
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 26 June 2024
Sec. Plant Genetics, Epigenetics and Chromosome Biology
Volume 15 - 2024 | https://doi.org/10.3389/fpls.2024.1372127
 Kuan Yan1,2†
Kuan Yan1,2† Hui Zhu3†
Hui Zhu3† Guiling Cao1,2
Guiling Cao1,2 Lina Meng1,2Junqiang Li1,2Jian Zhang1,2Sicen Liu1,2Yujie Wang1,2Ruizhang Feng1,2Salma A. Soaud4Mohamed A. Abd Elhamid4Rania M. Y. Heakel4Qin Wei1,2*
Lina Meng1,2Junqiang Li1,2Jian Zhang1,2Sicen Liu1,2Yujie Wang1,2Ruizhang Feng1,2Salma A. Soaud4Mohamed A. Abd Elhamid4Rania M. Y. Heakel4Qin Wei1,2* Ahmed H. El-Sappah1,2,4*Dafu Ru3*
Ahmed H. El-Sappah1,2,4*Dafu Ru3*Introduction: Camphora longepaniculata, a crucial commercial crop and a fundamental component of traditional Chinese medicine, is renowned for its abundant production of volatile terpenoids. However, the lack of available genomic information has hindered pertinent research efforts in the past.
Methods: To bridge this gap, the present study aimed to use PacBio HiFi, short-read, and highthroughput chromosome conformation capture sequencing to construct a chromosome-level assembly of the C. longepaniculata genome.
Results and discussion: With twelve chromosomes accounting for 99.82% (766.69 Mb) of the final genome assembly, which covered 768.10 Mb, it was very complete. Remarkably, the assembly’s contig and scaffold N50 values are exceptional as well—41.12 and 63.78 Mb, respectively—highlighting its excellent quality and intact structure. Furthermore, a total of 39,173 protein-coding genes were predicted, with 38,766 (98.96%) of them being functionally annotated. The completeness of the genome was confirmed by the Benchmarking Universal Single-Copy Ortholog evaluation, which revealed 99.01% of highly conserved plant genes. As the first comprehensive assembly of the C. longepaniculata genome, it provides a crucial starting point for deciphering the complex pathways involved in terpenoid production. Furthermore, this excellent genome serves as a vital resource for upcoming research on the breeding and genetics of C. longepaniculata.
A species of evergreen tree native to southwestern China, namely the Yibin region of Sichuan, Camphora longepaniculata (Gamble) Y. Yang, Bing Liu, and Zhi Yang, is important both culturally and industrially (Figure 1A; Wu et al., 2022). Because of its fragrant qualities, it has become a traditional plant of interest that is both extensively grown and highly valued in regional customs. Its potential to extract essential oils from a variety of parts, including roots, stems, leaves, and seeds, accounts for most of its industrial value. Notably, the bulk is made up of leaf essential oil, which has a high concentration of terpenoids (>85%) (Hu et al., 2012). Prior research has revealed 1,8-cineole, α-terpilenol, and γ-terpinen as important components of leaf essential oil (Li et al., 2012, 2014). These essential oils are prized for their outstanding antibacterial, anti-inflammatory, and antioxidant qualities (Boutanaev et al., 2015; Wei et al., 2016). As with the biological traits of C. longepaniculata, studies have been conducted to investigate the molecular underpinnings of monoterpene production. Previous research has discovered important genes linked to terpene production using transcriptome sequencing technologies (Yan et al., 2017). In addition, Yan et al. (2019) investigated the differential expression of C. longepaniculata genes induced by endophytic fungi, revealing significant regulation within the monoterpene synthesis pathway. However, a high-quality genome is required as the starting point for additional research in order to fully comprehend the fundamental molecular mechanisms driving synthesis and oil production.
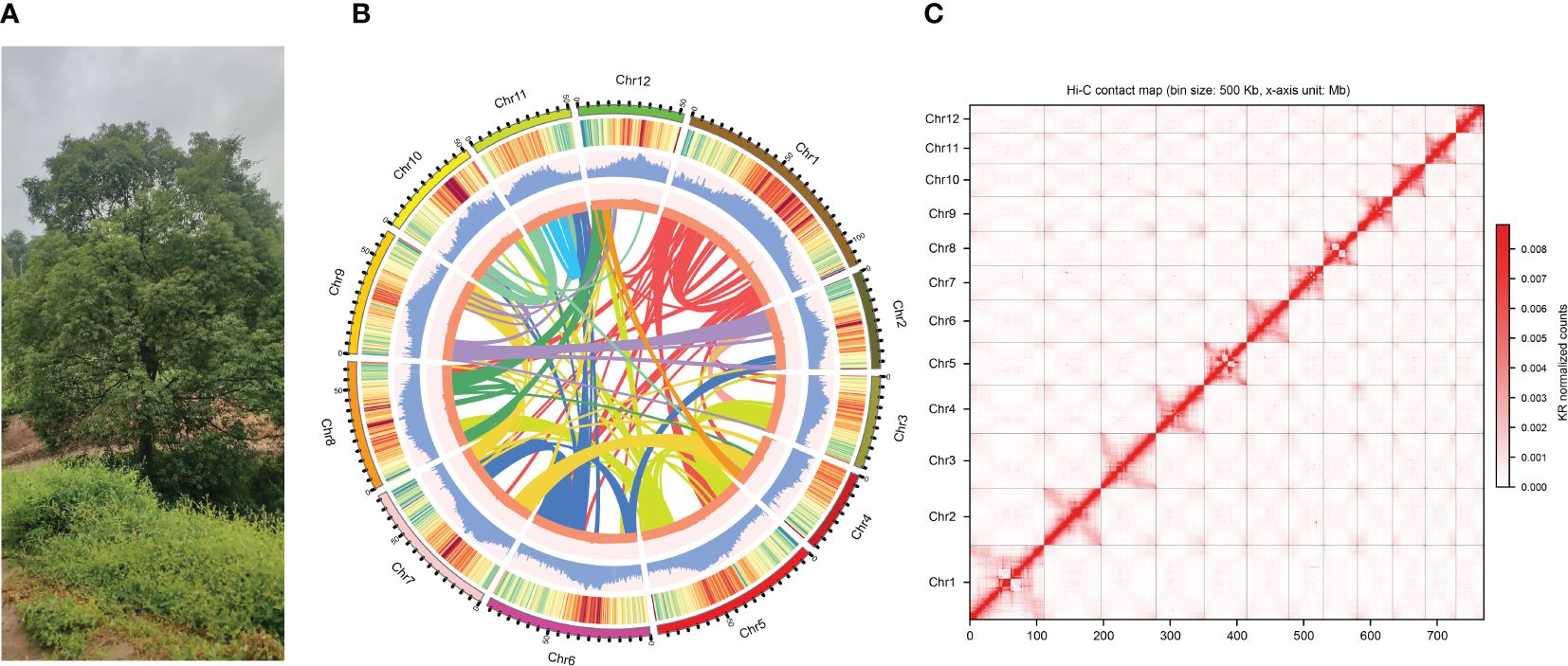
Figure 1 Sampling and genome assembly of Camphora longepaniculata. (A) The sequenced individual of C. longepaniculata leaves from Chinese province of Sichuan (27°50’ N, 105°20’ E). (B) Genome features from the C. longepaniculata assembly. From outer to inner: (1) genome chromosomes, (2) gene density, (3) repeat density, (4) GC (guanine-cytosine) content, and (5) synteny information. (C) Genome-wide chromosomal contact map of Camphora longepaniculata using Hi-C data. Interaction frequency distribution of Hi-C links among chromosomes shows in color key of heatmap ranging from white to dark red indicating the frequency of Hi-C interaction links from low to high.
The genomic terrain of C. longepaniculata is still unknown, despite advancements in genomic investigation within the Lauraceae family (Huang et al., 2016; Chaw et al., 2019; Song et al., 2019). Comprehensive and methodical study has been hampered by the lack of complete reference genomic material, particularly with regard to the unclear molecular mechanism of 1,8-cineole production. The goal of this work is to build a chromosome-level genome assembly of the C. longepaniculata by utilizing the quick development of high-throughput sequencing technologies including PacBio HiFi, short-read, and high-throughput chromosomal conformation capture (Hi-C) sequencing techniques. A wealth of genetic resources will be made available by this excellent genome assembly, allowing researchers to investigate the evolutionary background, gene functions, and regulatory mechanisms behind C. longepaniculata’s growth, development, and monoterpene synthesis. It will also provide a strong basis for the cultivation of high-yielding, high-quality cultivars of C. longepaniculata.
