 Shimei Yang1,2†
Shimei Yang1,2† Xirong Luo2†
Xirong Luo2† Jing Jin1
Jing Jin1 Ya Guo2
Ya Guo2 Lincheng Zhang1
Lincheng Zhang1 Jing Li2
Jing Li2 Shuoqiu Tong1
Shuoqiu Tong1 Yin Luo2Tangyan Li2
Yin Luo2Tangyan Li2 Xiaocui Chen3
Xiaocui Chen3 Yongjun Wu1*
Yongjun Wu1* Cheng Qin2,3*
Cheng Qin2,3*- 1Industrial Technology Institute of Pepper, Key Laboratory of Plant Resource Conservation and Germplasm Innovation in Mountainous Region (Ministry of Education), College of Life Sciences/Institute of Agro-bioengineering, Guizhou University, Guiyang, China
- 2Engineering Research Center of Zunyi Pepper Germplasm Resources Conservation and Breeding Cultivation of Guizhou Province, Department of Modern Agriculture, Zunyi Vocational and Technical College, Zunyi, China
- 3Key Lab of Zunyi Crop Gene Resource and Germplasm Innovation, Zunyi Academy of Agricultural Sciences, Zunyi, China
This study aimed to enhance the use of male sterility in pepper to select superior hybrid generations. Transcriptomic and proteomic analyses of fertile line 1933A and nucleic male sterility line 1933B of Capsicum annuum L. were performed to identify male sterility-related proteins and genes. The phylogenetic tree, physical and chemical characteristics, gene structure characteristics, collinearity and expression characteristics of candidate genes were analyzed. The study identified 2,357 differentially expressed genes, of which 1,145 and 229 were enriched in the Gene Ontology and Kyoto Encyclopedia of Genes and Genomes databases, respectively. A total of 7,628 quantifiable proteins were identified and 29 important proteins and genes were identified. It is worth noting that the existence of CaPRX genes has been found in both proteomics and transcriptomics, and 3 CaPRX genes have been identified through association analysis. A total of 66 CaPRX genes have been identified at the genome level, which are divided into 13 subfamilies, all containing typical CaPRX gene conformal domains. It is unevenly distributed across 12 chromosomes (including the virtual chromosome Chr00). Salt stress and co-expression analysis show that male sterility genes are expressed to varying degrees, and multiple transcription factors are co-expressed with CaPRXs, suggesting that they are involved in the induction of pepper salt stress. The study findings provide a theoretical foundation for genetic breeding by identifying genes, metabolic pathways, and molecular mechanisms involved in male sterility in pepper.
Introduction
Pepper (Capsicum annuum L.), an annual or perennial herb in the Solanaceae family, is native to the tropical and subtropical regions of Central and South America. It is cultivated globally and is a major vegetable species, especially in China (Wu et al., 2013) where Guizhou province has the largest pepper cultivation and processing area. Its products are distributed nationwide, rendering pepper cultivation a cornerstone industry in Guizhou (Liu et al., 2022). Consequently, pepper breeding has gained increased importance, and thus, it is imperative to develop new pepper germplasms, improve strain selection, and enhance breeding technology. With continuous research in pepper breeding, genes related to nuclear male sterility (NMS) and cytoplasmic male sterility (CMS) have been gradually discovered in recent years. Male sterility is abundant in flowering plants, offering a convenient pathway for the breeding of new food and economic crops. Presently, common crops such as corn, rice, and wheat rely exclusively on crossbreeding to provide growers with seed resources and maximize product quality and economic returns (Rajput and Kandalkar, 2018; Sadimantara et al., 2018; Lu et al., 2020). To develop high-quality germplasm resources and breeding methods for pepper varieties, efficient selection and breeding techniques are essential. Two primary methods for propagating hybrid pepper seeds include manual emasculation and manual pollination using male sterile lines (Lv et al., 2020). The latter is more efficient as it eliminates the need for manual emasculation, thereby enhancing purity and reducing hybrid seed costs. However, further research is required to enhance the use of male sterility in pepper to select elite hybrid generations.
The genetic control of male sterility can occur via CMS or NMS, also known as genetic male sterility (GMS) (Morales et al., 2022). CMS, a condition under which a plant is unable to produce functional pollen, is widespread among higher plants (Eckardt, 2006). However, NMS is caused by a defect in a nuclear gene, and it is usually inherited as a recessive trait (Budar and Pelletier, 2001). Meanwhile, NMS is important for understanding microspore development, and it could be used to facilitate the development of new strategies to control male sterility (Chen et al., 2019). In recent years, many key genes related to infertility have been precisely located and discovered. Wang et al. (2019) found that orf300a and orf314a were strong candidate genes for CMS in pepper via mitochondrial assembly of the sterile line 138A and maintainer line 138B. Another study documented more than 20 GMS mutants in pepper (Dong et al., 2022). Transcriptomic analysis of sterile (160S-male sterile (MS) and fertile (160S-MF) flowers of Brassica napus revealed that heat shock proteins, antioxidants, skeletal proteins, GTPase, and calmodulin might be involved in thermosensitive GMS (Tang et al., 2019). However, pollen sterilization is critical for male sterility. Jacobowitz et al. found that PRX9 and PRX40 affect the development of normal anthers of Arabidopsis thaliana. Using immunochemical and biochemical methods, the researchers demonstrated that PRX9 and PRX40 can crosslink extension proteins to promote the integrity of the tapete wall during anther development (Jacobowitz et al., 2019).
Peroxidase is an oxidoreductase that is widely distributed in nature and can facilitate the oxidation of various organic and inorganic substrates using hydrogen peroxide and related compounds (de Oliveira et al., 2021). In addition to animal peroxidases, hemoglobin peroxidases are categorized into three classes based on their sequence and catalytic properties: Class I, II, and III peroxidases (Yang et al., 2020). Peroxidases are further classified into three major types based on their protein structures and catalytic properties: Class I (ascorbate peroxidase), Class II (lignin peroxidase), and Class III (secretory peroxidase) (Bhatt and Tripathi, 2011). Class III peroxidases (PRXs, EC 1.11.1.7) are plant-specific enzymes that have been extensively studied in higher plants, particularly in terms of growth, development, and responses to various stresses such as salt, drought, and metal toxicity (Almagro et al., 2008; Chen et al., 2022). PRXs are widely distributed in the plant kingdom and have been reported in various plant groups, including Chlorophyta, Euglenophyta, Rhodophyta, Byophyta, Pteridophyta, and all the Spermatophyta studied to date (Passardi et al., 2007). Notably, 73 PRXs have been identified in Arabidopsis thaliana (Tognolli et al., 2002), 138 in Oryza sativa (Passardi et al., 2004), and 374, 159, and 169 in wheat (Triticum aestivum, Triticum urartu, and Aegilops tauschii, respectively) (Yan et al., 2019). Additionally, 102 have been identified in Solanum tuberosum and 210 in Nicotiana tabacum (Yang et al., 2020; Cheng et al., 2022). Increasingly, PRX genes were identified in multiple species, indicating their involvement in plant growth and development and their important roles in stress response.
Type III plant peroxidases (EC 1.11.1.7) are well-recognized for their crucial roles in various metabolic reactions during plant defense processes. These peroxidases are involved in a wide range of physiological processes, including lignification, suberization, auxin metabolism, crosslinking of cell wall proteins, defense against pathogen attacks, salt tolerance, and response to oxidative stress (Hiraga et al., 2001; Almagro et al., 2008; Pandey et al., 2017). Research has shown that OsPRX38 overexpression in transgenic Arabidopsis can enhance arsenic tolerance and increase lignification and cell wall–associated peroxidase activity in a positive correlation (Kidwai et al., 2019). In addition, it can enhance cold tolerance in overexpressing plants (Kim et al., 2012) and play a role in regulating plant hypocotyl elongation and auxin levels (Cosio et al., 2009). Furthermore, the Arabidopsis CIII PRX gene AtPRX71 is strongly expressed during the loss of cell wall integrity, affecting growth and cell size and positively regulating reactive oxygen species levels (Raggi et al., 2015).
Herein, a combination of proteomics and transcriptomics analysis was used to identify key genes in MS pepper lines by comparing the differential expression of proteins and genes. In addition, the study examined the differentially expressed gene, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment, differential metabolic pathways, and protein interactions in the sterile and fertile pepper lines. Furthermore, a systematic identification analysis of the key gene CaPRX was conducted to assess its structural characteristics, physicochemical properties, and collinearity. Finally, this study explored the network diagram and expression pattern of the CaPRX gene under salt stress conditions. By integrating transcriptome and proteome analysis of nucleic MS line and FL of pepper, candidate genes of male sterile line of capsicum were preliminarily identified. Thus, the study findings provide valuable insights into the selection and breeding of new pepper varieties and paves the way for further research in pepper breeding.
Materials and methods
Plant materials and processing
The fertile line (FL) (1933A) and nucleic male sterile line (NMSL) (1933B) of pepper were cultivated at the pepper-planting site of Zunyi Vocational and Technical College (Zunyi, Guizhou, 107°045’ E, 27°710’ N). Plants uniform in stage and height were selected. Flower buds (When the corolla is close to the dehiscent bud stage, peduncle 2–3 cm, corolla 2–3 cm. Buds, peduncle length 1–2 cm, transverse stem 0.5–0.8 cm, vertical stem 1–1.5 cm) from the FL 1933A and NMSL 1933B were collected in triplicate. Immediately after collection, the samples were stored in liquid nitrogen at −80°C for preservation.
Transcriptome sequencing
The complete genome of pepper (C. annuum L., Zunla-1) was obtained from NCBI (GenBank accession: GCA_000710875.1) (Qin et al., 2014). The pepper transcriptome data were filtered using Trimmomatic-0.39 software (Bolger et al., 2014). The genome index was constructed using Hisat2-2.2.1 software, which facilitated the alignment of the transcriptome to the reference genes in pepper (Kim et al., 2019). FeatureCounts v2.0.3 software was used to compute the expected number of fragments per kilobase of transcript sequence per million base pairs sequenced for each gene in each sample (Liao et al., 2014). Lastly, DESeq2 software was used to analyze the differentially expressed genes between samples, with a threshold at padj of <0.05 and |log2 FC| of >1 (Love et al., 2014).
