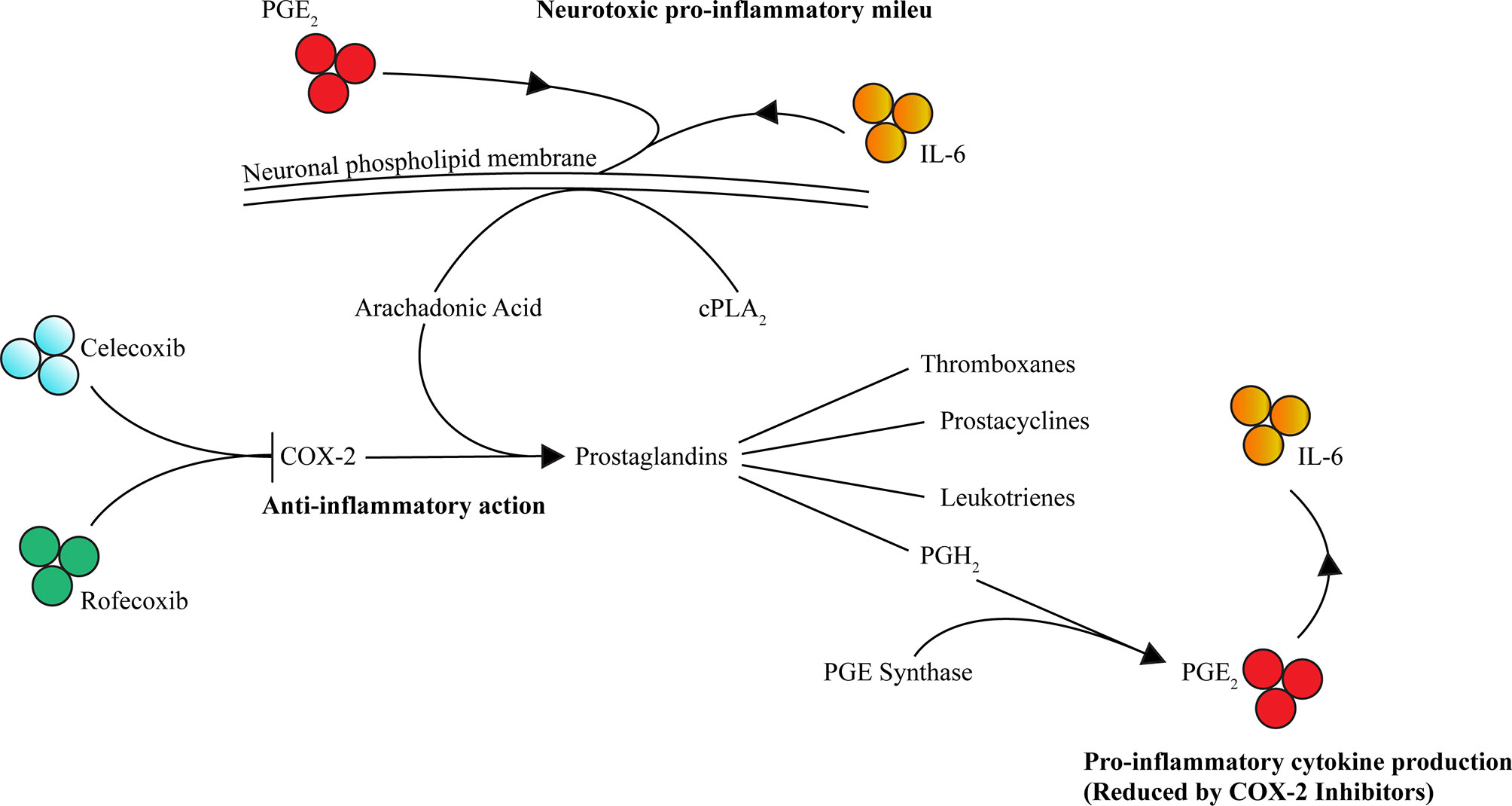
Figure 1 COX-2 contributes to inflammation through PGE2 and IL-6 and is selectively inhibited by celecoxib and rofecoxib. Activated by inflammatory cytokines including PGE2 and IL-6, AA (the precursor substrate of the COX-2 pathway) is extracted from phospholipid membranes by phospholipases such as cPLA2. COX-2 then drives production of prostaglandins including prostaglandin H2 (PGH2), which in turn is converted to PGE2 via PGE synthase. An accumulation of PGE2 leads to increased IL-6 (along with other cytokines) contributing to the inflammatory milieu, further potentiation of the pathway and neurotoxicity contributing to psychopathology. Celecoxib and rofecoxib exert selective inhibition of COX-2 reducing this pathway’s contribution to inflammation mediated neurotoxicity (→ = activates/increases, ⊥ = inhibits).
In 1995, the first generation of selective COX-2 inhibitors, celecoxib and rofecoxib, entered clinical trials (118). Over the next 4 years, numerous trials demonstrated selective COX-2 inhibitors could reduce pain and inflammation (119). From 2000 to 2004, larger trials such as CLASS, VIGOR, and TARGET identified the reduced gastrointestinal risks associated with COX-2 inhibitors, however, highlighted the increased cardiovascular risks leading to rofecoxib being pulled from the market (120–123). In 2005, valdecoxib was also withdrawn from the market for similar concerns by the Food and Drug Administration (FDA). In 2011, celecoxib was withdrawn from the market for the indication of cancer prevention while still being indicated for rheumatoid arthritis, osteoarthritis, and acute pain. Subsequently, this led to cautious prescribing due to concerns of cardiovascular side effects (124).
Preclinical trials have shown that rofecoxib can increase serotonin levels in the frontal and temporoparietal cortex (125). In addition, celecoxib was shown to potentiate the effects of reboxetine and fluoxetine on cortical noradrenaline and serotonin output (126).
Although ubiquitously expressed, COX-1 is typically described as gastro-protective and neuroprotective (127, 128), and pre-clinical data reveal some contradictory evidence regarding the inflammatory classifications of the COX isoforms. COX-2 may have anti-inflammatory and neuroprotective properties, aiding in neurotransmitter release, long-term potentiation, blood flow regulation, and memory consolidation (129). It was reported that COX-2 gene knockout mice were susceptible to inflammation compared to healthy mice (130, 131). Deletion of the COX-2 gene causes increased permeability of the blood–brain barrier and leukocyte infiltration (132). Another study showed that maximal COX-2 expression coincided with inflammatory resolution and was associated with minimal PGE2 synthesis (133).
Nevertheless, COX-2 inhibitors have proven to be beneficial in glutamate-mediated death prevention and suppression of pro-inflammatory cytokines (134, 135). Kainic acid (KA), a potent neurotoxin, elicits excitatory effects on N-methyl-D-aspartate (NMDA) receptors resulting in status epilepticus, neurodegeneration, and memory loss (134–138). KA has also been shown to increase COX-2 expression in the CNS (139); therefore, COX-2 inhibitors have been shown to prevent KA-induced neuronal death (140). There appears to be mounting evidence of therapeutic effect of COX-2 inhibitors in mediating glutamatergic processes (141–143). As well, there seems to be appropriate evidence for COX pathway in neuropsychiatric disorders.
Pathophysiology of Neuropsychiatric Disorders and the Efficacy of Cyclooxygenase-2 Inhibitors
We have now established there is substantive evidence for inflammation being a driver of neuropsychiatric symptoms by negatively impacting on neuronal proliferation, survival, and differentiation (144, 145). Furthermore, the COX pathway potentiates the inflammatory process and may exacerbate inflammation mediated neurodegeneration. COX-2 inhibition reduces this inflammatory load and thus the impact of these pathways on the brain.
In this section, we address the proposed mechanisms by which inflammatory states influence central monoamine effects, the hypothalamic–pituitary–adrenal (HPA) axis, and microglial activation, pathways at the center of neuropsychiatric pathogenesis. Discussion will focus on specific conditions including depression, bipolar disorder, schizophrenia, ASD, and OCD. Herein, a number of clinical trials investigating the efficacy of the cyclooxygenase-2 inhibitors for neuropsychiatric disorders are discussed, summarized in Table 1. Particular attention is being paid to the operative inflammatory pathways inherent to these conditions and the potential role for COX-2 inhibitors in their management.
Neurotransmitter Dysregulation Hypothesis
The dysregulation of the neurotransmitters such as serotonin, norepinephrine, dopamine, acetylcholine, and glutamate has been the foci of the biochemical etiology of neuropsychiatric illnesses. While treatment with antidepressants and neuroleptics aims to modulate monoamine signaling, there is a wealth of evidence supporting secondary mechanisms of action including effects on inflammatory pathways (27, 146). During inflammation, the pro-inflammatory cytokines IL-2 and IFNα have been shown to directly increase enzyme activity of the indoleamine-pyrrole 2,3-dioxygenase (IDO) enzyme of the kynurenine pathway, which promotes conversion of tryptophan to kynurenine, consequently depleting the antecedent supply to serotonin (147), in addition to direct catabolism of serotonin by IL-6 and IFNα (148, 149). This evidence supports the monoamine hypothesis regarding the hypoactive serotonin state featured in mood disorders (150–152). IFNα administration led to inflammation by increasing the concentration of kynurenine pathway metabolites in the CSF, namely kynurenine, kynurenic acid, and quinolinic acid (QUIN) (153). These metabolites have been presented as inducers of depressive and anxiety symptoms (154). Notably, QUIN can selectively activate NMDA receptors (155, 156) and has been associated with numerous neurological diseases, including: Alzheimer’s disease, anxiety, depression, epilepsy, human immunodeficiency virus-associated neurocognitive disorders, and Huntington’s disease (155, 157–161). QUIN has also been shown to cause neurodegeneration via multiple models (159).
HPA Axis Dysregulation
During a stress response, the HPA axis is activated (162). The hypothalamus secretes two hormones, corticotrophin releasing hormone (CRH) and arginine vasopressin, which act on the pituitary gland to increase adrenocorticotropic hormone (ACTH) release, subsequently accelerating the production of cortisol to aid in the homeostasis feedback loop (163). Studies have demonstrated an elevated inflammation state perpetuating cytokines such as IL-1, IL-6, TNFα, and IFNα; these in turn activate the HPA axis elevating levels of CRH, ACTH, and cortisol (162, 164–167). This relationship furthermore supports the feedback loop maintaining a hyperactive HPA system (163). Chronic elevation of endogenous glucocorticoids results in mood symptoms (163, 167). Additionally, it is proposed that cortisol increases the catabolizing enzyme tryptophan 2,3-dioxygenase (TDO) to deplete the precursor to serotonin implicating an association with the serotonin dysregulation (168, 169).
Contrary to advantageous effects of steroids in managing infections, there is evidence that glucocorticoid treatment duration for acute infections versus chronic infections results in changes of glucocorticoid receptor function and concentrations (170, 171). This subsequently influences HPA axis hyperactivity, which elevates cortisol and results in decreased function and quantity of glucocorticoid receptors resulting in impaired feedback and glucocorticoid resistance (170). Glucocorticoid resistance that is seen in depressed patients may also be a result of changes in expressed glucocorticoid receptors ratio via cellular phosphorylation (172–174). Patients with neuropsychiatric illnesses also exhibited heightened plasma, urine, and cerebrospinal fluid (CSF) levels of cortisol and anatomical changes in the pituitary and adrenal glands (175–178). Pavon et al. (179) reported elevated cortisol levels in depressed patients associated with elevated TNFα, in addition to decreased levels of IL-1β, suggesting that increased cortisol may influence inflammatory cytokines. However, it is important to bear in mind that while some subtypes of depression (namely melancholic or endogenous) are associated with hyperactive HPA axis, glucocorticoid resistance, and increased circulating cortisol levels, atypical and seasonal depression has been consistently reported to have normal or hypoactive HPA axis function (180, 181). Therefore, the hypothesis is supported regarding the impairment of the HPA axis through cellular mechanisms and dysfunctional feedback leading to HPA axis dysfunction, one of the most consistent findings in biological psychiatry, which is exhibited by patients with depression, bipolar disorder, and schizophrenia (169, 182).
Microglial Hyperactivation Hypothesis
Microglia function as macrophages of the CNS by clearing foreign particles and promoting healing after traumatic brain injury (183). They are also involved in the pruning process of neurons by tagging unutilized synapses for degradation to rebuild more active neurons during the maturation process (184). Pathological synaptic pruning may also contribute to prodromal, remittent and relapsing, and chronic stages of neuropsychiatric disorders (185–187). Prolonged microglial activation induces synaptic pruning subsequent to the accumulation of two pro-inflammatory cytokines, specifically TNFα and IL-1β, leading to neuronal apoptosis (188, 189). The subsequent dysfunctional neuronal pathways may be compensated by adaptive systems, which may resultantly produce and preserve maladaptive behaviors (190, 191). Individuals at an ultra-high risk for developing schizophrenia also appear to have significantly elevated activity of microglia (192). Histological changes in activated microglia have been observed in patients with schizophrenia who had committed suicide during an acute episode of psychosis (193). In contrast, conflicting data derived from post-mortem studies have reported reductions in microglial density and activation (194). These findings may indicate a difference of microglial activation depending on the stage of illness among other factors (194). Aberrant microglial activation is seen in other neuropsychiatric disorders including Alzheimer’s dementia, Parkinson’s disease, multiple sclerosis, herpes encephalitis, traumatic brain injury, and stroke (195, 196). Alterations in brain morphology have been described across the spectrum on neuropsychiatric conditions (197–201). Duration of mental illnesses also has evidence of significant brain morphologic changes (201). Mechanisms that may encourage these anatomical reductions include oxidative and nitrosative stress through activation of microglia (202).
Depression
Major depressive disorder (MDD) is highly prevalent throughout the world, and the prevalence has increased over time (73). The estimated lifetime prevalence of major depression and persistent depressive disorder in adults is 12% (203). In unipolar depression, inflammation and depressive symptoms share a bidirectional relationship. Immunological markers such as CRP, and cytokines IL-1, IL-6, and TNFα, are elevated in patients with depression (1, 204, 205). A recent meta-analysis comprised of 3,212 participants noted elevations in the concentrations of IL-6, TNFα, IL-10, the soluble IL-2 receptor, C-C chemokine ligand 2, IL-13, IL-18, IL-12, IL-1 receptor antagonist, and the soluble TNF receptor 2 in depression patients (1). Several groups of authors have reported that IL-6 may be a useful biomarker for predicting treatment response (115, 204–207). A prospective study revealed a correlation with elevated serum IL-6, as those with higher levels were more likely to be depressed by 18 years of age than individuals on the lower end levels (208). Pro-inflammatory cytokines have been shown to trend toward normalization with symptom improvement indicating treatment response (64).
Studies have shown biomarkers such as CRP and IL-6 may shed light on depression subtypes (55, 56). An interesting larger scale study, The Netherlands Study of Depression and Anxiety (NESDA), extrapolated gender variance while evaluating CRP and IL-6 (181). The authors described an increased level of CRP and IL-6 with normal levels of TNFα in male patients with depressive symptoms; however, there were no associations with the cytokines in women with depressive symptoms (209). Additionally, they noted a differential role of the HPA function, inflammatory markers, and metabolic variables between melancholic and atypical depression subtypes (181).
Elevated levels of kynurenine pathway toxic metabolites such as QUIN are also observed in patients with depression (210). Interestingly, in a 6-week RCT, Krause et al. (211) noted a correlation with kynurenine/tryptophan ratios that was predictive of celecoxib response to significant improvements in the Hamilton Depression Scale (HAMD-17) scores. Subsequently, the kynurenine/tryptophan ratio shows some promise as a potential biomarker for predicting response to COX-2 inhibitors.