C. longepaniculata leaves were obtained in the Chinese province of Sichuan (27°50’ N, 105°20’ E). Genomic DNA was then extracted and processed according to the recommended library preparation methodology by PacBio. Subsequently, the DNA was utilized to construct PacBio sequencing libraries using the standard single molecular real-time bell construction protocol provided by PacBio. The libraries were then sequenced on the PacBio CCS platform. Adapter sequences and short, low-quality reads were removed from the PacBio HiFi reads using SMRTLink (parameters: –min-passes = 3 –min-rq = 0.99). Using the Agencourt AMPure XP-Medium Kit, the same DNA was used to create an Illumina paired-end library with insert sizes ranging from 200 to 400 bp. The Illumina NovaSeq 6000 platform was then used to sequence the resulting library. On the MGISEQ2000 platform, a Hi-C library was sequenced in accordance with the proximo Hi-C plant methodology (https://info.phasegenomics.com/protocols) developed by Phase Genomics (Seattle, WA, United States). After removing adaptors and duplicate reads, raw reads were filtered with a sequencing quality of >Q30 (Supplementary Table S1).
Lastly, RNA sequencing was performed on a leaf sample obtained from the same individual. Using the Qiagen RNeasy Plant Mini kit, total RNA was extracted from each sample. Then, with the TruSeq RNA Library Preparation kit, RNA-seq libraries were created, and 11.74 Gb of RNA-seq sequences were obtained by sequencing on the Illumina NovaSeq 6000 platform (Belton et al., 2012) (Supplementary Table S1).
Utilizing FASTP v0.20.0 (Chen et al., 2018), the short-read sequences were filtered to exclude adaptor contamination and low-quality reads. Subsequently, genome size, repeat content, and heterozygosity rate were estimated using GCE v1.0.2 tool from 19-mer histograms (Liu et al., 2013).
With the parameters “‘–write-ec –write-paf -u -l 0’’, the PacBio HiFi reads were assembled using Hifiasm v.0.15 (Cheng et al., 2021) and redundant sequences were removed using Purge Haplotigs (Roach et al., 2018). Hi-C data was used to anchor the contigs to the chromosomes. In summary, genuine interaction pairs were obtained by filtering the Hi-C reads using FASTP v0.20.0 (Supplementary Table S5; Chen et al., 2018) and selecting unique mapped read pairs using the HiCUP v0.8.0 (Wingett et al., 2015) pipeline. Next, a chromosome-level assembly was created using 3D-DNA v201013 (https://github.com/aidenlab/3d-dna), after the sequence had been aligned against the draft genome assembly using Juicer (Dudchenko et al., 2017). The “embryophyta_odb10” ortholog set was used to assess the quality of the genome using BUSCO v4.05 (Simão et al., 2015).
To ensure the integrity of the C. longepaniculata genome assembly, high-quality short-read sequences were mapped back to the genome using the BWA v0.7.17 (Li and Durbin, 2010). This process helps to verify the correct alignment and orientation of the assembled sequences. Following the mapping, duplicate reads were removed using Picard v2.23.6 (broadinstitute.github.io/picard) to prevent redundancy that could skew variant identification results. Subsequently, SNPs (Single Nucleotide Polymorphisms) and InDels (insertions and deletions) were identified using SAMtools v1.10 (Li et al., 2009), which provides insights into potential genomic variations and polymorphisms.
In addition to the above steps, HiFi reads were mapped back to the genome using Minimap2 (Li, 2018). This step further ensures that the high-fidelity, long-read sequencing data aligns accurately with the assembled genome, confirming the assembly’s overall quality and completeness.
Repetitive sequences in the de novo repeat library were predicted using RepeatMasker v4.0.7 (Tarailo-Graovac and Chen, 2009), and a de novo repeat library was created using RepeatModeler (Flynn et al., 2020) and LTRfinder v1.07 (Xu and Wang, 2007) with default settings. In parallel, homologous repeat prediction was carried out using the Repbase v21.12 database and RepeatMasker v4.0.7 and RepeatProteinMasker v4.0.7 (http://www.repeatmasker.org/cgi-bin/RepeatProteinMaskRequest) (Bao et al., 2015). To create nonredundant repeated sequences, the two sets of anticipated repetitions were concatenated. With Tandem Repeat Finder v4.09 (Benson, 1999), tandem repetitions were found.
Gene prediction in the repeat-masked genome was done using three different methods: homology searching, reference-guided transcriptome assembly, and ab initio prediction. According to Keilwagen et al. (2016), homologous peptides from related species (Cinnamomum kanehirae, Phoebe bournei, Litsea cubeba, Phoebe kanehirae, and Phoebe zhennan) were aligned with the assembly using GeMoMa v1.6.1 (Keilwagen et al., 2016). Gene structure information was then collected for homolog prediction. Filtered mRNA-seq data were used to align STAR v2.7.3a (Dobin et al., 2013) to the reference genome in order to perform RNA-seq-based gene prediction (Dobin et al., 2013). PASA v2.3.3 (Haas et al., 2008) was used to predict open reading frames (ORFs), and StringTie v1.3.4d (Pertea et al., 2015) was used to assemble the transcripts. A training set was also produced for the de novo prediction. The training data was then used for ab initio gene prediction using Augustus v3.3.1 (Stanke et al., 2008) with default settings. After creating an integrated gene collection with EvidenceModeler (EVM, v1.1.1; Haas et al., 2008), miscoded genes were further filtered and genes containing TEs were eliminated using the TransposonPSI program (http://transposonpsi.sourceforge.net/) (Urasaki et al., 2017). PASA was used to identify alternative splicing regions and untranslated regions (UTRs) based on RNA-seq assemblies. For each locus, we kept the longest transcripts, and areas outside of the ORFs were called UTRs. By examining five protein/function databases, the projected protein-coding genes’ domains, motifs, and gene function information were found. Predicted protein-coding genes were thoroughly annotated using InterProScan v5.36 (Zdobnov and Apweiler, 2001), which included predicted signal peptides, transmembrane topologies, functional classifications, GO keywords, protein motifs and domains, and protein family identification. KO words were found using KaaS (https://www.genome.jp/kegg/kaas/) searching the KEGG database. A 1e-5 E value limit was utilized for searches against the Swiss-Prot (Bairoch and Apweiler, 2000), NR (Marchler-Bauer et al., 2011), TrEMBL (Bairoch and Apweiler, 2000), and COG databases (Galperin et al., 2015) using BLASTP v.2.7.1 (Altschul et al., 1997). From these database searches, the top hits were combined to get the final results.
Database searches and model-based prediction were the two methods employed to find noncoding RNA sequences in the genome. With eukaryote parameters, tRNAscan-SE v2.0.12 (Lowe and Eddy, 1997) was used to predict tRNAs. INFERNAL v1.1.5 (Nawrocki and Eddy, 2013) was utilized to search the Rfam database and identify sequences of microRNA, rRNA, small nuclear RNA, and small nucleolar RNA. RNAmmer v1.2 (Lagesen et al., 2007) was used to forecast the rRNAs and their components.