Protein analysis
The FL 1933A and NMSL 1933B were lysed using SDT (4% [w/v] sodium dodecyl sulfate, 100 mM Tris/HCl, pH 7.6, and 0.1 M DTT) for total protein extraction. Protein quantification was performed using the BCA assay. A specific amount of protein from each sample was collected and subjected to trypsinization via the filter-aided proteome preparation method. The peptides were desalted using a C18 cartridge, lyophilized, resolubilized in 40 μL 0.1% formic acid solution, and quantified (OD280). The peptides (100 μg) from each sample were collected and labeled using a TMT labeling kit (Thermo Fisher Scientific, Waltham, MA, USA) as per the manufacturer’s instructions. The samples were divided into two groups, each containing three biological replicates, totaling six samples.
Each sample was separated using an EASY nLC nanoflow high-performance liquid chromatography liquid phase system. Buffer A comprised a 0.1% formic acid aqueous solution and buffer B comprised a 0.1% formic acid/acetonitrile aqueous solution (84% acetonitrile). The chromatographic column was equilibrated using 95% buffer A. The samples were loaded onto the loading column (Acclaim PepMap100, 100 μm × 2 cm, nanofiber C18, Thermo Fisher Scientific) via an autosampler and passed through an analytical column (EASY column, 10 cm, ID 75 μm, 3 μm, C18-A2, Thermo Fisher Scientific) at a flow rate of 300 nL/min.
The samples were then separated using chromatography and analyzed via mass spectrometry using a Q-exactive mass spectrometer. The detection mode was positive ion, parent ion scan range was 300–1800 m/z, resolution of the primary mass spectrum was 70,000 at 200 m/z, automatic gain control target was 1e6, maximum IT was 50 ms, and dynamic exclusion time was 60.0 s. The mass-to-charge ratios of the peptides and peptide fragments were collected as follows: following each full scan, 20 fragmentation profiles (MS2 scan) were collected; MS2 activation type was higher-energy collision dissociation; isolation window was 2 m/z; secondary mass spectral resolution was 17,500 at 200 m/z; normalized collision energy was 30 eV; and underfill was 0.1%.
Transcriptome and proteome bioinformatics analyses
Protein clustering analysis was conducted using the Complexheatmap R package, which classified samples and protein expression into two dimensions (distance algorithm: Euclid, linkage: average linkage), yielding a hierarchical clustering heat map (Gu and Hübschmann, 2022). The subcellular localization of the proteins was predicted using CELLO (http://cello.life.nctu.edu.tw/) (Yu et al., 2004). The structural domains of the proteins were analyzed using Pfam data and compared with the InterProScan software package (Jones et al., 2014; El-Gebali et al., 2019). The target protein set was annotated for GO using Blast2GO and for KEGG using KAAS software (Conesa et al., 2005; Moriya et al., 2007). Enrichment analysis for GO and KEGG was performed using ClusterProfiler (Wu et al., 2021). The interaction relationships, direct and indirect, between the target proteins were identified using either the STRING (http://string-db.org/) databases. These interaction networks were generated and analyzed using Cytoscape v3.2.1 (Otasek et al., 2019).
Family identification and analysis
The whole-genome data (v2.0) of C. annuum L. (Zunla-1 and Chiltepin) were sourced from NCBI (GenBank: GCA_000950795.1) (Qin et al., 2014). The AtPRXs gene of Arabidopsis was downloaded from the Arabidopsis website (http://www.arabidopsis.org/index.jsp) with the sequence referenced from a study by Yang et al. (Yang et al., 2022). The PRXs model file (PF00141) was obtained from the Pfam database (http://pfam.xfam.org/) and used for model searching via HMMer v3.3.2 (El-Gebali et al., 2019). A Blastp comparison was performed on 73 Arabidopsis sequences with an E-value of e-20 using MEGA X, (https://megasoftware.net/), and an neighbor-joining phylogenetic tree was constructed using a bootstrap value of 1000 and default parameters (Kumar et al., 2018). The identified CaPRX gene family was visualized using iTol (Letunic and Bork, 2021). The physicochemical properties of the pepper CaPRXs protein were analyzed using Expasy-ProtParam (https://web.expasy.org/protparam/). The conserved domains of the proteins were analyzed using NCBI CDD with default parameters (https://www.ncbi.nlm.nih.gov/). Protein motifs were analyzed using the online tool meme (https://meme-suite.org/meme/tools/meme), with 10 motifs and default parameters (Bailey et al., 2015). The gene structure, conserved domain, and motif visualization of PRXs genes were performed using TBtools v1.106 (Chen et al., 2020). The MEGA X alignment output results were trimmed using TrimAL v1.4.rev22 (http://trimal.cgenomics.org) with default parameters, and a visual representation was created using WebLogo 3 (https://weblogo.threeplusone.com/) (Crooks et al., 2004; Capella-Gutiérrez et al., 2009). A collinearity analysis of pepper (Zunla-1 and Chiltepin) was performed using MCScanX and visualized via TBtools (Wang et al., 2013). The pepper plant materials, which included 40-day-old seedlings, were treated with NaCl (200 mM), and the treatment was subsequently applied to the roots and leaves. The gene expression network data and stress treatment were referenced from the study by Liu et al., and the visualization of gene expression data was performed using TBtools (Liu et al., 2017).
Pepper cDNA synthesis and quantitative real-time polymerase chain reaction analysis
The transcriptome samples designated for RNA sequencing underwent cDNA preparation. The quality of the RNA samples was confirmed through agarose gel electrophoresis and OD260/OD280 evaluation. The qualified samples were subsequently used for fluorescence quantitative experimentation to corroborate the transcriptome results. Initially, cDNA synthesis was performed using pepper RNA with K1622 from Thermo Fisher. A mixture of 2 μL pepper RNA, 1 μL Oligo(dT)18, and 9 μL ddH2O was prepared via low-speed centrifugation at 65°C for 5 min, followed by cooling on ice, another round of low-speed centrifugation, and additional cooling. In the second step, 10 μL of the product from the first step was uniformly mixed with 4 μL 5 × Reaction Buffer, 1 μL RiboLock™ RNA inhibitor, 2 μL 10 mM dNTP Mix, and 1 μL RevertAid™ M-MuLV reverse transcriptase via low-speed centrifugation. The mixture was subsequently incubated at 42°C for 60 min and then at 70°C for 5 min. Sample extraction and detection were conducted using the pepper housekeeping genes Ubiquitin (GenBank: AY486137.1) and Actin (GenBank: AY572427.1).
Primer3plus (http://www.primer3plus.com/cgi-bin/dev/primer3plus.cgi) online software was used to design primers for validating genes in the pepper transcriptome (Supplementary Table S1). quantitative real-time polymerase chain reaction (qRT-PCR)s were performed using the SYBR Premix ExTaq protocol (TaKaRa, Japan) via an Applied Biosystems 7500 real-time PCR system (Applied Biosystems, Foster City, CA). RNA samples from various types of pepper materials were selected with three biological replicates, and triplicate experiments were conducted using independent plant materials. PCR was conducted in the following cycles: 95°C for 30 s, 95°C for 15 s, 60°C for 30 s, and finally at 72°C for 1 min for 40 cycles. The qRT-PCR results were analyzed using the 2–△△Ct method (Livak and Schmittgen, 2001). The correlation between the transcriptome and qRT-PCR results was calculated using Pearson’s correlation coefficient. The fluorescence quantitative results were visualized using GraphPad Prism v6.
Results
Differentially expressed genes between samples
Six cDNA libraries were constructed for transcriptome sequencing and analysis. In total, 615,793,034 reads were sequenced, yielding 83.3 Gb clean data after quality control filtering, with the Q20 and Q30 values exceeding 97% and 92%, respectively (Table 1). The clean data were aligned with the pepper genome to identify the differentially expressed genes. This process detected 3,304 differentially expressed genes, including 947 new transcripts. Among these genes, 2,527 (including 757 new transcripts) were upregulated in the 1933A line and 777 genes (including 190 new transcripts) were upregulated in the 1933B line.
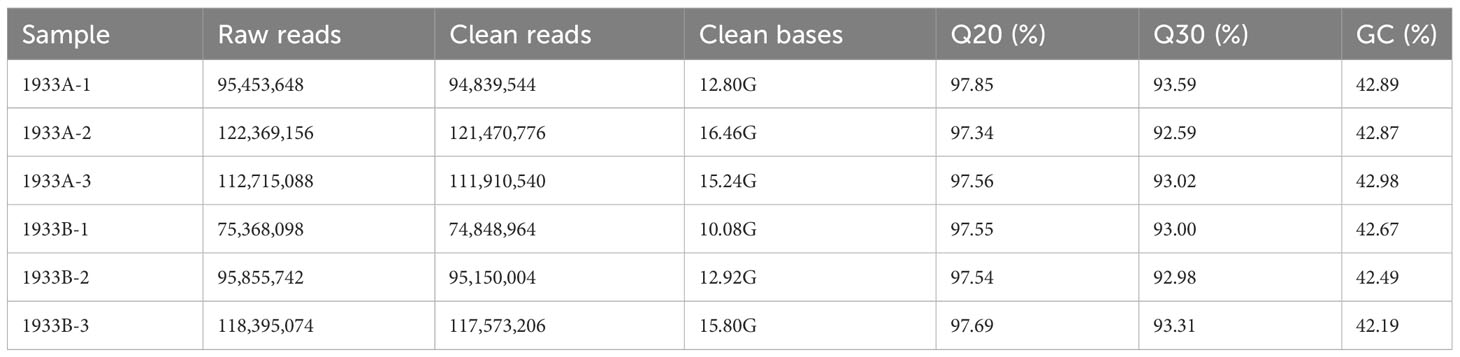
Table 1 Transcriptome sequencing information of pepper.