Gałecki et al. (212) reported an increase of non-coding micro ribonucleic acid (mRNA) expression of the COX-2 enzyme in recurrent depression. COX-2 inhibitors decrease IDO activity, subsequently decreasing glutamatergic-active by-products such as QUIN, which may add in neuro-stabilizing effects (4, 213, 214). Higher concentrations of QUIN and 3-hydroxykynurenine have been reported in depression (210). Preclinical studies have shown that celecoxib administration in rats was associated with reductions in PGE2 levels and a reversal of stress-induced depressive-like behaviors (215, 216). PGE2 had been shown to contribute to monoamine imbalance with decreased norepinephrine central neuronal release and dysregulation of the HPA axis (217). Consequently, this alters cortisol synthesis and subsequently suppresses serotonin (215, 218–220). Consistent with this assertion, an animal-based model of depression in rats demonstrated that celecoxib independently enhances the release of serotonin in the brain (126).
Celecoxib has also been shown to attenuate pro-inflammatory cytokines IL-1β and TNFα and significantly increase IL-10 levels in animal models (109, 221). IL-1β has been identified as a modulator of BDNF (144). Some evidence exists indicating that elevated levels of IL-6 results in a reduction in BDNF, implicating in imbalanced neurogenesis, resulting in neural circuitry dysfunction in depressive symptomology (144). In a preclinical study with rats with a depression-like phenotype, augmentation with acetylsalicylic acid, a non-selective COX inhibitor, enhanced the efficacy of fluoxetine (222).
To date, several reviews have suggested that celecoxib may be efficacious for the management of depressive symptoms (1, 223, 224); however, some have suggested it is clinically inadequate (225). Abbasi et al. assigned adjuvant celecoxib to MDD patients on sertraline and measured IL-6 levels in samples of their serum. They reported a correlation between decreasing IL-6 concentrations and improvement in the Hamilton Depression Rating Scale (HDRS) scores as outlined in Table 1, along with the key findings of all clinical trials assessing celecoxib in neuropsychiatric disorders appearing in this review. In another RCT (8) assessing celecoxib augmentation with sertraline in the treatment of drug-naïve women with depression, the authors reported an improvement in HDRS and Hamilton Anxiety Rating Scale (HAM-A) scores compared to the placebo group after 4 weeks of treatment. However, there were no significant differences between both groups at the end of the 8-week trial. Interestingly, the remission rates in the celecoxib group were statistically higher in comparison to the placebo group. Subsequently, Akhondzadeh et al. assessed the HDRS in 40 individuals in a 6-week RCT receiving fluoxetine plus celecoxib versus fluoxetine alone. They demonstrated significant improvements in depressive symptoms, and response and remission rates in the celecoxib group (5). Another RCT found more significant improvements in depressive symptoms in the adjuvant celecoxib group with reboxetine compared to reboxetine alone (4). However, Fields commented that there were no significant changes in late-life depressive symptoms in patients prescribed either placebo, celecoxib, or naproxen (6). These discrepancies in the efficacy of COX-2 inhibitors for this indication might be explained by methodological heterogeneity and variance in target sample characteristics. For example, the Geriatric Depression Scale (GDS) was used for the geriatric patients with a yearly frequency, which may not be a specific tool for detecting variations in depression diagnosis (226). However, the study did have strength in having a large sample size and median follow-up of 2 years with patients.
Inflammation may be the primary mechanism of pathogenesis in brucellosis (227, 228). Therefore, Jafari et al. assessed 40 individuals with celecoxib for treatment of mild to moderate depression due to acute brucellosis. They reported an improvement of the HDRS in the 8-week trial with the celecoxib with antibiotics group than placebo with antibiotics (9).
In patients with comorbid osteoarthritis, pooled data from five post-approval trials, each at 6 weeks in length, participants were randomized in placebo, ibuprofen or naproxen, or celecoxib groups while assessing the Patient Health Questionnaire-9 (PHQ-9). The authors report a trend toward a reduction in PHQ-9 depression scores. However, this lack of robust data is possibly due to the lack of efficacious dosing of celecoxib of 200 mg (229).
Interestingly, celecoxib may exhibit benefits in patients with colorectal cancer. Investigations have illustrated that celecoxib initiation in the head, neck, and gastrointestinal cancer population is associated with improvements in biological symptoms of depression, including an increase in appetite, body mass index, and quality of life (230, 231). A 6-week RCT included 40 colon cancer participants randomly assigned to either celecoxib monotherapy or a placebo group, which resulted in significant improvements in the HDRS among the former group, starting as early as week 2 and was sustained until the end of the trial (11).
Another cancer trial consisted of 52 outpatients with breast cancer undergoing 6 weeks of treatment with either celecoxib or diclofenac for mild to moderate depression. The outcome measures were scored using the HDRS to compare the COX-2 inhibitor with an indiscriminate COX-inhibitor. They reported significant improvements in depressive symptoms in both groups by week 3 and significantly more considerable improvements with the celecoxib group compared to diclofenac by week 6. None of the participants experienced remission HDRS less than or equal to 7 (10). A meta-analysis demonstrated with 150 participants showed that the adjunctive celecoxib cohort had better response rates and remissions compared to placebo (224). In summary, interactions between the immune system and neurotransmitters, the tryptophan/kynurenine system, and the glutamatergic system provide links between the immune system and depression; furthermore, data are suggestive of a role for celecoxib in treatment of depressive symptoms (115).
Bipolar Disorder
The estimated lifetime prevalence of bipolar disorder among adults worldwide is 1% to 3% (232). For many, bipolar disorder is a chronic and debilitating illness, with patients often experiencing poor inter-episodic remission (233, 234). Pro-inflammatory markers, such as IL-4, TNFα, IL-1β, and CCL2 cytokine, which have an established role of inflammation in neuronal damage and degeneration, have been observed to be elevated in patients with bipolar disorder (35, 59, 233–236). Elevated CRP levels were also identified in a meta-analysis of 730 patients with bipolar disorder (237).
Interestingly, during the euthymic phase of bipolar disorder, IL-4 has been shown to return to baseline levels; this apparent relationship between inflammatory and mood states provides an avenue for prospective biomarker investigations (65, 238). Furthermore, accumulating evidence is suggestive of chronic low-grade inflammation in bipolar patients (239, 240). Scans employing positron emission tomography (PET) have supported neuroanatomical changes and hyperactive microglial state in bipolar disorder (241–244). Gray matter reduction was observed in the anterior limbic region (197, 198) including ventricular enlargement (245). Lithium, a well-established mood stabilizer, has been shown to reduce IL-2, IL-6, IL-10, and IFNα levels after long-term use, possibly inferring its nebulous mechanism of action through anti-inflammatory processes (246). In addition, lithium has some potential in neurogenesis, which may be linked with particular anti-inflammatory mechanisms (202). Preclinical data also suggest that these mood stabilizing effects may downregulate the AA cascade, therefore decreasing COX-2 and prostaglandin levels (247–253).
A clinical trial investigated the efficacy of celecoxib in bipolar depression or mixed episode and found lower HAM-D scores initially after 1 week; however, no statistically significant difference was found at the end of the 6-week trial (12). In another study, celecoxib augmentation was trialed in individuals with acute mania without psychotic features alongside treatment-as-usual. This 6-week RCT demonstrated that adjuvant celecoxib with valproate was significantly effective for treatment in acute mania compared to valproate and placebo (14). The difference in trial outcomes may suggest greater inflammatory impact and therefore COX-2 inhibitor efficacy during the manic phase of illness, as opposed to the depressive phase; this is also supported by the relative increase in inflammatory markers in the former illness phase (66).
Electroconvulsive therapy (ECT) is an effective treatment modality for various phases of illness in bipolar disorder (254). ECT is reported to affect monoamines, hormones, in addition to the immune system, cytokines, ACTH, and cortisol (255–257). Immunomodulatory effects have also been reported, for example, effects on the kynurenine pathway via the decrease of QUIN concentrations in unipolar and bipolar depression (258, 259).
Kargar et al. (260) assessed cytokine and CRP changes in patients receiving both ECT and celecoxib, reporting a reduction in TNFα, but no significant changes in other inflammatory markers, such as IL-1β, IL-6, and CRP. However, they noted greater clinical improvement of depressive symptoms in the first week of celecoxib intervention but no persisting differences thereafter (260). Notably, the authors hypothesized that immunomodulatory effects associated with ECT might explain the baseline return of TNFα concentrations; however, no significant cytokine changes were observed. This may also be due to the post-ECT acute induction of cytokines hindering statistical significance (255, 261). Another RCT with 35 ECT participants with focus on BDNF levels in patients with mania concluded no statistical difference in BDNF levels or treatment efficacy with adjuvant celecoxib (13). The authors of this study suggested these effects were nearing statistical significance, and that their BDNF sampling protocol may have been a confounding factor (13). A longer multicenter trial has been proposed to assess augmentation with celecoxib and/or minocycline alongside treatment as usual (TAU) in bipolar I or bipolar II patients in depressive state; however, these results are yet to be published (262). Bipolar disorder has significant associations with inflammatory modulation resulting in aberrant brain changes that warrant further investigation of the roles of anti-inflammatory agents. In particular, celecoxib shows some promise requiring further investigation, particularly during a set phase of the bipolar illness.
Schizophrenia
Schizophrenia is a severe, chronic, and among the most disabling and economically catastrophic medical disorders. The World Health Organization ranks schizophrenia as one of the top 10 illnesses contributing to the global burden of disease (263). The dysregulation of dopamine and glutamatergic pathways in various brain regions are implicated in the positive, negative, and cognitive symptom domains of schizophrenia (264). Pro-inflammatory cytokines such as IL-1β and IL-6 can influence neuronal development, specifically on the dopaminergic and serotonergic systems (265–268). IL-1β administration after birth can influence the dopamine system in adulthood, which has been associated in dopaminergic and serotonergic neuronal moderation in rat models (266). In schizophrenia, elevated serum and CSF concentrations of kynurenine were reported (269). Developing findings are suggesting that infectious exposure during the prenatal period may contribute to the pathogenesis of schizophrenia (270). Raised maternal levels of IL-8 during pregnancy are associated with an increased risk for schizophrenia in offspring, in addition to decreased brain volumes, independent of the inflammatory etiology (271). Maternal immune activation in animal models generated oxidative stress in the fetus (264). Observed infectious agents including Toxoplasma gondii, Chlamydia, bornavirus cytomegalovirus, and influenza seem to increase the risk of schizophrenia. This may, however, occur as a result of the immune response rather than an infectious etiology (270). Several epidemiological studies have observed an elevated prevalence of schizophrenia in cohorts born during influenza epidemics (272) and significant association with immunological disorders (273). There is a higher prevalence of schizophrenia in individuals with celiac disease, bullous pemphigoid, interstitial cystitis, thyrotoxicosis, and acquired hemolytic anemia (273, 274). Surprisingly, rheumatoid arthritis reveals lower rates of co-morbid schizophrenia compared to the general population (275).
Post-mortem brain studies from schizophrenia patients have revealed significant inflammatory processes (276). In addition, PET imaging has signified microglial activation resulting in brain morphological changes in first-episode and chronic psychosis (277–280). These morphological changes have been expressed during prodromal phases of first-episode psychosis (281–283), suggestive of neurotoxic processes resulting in poor prognosis (281, 284). Moreover, some authors have shown a relationship between brain volume, IL-1, and IL-6 (285–287). Collectin inflammatory markers have also been implicated in patients with schizophrenia, C4A in particular, whose role is to influence microglial hyperactivity, neurodegeneration, and subsequent brain cortical volume reductions (288, 289). These microglial changes may derive from established elevations in serum pro-inflammatory factors, such as PGE2, CRP, IL-1β, IL- 6, IL-8, and TNFα (290–292). In addition, cytokines have shown some correlation with negative symptoms, cognitive deficits, and psychomotor retardation (293–295).
Positive correlations with cognitive severity and inflammatory markers have also been highlighted (296–298). In the first-episode and acutely relapsed patients with psychosis, an elevated level of pro-inflammatory cytokines, IL-6, TNFα, TGF-β, and IFNγ, was observed (61). In addition, IL-10 concentrations were decreased in acutely relapsed patients compared to controls (61). Cytokine concentrations at different stages of the disease and variable treatment agents may alter neuroprogression (115, 299).
Further support is provided from studies suggesting antipsychotics may exhibit immune-modulatory properties (264); however, there remains evidence that is contrary to this notion (300, 301). Patients undergoing long-term treatment with antipsychotics exhibit reduced pro-inflammatory cytokine levels (IL-1β, IL-6, sIL-6R, and TNFα) (302–304) and elevated anti-inflammatory markers (sIL-1RA, sIL-2R, and IL-10) (305–307). Second-generation antipsychotics may elicit more potent anti-inflammatory effects than to first-generation agents (303, 308).
Dysregulation of the tryptophan metabolism has been implicated with notable elevations of kynurenic acid in patients with schizophrenia (309). Furthermore, long-term antipsychotic treatments have an impact on kynurenic acid concentrations in rodent models (310). Preclinical studies suggest that COX-2 inhibitors protect against glutamate-mediated neurotoxicity, which may highlight an application to mediate neurodegeneration in kynurenine system (18, 135, 311).
One of the first RCTs evaluating the use of COX inhibitors for schizophrenia indication consisted of 50 patients with acute exacerbation of psychosis who were admitted and treated with risperidone with one group augmented with celecoxib versus placebo for 5 weeks (17). The celecoxib group revealed significant positive effects on the Positive and Negative Syndrome Scale (PANSS) (17). Post hoc analyses indicated an improvement in cognition parameters with augmentation of celecoxib in schizophrenia in this trial (312).
In contrast, Rapaport and colleagues assessed outpatients with schizophrenia on stable psychotropic regimens of olanzapine or risperidone, finding no significant changes with celecoxib augmentation in several of the psychometric parameters (18). This finding could be explained by differences in the study cohorts, given the participants in Müller’s study were acutely psychotic, whereas Rapaport’s sample consisted of patients in more stabilized psychotic states.
During an 8-week RCT, the treatment of 60 acutely psychotic patients was augmented with celecoxib (19). The risperidone and celecoxib combination was superior in the improvement of PANSS total scores over risperidone alone (19). Also, the Extrapyramidal Symptoms Rating Scale (ESRS) scores for the placebo group were higher than in the celecoxib group over the trial but not statistically significant (18). Müller et al. (20) completed a 6-week, RCT of 49 patients during their first-episode of schizophrenia. They were treated with amisulpride with random assignment of celecoxib or placebo (20). There was an improvement in the PANSS in the adjunct celecoxib group compared to the placebo group (20). The adjunct celecoxib group in this study also showed a significant improvement on the clinical global impression (CGI) scale (20). Overall, a superior therapeutic effect with augmentation with celecoxib was found, in particular a trend for improvement in negative symptoms.
A recent meta-analysis, including the above RCTs in addition to two inaccessible RCTs, revealed that adjunctive celecoxib did not prove efficacy over placebo in overall samples (313). However, with the sub-analysis, they discovered superior efficacy with celecoxib to placebo in first-episode patients (313). This may be explained by data from preclinical studies suggesting celecoxib’s effects on cytokines and behavioral symptoms are dependent on the stage of illness and time of intervention (217).
It is important to note the comorbid conditions that may contribute to inflammation and confound interpretation of outcomes, including trauma, stress, smoking, metabolic syndrome, diet, exercise, and poor dental hygiene (2). Nonetheless, there is an apparent association between inflammation and schizophrenia, with celecoxib demonstrating promise possibly during early disease onset.
Autism Spectrum Disorder
ASD is a neurodevelopmental disorder defined by impairments in two domains: 1) shortages in social communication and social interaction and 2) restricted repetitive patterns of behavior, interests, and activities (314). The prevalence of ASD in Western countries appears to have increased, possibly as a result of definition changes and heightened awareness. The pathogenesis of ASD remains idiopathic, although the consensus points to altered brain development leading to impairment in social and communication maturation, therefore resulting in restricted interests and repetitive behaviors (315). These brain morphologic aberrations appear to be a result of neural pruning processes and neuroinflammation (316–319).
An association with ASD and inflammatory response through the measles, mumps, and rubella (MMR) vaccine and enterocolitis were reported first in 1998 (320) and subsequently retracted. However, controversy still exists among these allegations in select groups, despite it being established that there is no causal association between MMR vaccine and ASD (320–324).