In order to investigate the evolutionary background of C. longepaniculata, we obtained the genomes of eleven different species from publicly accessible databases. These species include Aquilegia coerulea, Arabidopsis thaliana, Amborella trichopoda, Liriodendron chinense, Nymphaea colorata, Piper nigrum, Cinnamomum kanehirae, Persea americana, Chimonanthus salicifolius, and Litsea cubeba (Supplementary Table S2). Furthermore, genes with glaring mistakes were eliminated and the longest transcripts were selected as samples for genes having alternative splice variants. The homology matrix of the orthogroups (gene families) among these chosen species was also inferred using OrthoFinder v2.5.4 (Emms and Kelly, 2019). For the purpose of creating phylogenetic trees using the maximum-likelihood (ML) approach, 230 single-copy gene groupings were found. The single-copy gene groups were subjected to multiple sequence alignment using MAFFT v.7.453 (Katoh and Standley, 2013). Subsequently, the coding sequences (CDS sequences) were aligned using PAL2NAL v.14 (Suyama et al., 2006) in accordance with the alignments of the respective proteins. Each single-copy gene group was given a maximum likelihood phylogenetic tree using IQ-TREE v.2.1.4-beta (Nguyen et al., 2015). Ultimately, these 230 single-copy gene trees were combined using the ASTRAL v.5.6.3 (Zhang et al., 2022) software to create a species tree based on the multispecies coalescent model. We employed a Bayesian relaxed molecular clock technique to estimate the divergence periods between species using the MCMCTree tool in the PAML v.4.9 (Yang, 2007) package. Furthermore, we acquired four fossil constraints for divergence time calibration from the TimeTree website (http://www.timetree.org): Between A. trichopoda and N. colorata, 179–205 Mya; between A. coerulea and P. nigrum, 151–170 Mya; between A. coerulea and A. thaliana, 126–133 Mya; and between A. thaliana and P. tremuloides, 102–113 Mya. Moreover, gene families in phylogenetic tree-constructing species that had experienced expansion or contraction were identified using CAFÉ v.4.2.1 (De Bie et al., 2006). Genes belonging to particular extended gene families were then functionally analyzed using GO and KEGG enrichment with TBtools (Chen et al., 2023).
To find members of the gene families implicated in the pathways leading to terpen biosynthesis, we conducted searches using BLASTP and Hidden Markov Models (HMMs). In particular, we gathered genes that are known to be connected to this A. thaliana pathway. Then, BLASTP (e < 1e-5) was performed to discover pathway genes in the genome of C. longepaniculata using these genes as query sequences. HMM data for each gene family’s conserved protein domain were downloaded in the interim using the Pfam website (https://pfam.xfam.org/). These HMM files were used in batch searches, along with HMMER v3.2.1 (Johnson et al., 2010). These HMM files served as the basis for batch searches. The BLASTP-identified candidate genes that lacked the matching domain were eliminated. We utilized the PF02458 Pfam domain for BAHD searching and the PF01397 and PF03936 Pfam domains for HMMER searching in order to identify the TPS genes. In order to differentiate between distinct subfamilies, phylogenetic trees were created for each gene family’s candidate genes using IQTREE v.2.1.4-beta (Nguyen et al., 2015).
We used transcriptome data from the leaf to do RNA-seq analysis to look into the expression levels of genes involved in the terpene synthesis pathway. We first performed a raw data quality assessment for transcriptome analysis using FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Subsequently, quality control was performed using FASTP v0.20.0 to filter out raw reads containing poly-N sequences and low-quality reads (< Q30). We used the high-quality reads obtained from the quality control step for the subsequent expression analysis. After filtering the high-quality RNA-seq reads, we used HISAT2 v.2.1.0 (Kim et al., 2015) to map them to the genome of C. longepaniculata. Finally, using StringTie v.1.3.4d (Pertea et al., 2015), we quantified gene expression levels in transcripts per kilobase million (TPM).
In order to create the genome of C. longepaniculata (Figure 1A), we initially generated 49.43Gb of paired-end reads using the Illumina NovaSeq 6000 (Supplementary Table S1). Our 19-mer analysis indicated an estimated genome size of 702 Mb, with a heterozygosity rate of 3.31% (Supplementary Table S3). Building on this, we utilized 31.54 Gb of high-quality high-fidelity (HiFi) reads produced on the PacBio platform, achieving 44.91-fold coverage of the C. longepaniculata genome (Supplementary Table S1). Utilizing the Hifiasm tool, this sequencing effort yielded thirty contigs, totalling 768.10 Mb with an N50 size of 41.12 Mb (Supplementary Table S4). To enhance the assembly, we incorporated Hi-C data, employing Hi-C-Pro v2.8.1 to recognize and retain 89.18 Gb of valid interaction paired reads (Supplementary Table S5). This integration allowed the assembly to be further consolidated. Ultimately, we anchored 766.69 Mb (99.82%) of the contig sequences into 12 chromosomes (Figures 1B, C; Supplementary Table S6). The final scaffold N50 was improved to 63.78 Mb, with the longest scaffold measuring 111.09 Mb (Table 1; Supplementary Table S6). Additionally, a heatmap of chromosome interactions demonstrated the completeness and robustness of the genome assembly (Figure 1C).
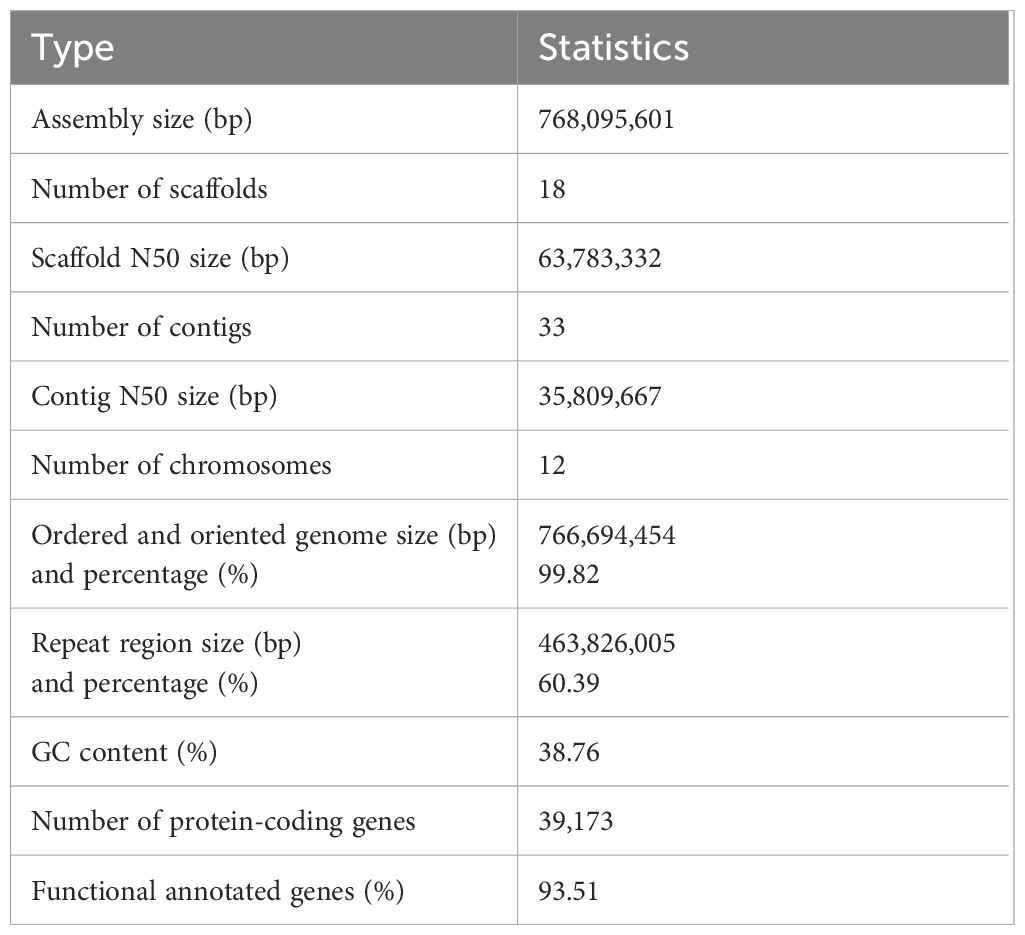
Table 1 Assembly and annotation features from the Camphora longepaniculata assembly.
A combination of homology-based, reference-guided transcriptome assembly, and ab initio gene approaches were used to predict gene models in the C. longepaniculata assembly. The consensus gene set was then generated by combining the gene prediction results with EVM software. To improve the quality of gene prediction, genes with transposable elements and miscoded genes were eliminated. In the end, 39,173 genes made up the final gene list that we acquired. These protein-coding genes had an average gene length of 7,152.40 bp and an average CDS length of 1,146.30 bp (Supplementary Tables S7, S8). There were 4.38 exons on average per gene, with an average exon length of 261.80 bp and intron length of 1608.33 bp (Supplementary Table S8).