Enrichment analysis of differentially expressed genes
The transcriptome data of FL 1933A and NMSL 1933B of Capsicum were analyzed, and 3304 differential genes were identified. To study the biological and metabolic processes of these genes, GO and KEGG enrichment analyses were performed. In total, 1145 and 229 enriched genes were found in the GO and KEGG databases, respectively. Concerning biological processes (Figure 1A), 29.61% of the genes were related to the establishment of localization, 29.19% were related to transport, and 6.42% were related to metabolic processes. Meanwhile, 2.07% of the genes were related to the process of pollen development. Regarding molecular functions, 33.71% of the genes were related to transporter activity, and 28.84% were related to transmembrane transporter activity. Concerning cell composition, 60.28% of the genes were associated with the membrane, 15.21% were related to the cell periphery, 7.32% were associated with extracellular regions, 0.99% were associated with lysosomes, and 0.42% were related to pollen tubes.
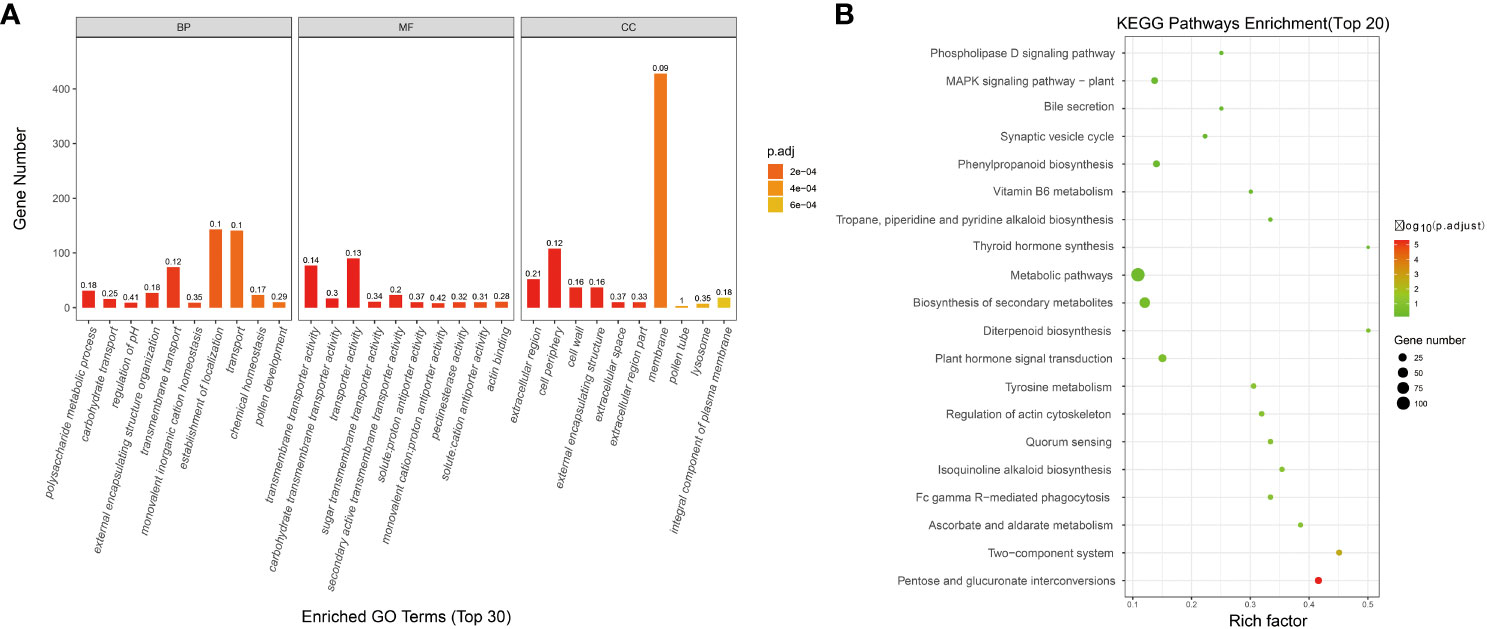
Figure 1 Enrichment analysis of differentially expressed genes in transcriptome, (A GO analysis; B KEGG analysis).
The developmental process of functional pollen involves many genes and pathways. In the KEGG enrichment analysis, 157 KEGG pathways were enriched. The top five pathways were metabolic pathways, biosynthesis of secondary metabolites, plant hormone signal transduction, pentose and glucuronate interconversions, and phenylpropanoid biosynthesis. Differential gene enrichment in several metabolic processes of the NMSL and FL of pepper provided a theoretical basis for the study of pepper NMSLs (Figure 1B).
Transcriptome, proteome association analysis, and candidate protein determination
Proteomic analysis of 1933A and 1933B uncovered 504 differentially expressed genes. Regarding the protein products of these genes, 351 proteins were upregulated in 1933A and 153 were upregulated in 1933B. Domain annotation, GO annotation, and KEGG annotation of these genes were performed, and the genes were annotated to 421, 364, and 133 pieces, respectively. Meanwhile, 130 proteins had protein–protein interactions, and 51 proteins were found in their intersections (Figure 2A). The analysis of significant differentially expressed proteins, GO and KEGG enrichment, and protein interactions in the FL and NMSL identified six candidate genes (Figures 2A, B) as follows: Capana02g002746, Capana04g000638 (CaPRXs, PF00141), Capana02g001798, Capana07g001786 (CP, PF08246; PF00112), Capana08g001522 (HSP70, PF00012), and Capana01g003855 (LOX, PF00305). Of these genes, Capana02g002764 and Capana02g001786 were upregulated in the NMSL, whereas the other four were downregulated. It is speculated that PRXs, HSP70, and CaLOX are important proteins involved in the NMSL process of pepper.
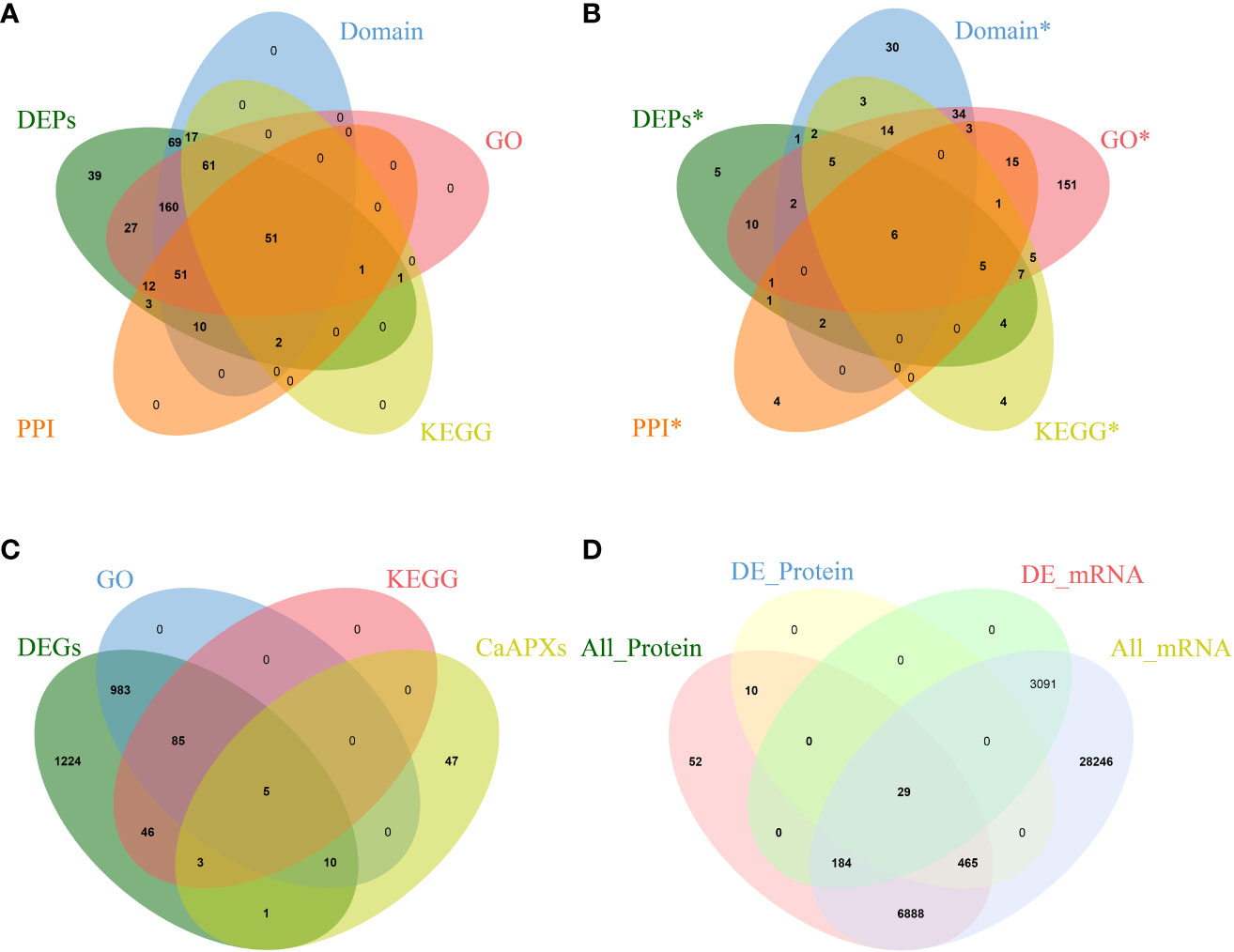
Figure 2 Venn diagrams of transcriptomics, proteomics, and correlation analyses, (A Venn diagram of proteomics; B Protein Venn diagram with significant difference; C Venn diagram of transcriptome and CaPRX genes family; D Combined analysis of Venn diagram).
In total, 2357 differentially expressed genes were identified via differential gene expression analysis, of which 1083 and 139 were significantly enriched in GO and KEGG pathways, respectively. Intersection analysis with PRX genes revealed that 19 genes overlapped with CaPRX genes (Figure 2C). Using integrated transcriptomics and proteomics, we conducted a comprehensive analysis of proteins and genes correlations in terms of quantification and differentiation at significant levels. This analysis identified 7,628 quantifiable proteins in the FL and NMSL, with 504 proteins exhibiting statistical significance. Additionally, 38,903 genes were identified, 3,304 of which exhibited significant differences. The most substantial correlation was observed among 29 proteins and genes, and within this correlation, three specific genes, Capana02g002746, Capana02g002990, and Capana04g000638, intersected with CaPRXs genes (Figure 2D).