Similar to discussions in Schizophrenia section with respect to schizophrenia, prenatal infections during early development may also be associated with the development of ASD (325–327). Moreover, there seem to be shared immune-related genetic abnormalities between the two disorders (325, 328). Aberrant activity of the glutamatergic system might play a role in neurotoxicity of both disorders. Kynurenine pathway abnormalities may also be linked to 16p11.2 mutations in ASD resulting in glutamatergic activity (329).
Associations with changes in the immunomodulatory system of ASD patients have been identified. Disruption in immunomodulatory proponents such as T-cells and monocytes have been noted (316, 330), in addition to changes in the concentration of immunoglobulins (331) and autoantibodies production (332). Furthermore, polymorphisms identified in macrophage migration inhibitory factor (MIF), seen in ASD-related abnormalities, seem to also activate the COX-2 system in microglia (333, 334).
Post-mortem studies revealed greater microglial densities in the visual cortex, cerebellum, anterior cingulate gyri, and dorsolateral prefrontal cortex (DLPFC) of ASD patients (335–338). Some studies have also shown elevated TNFα, IFNγ, IL-1, IL-6, IL-8, IL-12, CCL2, CCL5, and CCL11 in plasma and CSF of autistic subjects (317, 318, 338–340).
A study showed that repurposed anti-inflammatory agents such as pioglitazone resulted in moderation of irritability, lethargy, stereotypy, and hyperactivity symptoms in ASD (341). The pioglitazone class of drugs has been shown to inhibit COX-2 in LPS-stimulated microglia and neurons (342, 343). Celecoxib in the rat model has shown to also inhibit LPS-induced neuronal toxicity (344).
Our search yielded only one randomized, double-blind placebo-controlled trial of celecoxib combination with risperidone. Asadabadi et al. (21) assessed 40 patients diagnosed with ASD in a 10-week trial with the aberrant behavior checklist-community edition (ABC-C), which showed superior efficacy of adjuvant celecoxib with risperidone in the domains of irritability, social withdrawal, and stereotypy in children with ASD.
Paucity of evidence is opportune for further investigations with COX-2 inhibitors in neurodevelopmental disorders such as ASD as seen by preclinical and limited clinical data.
Obsessive-Compulsive Disorder
Obsessive-compulsive disorder (OCD) is a relatively common neuropsychiatric disorder with a reported lifetime prevalence of 1–3% in the general population (345, 346). At least one-third of individuals with OCD fail to adequately respond to current pharmacological treatment (347, 348). The cortico-striatal-thalamo-cortical (CSTC) circuit dysfunction is implicated in the pathophysiology of OCD (349).
There is evidence of early childhood infections, pediatric acute-onset neuropsychiatric syndrome (PANS), which encompasses pediatric autoimmune neuropsychiatric disorders associated with streptococcal (PANDAS), evoking OCD-like neuropsychiatric symptoms (15). This is suggestive of an inflammatory etiology to a subsect of this illness. Furthermore, immunomodulation treatment resulted in improvement of OCD-like neuropsychiatric symptoms (350).
Although the minority of cases of OCD results from PANS, it is speculated that the active inflammatory model may be relevant for the progression of OCD. As well, brain morphological changes have also been noted in OCD indicating progression in the neurodegenerative process (351). Rodent models exposed to LPS-induced inflammation exhibited increased anxiety with reduced exploration in the open field test (352, 353). The inducible chemokine, CXCL12, resulted in anxiety-like features in rat models (353), further supporting the role of inflammation in neuropsychiatric symptoms in anxiety disorders.
Animal models are suggestive of alternative microglial phenotypes, resulting in OCD-like behavior (354). Translocator protein distribution volume, a marker of increased microglial activation and thus neuroinflammation (355–358), was investigated after a prior study found increased expression in PANDAS patients (359). Kumar et al. discovered an increased translocator protein density in the CSTC circuit compared to healthy controls. Interestingly, this circuit involves multiple neuropsychiatric disorders aforementioned such as Huntington disease, cerebral vascular disease, Tourette disorders, and Sydenham chorea (360, 361). Repurposed microglial modulators such as minocycline have shown a reduction in the Yale-Brown Obsessive-Compulsive Scale (Y-BOCS) scores in a recent RCT in combination with fluvoxamine (362).
Most recently, Konuk et al. (86) reported significantly elevated levels of both IL-6 and TNFα in OCD patients compared to healthy controls. Furthermore, a correlation between elevated TNFα and onset with minimal association between IL-6 levels and duration of illness (86). A meta-analysis revealed no significant findings in TNFα and IL-6 plasma levels in OCD patients relative to controls; however, the authors did note reduced IL-1β in OCD patients (62). As celecoxib is shown to reduce levels of both IL-6 and TNFα, support for improving clinical symptoms of OCD is plausible (6).
Sayyah et al. (16) noted an improvement in the Y-BOCS for OCD patients with the augmentation of celecoxib with fluoxetine compared to fluoxetine alone. In addition, another recent RCT utilizing fluvoxamine with celecoxib augmentation compared to fluvoxamine alone noted an improvement in the celecoxib group (15). It is proposed that the notable efficacy in OCD, in addition to the microglial mechanisms, results from increased monoamines such as norepinephrine and serotonin via inhibition of prostaglandin synthesis by celecoxib (5, 306). However, further studies with larger sample sizes, longer duration, and measurements of pro-inflammatory markers may provide more robust evidence.
Conclusion
Evidence for the inflammatory hypothesis and the role of anti-inflammatory agents continues to accumulate suggesting etiological impact on the development of neuropsychiatric conditions, such as depression, bipolar disorder, schizophrenia, ASD, and OCD. A promising body of evidence suggests a role for COX-2 inhibition, in particular celecoxib, for phase-related interventions in bipolar disorder, schizophrenia, and possibly depression, ASD, and OCD. Despite the paucity of data for COX-2 inhibitors and investigated agents, including aspirin, minocycline, and statins, with purported pleiotropic anti-inflammatory mechanisms, further research is necessary to clarify the role of immunomodulation therapies and their comparative efficacies for integration of the psychiatric professions’ current paradigm of treatment modalities (146, 363).
Author Contributions
RS was the primary author of this manuscript, conducted initial literature search, review and synthesis of first draft and subsequent revisions. NG-C contributed to literature search and revisions. AW contributed to synthesis initial draft and revisions. OR contributed to manuscript draft, revisions and created figure. BA contributed to drafting and revision process. MB and SD were lead investigators providing initial concept and ongoing guidance throughout.
Funding
Berk A is supported by Stanley Medical Research Foundation NIH, MBF, NHMRC, NHMRC Senior Principal Research Fellowship (APP1059660 and APP1156072), Cooperative Research Centre, Simons Autism Foundation, Cancer Council of Victoria, MBF, Rotary Health, Meat and Livestock Board Woolworths, BeyondBlue, Geelong Medical Research Foundation Bristol Myers Squibb, Eli Lilly, Glaxo SmithKline, Organon, Novartis, Mayne Pharma, Servier. Berk is the speaker in Astra Zeneca, Bristol Myers Squibb Eli Lilly, Glaxo SmithKline Lundbeck, Pfizer, Sanofi Synthelabo Servier, Solvay, Wyeth; and consultant in AstraZeneca, Bristol Myers Squibb Eli Lilly, Bioadvantex, Merck, Glaxo SmithKline Lundbeck, Janssen Cilag Servier. Berk is a co-inventor of two provisional patents regarding the use of NAC and related compounds for psychiatric indications, which, while assigned to the Mental Health Research Institute, could lead to personal remuneration upon a commercialisation event.Walker A is supported by the Trisno Family Fellowship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
3MS-E, Modified Mini-Mental State Exam; AA, arachidonic acid; ABC-C, autism behavior checklist community edition; ACTH, adrenocorticotropic hormone; ASD, autism spectrum disorder; BDNF, brain derived neurotrophic factor; CCL2, C-C motif chemokine ligand 2; CGI-I, Clinical Global Impression: Improvement; CGI-S, Clinical Global Impression: Severity; CNS, central nervous system; COX, cyclooxygenase; CSF, cerebrospinal fluid; cPLA2, cytoplasmic phospholipase A2; CRH, corticotrophin releasing hormone; CRP, C-reactive protein; ECT, electroconvulsive therapy; ESRS, Extrapyramidal Symptoms Rating Scale; GDS, Geriatric Depression Scale; HAM-A, Hamilton Anxiety Rating Scale; HAM-D, Hamilton Depression Rating Scale; HDRS, Hamilton Depression Rating Scale; IDO, indolamine 2,3 dioxygenase; IL, interleukin; IL-1ra, interleukin-1 receptor antagonist; IFNα, interferon alpha; IFN-γ, interferon gamma; KA, kainic acid; LPS, lipopolysaccharide; MDD, major depressive disorder; mRNA, microribonucleic acid; NMDA, N-methyl-D-aspartate; NF, nuclear factor; OCD, obsessive compulsive disorder; PANS, pediatric acute-onset neuropsychiatric syndrome; PANDAS, pediatric autoimmune neuropsychiatric disorders associated with streptococcal; PANSS, Positive and Negative Syndrome Scale; PHQ-9, Patient Health Questionnaire-9; PGE2, prostaglandin E2; QUIN, quinolinic acid; RCT, randomized control trial; SANS, Scale for the Assessment of Negative Symptoms; SAS, Simpson–Angus Rating Scale of EPS; TAU, treatment as usual; TDO, tryptophan 2,3-dioxygenase; TNFα, tumor necrosis factor alpha
Acknowledgments
The team would like to thank the IMPACT Strategic Research Centre for their support in providing work spaces and accommodation for our international team during the initial stages of this manuscript. Without their support this work would not have been achievable and their generosity should not go without thanks.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpsyt.2019.00605/full#supplementary-material
References
1. Kohler CA, Freitas TH, Maes M, de Andrade NQ, Liu CS, Fernandes BS, et al. Peripheral cytokine and chemokine alterations in depression: a meta-analysis of 82 studies. Acta Psychiatr Scand (2017) 135(5):373–87. doi: 10.1111/acps.12698
2. Berk M, Williams LJ, Jacka FN, O’Neil A, Pasco JA, Moylan S, et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med (2013) 11(1):200. doi: 10.1186/1741-7015-11-200
3. Huynh NN, McIntyre RS. What are the implications of the STAR*D trial for primary care? A review and synthesis. Prim Care Companion J Clin Psychiatry (2008) 10(2):91–6. doi: 10.4088/PCC.v10n0201
4. Muller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Muller B, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry (2006) 11(7):680–4. doi: 10.1038/sj.mp.4001805
5. Akhondzadeh S, Jafari S, Raisi F, Nasehi AA, Ghoreishi A, Salehi B, et al. Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depress Anxiety (2009) 26(7):607–11. doi: 10.1002/da.20589
6. Fields C, Drye L, Vaidya V, Lyketsos C. Celecoxib or naproxen treatment does not benefit depressive symptoms in persons age 70 and older: findings from a randomized controlled trial. Am J Geriatr Psychiatry (2012) 20(6):505–13. doi: 10.1097/JGP.0b013e318227f4da
7. Abbasi SH, Hosseini F, Modabbernia A, Ashrafi M, Akhondzadeh S. Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: randomized double-blind placebo-controlled study. J Affect Disord (2012) 141(2–3):308–14. doi: 10.1016/j.jad.2012.03.033
8. Majd M, Hashemian F, Hosseini SM, Vahdat Shariatpanahi M, Sharifi A. A randomized, double-blind, placebo-controlled trial of celecoxib uugmentation of sertraline in treatment of drug-naive depressed women: a pilot study. IJPR (2015) 14(3):891–9.