Approximately sixty percent of the whole-genome assembly consisted of the detected repetitive sequences (463.83 Mb) (Supplementary Table S9). The most common repeat types were DNA elements (19.72%) and long terminal repeat (LTR) retrotransposons (31.96%) (Supplementary Table S9). LINE (3.19 Mb) made up 0.42% of the whole genome assembly, in contrast. With computed average lengths of 106.89, 74.99, 191.27, and 118.18 bp, respectively, 6,531 miRNAs, 582 transfer RNAs (tRNAs), 3,312 rRNAs, and 634 snRNAs were found (Supplementary Table S10).
With short reads an overall mapping rate of 99.20% was obtained, encompassing 99.92% of the assembly (Supplementary Table S11). Moreover, HiFi reads were mapped back using Minimap2 (Li, 2018), resulting in an assembly coverage of 100% and an overall mapping rate of 99.92% (Supplementary Table S11). These findings imply that the genetic information provided in our assembly was almost entirely complete. Moreover, SAMtools v1.4 (Li et al., 2009) was used to identify and filter single-nucleotide polymorphisms (SNPs), resulting in the identification of 11,919,258 heterozygous SNPs, 18,760 homozygous SNPs, and 9,182 homozygous INDELs with 5× sequencing depth. The high precision of the assembly (99.996385%) is supported by the low frequencies of homozygous SNPs and INDELs, which together make up 0.002442% and 0.001195% of the assembled genome, respectively. Ultimately, a scatter plot of the sequencing depth vs the GC-content based on 10-kb windows showed that the completed C. longepaniculata genome was free of foreign DNA contamination (Figure 1B, Supplementary Figure S1). Additionally, we used the embryophyta odb10 database (https://busco.ezlab.org/) to perform BUSCO analysis on the data (Simão et al., 2015). Out of 1,614 conserved plant genes, 4.52% of them contained duplicates, yet 99.01% of them had full coverage in the genome, 0.43% were fragmented, and only 0.56% were missing (Supplementary Table S12). These results clearly show that the genome assembly of C. longepaniculata we have produced is of excellent quality and has the potential to be useful for future research. The completeness of these anticipated genes was further evaluated using BUSCO analysis, yielding a BUSCO score of 93.31% (single = 89.34%, duplicated = 3.97%, fragmented = 4.96%, missing = 1.73%, genes = 1,614; Supplementary Table S13). Furthermore, functions for 65.80% (25,777), 35.86% (14,049), 71.29% (27,928), 36.48% (14,290), 92.78% (36,345), and 93.26% (36,533) of the genes were found by searching the Swiss-Prot, KEGG, COG, GO, TrEMBL, and NR databases, respectively (Supplementary Table S14). A total of 36,632 protein-coding genes, or 93.51% of them, had their conserved functional motifs or functional terms effectively annotated (Supplementary Table S14). These findings suggest that,the C. longepaniculata genome’s annotated gene set is rather comprehensive.
As described in the Materials and Methods section, 288 single-copy orthologous genes were discovered by analyzing the genomes of 12 different species. C. longepaniculata and C. camphora were found to have a closer relationship based on phylogenetic analyses of these genes. C. longepaniculata split from C. camphora and C. kanehirae between 6.01 million and 62.19 million years ago, according to projected divergence times (Figure 2A). Furthermore, the collinearity findings revealed a high level of collinearity among the three species (Figure 2B), and C. longepaniculata was discovered to have a WGD event shared by Lauraceae species. In addition, C. longepaniculata shared 10,384 gene families with four other species (C. camphora, C. kanehirae, C. salicifolius, and P. americana) and had 1,687 unique gene families (Figure 2C). We further investigated structural variations (SVs) among C. longepaniculata, C. camphra, and C. kanehhirae. We annotated a total of 695,731 SVs (including small insertions and deletions) between C. longepaniculata and C. camphra, and 801,107 SVs between C. longepaniculata and C. kanehhirae after whole-genome alignment using minimap2 and SV calling using SyRI (Supplementary Figure S3). After filtering, we focused on SVs longer than 10kb, resulting in the retention of 949 (between C. longepaniculata and C. camphora) and 701 (between C. longepaniculata and C. kanehirae) SVs, respectively. The analysis of functional enrichment of genes within 10kb of SV breakpoints showed significant enrichment in terms of terpene synthase activity (GO:0010333), terpenoid biosynthetic process (GO:0016114), monoterpene metabolic process (GO:0016098), and chloroplast RNA processing (GO:0031425), among others. This suggests that post-WGD diploidization may play a part in improving the terpenoid biosynthesis pathway (Supplementary Table S15).
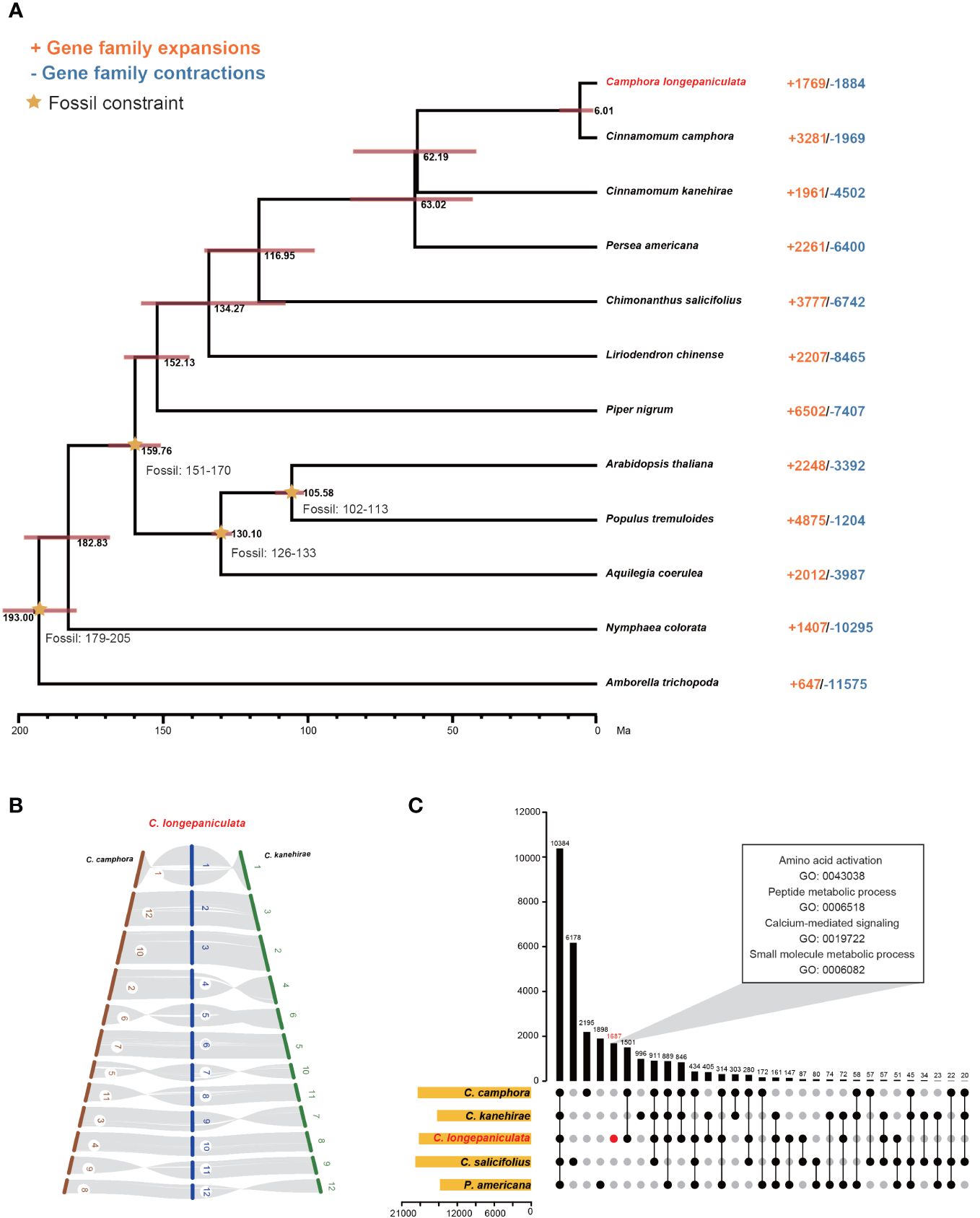
Figure 2 Comparative genomic, Co-linearity and Gene family sharing analysis, (A) Comparative genomic analysis of Camphora longepaniculata and other plant species. Annotated differentiation time, a 95% confidence interval for differentiation time, and the expansion or contraction of gene families in each species. (B) Co-linearity of Cinnamomum camphora, Cinnamomum kanehirae, and Camphora longepaniculata. (C) Gene family sharing in C. longepaniculata, C. camphora, C. kanehirae, C. salicifolius, and P. americana.