Of the 29 significantly associated genes, three were identified as CaPRX genes (Figure 3A). Of these genes, seven proteins and genes were upregulated in the pepper NMSL and the other 22 genes were upregulated in the FL (Figure 3B). GO and KEGG enrichment analyses of these 29 genes were performed (Figures 3C, D). The results illustrated that 14 proteins were enriched in 149 biological processes, 17 proteins were enriched in 83 molecular functions, and 12 proteins were enriched in cell composition. The top three enriched biological processes were biological process, metabolic process, and organic substance metabolic process. The top three molecular functions were molecular function, catalytic activity, and hydrolase activity. The top three enriched cell components were cellular component, cell, and cell part. Meanwhile, proteomic differences in protein concentration, pollen development, pollen wall assembly, hydrogen peroxide metabolic process, and gametophyte development were also enriched. These results indicate that in GO enrichment, more genes were involved in the response to pollen development in pepper.
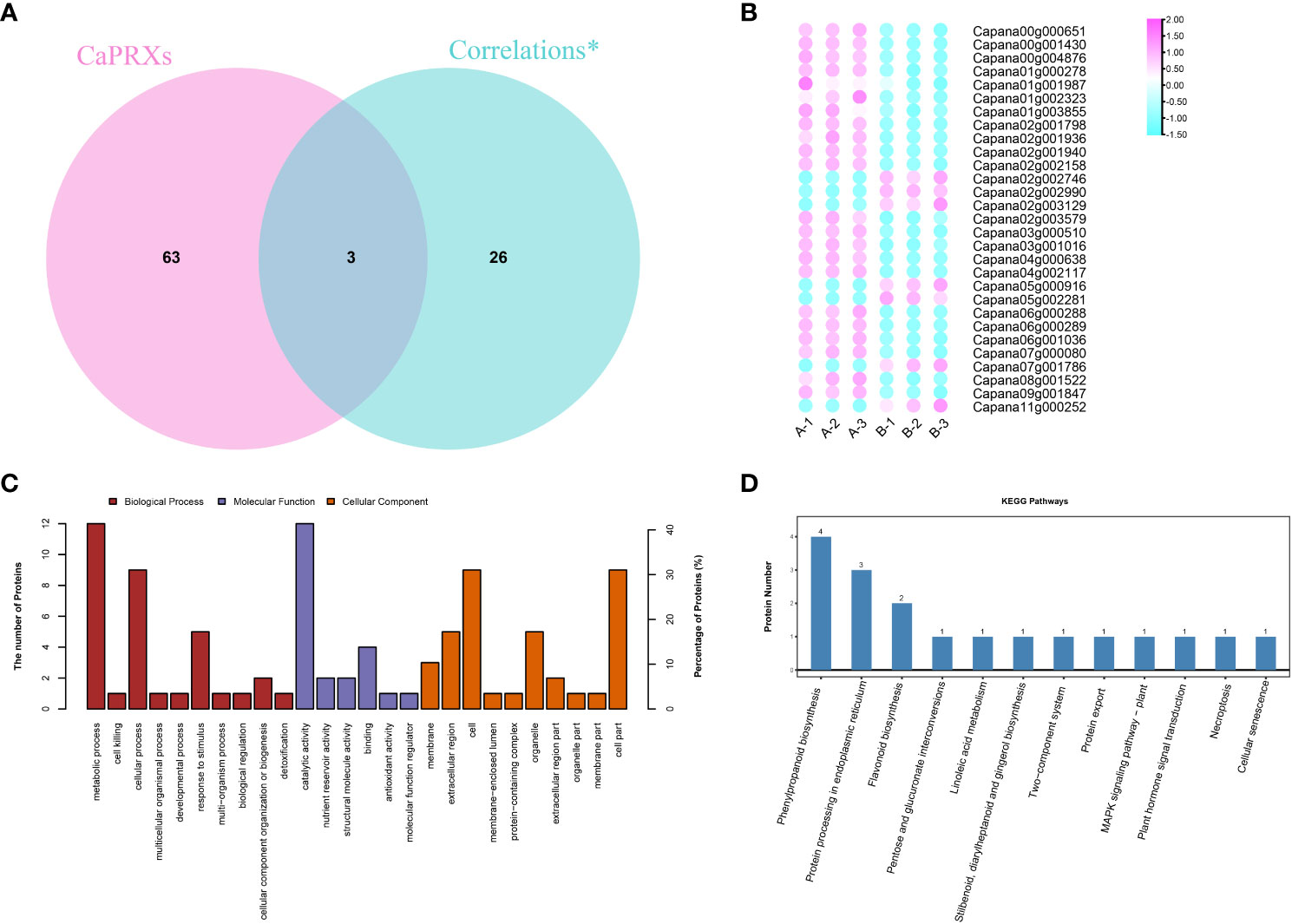
Figure 3 Identification and functional characterization of associated genes (A CaPRX genes family and combined analysis gene Venn diagram; B Combined analysis of gene expression; C GO comment; D KEGG comment).
Two of the six key proteins identified using proteomics were PRX protein (capan02g002746 and capan04g000638). Five of the key genes identified using transcriptomics were PRX genes (capan01g004380, Capana03g000271, Capana03g003406, Capana04g002827, and Capana11g001222). Transcriptomic and proteomic association analysis identified three significant associated genes, which are presumed to be the key candidate genes involved in the regulation of male sterility in pepper (Capan02g002746, Capan02g002990, and Capan04g000638).
CaPRXs identification and phylogenetic analysis
Through searching the pepper whole genome using Pfam model files, we successfully retrieved 107 sequences. Subsequently, we conducted a blast comparison by aligning these sequences with Arabidopsis AtPRX sequences, thereby discovering 66 sequences at their intersection. These 66 sequences were identified as candidate genes in subsequent analyses and were named CaPRXs based on their chromosome location and gene abbreviation. To investigate the evolutionary aspects of the pepper CaPRX gene family, we constructed a comprehensive phylogenetic tree by merging Arabidopsis AtPRX sequences with pepper CaPRX sequences, which revealed that pepper can be categorized into 13 subfamilies. Notably, the largest subfamily I contained 25 CaPRX genes, whereas the smallest subfamilies IX and XI each comprised only one CaPRX sequence (Figure 4). In contrast to the results of Yang et al.’s study on ClPRX identification in watermelon, which reported a total of 79 gene sequences categorized into 7 subfamilies and 11 subclasses, our study, focusing on pepper, identified a total of 66 sequences distributed across 13 subclasses (Yang et al., 2022).
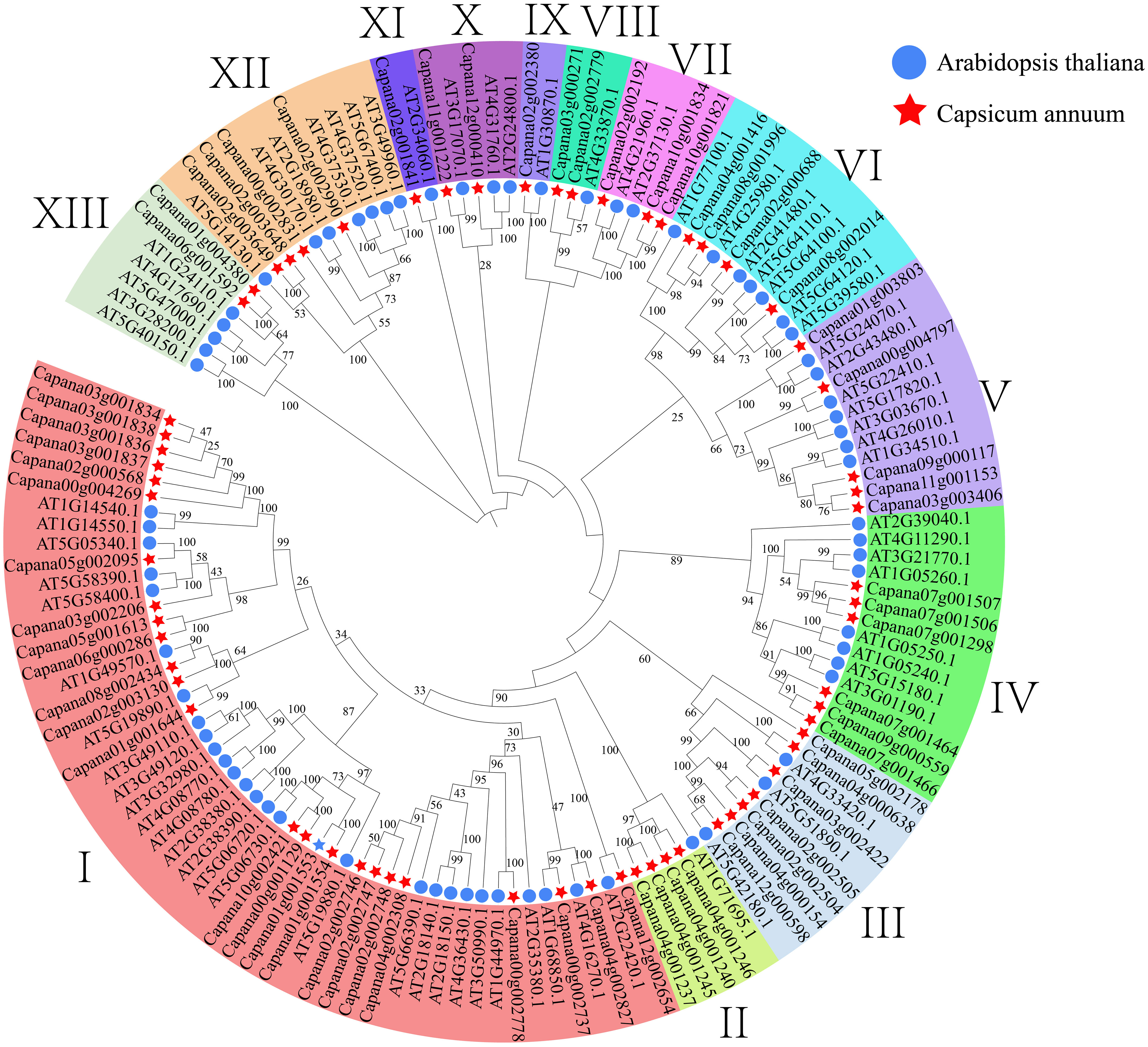
Figure 4 Phylogenetic tree of CaPRX genes in pepper (pentagons: pepper; circles: Arabidopsis).