9. Jafari S, Ashrafizadeh SG, Zeinoddini A, Rasoulinejad M, Entezari P, Seddighi S, et al. Celecoxib for the treatment of mild-to-moderate depression due to acute brucellosis: a double-blind, placebo-controlled, randomized trial. J Clin Pharm Ther (2015) 40(4):441–6. doi: 10.1111/jcpt.12287
10. Mohammadinejad P, Arya P, Esfandbod M, Kaviani A, Najafi M, Kashani L, et al. Celecoxib versus diclofenac in mild to moderate depression management among breast cancer patients: a double-blind, placebo-controlled, randomized trial. Ann Pharmacother (2015) 49(9):953–61. doi: 10.1177/1060028015592215
11. Alamdarsaravi M, Ghajar A, Noorbala A-A, Arbabi M, Emami A, Shahei F, et al. Efficacy and safety of celecoxib monotherapy for mild to moderate depression in patients with colorectal cancer: a randomized double-blind, placebo controlled trial. Psychiatry Res (2017) 255:59–65. doi: 10.1016/j.psychres.2017.05.029
12. Nery FG, Monkul ES, Hatch JP, Fonseca M, Zunta-Soares GB, Frey BN, et al. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol (2008) 23(2):87–94. doi: 10.1002/hup.912
13. Kargar M, Yoosefi A, Akhondzadeh S, Artonian V, Ashouri A, Ghaeli P. Effect of adjunctive celecoxib on BDNF in manic patients undergoing electroconvulsive therapy: a randomized double blind controlled trial. Pharmacopsychiatry (2015) 48(7):268–73. doi: 10.1055/s-0035-1559667
14. Arabzadeh S, Ameli N, Zeinoddini A, Rezaei F, Farokhnia M, Mohammadinejad P, et al. Celecoxib adjunctive therapy for acute bipolar mania: a randomized, double-blind, placebo-controlled trial. Bipolar Disord (2015) 17(6):606–14. doi: 10.1111/bdi.12324
15. Shalbafan M, Mohammadinejad P, Shariat SV, Alavi K, Zeinoddini A, Salehi M, et al. Celecoxib as an adjuvant to fluvoxamine in moderate to severe obsessive-compulsive disorder: a double-blind, placebo-controlled, randomized trial. Pharmacopsychiatry (2015) 48(4–5):136–40. doi: 10.1055/s-0035-1549929
16. Sayyah M, Boostani H, Pakseresht S, Malayeri A. A preliminary randomized double-blind clinical trial on the efficacy of celecoxib as an adjunct in the treatment of obsessive-compulsive disorder. Psychiatry Res (2011) 189(3):403–6. doi: 10.1016/j.psychres.2011.01.019
17. Muller N, Riedel M, Scheppach C, Brandstatter B, Sokullu S, Krampe K, et al. Beneficial antipsychotic effects of celecoxib add-on therapy compared to risperidone alone in schizophrenia. Am J Psychiatry (2002) 159(6):1029–34. doi: 10.1176/appi.ajp.159.6.1029
18. Rapaport MH, Delrahim KK, Bresee CJ, Maddux RE, Ahmadpour O, Dolnak D. Celecoxib augmentation of continuously ill patients with schizophrenia. Biol Psychiatry (2005) 57(12):1594–6. doi: 10.1016/j.biopsych.2005.02.024
19. Akhondzadeh S, Tabatabaee M, Amini H, Ahmadi Abhari SA, Abbasi SH, Behnam B. Celecoxib as adjunctive therapy in schizophrenia: a double-blind, randomized and placebo-controlled trial. Schizophr Res (2007) 90(1–3):179–85. doi: 10.1016/j.schres.2006.11.016
20. Müller N, Krause D, Dehning S, Musil R, Schennach-Wolff R, Obermeier M, et al. Celecoxib treatment in an early stage of schizophrenia: results of a randomized, double-blind, placebo-controlled trial of celecoxib augmentation of amisulpride treatment. Schizophr Res (2010) 121(1–3):118–24. doi: 10.1016/j.schres.2010.04.015
21. Asadabadi M, Mohammadi MR, Ghanizadeh A, Modabbernia A, Ashrafi M, Hassanzadeh E, et al. Celecoxib as adjunctive treatment to risperidone in children with autistic disorder: a randomized, double-blind, placebo-controlled trial. Psychopharmacology (2013) 225(1):51–9. doi: 10.1007/s00213-012-2796-8
22. Raju TN. Hot brains: manipulating body heat to save the brain. Pediatrics (2006) 117(2):e320–1. doi: 10.1542/peds.2005-1934
23. Lopez-Munoz F, Alamo C. Monoaminergic neurotransmission: the history of the discovery of antidepressants from 1950s until today. Curr Pharm Des (2009) 15(14):1563–86. doi: 10.2174/138161209788168001
24. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S3–23. doi: 10.1016/j.jaci.2009.12.980
25. Simon AK, Hollander GA, McMichael A. Evolution of the immune system in humans from infancy to old age. Proc R Soc B: Biol Sci (2015) 282(1821):20143085. doi: 10.1098/rspb.2014.3085
26. Jiang Z, Jiang JX, Zhang GX. Macrophages: a double-edged sword in experimental autoimmune encephalomyelitis. Immunol Lett (2014) 160(1):17–22. doi: 10.1016/j.imlet.2014.03.006
27. McNamara RK, Lotrich FE. Elevated immune-inflammatory signaling in mood disorders: a new therapeutic target? Expert Rev Neurother (2012) 12(9):1143–61. doi: 10.1586/ern.12.98
28. Kronfol Z, Remick DG. Cytokines and the brain: implications for clinical psychiatry. Am J Psychiatry (2000) 157(5):683–94. doi: 10.1176/appi.ajp.157.5.683
29. Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation (1995) 2(4):241–8. doi: 10.1159/000097202
30. Quan N, Whiteside M, Herkenham M. Time course and localization patterns of interleukin-1beta messenger RNA expression in brain and pituitary after peripheral administration of lipopolysaccharide. Neuroscience (1998) 83(1):281–93. doi: 10.1016/S0306-4522(97)00350-3
31. Morris G, Fernandes BS, Berk M. Leaky brain in neurological and psychiatric disorders: drivers and consequences. Aust N Z J Psychiatry (2018) 52(10):92448. doi: 10.1177/0004867418796955
32. Haroon E, Raison CL, Miller AH. Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology (2012) 37(1):137–62. doi: 10.1038/npp.2011.205
33. Bentivoglio M, Mariotti R, Bertini G. Neuroinflammation and brain infections: historical context and current perspectives. Brain Res Rev (2011) 66(1-2):152–73. doi: 10.1016/j.brainresrev.2010.09.008
34. Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci (2007) 8(1):57–69. doi: 10.1038/nrn2038
35. Goldstein BI, Kemp DE, Soczynska JK, McIntyre RS. Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J Clin Psychiatry (2009) 70(8):1078–90. doi: 10.4088/JCP.08r04505
36. Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O. Neuroinflammation and psychiatric illness. J Neuroinflammation (2013) 10:43. doi: 10.1186/1742-2094-10-43
37. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol (2016) 173(4):649–65. doi: 10.1111/bph.13139
38. Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci (2006) 29(2):68–74. doi: 10.1016/j.tins.2005.12.005
39. van Kesteren CF, Gremmels H, de Witte LD, Hol EM, Van Gool AR, Falkai PG, et al. Immune involvement in the pathogenesis of schizophrenia: a meta-analysis on postmortem brain studies. Transl Psychiatry (2017) 7(3):e1075. doi: 10.1038/tp.2017.4
40. Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation (2014) 11(1):98. doi: 10.1186/1742-2094-11-98
41. Wachholz S, Esslinger M, Plumper J, Manitz MP, Juckel G, Friebe A. Microglia activation is associated with IFN-alpha induced depressive-like behavior. Brain Behav Immun (2016) 55:105–13. doi: 10.1016/j.bbi.2015.09.016
42. Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol (2015) 7(5):a016303. doi: 10.1101/cshperspect.a016303
43. Kohler O, Krogh J, Mors O, Benros ME. Inflammation in depression and the potential for anti-inflammatory treatment. Curr Neuropharmacol (2016) 14(7):732–42. doi: 10.2174/1570159X14666151208113700
44. Kohler-Forsberg O, Buttenschon HN, Tansey KE, Maier W, Hauser J, Dernovsek MZ, et al. Association between C-reactive protein (CRP) with depression symptom severity and specific depressive symptoms in major depression. Brain Behav Immun (2017) 62:344–50. doi: 10.1016/j.bbi.2017.02.020
45. Inoshita M, Numata S, Tajima A, Kinoshita M, Umehara H, Nakataki M, et al. A significant causal association between C-reactive protein levels and schizophrenia. Sci Rep (2016) 6:26105. doi: 10.1038/srep26105
46. Wium-Andersen MK, Orsted DD, Nielsen SF, Nordestgaard BG. Elevated C-reactive protein levels, psychological distress, and depression in 73, 131 individuals. JAMA Psychiatry (2013) 70(2):176–84. doi: 10.1001/2013.jamapsychiatry.102
47. Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med (2004) 350(14):1387–97. doi: 10.1056/NEJMoa032804
48. Koenig W, Sund M, Frohlich M, Fischer HG, Lowel H, Doring A, et al. C-Reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation (1999) 99(2):237–42. doi: 10.1161/01.CIR.99.2.237
49. Lindahl B, Toss H, Siegbahn A, Venge P, Wallentin L. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. N Engl J Med (2000) 343(16):1139–47. doi: 10.1056/NEJM200010193431602
50. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur Heart J (2013) 34(45):3478–90. doi: 10.1093/eurheartj/eht273
51. Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest (2003) 111(12):1805–12. doi: 10.1172/JCI200318921
52. Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med (2000) 342(12):836–43. doi: 10.1056/NEJM200003233421202
53. Tracy RP, Lemaitre RN, Psaty BM, Ives DG, Evans RW, Cushman M, et al. Relationship of C-reactive protein to risk of cardiovascular disease in the elderly. Results from the Cardiovascular Health Study and the Rural Health Promotion Project. Arterioscler Thromb Vasc Biol (1997) 17(6):1121–7. doi: 10.1161/01.ATV.17.6.1121
54. Krogh J, Benros ME, Jorgensen MB, Vesterager L, Elfving B, Nordentoft M. The association between depressive symptoms, cognitive function, and inflammation in major depression. Brain Behav Immun (2014) 35:70–6. doi: 10.1016/j.bbi.2013.08.014
55. Furtado M, Katzman MA. Examining the role of neuroinflammation in major depression. Psychiatry Res (2015) 229(1–2):27–36. doi: 10.1016/j.psychres.2015.06.009
56. Karlovic D, Serretti A, Vrkic N, Martinac M, Marcinko D. Serum concentrations of CRP, IL-6, TNF-alpha and cortisol in major depressive disorder with melancholic or atypical features. Psychiatry Res (2012) 198(1):74–80. doi: 10.1016/j.psychres.2011.12.007
57. Felger JC, Lotrich FE. Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience (2013) 246:199–229. doi: 10.1016/j.neuroscience.2013.04.060
58. Goldsmith DR, Rapaport MH, Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry (2016) 21(12):1696–709. doi: 10.1038/mp.2016.3
59. Modabbernia A, Taslimi S, Brietzke E, Ashrafi M. Cytokine alterations in bipolar disorder: a meta-analysis of 30 studies. Biol Psychiatry (2013) 74(1):15–25. doi: 10.1016/j.biopsych.2013.01.007
60. Munkholm K, Vinberg M, Vedel Kessing L. Cytokines in bipolar disorder: a systematic review and meta-analysis. J Affect Disord (2013b) 144(1–2):16–27. doi: 10.1016/j.jad.2012.06.010
61. Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B. Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry (2011b) 70(7):663–71. doi: 10.1016/j.biopsych.2011.04.013
62. Gray SM, Bloch MH. Systematic review of proinflammatory cytokines in obsessive-compulsive disorder. Curr Psychiatry Rep (2012) 14(3):220–8. doi: 10.1007/s11920-012-0272-0
63. Masi A, Quintana DS, Glozier N, Lloyd AR, Hickie IB, Guastella AJ. Cytokine aberrations in autism spectrum disorder: a systematic review and meta-analysis. Mol Psychiatry (2015) 20(4):440–6. doi: 10.1038/mp.2014.59
64. Hannestad J, DellaGioia N, Bloch M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology (2011) 36(12):2452–9. doi: 10.1038/npp.2011.132
65. Licinio J, Wong ML. Pathways and mechanisms for cytokine signaling of the central nervous system. J Clin Invest (1997) 100(12):2941–7. doi: 10.1172/JCI119846
66. Brietzke E, Stertz L, Fernandes BS, Kauer-Sant’anna M, Mascarenhas M, Escosteguy Vargas A, et al. Comparison of cytokine levels in depressed, manic and euthymic patients with bipolar disorder. J Affect Disord (2009) 116(3):214–7. doi: 10.1016/j.jad.2008.12.001
67. de Oliveira GS, Cereser KM, Fernandes BS, Kauer-Sant’Anna M, Fries GR, Stertz L, et al. Decreased brain-derived neurotrophic factor in medicated and drug-free bipolar patients. J Psychiatr Res (2009) 43(14):1171–4. doi: 10.1016/j.jpsychires.2009.04.002
68. Fernandes BS, Molendijk ML, Kohler CA, Soares JC, Leite CM, Machado-Vieira R, et al. Peripheral brain-derived neurotrophic factor (BDNF) as a biomarker in bipolar disorder: a meta-analysis of 52 studies. BMC Med (2015a) 13:289. doi: 10.1186/s12916-015-0529-7
69. Fernandes BS, Berk M, Turck CW, Steiner J, Goncalves CA. Decreased peripheral brain-derived neurotrophic factor levels are a biomarker of disease activity in major psychiatric disorders: a comparative meta-analysis. Mol Psychiatry (2014) 19(7):750–1. doi: 10.1038/mp.2013.172
70. Fernandes BS, Steiner J, Berk M, Molendijk ML, Gonzalez-Pinto A, Turck CW, et al. Peripheral brain-derived neurotrophic factor in schizophrenia and the role of antipsychotics: meta-analysis and implications. Mol Psychiatry (2015b) 20(9):1108–19. doi: 10.1038/mp.2014.117
71. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci (2008) 9(1):46–56. doi: 10.1038/nrn2297
72. Leonard BE. The concept of depression as a dysfunction of the immune system. Curr Immunol Rev (2010) 6(3):205–12. doi: 10.2174/157339510791823835
73. Andrade L, Caraveo-Anduaga JJ, Berglund P, Bijl RV, De Graaf R, Vollebergh W, et al. The epidemiology of major depressive episodes: results from the International Consortium of Psychiatric Epidemiology (ICPE) Surveys. Int J Methods Psychiatr Res (2003) 12(1):3–21. doi: 10.1002/mpr.138
74. Wright CE, Strike PC, Brydon L, Steptoe A. Acute inflammation and negative mood: mediation by cytokine activation. Brain Behav Immun (2005) 19(4):345–50. doi: 10.1016/j.bbi.2004.10.003
75. O’Connor J, Lawson M, André C, Moreau M, Lestage J, Castanon N, et al. ‘Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry (2009) 14(5):511–22. doi: 10.1038/sj.mp.4002148
76. Reichenberg A, Yirmiya R, Schuld A, Kraus T, Haack M, Morag A, et al. Cytokine-associated emotional and cognitive disturbances in humans. Arch Gen Psychiatry (2001) 58(5):445–52. doi: 10.1001/archpsyc.58.5.445
77. Euteneuer F, Schwarz MJ, Hennings A, Riemer S, Stapf T, Selberdinger V, et al. Depression, cytokines and experimental pain: evidence for sex-related association patterns. J Affect Disord (2010) 131(1):143–9. doi: 10.1016/j.jad.2010.11.017
78. Brydon L, Walker C, Wawrzyniak A, Whitehead D, Okamura H, Yajima J, et al. Synergistic effects of psychological and immune stressors on inflammatory cytokine and sickness responses in humans. Brain Behav Immun (2009) 23(2):217–24. doi: 10.1016/j.bbi.2008.09.007
79. Dunn AJ, Swiergiel AH. Effects of interleukin-1 and endotoxin in the forced swim and tail suspension tests in mice. Pharmacol Biochem Behav (2005) 81(3):688–93. doi: 10.1016/j.pbb.2005.04.019
80. Eggermont AM, Suciu S, Santinami M, Testori A, Kruit WH, Marsden J, et al. Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. Lancet (2008) 372(9633):117–26. doi: 10.1016/S0140-6736(08)61033-8
81. Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med (2013) 368(20):1907–17. doi: 10.1056/NEJMra1213651
82. Friebe A, Horn M, Schmidt F, Janssen G, Schmid-Wendtner MH, Volkenandt M, et al. Dose-dependent development of depressive symptoms during adjuvant interferon-{alpha} treatment of patients with malignant melanoma. Psychosomatics (2010) 51(6):466–73. doi: 10.1176/appi.psy.51.6.466
83. Reichenberg A, Gorman JM, Dieterich DT. Interferon-induced depression and cognitive impairment in hepatitis C virus patients: a 72 week prospective study. Aids (2005) 19 Suppl 3:S174–8. doi: 10.1097/01.aids.0000192087.64432.ae
84. Udina M, Castellvi P, Moreno-Espana J, Navines R, Valdes M, Forns X, et al. Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J Clin Psychiatry (2012) 73(8):1128–38. doi: 10.4088/JCP.12r07694
85. Grigoleit J-S, Kullmann JS, Wolf OT, Hammes F, Wegner A, Jablonowski S, et al. Dose-dependent effects of endotoxin on neurobehavioral functions in humans. PLoS One (2011) 6(12):e28330. doi: 10.1371/journal.pone.0028330
86. Konuk N, Tekın IO, Ozturk U, Atik L, Atasoy N, Bektas S, et al. Plasma levels of tumor necrosis factor-alpha and interleukin-6 in obsessive compulsive disorder. Mediators Inflamm (2007) 2007:65704. doi: 10.1155/2007/65704
87. Dickens C, McGowan L, Clark-Carter D, Creed F. Depression in rheumatoid arthritis: a systematic review of the literature with meta-analysis. Psychosom Med (2002) 64(1):52–60. doi: 10.1097/00006842-200201000-00008
88. Korczak DJ, Pereira S, Koulajian K, Matejcek A, Giacca A. Type 1 diabetes mellitus and major depressive disorder: evidence for a biological link. Diabetologia (2011) 54(10):2483–93. doi: 10.1007/s00125-011-2240-3
89. Zhang L, Fu T, Yin R, Zhang Q, Shen B. Prevalence of depression and anxiety in systemic lupus erythematosus: a systematic review and meta-analysis. BMC Psychiatry (2017) 17(1):70. doi: 10.1186/s12888-017-1234-1
90. Benros ME, Waltoft BL, Nordentoft M, Ostergaard SD, Eaton WW, Krogh J, et al. Autoimmune diseases and severe infections as risk factors for mood disorders: a nationwide study. JAMA Psychiatry (2013) 70(8):812–20. doi: 10.1001/jamapsychiatry.2013.1111
91. Mardini HE, Kip KE, Wilson JW. Crohn’s disease: a two-year prospective study of the association between psychological distress and disease activity. Dig Dis Sci (2004) 49(3):492–7. doi: 10.1023/B:DDAS.0000020509.23162.cc
92. Langley RG, Feldman SR, Han C, Schenkel B, Szapary P, Hsu MC, et al. Ustekinumab significantly improves symptoms of anxiety, depression, and skin-related quality of life in patients with moderate-to-severe psoriasis: results from a randomized, double-blind, placebo-controlled phase III trial. J Am Acad Dermatol (2010) 63(3):457–65. doi: 10.1016/j.jaad.2009.09.014
93. Menter A, Augustin M, Signorovitch J, Yu AP, Wu EQ, Gupta SR, et al. The effect of adalimumab on reducing depression symptoms in patients with moderate to severe psoriasis: a randomized clinical trial. J Am Acad Dermatol (2010) 62(5):812–8. doi: 10.1016/j.jaad.2009.07.022
94. Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet (2006) 367(9504):29–35. doi: 10.1016/S0140-6736(05)67763-X
95. Kridin K, Zelber-Sagi S, Comaneshter D, Cohen AD. Bipolar disorder associated with another autoimmune disease-pemphigus: a population-based study. Can J Psychiatry (2017) 63(7):474–80. doi: 10.1177/0706743717740344
96. Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB. Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am J Psychiatry (2011) 168(12):1303–10. doi: 10.1176/appi.ajp.2011.11030516
97. Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, Breyer MD. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J Clin Invest (1994) 94(6):2504–10. doi: 10.1172/JCI117620
98. Hirst JJ, Teixeira FJ, Zakar T, Olson DM. Prostaglandin endoperoxide-H synthase-1 and -2 messenger ribonucleic acid levels in human amnion with spontaneous labor onset. J Clin Endocrinol Metab (1995) 80(2):517–23. doi: 10.1210/jc.80.2.517
99. Kirkby NS, Zaiss AK, Urquhart P, Jiao J, Austin PJ, Al-Yamani M, et al. LC-MS/MS confirms that COX-1 drives vascular prostacyclin whilst gene expression pattern reveals non-vascular sites of COX-2 expression. PLoS One (2013) 8(7):e69524. doi: 10.1371/journal.pone.0069524
100. Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene (1999) 18:7908. doi: 10.1038/sj.onc.1203286
101. Coyne DW, Nickols M, Bertrand W, Morrison AR. Regulation of mesangial cell cyclooxygenase synthesis by cytokines and glucocorticoids. Am J Physiol (1992) 263(1 Pt 2):F97–102. doi: 10.1152/ajprenal.1992.263.1.F97
102. DeWitt DL, Meade EA, Smith WL. PGH synthase isoenzyme selectivity: the potential for safer nonsteroidal antiinflammatory drugs. Am J Med (1993) 95(2a):40s–4s. doi: 10.1016/0002-9343(93)90396-7
103. Fu JY, Masferrer JL, Seibert K, Raz A, Needleman P. The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J Biol Chem (1990) 265(28):16737–40.