After a thorough evaluation of gene families in each of the 12 species, it was determined that 1,769 extended families and 1,884 contracted families are included in the genome of C. longepaniculata (Figure 2A). Statistical analysis revealed that 584 expanded and 497 contracted gene families were statistically significant (P < 0.05; Supplementary Tables S16, S17). These gene families were clarified by the Gene Ontology (GO) enrichment study. Significantly, gene families linked to the following were found to have expanded: “transferase activity (GO: 0016740)”, “catalytic activity (GO: 0003824)”, “response to oomycetes (GO: 0002239)”, “regulation of hydrogen peroxide metabolic process (GO: 0010310)”, and “secondary metabolic process (GO: 0019748)” (Supplementary Table S16). These gene families may help to explain the high levels of oil content and environmental resistance in C. longepaniculata. The gene families for “vesicle coating (GO: 0006901), Golgi vesicle transport (GO: 0048193), response to hydroperoxide (GO: 0033194), and channel activity (GO: 0015267)” on the other hand, showed shrinkage (Supplementary Table S17). Moreover, processes like “amino acid activation (GO: 0043038),” “peptide metabolic process (GO: 0006518),” “calcium-mediated signaling (GO: 0019722),” “organic acid metabolic process (GO: 0006082),” and “small molecule metabolic process (GO: 0044281)” were identified by GO enrichment analysis focused on C. longepaniculata specific gene families. These processes are all crucial to the process of oil biosynthesis (Figure 2C; Supplementary Table S18).
Due to the high concentration of terpenoids (volatile organic compounds) in the leaf essential oil of C. longepaniculata and their role as important sources of plant fragrance, we focused on the terpene biosynthesis pathway genes in the C. longepaniculata genome. Terpenoids (monoterpenes, sesquiterpenes and iridoids) are usually synthesized via the MVA and MEP pathways (Figure 3A). We identified a total of 37 relevant genes from these two pathways in the C. longepaniculata genome (Supplementary Table S19). The results showed that there was no significant difference in the copy number of these genes in the Lauraceae species overall. However, the MVD genes, which encodes diphosphomevalonate decarboxylase, showed significant expansion compared to other species, reaching up to 5 copies in C. longepaniculata (Supplementary Table S19). These genes are responsible for catalyzing the conversion of mevalonate-5-diphosphate to isopentenyl diphosphate (IPP), which is one of the important precursors for synthesizing various terpenoids. This reaction step links the early steps of terpene biosynthesis with the subsequent conversion steps. The expansion of the MVP gene family in the genome of C. longepaniculata may be of significance for its terpenoid biosynthesis.
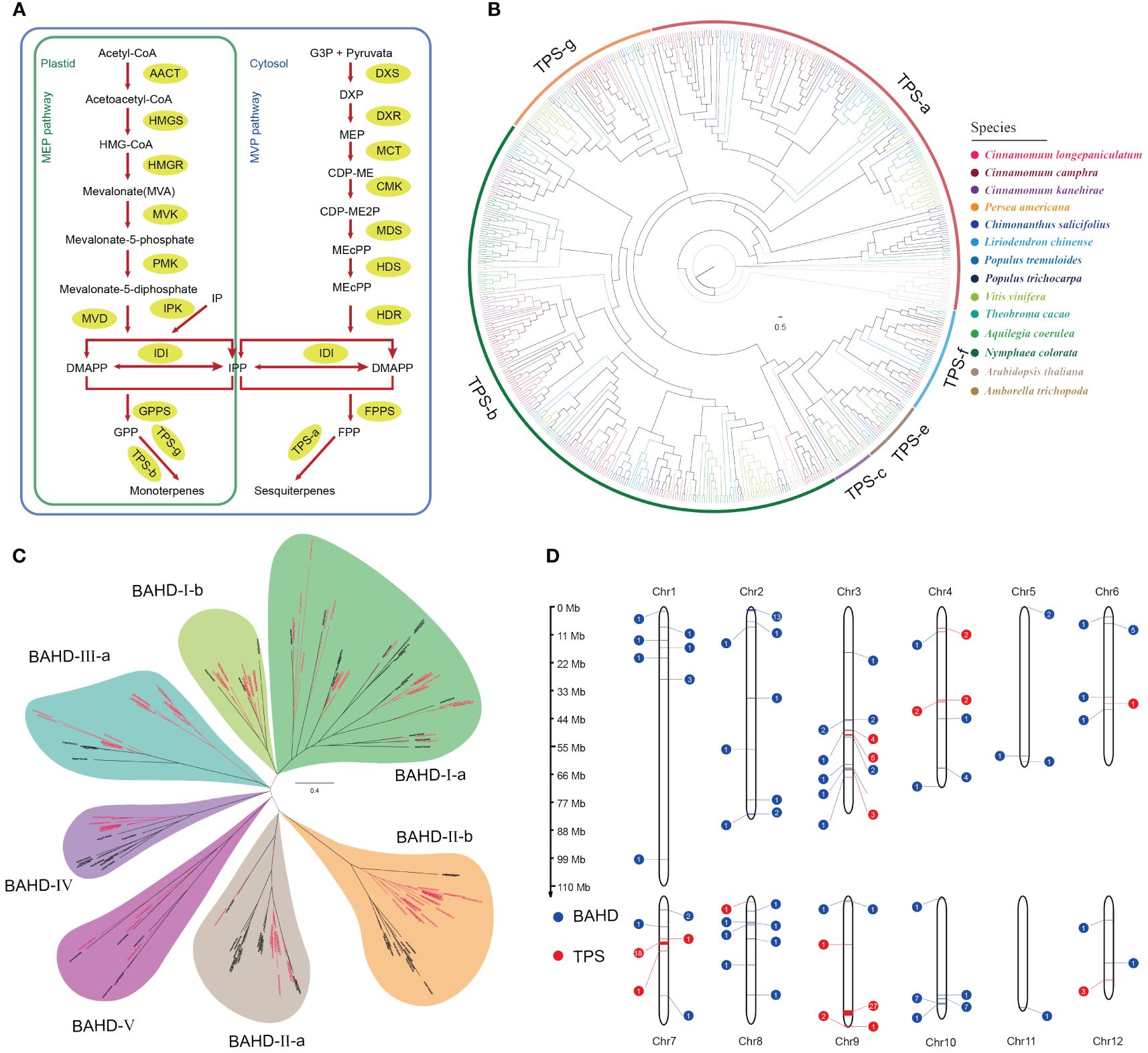
Figure 3 Genomic insights into terpenoid biosynthesis and distribution in Camphora longepaniculata. (A) MEP and MVP pathway for the biosynthesis of terpenoid. (B) Phylogenetic analysis of TPSs from Camphora longepaniculata and other plant species. (C) Phylogenetic analysis of BAHDs from Camphora longepaniculata and Arabidopsis thaliana. (D) Distribution of TPS and BAHD genes on chromosomes in Camphora longepaniculata.