Analysis of CaPRX gene family characteristics
An analysis of the physicochemical properties of the 66 identified CaPRX proteins yielded several interesting findings (Supplementary Table S2). The amino acid number within these genes varied from 222 to 493 and their molecular weights ranged from 24.84 to 53.63 kDa. The isoelectric point covered a spectrum from 4.45 to 9.85, indicating that CaPRX proteins exhibited amphipathic characteristics. The instability coefficient spanned from 21.73 to 53.08, suggesting that CaPRX proteins fell within a range of stability, with 21 proteins exhibitng an instability index of >40, thereby signifying their predicted instability. Conversely, the remaining 45 proteins demonstrated an instability index of <40, indicating their predicted stability. The hydrophilic–hydrophobic balance extended from −0.453 to 0.084, with only CaPRX19, CaPRX55, CaPRX45, CaPRX28, and CaPRX48 being classified as hydrophobic proteins, while the remaining CaPRXs exhibited hydrophilic properties.
Additionally, our investigation unveiled several motifs in the CaPRX proteins in peppers, with the majority containing 10 motifs. Notably, Motifs 3 and 9 were present in all the sequences. To further analyze the conserved domains, we observed that all the CaPRX proteins featured the highly conserved domain PRXs (PF00141). Gene structures were also visualized, revealing a range of exon numbers from 1 to 8 (Figure 5).
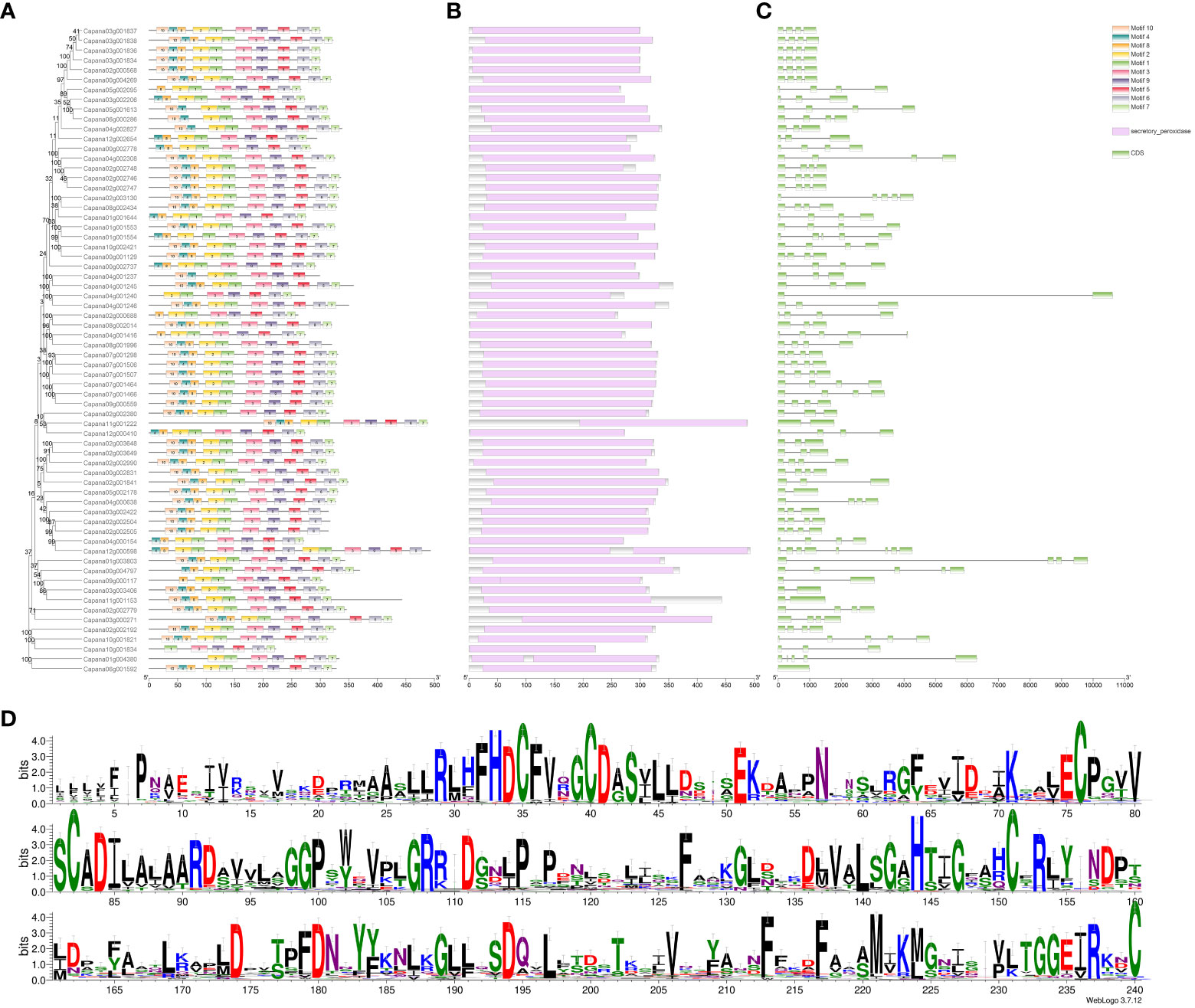
Figure 5 Characteristic analysis of the pepper CaPRX genes family (A Motif features; B Conserved domains; C Gene structure; D Sequence alignment results).
Analysis of CaPRXs chromosome location, collinearity, and expression characteristics
The chromosomal distribution of CaPRX genes in peppers was notably concentrated on chromosome Chr01, containing a total of 15 CaPRX genes, while the least representation was observed on chromosomes Chr06, Chr09, and Chr11, each containing only 2 CaPRX genes (Figure 6). This distribution pattern suggests that CaPRX genes are not evenly distributed across different chromosomes, with each chromosome containing at least one CaPRX target gene. Remarkably, six genes were identified on the Chr00 (Genes that are not attached to chromosomes) sequence chromosome. In the genomes of Zunla-1 and wild Chiltepin, collinear blocks were observed on each chromosome, with the exception of the virtual chromosomes Chr00 and Chr08, where no collinear blocks were detected. The analysis of collinear blocks revealed that 58 collinear genes in Zunla-1 and Chiltepin share a common ancestor. Furthermore, a more in-depth examination of the CaPRX genes in different pepper types revealed that they were predominantly upregulated in the 1933A line. Conversely, in the 1933B line, Capana02g002746 (CaPRX13) was upregulated, and Capana04g000638 (CaPRX30) was upregulated in the 1933A line. Notably, these two genes displayed a reverse expression pattern, adding an intriguing dimension to the analysis.
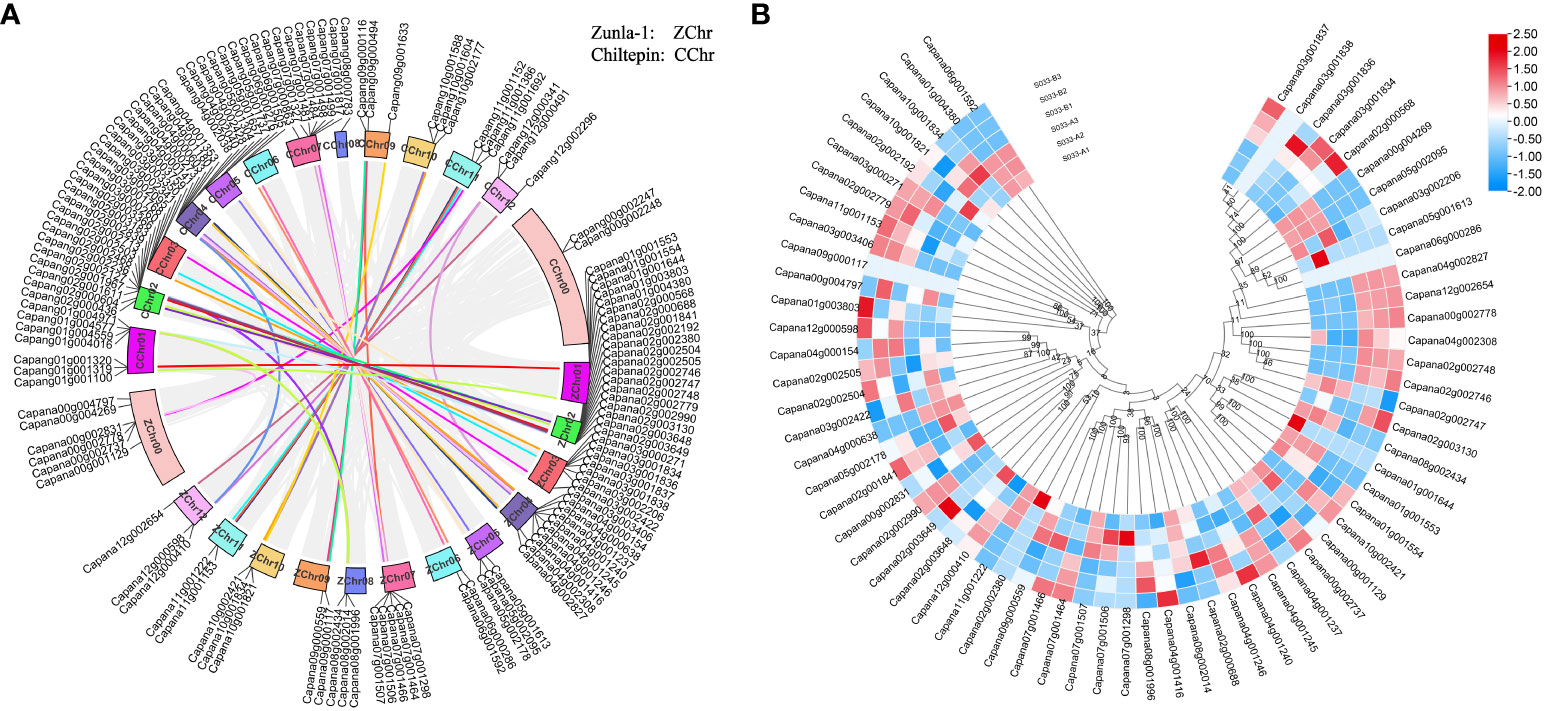
Figure 6 Analysis of chromosomal localization, collinearity, and expression characteristics of CaPRX genes in pepper (A Chromosomal localization and collinearity blocks of the CaPRX genes; B Expression patterns of CaPRXs in FL and NMSL).