104. Geng Y, Blanco FJ, Cornelisson M, Lotz M. Regulation of cyclooxygenase-2 expression in normal human articular chondrocytes. J Immunol (1995) 155(2):796–801.
105. Jones DA, Carlton DP, McIntyre TM, Zimmerman GA, Prescott SM. Molecular cloning of human prostaglandin endoperoxide synthase type II and demonstration of expression in response to cytokines. J Biol Chem (1993) 268(12):9049–54.
106. Lee SH, Soyoola E, Chanmugam P, Hart S, Sun W, Zhong H, et al. Selective expression of mitogen-inducible cyclooxygenase in macrophages stimulated with lipopolysaccharide. J Biol Chem (1992) 267(36):25934–8.
107. O’Sullivan MG, Huggins EM Jr., Meade EA, DeWitt DL, McCall CE. Lipopolysaccharide induces prostaglandin H synthase-2 in alveolar macrophages. Biochem Biophys Res Commun (1992) 187(2):1123–7. doi: 10.1016/0006-291X(92)91313-F
108. Riese J, Hoff T, Nordhoff A, DeWitt DL, Resch K, Kaever V. Transient expression of prostaglandin endoperoxide synthase-2 during mouse macrophage activation. J Leukoc Biol (1994) 55(4):476–82. doi: 10.1002/jlb.55.4.476
109. Maes M. Targeting cyclooxygenase-2 in depression is not a viable therapeutic approach and may even aggravate the pathophysiology underpinning depression. Metab Brain Dis (2012) 27(4):405–13. doi: 10.1007/s11011-012-9326-6
110. Minghetti L, Greco A, Potenza RL, Pezzola A, Blum D, Bantubungi K, et al. Effects of the adenosine A2A receptor antagonist SCH 58621 on cyclooxygenase-2 expression, glial activation, and brain-derived neurotrophic factor availability in a rat model of striatal neurodegeneration. J Neuropathol Exp Neurol (2007) 66(5):363–71. doi: 10.1097/nen.0b013e3180517477
111. Song C, Lin A, Bonaccorso S, Heide C, Verkerk R, Kenis G, et al. The inflammatory response system and the availability of plasma tryptophan in patients with primary sleep disorders and major depression. J Affect Disord (1998) 49(3):211–9. doi: 10.1016/S0165-0327(98)00025-1
112. Rapoport SI. Lithium and the other mood stabilizers effective in bipolar disorder target the rat brain arachidonic acid cascade. ACS Chem Neurosci (2014) 5(6):459–67. doi: 10.1021/cn500058v
113. Inoue H, Tanabe T, Umesono K. Feedback control of cyclooxygenase-2 expression through PPARgamma. J Biol Chem (2000) 275(36):28028–32. doi: 10.1074/jbc.M001387200
114. Sales KJ, Grant V, Jabbour HN. Prostaglandin E2 and F2alpha activate the FP receptor and up-regulate cyclooxygenase-2 expression via the cyclic AMP response element. Mol Cell Endocrinol (2008) 285(1–2):51–61. doi: 10.1016/j.mce.2008.01.016
115. Müller N. Immunological aspects of the treatment of depression and schizophrenia. Dialogues Clin Neurosci (2017) 19(1):55–63.
116. Choi SH, Langenbach R, Bosetti F. Cyclooxygenase-1 and -2 enzymes differentially regulate the brain upstream NF-kappa B pathway and downstream enzymes involved in prostaglandin biosynthesis. J Neurochem (2006) 98(3):801–11. doi: 10.1111/j.1471-4159.2006.03926.x
117. Rao JS, Langenbach R, Bosetti F. Down-regulation of brain nuclear factor-kappa B pathway in the cyclooxygenase-2 knockout mouse. Brain Res Mol Brain Res (2005) 139(2):217–24. doi: 10.1016/j.molbrainres.2005.05.008
119. FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med (2001) 345(6):433–42. doi: 10.1056/NEJM200108093450607
120. Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N Engl J Med (2000) 343(21):1520–8. doi: 10.1056/NEJM200011233432103
121. Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med (2005) 352(11):1092–102. doi: 10.1056/NEJMoa050493
122. Farkouh ME, Kirshner H, Harrington RA, Ruland S, Verheugt FW, Schnitzer TJ, et al. ‘Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), cardiovascular outcomes: randomised controlled trial. Lancet (2004) 364(9435):675–84. doi: 10.1016/S0140-6736(04)16894-3
123. Schnitzer TJ, Burmester GR, Mysler E, Hochberg MC, Doherty M, Ehrsam E, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised controlled trial. Lancet (2004) 364(9435):665–74. doi: 10.1016/S0140-6736(04)16893-1
124. Scarpignato C, Lanas A, Blandizzi C, Lems WF, Hermann M, Hunt RH. Safe prescribing of non-steroidal anti-inflammatory drugs in patients with osteoarthritis—an expert consensus addressing benefits as well as gastrointestinal and cardiovascular risks. BMC Med (2015) 13:55. doi: 10.1186/s12916-015-0285-8
125. Sandrini M, Vitale G, Pini LA. Effect of rofecoxib on nociception and the serotonin system in the rat brain. Inflamm Res (2002) 51(3):154–9. doi: 10.1007/PL00000287
126. Johansson D, Falk A, Marcus MM, Svensson TH. Celecoxib enhances the effect of reboxetine and fluoxetine on cortical noradrenaline and serotonin output in the rat. Prog Neuropsychopharmacol Biol Psychiatry (2012) 39(1):143–8. doi: 10.1016/j.pnpbp.2012.06.003
127. Choi S-H, Aid S, Bosetti F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol Sci (2009) 30(4):174–81. doi: 10.1016/j.tips.2009.01.002
128. Minghetti L, Pocchiari M. Cyclooxygenase-2, prostaglandin E2, and microglial activation in prion diseases. Int Rev Neurobiol (2007) 82:265–75. doi: 10.1016/S0074-7742(07)82014-9
129. Gilroy DW, Stables M, Newson J. In vivo models to study cyclooxygenase products in health and disease: introduction to part III. Methods Mol Biol (2010) 644:181–8. doi: 10.1007/978-1-59745-364-6_15
130. Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, et al. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature (1995) 378(6555):406–9. doi: 10.1038/378406a0
131. Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, et al. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell (1995) 83(3):473–82. doi: 10.1016/0092-8674(95)90125-6
132. Aid S, Silva AC, Candelario-Jalil E, Choi SH, Rosenberg GA, Bosetti F. Cyclooxygenase-1 and -2 differentially modulate lipopolysaccharide-induced blood–brain barrier disruption through matrix metalloproteinase activity. J Cereb Blood Flow Metab (2010) 30(2):370–80. doi: 10.1038/jcbfm.2009.223
133. Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med (1999) 5(6):698–701. doi: 10.1038/9550
134. Kimura T, Iwase M, Kondo G, Watanabe H, Ohashi M, Ito D, et al. Suppressive effect of selective cyclooxygenase-2 inhibitor on cytokine release in human neutrophils. Int Immunopharmacol (2003) 3(10–11):1519–28. doi: 10.1016/S1567-5769(03)00179-6
135. Strauss KI, Marini AM. Cyclooxygenase-2 inhibition protects cultured cerebellar granule neurons from glutamate-mediated cell death. J Neurotrauma (2002) 19(5):627–38. doi: 10.1089/089771502753754091
136. Izquierdo LA, Barros DM, Ardenghi PG, Pereira P, Rodrigues C, Choi H, et al. Different hippocampal molecular requirements for short- and long-term retrieval of one-trial avoidance learning. Behav Brain Res (2000) 111(1–2):93–8. doi: 10.1016/S0166-4328(00)00137-6
137. Zagulska-Szymczak S, Filipkowski RK, Kaczmarek L. Kainate-induced genes in the hippocampus: lessons from expression patterns. Neurochem Int (2001) 38(6):485–501. doi: 10.1016/S0197-0186(00)00101-7
138. Zheng X-Y, Zhang H-L, Luo Q, Zhu J. Kainic acid-induced neurodegenerative model: potentials and limitations. J Biomed Biotechnol (2011) 2011:457079. doi: 10.1155/2011/457079
139. Ciceri P, Zhang Y, Shaffer AF, Leahy KM, Woerner MB, Smith WG, et al. Pharmacology of celecoxib in rat brain after kainate administration. J Pharmacol Exp Ther (2002) 302(3):846. doi: 10.1124/jpet.302.3.846
140. Byun J-S, Cho S-Y, Kim S-I, Kwon Y-S, Jeon S-H, Kim M-J, et al. Celecoxib attenuates kainic acid-induced neuronal cell death through suppression of microglial c-Jun N-terminal kinase phosphorylation. Exp Neurobiol (2009) 18(1):13–8. doi: 10.5607/en.2009.18.1.13
141. Baik EJ, Kim EJ, Lee SH, Moon C-H. Cyclooxygenase-2 selective inhibitors aggravate kainic acid induced seizure and neuronal cell death in the hippocampus. Brain Res (1999) 843(1):118–29. doi: 10.1016/S0006-8993(99)01797-7
142. Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci (1997) 17(8):2746–55. doi: 10.1523/JNEUROSCI.17-08-02746.1997
143. Tian J, Kim SF, Hester L, Snyder SH. S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proc Natl Acad Sci U S A (2008) 105(30):10537–40. doi: 10.1073/pnas.0804852105
144. Calabrese F, Rossetti AC, Racagni G, Gass P, Riva MA, Molteni R. Brain-derived neurotrophic factor: a bridge between inflammation and neuroplasticity. Front Cell Neurosci (2014) 8:430. doi: 10.3389/fncel.2014.00430
145. Hayley S. The neuroimmune-neuroplasticity interface and brain pathology. Front Cell Neurosci (2014) 8:419. doi: 10.3389/fncel.2014.00419
146. Robertson OD, Coronado NG, Sethi R, Berk M, Dodd S . Putative neuroprotective pharmacotherapies to target the staged progression of mental illness. Early Intervention in Psychiatry (2019) 1–18. doi: 10.1111/eip.12775
147. Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther (2011) 130(2):226–38. doi: 10.1016/j.pharmthera.2011.01.014
148. Miller AH, Haroon E, Raison CL, Felger JC. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety (2013) 30(4):297–306. doi: 10.1002/da.22084
149. Peters JC. Tryptophan nutrition and metabolism: an overview. Adv Exp Med Biol (1991) 294:345–58. doi: 10.1007/978-1-4684-5952-4_32
150. Hirschfeld RM. History and evolution of the monoamine hypothesis of depression. J Clin Psychiatry (2000) 61(Suppl 6):4–6.
151. Maes M. The cytokine hypothesis of depression: inflammation, oxidative & nitrosative stress (IO&NS) and leaky gut as new targets for adjunctive treatments in depression. Neuro Endocrinol Lett (2008) 29(3):287–91.
152. Wang J, Dunn AJ. Mouse interleukin-6 stimulates the HPA axis and increases brain tryptophan and serotonin metabolism. Neurochem Int (1998) 33(2):143–54. doi: 10.1016/S0197-0186(98)00016-3
153. Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry (2010) 15(4):393–403. doi: 10.1038/mp.2009.116
154. Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein H-G, Sarnyai Z, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflammation (2011) 8(1):94. doi: 10.1186/1742-2094-8-94
155. Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov (2002) 1(8):609–20. doi: 10.1038/nrd870
156. Stone TW, Stoy N, Darlington LG. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci (2013) 34(2):136–43. doi: 10.1016/j.tips.2012.09.006
157. Beal MF, Matson WR, Swartz KJ, Gamache PH, Bird ED. Kynurenine pathway measurements in Huntington’s disease striatum: evidence for reduced formation of kynurenic acid. J Neurochem (1990) 55(4):1327–39. doi: 10.1111/j.1471-4159.1990.tb03143.x
158. Irwin MR, Miller AH. Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav Immun (2007) 21(4):374–83. doi: 10.1016/j.bbi.2007.01.010
159. Lugo-Huitrón R, Muñiz U, Pineda B, Pedraza-Chaverrí J, Ríos C, Pérez-de la Cruz V. Quinolinic acid: an endogenous neurotoxin with multiple targets. Oxid Med Cell Longev (2013) 2013:14. doi: 10.1155/2013/104024
160. Miller AH. Conceptual confluence: the kynurenine pathway as a common target for ketamine and the convergence of the inflammation and glutamate hypotheses of depression. Neuropsychopharmacology (2013) 38(9):1607–8. doi: 10.1038/npp.2013.140
161. Schwarcz R. ‘The kynurenine pathway of tryptophan degradation as a drug target. Curr Opin Pharmacol (2004) 4(1):12–7. doi: 10.1016/j.coph.2003.10.006
162. Herman JP, McKlveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, et al. Regulation of the hypothalamic–pituitary–adrenocortical stress response. Compr Physiol (2016) 6(2):603–21. doi: 10.1002/cphy.c150015
163. McKay MS, Zakzanis KK. The impact of treatment on HPA axis activity in unipolar major depression. J Psychiatr Res (2010) 44(3):183–92. doi: 10.1016/j.jpsychires.2009.07.012
164. Beishuizen A, Thijs LG. Endotoxin and the hypothalamo–pituitary–adrenal (HPA) axis. J Endotoxin Res (2003) 9(1):3–24. doi: 10.1179/096805103125001298
165. Dunn AJ. Cytokine activation of the HPA axis. Ann N Y Acad Sci (2000) 917:608–17. doi: 10.1111/j.1749-6632.2000.tb05426.x
166. Silverman MN, Pearce BD, Biron CA, Miller AH. Immune modulation of the hypothalamic–pituitary–adrenal (HPA) axis during viral infection. Viral Immunol (2005) 18(1):41–78. doi: 10.1089/vim.2005.18.41
167. Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its behavioral and metabolic correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci (2012) 1261:55–63. doi: 10.1111/j.1749-6632.2012.06633.x
168. Oxenkrug GF. Tryptophan–kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: the serotonin hypothesis revisited 40 years later. Isr J Psychiatry Relat Sci (2010) 47(1):56–63.