On the other hand, 1,8-Cineole, α-terpineol, and γ-terpinene are important constituents of the leaf essential oil in C. longepaniculata, and these three compounds all belong to the class of monoterpenes. Subsequently, we focused on terpene synthase (TPS), which is crucial in the biosynthesis of terpenes. It catalyzes a variety reactions in the MVA and MEP pathways to produce the basic skeletons of terpenoids. We identified 74 genes encoding terpene synthase in C. longepaniculata (Supplementary Table S20). Notably, the TPS gene family in Lauraceae species significantly expanded compared to other selected species (C. longepaniculata: 74; C. camphora: 75; C. kanehirae: 88; Supplementary Table S20). More tree-based study of these TPS genes showed that the TPS-b and TPS-g subfamilies have grown in Lauraceae species (Figure 3B; Supplementary Table S20). Members of these two subfamilies mainly synthesize non-cyclic monoterpenes, sesquiterpenes, and diterpenes. This partially clarifies the significant contribution of the TPS-b and TPS-g gene families’ expansion to the accumulation of monoterpenes in C. longepaniculata’s leaf essential oil. BAHD acyltransferases, which participate in the synthesis of various flavors and fragrances in plants, generally transform terpenes into esters. In the genome of C. longepaniculata, we identified 98 genes that belong to the BAHD gene family. Phylogenetic analysis showed that the growth of different subfamilies of the BAHD gene family was significantly different between species (Figure 3C; Supplementary Table S21). We observed expansion trend in several subfamilies of Lauraceae species, including BAHD-I-b, BAHD-II-b, and BAHD-III-a. Specifically, for C. longepaniculata, the number of genes in the BAHD-II-b subfamily reached up to 19 (Supplementary Table S21). Also, the BAHD-II-b and BAHD-III-a subfamilies of C. longepaniculata formed a separate sub-branch compared to the reference sequence of A. thaliana. This suggests that the lineage specificity of the BAHD-II-b and BAHD-III-a genes may have different functions, which may help create the unique smell of C. longepaniculata. Observation of the chromosomal locations of TPS and BAHD gene families revealed that, similar to previous results in other plants (Xu et al., 2022), many TPS genes in C. longepaniculata exhibited tandem repeats. Specifically, these TPS genes formed gene clusters on chromosomes 3, 4, 7, and 9, showing a distinct clustered distribution pattern. On chromosomes 7 and 9, we found two large gene clusters, one with 18 genes and the other with 27 genes. This suggests that tandem duplication events recently created these genes (Figure 3D). At the same time, we observed several gene clusters on chromosomes 2 and 10 for the distribution of BAHD genes their overall distribution did not follow the same clustered pattern as TPS.
In comparison, our findings show that, like C. camphora and C. kanehirae, C. longepaniculata has similar gene positions and good collinearity on the same chromosomes (Supplementary Figure S3). Tandem repeats, such as MVD1 and MVD2 in C. longepaniculata, as well as MVD3, MVD4, and MVD5 (Supplementary Figure S3), account for the majority of the differences in gene copy numbers between these genomes. Furthermore, structural arrangements lead to gene duplications, as seen in the cases of FPPS2 and FPPS3, which most likely resulted from chromosome segmental duplication events. Notably, an inversion on chromosome 9 reconfigured the TPS gene cluster, resulting in a more densely packed arrangement of these genes, which was also found in the C. camphora genome (Supplementary Figure S3). This data further demonstrates that pathway genes are highly expressed, with at least one gene from each family (Supplementary Figure S4).
Camphora longepaniculata’s chromosome-level genome assembly represents a significant step forward in Lauraceae genomic investigations. The assembly has the longest contig or scaffold N50 values reported for this family—41.11 Mb and 63.78 Mb, respectively—and an impressive genome completeness of 99.01% as assessed by BUSCO results (Table 1; Supplementary Tables S3, S7; Chaw et al., 2019; Han et al., 2022; Li et al., 2022; Shen et al., 2022; Xiong et al., 2022).With its high-quality metrics, it establishes a new benchmark. Besides, our mapping Illumina short reads, SNP validation, and Poisson distribution analysis (Supplementary Figure S1, Supplementary Table S11) collectively affirmed the reliability, accuracy, and completeness of C. longepaniculata genome assembly. This validation is critical for demonstrating our genomic data’s dependability in future biological studies. We need this high-quality genomic data to learn more about the genetic structure and metabolic abilities of C. longepaniculata, especially how it makes terpenoids, which are important parts of the leaf essential oil that have big economic and environmental values (Li et al., 2012, 2014). Our findings greatly increase our understanding of the Camphora genus, which is commonly mistaken for Cinnamomum in the Lauraceae family. New phylogenetic research and molecular data show that Cinnamomum is polyphyletic. Camphora is now a separate genus, containing 18 species that form a monophyletic group, according to Rohwer et al. (2019) and Yang et al. (2022). This differentiation is critical because Camphora species, found across Asia’s tropical and subtropical climates, have specific evergreen features that differ significantly from those of Cinnamomum. It’s important to know the differences between Camphora and other Cinnamomum species by looking at their genetic traits. For example, Camphora has alternating, pinnately veined leaves and fruiting tepals that don’t stay in place, while some Cinnamomum species have opposite, triplivined leaves and persistent tepals. Our genomic study backs up these results by showing that C. longepaniculata and C. camphora share more gene families and fewer structural variants (SVs) than C. kanehirae (Figure 2; Supplementary Figure S3). This genetic similarity supports taxonomic distinction and indicates evolutionary divergences tailored to specific ecological niches. Furthermore, our findings are consistent with morphological observations published by Gang et al. (2021) and Abeysinghe et al. (2020), giving a solid foundation for understanding the evolutionary processes of these taxa. Our comparative genomic analyses, which include divergence times and gene family compositions, not only show that Camphora has a unique genetic lineage, but they also help us learn more about its evolutionary history and how it has changed over time (Figure 2). This genomic clarity enables a more in-depth examination of Camphora’s distinctive features, as well as their ecological and evolutionary consequences, laying the groundwork for future research on biodiversity conservation and the sustainable usage of these species. The genetic divergence between Camphora and Cinnamomum emphasizes the necessity of proper taxonomic classifications in advancing our understanding of plant biology and informing conservation initiatives. Furthermore, the genome data from C. longepaniculata sheds light on magnoliid evolution, providing a more complete evolutionary context within the angiosperms. This closes a major gap in our evolutionary knowledge.
The identification of a robust ensemble of genes participating in the MVA and MEP pathways, particularly the expansion of the MVD genes, demonstrates a genetic propensity for increased terpenoid synthesis (Figure 3; Supplementary Table S19). This enlargement, which is suggestive of evolutionary adaptation, increases the plant’s ability to synthesize terpenoids. These chemicals are important for the plant’s survival and ability to interact with its environment because they help protect it and attract pollinators (Nerg et al., 2004; Della Rocca et al., 2020). Surprisingly, the discovery of 74 TPS genes—a substantially higher number than observed in other species—demonstrates the plant’s particular capacity for terpene synthesis (Supplementary Table S20). TPS enzymes are known for accelerating the production of the basic carbon skeletons of terpenoids, hence their quantity and diversity indicate the plant’s ability to synthesize a wide range of terpenoid molecules. The growth of specific TPS subfamilies, particularly TPS-b and TPS-g, which are primarily involved in the synthesis of non-cyclic monoterpenes, sesquiterpenes, and diterpenes, may contribute to the high concentration of these chemicals in C. longepaniculata essential oils (Chen et al., 2011). This association not only confirms the functional relevance of these gene expansions, but also shows their potential contribution to the species’ specific aromatic qualities. Furthermore, the expansion of the BAHD acyltransferase gene family in C. longepaniculata, as evidenced by lineage-specific expansions in subfamilies BAHD-II-b and BAHD-III-a, indicates evolutionary adaptation for more complex terpenoid modifications into aroma compounds (Figure 3C; Supplementary Table S21; Srivastava and Sangwan, 2012). This increase is consistent with the family’s recognized role in esterification, which is essential for producing varied flavors and scents and so contributes to the species’ distinct aromatic properties (Souleyre et al., 2005; Cumplido-Laso et al., 2012). The chromosomal mapping of TPS and BAHD genes improves our understanding of the genomic architecture that underpins terpenoid biosynthesis. The presence of large gene clusters, notably TPS genes on chromosomes 7 and 9, suggests recent tandem duplication events, which may be promoting the diversity of terpene biosynthesis enzymes in C. longepaniculata (Figure 3D). This genomic architecture is consistent with observations from C. camphora, indicating a shared evolutionary approach within the genus (Shen et al., 2022). This pattern of gene duplication and clustering is prevalent in plant genomes and is frequently linked to the rapid evolution of gene families that respond to ecological and environmental stressors (Yang et al., 2021; Lei et al., 2024).