Salt stress treatment and qRT-PCR analysis
Studies have observed that overexpression of CaPRX genes can promote salt tolerance in transgenic plants. To study the regulatory pathway of CaPRX genes in pepper, 66 CaPRX genes were analyzed under salt stress treatment. An analysis of tissue-specific expression patterns of CaPRX genes under salt stress treatment (Figure 7A) revealed that all 66 CaPRX genes exhibited varying degrees of upregulation in the roots. Conversely, their expression levels in the leaves were predominantly downregulated compared with those in the roots. These findings underscore the significant role of CaPRXs in the stress response of pepper roots under salt stress conditions. To investigate the expression characteristics of CaPRXs under salt stress, 178 network nodes and 378 network pairs were extracted from the coexpression network module (Figure 7B). Notably, among the genes with a degree of >10, five CaPRX genes were identified, namely, CaPRX14 (degree = 41), CaPRX15 (degree = 41), CaPRX46 (degree = 45), CaPRX47 (degree = 26), and CaPRX63 (degree = 64). CaPRX15 and CaPRX63 were upregulated in the NMSL, while CaPRX47 was upregulated in the FL. Within the coexpression network, the top five transcription factors were bHLH (18), ERF (14), LBD (13), MYB (12), and WRKY (12). The transcription factor with a degree of >20 was G2-like (degree = 22, Capana00g004643). In the transcriptome analysis, 13 transcription factors were upregulated, while 7 were downregulated in the sterile line. Six genes were randomly selected from the transcriptome, including one gene (Capana04g000638, CaPRX30) from the CaPRX gene family. The qRT-PCR results of the randomly selected six genes revealed a strong correlation with the transcriptome fragments per kilobase of transcript per million mapped reads results (PCCs > 0.9; Figure 7C). Among these genes, CaPRX30 and Capana07g000080 exhibited high expression levels in the NMSL, while the other four genes were upregulated in the FL.
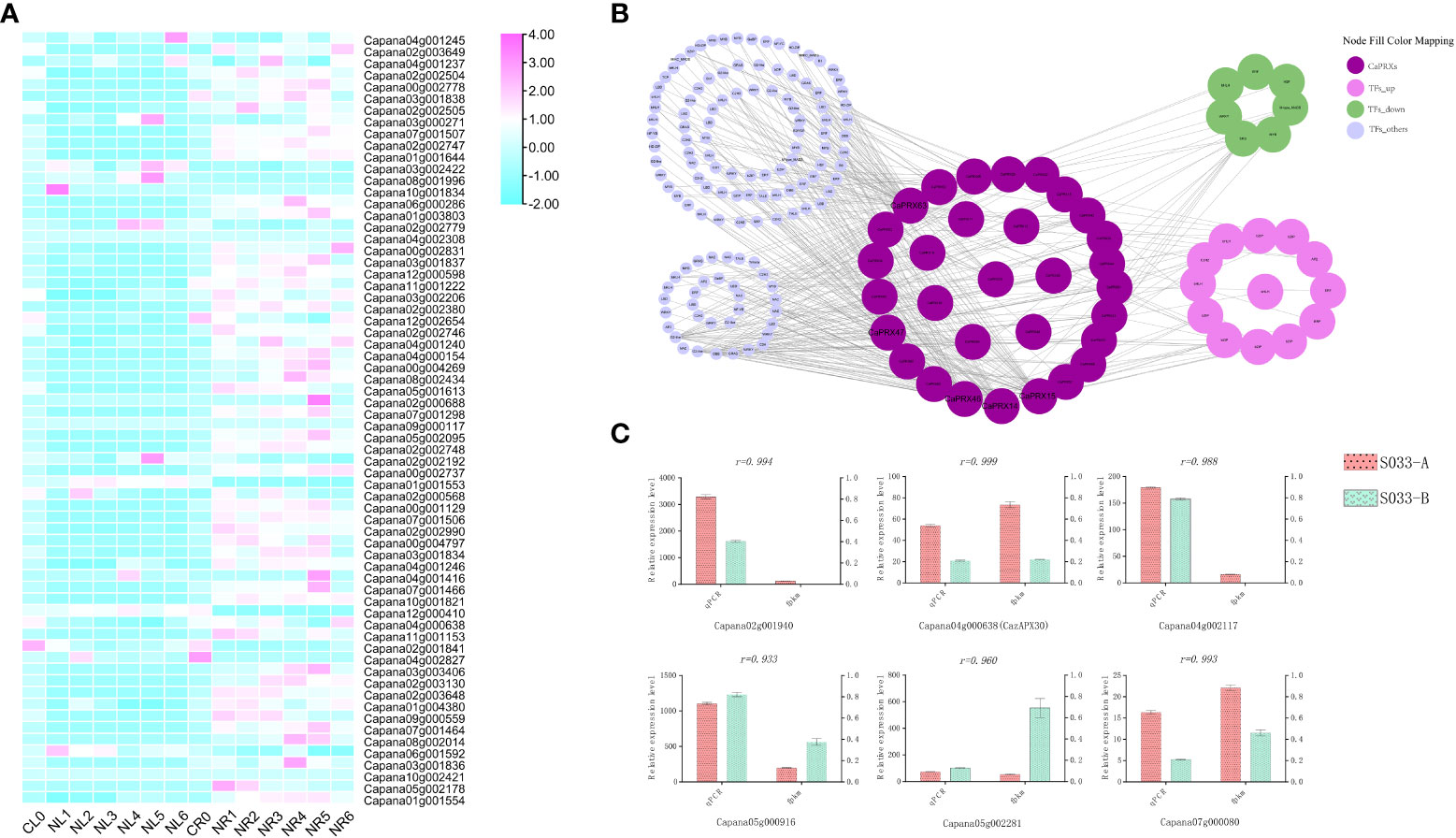
Figure 7 Expression characteristics, network module analysis, and qRT-PCR analysis of CaPRXs under salt stress (A Expression characteristics analysis of leaves and roots under salt stress; B Network module analysis under salt stress; C qRT-PCR analysis under salt stress).
Discussion
Owing to the continuous innovation and advancements in breeding methods, numerous novel germplasms and resources have been developed for various economic crops, effectively ensuring agricultural and food production. The MS mutant 1933B of the pepper plant, which lacks pollen, plays a crucial role in the hybrid breeding of pepper. Herein, we conducted a combined analysis of the pepper 1933A FL and 1933B NMSL at the molecular biology and protein levels. The transcriptome analysis identified 3,304 differentially expressed genes, and GO was primarily enriched in processes related to polysaccharide metabolism, carbohydrates, pH regulation, pollen development, transmembrane transport protein activity, extracellular zone, and pollen tubes. In the KO analysis, pathways such as the phospholipase D signaling pathway, MAPK signaling pathway in plants, bile secretion, synaptic vesicle cycle, and phenylpropanoid biosynthesis pathways were enriched. These findings in GO enrichment, particularly those related to pollen development and pollen tubes, underscore the significant influence of pollen development on male sterility in pepper. MS eggplants reportedly exhibit differentially expressed genes involved in redox, carbohydrate, and amino acid metabolism (Yang et al., 2018), whereas MS watermelons and broccoli have exhibited the involvement of phenylpropanoid biosynthesis and plant hormone signaling pathways (Shu et al., 2018; Wang et al., 2020). These differentially expressed genes may play direct or indirect roles in plant pollen development processes, including meiosis and cell wall formation, thereby affecting male sterility in pepper.
In the previous proteomic studies of pepper, 1645 differentially abundant proteins were found via proteomic analysis, 45 of which were related to the anther and pollen development of MS lines of pepper, indicating that many proteins were involved in the flowering process of pepper (Pei et al., 2022). Mutations in different strains can result from various factors, including natural environmental influences or external environmental conditions. For instance, MS lines have been observed in several crops, such as rice, and its presence has been associated with temperature sensitivity. Additionally, there have been instances of light-sensitive male sterility in rice plants (Fan et al., 2016). Wu et al. (2022) found a natural allele OsMS1 that can respond to temperature changes and imparts heat-sensitive male sterility. In the model plant Arabidopsis, Lox3 and Lox4 are pivotal for male fertility and play a significant role in pollen development (Caldelari et al., 2011). During our study, peppers were cultivated in Zunyi, Guizhou Province, an area with limited light and unstable temperatures from March to April. It is hypothesized that the low temperature and insufficient light during the planting process affected the growth and development of the peppers.
Herein, we discovered natural mutations leading to male sterility and identified candidate key genes, CaPRXs, involved in regulating male sterility in pepper through multiomics analysis. Our analysis delved into the structural, tissue-specific, and expression characteristics of CaPRXs genes, revealing a total of 66 Class III peroxidases in pepper with typical PRX conserved domains. Notably, we observed tissue-specific expression in roots and leaves under salt stress, with CaPRXs primarily upregulated in the roots. Coexpression network analysis revealed that CaPRXs are coexpressed with multiple transcription factors. This finding indicates that CaPRX, identified via proteomics and transcriptomics in multiomics analysis, indirectly influence the process of male sterility in pepper. Furthermore, Dong et al. identified CaMYB80 as a key regulator gene specifically expressed in anthers, with its downregulation causing male sterility in pepper (Dong et al., 2022). In wheat genetic breeding studies, CaPRXs were found to be upregulated during early pollen development (Hiraga et al., 2001; Liu et al., 2018). It is speculated that CaPRX genes are involved in the development of pepper pollen, which leads to its hypoplasia and the formation of MS line of pepper. Meanwhile, in this study, it was found that CaPRX genes co-express with several transcription factors via a co-expression network. Transcription factors such as bHLH, ERF, LBD, MYB, WRKY, and G2-like may play a role in male sterility of pepper via the indirect regulation of CaPRXs.