169. Rosenblat JD, Cha DS, Mansur RB, McIntyre RS. ‘Inflamed moods: a review of the interactions between inflammation and mood disorders. Prog Neuropsychopharmacol Biol Psychiatry (2014) 53:23–34. doi: 10.1016/j.pnpbp.2014.01.013
170. McQuade R, Young AH. Future therapeutic targets in mood disorders: the glucocorticoid receptor. Br J Psychiatry (2000) 177:390–5. doi: 10.1192/bjp.177.5.390
171. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry (2003) 160(9):1554–65. doi: 10.1176/appi.ajp.160.9.1554
172. Adzic M, Djordjevic J, Djordjevic A, Niciforovic A, Demonacos C, Radojcic M, et al. Acute or chronic stress induce cell compartment-specific phosphorylation of glucocorticoid receptor and alter its transcriptional activity in Wistar rat brain. J Endocrinol (2009) 202(1):87–97. doi: 10.1677/JOE-08-0509
173. Anacker C, Zunszain PA, Carvalho LA, Pariante CM. The glucocorticoid receptor: pivot of depression and of antidepressant treatment? Psychoneuroendocrinology (2011) 36(3):415–25. doi: 10.1016/j.psyneuen.2010.03.007
174. Pariante CM. Risk factors for development of depression and psychosis. Glucocorticoid receptors and pituitary implications for treatment with antidepressant and glucocorticoids. Ann N Y Acad Sci (2009) 1179:144–52. doi: 10.1111/j.1749-6632.2009.04978.x
175. Atmaca M, Yildirim H, Ozler S, Koc M, Kara B, Sec S. Smaller pituitary volume in adult patients with obsessive-compulsive disorder. Psychiatry Clin Neurosci (2009) 63(4):516–20. doi: 10.1111/j.1440-1819.2009.01981.x
176. Clark IA, Mackay CE, Goodwin GM. ‘Pituitary gland volumes in bipolar disorder. J Affect Disord (2014) 169:197–202. doi: 10.1016/j.jad.2014.08.022
177. Nemeroff CB, Vale WW. The neurobiology of depression: inroads to treatment and new drug discovery. J Clin Psychiatry (2005) 66(Suppl 7):5–13.
178. Nordholm D, Krogh J, Mondelli V, Dazzan P, Pariante C, Nordentoft M. Pituitary gland volume in patients with schizophrenia, subjects at ultra high-risk of developing psychosis and healthy controls: a systematic review and meta-analysis. Psychoneuroendocrinology (2013) 38(11):2394–404. doi: 10.1016/j.psyneuen.2013.06.030
179. Pavon L, Sandoval-Lopez G, Eugenia Hernandez M, Loria F, Estrada I, Perez M, et al. Th2 cytokine response in major depressive disorder patients before treatment. J Neuroimmunol (2006) 172(1–2):156–65. doi: 10.1016/j.jneuroim.2005.08.014
180. Bocharova M, Agustini B, Young A, Juruena MF. HPA axis function as biomarker for atypical and melancholic depression. Front Psychiatry (2019). doi: 10.3389/conf.fpsyt.2017.48.00006
181. Lamers F, Vogelzangs N, Merikangas KR, de Jonge P, Beekman AT, Penninx BW. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression. Mol Psychiatry (2013) 18(6):692–9. doi: 10.1038/mp.2012.144
182. Girshkin L, Matheson SL, Shepherd AM, Green MJ. Morning cortisol levels in schizophrenia and bipolar disorder: a meta-analysis. Psychoneuroendocrinology (2014) 49:187–206. doi: 10.1016/j.psyneuen.2014.07.013
183. Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science (2016) 353(6301):783–5. doi: 10.1126/science.aaf6260
184. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science (2011) 333(6048):1456–8. doi: 10.1126/science.1202529
185. Keshavan MS, Anderson S, Pettergrew JW. Is Schizophrenia due to excessive synaptic pruning in the prefrontal cortex? The Feinberg hypothesis revisited. J Psychiatr Res (1994) 28(3):239–65. doi: 10.1016/0022-3956(94)90009-4
186. Kim HJ, Cho MH, Shim WH, Kim JK, Jeon EY, Kim DH, et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry (2016) 22:1576. doi: 10.1038/mp.2016.103
187. Stertz L, Magalhaes PV, Kapczinski F. Is bipolar disorder an inflammatory condition? The relevance of microglial activation. Curr Opin Psychiatry (2013) 26(1):19–26. doi: 10.1097/YCO.0b013e32835aa4b4
188. Ekdahl CT. Microglial activation—tuning and pruning adult neurogenesis. Front Pharmacol (2012) 3:41. doi: 10.3389/fphar.2012.00041
189. Kraft AD, Harry GJ. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int J Environ Res Public Health (2011) 8(7):2980–3018. doi: 10.3390/ijerph8072980
190. Paradise MB, Naismith SL, Norrie LM, Graeber MB, Hickie IB. The role of glia in late-life depression. Int Psychogeriatr (2012) 24(12):1878–90. doi: 10.1017/S1041610212000828
191. Park KM, Bowers WJ. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell Signal (2010) 22(7):977–83. doi: 10.1016/j.cellsig.2010.01.010
192. Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, et al. Microglial activity in people at ultra high risk of psychosis and in schizophrenia; an [(11)C]PBR28 PET brain imaging study. Am J Psychiatry (2016) 173(1):44–52. doi: 10.1176/appi.ajp.2015.14101358
193. Laskaris LE, Di Biase MA, Everall I, Chana G, Christopoulos A, Skafidas E, et al. Microglial activation and progressive brain changes in schizophrenia. Br J Pharmacol (2016) 173(4):666–80. doi: 10.1111/bph.13364
194. Trépanier MO, Hopperton KE, Mizrahi R, Mechawar N, Bazinet RP. Postmortem evidence of cerebral inflammation in schizophrenia: a systematic review. Mol Psychiatry (2016) 21:1009. doi: 10.1038/mp.2016.90
195. Katsumoto A, Lu H, Miranda AS, Ransohoff RM. Ontogeny and functions of central nervous system macrophages. J Immunol (2014) 193(6):2615–21. doi: 10.4049/jimmunol.1400716
196. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest (2012) 122(4):1164–71. doi: 10.1172/JCI58644
197. Bora E, Fornito A, Pantelis C, Yucel M. Gray matter abnormalities in major depressive disorder: a meta-analysis of voxel based morphometry studies. J Affect Disord (2012a) 138(1–2):9–18. doi: 10.1016/j.jad.2011.03.049
198. Bora E, Fornito A, Yucel M, Pantelis C. Voxelwise meta-analysis of gray matter abnormalities in bipolar disorder. Biol Psychiatry (2010) 67(11):1097–105. doi: 10.1016/j.biopsych.2010.01.020
199. Bora E, Harrison BJ, Davey CG, Yucel M, Pantelis C. Meta-analysis of volumetric abnormalities in cortico-striatal-pallidal-thalamic circuits in major depressive disorder. Psychol Med (2012b) 42(4):671–81. doi: 10.1017/S0033291711001668
200. Goodkind M, Eickhoff SB, Oathes DJ, Jiang Y, Chang A, Jones-Hagata LB, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry (2015) 72(4):305–15. doi: 10.1001/jamapsychiatry.2014.2206
201. Takahashi T, Yucel M, Lorenzetti V, Nakamura K, Whittle S, Walterfang M, et al. Midline brain structures in patients with current and remitted major depression. Prog Neuropsychopharmacol Biol Psychiatry (2009) 33(6):1058–63. doi: 10.1016/j.pnpbp.2009.05.020
202. Dodd S, Maes M, Anderson G, Dean OM, Moylan S, Berk M. Putative neuroprotective agents in neuropsychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry (2013) 42:135–45. doi: 10.1016/j.pnpbp.2012.11.007
203. Kessler RC, Ormel J, Petukhova M, McLaughlin KA, Green JG, Russo LJ, et al. Development of lifetime comorbidity in the World Health Organization world mental health surveys. Arch Gen Psychiatry (2011) 68(1):90–100. doi: 10.1001/archgenpsychiatry.2010.180
204. Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry (2010) 67(5):446–57. doi: 10.1016/j.biopsych.2009.09.033
205. Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med (2009) 71(2):171–86. doi: 10.1097/PSY.0b013e3181907c1b
206. Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology (2000) 22(4):370–9. doi: 10.1016/S0893-133X(99)00134-7
207. O’Brien SM, Scully P, Fitzgerald P, Scott LV, Dinan TG. Plasma cytokine profiles in depressed patients who fail to respond to selective serotonin reuptake inhibitor therapy. J Psychiatr Res (2007) 41(3–4):326–31. doi: 10.1016/j.jpsychires.2006.05.013
208. Khandaker GM, Pearson RM, Zammit S, Lewis G, Jones PB. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiatry (2014) 71(10):1121–8. doi: 10.1001/jamapsychiatry.2014.1332
209. Penninx BW, Beekman AT, Smit JH, Zitman FG, Nolen WA, Spinhoven P, et al. The Netherlands Study of Depression and Anxiety (NESDA): rationale, objectives and methods. Int J Methods Psychiatr Res (2008) 17(3):121–40. doi: 10.1002/mpr.256
210. Savitz J, Drevets WC, Wurfel BE, Ford BN, Bellgowan PS, Victor TA, et al. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav Immun (2015) 46:55–9. doi: 10.1016/j.bbi.2015.02.007
211. Krause D, Myint AM, Schuett C, Musil R, Dehning S, Cerovecki A, et al. High kynurenine (a tryptophan metabolite) predicts remission in patients with major depression to add-on treatment with celecoxib. Front Psychiatry (2017) 8:16. doi: 10.3389/fpsyt.2017.00016
212. Gałecki P, Galecka E, Maes M, Chamielec M, Orzechowska A, Bobinska K, et al. The expression of genes encoding for COX-2, MPO, iNOS, and sPLA2-IIA in patients with recurrent depressive disorder. J Affect Disord (2012) 138(3):360–6. doi: 10.1016/j.jad.2012.01.016
213. Lee SY, Choi HK, Lee KJ, Jung JY, Hur GY, Jung KH, et al. The immune tolerance of cancer is mediated by IDO that is inhibited by COX-2 inhibitors through regulatory T cells. J Immunother (2009) 32(1):22–8. doi: 10.1097/CJI.0b013e31818ac2f7
214. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry (2009) 65(9):732–41. doi: 10.1016/j.biopsych.2008.11.029
215. Hu F, Wang X, Pace TWW, Wu H, Miller AH. Inhibition of COX-2 by celecoxib enhances glucocorticoid receptor function. Mol Psychiatry (2005) 10(5):426–8. doi: 10.1038/sj.mp.4001644
216. Song C, Zhang XY, Manku M. Increased phospholipase A2 activity and inflammatory response but decreased nerve growth factor expression in the olfactory bulbectomized rat model of depression: effects of chronic ethyl-eicosapentaenoate treatment. J Neurosci (2009a) 29(1):14–22. doi: 10.1523/JNEUROSCI.3569-08.2009
217. Hillier K, Templeton WW. Regulation of noradrenaline overflow in rat cerebral cortex by prostaglandin E2. Br J Pharmacol (1980) 70(3):469–73. doi: 10.1111/j.1476-5381.1980.tb08725.x
218. Casolini P, Catalani A, Zuena AR, Angelucci L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J Neurosci Res (2002) 68(3):337–43. doi: 10.1002/jnr.10192
219. Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci (2002) 25(3):154–9. doi: 10.1016/S0166-2236(00)02088-9
220. Schiepers OJ, Wichers MC, Maes M. Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry (2005) 29(2):201–17. doi: 10.1016/j.pnpbp.2004.11.003
221. Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B. Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord (2007) 98(1–2):143–51. doi: 10.1016/j.jad.2006.07.013
222. Brunello N, Alboni S, Capone G, Benatti C, Blom JM, Tascedda F, et al. Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in the chronic escape deficit model of depression. Int Clin Psychopharmacol (2006) 21(4):219–25. doi: 10.1097/00004850-200607000-00004
223. Kohler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O, et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry (2014) 71(12):1381–91. doi: 10.1001/jamapsychiatry.2014.1611
224. Na KS, Lee KJ, Lee JS, Cho YS, Jung HY. Efficacy of adjunctive celecoxib treatment for patients with major depressive disorder: a meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry (2014) 48:79–85. doi: 10.1016/j.pnpbp.2013.09.006
225. Eyre HA, Air T, Pradhan A, Johnston J, Lavretsky H, Stuart MJ, et al. A meta-analysis of chemokines in major depression. Prog Neuropsychopharmacol Biol Psychiatry (2016) 68:1–8. doi: 10.1016/j.pnpbp.2016.02.006
226. Kim JY, Park JH, Lee JJ, Huh Y, Lee SB, Han SK, et al. Standardization of the Korean version of the Geriatric Depression Scale: reliability, validity, and factor structure. Psychiatry Investig (2008) 5(4):232–8. doi: 10.4306/pi.2008.5.4.232
227. Baldi PC, Giambartolomei GH. Immunopathology of Brucella infection. Recent Pat Antiinfect Drug Discov (2013a) 8(1):18–26. doi: 10.2174/1574891X11308010005
228. Baldi PC, Giambartolomei GH. Pathogenesis and pathobiology of zoonotic brucellosis in humans. Rev Sci Tech (2013b) 32(1):117–25. doi: 10.20506/rst.32.1.2192
229. Iyengar RL, Gandhi S, Aneja A, Thorpe K, Razzouk L, Greenberg J, et al. NSAIDs are associated with lower depression scores in patients with osteoarthritis. Am J Med (2013) 126(11):1017.e11–8. doi: 10.1016/j.amjmed.2013.02.037
230. Lai V, George J, Richey L, Kim HJ, Cannon T, Shores C, et al. Results of a pilot study of the effects of celecoxib on cancer cachexia in patients with cancer of the head, neck, and gastrointestinal tract. Head Neck (2008) 30(1):67–74. doi: 10.1002/hed.20662
231. Lundholm K, Daneryd P, Korner U, Hyltander A, Bosaeus I. Evidence that long-term COX-treatment improves energy homeostasis and body composition in cancer patients with progressive cachexia. Int J Oncol (2004) 24(3):505–12. doi: 10.3892/ijo.24.3.505
232. Pedersen CB, Mors O, Bertelsen A, Waltoft BL, Agerbo E, McGrath JJ, et al. A comprehensive nationwide study of the incidence rate and lifetime risk for treated mental disorders. JAMA Psychiatry (2014) 71(5):573–81. doi: 10.1001/jamapsychiatry.2014.16
233. Malhi GS, Ivanovski B, Hadzi-Pavlovic D, Mitchell PB, Vieta E, Sachdev P. Neuropsychological deficits and functional impairment in bipolar depression, hypomania and euthymia. Bipolar Disord (2007) 9(1–2):114–25. doi: 10.1111/j.1399-5618.2007.00324.x
234. Marotta A, Chiaie RD, Spagna A, Bernabei L, Sciarretta M, Roca J, et al. Impaired conflict resolution and vigilance in euthymic bipolar disorder. Psychiatry Res (2015) 229(1–2):490–6. doi: 10.1016/j.psychres.2015.06.026
235. Baumeister D, Russell A, Pariante CM, Mondelli V. Inflammatory biomarker profiles of mental disorders and their relation to clinical, social and lifestyle factors. Soc Psychiatry Psychiatr Epidemiol (2014) 49(6):841–9. doi: 10.1007/s00127-014-0887-z