The identification of SVs associated with terpenoid synthesis genes, notably inside the greatest tandem repeat sections of TPS genes on chromosome 9 of the C. longepaniculata genome, provides deep insights into the plant’s evolutionary biology (Figure 3D; Supplementary Figure S3). These findings imply that SVs play a substantial role in the diversification and specialization of terpenoid biosynthesis pathways, emphasizing their importance as evolutionary tools that allow C. longepaniculata to tune chemical outputs to environmental stresses. The link between genetic rearrangements and metabolic pathway specialization improves our understanding of terpenoid production’s molecular dynamics and identifies prospective targets for biotechnological improvements targeted at improving terpenoid yield and variety. Our study highlights critical targets for genetic engineering to increase the production and diversity of terpenoids, which have significant commercial and ecological significance. Using full genomic knowledge could transform approaches for analyzing gene activity within organisms, hastening developments in agriculture and pharmaceuticals. C. longepaniculata’s unique biochemical capabilities provide opportunities to generate new products and improve existing ones by using its genetic resources. This genomic research sets the path for future studies that use C. longepaniculata’s inherent genetic variety to potentially increase terpenoid production, contributing to the fields of natural product chemistry and sustainable resource management. It emphasizes the significance of combining genomic data with ecological and evolutionary insights in order to fully realize plant species’ potential in a changing global context.
Our study presents a chromosome-level genome assembly of C. longepaniculata using HiFi sequencing, which was supplemented with short-read and Hi-C sequencing. C. longepaniculata genomic analysis has provided valuable insights into its genetic composition and functional characteristics. We identified key patterns of gene family sharing and divergence using comparative genomics with 11 other species, shedding light on the relationships between C. longepaniculata and closely related species like C. camphora. The estimated divergence times, collinearity results, and evidence of Whole Genome Duplication (WGD) provide a complete picture of the species’ evolutionary history. The fact that the TPS and BAHD gene families have grown so much in the genome shows how important secondary metabolite pathways have been for evolution as ways for Camphora species to adapt. This is in line with ecological and evolutionary theories that say secondary metabolites are very important for plants to adapt and survive. This genomic characterization of C. longepaniculata lays the groundwork for more biochemical and ecological studies that will look into how these genomic features affect function. This could pave the way for the biotechnological use of these natural products.
The data presented in the study are deposited in the NCBI repository, accession number PRJNA1068955. The genome assembly file, all the annotation files, and source data for phylogenetic and population analyses are available at Figshare (https://figshare.com/s/ff6a0f810527f61ef63c). All other data are available from the corresponding authors on reasonable request.
KY: Conceptualization, Formal analysis, Funding acquisition, Methodology, Project administration, Resources, Software, Writing – original draft, Writing – review & editing. HZ: Conceptualization, Data curation, Investigation, Methodology, Visualization, Writing – review & editing. GC: Data curation, Validation, Writing – review & editing. LM: Data curation, Formal analysis, Writing – review & editing. JL: Methodology, Validation, Writing – review & editing. JZ: Investigation, Software, Writing – review & editing. SL: Formal analysis, Validation, Writing – review & editing. YW: Data curation, Validation, Writing – review & editing. RF: Formal analysis, Investigation, Writing – review & editing. SS: Formal analysis, Software, Writing – review & editing. ME: Formal analysis, Software, Writing – review & editing. RH: Formal analysis, Software, Writing – review & editing. QW: Conceptualization, Methodology, Validation, Writing – original draft, Writing – review & editing. AE-S: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. DR: Conceptualization, Methodology, Validation, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was co-supported by the Yibin City Science and Technology Bureau Project (Grant No. 2022NY010), the Scientific Research Project of Yibin University (Grant No. 2022YY07) and the National Natural Science Foundation of China (Grant No. 32371900).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1372127/full#supplementary-material
Supplementary Figure 1 | Scatter plot illustrating the sequencing depth versus the GC-content based on 10-kilobase (kb) windows.
Supplementary Figure 2 | Intra-genomic comparison within Camphora longepaniculata.
Supplementary Figure 3 | SVs identified between Camphora longepaniculata and Cinnamomum camphora (top) and between Camphora longepaniculata and Cinnamomum kanehirae (bottom) across the 12 chromosomes.
Supplementary Figure 4 | Expression level of pathway genes.
Abeysinghe, P. D., Bandaranayake, P. C., Pathirana, R. (2020). Botany of endemic Cinnamomum species of Sri Lanka. Cinnamon: Botany Agronomy Chem. Ind. Appl., 85–118. doi: 10.1007/978-3-030-54426-3_4
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bairoch, A., Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi: 10.1093/nar/28.1.45
Bao, W., Kojima, K. K., Kohany, O. (2015). Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 1–6. doi: 10.1186/s13100-015-0041-9
Belton, J.-M., McCord, R. P., Gibcus, J. H., Naumova, N., Zhan, Y., Dekker, J. (2012). Hi–C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276. doi: 10.1016/j.ymeth.2012.05.001
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Boutanaev, A. M., Moses, T., Zi, J., Nelson, D. R., Mugford, S. T., Peters, R. J., et al. (2015). Investigation of terpene diversification across multiple sequenced plant genomes. Proc. Natl. Acad. Sci. U.S.A. 112, E81–E88. doi: 10.1073/pnas.1419547112
Chaw, S.-M., Liu, Y.-C., Wu, Y.-W., Wang, H.-Y., Lin, C.-Y. I., Wu, C.-S., et al. (2019). Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nat. Plants 5, 63–73. doi: 10.1038/s41477-018-0337-0
Chen, C., Wu, Y., Li, J., Wang, X., Zeng, Z., Xu, J., et al. (2023). TBtools-II: A “one for all, all for one“ bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742. doi: 10.1016/j.molp.2023.09.010
Chen, F., Tholl, D., Bohlmann, J., Pichersky, E. (2011). The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229. doi: 10.1111/j.1365-313X.2011.04520.x
Chen, S., Zhou, Y., Chen, Y., Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. doi: 10.1038/s41592-020-01056-5
Cumplido-Laso, G., Medina-Puche, L., Moyano, E., Hoffmann, T., Sinz, Q., Ring, L., et al. (2012). The fruit ripening-related gene FaAAT2 encodes an acyl transferase involved in strawberry aroma biogenesis. J. Exp. Bot. 63, 4275–4290. doi: 10.1093/jxb/ers120
De Bie, T., Cristianini, N., Demuth, J. P., Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Della Rocca, G., Danti, R., Hernando, C., Guijarro, M., Michelozzi, M., Carrillo, C., et al. (2020). Terpenoid accumulation links plant health and flammability in the cypress-bark canker pathosystem. Forests 11, 651. doi: 10.3390/f11060651
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Dudchenko, O., Batra, S. S., Omer, A. D., Nyquist, S. K., Hoeger, M., Durand, N. C., et al. (2017). De novo assembly of the Aedes aEgypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. doi: 10.1126/science.aal3327
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14. doi: 10.1186/s13059-019-1832-y
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457. doi: 10.1073/pnas.1921046117
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Gang, Z., Liu, B., Rohwer, J. G., Ferguson, D. K., Yang, Y. (2021). Leaf epidermal micromorphology defining the clades in Cinnamomum (Lauraceae). PhytoKeys 182, 125. doi: 10.3897/phytokeys.182.67289
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. doi: 10.1186/gb-2008-9-1-r7
Han, X., Zhang, J., Han, S., Chong, S., Meng, G., SOng, M., et al. (2022). The chromosome-scale genome of Phoebe bournei reveals contrasting fates of terpene synthase (TPS)-a and TPS-b subfamilies. Plant Commun. 3, 100410. doi: 10.1016/j.xplc.2022.100410
Hu, W., Gao, H., Jiang, X., Yang, H. (2012). Analysis on constituents and contents in leaf essential oil from three chemical types of Cinnamum camphora. J. Cent. South Univ. Forestry Technol. 32, 186–194.