Numerous studies have reported that Class III peroxidases are integral to plant growth, development, and physiological processes, especially for pollen production and stress responses. For example, in a study on male sterility in tomatoes, higher overall peroxidase activity was observed in MS stamens compared with that in male fertile stamens (Markova et al., 1992). Chen et al. cloned the Ghpod gene, which contained a conserved domain of Class III peroxidase, and found its expression in the reproductive organs of cotton, particularly pollen, suggesting the involvement of peroxidases in the male reproductive process of angiosperms (Chen et al., 2009). Similarly, hazelnut (Corylus avellana) specifically expresses CavPrx in pistils, indicating the crucial role of CaPRXs in causing male sterility in pepper. Moreover, PRXs serve vital functions in various plant physiological and developmental processes, acting as essential defenses against biotic and abiotic stresses (Kidwai et al., 2020). For example, PRXs are not only upregulated in pollen and pistils but also participate in nonbiological stress responses. Research has found that an example includes PRXs overexpression in wheat, which increases the salt tolerance of transgenic wheat (Su et al., 2020). Our study on pepper under salt stress revealed that CaPRXs are involved in abiotic stress responses and were upregulated in the roots, reaffirming the diverse roles of PRXs in plants.
The continuous advancement of biotechnology enables its increasing practical application. For instance, Jung et al. employed gene editing to create a MS phenotype by knocking out the SlMS10 gene encoding a bHLH transcription factor (Jung et al., 2020). Similarly, Du et al. utilized CRISPR/Cas9 gene editing technology to establish a specific MS tomato line and developed a fertility restoration system using this MS line (Du et al., 2020). It is anticipated that as research on male sterility in peppers progresses, biotechnology may offer innovative germplasm solutions.
Conclusions
This study conducted proteomics, transcriptomics, and association analyses to identify 29 key genes, highlighting a significant number of CaPRX genes in differential proteins and genes. The association analysis unveiled three intersections between the 29 genes and 66 CaPRX genes, underscoring the significant role of CaPRXs in male sterility in pepper and their involvement in the process. To explore the regulatory roles of CaPRXs, the study comprehensively identified pepper CaPRXs at the whole-genome level, revealing a total of 66 genes containing typical Class III peroxidase domains. The 66 CaPRX genes were categorized into 13 subfamilies, which were stable proteins. Salt stress showed that CaPRXs were mainly upregulated in roots and coexpressed with multiple transcription factors. This study speculated that the CaPRXs gene is involved in the development process of pepper NMSL, which is regulated by the CaPRXs gene, resulting in pollen agenesis of pepper NMSL. By exploring NMSL in pepper at the transcriptomic and proteomic levels, this research furnishes a theoretical foundation for pepper genetic breeding and offers practical insights for innovative breeding solutions.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
CQ: Funding acquisition, Writing – original draft, Writing – review & editing. SY: Visualization, Writing – original draft, Writing – review & editing, Software. XL: Writing – original draft, Software, Visualization. JJ: Data curation, Writing – review & editing. YG: Data curation, Writing – review & editing. LZ: Data curation, Writing – review & editing. JL: Data curation, Software, Writing – review & editing. ST: Data curation, Writing – review & editing. YL: Data curation, Writing – review & editing. TL: Data curation, Writing – review & editing. XC: Data curation, Writing – review & editing. YW: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Guizhou Province Science and technology plan program of China (Qian Ke He [2020]1Y088, [2019]2258, [2020]2102, [2021]yiban262, [2021]yiban267, [2022]yiban117 and Qian Ke He Achievement [2020]1Z005), the engineering center construction project of education department of Guizhou Province (Qian Jiao He KY [2021]006), and the Zunyi Excellent Young Scientific and Technological Innovative Talents Training Project (Zunyouqingke No. 201806).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2024.1334430/full#supplementary-material
References
Almagro, L., Gómez Ros, L. V., Belchi-Navarro, S., Bru, R., Ros Barceló, A., Pedreño, M. A. (2008). Class III peroxidases in plant defence reactions. J. Exp. Bot. 60 (2), 377–390. doi: 10.1093/jxb/ern277
Bailey, T. L., Johnson, J., Grant, C. E., Noble, W. S. (2015). The MEME suite. Nucleic. Acids Res. 43 (W1), W39–W49. doi: 10.1093/nar/gkv416
Bhatt, I., Tripathi, B. N. (2011). Plant peroxiredoxins: Catalytic mechanisms, functional significance and future perspectives. Biotechnol. Adv. 29 (6), 850–859. doi: 10.1016/j.biotechadv.2011.07.002
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Budar, F., Pelletier, G. (2001). Male sterility in plants: occurrence, determinism, significance and use. Comptes Rendus l’Académie Des. Sciences-Series III-Sciences la Vie 324, 543–550. doi: 10.1016/s0764-4469(01)01324-5
Caldelari, D., Wang, G., Farmer, E. E., Dong, X. (2011). Arabidopsis lox3 lox4 double mutants are male sterile and defective in global proliferative arrest. Plant Mol. Biol. 75 (1), 25–33. doi: 10.1007/s11103-010-9701-9
Capella-Gutiérrez, S., Silla-Martínez, J. M., Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25 (15), 1972–1973. doi: 10.1093/bioinformatics/btp348
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13 (8), 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, D., Ding, Y., Guo, W., Zhang, T. (2009). Molecular cloning and characterization of a flower-specific class III peroxidase gene in G. Hirsutum. Mol. Biol. Rep. 36 (3), 461–469. doi: 10.1007/s11033-007-9202-3
Chen, J., Jiao, Y., Laza, H., Payton, P., Ware, D., Xin, Z. (2019). Identification of the first nuclear male sterility gene (Male-sterile 9) in sorghum. Plant Genome 12 (3), 1–12. doi: 10.3835/plantgenome2019.03.0020
Chen, Y., Feng, J., Qu, Y., Zhang, J., Zhang, L., Liang, D., et al. (2022). Genome-wide identification and functional analysis of class III peroxidases in Gossypium hirsutum. PeerJ 10, e13635. doi: 10.7717/peerj.13635
Cheng, L., Ma, L., Meng, L., Shang, H., Cao, P., Jin, J. (2022). Genome-wide identification and analysis of the class III peroxidase gene family in tobacco (Nicotiana tabacum). Front. Genet. 13. doi: 10.3389/fgene.2022.916867
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21 (18), 3674–3676. doi: 10.1093/bioinformatics/bti610
Cosio, C., Vuillemin, L., De, Meyer, M., Kevers, C., Penel, C., Dunand, C. (2009). An anionic class III peroxidase from zucchini may regulate hypocotyl elongation through its auxin oxidase activity. Planta 229 (4), 823–836. doi: 10.1007/s00425-008-0876-0
Crooks, G. E., Hon, G., Chandonia, J. M., Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14 (6), 1188–1190. doi: 10.1101/gr.849004
de Oliveira, F. K., Santos, L. O., Buffon, J. G. (2021). Mechanism of action, sources, and application of peroxidases. Food Res. Int. 143, 110266. doi: 10.1016/j.foodres.2021.110266
Dong, J., Hu, F., Guan, W., Yuan, F., Lai, Z., Zhong, J., et al. (2022). A 163-bp insertion in the Capana10g000198 encoding a MYB transcription factor causes male sterility in pepper (Capsicum annuum L.). Plant J. 113 (3), 521–535. doi: 10.1111/tpj.16064
Du, M., Zhou, K., Liu, Y., Deng, L., Zhang, X., Lin, L., et al. (2020). A biotechnology-based male-sterility system for hybrid seed production in tomato. Plant J. 102 (5), 1090–1100. doi: 10.1111/tpj.14678
Eckardt, N. A. (2006). Cytoplasmic male sterility and fertility restoration. Plant Cell 18 (3), 515–517. doi: 10.1105/tpc.106.041830
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2019). The Pfam protein families database in 2019. Nucleic. Acids Res. 47 (D1), D427–D432. doi: 10.1093/nar/gky995
Fan, Y., Yang, J., Mathioni, S. M., Yu, J., Shen, J., Yang, X., et al. (2016). PMS1T, producing phased small-interfering RNAs, regulates photoperiod-sensitive male sterility in rice. P. Natl. Acad. Sci. U.S.A. 113 (52), 15144–15149. doi: 10.1073/pnas.1619159114
Gu, Z., Hübschmann, D. (2022). Make interactive complex heatmaps in R. Bioinformatics 38 (5), 1460–1462. doi: 10.1093/bioinformatics/btab806
Hiraga, S., Sasaki, K., Ito, H., Ohashi, Y., Matsui, H. (2001). A large family of class III plant peroxidases. Plant Cell Physiol. 42 (5), 462–468. doi: 10.1093/pcp/pce061
Jacobowitz, J. R., Doyle, W. C., Weng, J. K. (2019). PRX9 and PRX40 are extensin peroxidases essential for maintaining tapetum and microspore cell wall integrity during Arabidopsis anther development. Plant Cell 31 (4), 848–61.