236. Drexhage RC, van der Heul-Nieuwenhuijsen L, Padmos RC, van Beveren N, Cohen D, Versnel MA, et al. Inflammatory gene expression in monocytes of patients with schizophrenia: overlap and difference with bipolar disorder. A study in naturalistically treated patients. Int J Neuropsychopharmacol (2010b) 13(10):1369–81. doi: 10.1017/S1461145710000799
237. Dargel AA, Godin O, Kapczinski F, Kupfer DJ, Leboyer M. C-reactive protein alterations in bipolar disorder: a meta-analysis. J Clin Psychiatry (2015) 76(2):142–50. doi: 10.4088/JCP.14r09007
238. Munkholm K, Brauner JV, Kessing LV, Vinberg M. ‘Cytokines in bipolar disorder vs. healthy control subjects: a systematic review and meta-analysis. J Psychiatr Res (2013a) 47(9):1119–33. doi: 10.1016/j.jpsychires.2013.05.018
239. Barbosa IG, Bauer ME, Machado-Vieira R, Teixeira AL. Cytokines in bipolar disorder: paving the way for neuroprogression. Neural Plast (2014a) 2014:360481. doi: 10.1155/2014/360481
240. Barbosa IG, Machado-Vieira R, Soares JC, Teixeira AL. The immunology of bipolar disorder. Neuroimmunomodulation (2014b) 21(2-3):117–22. doi: 10.1159/000356539
241. Haarman BC, Riemersma-Van der Lek RF, de Groot JC, Ruhe HG, Klein HC, Zandstra TE, et al. Neuroinflammation in bipolar disorder - A [(11)C]-(R)-PK11195 positron emission tomography study. Brain Behav Immun (2014) 40:219–25. doi: 10.1016/j.bbi.2014.03.016
242. Jakobsson J, Bjerke M, Sahebi S, Isgren A, Ekman CJ, Sellgren C, et al. Monocyte and microglial activation in patients with mood-stabilized bipolar disorder. J Psychiatry Neurosci (2015) 40(4):250–8. doi: 10.1503/jpn.140183
243. Rao JS, Harry GJ, Rapoport SI, Kim HW. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry (2010) 15(4):384–92. doi: 10.1038/mp.2009.47
244. Schneider MR, DelBello MP, McNamara RK, Strakowski SM, Adler CM. Neuroprogression in bipolar disorder. Bipolar Disord (2012) 14(4):356–74. doi: 10.1111/j.1399-5618.2012.01024.x
245. Strakowski SM, Adler CM, Almeida J, Altshuler LL, Blumberg HP, Chang KD, et al. The functional neuroanatomy of bipolar disorder: a consensus model. Bipolar Disord (2012) 14(4):313–25. doi: 10.1111/j.1399-5618.2012.01022.x
246. Boufidou F, Nikolaou C, Alevizos B, Liappas IA, Christodoulou GN. Cytokine production in bipolar affective disorder patients under lithium treatment. J Affect Disord (2004) 82(2):309–13. doi: 10.1016/j.jad.2004.01.007
247. Basselin M, Kim H-W, Chen M, Ma K, Rapoport SI, Murphy RC, et al. Lithium modifies brain arachidonic and docosahexaenoic metabolism in rat lipopolysaccharide model of neuroinflammation. J Lipid Res (2010) 51(5):1049–56. doi: 10.1194/jlr.M002469
248. Breunis MN, Kupka RW, Nolen WA, Suppes T, Denicoff KD, Leverich GS, et al. High numbers of circulating activated T cells and raised levels of serum IL-2 receptor in bipolar disorder. Biol Psychiatry (2003) 53(2):157–65. doi: 10.1016/S0006-3223(02)01452-X
249. Chiu CT, Wang Z, Hunsberger JG, Chuang DM. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev (2013) 65(1):105–42. doi: 10.1124/pr.111.005512
250. Knijff EM, Breunis MN, Kupka RW, de Wit HJ, Ruwhof C, Akkerhuis GW, et al. An imbalance in the production of IL-1beta and IL-6 by monocytes of bipolar patients: restoration by lithium treatment. Bipolar Disord (2007) 9(7):743–53. doi: 10.1111/j.1399-5618.2007.00444.x
251. Lee HJ, Rao JS, Rapoport SI, Bazinet RP. Antimanic therapies target brain arachidonic acid signaling: lessons learned about the regulation of brain fatty acid metabolism. Prostaglandins Leukot Essent Fatty Acids (2007) 77(5–6):239–46. doi: 10.1016/j.plefa.2007.10.018
252. O’Brien SM, Scully P, Scott LV, Dinan TG. Cytokine profiles in bipolar affective disorder: focus on acutely ill patients. J Affect Disord (2006) 90(2–3):263–7. doi: 10.1016/j.jad.2005.11.015
253. Rapoport SI, Bosetti F. Do lithium and anticonvulsants target the brain arachidonic acid cascade in bipolar disorder? Arch Gen Psychiatry (2002) 59(7):592–6. doi: 10.1001/archpsyc.59.7.592
254. Perugi G, Medda P, Toni C, Mariani MG, Socci C, Mauri M. The role of electroconvulsive therapy (ect) in bipolar disorder: effectiveness in 522 patients with bipolar depression, mixed-state, mania and catatonic features. Curr Neuropharmacol (2017) 15(3):359–71. doi: 10.2174/1570159X14666161017233642
255. Fluitman SB, Heijnen CJ, Denys DA, Nolen WA, Balk FJ, Westenberg HG. Electroconvulsive therapy has acute immunological and neuroendocrine effects in patients with major depressive disorder. J Affect Disord (2011) 131(1–3):388–92. doi: 10.1016/j.jad.2010.11.035
256. Hestad KA, Tonseth S, Stoen CD, Ueland T, Aukrust P. Raised plasma levels of tumor necrosis factor alpha in patients with depression: normalization during electroconvulsive therapy. J ECT (2003) 19(4):183–8. doi: 10.1097/00124509-200312000-00002
257. Taylor S. Electroconvulsive therapy: a review of history, patient selection, technique, and medication management. South Med J (2007) 100(5):494–8. doi: 10.1097/SMJ.0b013e318038fce0
258. Guloksuz S, Arts B, Walter S, Drukker M, Rodriguez L, Myint AM, et al. The impact of electroconvulsive therapy on the tryptophan–kynurenine metabolic pathway. Brain Behav Immun (2015) 48:48–52. doi: 10.1016/j.bbi.2015.02.029
259. Schwieler L, Samuelsson M, Frye MA, Bhat M, Schuppe-Koistinen I, Jungholm O, et al. Electroconvulsive therapy suppresses the neurotoxic branch of the kynurenine pathway in treatment-resistant depressed patients. J Neuroinflammation (2016) 13(1):51. doi: 10.1186/s12974-016-0517-7
260. Kargar M, Yousefi A, Mojtahedzadeh M, Akhondzadeh S, Artounian V, Abdollahi A, et al. Effects of celecoxib on inflammatory markers in bipolar patients undergoing electroconvulsive therapy: a placebo-controlled, double-blind, randomised study. Swiss Med Wkly (2014) 144:w13880. doi: 10.4414/smw.2014.13880
261. Lehtimaki K, Keranen T, Huuhka M, Palmio J, Hurme M, Leinonen E, et al. Increase in plasma proinflammatory cytokines after electroconvulsive therapy in patients with depressive disorder. J ECT (2008) 24(1):88–91. doi: 10.1097/YCT.0b013e3181571abb
262. Husain MI, Chaudhry IB, Hamirani MM, Minhas FA, Kazmi A, Hodsoll J, et al. Minocycline and celecoxib as adjunctive treatments for bipolar depression: a study protocol for a multicenter factorial design randomized controlled trial. Neuropsychiatr Dis Treat (2016) 13:1–8. doi: 10.2147/NDT.S115002
263. van Os J, Kapur S. Schizophrenia. Lancet (2009) 374(9690):635–45. doi: 10.1016/S0140-6736(09)60995-8
264. Marini S, De Berardis D, Vellante F, Santacroce R, Orsolini L, Valchera A, et al. Celecoxib adjunctive treatment to antipsychotics in schizophrenia: a review of randomized clinical add-on trials. Mediators Inflamm (2016) 2016:3476240. doi: 10.1155/2016/3476240
265. Jarskog LF, Xiao H, Wilkie MB, Lauder JM, Gilmore JH. Cytokine regulation of embryonic rat dopamine and serotonin neuronal survival in vitro. Int J Dev Neurosci (1997) 15(6):711–6. doi: 10.1016/S0736-5748(97)00029-4
266. Kabiersch A, Furukawa H, del Rey A, Besedovsky HO. Administration of interleukin-1 at birth affects dopaminergic neurons in adult mice. Ann N Y Acad Sci (1998) 840:123–7. doi: 10.1111/j.1749-6632.1998.tb09556.x
267. Ling ZD, Potter ED, Lipton JW, Carvey PM. Differentiation of mesencephalic progenitor cells into dopaminergic neurons by cytokines. Exp Neurol (1998) 149(2):411–23. doi: 10.1006/exnr.1998.6715
268. Potter ED, Ling ZD, Carvey PM. Cytokine-induced conversion of mesencephalic-derived progenitor cells into dopamine neurons. Cell Tissue Res (1999) 296(2):235–46. doi: 10.1007/s004410051285
269. Kegel ME, Bhat M, Skogh E, Samuelsson M, Lundberg K, Dahl ML, et al. Imbalanced kynurenine pathway in schizophrenia. Int J Tryptophan Res (2014) 7:15–22. doi: 10.4137/IJTR.S16800
270. Zuckerman L, Weiner I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J Psychiatr Res (2005) 39(3):311–23. doi: 10.1016/j.jpsychires.2004.08.008
271. Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull (2006) 32(2):200–2. doi: 10.1093/schbul/sbj052
272. Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry (2010) 167(3):261–80. doi: 10.1176/appi.ajp.2009.09030361
273. Eaton WW, Byrne M, Ewald H, Mors O, Chen CY, Agerbo E, et al. Association of schizophrenia and autoimmune diseases: linkage of Danish national registers. Am J Psychiatry (2006) 163(3):521–8. doi: 10.1176/appi.ajp.163.3.521
274. Chen YJ, Wu CY, Lin MW, Chen TJ, Liao KK, Chen YC, et al. Comorbidity profiles among patients with bullous pemphigoid: a nationwide population-based study. Br J Dermatol (2011) 165(3):593–9. doi: 10.1111/j.1365-2133.2011.10386.x
275. Eaton WW, Hayward C, Ram R. Schizophrenia and rheumatoid arthritis: a review. Schizophr Res (1992) 6(3):181–92. doi: 10.1016/0920-9964(92)90001-L
276. Korschenhausen DA, Hampel HJ, Ackenheil M, Penning R, Muller N. Fibrin degradation products in post mortem brain tissue of schizophrenics: a possible marker for underlying inflammatory processes. Schizophr Res (1996) 19(2–3):103–9. doi: 10.1016/0920-9964(95)00073-9
277. Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med (2009) 50(11):1801–7. doi: 10.2967/jnumed.109.066647
278. Monji A, Kato T, Kanba S. Cytokines and schizophrenia: microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci (2009) 63(3):257–65. doi: 10.1111/j.1440-1819.2009.01945.x
279. Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R, et al. Peripheral benzodiazepine receptors in patients with chronic schizophrenia: a PET study with [11C]DAA1106. Int J Neuropsychopharmacol (2010) 13(7):943–50. doi: 10.1017/S1461145710000313
280. van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry (2008) 64(9):820–2. doi: 10.1016/j.biopsych.2008.04.025
281. Arango C, Rapado-Castro M, Reig S, Castro-Fornieles J, Gonzalez-Pinto A, Otero S, et al. Progressive brain changes in children and adolescents with first-episode psychosis. Arch Gen Psychiatry (2012) 69(1):16–26. doi: 10.1001/archgenpsychiatry.2011.150
282. DeLisi LE, Szulc KU, Bertisch HC, Majcher M, Brown K. Understanding structural brain changes in schizophrenia. Dialogues Clin Neurosci (2006) 8(1):71–8.
283. Steen RG, Mull C, McClure R, Hamer RM, Lieberman JA. Brain volume in first-episode schizophrenia: systematic review and meta-analysis of magnetic resonance imaging studies. Br J Psychiatry (2006) 188:510–8. doi: 10.1192/bjp.188.6.510
284. Kahn RS, Sommer IE. The neurobiology and treatment of first-episode schizophrenia. Mol Psychiatry (2015) 20(1):84–97. doi: 10.1038/mp.2014.66
285. Fillman SG, Cloonan N, Miller LC, Weickert CS. Markers of inflammation in the prefrontal cortex of individuals with schizophrenia. Mol Psychiatry (2013) 18(2):133. doi: 10.1038/mp.2012.199
286. Garver DL, Tamas RL, Holcomb JA. Elevated interleukin-6 in the cerebrospinal fluid of a previously delineated schizophrenia subtype. Neuropsychopharmacology (2003) 28(8):1515–20. doi: 10.1038/sj.npp.1300217
287. Meisenzahl EM, Rujescu D, Kirner A, Giegling I, Kathmann N, Leinsinger G, et al. Association of an interleukin-1beta genetic polymorphism with altered brain structure in patients with schizophrenia. Am J Psychiatry (2001) 158(8):1316–9. doi: 10.1176/appi.ajp.158.8.1316
288. Cannon TD, Chung Y, He G, Sun D, Jacobson A, van Erp TG, et al. Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry (2015) 77(2):147–57. doi: 10.1016/j.biopsych.2014.05.023
289. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature (2016) 530(7589):177–83. doi: 10.1038/nature16549
290. Drexhage RC, Knijff EM, Padmos RC, Heul-Nieuwenhuijzen L, Beumer W, Versnel MA, et al. The mononuclear phagocyte system and its cytokine inflammatory networks in schizophrenia and bipolar disorder. Expert Rev Neurother (2010a) 10(1):59–76. doi: 10.1586/ern.09.144
291. Fan X, Goff DC, Henderson DC. Inflammation and schizophrenia. Expert Rev Neurother (2007) 7(7):789–96. doi: 10.1586/14737175.7.7.789
292. Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry (2008) 63(8):801–8. doi: 10.1016/j.biopsych.2007.09.024
293. Holden JM, Meyers-Manor JE, Overmier JB, Gahtan E, Sweeney W, Miller H. Lipopolysaccharide-induced immune activation impairs attention but has little effect on short-term working memory. Behav Brain Res (2008) 194(2):138–45. doi: 10.1016/j.bbr.2008.06.032
294. Moller HJ. Clinical evaluation of negative symptoms in schizophrenia. Eur Psychiatry (2007) 22(6):380–6. doi: 10.1016/j.eurpsy.2007.03.010
295. Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr Res (2009) 110(1–3):1–23. doi: 10.1016/j.schres.2009.03.005
296. Dickerson F, Stallings C, Origoni A, Boronow J, Yolken R. C-reactive protein is associated with the severity of cognitive impairment but not of psychiatric symptoms in individuals with schizophrenia. Schizophr Res (2007) 93(1–3):261–5. doi: 10.1016/j.schres.2007.03.022
297. Liu L, Jia F, Yuan G, Chen Z, Yao J, Li H, et al. Tyrosine hydroxylase, interleukin-1beta and tumor necrosis factor-alpha are overexpressed in peripheral blood mononuclear cells from schizophrenia patients as determined by semi-quantitative analysis. Psychiatry Res (2010) 176(1):1–7. doi: 10.1016/j.psychres.2008.10.024
298. Pedersen A, Diedrich M, Kaestner F, Koelkebeck K, Ohrmann P, Ponath G, et al. Memory impairment correlates with increased S100B serum concentrations in patients with chronic schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry (2008) 32(8):1789–92. doi: 10.1016/j.pnpbp.2008.07.017
299. Miller B, Buckley PF, Mellor A, Kirkpatrick BW. Meta-analysis of cytokine alterations in schizophrenia: clinical status, symptoms, and antipsychotic effects. Schizophr Bull (2011a) 37:57.