Huang, J.-F., Li, L., van der Werff, H., Li, H.-W., Rohwer, J. G., Crayn, D. M., et al. (2016). Origins and evolution of cinnamon and camphor: A phylogenetic and historical biogeographical analysis of the Cinnamomum group (Lauraceae). Mol. Phylogenet. Evol. 96, 33–44. doi: 10.1016/j.ympev.2015.12.007
Johnson, L. S., Eddy, S. R., Portugaly, E. (2010). Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinf. 11, 431. doi: 10.1186/1471-2105-11-431
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Keilwagen, J., Wenk, M., Erickson, J. L., Schattat, M. H., Grau, J., Hartung, F. (2016). Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89. doi: 10.1093/nar/gkw092
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35 (9), 3100–3108. doi: 10.1093/nar/gkm160
Lei, W., Zhu, H., Cao, M., Zhang, F., Lai, Q., Lu, S., et al. (2024). From genomics to metabolomics: Deciphering sanguinarine biosynthesis in Dicranostigma leptopodum. Int. J. Biol. Macromol. 257, 128727. doi: 10.1016/j.ijbiomac.2023.128727
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34 (18), 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H., Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, F., Huang, S., Mei, Y., Wu, B., Hou, Z., Zhan, P., et al. (2022). Genome assembly provided new insights into the Cinnamomum burmannii evolution and D-borneol biosynthesis differences between chemotypes. Ind. Crops Prod. 186, 115181. doi: 10.1016/j.indcrop.2022.115181
Li, L., Li, Z.-W., Yin, Z.-Q., Wei, Q., Jia, R.-Y., Zhou, L.-J., et al. (2014). Antibacterial activity of leaf essential oil and its constituents from Cinnamomum longepaniculatum. Int. J. Clin. Exp. Med. 7, 1721.
Li, N., Zu, Y., Wang, W. (2012). Antibacterial and antioxidant of celery seed essential oil. Chin. Condiment 37, 28–30.
Liu, B., Shi, Y., Yuan, J., Hu, X., Zhang, H., Li, N., et al. (2013). Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv.org, arXiv: 1308.2012. vol. 22.
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Marchler-Bauer, A., Lu, S., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., DeWeese-Scott, C., et al. (2011). CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229. doi: 10.1093/nar/gkq1189
Nawrocki, E. P., Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Nerg, A. M., Heijari, J., Noldt, U., Viitanen, H., Vuorinen, M., Kainulainen, P., et al. (2004). Significance of wood terpenoids in the resistance of Scots pine provenances against the old house borer, Hylotrupes bajulus, and brown-rot fungus, Coniophora puteana. J. Chem. Ecol. 30, 125–141. doi: 10.1023/B:JOEC.0000013186.75496.68
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T.-C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Roach, M. J., Schmidt, S. A., Borneman, A. R. (2018). Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinf. 19, 460. doi: 10.1186/s12859-018-2485-7
Rohwer, J. G., Trofimov, D., Mayland-Quellhorst, E., Albach, D. (2019). Incongruence of morphological determinations and DNA barcode sequences: a case study in Cinnamomum (Lauraceae). Willdenowia 49, 383–400. doi: 10.3372/wi.49.49309
Shen, T., Qi, H., Luan, X., Xu, W., Yu, F., Zhong, Y., et al. (2022). The chromosome-level genome sequence of the camphor tree provides insights into Lauraceae evolution and terpene biosynthesis. Plant Biotechnol. J. 20, 244–246. doi: 10.1111/pbi.13749
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Song, K., He, M., Yu, J., Guan, Y., Bai, Y., Xin, S., et al. (2019). Characterization of the chloroplast genome of the family Lauraceae plant species, Cinnamomum cassia. Mitochondrial DNA Part B 4, 3906–3907. doi: 10.1080/23802359.2019.1687360
Souleyre, E. J., Greenwood, D. R., Friel, E. N., Karunairetnam, S., Newcomb, R. D. (2005). An alcohol acyl transferase from apple (cv. Royal Gala), MpAAT1, produces esters involved in apple fruit flavor. FEBS J. 272, 3132–3144. doi: 10.1111/j.1742-4658.2005.04732.x
Srivastava, S., Sangwan, R. S. (2012). Analysis of Artemisia annua transcriptome for BAHD alcohol acyltransferase genes: identification and diversity of expression in leaf, stem and root. J. Plant Biochem. Biotechnol. 21, S108–S118. doi: 10.1007/s13562-012-0141-2
Stanke, M., Diekhans, M., Baertsch, R., Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Suyama, M., Torrents, D., Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Tarailo-Graovac, M., Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4, 4.10.11–14.10.14. doi: 10.1002/0471250953.bi0410s25
Urasaki, N., Takagi, H., Natsume, S., Uemura, A., Taniai, N., Miyagi, N., et al. (2017). Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 24, 51–58. doi: 10.1093/dnares/dsw047
Wei, Q., Tan, Y., Li, Q., You, L., Wang, C., Wang, Y., et al. (2016). Effects of fungal endophytes on cell suspension culture of Cinnamomum longepaniculatum. Guangxi Zhiwu/Guihaia 36, 923–929.
Wingett, S., Ewels, P., Furlan-Magaril, M., Nagano, T., Schoenfelder, S., Fraser, P., et al. (2015). HiCUP: pipeline for mapping and processing Hi-C data. F1000Res. 4, 1310. doi: 10.12688/f1000research.7334.1
Wu, D., Zhu, P., Wu, H., Dai, H. (2022). Industry development status and prospect of Cinnamomum longepaniculatum. Open Access Library J. 9, 1–10. doi: 10.4236/oalib.1108616
Xiong, B., Zhang, L., Xie, L., Li, L., He, X., Niu, Y., et al. (2022). Genome of Lindera glauca provides insights into the evolution of biosynthesis genes for aromatic compounds. iScience 25 (8), 104761. doi: 10.1016/j.isci.2022.104761
Xu, S., Ding, Y., Sun, J., Zhang, Z., Wu, Z., Yang, T., et al. (2022). A high-quality genome assembly of Jasminum sambac provides insight into floral trait formation and Oleaceae genome evolution. Mol. Ecol. Resour. 22, 724–739. doi: 10.1111/1755-0998.13497
Xu, Z., Wang, H. (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. doi: 10.1093/nar/gkm286
Yan, K., Wei, Q., Feng, R., Zhou, W. (2019). Transcriptome analysis of the effects of endophytic fungi on the biosynthesis of essential oils in Cinnamomum longepaniculatum. Int. J. Agric. Biol. 21, 1301–1308. doi: 10.12688/f1000research.7334.1
Yan, K., Wei, Q., Feng, R., Zhou, W., Chen, F. (2017). Transcriptome analysis of Cinnamomum longepaniculatum by high-throughput sequencing. Electronic J. Biotechnol. 28, 58–66. doi: 10.1016/j.ejbt.2017.05.006
Yang, X., Gao, S., Guo, L., Wang, B., Jia, Y., Zhou, J., et al. (2021). Three chromosome-scale Papaver genomes reveal punctuated patchwork evolution of the morphinan and noscapine biosynthesis pathway. Nat. Commun. 12, 6030. doi: 10.1038/s41467-021-26330-8
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 (8), 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., Liu, B., Yang, Y., Ferguson, D. K. (2022). Phylogeny and taxonomy of cinnamomum (Lauraceae). Ecol. Evol. 12, e9378. doi: 10.1002/ece3.9378
Zdobnov, E. M., Apweiler, R. (2001). InterProScan—an integration platfor for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Keywords: Camphora longepaniculata, high-throughput sequencing, protein-coding genes, traditional Chinese medicine, terpenoid
Citation: Yan K, Zhu H, Cao G, Meng L, Li J, Zhang J, Liu S, Wang Y, Feng R, Soaud SA, Elhamid MAA, Heakel RMY, Wei Q, El-Sappah AH and Ru D (2024) Chromosome genome assembly of the Camphora longepaniculata (Gamble) with PacBio and Hi-C sequencing data. Front. Plant Sci. 15:1372127. doi: 10.3389/fpls.2024.1372127
Received: 17 January 2024; Accepted: 21 May 2024;
Published: 26 June 2024.
Edited by:
Sibum Sung, The University of Texas at Austin, United StatesReviewed by:
M. Kamran Azim, Mohammad Ali Jinnah University, PakistanCopyright © 2024 Yan, Zhu, Cao, Meng, Li, Zhang, Liu, Wang, Feng, Soaud, Elhamid, Heakel, Wei, El-Sappah and Ru. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qin Wei, d2VpcWluMjAwMS02N0AxNjMuY29t; Ahmed H. El-Sappah, YWhtZWRfZWxzYXBwYWgyMDA2QHlhaG9vLmNvbQ==; Dafu Ru, cnVkZkBsenUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.