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30 (9), 1236–1240. doi: 10.1093/bioinformatics/btu031
Jung, Y. J., Kim, D. H., Lee, H. J., Nam, K. H., Bae, S., Nou, I. S., et al. (2020). Knockout of SlMS10 Gene (Solyc02g079810) Encoding bHLH transcription factor using CRISPR/Cas9 System confers male sterility phenotype in tomato. Plants 9 (9), 1189. doi: 10.3390/plants9091189
Kidwai, M., Ahmad, I. Z., Chakrabarty, D. (2020). Class III peroxidase: an indispensable enzyme for biotic/abiotic stress tolerance and a potent candidate for crop improvement. Plant Cell Rep. 39 (11), 1381–1393. doi: 10.1007/s00299-020-02588-y
Kidwai, M., Dhar, Y. V., Gautam, N., Tiwari, M., Ahmad, I. Z., Asif, M. H., et al. (2019). Oryza sativa class III peroxidase (OsPRX38) overexpression in Arabidopsis thaliana reduces arsenic accumulation due to apoplastic lignification. J. Hazard Mater 362, 383–393. doi: 10.1016/j.jhazmat.2018.09.029
Kim, B. H., Kim, S. Y., Nam, K. H. (2012). Genes encoding plant-specific class III peroxidases are responsible for increased cold tolerance of the brassinosteroid-insensitive 1 mutant. Mol. Cells 34 (6), 539–548. doi: 10.1007/s10059-012-0230-z
Kim, D., Paggi, J. M., Park, C., Bennett, C., Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37 (8), 907–915. doi: 10.1038/s41587-019-0201-4
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35 (6), 1547–1549. doi: 10.1093/molbev/msy096
Letunic, I., Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic. Acids Res. 49 (W1), W293–W296. doi: 10.1093/nar/gkab301
Liao, Y., Smyth, G. K., Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30 (7), 923–930. doi: 10.1093/bioinformatics/btt656
Liu, K., Mu, Y., Chen, X., Ding, Z., Song, M., Xing, D., et al. (2022). Towards developing an epidemic monitoring and warning system for diseases and pests of hot peppers in Guizhou, China. Agronomy 12 (5), 1034. doi: 10.3390/agronomy12051034
Liu, Z., Shi, X., Li, S., Hu, G., Zhang, L., Song, X. (2018). Tapetal-delayed programmed cell death (PCD) and oxidative stress-induced male sterility of aegilops uniaristata cytoplasm in wheat. Int. J. Mol. Sci. 19 (6), 1708. doi: 10.3390/ijms19061708
Liu, F., Yu, H., Deng, Y., Zheng, J., Liu, M., Ou, L., et al. (2017). PepperHub, an informatics hub for the chili pepper research community. Mol. Plant 10 (8), 1129–1132. doi: 10.1016/j.molp.2017.03.005
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2– ΔΔCT method. methods 25 (4), 402–408. doi: 10.1006/meth.2001.1262
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 1–21. doi: 10.1186/s13059-014-0550-8
Lu, Y., Lauter, A. N., Makkena, S., Scott, M. P., Evans, M. (2020). Insights into the molecular control of cross-incompatibility in Zea mays. Plant Reprod. 33 (3), 117–128. doi: 10.1007/s00497-020-00394-w
Lv, J., Liu, Z., Liu, Y., Ou, L., Deng, M., Wang, J., et al. (2020). Comparative transcriptome analysis between cytoplasmic male-sterile line and its maintainer during the floral bud development of pepper. Hortic. Plant J. 6 (2), 89–98. doi: 10.1016/j.hpj.2020.01.004
Markova, M., Ancheva, M., Atanasova, B. (1992). Activity and isoenzyme composition of peroxidase and esterase in fertile and male-sterile lines of tomatoes (Lycopersicon esculentum Mill.). Biol. Plantarum. 34 (5), 387–394. doi: 10.1007/BF02923585
Morales, K. Y., Bridgeland, A. H., Hake, K. D., Udall, J. A., Thomson, M. J., Yu, J. Z. (2022). Homology-based identification of candidate genes for male sterility editing in upland cotton (Gossypium hirsutum L.). Front. Plant Sci. 13, 1006264. doi: 10.3389/fpls.2022.1006264
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35 (suppl_2), W182–W185. doi: 10.1093/nar/gkm321
Otasek, D., Morris, J. H., Bouças, J., Pico, A. R., Demchak, B. (2019). Cytoscape automation: empowering workflow-based network analysis. Genome Biol. 20 (1), 1–15. doi: 10.1186/s13059-019-1758-4
Pandey, V., Awasthi, M., Singh, S., Tiwari, S., Dwivedi, U. (2017). A comprehensive review on function and application of plant peroxidases. Biochem. Anal. Biochem. 6 (1), 308. doi: 10.4172/2161-1009.1000308
Passardi, F., Longet, D., Penel, C., Dunand, C. (2004). The class III peroxidase multigenic family in rice and its evolution in land plants. Phytochemistry 65 (13), 1879–1893. doi: 10.1016/j.phytochem.2004.06.023
Passardi, F., Zamocky, M., Favet, J., Jakopitsch, C., Penel, C., Obinger, C., et al. (2007). Phylogenetic distribution of catalase-peroxidases: Are there patches of order in chaos? Gene 397 (1), 101–113. doi: 10.1016/j.gene.2007.04.016
Pei, H., Xie, H., Wang, X., Yan, X., Wang, B., Feng, H., et al. (2022). Proteomic analysis of differential anther development from sterile/fertile lines in Capsicum annuum L. PeerJ 10, e13168. doi: 10.7717/peerj.13168
Qin, C., Yu, C., Shen, Y., Fang, X., Chen, L., Min, J., et al. (2014). Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. P. Natl. Acad. Sci. U.S.A. 111 (14), 5135–5140. doi: 10.1073/pnas.1400975111
Raggi, S., Ferrarini, A., Delledonne, M., Dunand, C., Ranocha, P., Lorenzo, G., et al. (2015). The arabidopsis class III peroxidase atPRX71 negatively regulates growth under physiological conditions and in response to cell wall damage. Plant Physiol. 169 (4), 2513–2525. doi: 10.1104/pp.15.01464
Rajput, R. S., Kandalkar, V. (2018). Combining ability and heterosis for grain yield and its attributing traits in bread wheat (Triticum aestivum L.). J. Pharmacogn. Phytochem. 7 (2), 113–119.
Sadimantara, G., Nuraida, W., Suliartini, N. (2018). Evaluation of some new plant type of upland rice (Oryza sativa L.) lines derived from cross breeding for the growth and yield characteristics. IOP Conference Series Earth Env. Sci. 157, 012048
Shu, J., Zhang, L., Liu, Y., Li, Z., Fang, Z., Yang, L., et al. (2018). Normal and abortive buds transcriptomic profiling of Broccoli ogu cytoplasmic male sterile line and its maintainer. Int. J. Mol. Sci. 19 (9), 2501. doi: 10.3390/ijms19092501
Su, P., Yan, J., Li, W., Wang, L., Zhao, J., Ma, X., et al. (2020). A member of wheat class III peroxidase gene family, TaPRX-2A, enhanced the tolerance of salt stress. BMC Plant Biol. 20 (1), 392. doi: 10.1186/s12870-020-02602-1
Tang, X., Hao, Y. J., Lu, J. X., Lu, G., Zhang, T. (2019). Transcriptomic analysis reveals the mechanism of thermosensitive genic male sterility (TGMS) of Brassica napus under the high temperature inducement. BMC Genomics 20 (1), 644. doi: 10.1186/s12864-019-6008-3
Tognolli, M., Penel, C., Greppin, H., Simon, P. (2002). Analysis and expression of the class III peroxidase large gene family in Arabidopsis thaliana. Gene 288 (1), 129–138. doi: 10.1016/S0378-1119(02)00465-1
Wang, Y., Li, J., Paterson, A. H. (2013). MCScanX-transposed: detecting transposed gene duplications based on multiple colinearity scans. Bioinformatics 29 (11), 1458–1460. doi: 10.1093/bioinformatics/btt150
Wang, P., Lu, Q., Ai, Y., Wang, Y., Li, T., Wu, L., et al. (2019). Candidate Gene Selection for Cytoplasmic Male Sterility in Pepper (Capsicum annuum L.) through Whole Mitochondrial Genome Sequencing. Int. J. Mol. Sci. 20 (3), 578. doi: 10.3390/ijms20030578
Wang, Y., Yang, X., Yadav, V., Mo, Y., Yang, Y., Zhang, R., et al. (2020). Analysis of differentially expressed genes and pathways associated with male sterility lines in watermelon via bulked segregant RNA-seq. 3 Biotech. 10 (5), 1–15. doi: 10.1007/s13205-020-02208-2
Wu, Z., Cheng, J., Qin, C., Hu, Z., Yin, C., Hu, K. (2013). Differential proteomic analysis of anthers between cytoplasmic male sterile and maintainer lines in Capsicum annuum L. Int. J. Mol. Sci. 14 (11), 22982–22996. doi: 10.3390/ijms141122982
Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2 (3), 100141. doi: 10.1016/j.xinn.2021.100141
Wu, L., Jing, X., Zhang, B., Chen, S., Xu, R., Duan, P., et al. (2022). A natural allele of OsMS1 responds to temperature changes and confers thermosensitive genic male sterility. Nat. Commun. 13 (1), 1–15. doi: 10.1038/s41467-022-29648-z
Yan, J., Su, P., Li, W., Xiao, G., Zhao, Y., Ma, X., et al. (2019). Genome-wide and evolutionary analysis of the class III peroxidase gene family in wheat and Aegilops tauschii reveals that some members are involved in stress responses. BMC Genomics 20 (1), 666. doi: 10.1186/s12864-019-6006-5
Yang, Y., Bao, S., Zhou, X., Liu, J., Zhuang, Y. (2018). The key genes and pathways related to male sterility of eggplant revealed by comparative transcriptome analysis. BMC Plant Biol. 18 (1), 209. doi: 10.1186/s12870-018-1430-2
Yang, X., Yuan, J., Luo, W., Qin, M., Yang, J., Wu, W., et al. (2020). Genome-wide identification and expression analysis of the class III peroxidase gene family in potato (Solanum tuberosum L.). Front. Genet. 11, 593577. doi: 10.3389/fgene.2020.593577
Yang, T., Zhang, P., Pan, J., Amanullah, S., Luan, F., Han, W., et al. (2022). Genome-wide analysis of the Peroxidase gene family and verification of lignin synthesis-related genes in watermelon. Int. J. Mol. Sci. 23 (2), 642. doi: 10.3390/ijms23020642
Keywords: pepper, proteomics, transcriptomic, male sterility, CaPRX genes, crop breeding
Citation: Yang S, Luo X, Jin J, Guo Y, Zhang L, Li J, Tong S, Luo Y, Li T, Chen X, Wu Y and Qin C (2024) Key candidate genes for male sterility in peppers unveiled via transcriptomic and proteomic analyses. Front. Plant Sci. 15:1334430. doi: 10.3389/fpls.2024.1334430
Received: 07 November 2023; Accepted: 12 January 2024;
Published: 06 February 2024.
Edited by:
Changlin Liu, Chinese Academy of Agricultural Sciences (CAAS), ChinaReviewed by:
Zhiwei Wang, Hainan University, ChinaZhi Gang Meng, Biotechnology Research institute of CAAS, China
Yunsong Lai, Sichuan Agricultural University, China
Copyright © 2024 Yang, Luo, Jin, Guo, Zhang, Li, Tong, Luo, Li, Chen, Wu and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongjun Wu, d3lqYmlvQDE2My5jb20=; Cheng Qin, cWluY2hlbmcxMDAxQDE2My5jb20=
†These authors have contributed equally to this work