300. Baumeister D, Ciufolini S, Mondelli V. Effects of psychotropic drugs on inflammation: consequence or mediator of therapeutic effects in psychiatric treatment? Psychopharmacology (Berl) (2016) 233(9):1575–89. doi: 10.1007/s00213-015-4044-5
301. Fernandes BS, Steiner J, Bernstein HG, Dodd S, Pasco JA, Dean OM, et al. C-reactive protein is increased in schizophrenia but is not altered by antipsychotics: meta-analysis and implications. Mol Psychiatry (2016) 21(4):554–64. doi: 10.1038/mp.2015.87
302. Kim YK, Myint AM, Verkerk R, Scharpe S, Steinbusch H, Leonard B. Cytokine changes and tryptophan metabolites in medication-naive and medication-free schizophrenic patients. Neuropsychobiology (2009) 59(2):123–9. doi: 10.1159/000213565
303. Pollmacher T, Haack M, Schuld A, Kraus T, Hinze-Selch D. Effects of antipsychotic drugs on cytokine networks. J Psychiatr Res (2000) 34(6):369–82. doi: 10.1016/S0022-3956(00)00032-7
304. Song XQ, Lv LX, Li WQ, Hao YH, Zhao JP. The interaction of nuclear factor-kappa B and cytokines is associated with schizophrenia. Biol Psychiatry (2009b) 65(6):481–8. doi: 10.1016/j.biopsych.2008.10.018
305. Cazzullo CL, Sacchetti E, Galluzzo A, Panariello A, Adorni A, Pegoraro M, et al. Cytokine profiles in schizophrenic patients treated with risperidone: a 3-month follow-up study. Prog Neuropsychopharmacol Biol Psychiatry (2002) 26(1):33–9. doi: 10.1016/S0278-5846(01)00221-4
306. Maes M, Bosmans E, Kenis G, De Jong R, Smith RS, Meltzer HY. In vivo immunomodulatory effects of clozapine in schizophrenia. Schizophr Res (1997) 26(2–3):221–5. doi: 10.1016/S0920-9964(97)00057-1
307. Song C, Lin A, Kenis G, Bosmans E, Maes M. Immunosuppressive effects of clozapine and haloperidol: enhanced production of the interleukin-1 receptor antagonist. Schizophr Res (2000) 42(2):157–64. doi: 10.1016/S0920-9964(99)00116-4
308. Drzyzga L, Obuchowicz E, Marcinowska A, Herman ZS. Cytokines in schizophrenia and the effects of antipsychotic drugs. Brain Behav Immun (2006) 20(6):532–45. doi: 10.1016/j.bbi.2006.02.002
309. Plitman E, Iwata Y, Caravaggio F, Nakajima S, Chung JK, Gerretsen P, et al. Kynurenic acid in schizophrenia: a systematic review and meta-analysis. Schizophr Bull (2017) 43(4):764–77. doi: 10.1093/schbul/sbw221
310. Ceresoli-Borroni G, Rassoulpour A, Wu HQ, Guidetti P, Schwarcz R. Chronic neuroleptic treatment reduces endogenous kynurenic acid levels in rat brain. J Neural Transm (Vienna) (2006) 113(10):1355–65. doi: 10.1007/s00702-005-0432-z
311. Mirjany M, Ho L, Pasinetti GM. Role of cyclooxygenase-2 in neuronal cell cycle activity and glutamate-mediated excitotoxicity. J Pharmacol Exp Ther (2002) 301(2):494–500. doi: 10.1124/jpet.301.2.494
312. Muller N, Riedel M, Schwarz MJ, Engel RR. Clinical effects of COX-2 inhibitors on cognition in schizophrenia. Eur Arch Psychiatry Clin Neurosci (2005) 255(2):149–51. doi: 10.1007/s00406-004-0548-4
313. Zheng W, Cai DB, Yang XH, Ungvari GS, Ng CH, Muller N, et al. Adjunctive celecoxib for schizophrenia: a meta-analysis of randomized, double-blind, placebo-controlled trials. J Psychiatr Res (2017) 92:139–46. doi: 10.1016/j.jpsychires.2017.04.004
314. Diagnostic and statistical manual of mental disorders: DSM-5. 2013, American Psychiatric Association, Arlington, VA.
315. Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatrics (2004) 113(5):e472–86. doi: 10.1542/peds.113.5.e472
316. Goines P, de Water J. The immune system’s role in the biology of autism. Curr Opin Neurol (2010) 23(2):111–7. doi: 10.1097/WCO.0b013e3283373514
317. Nakagawa Y, Chiba K. Involvement of neuroinflammation during brain development in social cognitive deficits in autism spectrum disorder and schizophrenia. J Pharmacol Exp Ther (2016) 358(3):504. doi: 10.1124/jpet.116.234476
318. Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun (2012) 26(3):383–92. doi: 10.1016/j.bbi.2011.08.007
319. Petrelli F, Pucci L, Bezzi P. Astrocytes and microglia and their potential link with autism spectrum disorders. Front Cell Neurosci (2016) 10:21. doi: 10.3389/fncel.2016.00021
320. Wakefield AJ, Murch SH, Anthony A, Linnell J, Casson DM, Malik M, et al. Ileal-lymphoid-nodular hyperplasia, non-specific colitis, and pervasive developmental disorder in children. Lancet (1998) 351(9103):637–41. doi: 10.1016/S0140-6736(97)11096-0
321. Fombonne E, Chakrabarti S. No evidence for a new variant of measles-mumps-rubella-induced autism. Pediatrics (2001) 108(4):E58. doi: 10.1542/peds.108.4.e58
322. Gerber JS, Offit PA. Vaccines and autism: a tale of shifting hypotheses. Clin Infect Dis (2009) 48(4):456–61. doi: 10.1086/596476
323. Offit PA, Coffin SE. Communicating science to the public: MMR vaccine and autism. Vaccine (2003) 22(1):1–6. doi: 10.1016/S0264-410X(03)00532-2
324. Taylor B, Miller E, Lingam R, Andrews N, Simmons A, Stowe J. Measles, mumps, and rubella vaccination and bowel problems or developmental regression in children with autism: population study. BMJ (2002) 324(7334):393–6. doi: 10.1136/bmj.324.7334.393
325. Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol (2012) 72(10):1272–6. doi: 10.1002/dneu.22024
326. Fang SY, Wang S, Huang N, Yeh HH, Chen CY. Prenatal Infection and autism spectrum disorders in childhood: a population-based case-control study in Taiwan. Paediatr Perinat Epidemiol (2015) 29(4):307–16. doi: 10.1111/ppe.12194
327. Jiang HY, Xu LL, Shao L, Xia RM, Yu ZH, Ling ZX, et al. Maternal infection during pregnancy and risk of autism spectrum disorders: A systematic review and meta-analysis. Brain Behav Immun (2016) 58:165–72. doi: 10.1016/j.bbi.2016.06.005
328. Ziats MN, Rennert OM. Expression profiling of autism candidate genes during human brain development implicates central immune signaling pathways. PLoS One (2011) 6(9):e24691. doi: 10.1371/journal.pone.0024691
329. Lim CK, Essa MM, de Paula Martins R, Lovejoy DB, Bilgin AA, Waly MI, et al. Altered kynurenine pathway metabolism in autism: Implication for immune-induced glutamatergic activity. Autism Res (2016) 9(6):621–31. doi: 10.1002/aur.1565
330. Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun (2011) 25(1):40–5. doi: 10.1016/j.bbi.2010.08.003
331. Heuer L, Ashwood P, Schauer J, Goines P, Krakowiak P, Hertz-Picciotto I, et al. Reduced levels of immunoglobulin in children with autism correlates with behavioral symptoms. Autism Res (2008) 1(5):275–83. doi: 10.1002/aur.42
332. Wills S, Cabanlit M, Bennett J, Ashwood P, Amaral D, de Water J. Autoantibodies in autism spectrum disorders (ASD). Ann N Y Acad Sci (2007) 1107:79–91. doi: 10.1196/annals.1381.009
333. Grigorenko EL, Han SS, Yrigollen CM, Leng L, Mizue Y, Anderson GM, et al. Macrophage migration inhibitory factor and autism spectrum disorders. Pediatrics (2008) 122(2):e438–45. doi: 10.1542/peds.2007-3604
334. Wang F, Wu H, Xu S, Guo X, Yang J, Shen X. Macrophage migration inhibitory factor activates cyclooxygenase 2-prostaglandin E2 in cultured spinal microglia. Neurosci Res (2011) 71(3):210–8. doi: 10.1016/j.neures.2011.07.1821
335. Blaylock RL, Strunecka A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr Med Chem (2009) 16(2):157–70. doi: 10.2174/092986709787002745
336. Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry (2010) 68(4):368–76. doi: 10.1016/j.biopsych.2010.05.024
337. Tetreault NA, Hakeem AY, Jiang S, Williams BA, Allman E, Wold BJ, et al. Microglia in the cerebral cortex in autism. J Autism Dev Disord (2012) 42(12):2569–84. doi: 10.1007/s10803-012-1513-0
338. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol (2005) 57(1):67–81. doi: 10.1002/ana.20315
339. Careaga M, Schwartzer J, Ashwood P. Inflammatory profiles in the BTBR mouse: how relevant are they to autism spectrum disorders? Brain Behav Immun (2015) 43:11–6. doi: 10.1016/j.bbi.2014.06.006
340. El-Ansary A, Al-Ayadhi L. Neuroinflammation in autism spectrum disorders. J Neuroinflammation (2012) 9:265. doi: 10.1186/1742-2094-9-265
341. Boris M, Kaiser CC, Goldblatt A, Elice MW, Edelson SM, Adams JB, et al. Effect of pioglitazone treatment on behavioral symptoms in autistic children. J Neuroinflammation (2007) 4:3. doi: 10.1186/1742-2094-4-3
342. Kim EJ, Kwon KJ, Park JY, Lee SH, Moon CH, Baik EJ. Effects of peroxisome proliferator-activated receptor agonists on LPS-induced neuronal death in mixed cortical neurons: associated with iNOS and COX-2. Brain Res (2002) 941(1–2):1–10. doi: 10.1016/S0006-8993(02)02480-0
343. Sepanjnia K, Modabbernia A, Ashrafi M, Modabbernia MJ, Akhondzadeh S. Pioglitazone adjunctive therapy for moderate-to-severe major depressive disorder: randomized double-blind placebo-controlled trial. Neuropsychopharmacology (2012) 37(9):2093–100. doi: 10.1038/npp.2012.58
344. Kaizaki A, Tien L-T, Pang Y, Cai Z, Tanaka S, Numazawa S, et al. Celecoxib reduces brain dopaminergic neuronal dysfunction, and improves sensorimotor behavioral performance in neonatal rats exposed to systemic lipopolysaccharide. J Neuroinflammation (2013) 10(1):818. doi: 10.1186/1742-2094-10-45
345. de Mathis MA, Diniz JB, Hounie AG, Shavitt RG, Fossaluza V, Ferrao Y, et al. Trajectory in obsessive-compulsive disorder comorbidities. Eur Neuropsychopharmacol (2013) 23(7):594–601. doi: 10.1016/j.euroneuro.2012.08.006
346. Heidari M, Zarei M, Hosseini SM, Taghvaei R, Maleki H, Tabrizi M, et al. Ondansetron or placebo in the augmentation of fluvoxamine response over 8 weeks in obsessive-compulsive disorder. Int Clin Psychopharmacol (2014) 29(6):344–50. doi: 10.1097/YIC.0000000000000043
347. Askari N, Moin M, Sanati M, Tajdini M, Hosseini SM, Modabbernia A, et al. Granisetron adjunct to fluvoxamine for moderate to severe obsessive-compulsive disorder: a randomized, double-blind, placebo-controlled trial. CNS Drugs (2012) 26(10):883–92. doi: 10.2165/11635850-000000000-00000
348. Erzegovesi S, Cavallini MC, Cavedini P, Diaferia G, Locatelli M, Bellodi L. Clinical predictors of drug response in obsessive-compulsive disorder. J Clin Psychopharmacol (2001) 21(5):488–92. doi: 10.1097/00004714-200110000-00006
349. Milad MR, Rauch SL. Obsessive-compulsive disorder: beyond segregated cortico-striatal pathways. Trends Cogn Sci (2012) 16(1):43–51. doi: 10.1016/j.tics.2011.11.003
350. Perlmutter SJ, Leitman SF, Garvey MA, Hamburger S, Feldman E, Leonard HL, et al. Therapeutic plasma exchange and intravenous immunoglobulin for obsessive-compulsive disorder and tic disorders in childhood. Lancet (1999) 354(9185):1153–8. doi: 10.1016/S0140-6736(98)12297-3
351. Boedhoe PS, Schmaal L, Abe Y, Ameis SH, Arnold PD, Batistuzzo MC, et al. Distinct subcortical volume alterations in pediatric and adult OCD: a worldwide meta- and mega-analysis. Am J Psychiatry (2017) 174(1):60–9.
352. Salazar A, Gonzalez-Rivera BL, Redus L, Parrott JM, O’Connor JC. Indoleamine 2,3-dioxygenase mediates anhedonia and anxiety-like behaviors caused by peripheral lipopolysaccharide immune challenge. Horm Behav (2012) 62(3):202–9. doi: 10.1016/j.yhbeh.2012.03.010
353. Yang L, Wang M, Guo YY, Sun T, Li YJ, Yang Q, et al. Systemic inflammation induces anxiety disorder through CXCL12/CXCR4 pathway. Brain Behav Immun (2016) 56:352–62. doi: 10.1016/j.bbi.2016.03.001
354. Antony JM. Grooming and Growing with Microglia. Sci Signal (2010) 3(147):jc8. doi: 10.1126/scisignal.3147jc8
355. Banati RB, Myers R, Kreutzberg GW. PK (‘peripheral benzodiazepine’)—binding sites in the CNS indicate early and discrete brain lesions: microautoradiographic detection of [3H]PK11195 binding to activated microglia. J Neurocytol (1997) 26(2):77–82. doi: 10.1023/A:1018567510105
356. Hannestad J, DellaGioia N, Gallezot JD, Lim K, Nabulsi N, Esterlis I, et al. The neuroinflammation marker translocator protein is not elevated in individuals with mild-to-moderate depression: a [(1)(1)C]PBR28 PET study. Brain Behav Immun (2013) 33:131–8. doi: 10.1016/j.bbi.2013.06.010
357. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci (1996) 19(8):312–8. doi: 10.1016/0166-2236(96)10049-7
358. Martin A, Boisgard R, Theze B, Van Camp N, Kuhnast B, Damont A, et al. Evaluation of the PBR/TSPO radioligand [(18)F]DPA-714 in a rat model of focal cerebral ischemia. J Cereb Blood Flow Metab (2010) 30(1):230–41. doi: 10.1038/jcbfm.2009.205
359. Kumar A, Williams MT, Chugani HT. Evaluation of basal ganglia and thalamic inflammation in children with pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection and Tourette syndrome: a positron emission tomographic (PET) study using 11C-[R]-PK11195. J Child Neurol (2015) 30(6):749–56. doi: 10.1177/0883073814543303
360. Fettes P, Schulze L, Downar J. Cortico-striatal-thalamic loop circuits of the orbitofrontal cortex: promising therapeutic targets in psychiatric illness. Front Syst Neurosci (2017) 11:25. doi: 10.3389/fnsys.2017.00025
361. Peters SK, Dunlop K, Downar J. Cortico-striatal-thalamic loop circuits of the salience network: a central pathway in psychiatric disease and treatment. Front Syst Neurosci (2016) 10:104. doi: 10.3389/fnsys.2016.00104
362. Esalatmanesh S, Abrishami Z, Zeinoddini A, Rahiminejad F, Sadeghi M, Najarzadegan MR, et al. Minocycline combination therapy with fluvoxamine in moderate-to-severe obsessive-compulsive disorder: a placebo-controlled, double-blind, randomized trial. Psychiatry Clin Neurosci (2016) 70(11):517–26. doi: 10.1111/pcn.12430
Keywords: depression, bipolar disorder, schizophrenia, obsessive compulsive disorder, autism spectrum disorder, psychiatry, inflammation, cyclooxygenase-2 inhibitors
Citation: Sethi R, Gómez-Coronado N, Walker AJ, Robertson OD’A, Agustini B, Berk M and Dodd S (2019) Neurobiology and Therapeutic Potential of Cyclooxygenase-2 (COX-2) Inhibitors for Inflammation in Neuropsychiatric Disorders. Front. Psychiatry 10:605. doi: 10.3389/fpsyt.2019.00605
Received: 17 September 2018; Accepted: 30 July 2019;
Published: 04 September 2019.
Edited by:
Richard G. Boles, Center for Neurological and Neurodevelopmental Health (CNNH), United StatesCopyright © 2019 Sethi, Gómez-Coronado, Walker, Robertson, Agustini, Berk and Dodd. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Berk bWljaGFlbC5iZXJrQGJhcndvbmhlYWx0aC5vcmcuYXU=